

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal cancers with patients having unresectable or metastatic disease at diagnosis, with poor prognosis and very short survival. Given that genetic variation within autophagy‐related genes influences autophagic flux and susceptibility to solid cancers, we decided to investigate whether 55,583 single nucleotide polymorphisms (SNPs) within 234 autophagy‐related genes could influence the risk of developing PDAC in three large independent cohorts of European ancestry including 12,754 PDAC cases and 324,926 controls. The meta‐analysis of these populations identified, for the first time, the association of the BID rs9604789 variant with an increased risk of developing the disease (ORMeta = 1.31, p = 9.67 × 10−6). We also confirmed the association of TP63 rs1515496 and TP63 rs35389543 variants with PDAC risk (OR = 0.89, p = 6.27 × 10−8 and OR = 1.16, p = 2.74 × 10−5). Although it is known that BID induces autophagy and TP63 promotes cell growth, cell motility and invasion, we also found that carriers of the TP63 rs1515496G allele had increased numbers of FOXP3+ Helios+ T regulatory cells and CD45RA+ T regulatory cells (p = 7.67 × 10−4 and p = 1.56 × 10−3), but also decreased levels of CD4+ T regulatory cells (p = 7.86 × 10−4). These results were in agreement with research suggesting that the TP63 rs1515496 variant alters binding sites for FOXA1 and CTCF, which are transcription factors involved in modulating specific subsets of regulatory T cells. In conclusion, this study identifies BID as new susceptibility locus for PDAC and confirms previous studies suggesting that the TP63 gene is involved in the development of PDAC. This study also suggests new pathogenic mechanisms of the TP63 locus in PDAC.
Keywords: autophagy, functional characterization, genetic variants, pancreatic cancer, polymorphisms, susceptibility
What's new?
The etiology of pancreatic ductal adenocarcinoma (PDAC), among the most aggressive and deadliest cancers worldwide, remains largely unknown. Here, using data from cohorts of European ancestry, the authors investigated the influence on PDAC risk of single nucleotide polymorphisms (SNPs) in genes associated with autophagy. Analyses identified multiple SNPs associated with PDAC risk, including variants within BID and TP63. Variants in BID potentially dysregulate BID‐dependent autophagy, while those in TP63 may influence PDAC risk by modulating levels of T regulatory cells involved in host immune responses against tumor cells. The variants warrant further study to better elucidate their involvement in PDAC.
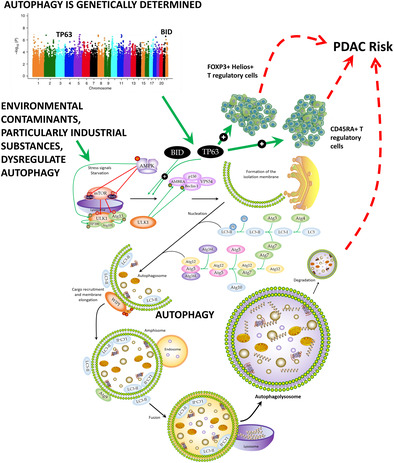
1. INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is the most common form of pancreatic cancer 1 , 2 , 3 with a worldwide incidence that has been increasing over decades and it is somewhat higher in males than females (6.7 vs. 6.0 cases per 100,000 people per year, respectively; https://gco.iarc.fr/today/home). Early diagnosis of PDAC is hampered by the lack of specific screening tests and by the fact that most of the patients do not have symptoms or if they do, they are very unspecific. 3 , 4 Consequently, PDAC is frequently diagnosed at late stages leading to an extremely poor prognosis and a 5‐year survival rate that ranges from 2% to 10%. 5 Several environmental factors (including exposure to pesticides, asbestos, benzene, and chlorinated hydrocarbons) and lifestyle risk factors (cigarette smoking, obesity, family history of cancer, pancreatitis, type 2 diabetes, heavy alcohol consumption, low physical activity, stress, mobile phone use, allergies, and poor oral health) have been identified for PDAC. 2 , 6 , 7 , 8 Furthermore, around 30 genetic susceptibility loci have been identified through genome wide association studies (GWAS) and large candidate gene or candidate region studies. 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 However, PDAC continues to be one of the most aggressive and lethal diseases with still a relatively unknown etiology. 17 , 18
Recent evidence suggests that autophagy, a lysosome‐dependent catabolic degradation process involved in removing toxic air pollutants, particulate particles and heavy metals, but also toxic aggregated cytosolic proteins and malfunctioning organelles from normal cells, might influence the onset of solid tumors and blood malignancies. 19 , 20 , 21 In PDAC, several studies have demonstrated that pancreatic tumoral tissues have an increased autophagy flux and a greater number of autophagosomes 22 in comparison to normal pancreatic cells. 23 , 24 Furthermore, it has been shown that autophagy participates in controlling multiple processes including tumor cell growth, metabolism, MHC‐I and MHC‐II presentation, 25 , 26 cell migration and metastasis. 27 These findings, along with the fact that treatment with chloroquine (an autophagy inhibitor) or the genetic ablation of autophagy initiation genes (ATG5 or ATG7) reduced the growth of human PDAC cell lines, 23 suggest that autophagy might represent a potential target for PDAC treatment. In support of this hypothesis, studies using xenografts or animal models have shown the benefit of inhibiting autophagy to induce tumor regression and prolong survival. 28 , 29 , 30 In addition, autophagy is implicated in controlling the resistance to apoptosis 31 and, therefore, it might represent a poor prognostic factor for PDAC. 32 Nonetheless, several studies have also suggested an anti‐tumorigenic effect of autophagy in PDAC 33 , 34 and some clinical trials using autophagy inhibitors have shown limited success. 35 , 36
Considering the above‐reported results, but also previous findings suggesting that genetic variation within autophagy‐related genes influences autophagic flux and the susceptibility to solid and hematological cancers, 37 , 38 we decided to comprehensively evaluate the impact of 55,583 common genetic variants within 234 autophagy‐related genes in determining the risk of developing PDAC. Understanding the impacts of environmental exposure through autophagy and how this catabolic process is genetically regulated might offer new approaches for risk assessment, protection and preventive actions against cancer. Given the known role of autophagy in shaping immune responses, inflammation and immune tolerance 39 and its potential role in regulating immunotherapy efficacy, 40 we also assessed the correlation of the most promising autophagy variants with cytokine production after in vitro stimulation of whole blood (WB), peripheral blood mononuclear cells (PBMCs), or monocyte‐derived macrophages (MDM) with lipopolysaccharide (LPS), phytohemagglutinin (PHA), Pam3Cys or CpG. Additionally, we assessed the impact of genetic variants in autophagy genes on the absolute numbers of 91 blood‐derived cell populations, 103 serum immunological proteins, and 7 steroid hormones in a large cohort of 408 healthy donors (500FG) from the Human Functional Genomic Project (HFGP). 41
2. MATERIALS AND METHODS
A workflow diagram of the study is included as Figure 1.
