

Abstract
Spinocerebellar ataxias (SCAs) comprise a heterogeneous group of autosomal dominant disorders. The relative frequency of the different SCA subtypes varies broadly among different geographical and ethnic groups as result of genetic drifts. This review aims to provide an update regarding SCA founders in the American continents and the Caribbean as well as to discuss characteristics of these populations. Clusters of SCAs were detected in Eastern regions of Cuba for SCA2, in South Brazil for SCA3/MJD, and in Southeast regions of Mexico for SCA7. Prevalence rates were obtained and reached 154 (municipality of Báguano, Cuba), 166 (General Câmara, Brazil), and 423 (Tlaltetela, Mexico) patients/100,000 for SCA2, SCA3/MJD, and SCA7, respectively. In contrast, the scattered families with spinocerebellar ataxia type 10 (SCA10) reported all over North and South Americas have been associated to a common Native American ancestry that may have risen in East Asia and migrated to Americas 10,000 to 20,000 years ago. The comprehensive review showed that for each of these SCAs corresponded at least the development of one study group with a large production of scientific evidence often generalizable to all carriers of these conditions. Clusters of SCA populations in the American continents and the Caribbean provide unusual opportunity to gain insights into clinical and genetic characteristics of these disorders. Furthermore, the presence of large populations of patients living close to study centers can favor the development of meaningful clinical trials, which will impact on therapies and on quality of life of SCA carriers worldwide.
Keywords: Founder effects, Latin America and the Caribbean, Machado-Joseph disease, Prevalence, SCA2, SCA3, MJD, SCA7, SCA10, Spinocerebellar ataxia, Spinocerebellar ataxia type 2, Spinocerebellar ataxia type 3, Spinocerebellar ataxia type 7, Spinocerebellar ataxia type 10
Introduction
Changes in allele frequency in a population, due to a random selection of certain alleles, are known as genetic drift. Genetic drift can be due to two kinds of events: (i) when a population is sharply reduced in size by a disaster (bottleneck effect) or (ii) when a small group splits off from the main population to find a colony (founder effect)[1].
Many genetic diseases show variations in prevalence among human populations due to genetic drift. Reductions in genetic diversity due to a bottleneck effect in humans are quite hard to trace, because the existing group has to be compared to the original population prior to the event. On the other hand, repeated episodes of forced or voluntary separation of small groups from a larger original population can be better registered and have occurred throughout human history
At least two singular colonization events characterize populations of the American continents. Archeological, linguistic, and genetic evidence have shown that the original human inhabitants of the Western Hemisphere arrived from Asia 20,000 to 10,000 years ago, during the Late Pleistocene. Founders were less variable than the original groups, since studies estimated that less than 1% of the ancestral Asian population left to populate the New World [2–4].
More than 9000 years later, systematic European colonization of the Americas began when Columbus reached Hispaniola, a Caribbean island, in 1492. Large-scale colonization followed hereafter. North and South American mainland fell to the conquistadors, with an estimated of 8,000,000 deaths of indigenous populations [5]. Economic growth related to colonial period relied on African slavery; the total slave trade to the New World being estimated to involve 12 million Africans [6]. When slavery ended, over 50 million people left Western Europe for the Americas during the nineteenth century [7]. As a result of large migratory waves in the last 5 centuries, plus the strong bottleneck events related to the Amerindian deaths, the American continents contain one of the most diverse and recent human populations in the world nowadays.
Autosomal dominant disorders may evade negative selection if the phenotype starts after the reproductive period, as in the case of late onset spinocerebellar ataxias (SCAs). For that reason, differences in SCA frequencies might escape negative selection and can undergo genetic drift phenomenon. Geographical isolation prevents the offspring from dispersing and the phenotypic trait from remaining undetected and can help the identification of autosomal dominant founders. SCA founder populations were particularly frequent in Latin America and the Caribbean (LAC) possibly due to the peculiar origins and characteristics of these populations. In recent years, SCA founder populations have been described in Eastern regions of Cuba (spinocerebellar ataxia type 2 or SCA2, MIM: #183090) [8, 9], in South Brazil (Rio Grande do Sul state) (spinocerebellar ataxia type 3, also known as Machado-Joseph disease, SCA3/MJD, MIM: #109150) [10, 11], in southeast regions of Mexico (spinocerebellar ataxia type 7 or SCA7, MIM: #164500) [12, 13], and all over North and South Americas in the case of spinocerebellar ataxia type 10 (SCA10, MIM: #603516) [14, 15].
This paper aimed to review the state of the art of SCA founder effects and/or isolates in the American continents and the Caribbean. Founder and isolates are important phenomena from the point of view of population genetics. These populations might allow researchers to study a large number of affected carriers of a given disease, with relatively homogeneous genetic and environmental backgrounds, speeding up some discoveries that could be elusive in other circumstances.
Methods
A panel of specialists in autosomal dominant ataxias met in Havana, Cuba, in March 2019, and identified that at least three clusters of SCAs in LAC, plus the unique phenomenon of SCA10 in these continents, were the best focus for the present report. Two procedures were done: estimation of prevalence of SCA2, SCA3/MJD, and SCA7 in regions of interest and comprehensive reviews of scientific contributions by each group. The number of subjects with clinical and molecular diagnosis of SCA2 in Cuba was obtained in a nationwide epidemiological survey carried out between March 2017 and June 2018. The data were retrieved from medical visits performed in the Centre for the Research and Rehabilitation of Hereditary Ataxias in Holguin, in nine provincial hospitals or in the patient’s houses. For those SCA2 patients that were not directly assessed, the demographic information was obtained by telephone calls and/or interviews with their affected or unaffected relatives. The number of symptomatic sub-jects alive and belonging to families with a molecular diagnosis was obtained for SCA3/MJD in Rio Grande do Sul and for SCA7 in seven communities from the central region of Veracruz, Mexico. For SCA3/MJD, data was retrieved from electronic records of families of subjects followed up at Hospital de Clinicas de Porto Alegre; the place where subjects were living was confirmed by telephone contact, in 2019. For SCA7, data was gathered from the records of the Rehabilitation and Social Inclusion Center of Veracruz (CRIS-DIF), Xalapa. Minimal prevalence of Brazilian and Mexican symptomatic carriers was estimated for a 5-year period finishing in June 2019.
Subsequently, each group reviewed the context of disease in focus in their study group, shedding light in the current knowledge of several aspects of research performed locally and showing the perspectives opened for future investigations and practical applications that can be generalized for all affected persons worldwide.
SCA2 and Cuba
SCA2 is probably the second most frequent SCA worldwide, after only to SCA3 [16]. The disease is caused by the abnormal expansion of the CAG repeat tract in the coding region of ATXN2 (MIM: * 601517), leading to expression of a toxic polyQ domain in the ataxin-2 protein [17]. Normal alleles vary from 13 to 31 triplet repeats, with a range of intermediate expansions between 28 and 33 repeats which may predispose to amyotrophic lateral sclerosis or Parkinson’s disease [18]. Expanded ATXN2 alleles have ≥ 32 triplet repeats with a large range of full penetrance above 35 units, which usually exhibit a pure CAG tract [19]. The disease is characterized by a progressive cerebellar syndrome, saccade slowing, peripheral neuropathy, and cognitive involvement [8, 9]. Some of these features start up to 15 years before the ataxia defining the SCA2 prodromal stage [20]. Similar to other polyQ diseases, the age at onset of SCA2 correlates significantly with the CAG repeat length [8].
Similar to other SCAs, the worldwide prevalence of SCA2 is biased toward few epidemiological studies performed in selected geographical regions [21]. This subtype has the highest prevalence and incidence rates in Cuba as result of a founder effect [8, 22]. In fact, in this country SCA2 is most prevalent than SCA3 [23]. Moreover, higher relative frequencies of SCA2 are found in Mexico [24], South Africa [25], India [26], Italy [27], and Venezuela [28].
The first descriptions of a large and homogeneous population with autosomal dominant cerebellar ataxia in Eastern Cuba were done in the 1960–1970s [29], setting the rationale for further studies identifying a large founder population of SCA2 families in Holguin province, which currently has spread throughout all the country. This population has been comprehensively assessed for many years, giving insights into disease natural history, prodromal stage, predictive testing, disease physiopathology, modifier genes, disease biomarkers, and therapeutic options [9]. An epidemiological survey performed between March 2017 and June 2018 in Cuba showed that the province with higher rates continues to be Holguín, where 497 SCA2 patients and 2754 at-risk descendants are living, for prevalence rates of 47.9 patients/ 100,000 inhabitants and 188.6 mutation carriers/100,000 inhabitants. In the central region of the province, there are municipalities with remarkable prevalence rates such as Baguanos (154.3/ 100,000 inhabitants) and Urbano Noris (87.20/100,000), whereas Cauto Cristo, a municipality bordering Holguin province, has a prevalence of 106 cases/100,000 inhabitants (LVP, unpublished data) (Fig. 1B and Supplemental material 1).
Fig. 1.
