

Abstract
The synthesis of primary amines from renewable biomass and its derivatives through reductive amination has garnered significant attention. How to construct efficient non-noble-metal catalysts that enable low-temperature catalysis still remains challenging. Herein, we report a Cu-doped Co@CoOx heterostructure catalyst that features structural Co–CoCuOx bifunctional sites, which enable room temperature reductive amination of various aldehydes with 1.57–45 times higher efficiency than Co@CoOx, outperforming many reported non-noble and even noble metal catalysts. Experiments and DFT calculations indicate that Cu doping leads to a phase transition of Co from hcp to fcc, while electrons are transferred from Cu to Co, forming a dual active site with electron-rich Co closely interacting with CoCuOx. These electron-rich Co sites demonstrate excellent activity in the activation and dissociation of hydrogen, while the CuOx component facilitates hydrogen spillover at the CoCuOx interface, thus resulting in a highly efficient cooperative effect for the furfural (FAL) reductive amination. This work provides general guidance for the rational design of high-performance reductive amination catalysts for biomass upgrading.
Cu doping induces a phase transition of Co from hcp to fcc and results in the formation of electron-rich Co, thereby facilitating hydrogen dissociation and the subsequent reductive amination of furfural (FAL) at room temperature.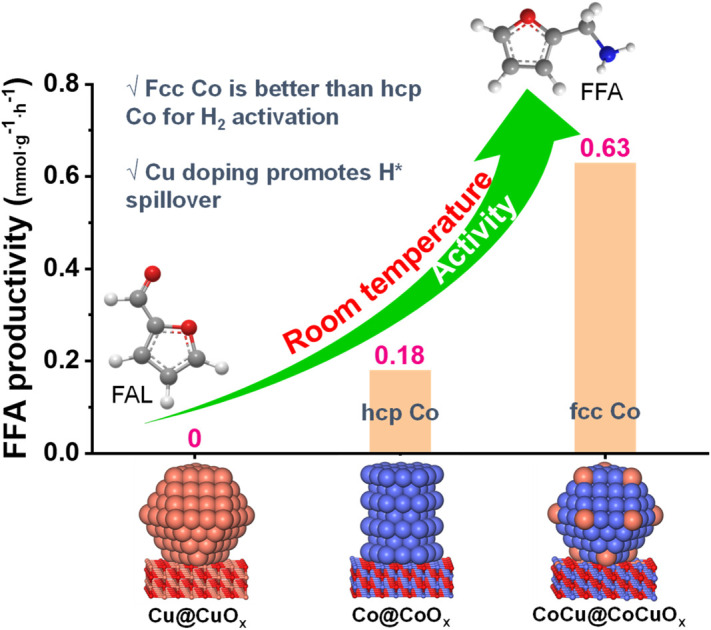
1. Introduction
The escalating energy and environmental crises caused by huge global fossil consumption have drawn significant attention to the search for renewable resources for chemical and fuel production.1–3 Biomass has emerged as a promising carbon source in the chemical industry due to its abundant reserves, environmental friendliness, and widespread distribution.4,5 Among them, FAL, dehydrated from hemicellulose, can be further catalytically upgraded into various chemicals via hydrogenation, oxidative amination, etc.6–9 In particular, reductive amination is one of the strategies for synthesizing high-value N-containing chemicals, and its product, furfuryl amine (FFA), is a promising intermediate for the polymer, agrochemical, dye, pigment, pesticide, and pharmaceutical industries.10,11 Nevertheless, the synthesis of primary amines with high selectivity poses a significant challenge due to the occurrence of numerous side reactions in the process.
Currently, significant efforts have been made to develop efficient and selective catalysts for the reductive amination of biomass-derived aldehydes and ketones. Researchers have developed homogeneous or heterogeneous catalysts based on noble metals such as Ru,12–17 Pd,18 Pt,19 and Rh.20,21 Although these catalysts have demonstrated good catalytic activity for reductive amination, their widespread application is limited by high cost and low abundance. In the case of non-noble metals, catalysts such as Ni,22–24 Co,25–30 and Cu31,32 have been widely studied. However, they usually require harsh reaction conditions (e.g., elevated temperatures or pressures), which can lead to catalyst challenges such as leaching, instability, and deactivation.33 Therefore, there is an urgent need to develop an efficient non-noble metal catalyst system that facilitates mild reductive amination of carbonyl compounds with high efficacy.