FIGURE 1.

Flow diagram of the study.
2.1. Study populations
The discovery population consisted of 15,776 subjects from previous GWAS on PDAC conducted by the Pancreatic Cancer Cohort (PanScan I–III), 42 , 43 , 44 and the Pancreatic Cancer Case–Control (PanC4) consortia 45 (Figure 1). In these studies, the PDAC cases presented an established diagnosis and controls were blood donors or healthy subjects randomly selected from the general population and with no history of previous cancers. As part of the discovery cohorts, we also included the FinnGen research genomic project, which arose from Finnish biobanks and digital health record data from Finnish health registries (https://www.finngen.fi/en). At the time this study was conducted, FinnGen (Data Freeze 10 or Ristey10) included genetic data of 314,924 individuals (731 PDAC cases and 314,193 controls).
2.2. SNP selection and meta‐analysis of discovery cohorts
A total of 234 autophagy‐related genes were selected based on their presence in the autophagy database (http://autophagy.lu/clustering/index.html; Table S1) and association of 55,583 genotyped or imputed SNPs within or near these genes (5 kb upstream and 3 kb downstream) 46 , 47 , 48 with PDAC risk was computed from a GWAS conducted in the PanScan I–III and the PanC4 population. GWAS datasets were downloaded from the NCBI database of genotypes and phenotypes (dbGaP; study accession numbers phs000206.v5.p3 and phs000648. v1.p1; project reference no. 12644), namely PanScan I, PanScan II and PanC4. These PanScan I–III + PanC4 GWAS were subjected to rigorous standard quality control protocols before imputation using the Michigan imputation server (based on the Haplotype Reference Consortium). 49 After imputation, GWAS data were filtered to include only high‐quality imputed variants (info score >0.8). Further quality‐control checks were implemented including checks for missingness, duplicates, abnormal heterozygosity, cryptic relatedness, population outliers (evaluated by principal components analyses using Eigenstrat software), and genomic inflation (λ = 1.00). 50 Detailed information about the genotyping technologies used, quality control, imputation, and ethnic composition of the discovery cohorts is provided as Data S1. A fixed‐effect meta‐analysis of the PanScan I–III + PanC4 and publicly available FinnGen GWAS data was conducted using METAL. 51 The I 2 statistic was used to assess statistical heterogeneity between the study cohorts and pooled odds ratios (ORs) were computed using the fixed‐effect model. Among the 55,583 selected variants, a total of 45,036 SNPs were shared by the GWAS platforms and were, therefore, available for association analysis. Of those 45,036 variants, a total of 2907 SNPs were considered independent (r 2 < .1) according to LDLink data for European cohorts (https://ldlink.nci.nih.gov/?tab=snpclip), and therefore, the multiple testing significance threshold for the study was set to p = 1.72 × 10−5 (0.05/2907 SNPs; Table S2). To select the most interesting markers for further validation, we excluded those SNPs that were previously reported as susceptibility markers for PDAC and we advanced for replication in the PANcreatic Disease ReseArch (PANDoRA) consortium only those markers that showed a significant association with PDAC risk after multiple testing correction (p < 1.72 × 10−5). Genotyping of the genetic markers included 3283 PDAC cases and 3697 controls.
2.3. Genotyping and meta‐analysis
Genotyping of genetic variants advanced for replication in PANDoRA was carried out at University of Pisa (Department of Biology, University of Pisa, Pisa, Italy) using KASPar (LGC Genomics, Hoddesdon, UK) according to previously reported protocols. 52 , 53 For internal quality control, ~5% of samples were randomly selected and included as duplicates. Concordance between the original and the duplicate samples for the SNPs tested was ≥99.0%. Selected SNPs showed genotype frequencies in the control population similar to those found in the 1000 Genomes database (data not shown) and were in Hardy–Weinberg equilibrium (HWE). After the genotyping of selected markers in PANDoRA, an overall meta‐analysis of the association estimates of PANDoRA with those from the PanScan I–III, PANC4, and FinnGen studies was conducted in R using the Meta package. 54
2.4. Functional effect of the autophagy‐related variants
To provide insight into the functional role of those SNPs that remained statistically significant after multiple correction (p = 1.72 × 10−5), but also those SNPs that are well‐established susceptibility markers for PDAC, we tested if any of them were associated with cytokine expression quantitative trait loci (cQTL) data from in vitro stimulation experiments. We also tested the association with the absolute numbers of 91 blood‐derived cell populations, 103 serum or plasmatic inflammatory proteins and 7 steroid hormones quantified in 408 volunteers from the 500 Functional Genomics (500FG) cohort from the HFGP.
2.4.1. Correlation of autophagy‐related SNPs with cQTL data
The cQTL data included cytokine levels (IFNγ, IL1β, IL6, TNFα, IL17 and IL22) measured when peripheral blood mononuclear cells (PBMCs), monocyte‐derived macro‐phages (MDM), or whole blood from 408 healthy subjects were left untreated or stimulated for 24 h with LPS (1 or 100 ng/mL; Sigma Aldrich, St. Louis, MO), PHA (10 μg/mL, Sigma, St. Louis, MO), Pam3Cys (10 μg/mL, EMC microcollections, Tübingen, Germany) or CpG (100 ng/mL, InvivoGen, San Diego, CA). Detailed protocols for PBMCs isolation, macrophage differentiation and stimulation assays have been reported elsewhere. 38 , 55 Briefly, PBMCs were washed twice in saline and suspended in medium (RPMI 1640) supplemented with gentamicin (10 mg/mL), l‐glutamine (10 mM) and pyruvate (10 mM). PBMC stimulations were performed with 5 × 105 cells/well in round‐bottom 96‐well plates (Greiner Bio‐one, Frickenhausen, Germany) for 24 h in the presence of 10% human pool serum at 37°C and 5% CO2. Supernatants were collected and stored at −20°C until used for ELISA. Concentrations of human IFNγ, IL1β, IL6, TNFα, IL17 and IL22 were determined using specific commercial ELISA kits (PeliKine Compact, Amsterdam or R&D Systems), in accordance with the manufacturers' instructions. When values were below or above the detection limit of the ELISA, the corresponding limit was used. After log transformation, linear regression analyses adjusted for age and sex were used to determine the correlation of the selected SNPs with cQTL data.