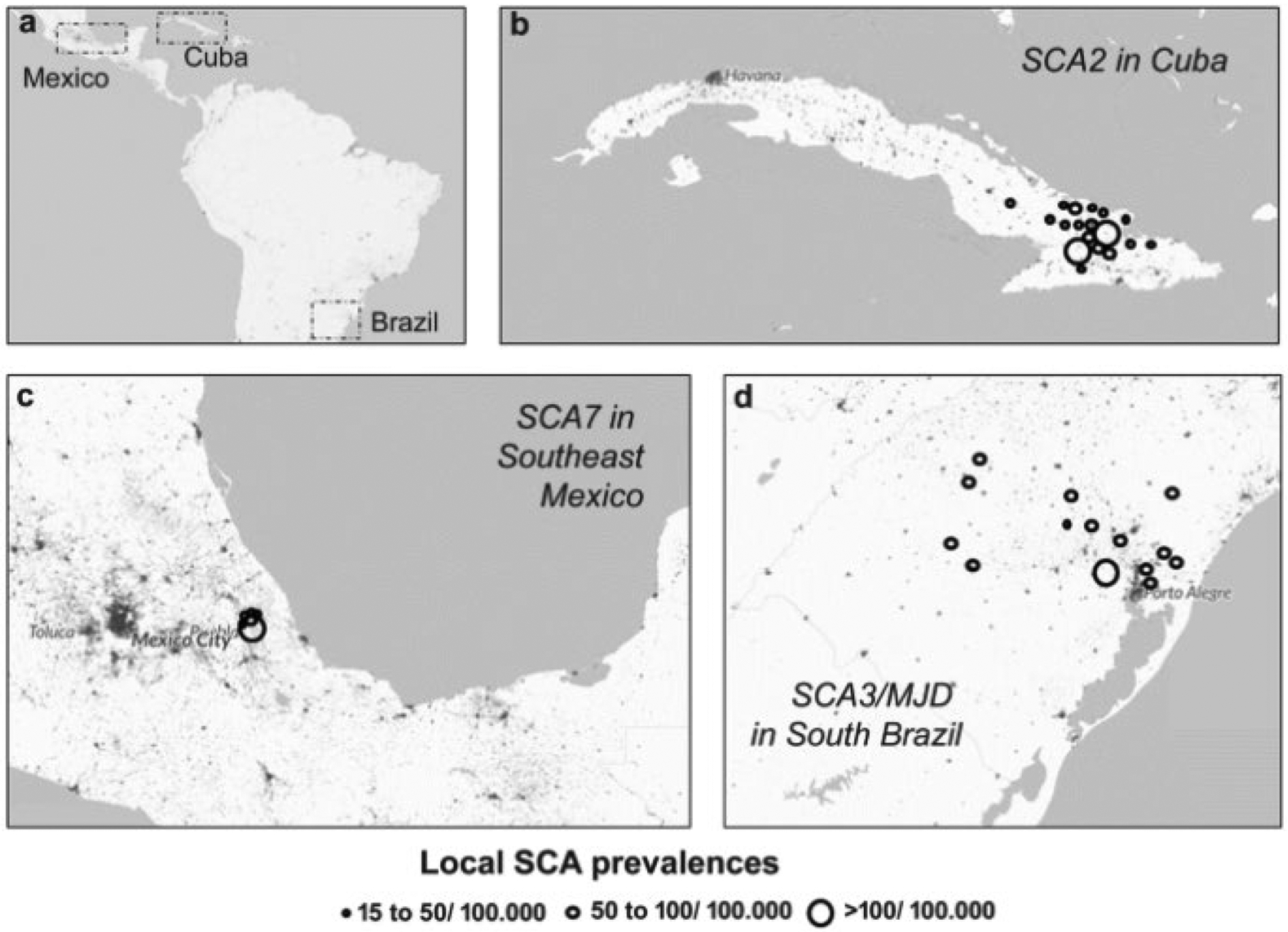
Prevalence rates of spinocerebellar ataxia clusters in Americas and the Caribbean. a The three main regions of interest in the American continents and the Caribbean. b Spinocerebellar ataxia type 2 (SCA2) in some Cuban municipalities. c Spinocerebellar ataxia type 7 (SCA7) in Southeast Mexico (Veracruz province). d Spinocerebellar ataxia type 3/Machado-Joseph disease (SCA3/MJD) in South Brazil (Rio Grande do Sul state). The background color represents population’s density. Adapted from Global Human Settlement Layer (GHSL), European Commission (http://luminocity3d.org/WorldPopDen/#6/-29.688/-61.282). Consulted in August, 2019
Evidence about the founder effect of SCA2 in Holguín was gained from molecular, clinic-genetic, and historiographical studies. Haplotype studies dating back from 1995 identified a common ancestral haplotype surrounding SCA2 mutation region that segregated with the disease in overall assessed pedigrees [30, 31]. A study assessing 132 chromosomes (43 expanded, 89 unexpanded) from 13 Cuban pedigrees found that 12 of them (92%) share the same haplotype [32].
Large normal alleles (LNAs) might contribute to generation of new expanded alleles in some polyQ SCAs [33]. A comprehensive study encompassing ~ 3.000 chromosomes from Cuban healthy controls revealed that the frequency of ataxin-2’ LNAs is higher in Cuba than in other six populations, which would be in agreement with a local founder effect. These alleles are highly polymorphic in the CAG sequence, characterized by the loss of the anchor CAA interruption(s) in the trinucleotide tract, under a predisposed haplotype. Moreover, Cuban LNAs show a prominent germ line instability, which provides important evidence about their role in the generation of pathological alleles in the Cuban population [34].
Regarding the origin of Cuban SCA2, historiographical and molecular data suggest a Spanish ancestry. The Cuban population has a well-documented story of Hispanic origin. Holguin was founded in 1545 and presented a slow socio-demographic development, characterized by the gradual arrival of Spanish and a high occurrence of endogamous and closed cycle marriages, which altogether with presumed restricted environmental unbalancing caused the rapid increase of SCA2 in Holguin. A Monte Carlo simulation suggested that the SCA2 founder mutation (or premutation) was probably introduced in Holguin at the first half of the seventeenth century. Haplotype studies comparing Cuban and Spanish DNA samples from SCA2 populations detected a high coincidence of haplotype markers between both populations, therefore supporting a common origin [32].
A distinctive characteristic of founder populations is the presence of clinical-genetic features that cannot be generalized to other populations. Indeed, several characteristics found in SCA2 Cuban subjects were not confirmed in other populations yet. The modifier effect of CACNA1A CAG repeats on the SCA2 age at onset was detected in Cuban [35], but not in other populations [36, 37]. The influence of the expanded CAG repeats in the ATXN2 gene on the age at onset is stronger in Cuba than other populations, suggesting a minor effect of modifier genetic and/or environmental factors on disease phenotype [8]. Cuban patients seem to have phenotypes more homogeneous than SCA2 subjects from other regions. The frequency of unusual movement disorders such as dystonia and chorea are lower in SCA2 Cuban patients than the European cohort. Clinical homogeneity of Cuban carriers is observed even during prodromal stage: Muscle cramps were referred by 80% of Cuban [20] and by 35% of European SCA2 preclinical carriers [38].
SCA3/MJD and South Brazil
SCA3/MJD is probably the most common polyQ SCA worldwide [16]. SCA3/MJD involves predominantly the cerebellar, pyramidal, extrapyramidal, motor neuron, and oculomotor systems and shows a mean age at onset (AO) between 34 and 40 years. An important correlation between AO and the repeat length is observed in SCA3/MJD, as in other polyQ disorders. The CAG repeat expansion in the ATXN3 gene (MIM: * 607047) ranges from 47 to 91 or more repeats, being larger than those tracts commonly seen in other polyQ SCAs. Clinical and molecular characteristics as well as management of SCA3/MJD have been recently reviewed elsewhere [39]. The close description of three different North American families carrying the disorder, all of them with Azorean ancestry, urged a survey that confirmed the existence of a geographical isolate of SCA3/MJD in Azores, an archipelago discovered by the Portuguese navigators in 1423 and colonized since then [16].
SCA3/MJD families living in Rio Grande do Sul (RS), the southernmost state of Brazil, began to be described in 2001 [40]. The epidemiological importance of this condition in this region became clear right after. In the present survey about living people from SCA3/MJD families, we detected 770 symptomatic and 1500 individuals at 50% risk in RS, an area inhabited by 11 million people, giving a prevalence of 7:100,000, a figure that updates former estimates [11]. The frequency reached 17 to 166:100,000 in some cities (Fig. 1D and Supplemental material 1). The number of people affected living close to the university center allowed several studies with different designs, including case-control, cross-sectional, longitudinal, and randomized clinical trial. Hence, local researchers and affected people contributed to boost knowledge about the disease natural history [41–44], modifier factors [45–48], biomarkers [49–51], and trial designs [52–54]. Of note was the observation that the CAG repeat expansion influences not only AO but also the progression rate of the disease [43]. Brazilian researchers have raised epidemiological evidence on CAG repeat expansion antagonistic pleiotropism – deleterious to neurons but advantageous to the rate of reproduction [55], to the carrier gametes [11], and to anti-tumor processes [56].
The history of RS colonization might explain this geographic “concentrate” of SCA3/MJD in this region. Until 1750, RS had almost no inhabitants: the few Amerindians who lived in this region were hunter-gatherers. Two to five thousand Azorean people were sent to RS by the Portuguese crown from 1750 to 1770 [57]. This effort of territory occupation initiated the entrance of Europeans in South Brazil. We presented the hypothesis that the SCA3/MJD cohort of RS had an Azorean ancestry a long time ago [10]. Whatever the real lineage, SCA3/MJD seems to be recent in RS and might not date more than 13 generations (260 years). Notably, RS does not have any characteristic that predisposes or facilitates the occurrence of population isolates. People can travel and emigrate easily.
Since 2001, informative ancestral haplotypes in SCA3/MJD have been built with three intragenic single nucleotide polymorphisms [58]. These variants are commonly described as the A669/G669 (A/G), C987/G987(C/G), and A1118/C1118 (A/C) polymorphisms. From all possible haplotype configurations linked to CAG repeat expansion, ACA (also known as Joseph) and GGC (also known as Machado) haplotypes were the most common worldwide. Remarkably, both were found in the Azorean archipelago. The widely distributed ACA haplotype seems to have an Asian origin dating back of about 9000 to 17,000 years ago [59, 60]. Although the inclusion of short tandem repeats (STR) might refine ATXN3 lineages, the ACA/ GGC polymorphisms remain as the minimally informative haplotype surrounding CAG repeat expansion. More recently, unpublished data were obtained on ancestral haplotypes of the SCA3/MJD lineages from RS. The ACA haplotype was present in 96% (178/185) of families from RS, while 85% of normal chromosomes carried GGC haplotype. Most importantly, 170 of 178 ACA SCA3/MJD families carried the extended haplotype (with STRs) 11–21-ACA-14–15 (ACA H1), suggesting that 92% (170/185) of SCA3/MJD pedigrees from RS might be closely related (ML. Saraiva-Pereira, personal communication). Since ACA H1 haplotype was also the most common among Azoreans [59], these results are in line with the hypothesis that a Joseph lineage that gave origin to the founder effect has an Azorean origin in RS.