The catalytic activity and selectivity of reductive amination can be tuned by creating efficient bifunctional active sites, with one site strategically positioned near the other.34–36 These metal bifunctional sites require appropriate spatial intimacy to meet the cooperative active site requirements for the activation and transformation of reactant molecules. Metal bifunctional sites can be constructed in various structures and forms, including synthesizing dual single atoms,37 dispersing bimetallic alloys on supports,38 and grafting metal particles onto metal oxides.16,39 Once the highly dispersed metal centers form intimate interfaces with the oxides, the synergistic effects between them enable these catalysts to exhibit excellent performance. For example, the Co–CoOx bifunctional site, composed of highly dispersed Co (center I) and CoOx (center II), demonstrates a reaction mechanism where the CoOx center activates NH3 to generate NH2δ− species, while H2 dissociates on the Co surface and flows toward CoOx.40 Despite significant advances in catalyst synthesis, achieving catalytically-efficient structures via precisely controlling the spatial intimacy of multiple active sites at the sub-nanoscale remains challenging.
Herein, we constructed a CoCu–CoCuOx heterojunction by modulating the reduction temperature of the crystal phase. Simultaneously, Cu doping induces the transformation of the Co crystal phase from hcp to fcc, resulting in a metal bifunctional site with electron-rich Co and oxygen-vacancy-rich CoCuOx. This heterojunction catalyst facilitated the reductive amination of FAL at room temperature with N2H4·H2O as the nitrogen source, displaying significantly higher activity compared to its mono Cu and Co counterparts. Compared with NH3, the utilization of N2H4·H2O as a nitrogen source completely eliminates the hydrogenation of aldehydes to alcohols, and the intermediate with moderate reactivity notably enhances amine selectivity and effectively prevents the occurrence of the imine-induced trimerization side reaction.12,41,42 The morphology and structure of the catalyst were thoroughly characterized, and a structure–performance relationship was established. Furthermore, the catalyst's recyclability and substrate tolerance were investigated. Finally, the reaction process and mechanism were explored through in situ FTIR, DFT, and kinetic studies. This study presents a novel approach for the efficient and selective synthesis of primary amines from carbonyl compounds under extremely mild conditions without the aid of noble metal catalysts.
2. Results and discussion
2.1. Catalyst characterization
We synthesized the bimetallic CoCu@CoCuOx catalyst (Co/Cu feeding ratio of 8/2) via a coprecipitation process (Fig. 1a). Typically, salts and base were dissolved separately in deionized water, and the two solutions were mixed to produce CoCu(CO3)x(OH)y precipitation. After calcination, the solid is transformed into oxidized CoCuOx, which can be partially reduced at 250 °C to form the CoCu@CoCuOx catalyst based on the H2-TPR profile (Fig. S1†), with a specific surface area of 20 m2 g−1 (Fig. S2†), 62.9% Co and 16.1% Cu loadings determined by ICP-OES (Table S1†), well consistent with the theoretical values.
Fig. 1. (a) Schematic illustration for the preparation procedure of CoCu@CoCuOx. (b) XRD patterns, (c) Raman spectra, (d) Co 2p XPS, and (e) Cu 2p XPS spectra of CoCu@CoCuOx, Co@CoOx and Cu catalysts. TEM and HRTEM images of (f and h) CoCu@CoCuOx, (j) Co@CoOx and (k) Cu@CuOx catalysts. (g) SEAD pattern and (i) EDS elemental mapping of CoCu@CoCuOx.

The crystalline structure of CoCu@CoCuOx was characterized using XRD (Fig. 1b). Co@CoOx exhibits four distinct diffraction peaks at 41.6°, 44.7°, 47.5°, and 75.9°, corresponding to (100), (002), (101), and (110) crystal planes of the Co hcp phase (PDF#05-0727). Additionally, a diffraction peak attributed to the (111) crystal plane of CoO (PDF#48-1719) appears at 36.5°, indicating the coexistence of Co and CoO in Co@CoOx. As for CoCu@CoCuOx, the diffraction peaks corresponding to the Co hcp phase disappear, and a new peak appears at 44.2°, corresponding to the (111) crystal plane of the Co fcc phase (PDF#15-0806). However, no diffraction peaks related to metallic Cu are observed, which may be due to more uniformly distributed Cu in the bimetallic catalyst. These results reveal that the Co phase transition from the hcp to fcc phase is promoted by Cu doping with H2 reduction at a temperature of 250 °C.