2.4.2. Correlation of autophagy SNPs and blood cell counts and serum/plasmatic proteomic profile
Next, we evaluated the impact of selected SNPs on cell‐level variation. A total of 91 blood‐derived cell populations were measured by 10‐color flow cytometry (Navios flow cytometer, Beck‐man Coulter, Miami, FL) after blood sampling (2–3 h), and cell count analysis was performed using Kaluza software (Beckman Coulter, v.1.3). To reduce inter‐experimental noise and increase statistical power, cell count analysis was performed by calculating parental and grandparental percentages, which were defined as the percentage of a certain cell type within the subpopulation of the cells from which it was isolated (Table S3). 55 Detailed laboratory protocols for cell isolation, reagents, gating, and flow cytometry analysis have been reported elsewhere 56 and raw flow cytometry data and analyzed data files are available upon reasonable request to the authors (http://hfgp.bbmri.nl, accessed on 13 February 2024). A proteomic analysis was also performed in serum and plasma samples from the HFGP study. Circulating proteins were measured using the proximity extension assay (Olink Inflammation panel, Olink, Sweden) that resulted in the measurement of 103 different biomarkers (Table S4). Protein levels were expressed on a log2‐scale as normalized protein expression values and normalized using bridging samples to correct for batch variation. Considering the number of proteins (n = 103), blood‐derived cell populations (n = 91), and SNPs (n = 4) tested, significance p‐values were set to be 1.21 × 10−4 and 1.37 × 10−4 for the proteomic and blood cell count analyses, respectively.
2.4.3. Correlation between autophagy‐related SNPs and serum steroid hormone levels
Besides the immunological experiments, we also evaluated the correlation of autophagy SNPs with serum steroid hormone levels (androstenedione, cortisol, 11‐deoxy‐cortisol, 17‐hydroxyprogesterone, progesterone, testosterone and 25 hydroxy vitamin D3) from 279 healthy controls of the 500FG cohort without hormone replacement or oral contraceptive therapies. Serum steroid hormone levels were determined by ELISA following the manufacturer's instructions. Correlation between levels of 7 serum steroid hormones and autophagy‐related SNPs was evaluated by linear regression analysis adjusted for age and sex. The significance threshold was set to p = .00178 considering the number of independent SNPs tested (n = 4) and the number of hormones determined (n = 7).
All analyses of functional data were performed using R software (http://www.r-project.org), using custom scripts in the R programming language based on existing functions such as lm (stats). Functional plots were displayed using the Prism software.
2.5. In silico functional analysis
Haploreg (http://www.broadinstitute.org/mammals/haploreg/haploreg.php) 57 was also used to predict the functional role of the autophagy SNPs. We also tested whether all these autophagy SNPs could represent expression quantitative trait loci (eQTL) for different cell types and tissues using publicly available GTex portal data (https://gtexportal.org/home/) and information available at the Blood eQTL browser (https://genenetwork.nl/bloodeqtlbrowser/).
3. RESULTS
This study included 337,680 individuals consisting of 12,754 cases of PDAC and 324,926 controls, 8740 cases and 7036 controls from the PanScan I–III and PanC4 cohort, 731 cases and 314,193 controls from the FinnGen study and 3283 patients and 3697 healthy controls from the PANDoRA cohort. A total of 45,036 SNPs showing no deviation from HWE (p < .001) neither in the PanScan I–III + PanC4 and FinnGen cohorts were selected for association analysis. After testing HWE in the control group by a standard observed‐expected chi‐square (χ 2) test (p < 10−5) and filtering by linkage disequilibrium (LD) values (r 2 < .1) and a minor allele frequency (MAF) of 0.01, the meta‐analysis of the association estimates for these SNPs in the PanScan I–III + PanC4 and FinnGen cohorts showed that nine polymorphisms were associated with the risk of developing PDAC at p ≤ 10−4 level (Table 1).
TABLE 1.
Association analysis of autophagy‐related SNPs in the discovery cohorts.
| SNP | Gene | Chr | A1 | MAF | PanScan + PanC4 (n = 15.776) | FinnGen R10 (n = 314.924) | Meta‐analysis (n = 330.700) a | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OR (95% CI) | p | OR (95% CI) | p | OR (95% CI) | p | Direction | p Het | |||||
| rs9604789 | BID | 22 | A | 0.017 | 1.33 (1.13–1.56) | 4.76E‐04 | 1.34 (1.07–1.68) | .0107 | 1.33 (1.17–1.52) | 1.52E‐05 | ++ | .9375 |
| rs11653998 | ERBB2 | 17 | C | 0.335 | 1.09 (1.04–1.14) | 7.34E‐04 | 1.08 (0.98–1.18) | .1196 | 1.09 (1.04–1.13) | 2.02E‐04 | ++ | .9283 |
| rs11540923 | MAP1LC3B | 16 | C | 0.029 | 0.73 (0.62–0.86) | 1.30E‐04 | 0.52 (0.23–1.16) | .1087 | 0.72 (0.61–0.84) | 4.72E‐05 | − | .4182 |
| rs72552382 | MTOR | 1 | C | 0.015 | 1.52 (1.23–1.88) | 1.33E‐04 | 0.67 (0.22–2.09) | .4911 | 1.48 (1.2–1.82) | 2.89E‐04 | +− | .1659 |
| rs75447274 | NBR1 | 17 | T | 0.031 | 0.77 (0.67–0.90) | 6.50E‐04 | 0.91 (0.67–1.24) | .5697 | 0.80 (0.70–0.91) | 8.94E‐04 | − | .3368 |
| rs78344309 | PEX14 | 1 | T | 0.070 | 0.81 (0.72–0.92) | 9.99E‐04 | 0.87 (0.69–1.10) | .2530 | 0.83 (0.74–0.92) | 5.67E‐04 | − | .6135 |
| rs1515496 | TP63 | 3 | G | 0.389 | 0.88 (0.84–0.92) | 3.50E‐08 | 1.05 (0.94–1.17) | .3907 | 0.89 (0.85–0.93) | 6.27E‐08 | ++ | .1725 |
| rs35389543 | TP63 | 3 | C | 0.099 | 1.17 (1.08–.26) | 1.48E‐04 | 1.15 (0.99–1.34) | .0727 | 1.16 (1.08–1.25) | 2.74E‐05 | − | .8619 |
| rs9882798 | TP63 | 3 | A | 0.096 | 1.17 (1.08–1.26) | 1.39E‐04 | 1.05 (0.90–1.22) | .5464 | 1.14 (1.06–1.22) | 2.61E‐04 | ++ | .2141 |
Note: The TP63 rs1515496 SNPs (or other SNPs in strong LD) is a well‐established susceptibility markers for PDAC. p < .05 in bold.
Abbreviations: A1, effect‐allele; SNP, single nucleotide polymorphism.
Meta‐analysis: PanScan + PanC4 + FinnGen; FinnGen releases DF10 according to a random effect model.
Importantly, among these 9 SNPs, we found a statistically significant association for a SNP within the BID locus with PDAC risk (p < 1.72 × 10−5). Therefore, it was advanced for replication and genotyped in PANDoRA. Association results for this SNP are reported in Table 2. After the meta‐analysis of the three European cohorts, we found that carriers of the BID rs9604789G allele had an increased risk of developing PDAC when compared with those carrying the most frequent allele (OR = 1.31, p = 9.67 × 10−6; Table 2). The association of the BID rs9604789 SNP with PDAC risk remained significant after correction for multiple testing, which suggested that this variant might have a functional role in modulating PDAC risk. In support of this hypothesis, it has been reported that the BID rs9604789 SNP affects chromatin states in multiple primary cell types including primary monocytes from peripheral blood, T regulatory cells, T helper cells, CD8+ T cells, B cells, NK cells, and neutrophils.
