

Abstract
Background
Passive smoke has a significant impact on lung function and constitutes a critical public health issue, as smoking generates free radicals that damage the lungs and other tissues. Currently, limited research exists on whether the antioxidant melatonin can mitigate lung damage caused by smoking. This study aims to investigate the mechanisms through which melatonin alleviates acute lung disease induced by passive smoking.
Methods
Rats were divided into five groups (n = 6): a control group and three groups exposed to low, medium, and high concentrations of smoke, and a melatonin treatment group.
Results
Data indicated that in the high concentration passive smoking group, the alveolar structure of the lung tissue was destroyed, and the total antioxidant capacity in lung tissue diminished as the concentration of smoke increased. The expressions of TNF-α, IL-6, and IL-1β exhibited similar results. The anti-apoptotic factors Bcl-2 and Bcl-xL mRNA level significantly decreased in the high concentration smoking group, while no significant changes were observed in the medium and low concentration groups. Conversely, the high concentration passive smoking increased the pro-apoptotic factors Bax and Caspase-3 mRNA levels. Additionally, endogenous melatonin levels in lung tissue gradually decreased following exposure to smoke, whereas the exogenous melatonin alleviated the changes in inflammatory factors and apoptosis-related factors in lung tissue. Furthermore, at high smoking concentrations, the mRNA levels of lung cancer-related genes vascular endothelial growth factor (VEGF), cytochromeP450 1A1 (CYP1A1), and cytochrome P450 1B1 (CYP1B1) were significantly increased, while exogenous melatonin reduced the expression of these genes in lung tissue.
Conclusions
These findings suggest that melatonin can diminish lung tissue damage, apoptosis, and inflammatory responses induced by passive smoking, as well as decrease the expression of lung cancer-related genes. Further experimental investigations involving exogenous melatonin treatments will be needed.
Keywords: Melatonin, Passive smoking, Lung, Oxidative stress
Introduction
As we know, people in many countries spend most of their time indoors, so a clean indoor environment is important for human health. Environmental tobacco smoke is known as an important source of multiple pollutants, especially in indoor environments [1]. Tobacco smoke is similar to other combustion-sourced pollutants, it has a direct effect on indoor PM2.5 particles. Studies have demonstrated that PM2.5 is closely correlated with chronic lung diseases [2]. In fact, tobacco smoke residues can remain airborne or be absorbed by surfaces in the room, and become a source of long-term exposure to harmful pollutants even smoking has ended. Ni systematically summarized the relationship of tobacco smoking and indoor PM2.5 and the mechanism that underpin the link of tobacco smoke, PM2.5 and chronic lung diseases [3].
Cigarette smoke (CS), a highly complex mixture containing more than 4,000 compounds, causes aberrant cell responses leading to tissue damage around the airways and alveoli, which underlies various lung diseases [4]. The ingredients in cigarette smoke can cause DNA damage in the body, electrophilic compounds from tobacco smoke bind to the nucleophilic sites of DNA nucleotides and lead to the DNA adducts formation, which cause gene mutations [5]. In addition to that, smoking altered microbial diversities and communities in the lower respiratory tract of mice which should be considered in future studies focusing on smoking-induced inflammatory disease [6]. Also, CS is the most important risk factor for chronic obstructive pulmonary disease (COPD) [7]. Accumulating studies have demonstrated that the apoptosis of bronchial and alveolar epithelial cells provoked by CS plays a key role in the pathogenesis of COPD [8, 9].
In individuals who smoke, the immune system is greatly altered and homeostasis is changed. Exposure to tobacco smoke can skew immune responses by impairing Th1-type and augmenting Th2-dependent responses, mainly by altering the immune functions of a variety of immune cells and aggravating the allergic inflammation and sensitization [10, 11]. Research has demonstrated that the phagocytic ability of macrophages was significantly decreased from healthy smokers compared with never smokers [12]. Macrophages representing over 90% of the alveolar immune cells. Macrophages classically exert regulatory effects via the expression of inflammatory cytokines such as interleukin (IL)-1, IL‐2, IL‐4, IL‐6, IL‐8, tumor necrosis factor‐α (TNF-α) and interferon gamma (IFN-γ) [13]. Inflammation is triggered when lung tissue is damaged, leading to a marked increase in the expression of inflammatory factors such as IL-6, INF-γ, and TNF-α [13, 14]. CS exposure has been shown to elevate levels of TNF-α, a key mediator in acute smoke-induced inflammation and subsequent connective tissue degradation [15]. Moreover, CS can also induce apoptosis in a number of cell types [16]. For instance, smoking induces apoptosis of the oocytes, accelerating ovarian aging and inducing earlier menopause [17]. Furthermore, studies showed that smoking induced anti-apoptosis factors that can inhibit apoptosis in both caspase-dependent and -independent pathways [18]. In addition to that, studies have shown a significant increase in lung cancer caused by smoking. CYP1 family members, including CYP1A1, CYP1A2, and CYP1B1, are induced by aryl hydrocarbon receptors (AhRs). The ligands that induce CYP1 expression are reported to be carcinogenic xenobiotics [19]. Cigarette smoke exposure was found to induce the expression of CYP enzymes in the lungs and liver of rats, which, in turn, increased oxidative stress and inflammation [20]. The levels of inflammatory cells and VEGF were positively correlated with the number of cancer cells in bronchoalveolar lavage fluid (BALF) from lung cancer patients [21].