As noted, founder effects produce more homogeneous populations than the original ones. Indeed, the recent forthcoming and homogeneity of the SCA3/MJD in RS may be related to two intriguing phenomena detected by recent observations. The CAG repeat expansion length in RS cohort – from 68 to 91 repeats – is shifted to the right, when compared to the 47 to 87 repeats range seen in other SCA3/MJD populations [61]. It remains to be established whether this distribution was due to the founders or to an increased rate of breeding as a way to occupy the “empty space” (demographic transition) in South Brazil. Contradictorily, age at onset as predicted by the CAG repeat expansion is later in RS than the expectation for Europeans. This last finding strongly supports the occurrence of protective factors in the SCA3/MJD population of RS [62]. Due to these findings, we believe that comparative studies looking for environmental and genetic modifiers, and including SCA3/MJD population from RS and from other regions, will have good chances of finding candidates for neuroprotection and prevention against the burden usually associated with this disease. Then, GWAS studies would be a powerful approach to identify genetic modifiers of the age at onset. However, it requires a large cohort of SCA3 patients from different parts of the world. The PAHAN may collaborate with the newly organized SCA Global to create a powerful platform to address this question.
SCA7 on Southeast Mexico
SCA7 is a polyQ disease characterized by cerebellar dysfunction, hyperreflexia, postural tremor, pigmentary retinal degeneration, and ophthalmoplegia [63–65]. The disease is caused by the progressive loss of neurons and glial cells within the cerebellum, retina, and brainstem [66]. Clinically, SCA7 was first described in 1937 [67] due to the severe retinopathy observed in most patients; SCA7 was originally considered as a separate entity from the autosomal dominantly inherited cerebellar ataxias (ADCA) [68]. The first evidence indicating that SCA7 might likely be caused by the expansion of unstable microsatellite was the observation of the anticipation phenomenon in affected families [69]. In 1995 three independent groups mapped the gene responsible for SCA7 (ATXN7; MIM: * 607047), which was localized in chromosome 3p21.1-p12 [70–72], and then the unstable CAG repeat region was identified [73]. In unaffected individuals the length of CAG repeats ranges from 4 to 18, while affected individuals have from 36 to up to 460 CAG repeats in the expanded chromosome [65]. SCA7 presents also a range of intermediate alleles (from 28 to 33 CAG repeats), as well as alleles with reduced penetrance (from 34 to 36 CAG repeats) [64, 65]. Although different molecular mechanisms appear to underlie SCA7, it is thought that the incorporation of an expanded polyglutamine (polyQ) tract confers a toxic gain-of-function on the mutant protein (ataxin-7), which in turn impairs multiple cellular processes, including transcriptional regulation, mitochondrial function, and oxidative stress, leading ultimately to cell dysfunction and death of target cells [74].
Worldwide prevalence of SCA7 is estimated in less than 1: 100,000 [75], representing 2% of all SCAs [76]. SCA7 was identified in families from various ethnic backgrounds, including kindreds from Europe (France, Belgium, Germany, and the UK), Africa (Algeria, Morocco, Libya, Tunisia, Zambia, and South Africa), Asia and Oceania (Israel, Korea, Philippines, and Australia), North America (USA and Mexico), South America (Brazil), and the Caribbean (Jamaica) [12, 64, 77–83]. SCA7 is the most prevalent SCA in certain countries, including Sweden, Finland, and Mexico [12, 13], and several founding populations have been reported in Scandinavia (Sweden and Finland) [82] and Africa (South Africa and Zambia) [84]. Recently, a Southeast Mexican population with high prevalence of SCA7 was identified, comprising five different communities distributed geographically in a small region of 1200 km2 [12, 13]. Currently, new cases have been identified in nearby communities, reaching in the central region of Veracruz prevalence higher than other regions of the world (Fig. 1C and Supplemental material 1). Interestingly all SCA7 carriers share a common informative haplotype A-254-82-98, corresponding to the intragenic (3145G/A) and centromeric (D3S1287, D3S1228, and D3S3635) genetic markers, respectively, which suggests a founder effect in this population [13]. Further genomic analysis allowed us to trace the ancestral mutation of SCA7 Mexican patients to Western European, specifically to the French and Spanish populations of Basque origin [13]. Consistent with this hypothesis, historic and anthropological findings documented two migratory events of French Basque descents from Eastern Pyrenees to Mexico during the eighteenth century, which could originate settlements in the central region of Veracruz. This geographic region is surrounded by a chain of mountains and hills, which could have undermined communication with other populations and limit further human migration, contributing then to spread the ancestral mutation only within the original settler descendants and promoting the founder effect [12].
The above-mentioned findings provide one of the largest series of patients with SCA7 worldwide, which has allowed to perform a comprehensive clinical, neuroimaging, and genetic characterization of the disease and to identify sensitive biomarkers. In fact, novel neuropathological, electrophysiological, and neuro-acoustical features of the disease have been described [65, 85–87]. The initial neuroimaging findings of patients show the extent of the white and gray matter neurodegeneration, including the cerebellum, middle, medial, and inferior frontal gyri, precuneus, parahippocampal cortex, inferior parietal lobe, and the lingual gyrus [88]. Additional analyses found correlations between clinical and cognitive impairment with degeneration of specific areas, like the correlation between auditory learning rate and the remaining of the parahippocampal cortex [89, 90]. Currently, a cerebellar neurodegeneration signature is being characterized for SCA7 and other SCAs [91].
Furthermore, in this population for the first time, the status of oxidative state and the expression profile of circulating microRNAs (miRNAs) in the plasma of patients has been reported, and a signature of four miRNAs was associated with disease severity [92, 93], which would be used in clinical studies to surveil the natural history of SCA7 and to monitor disease progression in patients undergoing clinical trials. Clinical approaches have also been undertaken in Mexican SCA7 patients. A recent study showed that a physical rehabilitation regime improved some cerebellar characteristics as well as the oxidative state of patients, which suggests that physical intervention could improve their general health condition [94]. Finally, the characterization of a Purkinje cell-based model for SCA7 is currently underway by local researchers. Overall these studies will help to better design therapeutic approaches aimed at ameliorating SCA7 symptomatology [95].
SCA10 and the Native American Populations
First two families of SCA10 reported in the literature were Mexican Americans with the phenotype characterized by cerebellar ataxia and epilepsy with autosomal dominant inheritance [96, 97]. Supplemented by several additional families with dominantly inherited ataxia and epilepsy in the Mexico
City area from where the two original SCA10 families immigrated [98], investigators used positional cloning to discover the pathogenic expansion of a pentanucleotide repeat in intron 9 of the E46L gene, now known as ATXN10 (MIM: * 611150) [99]. In the normal population, the number of repeat units ranges from 19 to 32. In large normal alleles (repeat units ≥ 17), ATTGT and TTTCT units often interrupt the ATTCT repeat [100]. SCA10 pathological expansions have 800 to 4500 pentanucleotide repeats [99]. The putative pathogenic mechanism is a gain of toxic function by the expanded intronic RNA repeat [99, 101, 102]. Subsequent genotyping of families with pure cerebellar ataxia from the Parana and Santa Catarina states in Southern Brazil led to the recognition of additional SCA10 families [103, 104]. Since then, SCA10 has been found in various Latin American countries, including Peru [105, 106], Bolivia [107], Argentina, [108], Venezuela [28], and Guatemala [109] (Fig. 2).
Fig. 2.
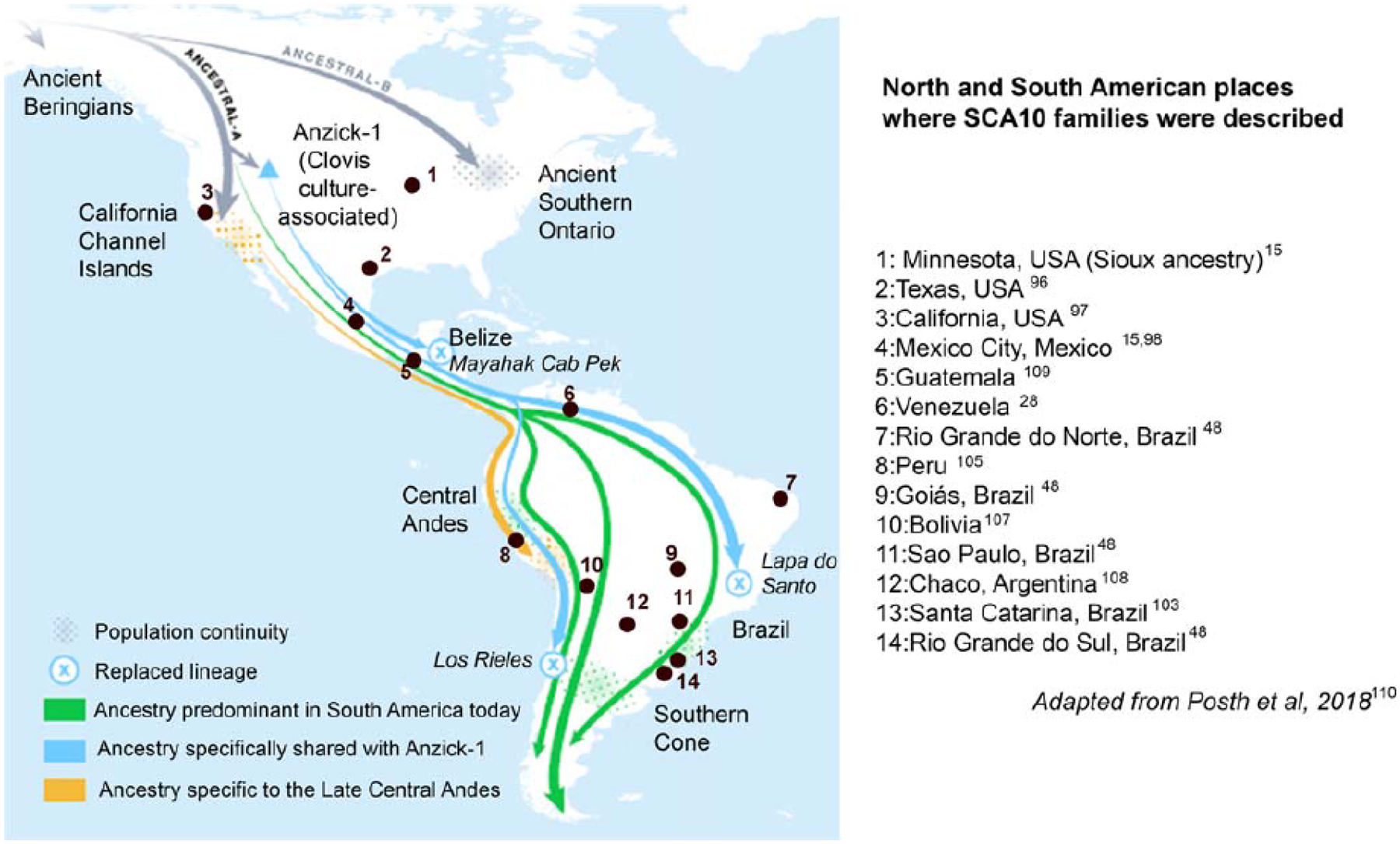
Occurrences of spinocerebellar ataxia type 10 (SCA10) in the American continents, according to the literature. Adapted from Posch et al. (2018) [110]
Recognizing the conspicuous absence of SCA10 in Europe, investigators looked into the Native American ancestry of patients with SCA10. All patients with SCA10 from Mexico and the Paraná state of Brazil had either an oral history or physical characteristics suggestive of Native American admixture, raising a hypothesis that the original SCA10 mutation occurred within a Native American population [104]. The report of SCA10 traced back to a Sioux individual in Minnesota with no Latin American connection endorsed the hypothesis that SCA10 is a genetic disease of Native Americans rather than that brought by European immigrants to American continents [15] (Fig. 2). To date all SCA10 patients share a common SNP haplotype, C-expansion-G-G-C, suggesting a single ancestral origin for SCA10 [14, 106]. Furthermore, little or null genetic distance in small normal alleles of different repeat sizes, from the same SNP lineage, indicates that they originated by a single-step mechanism [14].
