

Abstract
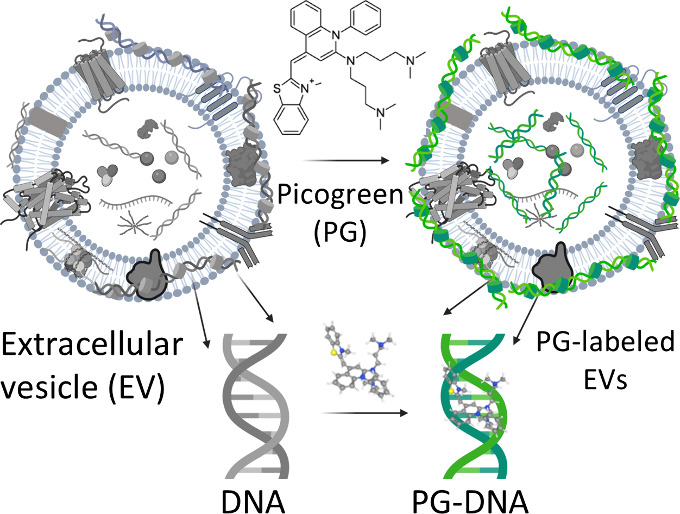
Extracellular vesicles (EVs) are cell-secreted lipid bilayer delimited particles that mediate cellular communication. These tiny sacs of cellular information play an important role in cell communication and alter the physiological process under both normal and pathological conditions. As such, tracking EVs can provide valuable information regarding the basic understanding of cell communication, the onset of early malignancy, and biomarker discovery. Most of the current EV-tracking strategies are invasive, altering the natural characteristics of EVs by modifying the lipid bilayer with lipophilic dyes or surface proteins with fluorescent reporters. The invasive labeling strategies could alter the natural processes of EVs and thereby have major limitations for functional studies. Here, we report an alternative minimally invasive EV labeling strategy using PicoGreen (PG), a small molecule that fluoresces at 520 nm when bound to dsDNA. We show that PG binds to dsDNA associated with small EVs (50–200 nm), forming a stable and highly fluorescent PG-DNA complex in EVs (PG-EVs). In both 2D cell culture and 3D organoid models, PG-EV showed efficient tracking properties, including a high signal-to-noise ratio, time- and concentration-dependent uptake, and the ability to traverse a 3D environment. We further validated PG-EV tracking using dual-labeled EVs following two orthogonal labeling strategies: (1) Bioconjugation via surface amine labeling and (2) donor cell engineering via endogenously expressing mCherry-tetraspanin (CD9/CD63/CD81) reporter proteins. Our study has shown the feasibility of using PG-EV as an effective EV tracking strategy that can be applied for studying the functional role of EVs across multiple model systems.
Keywords: EV-tracking, PicoGreen-based EV-labeling, dsDNA intercalator, minimally invasive EV-labeling, mCherry EVs, dual-labeled EVs
Introduction
Extracellular vesicles (EVs) are lipid-bilayer-delimited particles secreted by cells that play an important role in cellular communication and alter physiological processes. EVs mediate cell communications both locally and systemically by autocrine, paracrine, and endocrine routes.1 The ability of EVs to navigate the extracellular matrix, traverse the biological barrier, reach into circulation, and distribute to distant organs makes them the mediators of cellular information.1−4 As such, they are involved in different pathological conditions, including cancer. In cancer, EV cargo can mediate organotropism and homing to a particular tissue microenvironment.5,6 We and others have shown that exogenously labeled EVs distribute to different organs including tumor sites in animal models.7−9 Moreover, EVs are stable and abundant in body fluids like blood, urine, and saliva and could be used as biomarkers for early detection of diseases.10−12 These and many ongoing studies have shown that EVs play an important role in cellular communication and can be exploited in understanding the pathogenesis of disease and biomarker development. Therefore, tracking these small particles released from cells could provide valuable information regarding a basic understanding of cellular communication, the onset of early malignancy, and biomarker discovery.
Various strategies to label extracellular vesicles exist including lipid insertion approaches, labeling free amines of EV surface proteins, nucleic acid labeling, use of EV-permeable dyes, and endogenous labeling of EV-secreting cells using fluorescent fusion proteins.9,13,14 Widely reported lipid insertion approaches that use lipophilic dyes (e.g., DiI, DiO, and PKH) are easy-to-use pan-EV labeling strategies. However, these methods can be limited due to the formation of EV-like micellular lipid dye aggregates, resulting in a false signal.15,16 Alternative methods such as pan-EV labeling targeting free amines and endogenously engineering cells to produce reporter EVs with fluorescent fusion proteins (mCherry-tetraspanin, GFP-tetraspanin) offer true tracking ability of EVs.17,18 However, these methods alter the surface protein of EVs and thus can potentially change the EV functionality. For example, a recent study of EVs derived from triple-negative breast cancer cell lines showed that EV labeling strategies altered the detection of EV-cell interactions and EV content delivery to particular cell types.19 Therefore, careful consideration should be given to EV labeling strategies based on downstream applications. A minimally invasive EV-labeling method can label EVs with minimal changes in the native EV properties. In this regard, labeling the nucleic acids present in EVs with small molecules offers an alternative, simple, and efficient minimally invasive EV-labeling strategy. Here, we report the use of PicoGreen (PG), a small molecule that intercalates specifically with double-stranded DNA resulting in an exponential increase in fluorescence, as a minimally invasive EV-labeling method. We focus on small EVs (sEVs), with a size range of ∼50–200 nm.
EVs cargo consists of nucleic acids, both DNAs and RNAs.20−22 Although controversial regarding its origin in EVs, studies have suggested that the DNA cargo in EVs includes single-stranded and double-stranded DNA, retrotransposon elements, mitochondrial DNA, and genomic DNA.4,23 While these nucleic acids play a functional role in educating recipient cells,24−26 they can also be exploited as labeling targets for tracking EVs. The presence of DNA (both nuclear and mitochondrial) in EVs has been reported in multiple studies.27−29 DNA is reported to be present in both EV surfaces (potentially associated with membrane proteins) and EV-lumen.28,29 Using single vesicle analysis by nanoflow cytometry, Liu et al. showed that HCT-15 cells (human colorectal cancer cell line) secrete DNA-positive EVs with 33% present in EV-surface and 28% in EV-lumen.30 Further, they reported EVs with <100 nm size mainly possesses DNA on the EV surface, whereas EVs with a size range of 80–200 nm mainly have DNA localized in the lumen. Licha et al. reported the presence of DNA on the surface of 60–75% of plasma-derived EVs.31 The mechanism of DNA loading in EVs is poorly understood. It has been proposed that stress-induced DNA damage causes leakage of DNA in the cytoplasm and shuttling to extracellular vesicles.28,29 More study is required to explore the mechanism of DNA loading into EVs and understand the significance of DNA-positive EV subpopulation. Currently, the presence of DNA in EVs can be exploited as labeling targets by a nucleic acid binding dye.
PicoGreen (PG), or (2-(n-bis(3-dimethylaminopro pyl)-amino)-4-(2,3-dihydro-3-methyl-(benzo-1,3-thiazol-2- yl)-methylidene)-1-phenyl-quinolinium), is a small molecule intercalator that preferentially intercalates to double-stranded DNA forming a highly fluorescent PG-DNA complex compared to the free PG.32,33 The excitation and emission maxima are 480 and 520 nm, respectively. Compared to similar DNA binding dyes like Hoechst, DAPI, and SYBR green, PG offers better sensitivity and superior range of detection, with a 4 orders of magnitude range of detection (25 pg/mL to 1 μg/mL).33,34 PG has a high quantum yield (0.5 bound to dsDNA) and large extinction coefficient (70,000 cm–1 M–1) contributing to an intense fluorescent signal upon binding to dsDNA.33 The dissociation constant (Kd) of the PG-dsDNA complex is reported to be 5.0 ± 0.3 nM (in Tris-EDTA buffer, pH 7.6), which suggests a strong binding affinity.32 In the free state, PG fluorescence is quenched due to intramolecular dynamic fluctuations. Immobilization of PG in DNA via intercalation and electrostatic interaction results in a > 1000-fold enhancement in its fluorescence.32 This unique property of PG allows for specifically tracking the PG-DNA complex and not free PG, which we envision applying to label DNA present in EVs and precise tracking of EVs. PG has been widely used in DNA quantification assay including quantifying DNA present in EVs.35,36 However, PG has not been explored to track EV internalization specifically. Here, we report PG-based labeling of EVs (PG-EV) as an effective means of labeling EVs for biological tracking studies. We show PG-EV internalization in cell-line and organoid models as a proof-of-concept of using a PG-labeling strategy for tracking EVs. Further, we explore the fate of PG-EVs following cellular internalization using two orthogonal dual-labeling approaches: (1) AF647 and PG dual-labeled EVs, and (2) tetraspanins (CD9/CD63/CD81)-mCherry and PG dual-labeled EVs. Our study has shown the feasibility of using PG as a minimally invasive EV tracking strategy that can be applied for precision tracking of EVs across multiple model systems.
Material and Methods
Cell Culture
This study uses an immortalized, nontumorigenic human ovarian surface epithelium cell line (HIO-80), an immortalized human fallopian tube cell line (FT240), an immortalized human embryonic kidney cell line (293T), and two established high-grade serous ovarian cancer cell lines (OVCAR3, OVSAHO). The HIO-80 cells were previously derived by Godwin and colleagues from normal ovarian epithelial cells and immortalized with SV40 large T antigen.37−39 FT240 cell was kindly provided by Dr. Ronny Drapkin (University of Pennsylvania).40 FT240 cells were cultured with DMEM/F-12 (1:1) media without l-glutamine (Gibco 21331020) supplemented with GlutaMAXTm (Gibco 35050061) at a final concentration of 1×, 10% v/v fetal bovine serum (FBS, R&D System, S11150) following EV-depletion (spun at 100 000g at 4 °C for at least 18 h and filtered using a 0.2 μm filter), and 1% v/v penicillin-streptomycin (Gibco 15140122) at 37 °C and 5% CO2. OVCAR3, OVSAHO, and HIO-80 cell lines were cultured with RPMI-medium 1640 (Gibco 11875093) supplemented with 0.3% of 2.5 mg/mL insulin, 10% v/v fetal bovine serum following EV-depletion, and 1% v/v penicillin-streptomycin at 37 °C and 5% CO2.