Melatonin, synthesized by the pineal gland in mammals as well as by non-endocrine organs such as the skin, gut, and immune system, has been identified in experimental studies as possessing antioxidant and anti-inflammatory properties under conditions of exacerbated inflammation [22]. Simultaneously, numerous studies have documented the protective effects of melatonin on lung tissue [23]. Dong et al. investigated the molecular mechanisms underlying melatonin’s inhibitory effects on lipopolysaccharide (LPS)-induced acute lung injury in Sprague-Dawley rats, focusing on the suppression of p38 MAPK overactivation [24]. Smoking is easy to induce the occurrence of COPD, but He et al. observed melatonin administration significantly suppressed apoptosis and endoplasmic reticulum stress, and increased the expression of SIRT1 in rat lung tissues to protects against COPD [7]. Additionally, the NLRP3 inflammasome is markedly activated in the lungs, leading to elevated levels of IL-1β and caspase-1. Melatonin has been shown to inhibit the activation of the NLRP3 inflammasome, thereby mitigating acute lung injury [25].
Considering the above, this study investigates the impact of passive smoking on rat lung tissue and examines whether melatonin can mitigate this damage by influencing the expression of inflammatory and apoptosis factors in immune cells, or change the expression of lung cancer-associated genes.
Materials and methods
Animals
A total of 30 male Sprague-Dawley rats (mean weight: 350 g) were obtained for the experiments. All rats were randomly divided into 5 groups (n = 6 in each group): Group A (control group; the rats were subjected to air exposure), Group B-D (smoking group; the rats were subjected to cigarette smoking), and Group E [smoking + melatonin group; the rats were subjected to cigarette smoking, melatonin (50 mg/kg; Sigma-Aldrich, St. Louis, MO, USA) injection].
In the rat smoke exposure experiment, a modular smoking apparatus supplied by China Tobacco Jiangsu Industrial Co., LTD was utilized. The device can automatically smoke four cigarettes at one time and deliver the smoke to the rat nasal cavity through a hose. The brand of cigarettes used in the study was Marlboro (nicotine 0.5 mg, Philip Morris Companies Inc, USA). The rats in the low-dose group (Group B) were exposed 1 cigarette each time and burned a total of 5 cigarettes. The rats in the medium dose group (Group C) were exposed 2 cigarettes each time and burned a total of 10 cigarettes. The high-dose group (Group D/E) were exposed 4 cigarettes each time and burned 20 cigarettes in total. The rats in each group were aspirated for 1 h without interruption. Cigarette exposure will last for 7 days. Meanwhile, the rats in Group E were intraperitoneally administered with 40 mg/kg melatonin each day during cigarette smoking, while those in Group A-D were injected with equivalent volumes of saline solution. The steps for smoke exposure and melatonin injection were repeated according to the aforementioned methods. At the end of the experiment day, the animals were intraperitoneally anaesthetized with pentobarbital sodium (Sunbiotech, Beijing, China). Finally, the lung tissue was excised, then weighed, and stored in 4% paraformaldehyde or liquid nitrogen temperatures until further analysis. All experimental procedures were approved by the local ethical committee for animal experiments (Jinling Institute of Science and Technology, Nanjing).
qRT-PCR
The total RNA of lung tissue removed from rat was extracted using TRIzol (Invitrogen, Carlsbad, CA, USA). The concentration and purity of the RNA were measured using a spectrophotometer (NanoDrop 2000). For each sample, 2 µg of the total RNA was taken for reverse transcription according to the manufacturer’s instructions. The transcribed cDNA samples were stored in a -20 °C in refrigerator.
PCR amplification test in briefly. PCR amplification system contains 2 µL cDNA sample, 0.4 µL target primer, 10 µL AceQ® qPCR SYBR® Green Master Mix (Vazyme, Nanjing, China), and 7.2 µL ddH2O; mixed system and centrifugation in 4 °C. Put the mixture into the instrument for amplification (Roche, LightCycler® 480 System, Switzerland). The program set as 95 °C for 10 min, 95 °C for 30 s by 40 cycles, annealing at 57 °C for 30 s, and extension at 72 °C for 30 s. The GAPDH gene was used as an internal control. Relative quantification was determined using the 2−ΔΔCt method. The PCR primers are listed in Table 1.
Table 1.
The primer sequence
| Genes | Primer sequences (5’-3’) | Product size (bp) | ||
|---|---|---|---|---|
| Bax | F: ACA TGG AGC TGC AGA GGA TG | 127 | ||
| R: TAG AAA AGG GCA ACC ACC CG | ||||
| Caspase-3 | F: GGA GCT TGG AAC GCG AAG AA | 169 | ||
| R: ACA CAA GCC CAT TTC AGG GT | ||||
| Bcl-2 | F: TGT GTG GAG AGC GTC AAC AG | 132 | ||
| R: TAG TTC CAC AAA GGC ATC CCA G | ||||
| Bcl-xl | F: TCG CAT TGT GGC CTT CTT CT | 165 | ||
| R: TCC ACA AAA GTG TCC CAG CC | ||||
| VEGF | F: AGC AGA TGT GAA TGC AGA CCA | 138 | ||
| R: ACG CGA GTC TGT GTT TTT GC | ||||
| CYP1A1 | F: TCA CCC TAA CCA TGA AGG GG | 111 | ||
| R: ATG ATC TAG GTG GCT TTG GCT | ||||
| CYP1B1 | F: GAA AAG AAG GCG ACT GGG GA | 158 | ||
| R: TGC ACA TCC GGG TAT CTG GT | ||||
| GAPDH | F: AGG TCG GTG TGA ACG GAT TT | 178 | ||
| R: TTC TCA GCC TTG ACT GTG CC |
H.E staining
Lung tissues were fixed with 4% paraformaldehyde for 48 h, then dehydrated, embedded in paraffin. Then, the lung tissues were sectioned at 5 μm thickness and deparaffinized with xylene. After a series of gradient alcohol dehydration, tissues were stained with hematoxylin and eosin (H&E) and were viewed under the light microscope.