SCA10 is a common SCA in Peru [111]. In Mexico SCA10 is one of the most common SCA after SCA2 [22]. SCA10 families in eastern Parana/Santa Catarina regions of Brazil appear to have a similar founder effect which may have resulted in the high prevalence of SCA10 second to SCA3, while the prevalence of SCA10 in other regions of Brazil appears to be lower [47, 112]. It is noteworthy that the frequency of epilepsy in this Brazilian SCA10 cohort is much lower than other regions [47], which could be explained by variations in the internal repeat sequence structure [113].
After negative searches for the SCA10 in China [114, 115], a single family of northern Chinese Han family was found to have SCA10 repeat expansions [116], followed by a discovery of a family from Western Japan with SCA10 [117]. These East Asian and American SCA10 families that have been tested all shared the same SCA10 haplotype, suggesting that the original SCA10 mutation may have occurred before the divergence of Proto-Amerinds from ancestral Asians. However, as the shared SCA10 haplotype is relatively common on both sides of the Pacific, independent expansion at two occasions across the Pacific could not be excluded. Individuals with the SCA10 expansion are rare in North America, while in South America, their distribution is widespread (TA, unpublished data). This is consistent with a major pattern of peopling Americas, i.e., a subgroup of East Asians went through the population bottleneck around the Bering land mass 15,000–20,000 years ago and then went through North and Central America and finally spread into South America [3, 4].
There has been only one neuropathological report on SCA10 brain of a Mexican American patient who had severe cerebellar ataxia and poorly controlled complex partial seizures with secondary generalization [118]. Despite the clinical severity, the neuronal loss was confined to cerebellar Purkinje cells. In contrast, a recent report of MRI findings of 18 patients with SCA10 showed degenerative changes in not only cerebellum and brainstem but also other parts of the brain, especially the putamen and thalamus whose volume loss is strongly associated with seizures [119]. The single-molecule real-time (SMRT) sequencing, which allows for obtaining long reads of continuous DNA sequence of SCA10 expansion alleles, led to the identification of three types of expansion: type A is mostly pure ATTCT repeats with periodic insertions of AT, TT, and CT dinucleotides; type B starts with a stretch of ATTCT repeat and then switches to ATTCC repeat; and type C consists of ATTCT repeat, ATCCT repeat, and then ATCCC repeat from 5′ to 3′ [120]. Limited genotype-phenotype correlations showed that type A is associated with late-onset ataxia or reduced penetrance, type B with ataxia and, in some patients, epilepsy, and type C always with ataxia and epilepsy. In some families, type A and type B expansions coexist in different siblings, and type A is associated with reduced penetrance or atypical phenotype [121]. However, the genotype-phenotype correlation is further confounded by the repeat length. Expansion alleles of 280 [99], 360, 370 [122], and 850 repeats [123] have shown reduced penetrance. The presence of ATCCT repeat, which resides between the ATTCT repeat and the ATCCC repeat in type C expansion, increases the probability of epilepsy by sixfold [113] and affects the pattern of clinical anticipation [124]. Further studies of long repeat sequence reads of SCA10 repeats would clarify the genotype-phenotype correlations and geographic disparities of clinical phenotype.
In summary, SCA10 is a SCA of Native Americans that may have risen in East Asia and migrated to Americas through Bering land mass 15,000 and 20,000 years ago. The repeat expansion motifs differ from family to family and may determine the penetrance and phenotypic characteristics.
Discussion
Founder events are usually determined through haplotype studies aimed at describing ancestral lineages of a given disease. The question whether SCAs that are due to an expanded microsatellite have multiple or few common ancestors is of interest, since the answer would clarify mutational mechanisms. The study of SCA clusters and geographical isolates, in contrast, is far less frequent in the literature. New World territories harbor a varied number of neurogenetic disease clusters, including SCAs. We reviewed here evidences concerning four SCA founders in the American continents and the Caribbean and discussed the characteristics of these populations. Moreover, we showed that the studies performed in these SCA populations have resulted in major scientific advances worldwide.
Disease cluster can be defined as an area and/or period of time where the prevalence of a particular disease exceeds expectations. Inherited disease clusters are due to a combination of two main vectors: a founder mutational event, plus isolation of geographical, cultural, religious, or even linguistic nature [125, 126]. The American continent was the latest piece of land to be occupied by Homo sapiens, and the occurrence of SCA10 in the American populations practically recapitulates mankind’s entrance and migration through these continents. This fact raises the important question about why SCA10 has been kept in the American populations for the last 10,000– 20,000 years. How does a trait associated with so much disability remain in populations? Even if the onset of symptoms occurs in the post-reproductive phase, the phenomenon is surprising, and the underlying mechanisms are unknown in SCA10. However, there are a couple of hypotheses. One is the de novo mutation which has been demonstrated in a handful of repeat expansion disorders, where alleles that contain a large normal-size repeat in the general population serve as a reservoir for the new full mutation (127). This phenomenon is based on microsatellite reconfigurations due to instabilities. Expanded microsatellites have variable penetrance and expressivity. According to this rationale, the expanded repeat could conceal itself in the general population after a contraction, to reappear phenotypically in a future generation. An alternative hypothesis is that an ancestral predisposition haplotype is present in the population, in which additional mutational events are necessary to trigger the disease. This may include a change in the repeat unit composition of expanded SCA10 allele affecting the penetrance. In fact, reduced penetrance has been found in SCA8 with such repeat sequence variations [128]. Whatever the reason SCA10 remains in Amerindian populations, this answer will have relevant consequences for the understanding of the pathophysiology of the disease – and therefore for its future clinical management – including treatment and prevention.
Much more recently, over the past five centuries, immigration waves from Europe and Africa reconfigured American populations again. These migratory waves brought with them new founder events. At the same time, American continents have a number of factors related to a high frequency of geographical and/or socioeconomic isolates. The high number (144) of potential clusters/isolates of inherited diseases described recently in Brazil can illustrate this phenomenon [129].
One of the most important clusters of inherited diseases related to post-Colombian migrations is the Huntington disease (HD; MIM: # 143100) isolate of Maracaibo, Venezuela. After the description by Americo Negrette, a research team from the USA-Venezuela Collaborative Research Project began making annual visits in 1981. The samples collected in Maracaibo led to the identification of HD locus in 1983 and gene identification 10 years later [130, 131]. Many HD features were discovered by the study of Venezuelan families, such as replication initiation regions at human trinucleotide repeat disease loci [132], soluble htt in the brain [133], and modifiers of the phenotype [134]. Therefore, the Maracaibo HD population is a very important example of how the study of an isolate/cluster can foster and speed up knowledge about a rare disease.
We reported here the LAC clusters of the polyQ SCA2, SCA3/MJD, and SCA7, all apparently related to European origins. The identification of these clusters may help to understand the selective forces at play in each of these conditions. The actual prevalence rates were determined in the reported geographical clusters. The current prevalence is also important because they will allow comparisons with estimates made in the future, for example, in 10 years time. Unstable repeats related to SCA2, SCA3/MJD, and SCA7 may contract or, more frequently, expand upon gamete generation. If expansions prevail, anticipation will produce a reduction in local SCA2, SCA3/MJD, and/or SCA7 prevalence rates. In contrast, SCA expanded repeats were also associated to positive selection forces such as the increased reproductive success of the carriers in SCA2 [135] and SCA3/MJD [55] and segregation distortion favoring the expanded allele in SCA3/MJD [11]. These phenomena can compensate for the effect of negative selection related to the disability, over the resultant prevalence of these disorders. On the other hand, haplotype studies help to clarify the “resilience” of a given mutation in time. SCA3/MJD has quite a few ancestral haplotypes. The most common is present worldwide and is 9,000 to 17,000 years old, favoring the hypothesis that the CAG repeat expansion at ATXN3 has some selective advantage [59, 60]. More studies on SCA2 and SCA7 haplotypes will brighten this theme in the near future [136, 137].
We also aimed to summarize the contributions that studies on SCA2 in Cuba, SCA3/MJD in Brazil, SCA7 in Mexico, and SCA10 in the Americas have brought to the scientific field. Of note, research in LAC has been mainly focused on one SCA at a time. One reason for this kind of approach was the detection of large number of affected carriers of only one SCA in certain regions. This urged the development of healthcare facilities near these populations and prompted the development of clinical research groups dedicated to them.