Human Specimen Collection Procedure
Human specimens were collected following the guidelines of the University of Kansas Medical Center (KUMC) research ethics committee. The studies were performed in accordance with the ethical standards, as laid down in the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards. Deidentified fallopian tube tissues were obtained from women who provided informed consent. The use of human samples was approved by the KUMC Institutional Review Board under the existing Biospecimen Repository Core Facility protocol (HSC #5929).
Isolation and Characterization of Small EVs
Small extracellular vesicles (sEVs) were isolated from cell-culture-conditioned media using differential ultracentrifugation. Briefly, cells were grown in EV-depleted serum media, and conditioned media was collected, centrifuged at 500g for 10 min followed by 2000g for 20 min, and stored at −20 °C until further use. 500 mL of conditioned media was ultracentrifuged at 10 000g for 75 min at 4 °C to remove large extracellular vesicles. The supernatant was ultracentrifuged at 100 000g for 90 min at 4 °C to pellet sEVs. To wash EVs, the sEV pellets were resuspended in filtered PBS, then pelleted a second time at 100 000g for 90 min at 4 °C. sEV pellets were resuspended in 200–300 μL of filtered PBS and stored at −80 °C until further use. The total protein content of sEVs was quantified using a Bradford assay. Particle concentration was quantified using the NanoSight LM10 system and accompanying NTA software v2.3 (NanoSight Ltd., Salisbury, UK) with the following parameters: temperature: 20 °C; camera level:12, and detection threshold:4.
Purification of Nonvesicular DNA from sEVs
Nonvesicular DNA present in sEV suspension was purified using size exclusion chromatography (SEC, Cell Guidance System) following the manufacturer’s protocol. Briefly, the preservative in the column was discarded and the column was equilibrated with 2 × 10 mL filtered PBS. The column was run manually under gravity. sEV isolated from differential ultracentrifugation was made up to a 1 mL volume by adding filtered PBS and added to the column. The volume was allowed to drain, and 2 mL of filtered PBS was added. Once the PBS was drained, the column was changed to a collection tube. Five mL of filtered PBS was added to the column and collected as EV-rich elution. Next, the isolated EV suspension was concentrated to 200–300 μL in PBS using 10000 MWCO Amicon filters (3000g, 20 min, 4 °C). The purified sEV solution was stored at −80 °C until further use.
Labeling sEVs Using PicoGreen Dye
sEVs were labeled with PicoGreen (PG) using incubation. Ten μL of PicoGreen (50× diluted from manufacturer’s concentration, ThermoFisher P7581) was added to 100 μL of sEV solution (containing up to 10 μg of sEVs) in Eppendorf tubes and incubated for 1 h at room temperature. Excess unlabeled dye was washed 3 times using 10K MWCO Amicon filters with 1 mL of filtered PBS. Dye-only controls were prepared following the same methodology by replacing sEVs with PBS. Fluorescence of PG-labeled sEVs (PG-EV) was quantified using fluorescence spectroscopy at 480 nm emission and 520 nm excitation wavelength. Dye controls were prepared by following the same protocol using PBS instead of sEVs.
Cell Viability Assay
Cell viability following PG-EV treatment was assessed using a CellTiter-Glo Luminescent Cell Viability Assay (Promega G7572) to evaluate the biocompatibility of PG-EVs. The assay quantifies the number of viable cells based on the quantitation of ATP present and relies on the ATP-dependent luciferase reaction which gives luminescent product proportional to the viable cells.41 Briefly, 500 FT-240 cells per well (80 μL) were plated in 96 well plates. After 24 h, OVCAR3 PG-EVs or OVCAR3 sEVs (isolated via differential ultracentrifugation) were treated at final concentrations of 10, 5, and 0.5 μg/mL (100 μL) for 72 h. Control cells were maintained with media only. CellTiter-Glo reagents were mixed with Glo lysis buffer (Promega E2661) in a 1:2 ratio and added to 96-well plates in a 1:1 ratio of media volume to cell titer reagent. The reagents were mixed well and incubated for 10 min at room temperature. The luminescence was read by using a TECAN Infinite 200 PRO plate reader (RRID:SCR_019033). Cell viability is reported in terms of the percentage cell viability relative to untreated control cells.
Surface Amine Labeling of sEV Using AF647 Dye
Surface amine labeling of sEV was carried out using Alexa Fluor-647 (Invitrogen, A20173) according to the manufacturer’s instructions.42 sEV solution was made alkaline by adding 1 M Na2CO3, added to the dye tube, and incubated for 1.5 h at room temperature in stirring condition. AF647-labeled sEVs were filtered using the column provided in the kit to remove the free unconjugated dye. For dual labeling, AF647-labeled sEVs were further labeled with PicoGreen as detailed previously. Dye controls were prepared following the same protocol using PBS instead of sEVs.
Lentivirus Packaging
Third-generation lentivirus transfer vectors encoding codon-optimized hCD9–3xGGGGS-mCherry, hCD63–3xGGGGS-mCherry, and hCD81–3xGGGGS-mCherry were purchased from Vectorbuilder.com. These vectors encoded hygromycin, blasticidin, and bleomycin resistance, respectively. Lentivirus was packaged by seeding 293T cells (ATCC, CRL-3216) to 90% confluency in a 15 cm cell culture dish. Once at 90% confluency, 293T cells were transfected with 22.4 μg of psPAX2 (Addgene, #12259), 12.2 μg of pMD2.g (Addgene, #12259), and 13.6 μg of transfer plasmid using Lipofectamine 3000 (ThermoFisher, L3000015) according to the manufacturer’s protocol. In brief, 90 μL of P3000 was added to 1300 μL of Opti-MEM I Reduced Serum Medium (Gibco, 31985062), and plasmid DNA was added to this solution. In another tube, 112 μL of Lipofectamine 3000 was added to 1300 μL of the Opti-MEM I Reduced Serum Medium. The DNA/P3000/Opti-MEM solution was added dropwise to the Lipofectamine 3000/Opti-MEM solution and allowed to form DNA-lipid complexes for 15 min at room temperature. The DNA-lipid complexes were then added to 293T cells and incubated overnight at 37 °C in 5% CO2.
After 24 h, the media was replaced with RPMI-1640 supplemented with 10% FBS and 2 mM Glutamine without antibiotics. Cells were incubated for an additional 48 h. The media was collected and kept at 4 °C after depleting cells by centrifugation at 300g. The medium was replaced and harvested once more after 24 h. LentiX-Concentrator (4×, Takara, 631232) was added to cell-depleted conditioned media and allowed to precipitate viral particles overnight at 4 °C. Precipitated viral particles were centrifuged at 4000g for 30 min. The supernatant was removed, and the pellet was resuspended in 1/100th of the original volume of the collected media in 1× DPBS. Viral particles were allowed to solvate overnight at 4 °C. Redissolved viral particles were aliquoted in 50 μL portions and stored at −80 °C for future use.
Creation of CD9-CD63-CD81 mCherry Expressing FT240 Cell Line
To create a cell line derivative of FT240 expressing CD9-mCherry, CD63-mCherry, and CD81-mCherry, FT240-CD9 cells were first generated. One 50 μL aliquot of CD9 lentiviral particles was added to 50% confluent FT240 cells in a 6-well dish supplemented with 8 μg/mL Polybrene. The cells were expanded over time to eventually fill a T-175 flask. Since FT240 cells are already resistant to hygromycin, cells were sorted for the top 10% of mCherry-positive cells using a Cytek Aurora CS system.
The CD9-positive cells were subsequently transduced with CD63 lentiviral particles using the same technique and selected using 10 μg/mL blasticidin S HCl (Gibco, A1113903). After again sorting for the top 10% of mCherry-positive cells, FT240-CD9/CD63 cells were transduced with CD81 viral particles and selected with 100 μg/mL Zeocin Selection Reagent (Invitrogen, R25001). After 90% confluence was reached in a T-75 flask, cells were sorted into single-cell colonies in a 96-well plate, selecting the top 10% of mCherry-expressing cells. Monoclonal FT240-CD9/CD63/CD81-mCherry cells were confirmed to express all three markers by flow cytometry.
Validation of mCherry Expression in Cell Lines and EVs Using Flow Cytometry and Capillary Western Blot
FT240-CD9/CD63/CD81 expression profiling was conducted using an Aurora Spectral Flow Cytometer (Cytek Biosciences, Fremont, CA) and analyzed with FlowJo 10.10 (BD, Ashland, OR). Samples were stained with extracellular markers CD9 (FITC, Biolegend, 312104, San Diego, CA), CD63 (Pacific Blue, Biolegend, 353012, San Diego, CA), and CD81 (Alexa Fluor 700, Biolegend, 349518, San Diego, CA), as well as a Live/Dead fixable viability dye (violet 510, Tonbo Biosciences, 13–0870-T100, San Diego, CA, USA). Each sample was incubated with 1 μL of antibody in 50 μL of flow cytometry buffer (1x PBS + 2 mM EDTA & 0.1% BSA) prior to inactivation of the Live/Dead stain using 1× PBS + 2 mM EDTA + 1.0% BSA. Cells were resuspended in 50 μL of BD Cytofix/Cytoperm (554714, BD, Franklin Lakes, NJ) and incubated for 30 min at 4 °C, protected from light. Following fixation, 450 μL of flow cytometry buffer was added, and the cells were washed once with the same buffer. Subsequently, the cells were resuspended in 200 μL of flow cytometry buffer. Colonies derived from single cells were then analyzed for uniform and robust overexpression of all three markers.