Surface oxides were characterized by Raman spectroscopy, as shown in Fig. 1c. Co@CoOx exhibits five typical Raman peaks at 180, 457, 503, 585, and 660 cm−1, corresponding to the F12g, Eg, F22g, F32g, and A1g modes of CoO.43 Cu@CuOx mainly shows three characteristic Raman peaks at 277, 323, and 602 cm−1, corresponding to the Ag, B1g, and B2g vibration modes of CuO. CoCu@CoCuOx primarily displays two peaks, with the peak at 531 cm−1 corresponding to the vibration of Cu2O, and the peak at 676 cm−1 corresponding to the A1g vibration mode of CoO.44 It is worth noting that the A1g mode Raman peak of CoO in CoCu@CoCuOx shows a pronounced redshift compared to Co@CoOx, which is attributed to lattice distortion induced by abundant oxygen vacancies (Fig. S3, S4 and Table S2†).45
The composition and chemical states of the catalysts were characterized using XPS spectra (Fig. 1d, e and S5†). Co 2p XPS spectra (Fig. 1d) confirm the coexistence of Co0 and Co2+ for both Co@CoOx and CoCu@CoCuOx. Compared to Co@CoOx, the Co 2p spectrum of CoCu@CoCuOx shifts towards lower binding energies, and the ratio of Co0/Co2+ in CoCu@CoCuOx is 1.49, higher than that in Co@CoOx (0.47) (Table S2†), indicating electron transfer from Cu to Co and the generation of more Co0 species.46 As for Cu 2p spectra (Fig. 1e), Cu0/+ and Cu2+ are detected for both Cu@CuOx and CoCu@CoCuOx. Compared to Cu@CuOx, the Cu 2p spectrum of CoCu@CoCuOx shifts towards higher binding energies, indicating electron loss from Cu. The shift of Cu 2p towards higher binding energy corresponds to the shift of Co 2p towards lower binding energy, suggesting that electron transfer occurs from Cu to Co, as confirmed by DFT calculations (Fig. S6†). This electron redistribution contributes to the interaction with the substrate (Fig. S7 and Table S3†) and improves the catalytic activity of the CoCu@CoCuOx catalyst.47
Fig. 1f–k show the low-resolution TEM images of CoCu@CoCuOx, Co@CoOx, and Cu@CuOx, respectively, all of which display porous particles with a size of around 50 nm. To gain a deeper understanding of the surface microstructure, high-resolution TEM imaging was performed. As for Co@CoOx (Fig. 1j), lattice fringe spacings of 0.203 nm and 0.246 nm can be observed, corresponding to the (002) and (111) crystal planes of hcp Co and CoO, respectively. In the case of Cu (Fig. 1k), Cu species exist mainly in the form of Cu0, with a lattice fringe spacing of 0.208 nm corresponding to the (111) crystal plane of Cu. As shown in Fig. 1g and h, the HR-TEM image and SAED patterns of CoCu@CoCuOx show lattice fringe spacings of 0.207 nm and 0.246 nm, corresponding to the (111) crystal planes of fcc Co and CoO, respectively, confirming the Co phase transition promoted by Cu doping. Furthermore, the high-angle annular dark-field (HAADF) image and the corresponding EDS elemental mapping of CoCu@CoCuOx (Fig. 1i) demonstrate the uniform distribution of Co, Cu, and O elements on the catalyst surface.
2.2. Catalyst performance for FAL reductive amination
Initially, with methanol as the optimal solvent (Fig. S8†), the catalytic performance of CoCu@CoCuOx with different levels of Cu doping in the FAL reductive amination was compared at 30 °C (Fig. 2a). The FAL conversion reaches 100% for all catalysts at 12 h. When employing the Co@CoOx catalyst, the FFA yield is only 27.5%. As the Cu doping level increases, the FFA yield initially increases and then decreases. At a n(Co) : n(Cu) ratio of 8 : 2, CoCu@CoCuOx (8 : 2) exhibits the highest FFA yield (94.5%). However, at a n(Co) : n(Cu) ratio of 7 : 3, the selectivity of int. 3 and int. 4 increases, while the FFA yield decreases to 68%. In the case of the Cu@CuOx catalyst, no FFA is observed, probably due to the lack of active sites for imine hydrogenation. In contrast the commercial RANEY® Ni catalyst achieved only a 24.8% yield of FFA under the same reaction conditions. Therefore, CoCu@CoCuOx (8 : 2) is identified as the optimal catalyst.
Fig. 2. (a) Effects of Cu doping on the FAL reductive amination. Time curves of FAL amination reaction over (b) Co@CoOx and (c) CoCu@CoCuOx. (d) FFA productivity rate on three catalysts. (e) Effects of hydrogen pressure on the FAL reductive amination. Reaction conditions: FAL 0.24 mmol, N2H4·H2O 2 mmol, methanol 4 mL, H2 3 MPa, 30 °C, catalyst 30 mg, 12 h. Note: FAL was fully converted in this figure.