ELISA
T-AOC (A015-2-1, Nanjing Jiancheng Bioengineering Institue), TNF-α (CSB-E11987r, CUSABIO; detection limit, 1.56 pg/mL), IL-6 (CSB-E04640r, CUSABIO; detection limit, 0.078 pg/mL) and IL-1β (CSB-E08055r, CUSABIO; detection limit, 0.039 ng/mL) were measured using a standard quantitative ELISA kit. All ELISAs were performed according to the manufacturer’s recommendations. A commercial ELISA kit was used to detect the melatonin concentration in lung tissue (CSB-E13433r, CUSABIO; detection limit, 12.5 pg/mL).
Statistical analysis
Statistical analysis was performed using the SPSS 17.0.1 (SPSS Inc., Michigan, IL, USA). For comparative studies, one-way ANOVA followed by Dunnett’s post hoc tests was used, respectively. P values < 0.05 were considered significant. Data are expressed as the mean ± S.E.M. (standard error of the mean). All graphs were generated using GraphPad Prism 5.0 (GraphPad Software Inc., La Jolla, CA, USA).
Results
Melatonin treatment could alleviate smoking-induced lung injury
As illustrated in Fig. 1, the alterations in melatonin concentration within lung tissue are depicted. The melatonin content in the lung tissue of rat exhibits variation across different smoking concentrations. Compared to the control group, the melatonin concentrations in the low, medium, and high smoking concentration groups decreased by 32.81% (P = 0.046), 48.39% (P = 0.006), and 61.52% (P = 0.001), respectively. Following melatonin treatment, the melatonin levels in the lung tissue increased by 109.39% (P = 0.014) when compared to the high concentration smoking group.
Fig. 1.
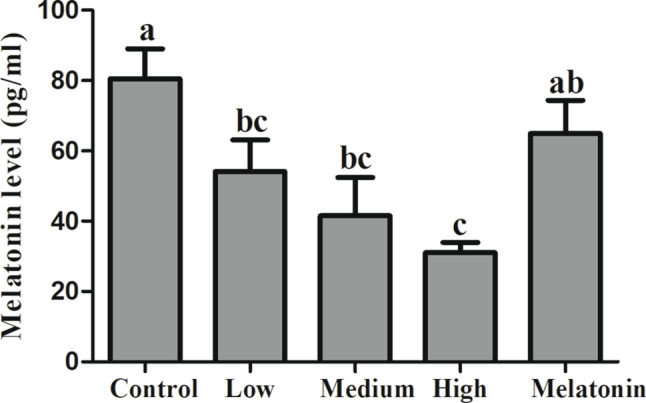
Melatonin concentration in rat lung tissue. Different letters on the column indicate significant differences (P < 0.05) between treatment groups. Statistical significance was assessed using analysis of variance (ANOVA) followed by a multiple comparison test with Dunnett adjustment
Figure 2 demonstrated significant acute pathological changes in the lungs of rats exposed to varying smoking concentrations. Relative to the control group, the lung tissue sections from the passive smoking cohort exhibited disrupted alveolar architecture, expanded alveolar spaces, and attenuated alveolar walls. The severity of alveolar damage was directly proportional to the concentration of smoke exposure, with higher concentrations resulting in more pronounced damage (Fig. 2B and C). Conversely, the group treated with melatonin demonstrated a marked improvement in alveolar structure (Fig. 2D).
Fig. 2.

Hematoxylin eosin (HE) staining shows alveolar damage caused by cigarette smoke in rats. A: control group; B: low concentration passive smoking group; C: high concentration passive smoking group; D: melatonin treatment group. Scale bar = 100 μm
Melatonin alleviates oxidative stress in the lung tissue of rats induced by smoking
Figure 3A shows the variations in total antioxidant levels within lung tissue. Statistical analysis revealed no significant difference in total antioxidant capacity between the low concentration group and the control group (P = 0.635). In contrast, the medium and high concentration groups exhibited reductions of 20.08% (P = 0.004) and 37.74% (P < 0.001), respectively, relative to the control group. Notably, the melatonin treatment group demonstrated a substantial increase of 70.28% in total antioxidant capacity compared to the high concentration smoking group (P < 0.001).
Fig. 3.

Melatonin alleviates oxidative stress induced by passive smoking in rat lung. A: Total antioxidant capacity; B: Tumor necrosis factor alpha (TNF-α); C: Interleukin 6 (IL-6). D: Interleukin 1 beta (IL-1β). Different letters on the column indicate significant differences (P < 0.05) between treatment groups. Statistical significance was assessed using analysis of variance (ANOVA) followed by a multiple comparison test with Dunnett adjustment
Figure 3B-D illustrate the alterations in the expression levels of the inflammatory factors TNF-α, IL-6, and IL-1β following exposure of rats to passive smoking. The data indicate a significant elevation in TNF-α levels, with negligible changes observed in the low concentration group (P = 0.647), a 67.92% increase in the medium concentration group (P < 0.001), and a 114.47% increase in the high concentration group (P < 0.001) compared to the control group. Notably, administration of melatonin resulted in a substantial reduction of TNF-α levels in lung tissue by 33.00% (P < 0.001) compared to the high concentration smoking group.