Expression of CD9, CD63, CD81, and mCherry proteins was analyzed using a capillary-based Simple Western assay (Wes, ProteinSimple) in engineered FT240-CD9/CD63/CD81-mCherry cell lysate and EVs. FT240 cell lysate and EVs were used as controls. Cell lysates at 0.6 and EVs at 0.4 mg/mL concentrations were used for the assay. The 12–230 kDa Separation module with a capillary cartridge (Protein Simple #SM-W004) was used for the separation of proteins and immunodetection, which takes place in a fully automated capillary system. A secondary antirabbit module was used for detection following the manufacturer’s protocol. The details of primary antibodies used for detection are provided in Table S1. Blot images were taken using Compass software, version 6.0.0. (ProteinSimple) using High Dynamic Range 4.0, and contrast was manually adjusted for each set of samples. PBS control was used for each detection antibody as a negative control for nonspecific signals.
Tracking Dye-Labeled sEVs Using Confocal Microscopy
sEV internalization was tracked in vitro in one-dimensional cell culture and three-dimensional organoid and tissue culture. For cell culture, 5000–10 000 cells were seeded in 8 well plates (Cellvis C8–1.5H–N) in 300 μL media. After 24–48 h, dye-labeled sEVs were treated for 24 h or more based on experimental conditions. Following the treatment, media was removed, and cells were washed with PBS 3 times and fixed with 200 μL of 4% paraformaldehyde for 15 min. Fixed cells were washed with PBS once and incubated with 200 μL of DAPI (1 μg/mL) for 10 min for nuclear staining. DAPI solution was then discarded, and cells were washed with PBS twice. Finally, 5 drops of Fluoromount-G mounting medium (Invitrogen 00–4958–02) were added to cells and imaged using confocal microscopy (2037a inverted Eclipse TiE with A1R confocal). Quantification of internalization was done using ImageJ in terms of corrected total cell fluorescence (CTCF).43 CTCF was calculated by using the following equation: CTCF = Integrated density of cell − (Area of selected cell x Mean fluorescence of background reading).
For time-dependent EV uptake analysis, fixed-time end point imaging was performed. Cells were grown in eight-well plates, and PG-EVs were added to cells at different time points before the end of the experiment: 120, 48, 14, 6, 3, and 1 h before the end of the experiment. At the end point, all cells were fixed, incubated with DAPI for nuclear staining, and imaged.
Patient-Derived Organoid Culture
Organoid cultures were established from patient-derived fallopian tubes using the protocol described by Maenhoudt and Vankelecom with slight optimization.44 Normal fallopian tube tissue samples were obtained from the University of Kansas Medical Center’s Biospecimen Repository Core Facility (BRCF). Samples were collected in DMEM/F12 media supplemented with 10% FBS and 1% Penicillin/Streptomycin and stored at 4 °C prior to processing. The tissue was washed with 25 mL of ADF+ medium with Rho Kinase Inhibitor (Y27632- Rho Kinase Inhibitor 5 μM final concentration). For the full composition of organoid media, see Table S2. The tissue was then cut into small, ∼10–20 mm3 pieces.
Tissue dissociation was performed using the MACS human Tumor Dissociation Kit (Miltenyi Biotec). Briefly, all tissue pieces were placed in a gentle MACs C Tube containing about 5 mL of ADF+ media with the Rho Kinase inhibitor. Aliquots of 100 μL of Enzyme H, 50 μL of Enzyme R, and 12.5 μL of Enzyme H (Tumor dissociation kit, MIltenyi Biotec #130–095–929) were added. The tube was then mixed in the gentleMACS dissociator for 5 min (h_tumor_01.01 30-s program). Following the end of the program, the C tube was detached, and the sample was incubated at 37 °C for 30 min under continuous shaking. The C Tube was again mixed in the gentleMACS dissociator for five min, followed by a second incubation at 37 °C for 30 min under continuous shaking.
The resulting solution was filtered using a 70 μm filter (Falcon Cell Strainer 70 μm nylon, ref 352350) into a 50 mL tornado tube. ADF+ media supplemented with Rho Kinase inhibitor was added to a final volume of 20 mL. The tube was then centrifuged at 300g for 5 min to pellet cells. If the pellet contained any red color (due to red blood cells), cells were resuspended in 20 mL 1× Red Blood Cell Lysis Buffer (BioLegend Cat. No. 420301) and recentrifuged. These steps were repeated until RBCs were no longer apparent in the pellet. The final pellet was resuspended to the desired cell concentration in Matrigel (Cultrex, RGF BME Type 2, Select, Catalog # 3526–005–02) kept on ice. 40 μL of Matrigel cell suspension was added to a 24-well plate as a single drop in the middle of each well and then allowed to solidify at 37 °C for 30 min. After solidification, 500 μL of FT-organoid media was added to each well.
To passage organoids, Matrigel drops were digested using TrypLE express (Thermo Fisher Scientific) containing Rho Kinase inhibitor (Y27632, 5 μM) at 37 °C for 5–10 min. The TrypLE was inactivated by mixing with FT-organoid media, and then the suspension was pelleted at 500g for 5 min. Pellets were resuspended into a single-cell suspension via pipetting and plated as above.
Statistical Analysis
Statistical analysis was performed in GraphPad Prism (ver. 9.0) and Microsoft Excel. Data represent mean ± standard deviation. For the quantification of internalization, each data point represents a single cell. Data include the mean signal from at least 20 cells.
Results
PicoGreen Intercalates with dsDNA in Extracellular Vesicles Giving Stable Fluorescence
PicoGreen forms a stable interaction with dsDNA present in small EVs (sEVs, mean size: 50–200 nm) with a simple incubation protocol. Immobilization of PG in dsDNA gives rise to significant enhancement of fluorescence (more than 100-fold) compared to free PG, making it ideal for labeling and tracking EVs (Figure 1A). The signal is specific to dsDNA as PicoGreen preferentially intercalates with dsDNA compared to single-stranded DNA and RNA giving rise to a dramatic increment of fluorescent signal (Figure S1A & B). We isolated sEVs from the conditioned media of four different cell lines including immortalized nontumorigenic epithelial cells from the ovarian surface or fallopian tube (HIO-80 and FT240, respectively) and two established high-grade serous ovarian cancer cell lines (OVCAR3 and OVSAHO) and tested the feasibility of the PicoGreen labeling approach. sEVs were isolated using differential ultracentrifugation, which showed a mean size range of 50–200 nm (Figure S2A). All sEVs showed a dramatic increment (50–200-fold) in fluorescent signal following PicoGreen labeling compared with free PicoGreen (Figure 1B). Furthermore, the intensity of the PG-EV fluorescent signal decreased significantly (by ∼44%) following DNase treatment, which degrades and breaks dsDNA and disrupts the PG-DNA complex (Figure S2B).
Figure 1.

PicoGreen intercalates with dsDNA in sEVs. (A) Schematic showing intercalation of PicoGreen with dsDNA present in sEVs. (B) Fluorescent measurement of PG-EVs in sEVs derived from four different cancer and noncancer cell lines: OVSAHO, OVCAR3, HIO-80, and FT240 cells. Fluorescent measurement carried out at Ex480/Em 520 nm with 2 μg PG-EVs. sEVs are isolated from differential ultracentrifugation. PG-EV fluorescent increases 50- to 200-fold compared to free PG dye. The immobilization of PG in dsDNA present in EVs results in a dramatic increment in the fluorescence. (C) Titration of PG-EV fluorescence with respect to varied OVCAR3 EV protein content. Fluorescence shows dose–response behavior. (D) Stability of PG-EV over time stored at 4 °C and −80 °C. (E) Cell viability of OVCAR3 PG-EVs and OVCAR3 EVs in FT240 cells. EVs were treated at different concentrations for 72 h and percentage cell viability was reported relative to untreated control cells. (F) The average hydrodynamic size (mean) and mode size of OVCAR3 EV and OVCAR3 PG-EV were quantified by nanoparticle tracking analysis, which shows nominal changes in size (ns: nonsignificant, unpaired t test). (G) The hydrodynamic size of OVCAR3 EV and OVCAR3 PG-EV plotted as a percentile of the EV population. Both EVs show an overlapping distribution profile with around 71 percentiles of EVs below 200 nm size. (H) Average hydrodynamic size (mean) and mode size of FT240 EV and FT240 PG-EV showing nominal changes in size (ns: nonsignificant, unpaired t test). (I) The hydrodynamic size of FT240 EV and FT240 PG-EV plotted as a percentile of the EV population. Both EVs show an overlapping distribution profile with around 80 percentiles of EVs below 200 nm size.
We observed significant variation in the fluorescent signal among different sEVs, which can be attributed to the varied amount of dsDNA present in sEVs. Moreover, we observed a linear increment of fluorescent signal with respect to sEV number and protein content (R-square: 0.9924), suggesting PicoGreen can be used for quantitative tracking of sEVs (Figure 1C and Figure S2C). The stability of the PicoGreen labeled EVs (PG-EVs) was analyzed in terms of fluorescent signal over the period of 90 days at different storage conditions (4 and −80 °C, stored in dark). PG-EV fluorescence showed excellent stability with 100% retention of fluorescence for up to 10 days and over 85% fluorescence over the period of 90 days (Figure 1D). Biocompatibility of PG-EV was assessed in terms of the cell viability of treated cells. Different concentrations of OVCAR3 PG-EV and OVCAR3 EV (10–2.5 μg/mL) treatment did not alter the cell viability of FT240 cells when tested for a 72 h treatment period (Figure 1E). This shows that the PG labeling of EVs is well tolerated by cells and could be used for tracking studies.