The time profiles of FAL reductive amination for Co@CoOx, CoCu@CoCuOx, and Cu@CuOx were further studied at 30 °C (Fig. 2b–d). As for Co@CoOx (Fig. 2b), no FFA is observed within 2 h, with yields of int. 3 and int. 4 being 82.9% and 17.1%, respectively. As the reaction time extends to 4 h, FFA gradually forms with a 1.6% yield, while the yields of int. 3 and int. 4 are 58.4% and 40%, respectively. Further prolonging reaction time to 12 h, the FFA yield increases to 27.5%, while the yields of int. 3 and int. 4 decrease to 39.9% and 32.6%, respectively. The CoCu@CoCuOx catalyst demonstrates significantly improved activity in comparison to Co@CoOx (Fig. 2c). After 4 h, the FFA yield reaches 29.2%, much higher than that observed with Co@CoOx (1.6%). When the reaction time reaches 12 h, the FFA yield increases to 94.5%, with the yield of int. 4 at 2.7% and the generation of 2.8% THFOL, mainly due to the over-hydrogenation of the furan ring. Comparatively, no FFA was observed throughout the entire reaction process for Cu@CuOx (Fig. S9†). Furthermore, the FFA production rates for different catalysts were calculated (Fig. 2d), and it was found that the value is 0.63 mmol g−1 h−1 when using the CoCu@CoCuOx catalyst, which is 3.5 times higher than that of Co@CoOx (0.18 mmol g−1 h−1), further confirming the unique superiority of the bimetallic catalyst CoCu@CoCuOx for reductive amination.
Next, the influence of hydrogen pressure on the reaction was studied (Fig. 2e). In the absence of H2, only int. 3 is detected. With an increase in hydrogen pressure, the FFA yield gradually increases, reaching a maximum yield of 94.5% at 3 MPa, and then decreases to 86.1% at 4 MPa, accompanied by 13.9% THFOL due to excessive hydrogenation. The type of nitrogen source also significantly affects the reaction (Fig. S10†). When ammonia and hydroxylamine are used as nitrogen sources, the FFA yields are 19.7% and 0%, respectively, which are much lower than that achieved with hydrazine hydrate, mainly due to the less active int. 3 and a simplified reaction network when using hydrazine hydrate. Moreover, both the hydrazine hydrate dosage and catalyst dosage also affect the reaction rate and product distribution (Fig. S11 and S12†). Based on the corresponding optimization, 2 mmol of hydrazine hydrate and 30 mg of catalyst are determined to be the optimal reaction conditions for room-temperature reductive amination. Compared with the reported literature (Fig. 2a and Table S4†), CoCu@CoCuOx demonstrates comparable FFA yield and productivity for FAL reductive amination under much milder conditions compared to non-noble Co and Ni catalysts (≥60 °C), and it even surpasses the performance of noble Ru and Pt catalysts, particularly at high temperatures exceeding 80 °C.16,48
CoCu@CoCuOx exhibited exceptional activity in selective reductive amination involving FAL, benzaldehyde, 2-octanone, 3-phenylpropanal, and cyclohexanone, with primary amine yields exceeding 95% (Fig. 3a–e). Notably, CoCu@CoCuOx is much effective than Co@CoOx and Cu@CuOx. For instance, Cu@CuOx is nearly inactive for all substrates, while Co@CoOx exhibits 1.57–45 times lower primary amine yields regardless of whether the substrate is an aldehyde or a ketone. These results further confirm the remarkable versatility of CoCu@CoCuOx across various carbonyl compounds (Fig. S13†).
Fig. 3. (a–e) Selective reductive amination of various aldehydes and ketones over Co@CoOx, CoCu@CoCuOx and Cu@CuOx catalysts. (f) Catalyst recycling for CoCu@CoCuOx. Reaction conditions: substrate 0.24 mmol, N2H4·H2O 2 mmol, catalyst 30 mg, methanol 4 mL, 3 MPa H2, 30 °C.

The stability and recyclability of the catalyst are paramount for its potential industrial applications. First, a heat filtration experiment (Fig. S14†) confirms that the FAL reductive amination catalyzed by CoCu@CoCuOx follows a heterogeneous catalytic reaction pathway. Subsequently, the catalyst was further assessed over five consecutive cycles (Fig. 3f), with the FFA yield decreasing from the initial 94.5% to 82.4%, indicating that the catalyst is recyclable over several cycles. Characterization of the spent catalyst reveals an increase in the proportion of Co2+ and Cu2+ (Fig. S15–S17†), likely due to partial oxidation during the post-treatment; however, this can be partially regenerated through an in situ reduction strategy (Fig. S18†).