Similarly to TNF-α, IL-6 levels in lung tissue exhibited an increase of 2.97% (P = 0.822), 13.63% (P = 0.307), and 59.35% (P < 0.001) at varying smoking concentrations when compared to the control group (Fig. 3C). Similarly, IL-1β concentrations increased by 20.27% (P = 0.099), 73.86% (P < 0.001), and 70.20% (P < 0.001) compared to the control group (Fig. 3D). After melatonin treatment, IL-6 concentration in lung tissue decreased by 46.49% (P = 0.001), while IL-1β concentration decreased by 20.99% (P = 0.005) compared to the high concentration smoking group.
Melatonin treatment could inhibit apoptosis-related genes response in smoking-treated lung tissues
Figure 4 illustrates the effects of passive smoking on the expression levels of apoptotic genes in rat lung tissue. No statistically significant differences were observed in the anti-apoptotic factors Bcl-2 and Bcl-xl between the low and medium concentration smoking groups and the control group (P > 0.05). However, in the high concentration smoking group, the Bcl-2 gene expression level decreased by 32.85% relative to the control group (P = 0.029), while the Bcl-xl gene expression level decreased by 41.74% (P = 0.001). Following melatonin treatment, the expression levels of Bcl-2 and Bcl-xl increased by 44.61% (P = 0.046) and 62.18% (P = 0.002), respectively (Fig. 4A/B).
Fig. 4.

Melatonin alleviates passive smoking-induced apoptotic genes in rat lung tissue. A: mRNA level of anti-apoptotic factor Bcl-2; B: mRNA level of anti-apoptotic factor Bcl-xl; C: mRNA level of pro-apoptotic factor Bax; D: mRNA level of pro-apoptotic factor Caspase-3. Different letters on the column indicate significant differences (P < 0.05) between treatment groups. Statistical significance was assessed using analysis of variance (ANOVA) followed by a multiple comparison test with Dunnett adjustment
In addition, the gene levels of the pro-apoptotic factors Bax and Caspase-3 also exhibited corresponding alterations. Compared to the control group, the Bax gene expression did not show a significant change in the low and medium concentration passive smoking groups. However, high concentration passive smoking resulted in a substantial increase in Bax gene expression in rat lung tissue by 112.00% (P < 0.001) (Fig. 4C). Following melatonin treatment, Bax expression exhibited a significant reduction of 38.75% (P < 0.001). In contrast, Caspase-3 mRNA levels showed a substantial increase of 100.07% (P < 0.001) in the medium concentration group and 68.83% (P = 0.002) in the high concentration group, relative to the control group. Additionally, the melatonin treatment group demonstrated a 31.13% decrease in Caspase-3 expression compared to the smoking group (P = 0.015) (Fig. 4D).
Melatonin suppressed the lung cancer related gene expression in smoking-treated lung tissues
Figure 5 illustrates the alterations in the expression of lung cancer-associated genes in rats subjected to passive smoking. The data reveal that the expression levels of VEGF and CYP1A1 in the low and medium concentration groups did not exhibit significant differences compared to the control group (P < 0.001). Conversely, in the high concentration group, the expression of VEGF was markedly elevated by 50.08% (P < 0.001), and the expression of CYP1A1 was significantly increased by 139.60% (P < 0.001). After melatonin treatment, compared to the high concentration smoking group, the gene level of VEGF significantly decreased by 26.47% (P = 0.001). Similarly, the gene expression level of CYP1A1 exhibited a significant decrease of 40.49% (P < 0.001). Conversely, the expression level of CYP1B1 demonstrated an increase of 11.02% (P = 0.405), 53.88% (P < 0.001), and 139.60% (P < 0.001) across three different smoking concentrations relative to the control group. However, melatonin treatment significantly attenuated the expression level of CYP1B1 in the lung tissue of rats subjected to passive smoking, showing a reduction of 40.49% (P < 0.001).
Fig. 5.

Melatonin alleviates the effect of passive smoking on oncogenes in rat lung tissue. A: mRNA level of vascular endothelial growth factor (VEGF); B: mRNA level of cytochrome P4501A1 (CYP1A1); C: mRNA level of Cytochrome P4501B1 (CYP1B1); Different letters on the column indicate significant differences (P < 0.05) between treatment groups. Statistical significance was assessed using analysis of variance (ANOVA) followed by a multiple comparison test with Dunnett adjustment
Discussion
The current study aimed to elucidate the mechanisms by which melatonin mitigates smoking-induced lung injury in rats. Existing research suggests that the toxic constituents of cigarette smoke can precipitate a range of pathological alterations in lung tissue, including damage to the alveolar architecture and disruption of bronchial morphology and function [26]. Additionally, cigarette smoke exposure promotes the accumulation of immune cells, such as macrophages and lymphocytes, culminating in localized inflammation [27].
The results of this study indicate that exposure to secondhand smoke induces significant alterations in the alveolar architecture of rats, characterized by the thinning of alveolar walls. Given that alveoli serve as the fundamental units for gas exchange, such structural compromise inflicts substantial harm on the organism and has the potential to precipitate respiratory failure. Furthermore, prolonged exposure to tobacco smoke is associated with pathological modifications in the lung interstitium, notably interstitial fibrosis. These alterations diminish pulmonary elasticity, impair normal respiratory function, and may ultimately culminate in chronic obstructive pulmonary disease [28].