Next, we analyzed the impact of PicoGreen labeling on the structural properties of sEVs by analyzing the hydrodynamic size of EVs following the labeling. The hydrodynamic size of vesicles measures the spherical structure of the solvated vesicles in the solution which depends upon the diffusion coefficient of vesicles in suspension.45 Therefore, changes in the surface properties of vesicles can lead to changes in hydrodynamic size. Nanoparticle tracking analysis revealed no significant changes in the overall hydrodynamic size distribution of PG-EV compared with unlabeled EVs for both OVCAR3 and FT240-derived sEVs. OVCAR3 EVs and OVCAR3 PG-EVs showed similar size distribution profiles. The mean hydrodynamic size of OVCAR3 EV and OVCAR3 PG-EV were 180.5 and 188.3 nm respectively, and the mode hydrodynamic size of OVCAR3 EV and OVCAR3 PG-EV were 137.9 and 128.4 nm (Figure 1F). Additionally, the hydrodynamic size was plotted as a percentile for comparison, which showed overlap between OVCAR3 EV and OVCAR3 PG-EV, with around 71 percentile EVs below the size of 200 nm (Figure 1G). A similar observation was seen in FT240-derived sEVs. FT240 EVs and FT240 PG-EVs showed similar hydrodynamic size distribution profiles. There were nonsignificant changes between the mean and mode size of FT240 EV compared to FT240 PG-EVS: 163.0 vs 157.8 nm for mean size and 122.4 vs 121.9 nm for mode size (Figure 1H). Additionally, the hydrodynamic size was plotted as a percentile for comparison, which showed overlap between FT24O EV and FT240 PG-EV with around 80 percentile EVs below the size of 200 nm (Figure 1I). This analysis confirmed PG-EV labeling approach has minimal altercation in the structural properties of naïve EVs.
Purification of sEVs to Remove Non-EV DNA
Different sEV isolation methods can result in sEV suspensions containing free DNA not associated with sEVs (non-EV DNA). For sEV tracking purposes using PG, it is important to remove non-EV DNA from sEV-associated DNA. DNase treatment method, often reported to remove naked DNA from EV can also degrade membrane-associated DNA, a major proportion of DNA-positive EV population.30 Therefore, the DNase treatment method to remove naked DNA is not ideal for EV-tracking applications. Here, we used size exclusion chromatography (SEC) to purify sEV from non-EV DNA (Figure 2A). sEVs were isolated using the differential ultracentrifugation (dUC) method from cell-culture-derived conditioned media and further purified via SEC. Size distribution analysis by NTA showed a similar size distribution profile of sEV before and after SEC purification (Figure S3A, B). DNA content in sEV was quantified using the lambda DNA standard curve (Figure S3C). Following SEC, we observed a drastic reduction in the sEV protein concentration by 7.7-fold (Figure 2B), whereas the sEV number decreased by only 1.35-fold (Figure 2C). This finding suggests soluble protein contamination in EV preparation from dUC which was removed following the SEC method. Likewise, the DNA content in sEV also decreased by 2.87-fold following the SEC-based purification method (Figure 2D, E). A portion of this loss can be attributed to technical issues, while most of the decreases in sEV protein and DNA levels is due to the removal of free proteins and non-EV DNA. We then analyzed if the purification approach has any impact on EV tracking applications using cellular internalization assay. We did a time-dependent EV-uptake assay in OVCAR3 cells using PG-EV isolated from (1) UC (UC PG-EV), and (2) UC followed by SEC purification (UC-SEC PG-EV). UC PG-EV contains both non-EV DNA and sEV-associated DNA whereas UC-SEC PG-EV contains sEV-associated DNA only. For comparison, cells were treated with a similar DNA-equivalent of EVs (∼255 ng of DNA) in both cases and imaged using confocal microscopy (Figure 2F). Quantification of cellular internalization was done in terms of corrected total cell fluorescence (CTCF) which is based upon background subtracted integrated density of cell.43 EV uptake was similar at the 1 and 3 h time points; however, there was significantly more signal in UC PG-EVs compared to that in UC-SEC PG-EVs at 6 h time points (Figure 2G). This discrepancy in signal observed following the same DNA-equivalent sEV treatment suggests that the presence of non-EV DNA in EV preparation can impact the result of EV-tracking and internalization studies. Therefore, our results suggest that it is important to remove non-EV DNA for PG-based sEV tracking strategy. Moving forward, we used SEC-purified PG-EVs for all experiments, which are referred to as “PG-EVs” for simplicity.
Figure 2.

Purification of sEVs to remove non-EV dsDNA. (A) Schematic showing purification of OVCAR3 sEVs isolated from dUC (UC-EV) following SEC to remove non-EV associated dsDNA. 700 μL of 699 μg/mL OVCAR3 EVs isolated from dUC was further purified using SEC and the final volume of isolated sEVs (UC-SEC EV) was made to 700 μL. EV characterization before and after SEC showed reduced (B) protein content, (C) EV number, (D) DNA amount, and (E) PG-EV fluorescence following SEC-based purification. (F) UC-EV and UC-SEC EV were labeled with PG and internalization was assessed in FT240 cells (Scale bar: 100 μm). EV treatment was normalized based on the same DNA amount (255 ng). UC EV showed a higher signal at a 6h time point compared to UC-SEC EV suggesting the contribution of non-EV associated DNA in the observed signal. (G) Quantification of EV internalization in terms of corrected total cell fluorescence using ImageJ (***: P-value <0.001, ns: not significant, Unpaired t test).
PG-EV Internalization Shows Time-Dependent and Concentration-Dependent Uptake
We tracked PG-EV at different time points to evaluate the time-dependent uptake behavior. FT240 cells were exposed to OVCAR3 PG-EVs for varied periods (1, 3, 6, 14, 24, 48, and 120 h) and images were taken using confocal microscopy (Figure 3A). PG-EV internalization in cells was shown by green fluorescence. We observed a linear increment in signal up to 14 h, which maximized at 48 h and decreased at 120 h. While the signal decreases at 120 h from its peak at the 48 h time point, the observed signal is still above the background at the 120 h time point. The data show that PG-EV are fully internalized by 48 h and the signal is retained up to 120 h, suggesting that this PG-EV tracking method is useful for extended time-course studies (Figure 3B). Quantification of the fluorescent signal in cells was reported in terms of corrected total cell fluorescence (CTCF). In the dye control treatment, sEVs were replaced with PBS with dye and washed to remove the free dye following the same protocol as for sEV labeling. No or minimal signal in the dye control panel suggests the washing step effectively removed free dye.
Figure 3.

Time-dependent PG-EV internalization in FT-240 cells. (A) 2 μg of OVCAR3 sEVs were treated to FT-240 cells with varied time points (1, 3, 6, 14, 48, and 120 h) and internalization was assessed using confocal microscopy (40× with oil). Images shown are maximum intensity projections of Z-stacked images. PG-EV showed time-dependent uptake characteristics with the maximum signal at a 48 h time point. In the dye control treatment, PG-EVs were replaced by PBS with dye and washed to remove the free dye following the same protocol as EV labeling. No or minimal signal in the dye control panel suggests washing step effectively removed free dye. The scale bar represents 100 μm. (B) Quantification of internalization is reported in terms of corrected total cell fluorescence using ImageJ (****: P-value <0.0001,***: P-value <0.001,*: P-value <0.05, Unpaired t test).
Next, we tracked sEVs using varied concentrations of input of intact EVs based on protein levels. We analyzed the internalization characteristic of the PG-EV using various amounts based on EV protein levels (i.e., 0.01 to 2 μg of protein per well) in three different cell lines: HIO-80, OVCAR3, and FT-240 (Figure 4A-C). PG-EV showed a concentration-dependent uptake behavior. Different cell lines showed varied degrees of uptake at the same sEV dose, which may be cell dependent. Fluorescent uptake was observed starting at 0.05 μg equivalent sEV protein treatment for all cell lines, which increased gradually as the concentration increased showing a dose–response behavior (Figure 4D). The detection of fluorescence at a low dose of 0.05 μg of EV-protein input demonstrates the capacity of PicoGreen to track small amounts of sEVs. Furthermore, the correlation of observed fluorescence with the dose of sEVs suggests that PG-EV can be used as a quantitative approach for sEV internalization in cells.
Figure 4.

Concentration-dependent PG-EV internalization. (A) HIO-80, (B) OVCAR3, and (C) FT-240 cells. OVCAR3 PG-EVs at different amounts (0.01 to 2 μg protein) are treated in 3 different cell lines for 24 h. Confocal microscopy images (40× with oil, maximum intensity projection) showed concentration-dependent uptake of PG-EVs. (D) Quantification of internalization is reported in terms of corrected total cell fluorescence using ImageJ (****: P-value <0.0001,***: P-value <0.001,**: P-value <0.01, ns: nonsignificant, Unpaired t test). The scale bar represents 100 μm.
Tracking of PG-EV in Three-Dimensional Organoid Models
We assessed the feasibility of the PG tracking system to assess sEVs uptake in a three-dimensional model using human fallopian tube-derived organoids (Figure 5A). The fallopian tube organoid forms a three-dimensional lobular structure representing fallopian tube tissue morphology.46 The PG-EVs were able to penetrate and traverse cells within the organoids (∼117 μm depth). We were able to visualize the internalization of these PG-EVs using Z-stacked images from a confocal microscope (Figure S4A). Maximum intensity projection of Z-stacked images allows focusing individual cells in the 3D structure (Figure S4B). For better visualization, we treated PG-EV organoids with Phalloidin dye that labels the cytoskeleton of cells. Merged images show colocalization of blue (nucleus), green (PG-EVs), and red (cytoskeletal), confirming PG-EV localization in the entire organoid structure (Figure 5B). Dye only control showed no signal. These data confirm the PG-EV can be used to track sEV in a three-dimensional model.
Figure 5.

PG-EV internalization in the 3D organoid model. (A) Schematic showing the development of patient-derived human fallopian tube organoid models. Images show organoid growth at days 3, 5, and 7 following seeding in matrigel. (B) Assessment of PG-EV internalization in organoid model. Two μg OVCAR3 PG–EV were treated to patient-derived fallopian tube organoid model for 24 h and imaged using confocal microscopy (40X with oil, maximum intensity projection). The scale bar represents 100 μm (for stitched images) and 1000 μm (for maximum intensity projection). In the dye control treatment, PG-EVs were replaced by PBS with dye and washed to remove the free dye following the same protocol as EV labeling. No or minimal signal in the dye control panel suggests the washing step effectively removed free dye.