2.3. Study of the reaction mechanism
The in situ FTIR spectra of FAL on different catalysts are shown in Fig. S19.† The absorption peaks at 1440, 1473, and 1570 cm−1 correspond to the vibration of the ν(C C) bond, while the peak at 1671 cm−1 corresponds to the vibration of the ν(C O) bond.49 Both CoCu@CoCuOx, and Co@CoOx exhibit a strong adsorption capacity for FAL. To further monitor the FAL evolution on the catalysts, the in situ FTIR spectra of FAL in the presence of NH3/H2 were recorded, as shown in Fig. 4a and b. After introducing NH3/H2, the ν(C O) bond vibration immediately disappeared, and vibrations corresponding to theν(C N) bonds were also observed at 1627 and 1647 cm−1, indicating the formation of an imine int. from FAL and NH3. With the passage of reaction time, infrared vibration peaks attributed to δ(NH2), ν(C–N), and ν(N–H) appear at 1445, 1557, and 3334 cm−1, respectively,50 indicating that the imine int. is further reduced to FFA. Furthermore, the intensity changes of the ν(N–H) (3334 cm−1) vibration related to FFA as a function of time are recorded for Co@CoOx and CoCu@CoCuOx (Fig. 4c). It is found the signal intensity of ν(N–H) on CoCu@CoCuOx increases rapidly with time, indicating that CoCu@CoCuOx is more active for FAL reductive amination than Co@CoOx at ambient temperature and pressure.
Fig. 4. In situ FAL-absorbed FTIR spectra in the presence of NH3/H2 on (a) CoCu@CoCuOx and (b) Co@CoOx catalysts. (c) Intensity evolution of the N–H bond (at 3334 cm−1) over CoCu@CoCuOx and Co@CoOx catalysts. (d) H2-TPD profiles and (e) hydrogen spillover experiments of Co@CoOx, Cu@CuOx and CoCu@CoCuOx catalysts. (f) Reaction rate comparison of FAL hydrogenation over Co@CoOx, Cu@CuOx and CoCu@CoCuOx catalysts. Reaction conditions: FAL 0.24 mmol, catalyst 30 mg, methanol 4.0 mL, H2 3 MPa, 30 °C, 6 h.

To determine whether the substrate adsorption is the most crucial factor or not, the acid properties of the catalysts were studied by NH3-TPD (Fig. S20†) and pyridine-adsorbed IR (Fig. S21†). It was found that Co@CoOx, CoCu@CoCuOx, and Cu@CuOx exhibit the same Lewis acid sites, with the acid strength following the trend: Co@CoOx > CoCu@CoCuOx > Cu@CuOx; this indicates that acid properties cannot explain the differences in their performance difference. It has beeen reported that oxygen defects play a crucial role in substrate adsorption and activation due to their special electronic structure.51 However, Co@CoOx, CoCu@CoCuOx, and Cu@CuOx exhibit comparative EPR signal intensities at g = 2.00 (Fig. S3†), indicating similar concentrations of oxygen vacancies in all three catalysts, which is also verified using O 1s XPS spectra (Fig. S4†), where vacancy oxygen accounts for 42–62% of all oxygen species. Especially, although Cu@CuOx contains abundant oxygen defects, no desired amine was detected during the reductive amination, further excluding the vital role for determining the activity differences.
Hydrogen activation, dissociation and migration are also of importance for catalytic reductive amination.52 H2-TPD profiles (Fig. 4d) show that Co@CoOx exhibits two H2 desorption peaks located at 123 and 302 °C. In contrast, the desorption peak of CoCu@CoCuOx shifts to a lower temperature (91 °C) and exhibits significantly enhanced intensity, primarily attributed to the enhanced hydrogen spillover on the catalyst. However, for the Cu@CuOx catalyst, no significant peaks are observed, indicating the lack of hydrogen activation ability for the monometallic Cu catalyst. The WO3 coloration experiment shows that the color of WO3 changes to green and pale green when Co@CoOx and Cu@CuOx are added (Fig. 4e), respectively; while the color change of WO3 is more pronounced, turning almost dark green in the presence of CoCu@CoCuOx, indicating a significant amount of active hydrogen spillover.53
To gain a deeper understanding of the impact of Cu doping on hydrogen activation, FAL hydrogenation was used as a probe reaction. As shown in Fig. 4f, Co@CoOx and Cu@CuOx exhibit weaker hydrogenation ability toward C O, with FOL yields of 2.3% and 0.2%, respectively. In comparison, when CoCu@CoCuOx is used as the catalyst, the FOL yield reaches 72.3%. Through calculations, it is found that the FOL production rate of CoCu@CoCuOx is 1.00 mmol g−1 h−1, much higher than those of Co@CoOx (0.3 mmol g−1 h−1) and Cu@CuOx (0 mmol g−1 h−1). These confirm the synergistic effect between Co and Cu bimetallic species, which effectively promotes the activation of H2 on the catalyst surface, which accelerates hydrogen migration and favors the subsequent hydrogenation.