The study of genetic isolates has advantages and limitations. Characterization of a disease and evidence on efficacy of treatments usually requires a large number of study participants. In this sense, populations affected by SCA2, SCA3/MJD, and SCA7 in certain LAC regions will allow for suitable clinical trials in local institutions. However, the relatively homogeneous genetic and environmental backgrounds might produce some drawbacks. On one hand, they might allow a more controlled background, over which a study factor can outdo and be better investigated. On the other hand, local characteristics can prevent general conclusions to other SCA populations.
The future of SCA studies is promising in the American continents, especially with the creation of the Pan-American Hereditary Ataxia Network (PAHAN) in March 2019, in Havana. This network aims to bring together Latin American researchers interested in SCAs in order to stimulate collaboration and to build local consortiums for clinical and genetic studies. The present paper is the first scientific contribution of this network for increasing knowledge about SCAs, taking advantage of LAC potentials and characteristics.
In conclusion, clusters of SCA2, SCA3/MJD, and SCA7 and the almost exclusive occurrence of SCA10 in the American continents and the Caribbean present a unique opportunity for improving the understanding of clinical and genetic characteristics of these disorders. Moreover, we believe that the existence of large populations of carriers living close to study centers will allow researchers to perform worthwhile clinical trials and to speed up discoveries that will impact on therapies and on the quality of life of SCA carriers worldwide.
Supplementary Material
Acknowledgments
ACM, MLSP, and LBJ were supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). JJM, JFR, BC, and CMCZ were supported by CONACyT (grant no. 258043). JFR was also supported by CONACYT (grant no. A1-S-10669). LVP, RRL, YVM and JMM were supported by the Cuban Ministry of Public Health. TA, LBJ, MLS-P were supported by NIH NS115002, and TA and KNM were supported by NIH NS083564.
Footnotes
Electronic supplementary material: The online version of this article (https://doi.org/10.1007/s12311-020-01109-7) contains supplementary material, which is available to authorized users.
Conflict of Interest The authors declare no conflict of interest that could be perceived as prejudicial to the impartiality of the reported research.
References
- 1.Bittles AH. Consanguinity, genetic drift, and genetic diseases in populations with reduced numbers of founders. In: Speicher M, Antonarakis SE, Motulsky AG. (Eds.). Vogel and Motulsky’s human genetics. Problems and Approaches. Springer-Verlag Berlin Heidelberg, 2010, p. 507. [Google Scholar]
- 2.Reich D, Patterson N, Campbell D, Tandon A, Mazieres S, Ray N, et al. Reconstructing native American population history. Nature. 2012;488(7411):370–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Silva WA, Bonatto SL, Holanda AJ, Ribeiro-Dos-Santos AK, Paixao BM, et al. Mitochondrial genome diversity of Native Americans supports a single early entry of founder populations into America. Am J Hum Genet. 2002;71:187–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moreno-Mayar JV, Vinner L, de Barros Damgaard P, de la Fuente C, Chan J, et al. Early human dispersals within the Americas. Science. 2018;362(6419):eaav2621. [DOI] [PubMed] [Google Scholar]
- 5.Forsythe DP. Encyclopedia of Human Rights, Volume 4. Oxford University Press. 2009; p. 297. ISBN 978-0-19-533402-9. [Google Scholar]
- 6.Segal R The black diaspora: five centuries of the black experience outside Africa. New York: Farrar, Straus and Giroux; 1985. [Google Scholar]
- 7.Eltis D Economic growth and the ending of the transatlantic slave trade: Oxford University Press; 1987. [Google Scholar]
- 8.Velázquez-Pérez L, Cruz GS, Santos Falcon N, Enrique Almaguer Mederos L, Escalona Batallan K, Rodríguez Labrada R, et al. Molecular epidemiology of spinocerebellar ataxias in Cuba: insights into SCA2 founder effect in Holguin. Neurosci Lett. 2009;454(2):157–60. [DOI] [PubMed] [Google Scholar]
- 9.Velázquez-Pérez LC, Rodríguez-Labrada R, Fernandez-Ruiz J. Spinocerebellar Ataxia type 2: clinicogenetic aspects, mechanistic insights, and management approaches. Front Neurol. 2017;8:472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jardim LB, Silveira I, Pereira ML, Ferro A, Alonso I, Do Céu Moreira M, et al. A survey of spinocerebellar ataxia in South Brazil - 66 new cases with Machado-Joseph disease, SCA7, SCA8, or unidentified disease-causing mutations. J Neurol. 2001;248(10):870–6. [DOI] [PubMed] [Google Scholar]
- 11.Souza GN, Kersting N, Krum-Santos AC, Santos AS, Furtado GV, Pacheco D, et al. Spinocerebellar ataxia type 3/Machado-Joseph disease: segregation patterns and factors influencing instability of expanded CAG transmissions. Clin Genet. 2016;90(2):134–40. [DOI] [PubMed] [Google Scholar]
- 12.Magaña JJ, Gómez R, Maldonado-Rodríguez M, Velázquez-Pérez L, Tapia-Guerrero YS, Cortés H, et al. Origin of the spinocerebellar ataxia type 7 gene mutation in Mexican population. Cerebellum. 2013;12(6):902–5. [DOI] [PubMed] [Google Scholar]
- 13.Magaña JJ, Tapia-Guerrero YS, Velázquez-Pérez L, Cerecedo-Zapata CM, Maldonado-Rodríguez M, Jano-Ito JS, et al. Analysis of CAG repeats in five SCA loci in Mexican population: epidemiological evidence of a SCA7 founder effect. Clin Genet. 2014;85(2):159–65. [DOI] [PubMed] [Google Scholar]
- 14.Almeida T, Alonso I, Martins S, Ramos EM, Azevedo L, Ohno K, et al. Ancestral origin of the ATTCT repeat expansion in spinocerebellar ataxia type 10 (SCA10). PLoS One. 2009;4(2): e4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bushara K, Bower M, Liu J, McFarland KN, Landrian I, Hutter D, et al. Expansion of the Spinocerebellar ataxia type 10 (SCA10) repeat in a patient with Sioux Native American ancestry. PLoS One. 2013;8(11):e81342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sequeiros J, Martins S, Silveira I. Epidemiology and population genetics of degenerative ataxias. Handb Clin Neurol. 2012;103: 227–51. [DOI] [PubMed] [Google Scholar]
- 17.Pulst MS, Nechiporuk A, Nechiporuk T, Gispert S, Chen XN, Lopes-Cendes I, et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet. 1996;14:269–76. [DOI] [PubMed] [Google Scholar]
- 18.Ross O, Rutherford N, Baker M, Soto-Ortolaza AI, Carrasquillo MM, DeJesus-Hernandez M, et al. Ataxin-2 repeat-length variation and neurodegeneration. Hum Mol Genet. 2011;20:3207–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sequeiros J, Seneca S, Martindale J. Consensus and controversies in best practices for molecular genetic testing of spinocerebellar ataxias. Eur J Hum Genet. 2010;18(11):1188–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Velázquez-Pérez L, Rodríguez-Labrada R, Canales-Ochoa N, Medrano-Montero J, Sanchez-Cruz G, Aguilera-Rodriguez R, et al. Progression of early features of spinocerebellar ataxia type 2 in individuals at risk: a longitudinal study. Lancet Neurol. 2014;13(5):482–9. [DOI] [PubMed] [Google Scholar]
- 21.Durr A Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 2010;9(9):885–94. [DOI] [PubMed] [Google Scholar]
- 22.Auburger G, Diaz GO, Capote RF, Sanchez SG, Pérez MP, del Cueto ME, et al. Autosomal dominant ataxia: genetic evidence for locus heterogeneity from a Cuban founder-effect population. Am J Hum Genet. 1990;46(6):1163–77. [PMC free article] [PubMed] [Google Scholar]
- 23.González-Zaldívar Y, Vázquez-Mojena Y, Laffita-Mesa JM, Almaguer-Mederos LE, Rodríguez-Labrada R, Sánchez-Cruz G, et al. Epidemiological, clinical, and molecular characterization of Cuban families with spinocerebellar ataxia type 3/Machado-Joseph disease. Cerebellum Ataxias. 2015;2:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alonso E, Martínez-Ruano L, De Biase I, Mader C, Ochoa A, Yescas P, et al. Distinct distribution of autosomal dominant spinocerebellar ataxia in the Mexican population. Mov Disord. 2007;22(7):1050–3. [DOI] [PubMed] [Google Scholar]
- 25.Bryer A, Krause A, Bill P, Davids V, Bryant D, Butler J, et al. The hereditary adult-onset ataxias in South Africa. J Neurol Sci. 2003;216:47–54. [DOI] [PubMed] [Google Scholar]
- 26.Faruq M, Scaria V, Singh I, Tyagi S, Srivastava AK, Mukerji M. SCA-LSVD: a repeat-oriented locus-specific variation database for genotype to phenotype correlations in spinocerebellar ataxias. Hum Mutat. 2009;30:1037–42. [DOI] [PubMed] [Google Scholar]
- 27.Brusco A, Gellera C, Cagnoli C, Saluto A, Castucci A, Michielotto C, et al. Molecular genetics of hereditary spinocerebellar ataxia: mutation analysis of spinocerebellar ataxia genes and CAG/CTG repeat expansion detection in 225 Italian families. Arch Neurol. 2004;61:727–33. [DOI] [PubMed] [Google Scholar]