Validation of PG-EVs Tracking by Dual-Labeling of EVs
PG-EV internalization was validated using secondary EV labeling to create dual-dye labeled EVs. For this purpose, the surface amine of the sEV protein was labeled using AF647 dye (red) followed by PicoGreen labeling (green). FT240 and OVCAR3 cells were treated with the dual-labeled AF647-PG sEVs for 24 h and internalization was assessed using imaging (Figure 6A, B). AF647-PG sEVs treated cells showed signals for both dyes (green and red) in both cells tested. This observation validates that the green signal observed in PG-EV treated cells corresponds to sEV internalization. AF647 dye demonstrated no interference with the PG signal, as shown by the absence of signal in the green channel with AF647 EV treatment (Figures S5 and Figure S6). This finding suggests that dual-labeled AF647-PG EVs are ideal for colocalization analysis. Notably, there were differences in the signal pattern observed in the sEV internalized cells. The PicoGreen signal showed whole-cell staining while the AF647 signal showed distributed puncta (Figure 6C). Our results suggest different mechanisms of signal presentation for the PicoGreen labeling strategy.
Figure 6.

Validation of PG-EV internalization by dual-labeled AF647-PG EVs. OVCAR3 sEVs were labeled by PG and AF647 dye and exposed to (A) OVCAR3 and (B) FT240 cells. Two μg of AF647-PG EVs were treated to cells for 24 h and imaged using confocal microscopy (40× with oil and 20×, maximum intensity projection). (C) Schematic depiction of dual-labeled AF647-PG EVs and enlarged overlay image showing AF647-PG EV internalization in OVCAR3 and FT240 cells. The image shows the signal for both dyes. In AF647-PG dye control treatment, PG-EVs were replaced by PBS with dye and washed to remove the free dye following the same protocol as EV labeling. No or minimal signal in the dye control panel suggests the washing step effectively removed free dye. The scale bar for panel A represents 50 μm and panel B represents 100 μm.
To further confirm the above observation, we used dual-labeled sEVs following another approach that does not interfere with the surface amine of sEV proteins. For this purpose, we engineered FT-240 cells to endogenously express fluorescent reporter protein using a fusion construct of mCherry and three EV-sorting tetraspanins (i.e., CD9, CD63, and CD81) (Figure S7A). A monoclonal cell lineage of FT240 cells uniformly overexpressing three mCherry-tetraspanin fusion constructs (FT240-mCherry) were selected using flow cytometry (Figure S7B). The engineered cells produce fluorescent CD9-mCherry, CD63-mCherry, and CD81-mCherry proteins that are sorted into sEVs, creating endogenously labeled fluorescent sEVs (Figure S8). As such, these EVs do not require any post-isolation labeling and can be tracked in their native form. FT240-mCherry EVs were further labeled with PicoGreen and tracked in the cell culture (Figure 7). The dual-labeled mCherry-PG sEVs showed signals for both PicoGreen and mCherry in FT240 cells, further validating the PG-EV labeling strategy to track sEVs. Like the observation in AF647-PG EVs, mCherry-PG sEV also showed a distinct difference in signal pattern: Whole-cell staining by PicoGreen and distributed puncta by mCherry. We observed slight bleeding of the mCherry signal in the green channel as shown by the minimal signal observed in mCherry EV treatment (Figure 7). Thus, the limitation of this dual-labeling of EVs is that it can potentially interfere with the colocalization analysis. Nonetheless, the dual-labeled EV confirms the internalization of EVs using two different orthogonal EV-labeling strategies and thus validates the PG labeling approach used in this study to track sEV in the cell and organoid models.
Figure 7.

Validation of sEV internalization by dual-labeled mCherry-PG EVs. Two μg of CD9-CD63-CD81-triple mCherry-PG EVs and PG-EVs from FT240 cells were treated to FT240 cells for 24 h. Images were taken using confocal microscopy (40× with oil, maximum intensity projection). The scale bar represents 50 μm.
Discussion
EV tracking strategies can provide a powerful approach to understanding the functional role of EVs in cell communication, the onset of malignancy, and disease progression. This information is pivotal to realizing the potential of EVs as therapeutics, drug delivery vehicles, and biomarkers. Current EV labeling strategy can be broadly divided into two groups: (1) Direct labeling: labeling of EVs after isolation, and (2) indirect labeling: Using EV producing cells to endogenously label cellular cargo that is sorted in EVs.47−49 Direct labeling techniques include lipophilic fluorescent dye labeling (intercalation of dye in lipid bilayer),50 bioconjugation labeling (covalent conjugation of dyes with EV surface proteins),42 immunolabeling (using fluorophore conjugate antibodies targeting EV surface proteins),51 and incubation labeling (incubation of EV with small molecule fluorophore or nanoparticles).52 Indirect labeling of EVs includes genetically engineering donor cells with EV-sorting fluorescent reporter proteins,53 metabolic labeling of donor cells to add functional tags to EV-sorting cargo,54 and staining of donor cells.55 These various EV labeling strategies generally target one of the 3 components of EVs: (1) Lipid bilayer, (2) proteins, and (3) EV lumen. The DNA present in EVs is largely not explored as a labeling target, which we have explored in this study.
Important consideration should be given to the EV labeling strategy while tracking EVs for functional studies including internalization in cells or homing to certain organs. Does the labeling strategy affect the functional role of EVs? The lipid bilayer and EV surface proteins play a role in cellular internalization as EVs are predominantly internalized through endocytosis.9,56,57 As such, modification of the EV lipid content by lipophilic dyes and EV surface protein by protein conjugation dyes could alter the internalization behavior of EVs. Loconte et al. showed that different EV labeling strategies ((1) Lipophilic dye, (2) EV lumen soluble small molecule dye and (3) membrane-bound fluorescent reporter proteins) influenced the interaction of tumor-derived EVs with immune cells.19 Hence, it is important to design an EV labeling strategy with minimal alteration of native EV properties for functional studies of EVs. The approach of using a small molecule dye PicoGreen (Molecular weight: 553 Da) to label the dsDNA present in EVs offers the advantage of labeling EVs without altering the lipid bilayer or protein content of EVs. Further, due to the strong binding interaction of PG with dsDNA, this approach is superior to the incubation labeling approach with small molecule dyes, which are nonspecifically retained in EV lumen.32
Another important aspect of EV labeling strategies is the incorporation of efficient purification steps to remove the unlabeled dye. This is important to track dye-labeled EVs and avoid false positive signals from unbound/free dye. In this regard, lipophilic dyes have major limitations due to the formation of EV-like micelles which are difficult to remove from dye-labeled EVs.50,58 Lipophilic dye tends to self-aggregate and form micelles that resemble the size of the EVs leading to the false positive signal.16,59 In this study, the PG-EV labeling strategy showed efficient removal of unbound dye using simple molecular-weight cutoff filtration (amicon 10k MWCO). Imaging studies showing minimal or no signal observed in the dye control treatment confirm free unbound PG is removed. Since PicoGreen is a small molecule of 553 Da molecular weight, PG removal is easy and efficient.32
Significant to the field of EV tracking, we observed a linear increment of PG-EV fluorescence with sEV protein amount with high correlation (Pearson r value of 0.9975) suggesting the assay is highly quantitative and therefore can also be used as an alternative EV quantitation method. We also observed that the quantitative characteristic of PG-EV translates when internalized into the cell, based on our time-dependent and dose-dependent EV uptake studies. This further adds to the advantage of the PG-EV tracking strategy. We can potentially use DNA present in EVs as an EV quantitation method, similar to protein-based or EV-number based quantitation method currently used in EV studies.60−62
We validated the internalization of PG-EV using dual labeled EVs following two different labeling strategies: Surface amine labeling (AF647 EVs) and endogenous labeling of tetraspanins with mCherry fusion proteins (mCherry CD9/CD63/CD81 EVs). The validation experiments revealed different staining patterns of PG compared with AF647 and mCherry dyes. Specifically, PG demonstrated whole cell staining compared to distributed puncta for AF647 and mCherry dyes. Following internalization in the cell, PG-EV could be cleaved by intracellular DNase to release free PG. This released free PG can stain dsDNA present in the cytoplasm and nucleus, resulting in broad intracellular staining. This hypothesis is further supported by the observation that PG shows a consistent signal in the entire nucleus, while AF647 and mCherry show minimally distributed signal. Staining whole cells with PG-EV labeling strategies has both advantages and shortcomings. On the plus side, whole cell staining could help to enhance the observed signal, resulting in improved assay sensitivity. This will be particularly advantageous to confirm if EVs have been internalized by cells through processes such as phagocytosis and clathrin- and caveolin-mediated endocytosis or not. On the negative side, this approach fails to track the EV localization inside cells after internalization. Nonetheless, PG-EVs can be used to assess the cellular uptake.
The presence of dsDNA in EVs is now well established, although the percentage of ds-DNA positive EVs and the surface versus lumen location need to be explored more. dsDNA-positive EVs can be considered a subpopulation of functional EVs that can alter the behavior of recipient cells.24−26 As such, the PG-EV tracking strategy is ideal for tracking subpopulations of functional EVs. For PG-EV tracking, it is important to obtain EVs with minimal soluble impurities to avoid interference from nonvesicular DNA. DNase treatment to remove nonvesicular DNA is not ideal as it can cleave the EV surface-associated DNA as well. In this study, we combined two orthogonal EV isolation methods: Differential ultracentrifugation followed by SEC was used to isolate “clean” EVs with minimal soluble impurities. Differential ultracentrifugation isolates small EVs with most of the size between 50 and 200 nm, and SEC (30 nm pore size) separates soluble impurities like free proteins and nucleic acid. Other studies have also shown that combining two orthogonal EV-isolation methods increases the purity of isolated EVs.63,64
PG is a known molecular rotor.65,66 Molecular rotors are nonfluorescent in freely rotating confirmation and become fluorescent when restricted to the planar confirmation. When PG intercalates between DNA base pairs and is restricted in planar confirmation, it becomes fluorescent.32,65 As such, other similar molecular rotors could potentially replace PG to track EVs as the mechanism of the fluorescence gain would be similar. For example, Goh et al. reported a readily synthesizable molecular rotor incorporating an acridine orange DNA intercalating group (AO-R) which showed enhanced fluorescent signals when bound to dsDNA.67 Likewise, Singh et al. reported benzothiazole-based molecular rotor, Thioflavin-T, which showed fluorescent enhancement by ∼210 times upon binding to dsDNA.68 These molecular rotors, which show dramatic enhancement of fluorescence upon binding to dsDNA can be explored to track EVs. Our study has provided a foundation to explore other molecular rotors like PG to explore their potential as EV tracking probe.