Periodic density functional theory (DFT) calculations were further performed on CoCu@CoCuOx to elucidate active sites (Fig. 5a, b, S6 and S7†). Based on the XRD pattern (Fig. 1b) and HRTEM images (Fig. 1f–k), hcp Co (101), fcc Co (111) and Cu (111) crystals were used to simulate the hydrogen activation sites in Co@CoOx, CoCu@CoCuOx and Cu@CuOx, respectively. It is found that the adsorption energies of H2 on hcp Co (101) and fcc Co (111) are similar, at −0.547 eV and −0.516 eV, respectively (Fig. 5a), but significantly higher than that on Cu (111) (−0.241 eV). Further investigation (Fig. 5b) revealed that the dissociation energy of H2 on fcc Co (111) is 0.162 eV, much lower than that on hcp Co (101) (0.304 eV) and Cu (111) (0.998 eV). Additionally, the reaction energy for the dissociation of active hydrogen on fcc Co (111) is much larger (0.799 eV). These observations, combined with the low activity of Cu@CuOx, suggest that the fcc Co phase present in CoCu@CoCuOx has stronger hydrogen activation and dissociation capabilities.
Fig. 5. (a) Calculated adsorption energies of H2 and (b) dissociation energy of H2 on hcp Co (101), fcc Co (111), and Cu (111) surfaces and the (c) reaction mechanism of FAL reductive amination on CoCu@CoCuOx.

Based on the preceding results, a plausible reaction pathway for FAL reductive amination on CoCu@CoCuOx is proposed (Fig. 5c). Initially, FAL undergoes condensation with hydrazine hydrate to form int. 3. Then the C N bond of int. 3 is adsorbed and activated by the oxygen vacancies on CoCuOx, while H2 is adsorbed onto the electron-rich fcc Co site, generating active hydrogen species. Moreover, the presence of CuOx facilitates the migration of active hydrogen from the Co surface to the CoCuOx surface. Ultimately, the C N bond in int. 3 is reduced by the active hydrogen on the catalyst, leading to the production of FFA.
3. Conclusions
In summary, we demonstrated that the Cu-doped Co@CoOx heterogeneous interface catalyst contains CoCu–CoCuOx bifunctional sites, exhibiting highly active reductive amination capabilities at room temperature, outperforming reported non-noble metal catalysts and even noble metal catalysts. Experimental and DFT calculation results indicate that Cu doping leads to a phase transition of Co from hcp to fcc, while electrons are transferred from Cu to Co, forming a dual active site with electron-rich Co closely interacting with CoCuOx. These electron-rich Co sites show excellent activity in the activation and dissociation of hydrogen, while the CuOx component facilitates hydrogen spillover on the CoCuOx interface. The efficient cooperative effect of CoCu–CoCuOx enhances the high activity of Co in the room-temperature reductive amination of furfural. This study offers valuable insights into the catalytic mechanism at bifunctional sites and paves the way for developing efficient bifunctional catalysts with a structurally heterogeneous interface for biomass upgrading.
Data availability
The data supporting this study's findings are available from the corresponding author upon reasonable request.
Author contributions
R. N. designed the experiments and supervised the project. Y. X. Z. conducted the DFT calculations. Y. W. and Z. M. performed the experiments. All authors discussed the experiments and results. R. N., Y. W. and Z. M. prepared and revised the manuscript.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
The authors acknowledge the financial support from the NSFC (22478366 and U23A20124), the Science and Technology Development Plan Joint Fund Project of Henan Province (232301420048), the Distinguished Young Researchers' Program of Zhengzhou University in China, the Open Funding Project of the State Key Laboratory of Biocatalysis and Enzyme Engineering (SKLBEE20220020) and the Program of Processing and Efficient Utilization of Biomass Resources of Henan Center for Outstanding Overseas Scientists (GZS2022007).