TABLE 2.
Meta‐analysis of the association between the BID SNP and PDAC risk.
| SNP | Gene | Chr. | EA | MAF | PanScan + PanC4 (n = 15.776) | FinnGen# (n = 314.924) | PANDoRA (n = 6.980) | Meta‐analysis (n = 337.680) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OR (95% CI) | p | OR (95% CI) | p | OR (95% CI) | p | OR (95% CI) | p Meta | p Het | |||||
| rs9604789 | BID | 22 | A | 0.02 | 1.33 (1.13–1.56) | 4.70E‐04 | 1.34 (1.07–1.68) | .0107 | 1.19 (0.89–1.60) | .251 | 1.31 (1.16–1.47) | 9.674E‐06 | .7868 |
Note: p < .05 in bold.
Abbreviations: SNP, single nucleotide polymorphism; EA, effect allele; MAF, minor allele frequency according to the 1000genome project.
Besides these results, we found three autophagy‐related SNPs within the TP63 locus that have been previously identified as susceptibility markers for PDAC. Our results confirmed that carriers of the TP63 rs1515496G TP63 rs35389543C alleles had decreased risk of developing PDAC (OR = 0.89, p = 6.27 × 10−8 and OR = 1.16, p = 2.74 × 10−5; Table 1). As expected, the association of the TP63 rs1515496 SNP remained significant after correction for multiple testing (with a threshold of p Bonferroni = 1.72 × 10−5) whereas the TP63 rs35389543 SNP remained borderline significant.
Given that the TP63 rs1515496 SNP is a well‐established risk marker for PDAC, we investigated, for the first time, its correlation with host immune responses. Of note, we found a potentially interesting correlation of the TP63 rs1515496 SNP with increased numbers of FOXP3+ Helios+ T regulatory cells (p = 7.67 × 10−4; Figure 2A) and CD45RA+ T regulatory cells (p = 1.56 × 10−3; Figure 2B) but decreased levels of CD4+ T regulatory cells (p = 7.86 × 10−4; Figure 2C). Although these associations did not remain significant after correction for multiple testing (p Bonferroni = p = 6.1 × 10−5, 0.05/9SNPs/91 blood‐derived cell populations), these results, together with our genetic findings, might suggest a modest but still functional effect of the TP63 locus on determining PDAC risk by regulating absolute numbers of specific subpopulations of regulatory T cells.
FIGURE 2.
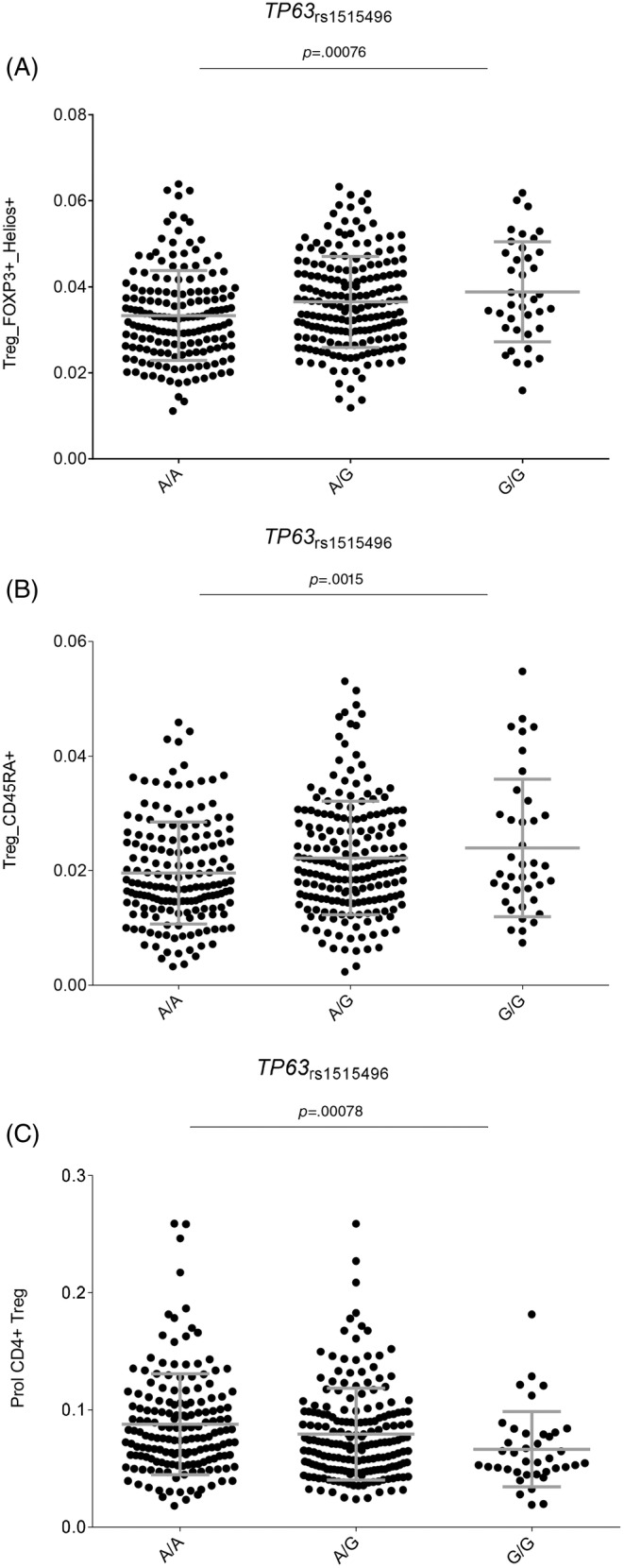
(A–C) Correlation of the TP63 rs1515496 SNP with numbers of specific T regulatory T cell subsets.
Interestingly, we could not find significant correlations between the BID and TP63 variants and levels of circulating inflammatory proteins or steroid hormones, which also suggests that these loci do not impact PDAC risk through the modulation of circulating hormone and inflammatory protein levels. However, further investigation into these aspects is warranted to fully understand their roles in PDAC development.