The significance of the PG-EV tracking approach is (1) it offers a simple incubation-based labeling method combined with the efficient removal of nonfluorescent free dye, and (2) it has the ability to label and track ds-DNA containing specific subpopulations of EVs. These features contribute to the efficient tracking of naïve EVs while avoiding nonspecific dye-related signals for cellular tracking of EV internalization. While PG-EVs offer a simple and efficient tracking method of naïve EVs, some limitations of the assay should also be considered based on downstream application. Two specific limitations of the PG-EV labeling strategy are (1) The inability to label and track dsDNA negative EVs, and (2) the inability to track cellular localization following internalization due to whole-cell staining behavior.
Conclusion
To summarize, we report a small molecule DNA intercalating agent PG-based sEV labeling strategy as a minimally invasive tracking system to track dsDNA positive sEVs. PG-EV showed a dramatic (50- to 200-fold) increase of fluorescence compared to free dye, superior stability, and biocompatibility. Further, PG-EV fluorescence showed dose–response behavior with a high correlation with sEV number or protein content. PG-EVs show time- and concentration-dependent uptake behavior in 2D cell culture and 3D organoid models with no/minimal signal in dye-only control. Finally, validation experiments using dual-labeled sEVs reveal and confirm distinctive labeling characteristics of PG-EVs: “whole-cell staining”. The reported PicoGreen labeling strategy provides an ideal approach to track sEV trafficking for functional studies, which can provide important information about the fundamental role of sEVs in cellular communication and the pathology of the disease.
Acknowledgments
The authors would like to acknowledge Sarah Tague, Ph.D. (Senior Scientist, Integrative Imaging Core, KUMC), and Integrative Imaging Core of KUMC for their support with confocal microscopy. The core is supported by the KIDDRC (NIH U54 HD 090216) at the University of Kansas Medical Center, Kansas City, KS. We also thank the University of Kansas Medical Center’s/University of Kansas Cancer Center’s Biospecimen Repository Core Facility (BRCF) staff for providing human specimens. The BRCF is supported in part by the KU Cancer Center (NCI CCSG-P30 CA168524) and the Kansas Institute for Precision Medicine (NIGMS COBRE-P20 GM130423). Figure schematics were created with Biorender.com.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsabm.4c01500.
Table S1: List of antibodies used for capillary-based Simple Western Assay (WES); Table S2: Table with components of fallopian tube organoid media; Figure S1. PicoGreen preferentially intercalates with dsDNA to give a fluorescent complex; Figure S2: Size distribution profile of sEVs from different cell sources and analysis of PG-EV fluorescence; Figure S3: Size distribution profile of sEV before and after SEC purification and quantification of DNA amount using lambda DNA standard curve; Figure S4: 3D view of organoid showing PG-EV traversing to an entire organoid; Figure S5: OVCAR3 EV tracking in OVCAR3 cells using different labeling approaches; Figure S6: OVCAR3 EV tracking in FT240 cells using different labeling approaches; Figure S7: Transfer plasmid construct for mCherry expression and selection of monoclonal cell lineage of FT240 CD9/CD63/CD81 mCherry cell line; and Figure S8: Capillary Western blot of cell lysates and EVs showing CD9/CD63/CD81-mCherry expression (PDF)
Author Contributions
S.R., S.H.B., A.K.G.: Conceptualization and Investigation. S.R., B.K.G., J.S.: Methodology, Investigation, Formal Analysis, Data Curation, and Visualization. S.R. and B.K.G.: Writing- Original Draft. J.S., H.P., A.K.G.: Writing- Review and Editing. A.K.G., H.B.P., S.H.B.: Supervision. A.K.G.: Project administration and Funding acquisition.
This study was supported in part by grants from the NIH National Cancer Institute (R01 CA260132 to A.K.G.), the Kansas Institute for Precision Medicine (GM130423 to A.K.G.), graduate student fellowship awards from the OVERRUN Ovarian Cancer Foundation (to B.G. and J.S.), a grant from the Ovarian Cancer Research Alliance (to A.K.G.), a grant from the Basser Center for BRCA (to A.K.G.), and a grant from the Honorable Tina Brozman Foundation, Inc. (Tina’s Wish to A.K.G.), and Predicine Inc. S.H.B. was supported in part by the KU Cancer Center (NCI CCSG-P30 CA168524). A.K.G. is the Chancellors Distinguished Chair in Biomedical Sciences Endowed Professor.
The authors declare the following competing financial interest(s): A.K.G. is a co-founder of Sinochips Diagnostics, serves as a scientific advisory board member to Biovica, Clara Biotech, EXOKRYX, VITRAC Therapeutics, and Sinochips Diagnostics, and receives research funding from Predicine and VITRAC Therapeutics. S.H.B. serves as a consultant to Hawkeye Bio, Deltamed, and Dynatek and as a lead chemist to Hydrograph Clean Power. The other authors report no conflict of interest.
Supplementary Material
References
- van Niel G.; Carter D. R. F.; Clayton A.; Lambert D. W.; Raposo G.; Vader P. Challenges and Directions in Studying Cell–Cell Communication by Extracellular Vesicles. Nat. Rev. Mol. Cell Biol. 2022, 23 (5), 369–382. 10.1038/s41580-022-00460-3. [DOI] [PubMed] [Google Scholar]
- Rani S.; Lai A.; Nair S.; Sharma S.; Handberg A.; Carrion F.; Möller A.; Salomon C. Extracellular Vesicles as Mediators of Cell-Cell Communication in Ovarian Cancer and beyond – A Lipids Focus. Cytokine & Growth Factor Reviews 2023, 73, 52–68. 10.1016/j.cytogfr.2023.06.004. [DOI] [PubMed] [Google Scholar]
- Milane L.; Singh A.; Mattheolabakis G.; Suresh M.; Amiji M. M. Exosome Mediated Communication within the Tumor Microenvironment. J. Controlled Release 2015, 219, 278–294. 10.1016/j.jconrel.2015.06.029. [DOI] [PubMed] [Google Scholar]
- Colombo M.; Raposo G.; Thery C. Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annual Reviews 2014, 30, 255–289. 10.1146/annurev-cellbio-101512-122326. [DOI] [PubMed] [Google Scholar]
- Atay S.; Banskota S.; Crow J.; Sethi G.; Rink L.; Godwin A. K. Oncogenic KIT-Containing Exosomes Increase Gastrointestinal Stromal Tumor Cell Invasion. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (2), 711–716. 10.1073/pnas.1310501111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino A.; Costa-Silva B.; Shen T.-L.; Rodrigues G.; Hashimoto A.; Tesic Mark M.; Molina H.; Kohsaka S.; Di Giannatale A.; Ceder S.; Singh S.; Williams C.; Soplop N.; Uryu K.; Pharmer L.; King T.; Bojmar L.; Davies A. E.; Ararso Y.; Zhang T.; Zhang H.; Hernandez J.; Weiss J. M.; Dumont-Cole V. D.; Kramer K.; Wexler L. H.; Narendran A.; Schwartz G. K.; Healey J. H.; Sandstrom P.; Jørgen Labori K.; Kure E. H.; Grandgenett P. M.; Hollingsworth M. A.; de Sousa M.; Kaur S.; Jain M.; Mallya K.; Batra S. K.; Jarnagin W. R.; Brady M. S.; Fodstad O.; Muller V.; Pantel K.; Minn A. J.; Bissell M. J.; Garcia B. A.; Kang Y.; Rajasekhar V. K.; Ghajar C. M.; Matei I.; Peinado H.; Bromberg J.; Lyden D. Tumour Exosome Integrins Determine Organotropic Metastasis. Nature 2015, 527 (7578), 329–335. 10.1038/nature15756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang M.; Jordan V.; Blenkiron C.; Chamley L. W. Biodistribution of Extracellular Vesicles Following Administration into Animals: A Systematic Review. Journal of Extracellular Vesicles 2021, 10 (8), e12085 10.1002/jev2.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayamajhi S.; Marasini R.; Nguyen T. D. T.; Plattner B. L.; Biller D.; Aryal S. Strategic Reconstruction of Macrophage-Derived Extracellular Vesicles as a Magnetic Resonance Imaging Contrast Agent. Biomater. Sci. 2020, 8 (10), 2887–2904. 10.1039/D0BM00128G. [DOI] [PubMed] [Google Scholar]
- Verweij F. J.; Balaj L.; Boulanger C. M.; Carter D. R. F.; Compeer E. B.; D’Angelo G.; El Andaloussi S.; Goetz J. G.; Gross J. C.; Hyenne V.; et al. The Power of Imaging to Understand Extracellular Vesicle Biology in Vivo. Nat. Methods 2021, 18 (9), 1013–1026. 10.1038/s41592-021-01206-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayamajhi S.; Sipes J.; Tetlow A. L.; Saha S.; Bansal A.; Godwin A. K. Extracellular Vesicles as Liquid Biopsy Biomarkers across the Cancer Journey: From Early Detection to Recurrence. Clinical Chemistry 2024, 70 (1), 206–219. 10.1093/clinchem/hvad176. [DOI] [PubMed] [Google Scholar]