Electronic supplementary information (ESI) available: Additional experimental results. See DOI: https://doi.org/10.1039/d4sc05885b
References
- Resasco D. E. Wang B. Sabatini D. Nat. Catal. 2018;1:731–735. [Google Scholar]
- Liu W.-J. Jiang H. Yu H.-Q. Energy Environ. Sci. 2019;12:1751–1779. [Google Scholar]
- Huber G. W. Chheda J. N. Barrett C. J. Dumesic J. A. Sci. Adv. 2005;308:1446–1450. doi: 10.1126/science.1111166. [DOI] [PubMed] [Google Scholar]
- Jacobson M. Z. Delucchi M. A. Energy Policy. 2011;39:1154–1169. [Google Scholar]
- Mika L. T. Cséfalvay E. Németh Á. Chem. Rev. 2018;118:505–613. doi: 10.1021/acs.chemrev.7b00395. [DOI] [PubMed] [Google Scholar]
- Chen S. Wojcieszak R. Dumeignil F. Marceau E. Royer S. Chem. Rev. 2018;118:11023–11117. doi: 10.1021/acs.chemrev.8b00134. [DOI] [PubMed] [Google Scholar]
- Li X. Jia P. Wang T. ACS Catal. 2016;6:7621–7640. [Google Scholar]
- Liang G. Wang A. Li L. Xu G. Yan N. Zhang T. Angew. Chem., Int. Ed. 2017;56:3050–3054. doi: 10.1002/anie.201610964. [DOI] [PubMed] [Google Scholar]
- Fang L. Yan Z. Wu J. Bugaev A. Lamberti C. Pera-Titus M. Appl. Catal., B. 2021;286:119942. [Google Scholar]
- Froidevaux V. Negrell C. Caillol S. Pascault J.-P. Boutevin B. Chem. Rev. 2016;116:14181–14224. doi: 10.1021/acs.chemrev.6b00486. [DOI] [PubMed] [Google Scholar]
- Chandrashekhar V. G. Baumann W. Beller M. Jagadeesh R. V. Chem. Rev. 2022;376:1433–1441. doi: 10.1126/science.abn7565. [DOI] [PubMed] [Google Scholar]
- Zou H. Chen J. Appl. Catal., B. 2022;309:121262. [Google Scholar]
- Boosa V. Varimalla S. Dumpalapally M. Gutta N. Velisoju V. K. Nama N. Akula V. Appl. Catal., B. 2021;292:120177. [Google Scholar]
- Deng D. Kita Y. Kamata K. Hara M. ACS Sustainable Chem. Eng. 2019;7:4692–4698. [Google Scholar]
- Chandra D. Inoue Y. Sasase M. Kitano M. Bhaumik A. Kamata K. Hosono H. Hara M. Chem. Sci. 2018;9:5949–5956. doi: 10.1039/c8sc01197d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C. Song J. Hua M. Hu Y. Huang X. Wu H. Yang G. Han B. ACS Catal. 2020;10:7763–7772. [Google Scholar]
- Komanoya T. Kinemura T. Kita Y. Kamata K. Hara M. J. Am. Chem. Soc. 2017;139:11493–11499. doi: 10.1021/jacs.7b04481. [DOI] [PubMed] [Google Scholar]
- Jv X. Sun S. Zhang Q. Du M. Wang L. Wang B. ACS Sustainable Chem. Eng. 2020;8:1618–1626. [Google Scholar]
- Nakamura Y. Kon K. Touchy A. S. Shimizu K.-i. Ueda W. ChemSusChem. 2015;7:921–924. [Google Scholar]
- Chatterjee M. Ishizaka T. Kawanami H. Green Chem. 2016;18:487–496. [Google Scholar]
- Gross T. Seayad A. M. Ahmad M. Beller M. Org. Lett. 2002;4:2055–2058. doi: 10.1021/ol0200605. [DOI] [PubMed] [Google Scholar]
- Hahn G. Kunnas P. de Jonge N. Kempe R. Nat. Catal. 2019;2:71–77. [Google Scholar]
- Murugesan K. Beller M. Jagadeesh R. V. Angew. Chem., Int. Ed. 2019;58:5064–5068. doi: 10.1002/anie.201812100. [DOI] [PubMed] [Google Scholar]
- Dong C. Wu Y. Wang H. Peng J. Li Y. Samart C. Ding M. ACS Sustainable Chem. Eng. 2021;9:7318–7327. [Google Scholar]
- Jagadeesh R. V. Murugesan K. Alshammari A. S. Neumann H. Pohl M.-M. Radnik J. Beller M. Science. 2017;358:326–332. doi: 10.1126/science.aan6245. [DOI] [PubMed] [Google Scholar]
- Bhunia M. K. Chandra D. Abe H. Niwa Y. Hara M. ACS Appl. Mater. Interfaces. 2022;14:4144–4154. doi: 10.1021/acsami.1c21157. [DOI] [PubMed] [Google Scholar]
- Zhuang X. Liu J. Zhong S. Ma L. Green Chem. 2022;24:271–284. [Google Scholar]
- Elfinger M. Schönauer T. Thomä S. L. J. Stäglich R. Drechsler M. Zobel M. Senker J. Kempe R. Chem. Sci. 2021;14:2360–2366. doi: 10.1002/cssc.202100553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z. Liu B. Zhou P. Zhang Z. Chi Q. J. Catal. 2019;307:347–356. [Google Scholar]