4. DISCUSSION
This study comprehensively evaluated the role of autophagy‐related variants in modulating PDAC risk in three large and independent cohorts including a total of 13,215 PDAC cases and 270,274 healthy controls. The meta‐analysis of these European populations identified for the first time that the BID rs9604789 SNP was significantly associated with an increased risk of PDAC. This association remained significant after Bonferroni correction (multiple testing). The BID gene (BH3 Interacting Domain Death Agonist) is located on chromosome 22 and it encodes for a Bcl‐2 family member that promotes autophagy‐mediated cell death and apoptosis through different mechanisms involving caspase‐8, but also Bak and Bax proteins. 58 , 59 , 60 Interestingly, it has been proposed that BID also acts as a mediator in modulating the autophagic flux and host immune responses, 61 , 62 which underlines the central role of this gene in tumorigenesis. In this regard, it has been described that the BID rs9604789 SNP alters binding motifs for multiple transcription factors including NFκB, POL2, TAF1, TCF12, EGR1, GABP, TBP, POL24H8 and PAX5N19 that are very well‐known regulators of key immune processes involving multiple immune cell types. 63 Furthermore, it has been shown that the BID rs9604789 SNP affect chromatin states and modulates histone marks in multiple primary immune cell types, 57 which might suggest a role of this SNP in modulating BID expression in specific immune cells. Likewise, it has been reported that the BID protein interacts with NOD1, NOD2 and IKK complex to promote the activation of NFκB and the induction of the extracellular signal‐regulated kinase (ERK) signaling pathway. Previous studies have reported that the constitutive activation of ERK/MAPK signaling pathway (resulting from KRAS mutation) has a relevant role in modulating autophagy, which confirms that BID is an autophagy regulator gene that has a relevant role in PDAC.
This activation is linked to macrophage cell survival 64 and it is independent of the BID‐induced apoptosis. 61 Furthermore, it has been demonstrated that macrophages from BID −/− mice are markedly defective in the production of IL6 and TNFα after stimulation with a NOD2 agonist, 61 which suggests that any disruption of BID function might modulate cytokine production. In agreement with these findings, it has been reported that different mutant forms of the BID gene affecting protein phosphorylation regulated IL‐6 production after stimulation of macrophages with muramyl‐dipeptide, a NOD2 agonist. 65 Importantly, it has been also shown that BID modulates myeloid homeostasis and tumor suppression 66 and acts along with BIM to regulate T cell expansion following acute and persistent infection. 62 Therefore, considering the above information, it seems plausible to suggest that the BID rs9604789 SNP might dysregulate BID‐dependent autophagy, host innate immune responses and survival of macrophages and T cells and, thereby, influence cancer development. However, although tempting, we could not demonstrate the immunological role of this marker to modulate PDAC risk as our functional experiments did not show any significant effect on cytokine production, absolute numbers of blood‐derived cell populations and serum inflammatory mediators.
Considering these results, it seems more plausible to suggest that the role of BID rs9604789 SNP in modulating the risk of PDAC is mediated by its effect on the modulating of autophagy‐mediated cell death and/or apoptosis. 67 In support of this hypothesis, Li et al. (1998) reported that BID is a specific proximal substrate of CASP8 in the Fas apoptotic signaling pathway that mediates mitochondrial damage induced by CASP8. 68 Conversely, although BID is mainly known as a pro‐apoptotic protein, 65 it has been reported that it may also lead to the inhibition of apoptosis and induce a shift of toward autophagy‐mediated cell death especially in cells resistant to apoptosis. 69 In addition, it has been reported that BID is a target gene of p53, 70 which suggests its implication in the regulation of autophagy‐dependent cell death. However, despite the above‐reported information, it is difficult to draw definitive conclusions on how the BID rs9604789 SNP within the BID gene might determine BID function and, thereby, cancer risk. Therefore, additional functional studies are now warranted to decipher the specific role of this SNP in modulating the risk of PDAC.
Besides the identification of the BID SNP in determining PDAC risk, the meta‐analysis of the three cohorts validated results from previous studies demonstrating that autophagy‐related variants within the TP63 locus (or specific SNPs in strong LD with them) are susceptibility biomarkers for PDAC risk. 16 , 45 , 71 The strongest effect was found for the TP63 rs1515496 SNP that survived correction for multiple testing (p = 5.0 × 10−8). Although the association of the TP63 rs1515496 SNP with PDAC risk has been previously established using among others PanScan, PanC4 and PANDoRA populations, 45 , 72 its specific biological function has not been completely elucidated. In this regard, we could demonstrate that the TP63 rs1515496 SNP was associated with increased absolute numbers of FOXP3+ Helios+ T regulatory cells, and CD45RA+ T regulatory cells, but also with decreased numbers of CD4+ T regulatory cells, the latter being a subset of regulatory T cells with potentially suppressing immunological activities. These findings are in agreement with previous studies demonstrating that higher counts of T regulatory cells were significantly associated with an increased risk of PDAC in participants diagnosed within the first 5 years of follow‐up 73 and even with poor prognosis. 74 , 75 Furthermore, functional data from Haploreg showed that the TP63 rs1515496 SNP alters binding sites for FOXA1 and CTCF transcription factors, which are involved in modulating T regulatory cell numbers and, therefore, host immune responses. 63 In addition to this, it has also been shown that TP63 is frequently overexpressed in PDAC tissues 76 and that its expression correlates with disease aggressiveness. 76 Likewise, besides the role of the TP63 rs1515496 variant in determining absolute numbers of regulatory T cell subsets and host immune responses, it is conceivable that this intronic variant (or any other polymorphism in strong LD with it) could influence TP63 function through alteration of splicing sites and thus give rise to functionally different TP63 isoforms. In this regard, a recent study has demonstrated that whereas the TAp63 isoform induces cell death and cell cycle arrest with tumor suppressor features, 65 the DNp63 isoform, which is the most common isoform in PDAC cell lines, has an opposite effect inducing pancreatic cancer growth, motility and invasion. 77 , 78
Finally, even though the association of genetic markers within the MAP1LC3B, ERBB2, MTOR, PEX14 and NRB1 loci did not reach the multiple testing significance threshold and, therefore, were not advanced for replication in the PANDORA cohort, we think that they are potentially interesting markers that need to be further analyzed in future studies. In particular, it has been reported that the MAP1LC3B and ERBB2 genes, which showed SNPs associated with PDAC risk close to the Bonferroni significance threshold, may play a role in modulating tumor cell survival and resistance to treatments. Rouschop et al. (2010) demonstrated that the unfolded protein response protects human tumor cells during hypoxia by regulating MAP1LC3B, which is involved in phagophore expansion and autophagosome formation. 79 In addition, expression of the MAP1LC3B protein in carcinoma‐associated fibroblasts (CAFs) has been linked to poor survival in PDAC patients, 80 suggesting its involvement in modulating adaptative immune resistance of tumor cells.