- Crow J.; Samuel G.; Godwin A. K. Beyond Tumor Mutational Burden: Potential and Limitations in Using Exosomes to Predict Response to Immunotherapy. Expert Review of Molecular Diagnostics 2019, 19 (12), 1079–1088. 10.1080/14737159.2020.1688144. [DOI] [PubMed] [Google Scholar]
- Atay S.; Wilkey D. W.; Milhem M.; Merchant M.; Godwin A. K. Insights into the Proteome of Gastrointestinal Stromal Tumors-Derived Exosomes Reveals New Potential Diagnostic Biomarkers. Molecular & Cellular Proteomics 2018, 17 (3), 495–515. 10.1074/mcp.RA117.000267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.-J.; Wu J.-Y.; Wang J.-M.; Hu X.-B.; Xiang D.-X. Emerging Strategies for Labeling and Tracking of Extracellular Vesicles. J. Controlled Release 2020, 328, 141–159. 10.1016/j.jconrel.2020.08.056. [DOI] [PubMed] [Google Scholar]
- Boudna M.; Campos A. D.; Vychytilova-Faltejskova P.; Machackova T.; Slaby O.; Souckova K. Strategies for Labelling of Exogenous and Endogenous Extracellular Vesicles and Their Application for in Vitro and in Vivo Functional Studies. Cell Communication and Signaling 2024, 22 (1), 171. 10.1186/s12964-024-01548-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson S.; Yang K. S.; Zelga P.; Liss A. S.; Carlson J. C. T.; del Castillo C. F.; Weissleder R. Single-EV Analysis (sEVA) of Mutated Proteins Allows Detection of Stage 1 Pancreatic Cancer. Science Advances 2022, 8 (16), eabm3453 10.1126/sciadv.abm3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melling G. E.; Conlon R.; Pantazi P.; Dellar E. R.; Samuel P.; Baena-Lopez L. A.; Simpson J. C.; Carter D. R. F. Confocal Microscopy Analysis Reveals That Only a Small Proportion of Extracellular Vesicles Are Successfully Labelled with Commonly Utilised Staining Methods. Sci. Rep 2022, 12 (1), 262. 10.1038/s41598-021-04225-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stajano D.; Lombino F. L.; Schweizer M.; Glatzel M.; Saftig P.; Gromova K. V.; Kneussel M. Tetraspanin 15 Depletion Impairs Extracellular Vesicle Docking at Target Neurons. Journal of Extracellular Biology 2023, 2 (9), e113 10.1002/jex2.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rufino-Ramos D.; Lule S.; Mahjoum S.; Ughetto S.; Cristopher Bragg D.; Pereira de Almeida L.; Breakefield X. O.; Breyne K. Using Genetically Modified Extracellular Vesicles as a Non-Invasive Strategy to Evaluate Brain-Specific Cargo. Biomaterials 2022, 281, 121366 10.1016/j.biomaterials.2022.121366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loconte L.; Arguedas D.; El R.; Zhou A.; Chipont A.; Guyonnet L.; Guerin C.; Piovesana E.; Vázquez-Ibar J. L.; Joliot A.; Théry C.; Martín-Jaular L. Detection of the Interactions of Tumour Derived Extracellular Vesicles with Immune Cells Is Dependent on EV-Labelling Methods. Journal of Extracellular Vesicles 2023, 12 (12), 12384 10.1002/jev2.12384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen D. K.; Fenix A. M.; Franklin J. L.; Higginbotham J. N.; Zhang Q.; Zimmerman L. J.; Liebler D. C.; Ping J.; Liu Q.; Evans R.; et al. Reassessment of Exosome Composition. Cell 2019, 177 (2), 428–445.e18. 10.1016/j.cell.2019.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shurtleff M. J.; Temoche-Diaz M. M.; Schekman R. Extracellular Vesicles and Cancer: Caveat Lector. Annual Review of Cancer Biology 2018, 2, 395–411. 10.1146/annurev-cancerbio-030617-050519. [DOI] [Google Scholar]
- Lee Y. J.; Shin K. J.; Chae Y. C. Regulation of Cargo Selection in Exosome Biogenesis and Its Biomedical Applications in Cancer. Exp Mol. Med. 2024, 56 (4), 877–889. 10.1038/s12276-024-01209-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy J. W.; D’Souza-Schorey C. Tumor-Derived Extracellular Vesicles: Multifunctional Entities in the Tumor Microenvironment. Annual Review of Pathology: Mechanisms of Disease 2023, 18, 205–229. 10.1146/annurev-pathmechdis-031521-022116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao F.; Zheng T.; Gong W.; Wu J.; Xie H.; Li W.; Zhang R.; Liu P.; Liu J.; Wu X.; Zhao Y.; Ren J. Extracellular Vesicles Package dsDNA to Aggravate Crohn’s Disease by Activating the STING Pathway. Cell Death Dis 2021, 12 (9), 1–12. 10.1038/s41419-021-04101-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy J. W.; Sheehan C. S.; Boomgarden A. C.; D’Souza-Schorey C. Recruitment of DNA to Tumor-Derived Microvesicles. Cell Reports 2022, 38 (9), 110443 10.1016/j.celrep.2022.110443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolumar D.; Moncayo-Arlandi J.; Gonzalez-Fernandez J.; Ochando A.; Moreno I.; Monteagudo-Sanchez A.; Marin C.; Diez A.; Fabra P.; Checa M. A.; Espinos J. J.; Gardner D. K.; Simon C.; Vilella F. Vertical Transmission of Maternal DNA through Extracellular Vesicles Associates with Altered Embryo Bioenergetics during the Periconception Period. eLife 2023, 12, RP88008 10.7554/eLife.88008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsering T.; Li M.; Chen Y.; Nadeau A.; Laskaris A.; Abdouh M.; Bustamante P.; Burnier J. V. EV-ADD, a Database for EV-Associated DNA in Human Liquid Biopsy Samples. Journal of Extracellular Vesicles 2022, 11 (10), e12270 10.1002/jev2.12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elzanowska J.; Semira C.; Costa-Silva B. DNA in Extracellular Vesicles: Biological and Clinical Aspects. Molecular Oncology 2021, 15 (6), 1701–1714. 10.1002/1878-0261.12777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkin E. Z.; Bratman S. V. Bioactive DNA from Extracellular Vesicles and Particles. Cell Death Dis 2020, 11 (7), 1–13. 10.1038/s41419-020-02803-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Tian Y.; Xue C.; Niu Q.; Chen C.; Yan X. Analysis of Extracellular Vesicle DNA at the Single-Vesicle Level by Nano-Flow Cytometry. Journal of Extracellular Vesicles 2022, 11 (4), e12206 10.1002/jev2.12206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichá K.; Pastorek M.; Repiská G.; Celec P.; Konečná B. Investigation of the Presence of DNA in Human Blood Plasma Small Extracellular Vesicles. International Journal of Molecular Sciences 2023, 24 (6), 5915. 10.3390/ijms24065915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragan A. I.; Casas-Finet J. R.; Bishop E. S.; Strouse R. J.; Schenerman M. A.; Geddes C. D. Characterization of PicoGreen Interaction with dsDNA and the Origin of Its Fluorescence Enhancement upon Binding. Biophys. J. 2010, 99 (9), 3010–3019. 10.1016/j.bpj.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer V. L.; Jones L. J.; Yue S. T.; Haugland R. P. Characterization of PicoGreen Reagent and Development of a Fluorescence-Based Solution Assay for Double-Stranded DNA Quantitation. Anal. Biochem. 1997, 249 (2), 228–238. 10.1006/abio.1997.2177. [DOI] [PubMed] [Google Scholar]
- Ahn S. J.; Costa J.; Rettig Emanuel J. PicoGreen Quantitation of DNA: Effective Evaluation of Samples Pre-or Psost-PCR. Nucleic Acids Res. 1996, 24 (13), 2623–2625. 10.1093/nar/24.13.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcatti M.; Saada J.; Okereke I.; Wade C. E.; Bossmann S. H.; Motamedi M.; Szczesny B. Quantification of Circulating Cell Free Mitochondrial DNA in Extracellular Vesicles with PicoGreenTM in Liquid Biopsies: Fast Assessment of Disease/Trauma Severity. Cells 2021, 10 (4), 819. 10.3390/cells10040819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando M. R.; Jiang C.; Krzyzanowski G. D.; Ryan W. L. New Evidence That a Large Proportion of Human Blood Plasma Cell-Free DNA Is Localized in Exosomes. PLoS One 2017, 12 (8), e0183915 10.1371/journal.pone.0183915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grobelny J. V.; Godwin A. K.; Broccoli D. ALT-Associated PML Bodies Are Present in Viable Cells and Are Enriched in Cells in the G2/M Phase of the Cell Cycle. Journal of Cell Science 2000, 113 (24), 4577–4585. 10.1242/jcs.113.24.4577. [DOI] [PubMed] [Google Scholar]
- Kruk P. A.; Godwin A. K.; Hamilton T. C.; Auersperg N. Telomeric Instability and Reduced Proliferative Potential in Ovarian Surface Epithelial Cells from Women with a Family History of Ovarian Cancer. Gynecologic Oncology 1999, 73 (2), 229–236. 10.1006/gyno.1999.5348. [DOI] [PubMed] [Google Scholar]
- Auersperg N.; Maines-Bandiera S.; Booth J. H.; Lynch H. T.; Godwin A. K.; Hamilton T. C. Expression of Two Mucin Antigens in Cultured Human Ovarian Surface Epithelium: Influence of a Family Hostory of Ovarian Cancer. American Journal of Obstetrics & Gynecology 1995, 173 (2), 558–565. 10.1016/0002-9378(95)90282-1. [DOI] [PubMed] [Google Scholar]