Secondhand smoke not only compromises the structural integrity of pulmonary morphology but also induces acute inflammatory responses, which constitute a primary mechanism underlying pathological alterations in lung tissue. This study demonstrates that, following exposure to secondhand smoke in rats, the expression levels of inflammation-related cytokines TNF-α, IL-6, and IL-1β in lung tissue were significantly elevated compared to the control group. This elevation is attributable to the increased infiltration of pro-inflammatory cells, which secrete substantial quantities of inflammatory mediators, thereby exacerbating pulmonary damage [29]. On one hand, cigarette smoke induces a high concentration of reactive oxygen species (ROS), including hydroxyl radicals (•OH) and superoxide anions (O2•-), which are transmitted to the lungs and potentially throughout the body in exposed individuals [30]. On the other hand, CS depletes antioxidant defenses, comprising both enzymatic antioxidants such as superoxide dismutase (SOD), catalase, and glutathione peroxidase (GSH-Px), and non-enzymatic antioxidants like glutathione, thereby perpetuating the continuous generation of ROS [29]. Concurrent findings from this study indicate a marked decline in the overall antioxidant capacity within lung tissue following exposure to secondhand smoke.
Additionally, Furthermore, smoking has been shown to induce apoptosis in lung tissue cells, specifically targeting epithelial and endothelial cells. This apoptotic cell death not only diminishes the cellular population but also impairs the lung’s reparative mechanisms, thereby exacerbating pathological alterations [27]. In this study, the expression levels of the anti-apoptotic genes Bcl-2 and Bcl-xl were significantly decreased in the lung tissue of rats exposed to high concentrations of cigarette smoke. Conversely, the expression levels of the pro-apoptotic genes Bax and Caspase-3 were significantly increased. These findings suggest that passive smoking induces apoptosis in lung tissue through various mechanisms. Our results corroborate previous research, indicating that passive smoking facilitates cell apoptosis via apoptotic signaling pathways [31]. Several studies indicate that elevated levels of free radicals and oxidants present in smoke can induce oxidative stress within lung tissue, resulting in damage to cellular membranes and DNA, ultimately leading to cell death [32].
Smoking has been shown to alter the expression of genes associated with lung cancer. These alterations in gene expression are strongly correlated with the duration and frequency of smoking. Research has demonstrated that numerous genes are either upregulated or downregulated in the airway epithelial cells of smokers, potentially contributing to the pathogenesis of lung diseases [33]. Research has demonstrated that smoking induces the expression of various exogenous and redox-regulating genes, with the upregulation of these genes being closely associated with tumorigenesis. Furthermore, smoking has been shown to suppress the expression of certain tumor suppressor genes, thereby facilitating tumor development [34]. This study investigated the expression of pertinent oncogenes in lung tissue and observed that smoking elevated the mRNA levels of three lung cancer-related genes—VEGF, CYP1A1, and CYP1B1—in rat lung tissue. Liu et al. demonstrated that smoking could enhance the risk of oxidative DNA damage through a mechanism partially involving CYP1A1 hypomethylation [35].
In summary, exposure to secondhand smoke induces acute pathological alterations in rat lungs. Our measurements of melatonin levels in lung tissue post-exposure revealed a significant decrease in melatonin concentration across three different levels. This finding suggests that secondhand smoke results in a reduction of endogenous melatonin, corroborating previous reports in similar studies [36]. This underscores the significant role of melatonin in mitigating lung damage induced by passive smoking. Passive smoking, a prevalent environmental pollutant, is associated with the onset of conditions such as lung inflammation. Notably, melatonin exhibits potent anti-inflammatory properties that may mitigate lung damage induced by smoking. Nevertheless, the body of research investigating the efficacy of melatonin treatment specifically for smoking-related lung damage remains limited [37]. Most research has focused on cigarette smoke as a cause of COPD, with melatonin playing a mitigating role. For instance, under smoke stimulation, melatonin alleviates the inflammatory response in COPD by affecting the expression of SIRT1 [38]. These findings offer compelling evidence supporting the clinical application of melatonin in reducing smoking-related bodily harm. However, in different models of passive smoking, the molecular mechanisms of melatonin’s action vary. Zhao et al. conducted an in-depth investigation into the potential of melatonin to mitigate smoking-induced vascular damage and atherosclerosis via the Nrf2/ROS/NLRP3 signaling pathway [39]. Melatonin can also inhibit ROS production and the activation of the NLRP3 inflammasome induced by cigarette smoke extract, thereby alleviating endothelial cell pyroptosis [40]. In cigarette smoke exposure-induced pulmonary fibrosis responses, melatonin reduces levels of transforming growth factor (TGF-β1) [41].