Similarly, ERBB2 has also been consistently implicated in PDAC onset and patient survival. Previous studies have demonstrated that this gene is frequently amplified in PDAC patients, 81 and its oncogenic effect is mediated not only by gene amplification but also by overexpression, 82 highlighting the value of studying this component from a genetic perspective. Additionally, Ortega et al. (2015) recently demonstrated that ERBB2 is upregulated in a high proportion of PDAC patients, 83 suggesting its involvement not only in disease onset but also in disease progression 84 , 85 , 86 and patient survival. 83 However, further studies are needed to delineate the precise roles of MAP1LC3B and ERBB2 in PDAC pathogenesis, as some previous studies have indicated a lack of influence of these genes on disease progression. 87 , 88
This study has both strengths and limitations. The most important strengths of our study are the comprehensive analysis of autophagy‐related SNPs and the inclusion of three large independent populations of European ancestry for a total of 276,608 study participants. Furthermore, we comprehensively analyzed the functional impact of autophagy‐related SNPs in modulating host immune responses, absolute numbers of blood‐derived cell populations, serum and plasma metabolites, and steroid hormones in a large study of healthy subjects ascertained through the HFGP. However, this study also has limitations. Even though the 5 kb upstream and 3 kb downstream range used to select SNPs is generally sufficient to capture most relevant variants, including those in promoter regions and untranslated regions that can impact gene function, we acknowledge that regulatory elements, such as enhancers and repressors, can exist outside these regions. Therefore, our study might miss some potentially interesting and functional markers that affect the regulation of autophagy genes. Another limitation of our study is that it included only populations of European ancestry, which restricted the translation of the above‐reported results to other ethnicity groups. Although we attempted to validate the association of the BID SNP with PDAC risk in the JaPAN consortium, this cohort did not have genotyping information of the BID marker (or its proxies). 89 Additionally, the lack of environmental variables in the PANDORA database prevented us from conducting gene‐environmental interaction analyses to explore the relationship between genetic factors and environmental influences in PDAC development.
5. CONCLUSION
In summary, our study has identified, for the first time, the association of the BID rs9604789 SNP with an increased risk of developing the disease. In addition, it has confirmed the association of TP63 SNPs with the risk of developing PDAC. This study points to a functional role of the BID and TP63 loci in modulating PDAC onset likely through the regulation autophagy and host immune responses mediated by different subsets of T regulatory cells. Finally, this work has underlined the need of additional studies to elucidate the functional role of the MAP1LC3B, ERBB2, MTOR, PEX14 and NRB1 loci to determine PDAC risk.
AUTHOR CONTRIBUTIONS
Fernando Gálvez‐Montosa: Formal analysis; investigation; resources; writing – original draft. Giulia Peduzzi: Data curation; formal analysis; investigation; resources; writing – original draft. José Manuel Sanchez‐Maldonado: Formal analysis; investigation; writing – original draft. Rob ter Horst: Data curation; formal analysis; investigation; resources; writing – review and editing. Antonio J. Cabrera‐Serrano: Formal analysis; investigation; writing – review and editing. Manuel Gentiluomo: Resources; writing – review and editing. Angelica Macauda: Resources; writing – review and editing. Natalia Luque: Resources; writing – review and editing. Pelin Ünal: Resources; writing – review and editing. Francisco José García‐Verdejo: Resources; writing – review and editing. Yang Li: Resources; writing – review and editing. José Antonio López López: Resources; writing – review and editing. Angelika Stein: Resources; writing – review and editing. H. Bas Bueno‐de‐Mesquita: Resources; writing – review and editing. Paolo Giorgio Arcidiacono: Resources; writing – review and editing. Dalila Luciola Zanette: Resources; writing – review and editing. Christoph Kahlert: Resources; writing – review and editing. Francesco Perri: Resources; writing – review and editing. Pavel Soucek: Resources; writing – review and editing. Renata Talar‐Wojnarowska: Resources; writing – review and editing. George E. Theodoropoulos: Resources; writing – review and editing. Jakob R. Izbicki: Resources; writing – review and editing. Hussein Tamás: Resources; writing – review and editing. Hanneke Van Laarhoven: Resources; writing – review and editing. Gennaro Nappo: Resources; writing – review and editing. Maria Chiara Petrone: Resources; writing – review and editing. Martin Lovecek: Resources; writing – review and editing. Roel C. H. Vermeulen: Resources; writing – review and editing. Kestutis Adamonis: Resources; writing – review and editing. Fernando Jesus Reyes‐Zurita: Resources; writing – review and editing. Bernd Holleczek: Resources; writing – review and editing. Jolanta Sumskiene: Resources; writing – review and editing. Beatrice Mohelníková‐Duchoňová: Resources; writing – review and editing. Rita T. Lawlor: Resources; writing – review and editing. Raffaele Pezzilli: Resources; writing – review and editing. Mateus Nobrega Aoki: Resources; writing – review and editing. Claudio Pasquali: Resources; writing – review and editing. Vitalija Petrenkiene: Resources; writing – review and editing. Daniela Basso: Resources; writing – review and editing. Stefania Bunduc: Resources; writing – review and editing. Annalisa Comandatore: Resources; writing – review and editing. Hermann Brenner: Resources; writing – review and editing. Stefano Ermini: Resources; writing – review and editing. Giuseppe Vanella: Resources; writing – review and editing. Mara R. Goetz: Resources; writing – review and editing. Livia Archibugi: Resources; writing – review and editing. Maurizio Lucchesi: Resources; writing – review and editing. Faik Guntac Uzunoglu: Resources; writing – review and editing. Olivier Busch: Resources; writing – review and editing. Anna Caterina Milanetto: Resources; writing – review and editing. Marta Puzzono: Resources; writing – review and editing. Juozas Kupcinskas: Resources; writing – review and editing. Luca Morelli: Resources; writing – review and editing. Cosimo Sperti: Resources; writing – review and editing. Silvia Carrara: Resources; writing – review and editing. Gabriele Capurso: Resources; writing – review and editing. Casper H. J. van Eijck: Resources; writing – review and editing. Martin Oliverius: Resources; writing – review and editing. Susanne Roth: Resources; writing – review and editing. Francesca Tavano: Resources; writing – review and editing. Rudolf Kaaks: Resources; writing – review and editing. Andrea Szentesi: Resources; writing – review and editing. Ludmila Vodickova: Resources; writing – review and editing. Claudio Luchini: Resources; writing – review and editing. Ben Schöttker: Resources; writing – review and editing. Stefano Landi: Resources; writing – review and editing. Orsolya Dohan: Resources; writing – review and editing. Matteo Tacelli: Resources; writing – review and editing. William Greenhalf: Resources; writing – review and editing. Maria Gazouli: Resources; writing – review and editing. John P. Neoptolemos: Resources; writing – review and editing. Giulia Martina Cavestro: Resources; writing – review and editing. Ugo Boggi: Resources; writing – review and editing. Anna Latiano: Resources; writing – review and editing. Péter Hegyi: Resources; writing – review and editing. Laura Ginocchi: Resources; writing – review and editing. Mihai G. Netea: Resources; writing – review and editing. Pedro Sánchez‐Rovira: Resources; writing – review and editing. Federico Canzian: Conceptualization; investigation; resources; writing – review and editing. Daniele Campa: Conceptualization; funding acquisition; resources; writing – original draft; writing – review and editing. Juan Sainz: Conceptualization; formal analysis; funding acquisition; investigation; resources; writing – original draft; writing – review and editing.