- Zheng B. Xu J. Song J. Wu H. Mei X. Zhang K. Han W. Wu W. He M. Han B. Chem. Sci. 2022;13:9047–9055. doi: 10.1039/d2sc01596j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chary K. V. R. Seela K. K. Naresh D. Ramakanth P. Catal. Commun. 2008;9:75–81. [Google Scholar]
- Yuan H. Kusema B. T. Yan Z. Streiff S. Shi F. RSC Adv. 2019;9:38877–38881. doi: 10.1039/c9ra08560b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. Zhou K. Shu H. Liu H. Lou J. Guo D. Wei Z. Li X. Catal. Sci. Technol. 2017;7:4129–4135. [Google Scholar]
- Ren Y. Yang Y. Wei M. ACS Catal. 2023;13:8902–8924. [Google Scholar]
- Cao C. Guan W. Liu Q. Li L. Su Y. Liu F. Wang A. Zhang T. Green Chem. 2024;26:6511–6519. [Google Scholar]
- Wan C. Li R. Wang J. Cheng D.-g. Chen F. Xu L. Gao M. Kang Y. Eguchi M. Yamauchi Y. Angew. Chem., Int. Ed. 2024;63:e202404505. doi: 10.1002/anie.202404505. [DOI] [PubMed] [Google Scholar]
- Li R. Zhang Z. Liang X. Shen J. Wang J. Sun W. Wang D. Jiang J. Li Y. J. Am. Chem. Soc. 2023;145:16218–16227. doi: 10.1021/jacs.3c05184. [DOI] [PubMed] [Google Scholar]
- Hong E. Bang S. Cho J. H. Jung K.-D. Shin C.-H. Appl. Catal., A. 2017;542:146–153. [Google Scholar]
- Ma L. Sun K. Luo M. Yan L. Jiang Z. Lu A.-H. Ding Y. J. Phys. Chem. C. 2018;122:23011–23025. [Google Scholar]
- Guo W. Wang Z.-Q. Xiang S. Jing Y. Liu X. Guo Y. Gong X.-Q. Wang Y. Chin. J. Catal. 2023;47:181–190. [Google Scholar]
- Lv L. Yu L. Qiu Z. Li C.-J. Angew. Chem., Int. Ed. 2020;59:6466–6472. doi: 10.1002/anie.201915875. [DOI] [PubMed] [Google Scholar]
- Qiu Z. Lv L. Li J. Li C.-C. Li C.-J. Chem. Sci. 2019;10:4775–4781. doi: 10.1039/c9sc00595a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. Zheng Y. Feng D. Zhang L. Zhang L. Song X. Qiao Z.-A. Angew. Chem., Int. Ed. 2023;62:e202306261. doi: 10.1002/anie.202306261. [DOI] [PubMed] [Google Scholar]
- Zheng Y. Su Y. Pang C. Yang L. Song C. Ji N. Ma D. Lu X. Han R. Liu Q. Environ. Sci. Technol. 2022;56:1905–1916. doi: 10.1021/acs.est.1c05855. [DOI] [PubMed] [Google Scholar]
- Shen Y. Deng J. Impeng S. Li S. Yan T. Zhang J. Shi L. Zhang D. Environ. Sci. Technol. 2020;54:10342–10350. doi: 10.1021/acs.est.0c02680. [DOI] [PubMed] [Google Scholar]
- Shi Y. Luo B. Liu R. Sang R. Cui D. Junge H. Du Y. Zhu T. Beller M. Li X. Angew. Chem., Int. Ed. 2023;62:e202313099. doi: 10.1002/anie.202313099. [DOI] [PubMed] [Google Scholar]
- Sanad M. F. Puente Santiago A. R. Tolba S. A. Ahsan M. A. Fernandez-Delgado O. Shawky Adly M. Hashem E. M. Mahrous Abodouh M. El-Shall M. S. Sreenivasan S. T. Allam N. K. Echegoyen L. J. Am. Chem. Soc. 2021;143:4064–4073. doi: 10.1021/jacs.1c01096. [DOI] [PubMed] [Google Scholar]
- Qi H. Yang J. K. Liu F. Zhang L. Yang J. Liu X. Li L. Su Y. Liu Y. Hao R. Wang A. Zhang T. Nat. Commun. 2021;12:3295. doi: 10.1038/s41467-021-23429-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Z. Wu S. Fu Y. Luo L. Li M. Li Z. Shao M. Zheng L. Xu M. Duan H. J. Energy Chem. 2023;76:239–248. [Google Scholar]
- Ma X. An Z. Song H. Shu X. Xiang X. He J. J. Am. Chem. Soc. 2020;142:9017–9027. doi: 10.1021/jacs.0c02997. [DOI] [PubMed] [Google Scholar]
- Wei Y. Sun Z. Li Q. Wu D. Wang J. Zhang Y. Xu C. C. Nie R. Fuel. 2024;369:131703. [Google Scholar]
- Li B. Xu Z. J. Am. Chem. Soc. 2009;131:16380–16382. doi: 10.1021/ja9061097. [DOI] [PubMed] [Google Scholar]
- Deng K. Lian Z. Wang W. Yu J. Mao Q. Yu H. Wang Z. Wang L. Wang H. Appl. Catal., B. 2024;352:124047. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting this study's findings are available from the corresponding author upon reasonable request.