In our experimental results indicate that, relative to the high-dose smoking group, the melatonin-treated rats exhibited reduced levels of total antioxidant capacity, as well as inflammatory markers TNF-α, IL-6, and IL-1β. In accordance with the present results, previous studies have shown that in mice exposed to cigarette smoke, melatonin significantly reduces levels of inflammatory cells and cytokines (IL-6 and TNF-α), possibly by inhibiting phosphodiesterase 4, and is more effective when combined with roflunomide [42]. This finding suggested the potential for further investigation into optimal drug combinations involving melatonin. In addition, gene levels of the pro-apoptotic factors Bax and Caspase-3 were significantly increased in the lung tissue of rats in the high-concentration smoking group, while melatonin significantly decreased their levels. Conversely, levels of the anti-apoptotic factors Bcl-2 and Bcl-xL increased significantly in the melatonin-treated group compared to the high-concentration group. These results suggest that melatonin alleviates lung tissue damage caused by smoke exposure by regulating the expression of apoptosis-related factors. Xu Mengmeng pointed out that melatonin alleviates the acute exacerbation of COPD induced by influenza virus infection by inhibiting the apoptosis of lung immune cells [43]. Lung cancer and COPD often co-occur, and individuals with COPD are at a higher risk of developing lung cancer [44]. In recent years, an increasing number of studies have shown that melatonin plays an important role in the development of cancer. Additionally, melatonin prevents the progression of non-small cell lung cancer by inducing cell apoptosis and inhibiting uncontrolled cell proliferation [45]. In this study, melatonin also significantly reduced the expression of lung cancer-associated genes VEGF, CYP1A1, and CYP1B1 in rat lung tissue. However, this result has not previously been described. The inhibitory effect of melatonin on the development of lung cancer induced by cigarette smoke has been demonstrated.
In conclusion, the effects of smoking on lung tissue are multifaceted and extensive. The deleterious constituents of smoke induce cellular damage through a variety of mechanisms, including direct cytotoxicity, inflammatory responses, apoptosis, and immune system suppression. In this study, melatonin, recognized as a highly effective antioxidant, significantly mitigated acute passive smoke-induced alveolar damage, lung tissue inflammation, cell apoptosis, and the expression of lung cancer-related genes in rats. Despite certain limitations inherent in our research, for example, this study was conducted only in a rat model, and there is a lack of clinical data on the effects of melatonin treatment on lung tissue damage caused by smoking exposure. In addition, the receptor signaling and downstream molecular signaling mechanisms of melatonin regulation in rat lung tissue are not clarified in this study. We believe that our findings could serve as a foundation for future investigations. Further experimental studies are warranted to elucidate the signaling mechanisms of melatonin in smoking-induced lung injury.
Conclusions
In summary, passive smoking can cause changes in lung tissue structure, the total antioxidant capacity, inflammatory factors, and apoptosis factors. With increasing concentrations of cigarette smoke, there is a significant alteration in the expression of lung tissue-related factors. Melatonin, functioning as an antioxidant, substantially mitigates the damage inflicted by passive smoking on rat lung tissue. These findings offer a theoretical foundation for the clinical management of environmental passive smoking.
Acknowledgements
We thank Home for Researchers editorial team (www.home-for-researchers.com) for language editing service.
Abbreviations
- VEGF
Vascular endothelial growth factor
- CYP1A1
CytochromeP450 1A1
- CYP1B1
Cytochrome P450 1B1
- CS
Cigarette smoke
- PM2.5
Particulate matter with a diameter of < 2.5 μm
- TNF-α
Tumor necrosis factor-α
- IFN-γ
Interferon gamma
- ROS
Reactive oxygen species
- SOD
Superoxide dismutase
- GSH-Px
Glutathione peroxidase
- TGF-β1
Transforming growth factor
Author contributions
JJX: Data curation, Investigation, Visualization, Writing – original draft, and Writing – review and editing. LX, YRH and JHZ: Investigation, Visualization, Statistical analyses. ZYH, HYQ: Experimental operation, Statistical analyses. JJL: Revise the article.All authors read and approved the final manuscript.
Funding
This work was supported by High-level talent research project at Jinling Institute of Technology (jit-6-202122, jit-6-202123); Jinling Institute of Technology’s ‘Maker’ Virtual Class Construction Project (2023006).
Data availability
No datasets were generated or analysed during the current study.
Declarations
Ethics approval and consent to participate
The study was approved by the Ethics Committee of Jinling Institute of Technology.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Kaunelienė V, Meišutovič-Akhtarieva M, Martuzevičius D. A review of the impacts of tobacco heating system on indoor air quality versus conventional pollution sources. Chemosphere. 2018;206:568–78. [DOI] [PubMed] [Google Scholar]