FUNDING INFORMATION
This work has been funded by multiple institutions: Italian Minister of Health, Ricerca Corrente program 2022‐2024, to Fondazione IRCCS “Casa Sollievo della Sofferenza” Hospital, San Giovanni Rotondo (FG), Italy; National Institute for Cancer Research (Programme EXCELES, No. LX22NPO5102), Czech Republic Ministry of Health (GACR 21‐27902 and AZV NU21‐03‐00145) and Charles University (UNCE/MED/006); Associazione Italiana Ricerca Cancro (AIRC IG n. 26343); The Instituto de Salud Carlos III and FEDER (Madrid, Spain; PI17/02256 and PI20/01845), Consejería de Transformación Económica, Industria, Conocimiento y Universidades and FEDER (PY20/01282) and the voluntary economical contribution of patients.
CONFLICT OF INTEREST STATEMENT
All authors have no competing interests to disclose.
ETHICS STATEMENT
Each participant in the PanScan and PanC4 studies obtained approval from the responsible institutional review board (IRB) and IRB certification permitting data sharing in accordance with the NIH Policy for sharing of Data Obtained in NIH‐Supported or NIH‐Conducted Genome Wide Association Studies. The PANDoRA study protocol was approved by the Ethics Commission of the Medical Faculty of the University of Heidelberg (S‐565/2015, last update on April 3, 2017). In accordance with the Declaration of Helsinki, written informed consent was obtained from each participant. The PANDoRA cohort was set up in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki) for experiments involving humans. Ethical approval for this study was obtained from each participating institution in PANDoRA and all participating subjects provided written informed consent (Data S1). 90 , 91 The HFGP study was also approved by the Arnhem‐Nijmegen Ethical Committee (no. 42561.091.12) and biological specimens were collected after informed consent was obtained.
Supporting information
Data S1. Supporting Information.
Table S1. List of selected genes.
Table S2. List of autophagy‐related SNPs.
Gálvez‐Montosa F, Peduzzi G, Sanchez‐Maldonado JM, et al. Polymorphisms within autophagy‐related genes as susceptibility biomarkers for pancreatic cancer: A meta‐analysis of three large European cohorts and functional characterization. Int J Cancer. 2025;156(2):339‐352. doi: 10.1002/ijc.35196
[Correction added on 07 November 2024, after first online publication: The name of the author “Francisco José García‐Verdejo” has been corrected.]
Fernando Gálvez‐Montosa, Giulia Peduzzi, and José Manuel Sanchez‐Maldonado share first authorship.
Daniele Campa and Juan Sainz share last authorship.
DATA AVAILABILITY STATEMENT
The genotype data used in the present study come from three pancreatic cancer GWAS datasets downloaded from the NCBI database of genotypes and phenotypes (dbGaP; study accession numbers phs000206.v5.p3 and phs000648. v1.p1; project reference #12644), namely PanScan I, PanScan II and PanC4. Genotyping data of the genetic markers analyzed in the PANDORA cohort are available on request from the corresponding author. Functional data used in this project have been meticulously catalogued and archived in the BBMRI‐NL data infrastructure (https://hfgp.bbmri.nl/) using the MOLGENIS open‐source platform for scientific data. This allows flexible data querying and download, including sufficiently rich metadata and interfaces for machine processing (R statistics, REST API) and using FAIR principles to optimize Findability, Accessibility, Interoperability and Reusability. Further information is available from the corresponding author upon request.
REFERENCES
- 1. Kleeff J, Korc M, Apte M, et al. Pancreatic cancer. Nat Rev Dis Primers. 2016;2:16022. [DOI] [PubMed] [Google Scholar]
- 2. Hu JX, Zhao CF, Chen WB, et al. Pancreatic cancer: a review of epidemiology, trend, and risk factors. World J Gastroenterol. 2021;27:4298‐4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014;371:2140‐2141. [DOI] [PubMed] [Google Scholar]
- 4. De La Cruz MS, Young AP, Ruffin MT. Diagnosis and management of pancreatic cancer. Am Fam Physician. 2014;89:626‐632. [PubMed] [Google Scholar]
- 5. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73:17‐48. [DOI] [PubMed] [Google Scholar]
- 6. Simoes PK, Olson SH, Saldia A, Kurtz RC. Epidemiology of pancreatic adenocarcinoma. Chin Clin Oncol. 2017;6:24. [DOI] [PubMed] [Google Scholar]
- 7. Lu Y, Gentiluomo M, Lorenzo‐Bermejo J, et al. Mendelian randomisation study of the effects of known and putative risk factors on pancreatic cancer. J Med Genet. 2020;57:820‐828. [DOI] [PubMed] [Google Scholar]
- 8. Peduzzi G, Felici A, Pellungrini R, et al. Analysis of exposome and genetic variability suggests stress as a major contributor for development of pancreatic ductal adenocarcinoma. Dig Liver Dis. 2023;56:1054‐1063. [DOI] [PubMed] [Google Scholar]
- 9. Galeotti AA, Gentiluomo M, Rizzato C, et al. Polygenic and multifactorial scores for pancreatic ductal adenocarcinoma risk prediction. J Med Genet. 2021;58:369‐377. [DOI] [PubMed] [Google Scholar]
- 10. Pistoni L, Gentiluomo M, Lu Y, et al. Associations between pancreatic expression quantitative traits and risk of pancreatic ductal adenocarcinoma. Carcinogenesis. 2021;42:1037‐1045. [DOI] [PubMed] [Google Scholar]
- 11. Lu Y, Corradi C, Gentiluomo M, et al. Association of genetic variants affecting microRNAs and pancreatic cancer risk. Front Genet. 2021;12:693933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gentiluomo M, Canzian F, Nicolini A, Gemignani F, Landi S, Campa D. Germline genetic variability in pancreatic cancer risk and prognosis. Semin Cancer Biol. 2022;79:105‐131. [DOI] [PubMed] [Google Scholar]
- 13. Nodari Y, Gentiluomo M, Mohelnikova‐Duchonova B, et al. Genetic and non‐genetic risk factors for early‐onset pancreatic cancer. Dig Liver Dis. 2023;55:1417‐1425. [DOI] [PubMed] [Google Scholar]
- 14. Corradi C, Lencioni G, Gentiluomo M, et al. Polymorphic variants involved in methylation regulation: a strategy to discover risk loci for pancreatic ductal adenocarcinoma. J Med Genet. 2023;60:980‐986. [DOI] [PubMed] [Google Scholar]
- 15. Giaccherini M, Farinella R, Gentiluomo M, et al. Association between a polymorphic variant in the CDKN2B‐AS1/ANRIL gene and pancreatic cancer risk. Int J Cancer. 2023;153:373‐379. [DOI] [PubMed] [Google Scholar]
- 16. Campa D, Matarazzi M, Greenhalf W, et al. Genetic determinants of telomere length and risk of pancreatic cancer: a PANDoRA study. Int J Cancer. 2019;144:1275‐1283. [DOI] [PubMed] [Google Scholar]
- 17. Luo W, Tao J, Zheng L, Zhang T. Current epidemiology of pancreatic cancer: challenges and opportunities. Chin J Cancer Res. 2020;32:705‐719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nipp R, Tramontano AC, Kong CY, et al. Disparities in cancer outcomes across age, sex, and race/ethnicity among patients with pancreatic cancer. Cancer Med. 2018;7:525‐535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yun Z, Zhichao J, Hao Y, et al. Targeting autophagy in multiple myeloma. Leuk Res. 2017;59:97‐104. [DOI] [PubMed] [Google Scholar]