- 28.Paradisi I, Ikonomu V, Arias S. Spinocerebellar ataxias in Venezuela: genetic epidemiology and their most likely ethnic descent. J Hum Genet. 2016;61(3):215–22. [DOI] [PubMed] [Google Scholar]
- 29.Vallés L, Estrada GL, Bastecherrea SL. Algunas formas de heredoataxia en una región de Cuba. Rev Neurol (Cubana). 1978;27:163–76. [Google Scholar]
- 30.Hernandez A, Magarino C, Gispert S, Santos N, Lunkes A, Orozco G, et al. Genetic mapping of the spinocerebellar ataxia 2 (SCA2) locus on chromosome 12q23-q24.1. Genomics. 1995;25: 433–5. [DOI] [PubMed] [Google Scholar]
- 31.Allotey R, Twells R, Cemal C, Norte BS, Weissenbach J, Pook M, et al. The spinocerebellar ataxia 2 locus is located within a 3-cM interval on chromosome 12q23–24.1. Am J Hum Genet. 1995;57:185–9. [PMC free article] [PubMed] [Google Scholar]
- 32.Laffita-Mesa J. Genetics and molecular investigations on SCA2: from genetic predisposition to genetic and epigenetic modifying mechanisms acting in a very frequent disease in Holguín. In: Velazquez-Pérez L, editor. III International Symposium of Hereditary Ataxias. Cuba: Holguín; 2008. [Google Scholar]
- 33.Takano H, Cancel G, Ikeuchi T, Lorenzetti D, Mawad R, Stevanin G, et al. Close associations between prevalences of dominantly inherited spinocerebellar ataxias with CAG-repeat expansions and frequencies of large normal CAG alleles in Japanese and Caucasian populations. Am J Hum Genet. 1998;63:1060–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laffita-Mesa JM, Velázquez-Pérez LC, Santos Falcón N, Cruz-Mariño T, González Zaldívar Y, Vázquez Mojena Y, et al. Unexpanded and intermediate CAG polymorphisms at the SCA2 locus (ATXN2) in the Cuban population: evidence about the origin of expanded SCA2 alleles. Eur J Hum Genet. 2012;20(1):41–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pulst S, Santos N, Wang D, Yang H, Huynh D, Velázquez L, et al. Spinocerebellar ataxia type 2: polyQ repeat variation in the CACNA1A calcium channel modifies age of onset. Brain. 2005;128(Pt 10):2297–303. [DOI] [PubMed] [Google Scholar]
- 36.Tezenas du Montcel S, Durr A, Bauer P, Figueroa KP, Ichikawa Y, Brussino A, et al. Modulation of the age at onset in spinocerebellar ataxia by CAG tracts in various genes. Brain. 2014;137:2444–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pereira FS, Monte TL, Locks-Coelho LD, Silva AS, Barsottini O, Pedroso JL, et al. ATXN3, ATXN7, CACNA1A, and RAI1 genes and mitochondrial polymorphism A10398G did not modify age at onset in spinocerebellar Ataxia type 2 patients from South America. Cerebellum. 2015;14(6):728–30. [DOI] [PubMed] [Google Scholar]
- 38.Jacobi H, Reetz K, du Montcel ST, Bauer P, Mariotti C, Nanetti L, et al. Biological and clinical characteristics of individuals at risk for spinocerebellar ataxia types 1, 2, 3, and 6 in the longitudinal RISCA study: analysis of baseline data. Lancet Neurol. 2013;12(7):650–8. [DOI] [PubMed] [Google Scholar]
- 39.Saute AM, Jardim LB. Machado–Joseph disease: clinical and genetic aspects, and current treatment. Expert, Opin, Orphan. 2015;3:517–35. [Google Scholar]
- 40.Jardim LB, Pereira ML, Silveira I, Ferro A, Sequeiros J, Giugliani R. Machado–Joseph disease in South Brazil: clinical and molecular characterization of kindreds. Acta Neurol Scand. 2001;104: 224–31. [DOI] [PubMed] [Google Scholar]
- 41.Jardim LB, Pereira ML, Silveira I, Ferro A, Sequeiros J, Giugliani R. Neurologic findings in Machado-Joseph disease: relation with disease duration, subtypes, and (CAG)n. Arch Neurol. 2001;58(6):899–904. [DOI] [PubMed] [Google Scholar]
- 42.Kieling C, Prestes PR, Saraiva-Pereira ML, Jardim LB. Survival estimates for patients with Machado-Joseph disease (SCA3). Clin Genet. 2007;72(6):543–5. [DOI] [PubMed] [Google Scholar]
- 43.Jardim LB, Hauser L, Kieling C, Saute JA, Xavier R, Rieder CR, et al. Progression rate of neurological deficits in a 10-year cohort of SCA3 patients. Cerebellum. 2007;9(3):419–28. [DOI] [PubMed] [Google Scholar]
- 44.Donis KC, Saute JA, Krum-Santos AC, Furtado GV, Mattos EP, Saraiva-Pereira ML, et al. Spinocerebellar ataxia type 3/Machado-Joseph disease starting before adolescence. Neurogenetics. 2016;17(2):107–13. [DOI] [PubMed] [Google Scholar]
- 45.Jardim L, Silveira I, Pereira ML, Do Céu Moreira M, Mendonça P, Sequeiros J, et al. Searching for modulating effects of SCA2, SCA6 and DRPLA CAG tracts on the Machado-Joseph disease (SCA3) phenotype. Acta Neurol Scand. 2003;107(3):211–4. [DOI] [PubMed] [Google Scholar]
- 46.Emmel VE, Alonso I, Jardim LB, Saraiva-Pereira ML, Sequeiros J. Does DNA methylation in the promoter region of the ATXN3 gene modify age at onset in MJD (SCA3) patients? Clin Genet. 2011;79(1):100–2. [DOI] [PubMed] [Google Scholar]
- 47.Siebert M, Donis KC, Socal M, Rieder CR, Emmel VE, Vairo F, et al. Glucocerebrosidase gene variants in parkinsonian patients with Machado Joseph/spinocerebellar ataxia 3. Parkinsonism Relat Disord. 2012;18(2):185–90. [DOI] [PubMed] [Google Scholar]
- 48.de Castilhos RM, Furtado GV, Gheno TC, Schaeffer P, Russo A, Barsottini O, et al. Spinocerebellar ataxias in Brazil–frequencies and modulating effects of related genes. Cerebellum. 2014;13(1): 17–28. [DOI] [PubMed] [Google Scholar]
- 49.Tort AB, Portela LV, Rockenbach IC, Monte TL, Pereira ML, Souza DO, et al. S100B and NSE serum concentrations in Machado Joseph disease. Clin Chim Acta. 2005;351(1–2):143–8. [DOI] [PubMed] [Google Scholar]
- 50.da Silva Carvalho G, Saute JA, Haas CB, Torrez VR, Brochier AW, Souza GN, et al. Cytokines in Machado Joseph disease/ spinocerebellar ataxia 3. Cerebellum. 2016;15(4):518–25. [DOI] [PubMed] [Google Scholar]
- 51.de Assis AM, Saute JAM, Longoni A, Haas CB, Torrez VR, Brochier AW, et al. Peripheral oxidative stress biomarkers in spinocerebellar ataxia type 3/Machado-Joseph disease. Front Neurol. 2017;8:485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saute JA, de Castilhos RM, Monte TL, Schumacher-Schuh AF, Donis KC, D’Ávila R, et al. A randomized, phase 2 clinical trial of lithium carbonate in Machado-Joseph disease. Mov Disord. 2014;29(4):568–73. [DOI] [PubMed] [Google Scholar]
- 53.Saute JA, Rieder CR, Castilhos RM, Monte TL, Schumacher-Schuh AF, Donis KC, et al. Planning future clinical trials in Machado Joseph disease: lessons from a phase 2 trial. J Neurol Sci. 2015;358(1–2):72–6. [DOI] [PubMed] [Google Scholar]
- 54.Saute JAM, Jardim LB. Planning future clinical trials for Machado-Joseph disease. Adv Exp Med Biol. 2018;1049:321–48. [DOI] [PubMed] [Google Scholar]
- 55.Prestes PR, Saraiva-Pereira ML, Silveira I, Sequeiros J, Jardim LB. Machado-Joseph disease enhances genetic fitness: a comparison between affected and unaffected women and between MJD and the general population. Ann Hum Genet. 2008;72(Pt 1):57–64. [DOI] [PubMed] [Google Scholar]
- 56.Souza GN, Kersting N, Gonçalves TA, Pacheco DLO, Saraiva-Pereira ML, Camey SA, et al. Cancer in Machado-Joseph disease patients - low frequency as a cause of death. Cancer Gene Ther. 2017;212–213:19–23. [DOI] [PubMed] [Google Scholar]
- 57.Piazza WF. A epopéia açórico-madeirense 1748–1756. Florianopolis: Editora da UFSC, 1992. [Google Scholar]
- 58.Gaspar C, Lopes-Cendes I, Hayes S, Goto J, Arvidsson K, Dias A, et al. Ancestral origins of the Machado-Joseph disease mutation: a worldwide haplotype study. Am J Hum Genet. 2001;68(2):523–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martins S, Calafell F, Gaspar C, Wong VC, Silveira I, Nicholson GA, et al. Asian origin for the worldwide-spread mutational event in Machado-Joseph disease. Arch Neurol. 2007;64(10):1502–8. [DOI] [PubMed] [Google Scholar]
- 60.Li T, Martins S, Peng Y, Wang P, Hou X, Chen Z, et al. Is the high frequency of Machado-Joseph disease in China due to new mutational origins? Front Genet. 2019;9:740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.de Mattos EP, Leotti VB, Soong BW, Raposo M, Lima M, Vasconcelos J, et al. Age at onset prediction in spinocerebellar ataxia type 3 changes according to population of origin. Eur J Neurol. 2019;26(1):113–20. [DOI] [PubMed] [Google Scholar]
- 62.de Mattos EP, Kolbe Musskopf M, Bielefeldt Leotti V, Saraiva-Pereira ML, Jardim LB. Genetic risk factors for modulation of age at onset in Machado-Joseph disease/spinocerebellar ataxia type 3: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2019;90(2):203–10. [DOI] [PubMed] [Google Scholar]
- 63.David G, Durr A, Stevanin G, Cancel G, Abbas N, Benomar A, et al. Molecular and clinical correlations in autosomal dominant cerebellar ataxia with progressive macular dystrophy (SCA7). Hum Mol Genet. 1998;7:165–70. [DOI] [PubMed] [Google Scholar]
- 64.Michalik A, Martin JJ, Van Broeckhoven C. Spinocerebellar ataxia type 7 associated with pigmentary retinal dystrophy. Eur J Hum Genet. 2004;12(1):2–15. [DOI] [PubMed] [Google Scholar]