- 2.Schober W, Fembacher L, Frenzen A, Fromme H. Passive exposure to pollutants from conventional cigarettes and new electronic smoking devices (IQOS, e-cigarette) in passenger cars. Int J Hyg Environ Health. 2019;222:486–93. [DOI] [PubMed] [Google Scholar]
- 3.Ni Y, Shi G, Qu J. Indoor PM(2.5), tobacco smoking and chronic lung diseases: a narrative review. Environ Res. 2020;181:108910. [DOI] [PubMed] [Google Scholar]
- 4.Zuo H, Faiz A, van den Berge M, Mudiyanselage S, Borghuis T, Timens W, Nikolaev VO, Burgess JK, Schmidt M. Cigarette smoke exposure alters phosphodiesterases in human structural lung cells. Am J Physiol Lung Cell Mol Physiol. 2020;318:L59–64. [DOI] [PubMed] [Google Scholar]
- 5.Izzotti A, Pulliero A. Molecular damage and lung tumors in cigarette smoke-exposed mice. Ann N Y Acad Sci. 2015;1340:75–83. [DOI] [PubMed] [Google Scholar]
- 6.Zhang R, Chen L, Cao L, Li KJ, Huang Y, Luan XQ, Li G. Effects of smoking on the lower respiratory tract microbiome in mice. Respir Res. 2018;19:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He B, Zhang W, Qiao J, Peng Z, Chai X. Melatonin protects against COPD by attenuating apoptosis and endoplasmic reticulum stress via upregulating SIRT1 expression in rats. Can J Physiol Pharmacol. 2019;97:386–91. [DOI] [PubMed] [Google Scholar]
- 8.Gogebakan B, Bayraktar R, Ulaslı M, Oztuzcu S, Tasdemir D, Bayram H. The role of bronchial epithelial cell apoptosis in the pathogenesis of COPD. Mol Biol Rep. 2014;41:5321–7. [DOI] [PubMed] [Google Scholar]
- 9.Comer DM, Kidney JC, Ennis M, Elborn JS. Airway epithelial cell apoptosis and inflammation in COPD, smokers and nonsmokers. Eur Respir J. 2013;41:1058–67. [DOI] [PubMed] [Google Scholar]
- 10.Van Hove CL, Moerloose K, Maes T, Joos GF, Tournoy KG. Cigarette smoke enhances Th-2 driven airway inflammation and delays inhalational tolerance. Respir Res. 2008;9:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moerloose KB, Pauwels RA, Joos GF. Short-term cigarette smoke exposure enhances allergic airway inflammation in mice. Am J Respir Crit Care Med. 2005;172:168–72. [DOI] [PubMed] [Google Scholar]
- 12.Hodge S, Hodge G, Ahern J, Jersmann H, Holmes M, Reynolds PN. Smoking alters alveolar macrophage recognition and phagocytic ability: implications in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2007;37:748–55. [DOI] [PubMed] [Google Scholar]
- 13.Lugg ST, Scott A, Parekh D, Naidu B, Thickett DR. Cigarette smoke exposure and alveolar macrophages: mechanisms for lung disease. Thorax. 2022;77:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huai JP, Sun XC, Chen MJ, Jin Y, Ye XH, Wu JS, Huang ZM. Melatonin attenuates acute pancreatitis-associated lung injury in rats by modulating interleukin 22. World J Gastroenterol. 2012;18:5122–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Churg A, Dai J, Tai H, Xie C, Wright JL. Tumor necrosis factor-alpha is central to acute cigarette smoke-induced inflammation and connective tissue breakdown. Am J Respir Crit Care Med. 2002;166:849–54. [DOI] [PubMed] [Google Scholar]
- 16.Zhai R, Chen F, Liu G, Su L, Kulke MH, Asomaning K, Lin X, Heist RS, Nishioka NS, Sheu CC, et al. Interactions among genetic variants in apoptosis pathway genes, reflux symptoms, body mass index, and smoking indicate two distinct etiologic patterns of esophageal adenocarcinoma. J Clin Oncol. 2010;28:2445–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen CT, Fernández-Rhodes L, Brzyski RG, Carlson CS, Chen Z, Heiss G, North KE, Woods NF, Rajkovic A, Kooperberg C, Franceschini N. Replication of loci influencing ages at menarche and menopause in hispanic women: the women’s Health Initiative SHARe Study. Hum Mol Genet. 2012;21:1419–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Etehad Roodi N, Karkuki Osguei N, Hasanzadeh Daloee M, Pasdar A, Ghayour-Mobarhan M, Ferns G, Samadi Kuchaksaraei A. Association of Endonuclease G Gene Variants with Cardiovascular Disease Risk factors. Rep Biochem Mol Biol. 2019;8:147–52. [PMC free article] [PubMed] [Google Scholar]
- 19.Kwon YJ, Shin S, Chun YJ. Biological roles of cytochrome P450 1A1, 1A2, and 1B1 enzymes. Arch Pharm Res. 2021;44:63–83. [DOI] [PubMed] [Google Scholar]
- 20.Hussain T, Al-Attas OS, Al-Daghri NM, Mohammed AA, De Rosas E, Ibrahim S, Vinodson B, Ansari MG, El-Din KI. Induction of CYP1A1, CYP1A2, CYP1B1, increased oxidative stress and inflammation in the lung and liver tissues of rats exposed to incense smoke. Mol Cell Biochem. 2014;391:127–36. [DOI] [PubMed] [Google Scholar]
- 21.Chen L, Li Q, Zhou XD, Shi Y, Yang L, Xu SL, Chen C, Cui YH, Zhang X, Bian XW. Increased pro-angiogenic factors, infiltrating neutrophils and CD163(+) macrophages in bronchoalveolar lavage fluid from lung cancer patients. Int Immunopharmacol. 2014;20:74–80. [DOI] [PubMed] [Google Scholar]
- 22.Mauriz JL, Collado PS, Veneroso C, Reiter RJ, González-Gallego J. A review of the molecular aspects of melatonin’s anti-inflammatory actions: recent insights and new perspectives. J Pineal Res. 2013;54:1–14. [DOI] [PubMed] [Google Scholar]
- 23.Wang W, Gao J. Effects of melatonin on protecting against lung injury (review). Exp Ther Med. 2021;21:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dong YJ, Ding CH, Zhang Z, Gu WW, Ma YL. [Protective effects of melatonin in acute lung injury rats caused by LPS]. Zhongguo Ying Yong Sheng Li Xue Za Zhi. 2010;26:481–4. [PubMed] [Google Scholar]