- Karst A. M.; Drapkin R. Primary Culture and Immortalization of Human Fallopian Tube Secretory Epithelial Cells. Nat. Protoc 2012, 7 (9), 1755–1764. 10.1038/nprot.2012.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abebe F. A.; Hopkins M. D.; Vodnala S. N.; Sheaff R. J.; Lamar A. A. Development of a Rapid In Vitro Screening Assay Using Metabolic Inhibitors to Detect Highly Selective Anticancer Agents. ACS Omega 2021, 6 (28), 18333–18343. 10.1021/acsomega.1c02203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Jeppesen D. K.; Higginbotham J. N.; Graves-Deal R.; Trinh V. Q.; Ramirez M. A.; Sohn Y.; Neininger A. C.; Taneja N.; McKinley E. T.; Niitsu H.; Cao Z.; Evans R.; Glass S. E.; Ray K. C.; Fissell W. H.; Hill S.; Rose K. L.; Huh W. J.; Washington M. K.; Ayers G. D.; Burnette D. T.; Sharma S.; Rome L. H.; Franklin J. L.; Lee Y. A.; Liu Q.; Coffey R. J. Supermeres Are Functional Extracellular Nanoparticles Replete with Disease Biomarkers and Therapeutic Targets. Nat. Cell Biol. 2021, 23 (12), 1240–1254. 10.1038/s41556-021-00805-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayamajhi S.; Nguyen T. D. T.; Marasini R.; Aryal S. Macrophage-Derived Exosome-Mimetic Hybrid Vesicles for Tumor Targeted Drug Delivery. Acta Biomaterialia 2019, 94, 482–494. 10.1016/j.actbio.2019.05.054. [DOI] [PubMed] [Google Scholar]
- Maenhoudt N.; Vankelecom H. Protocol for Establishing Organoids from Human Ovarian Cancer Biopsies. STAR Protocols 2021, 2 (2), 100429 10.1016/j.xpro.2021.100429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire C. M.; Rösslein M.; Wick P.; Prina-Mello A. Characterisation of Particles in Solution – a Perspective on Light Scattering and Comparative Technologies. Sci. Technol. Adv. Mater. 2018, 19 (1), 732–745. 10.1080/14686996.2018.1517587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler M.; Hoffmann K.; Brinkmann V.; Thieck O.; Jackisch S.; Toelle B.; Berger H.; Mollenkopf H.-J.; Mangler M.; Sehouli J.; Fotopoulou C.; Meyer T. F. The Notch and Wnt Pathways Regulate Stemness and Differentiation in Human Fallopian Tube Organoids. Nat. Commun. 2015, 6 (1), 8989. 10.1038/ncomms9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayamajhi S.; Aryal S. Surface Functionalization Strategies of Extracellular Vesicles. J. Mater. Chem. B 2020, 8 (21), 4552–4569. 10.1039/D0TB00744G. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Huang J.; Xia J.; Liang Y.; Li G.. Tracking Tools of Extracellular Vesicles for Biomedical Research. Front. Bioeng. Biotechnol. 2022, 10, 10.3389/fbioe.2022.943712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao C.; Xiang H.; Chen Q.; Zhao Y.; Gao Q.; Huang F.; Mao L. A Review of Labeling Approaches Used in Small Extracellular Vesicles Tracing and Imaging. IJN 2023, 18, 4567–4588. 10.2147/IJN.S416131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.; Cai N.; Niu Q.; Tian Y.; Hu Y.; Yan X. Quantitative Assessment of Lipophilic Membrane Dye-Based Labelling of Extracellular Vesicles by Nano-Flow Cytometry. Journal of Extracellular Vesicles 2023, 12 (8), 12351 10.1002/jev2.12351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.; Zong S.; Wang Z.; Lu J.; Zhu D.; Zhang Y.; Cui Y. Imaging and Intracellular Tracking of Cancer-Derived Exosomes Using Single-Molecule Localization-Based Super-Resolution Microscope. ACS Appl. Mater. Interfaces 2016, 8 (39), 25825–25833. 10.1021/acsami.6b09442. [DOI] [PubMed] [Google Scholar]
- Betzer O.; Perets N.; Angel A.; Motiei M.; Sadan T.; Yadid G.; Offen D.; Popovtzer R. In Vivo Neuroimaging of Exosomes Using Gold Nanoparticles. ACS Nano 2017, 11 (11), 10883–10893. 10.1021/acsnano.7b04495. [DOI] [PubMed] [Google Scholar]
- Gupta D.; Wiklander O. P. B.; Görgens A.; Conceição M.; Corso G.; Liang X.; Seow Y.; Balusu S.; Feldin U.; Bostancioglu B.; Jawad R.; Mamand D. R.; Lee Y. X. F.; Hean J.; Mäger I.; Roberts T. C.; Gustafsson M.; Mohammad D. K.; Sork H.; Backlund A.; Lundin P.; de Fougerolles A.; Smith C. I. E.; Wood M. J. A.; Vandenbroucke R. E.; Nordin J. Z.; El-Andaloussi S. Amelioration of Systemic Inflammation via the Display of Two Different Decoy Protein Receptors on Extracellular Vesicles. Nat. Biomed Eng. 2021, 5 (9), 1084–1098. 10.1038/s41551-021-00792-z. [DOI] [PubMed] [Google Scholar]
- Lee T. S.; Kim Y.; Zhang W.; Song I. H.; Tung C.-H. Facile Metabolic Glycan Labeling Strategy for Exosome Tracking. Biochimica et Biophysica Acta (BBA) - General Subjects 2018, 1862 (5), 1091–1100. 10.1016/j.bbagen.2018.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monopoli M. P.; Zendrini A.; Wu D.; Cheung S.; Sampedro G.; Ffrench B.; Nolan J.; Piskareva O.; Stalings R. L.; Ducoli S.; Bergese P.; O’Shea D. F. Endogenous Exosome Labelling with an Amphiphilic NIR-Fluorescent Probe. Chem. Commun. 2018, 54 (52), 7219–7222. 10.1039/C8CC02135J. [DOI] [PubMed] [Google Scholar]
- Mulcahy L. A.; Pink R. C.; Carter D. R. F. Routes and Mechanisms of Extracellular Vesicle Uptake. Journal of Extracellular Vesicles 2014, 3 (1), 24641 10.3402/jev.v3.24641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzás E. I.; Tóth E. Á.; Sódar B. W.; Szabó-Taylor K. É. Molecular Interactions at the Surface of Extracellular Vesicles. Semin Immunopathol 2018, 40 (5), 453–464. 10.1007/s00281-018-0682-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen J. B. Pitfalls Associated with Lipophilic Fluorophore Staining of Extracellular Vesicles for Uptake Studies. Journal of Extracellular Vesicles 2019, 8 (1), 1582237 10.1080/20013078.2019.1582237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehghani M.; Gulvin S. M.; Flax J.; Gaborski T. R. Systematic Evaluation of PKH Labelling on Extracellular Vesicle Size by Nanoparticle Tracking Analysis. Sci. Rep 2020, 10 (1), 9533. 10.1038/s41598-020-66434-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toribio V.; Morales S.; López-Martín S.; Cardeñes B.; Cabañas C.; Yáñez-Mó M. Development of a Quantitative Method to Measure EV Uptake. Sci. Rep 2019, 9 (1), 10522 10.1038/s41598-019-47023-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta D.; Zickler A. M.; El Andaloussi S. Dosing Extracellular Vesicles. Adv. Drug Delivery Rev. 2021, 178, 113961 10.1016/j.addr.2021.113961. [DOI] [PubMed] [Google Scholar]
- Bonsergent E.; Grisard E.; Buchrieser J.; Schwartz O.; Théry C.; Lavieu G. Quantitative Characterization of Extracellular Vesicle Uptake and Content Delivery within Mammalian Cells. Nat. Commun. 2021, 12 (1), 1864. 10.1038/s41467-021-22126-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan K.; Martin K.; FitzGerald S. P.; O’Sullivan J.; Wu Y.; Blanco A.; Richardson C.; Mc Gee M. M. A Comparison of Methods for the Isolation and Separation of Extracellular Vesicles from Protein and Lipid Particles in Human Serum. Sci. Rep 2020, 10 (1), 1039. 10.1038/s41598-020-57497-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyhlídalová Kotrbová A.; Gömöryová K.; Mikulová A.; Plešingerová H.; Sladeček S.; Kravec M.; Hrachovinová Š.; Potěšil D.; Dunsmore G.; Blériot C.; Bied M.; Kotouček J.; Bednaříková M.; Hausnerová J.; Minář L.; Crha I.; Felsinger M.; Zdráhal Z.; Ginhoux F.; Weinberger V.; Bryja V.; Pospíchalová V. Proteomic Analysis of Ascitic Extracellular Vesicles Describes Tumour Microenvironment and Predicts Patient Survival in Ovarian Cancer. Journal of Extracellular Vesicles 2024, 13 (3), e12420 10.1002/jev2.12420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora A. K.; Singh P. K.; Patro B. S.; Nath S. PicoGreen: A Better Amyloid Probe than Thioflavin-T. Chem. Commun. 2016, 52 (82), 12163–12166. 10.1039/C6CC05600H. [DOI] [PubMed] [Google Scholar]
- Mora A. K.; Nath S. Ultrafast Dynamics of a Molecular Rotor-Based Bioprobe-PicoGreen: Understanding toward Fibril Sensing Mechanism. J. Phys. Chem. B 2019, 123 (41), 8767–8776. 10.1021/acs.jpcb.9b05922. [DOI] [PubMed] [Google Scholar]
- Goh W. L.; Lee M. Y.; Lim T. X.; Chua J. S.; Brenner S.; Ghadessy F. J.; Teo Y. N. A Novel Molecular Rotor Facilitates Detection of P53-DNA Interactions Using the Fluorescent Intercalator Displacement Assay. Sci. Rep 2018, 8 (1), 12946 10.1038/s41598-018-31197-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P. K.; Sujana J.; Mora A. K.; Nath S. Probing the DNA–Ionic Liquid Interaction Using an Ultrafast Molecular Rotor. J. Photochem. Photobiol., A 2012, 246, 16–22. 10.1016/j.jphotochem.2012.07.006. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.