- 65.Velázquez-Pérez L, Cerecedo-Zapata CM, Hernandez-Hernandez O, Martinez-Cruz E, Tapia-Guerrero YS, Gonzalez-Pina R, et al. A comprehensive clinical and genetic study of a large Mexican population with spinocerebellar ataxia type 7. Neurogenetics. 2015;16(1):11–21. [DOI] [PubMed] [Google Scholar]
- 66.Garden GA, La Spada AR. Molecular pathogenesis and cellular pathology of spinocerebellar ataxia type 7 neurodegeneration. Cerebellum. 2008;7(2):138–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Froment J, Bonnet P, Colrat A. Heredo-degenerations retinienne et spino-cerebelleuse: variantes ophtalmoscopiques et neurologiques presentees par trois generations successives. J Med Lyon. 1937;1937:153–63. [Google Scholar]
- 68.Harding AE. Clinical features and classification of inherited ataxias. Adv Neurol. 1993;61:1–14. [PubMed] [Google Scholar]
- 69.Trottier Y, Lutz Y, Stevanin G, Imbert G, Devys D, Cancel G, et al. Polyglutamine expansion as a pathological epitope in Huntington’s disease and four dominant cerebellar ataxias. Nature. 1995;378:403–6. [DOI] [PubMed] [Google Scholar]
- 70.Benomar A, Krols L, Stevanin G, Cancel G, Le Guern E, David G, et al. The gene for autosomal dominant cerebellar ataxia with pigmentary macular dystrophy maps to chromosome 3p12–p21.1. Nat Genet. 1995;10:84–8. [DOI] [PubMed] [Google Scholar]
- 71.Gouw LG, Kaplan CD, Haines JH, Digre KB, Rutledge SL, Matilla A, et al. Retinal degeneration characterizes a spinocerebellar ataxia mapping to 147chromosome 3p. Nat Genet. 1995;10:89–93. [DOI] [PubMed] [Google Scholar]
- 72.Holmberg M, Johansson J, Forsgren L, Heijbel J, Sandgren O, Holmgren G. Localization of autosomal dominant cerebellar ataxia associated with retinal degeneration and anticipation to chromosome 3p12–p21.1. Hum Mol Genet. 1995;4:1441–5. [DOI] [PubMed] [Google Scholar]
- 73.David G, Abbas N, Stevanin G, Durr A, Yvert G, Cancel G, et al. Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet. 1997;17:65–70. [DOI] [PubMed] [Google Scholar]
- 74.Karam A, Trottier Y. Molecular mechanisms and therapeutic strategies in spinocerebellar ataxia type 7. Adv Exp Med Biol. 2018;1049:197–218. [DOI] [PubMed] [Google Scholar]
- 75.Moseley ML, Benzow KA, Shut LJ, Bird TD, Gomez CM, Barkhaus PE, et al. Incidence of dominant spinocerebellar and Friedreich triplet repeats among 361 families. Neurology. 1998;51(6):1666–71. [DOI] [PubMed] [Google Scholar]
- 76.Storey E, du Sart D, Shaw JH, Lorentzos P, Kelly L, McKinley Gardner RJ. Frequency of spinocerebellar ataxia types 1, 2, 3, 6, and 7 in Australian patients with spinocerebellar ataxia. Am J Med Genet. 2000;95:351–7. [DOI] [PubMed] [Google Scholar]
- 77.Stevanin G, David G, Durr A, Giunti P, Benomar A, Abada-Bendib M, et al. Multiple origins of the spinocerebellar ataxia7 (SCA7) mutation revealed by linkage disequilibrium studies with closely flanking markers, including an intragenic polymorphism (G3145TG/A3145TG). Eur J Hum Genet. 1999;7:889–96. [DOI] [PubMed] [Google Scholar]
- 78.Atadzhanov M, Smith DC, Mwaba MH, Siddiqi OK, Bryer A, Greenberg LJ. Clinical and genetic analysis of spinocerebellar ataxia type 7 (SCA7) in Zambian families. Cerebellum Ataxias. 2017;4:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Azevedo PB, Rocha AG, Keim LMN, Lavinsky D, Furtado GV, De Mattos EP, et al. Ophthalmological and neurologic manifestations in pre-clinical and clinical phases of spinocerebellar ataxia type 7. Cerebellum. 2019;18(3):388–96. [DOI] [PubMed] [Google Scholar]
- 80.Watson L, Smith DC, Scholefield J, Ballo R, Kidson S, Greenberg LJ, et al. Spinocerebellar ataxia type 7 in South Africa: epidemiology, pathogenesis and therapy. S Afr Med J. 2016;106:S107–9. [DOI] [PubMed] [Google Scholar]
- 81.Kim JY, Park SS, Joo SI, Kim JM, Jeon BS. Molecular analysis of spinocerebellar ataxias in Koreans: frequencies and reference ranges of SCA1, SCA2, SCA3, SCA6 and SCA7. Mol Cell. 2001;12(3):336–41. [PubMed] [Google Scholar]
- 82.Jonasson J, Juvonen V, Sistonen P, Ignatius J, Johansson D, Bjorck EJ. Evidence for a common spinocerebellar ataxia type 7 (SCA7) founder mutation in Scandinavia. Eur J Hum Genet. 2000;8:918–22. [DOI] [PubMed] [Google Scholar]
- 83.Smith DC, Atadzhanov M, Mwaba M, Greenberj LJ. Evidence of common founder effect amongst South Africa and Zambian individuals with spinocerebellar ataxia type 7. J Neurol Sci. 2015;354(1–2):75–8. [DOI] [PubMed] [Google Scholar]
- 84.Demard JC. Émigration française au Mexique: 1, Les communautÉs agricoles (1828–1900). 1st edn. Dominique GuÉniot, Éditeur. Langres; 1995. p. 91. [Google Scholar]
- 85.Magaña JJ, Tapia-Guerrero YS, Velázquez-Pérez L, Cruz-Mariño T, Cerecedo-Zapata CM, Gómez R, et al. Clinical and molecular effect on offspring of a marriage of consanguineous spinocerebellar ataxia type 7 mutation carriers: a family case report. Int J Clin Exp Med. 2014;7(12):5896–903. [PMC free article] [PubMed] [Google Scholar]
- 86.Hernández-Castillo CR, Alcauter S, Galvez V, Barrios FA, Barrios FA, Yescas P, et al. Disruption of visual and motor connectivity in spinocerebellar ataxia type 7. Mov Disord. 2013;28(12):1708–16. [DOI] [PubMed] [Google Scholar]
- 87.Gomez-Coello A, Valadez-Jimenez VM, Cisneros B, Carrillo-Mora P, Parra-Cardenas M, Hernandez-Hernandez O, et al. Voice alterations in patients with spinocerebellar ataxia type 7 (SCA7): clinical-genetic correlations. J Voice. 2017;31(1):123: e1–5. [DOI] [PubMed] [Google Scholar]
- 88.Hernandez-Castillo CR, Vaca-Palomares I, Barrios F, Martinez L, Boll MC, Fernandez-Ruiz J. Ataxia severity correlates with White matter degeneration in spinocerebellar ataxia type 7. AJNR Am J Neuroradiol. 2016;37(11):2050–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hernandez-Castillo CR, Galvez V, Diaz R, Fernandez-Ruiz J. Specific cerebellar and cortical degeneration correlates with ataxia severity in spinocerebellar ataxia type 7. Brain Imaging Behav. 2016;10(1):252–7. [DOI] [PubMed] [Google Scholar]
- 90.Chirino A, Hernandez-Castillo CR, Galvez V, Contreras A, Diaz R, Beltran-Parrazal L, et al. Motor and cognitive impairments in spinocerebellar ataxia type 7 and its correlations with cortical volumes. Eur J Neurosci. 2018;48(10):3199–211. [DOI] [PubMed] [Google Scholar]
- 91.Hernandez-Castillo CR, King M, Diedrichsen J, Fernandez-Ruiz J. Unique degeneration signatures in the cerebellar cortex for spinocerebellar ataxias 2, 3, and 7. NeuroImage Clin. 2018;20: 931–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Torres-Ramos Y, Montoya-Estrada A, Cisneros B, Tercero-Pérez K, León-Reyes G, Leyva-García N, et al. Oxidative stress in spinocerebellar ataxia type 7 is associated with disease severity. Cerebellum. 2018;17(5):601–9. [DOI] [PubMed] [Google Scholar]
- 93.Borgonio-Cuadra VM, Valdes-Vargas C, Romero-Córdoba S, Hiodalgo-Miranda A, Tapia-Guerrero Y, Cerecedo-Zapata CM, et al. Wide profiling of circulating MicroRNAs in spinocerebellar ataxia type 7. Mol Neurobiol. 2019;56(9):6106–20. [DOI] [PubMed] [Google Scholar]
- 94.Tercero-Pérez K, Cortés H, Torres-Ramos Y, Rodríguez-Labrada R, Cerecedo-Zapata CM, Hernández-Hernández O, et al. Effects of physical rehabilitation in patients with spinocerebellar ataxia type 7. Cerebellum. 2019;18(3):397–405. [DOI] [PubMed] [Google Scholar]
- 95.Escalona-Rayo O, Fuentes-Vázquez P, Leyva-Gómez G, Cisneros B, Villalobos R, Magaña JJ, et al. Nanoparticulate strategies for the treatment of polyglutamine diseases by halting the protein aggregation process. Drug Dev Ind Pharm. 2017;43(6):871–88. [DOI] [PubMed] [Google Scholar]
- 96.Matsuura T, Achari M, Khajavi M, Bachinski LL, Zoghbi HY, Ashizawa T. Mapping of the gene for a novel spinocerebellar ataxia with pure cerebellar signs and epilepsy. Ann Neurol. 1999;45(3):407–11. [DOI] [PubMed] [Google Scholar]
- 97.Zu L, Figueroa KP, Grewal R, Pulst SM. Mapping of a new autosomal dominant spinocerebellar ataxia to chromosome 22. Am J Hum Genet. 1999;64(2):594–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rasmussen A, Matsuura T, Ruano L, Yescas P, Ochoa A, Ashizawa T, et al. Clinical and genetic analysis of four Mexican families with spinocerebellar ataxia type 10. Ann Neurol. 2001;50(2):234–9. [DOI] [PubMed] [Google Scholar]
- 99.Matsuura T, Yamagata T, Burgess DL, Rasmussen A, Grewal RP, Watase K, et al. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat Genet. 2000;26(2): 191–4. [DOI] [PubMed] [Google Scholar]
- 100.Matsuura T, Fang P, Pearson CE, Jayakar P, Ashizawa T, Roa BB, et al. Interruptions in the expanded ATTCT repeat of spinocerebellar ataxia type 10: repeat purity as a disease modifier? Am J Hum Genet. 2006;78(1):125–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wakamiya M, Matsuura T, Liu Y, Schuster GC, Gao R, Xu W. The role of ataxin 10 in the pathogenesis of spinocerebellar ataxia type 10. Neurology. 2006;67(4):607–13. [DOI] [PubMed] [Google Scholar]