- 25.Zhang Y, Li X, Grailer JJ, Wang N, Wang M, Yao J, Zhong R, Gao GF, Ward PA, Tan DX, Li X. Melatonin alleviates acute lung injury through inhibiting the NLRP3 inflammasome. J Pineal Res. 2016;60:405–14. [DOI] [PubMed] [Google Scholar]
- 26.Vasconcelos TB, Araújo FY, Pinho JP, Soares PM, Bastos VP. Effects of passive inhalation of cigarette smoke on structural and functional parameters in the respiratory system of guinea pigs. J Bras Pneumol. 2016;42:333–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Obernolte H, Niehof M, Braubach P, Fieguth HG, Jonigk D, Pfennig O, Tschernig T, Warnecke G, Braun A, Sewald K. Cigarette smoke alters inflammatory genes and the extracellular matrix - investigations on viable sections of peripheral human lungs. Cell Tissue Res. 2022;387:249–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Escalon JG, Girvin F. Smoking-related interstitial lung Disease and Emphysema. Clin Chest Med. 2024;45:461–73. [DOI] [PubMed] [Google Scholar]
- 29.Liang Y, Du R, Chen R, Chu PH, Ip MSM, Zhang KYB, Mak JCW. Therapeutic potential and mechanism of Dendrobium Officinale polysaccharides on cigarette smoke-induced airway inflammation in rat. Biomed Pharmacother. 2021;143:112101. [DOI] [PubMed] [Google Scholar]
- 30.Valavanidis A, Vlachogianni T, Fiotakis K. Tobacco smoke: involvement of reactive oxygen species and stable free radicals in mechanisms of oxidative damage, carcinogenesis and synergistic effects with other respirable particles. Int J Environ Res Public Health. 2009;6:445–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zeng H, Kong X, Zhang H, Chen Y, Cai S, Luo H, Chen P. Inhibiting DNA methylation alleviates cigarette smoke extract-induced dysregulation of Bcl-2 and endothelial apoptosis. Tob Induc Dis. 2020;18:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wick MR. Pathologic features of smoking-related lung diseases, with emphasis on smoking-related interstitial fibrosis and a consideration of differential diagnoses. Semin Diagn Pathol. 2018;35:315–23. [DOI] [PubMed] [Google Scholar]
- 33.Li JL, Jain N, Tamayo LI, Tong L, Jasmine F, Kibriya MG, Demanelis K, Oliva M, Chen LS, Pierce BL. The association of cigarette smoking with DNA methylation and gene expression in human tissue samples. Am J Hum Genet. 2024;111:636–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spira A, Beane J, Shah V, Liu G, Schembri F, Yang X, Palma J, Brody JS. Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proc Natl Acad Sci U S A. 2004;101:10143–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Y, Li X, Zhang B, Fu Y, Yang A, Zhang H, Zhang H, Niu Y, Nie J, Yang J. CYP1A1 methylation mediates the effect of smoking and occupational polycyclic aromatic hydrocarbons co-exposure on oxidative DNA damage among Chinese coke-oven workers. Environ Health. 2019;18:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ozguner F, Koyu A, Cesur G. Active smoking causes oxidative stress and decreases blood melatonin levels. Toxicol Ind Health. 2005;21:21–6. [DOI] [PubMed] [Google Scholar]
- 37.Kole E, Ozkan SO, Eraldemir C, Akar FY, Ozbek SK, Kole MC, Kum T, Filiz PC. Effects of melatonin on ovarian reserve in cigarette smoking: an experimental study. Arch Med Sci. 2020;16:1376–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shin NR, Ko JW, Kim JC, Park G, Kim SH, Kim MS, Kim JS, Shin IS. Role of melatonin as an SIRT1 enhancer in chronic obstructive pulmonary disease induced by cigarette smoke. J Cell Mol Med. 2020;24:1151–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao Z, Wang X, Zhang R, Ma B, Niu S, Di X, Ni L, Liu C. Melatonin attenuates smoking-induced atherosclerosis by activating the Nrf2 pathway via NLRP3 inflammasomes in endothelial cells. Aging. 2021;13:11363–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang X, Bian Y, Zhang R, Liu X, Ni L, Ma B, Zeng R, Zhao Z, Song X, Liu C. Melatonin alleviates cigarette smoke-induced endothelial cell pyroptosis through inhibiting ROS/NLRP3 axis. Biochem Biophys Res Commun. 2019;519:402–8. [DOI] [PubMed] [Google Scholar]
- 41.Shin NR, Park JW, Lee IC, Ko JW, Park SH, Kim JS, Kim JC, Ahn KS, Shin IS. Melatonin suppresses fibrotic responses induced by cigarette smoke via downregulation of TGF-β1. Oncotarget. 2017;8:95692–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lim JO, Kim WI, Lee SJ, Pak SW, Cho YK, Kim JC, Kim JS, Shin IS. The involvement of PDE4 in the Protective effects of Melatonin on cigarette-smoke-Induced Chronic Obstructive Pulmonary Disease. Molecules 2021, 26. [DOI] [PMC free article] [PubMed]
- 43.Xu MM, Kang JY, Wang QY, Zuo X, Tan YY, Wei YY, Zhang DW, Zhang L, Wu HM, Fei GH. Melatonin improves influenza virus infection-induced acute exacerbation of COPD by suppressing macrophage M1 polarization and apoptosis. Respir Res. 2024;25:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Forder A, Zhuang R, Souza VGP, Brockley LJ, Pewarchuk ME, Telkar N, Stewart GL, Benard K, Marshall EA, Reis PP, Lam WL. Mechanisms contributing to the Comorbidity of COPD and Lung Cancer. Int J Mol Sci 2023, 24. [DOI] [PMC free article] [PubMed]
- 45.Pourhanifeh MH, Sharifi M, Reiter RJ, Davoodabadi A, Asemi Z. Melatonin and non-small cell lung cancer: new insights into signaling pathways. Cancer Cell Int. 2019;19:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No datasets were generated or analysed during the current study.