- 102.White M, Xia G, Gao R, Wakamiya M, Sarkar PS, McFarland K, et al. Transgenic mice with SCA10 pentanucleotide repeats show motor phenotype and susceptibility to seizure: a toxic RNA gain-of-function model. J Neurosci Res. 2012;90(3):706–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Teive HA, Roa BB, Raskin S, Fang P, Arruda WO, Neto YC. Clinical phenotype of Brazilian families with spinocerebellar ataxia 10. Neurology. 2004;63(8):1509–12. [DOI] [PubMed] [Google Scholar]
- 104.Teive HA, Moro A, Moscovich M, Arruda WO, Munhoz RP, Raskin S, et al. Spinocerebellar ataxia type 10 in the south of Brazil: the Amerindian-Belgian connection. Arq Neuropsiquiatr. 2015;73(8):725–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Leonardi L, Marcotulli C, McFarland KN, Tessa A, DiFabio R, Santorelli FM, et al. Spinocerebellar ataxia type 10 in Peru: the missing link in the Amerindian origin of the disease. J Neurol. 2014;261(9):1691–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bampi GB, Bisso-Machado R, Hünemeier T, Gheno TC, Furtado GV, Veliz-Otani D, et al. Haplotype study in SCA10 families provides further evidence for a common ancestral origin of the mutation. NeuroMolecular Med. 2017;19(4):501–9. [DOI] [PubMed] [Google Scholar]
- 107.Baizabal-Carvallo JF, Xia G, Botros P, Laguna J, Ashizawa T, Jankovic J. Bolivian kindred with combined spinocerebellar ataxia types 2 and 10. Acta Neurol Scand. 2015;132(2):139–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gatto EM, Gao R, White MC, Uribe Roca MC, Etcheverry JL, Persi G, et al. Ethnic origin and extrapyramidal signs in an Argentinean spinocerebellar ataxia type 10 family. Neurology. 2007;69(2):216–8. [DOI] [PubMed] [Google Scholar]
- 109.Trikamji B, Singh P, Mishra S. Spinocerebellar ataxia-10 with paranoid schizophrenia. Ann Indian Acad Neurol. 2015;18(1):93–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Posth C, Nakatsuka N, Lazaridis I, Skoglund P, Mallick S, Lamnidis TC, et al. Reconstructing the deep population history of Central and South America. Cell. 2018;175(5):1185–1197.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Guio H, Poterico JA, Levano KS, Cornejo-Olivas M, Mazzetti P, Manassero-Morales G, et al. Genetics and genomics in Peru: clinical and research perspective. Mol Genet Genomic Med. 2018;6(6):873–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cintra VP, Lourenço CM, Marques SE, de Oliveira LM, Tumas V, Marques W Jr. Mutational screening of 320 Brazilian patients with autosomal dominant spinocerebellar ataxia. J Neurol Sci. 2014;347(1–2):375–9. [DOI] [PubMed] [Google Scholar]
- 113.McFarland KN, Liu J, Landrian I, Zeng D, Raskin S, Moscovich M, et al. Repeat interruptions in spinocerebellar ataxia type 10 expansions are strongly associated with epileptic seizures. Neurogenetics. 2014;15(1):59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang J, Shen L, Lei L, Xu Q, Zhou J, Liu Y, et al. Spinocerebellar ataxias in mainland China: an updated genetic analysis among a large cohort of familial and sporadic cases. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2011;36(6):482–9. [DOI] [PubMed] [Google Scholar]
- 115.Jiang H, Tang BS, Xu B, Zhao GH, Shen L, Tang JG, et al. Frequency analysis of autosomal dominant spinocerebellar ataxias in mainland Chinese patients and clinical and molecular characterization of spinocerebellar ataxia type 6. Chin Med J. 2005;118(10):837–43. [PubMed] [Google Scholar]
- 116.Wang K, McFarland KN, Liu J, Zeng D, Landrian I, Xia G, et al. Spinocerebellar ataxia type 10 in Chinese Han. Neurol Genet. 2015;1(3):e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Naito H, Takahashi T, Kamada M, Morino H, Yoshino H, Hattori N, et al. First report of a Japanese family with spinocerebellar ataxia type 10: the second report from Asia after a report from China. PLoS One. 2017;12(5):e0177955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Xia G, McFarland KN, Wang K, Sarkar PS, Yachnis AT, Ashizawa T. Purkinje cell loss is the major brain pathology of spinocerebellar ataxia type 10. J Neurol Neurosurg Psychiatry. 2013;84(12):1409–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hernandez-Castillo CR, Diaz R, Vaca-Palomares I, Torres DL, Chirino A, Campos-Romo A, et al. Extensive cerebellar and thalamic degeneration in spinocerebellar ataxia type 10. Parkinsonism Relat Disord. 2019. 10.1016/j.parkreldis.2019.08.011inpress. [DOI] [PubMed] [Google Scholar]
- 120.McFarland KN, Liu J, Landrian I, Godiska R, Shanker S, Yu F, et al. SMRT sequencing of long tandem nucleotide repeats in SCA10 reveals unique insight of repeat expansion structure. PLoS One. 2015;10(8):e0135906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Schüle B, McFarland KN, Lee K, Tsai YC, Nguyen KD, Sun C, et al. Parkinson’s disease associated with pure ATXN10 repeat expansion. NPJ Parkinsons Dis. 2017;3:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Alonso I, Jardim LB, Artigalas O, Saraiva-Pereira ML, Matsuura T, Ashizawa T, et al. Reduced penetrance of intermediate size alleles in spinocerebellar ataxia type 10. Neurology. 2006;66(10):1602–4. [DOI] [PubMed] [Google Scholar]
- 123.Raskin S, Ashizawa T, Teive HA, Arruda WO, Fang P, Gao R, et al. Reduced penetrance in a Brazilian family with spinocerebellar ataxia type 10. Arch Neurol. 2007;64(4):591–4. [DOI] [PubMed] [Google Scholar]
- 124.McFarland KN, Liu J, Landrian I, Gao R, Sarkar PS, Raskin S, et al. Paradoxical effects of repeat interruptions on spinocerebellar ataxia type 10 expansions and repeat instability. Eur J Hum Genet. 2013;21(11):1272–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Elliot P, Wakefield J. Disease clusters: should they be investigated, and, if so, when and how? J R Statist Soc A. 2001;164(1):3–12. [Google Scholar]
- 126.Zlotogora J Multiple mutations responsible for frequent genetic diseases in isolated populations. Eur J Hum Genet. 2007;15(3):272–8. [DOI] [PubMed] [Google Scholar]
- 127.Nicolas G, Veltman JA. The role of de novo mutations in adult-onset neurodegenerative disorders. Acta Neuropathol. 2019;137: 183–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ayhan F, Ikeda Y, Dalton JC, Day JW, Ranum LPW. Spinocerebellar Ataxia Type 8. 2001 Nov 27 [updated 2014 Apr 3]. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K,Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2019. Available from http://www.ncbi.nlm.nih.gov/books/NBK1268/. Accessed 18 Jan. 2020 [Google Scholar]
- 129.Cardoso GC, de Oliveira MZ, Paixão-Côrtes VR, Castilla EE, Schuler-Faccini L. Clusters of genetic diseases in Brazil. J Community Genet. 2019;10(1):121–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gusella JF, Wexler NS, Conneally PM, Naylor SL, Anderson MA, Tanzi RE, et al. A polymorphic DNA marker genetically linked to Huntington’s disease. Nature. 1983;306(5940):234–8. [DOI] [PubMed] [Google Scholar]
- 131.Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72(6):971–83. [DOI] [PubMed] [Google Scholar]
- 132.Nenguke T, Aladjem MI, Gusella JF, Wexler NS. Arnheim N; Venezuela HD project. Candidate DNA replication initiation regions at human trinucleotide repeat disease loci. Hum Mol Genet. 2003;12(9):1021–8. [DOI] [PubMed] [Google Scholar]
- 133.Sapp E, Valencia A, Li X, Aronin N, Kegel KB, Vonsattel JP, et al. Native mutant huntingtin in human brain: evidence for prevalence of full-length monomer. J Biol Chem. 2012;287(16):13487–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gayan J, Brocklebank D, Andresen JM Alkorta-Aranburu G; US-Venezuela collaborative research group, Zameel Cader M, et al. Genomewide linkage scan reveals novel loci modifying age of onset of Huntington’s disease in the Venezuelan HD kindreds. Genet Epidemiol. 2008;32(5):445–53. [DOI] [PubMed] [Google Scholar]
- 135.Sena LS, Castilhos RM, Mattos EP, Furtado GV, Pedroso JL, Barsottini O, et al. Selective forces related to spinocerebellar ataxia type 2. Cerebellum. 2019;18(2):188–94. [DOI] [PubMed] [Google Scholar]
- 136.Faruq M, Magaña JJ, Suroliya V, Narang A, Murillo-Melo NM, Hernández-Hernández O, et al. A complete association of an intronic SNP rs6798742 with origin of spinocerebellar ataxia type 7-CAG expansion loci in the Indian and Mexican population. Ann Hum Genet. 2017;81(5):197–204. [DOI] [PubMed] [Google Scholar]
- 137.Ramos EM, Martins S, Alonso I, Emmel VE, Saraiva-Pereira ML, Jardim LB, et al. Common origin of pure and interrupted repeat expansions in spinocerebellar ataxia type 2 (SCA2). Am J Med Genet B Neuropsychiatr Genet. 2010;153B(2):524–31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.