- 20. Dykstra KM, Allen C, Born EJ, Tong H, Holstein SA. Mechanisms for autophagy modulation by isoprenoid biosynthetic pathway inhibitors in multiple myeloma cells. Oncotarget. 2015;6:41535‐41549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Garcia Ruiz O, Sanchez‐Maldonado JM, Lopez‐Nevot MA, et al. Autophagy in hematological malignancies. Cancers. 2022;14:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eng CH, Wang Z, Tkach D, et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc Natl Acad Sci U S A. 2016;113:182‐187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang S, Wang X, Contino G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717‐729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grasso D, Garcia MN, Iovanna JL. Autophagy in pancreatic cancer. Int J Cell Biol. 2012;2012:760498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dengjel J, Schoor O, Fischer R, et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci U S A. 2005;102:7922‐7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yamamoto K, Venida A, Yano J, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC‐I. Nature. 2020;581:100‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gillson J, Abd El‐Aziz YS, Leck LYW, et al. Autophagy: a key player in pancreatic cancer progression and a potential drug target. Cancers. 2022;14:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li J, Chen X, Kang R, Zeh H, Klionsky DJ, Tang D. Regulation and function of autophagy in pancreatic cancer. Autophagy. 2021;17:3275‐3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17:528‐542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Levy JM, Thorburn A. Targeting autophagy during cancer therapy to improve clinical outcomes. Pharmacol Ther. 2011;131:130‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stromnes IM, DelGiorno KE, Greenberg PD, Hingorani SR. Stromal reengineering to treat pancreas cancer. Carcinogenesis. 2014;35:1451‐1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang W, He R, Yang W, et al. Autophagic Schwann cells promote perineural invasion mediated by the NGF/ATG7 paracrine pathway in pancreatic cancer. J Exp Clin Cancer Res. 2022;41:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gorgulu K, Diakopoulos KN, Ai J, et al. Levels of the autophagy‐related 5 protein affect progression and metastasis of pancreatic tumors in mice. Gastroenterology. 2019;156:203‐217. [DOI] [PubMed] [Google Scholar]
- 34. Akar U, Ozpolat B, Mehta K, Fok J, Kondo Y, Lopez‐Berestein G. Tissue transglutaminase inhibits autophagy in pancreatic cancer cells. Mol Cancer Res. 2007;5:241‐249. [DOI] [PubMed] [Google Scholar]
- 35. Karasic TB, O'Hara MH, Loaiza‐Bonilla A, et al. Effect of gemcitabine and nab‐paclitaxel with or without hydroxychloroquine on patients with advanced pancreatic cancer: a phase 2 randomized clinical trial. JAMA Oncol. 2019;5:993‐998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wolpin BM, Rubinson DA, Wang X, et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist. 2014;19:637‐638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sainz J, Garcia‐Verdejo FJ, Martinez‐Bueno M, et al. Polymorphisms within autophagy‐related genes influence the risk of developing colorectal cancer: a meta‐analysis of four large cohorts. Cancers. 2021;13:1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Clavero E, Sanchez‐Maldonado JM, Macauda A, et al. Polymorphisms within autophagy‐related genes as susceptibility biomarkers for multiple myeloma: a meta‐analysis of three large cohorts and functional characterization. Int J Mol Sci. 2023;24:8500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kuballa P, Nolte WM, Castoreno AB, Xavier RJ. Autophagy and the immune system. Annu Rev Immunol. 2012;30:611‐646. [DOI] [PubMed] [Google Scholar]
- 40. Jiang GM, Tan Y, Wang H, et al. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol Cancer. 2019;18:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bakker OB, Aguirre‐Gamboa R, Sanna S, et al. Integration of multi‐omics data and deep phenotyping enables prediction of cytokine responses. Nat Immunol. 2018;19:776‐786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Amundadottir L, Kraft P, Stolzenberg‐Solomon RZ, et al. Genome‐wide association study identifies variants in the ABO locus associated with susceptibility to pancreatic cancer. Nat Genet. 2009;41:986‐990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Petersen GM, Amundadottir L, Fuchs CS, et al. A genome‐wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p15.33. Nat Genet. 2010;42:224‐228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wolpin BM, Rizzato C, Kraft P, et al. Genome‐wide association study identifies multiple susceptibility loci for pancreatic cancer. Nat Genet. 2014;46:994‐1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Childs EJ, Mocci E, Campa D, et al. Common variation at 2p13.3, 3q29, 7p13 and 17q25.1 associated with susceptibility to pancreatic cancer. Nat Genet. 2015;47:911‐916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Khurana E, Fu Y, Colonna V, et al. Integrative annotation of variants from 1092 humans: application to cancer genomics. Science. 2013;342:1235587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Genomes Project Consortium , Abecasis GR, Auton A, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Romo L, Findlay SD, Burge CB. Regulatory features aid interpretation of 3'UTR variants. Am J Hum Genet. 2024;111:350‐363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Das S, Forer L, Schonherr S, et al. Next‐generation genotype imputation service and methods. Nat Genet. 2016;48:1284‐1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome‐wide association studies. Nat Genet. 2006;38:904‐909. [DOI] [PubMed] [Google Scholar]
- 51. Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta‐analysis of genomewide association scans. Bioinformatics. 2010;26:2190‐2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Manuel Sanchez‐Maldonado J, Martinez‐Bueno M, Canhao H, et al. NFKB2 polymorphisms associate with the risk of developing rheumatoid arthritis and response to TNF inhibitors: results from the REPAIR consortium. Sci Rep. 2020;10:4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sanchez‐Maldonado JM, Cabrera‐Serrano AJ, Chattopadhyay S, et al. GWAS‐identified variants for obesity do not influence the risk of developing multiple myeloma: a population‐based study and meta‐analysis. Int J Mol Sci. 2023;24:6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Balduzzi S, Rucker G, Schwarzer G. How to perform a meta‐analysis with R: a practical tutorial. Evid Based Ment Health. 2019;22:153‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Li Y, Oosting M, Smeekens SP, et al. A functional genomics approach to understand variation in cytokine production in humans. Cell. 2016;167:1099‐1110. [DOI] [PubMed] [Google Scholar]
- 56. Rios‐Tamayo R, Lupianez CB, Campa D, et al. A common variant within the HNF1B gene is associated with overall survival of multiple myeloma patients: results from the IMMEnSE consortium and meta‐analysis. Oncotarget. 2016;7:59029‐59048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Westra HJ, Peters MJ, Esko T, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet. 2013;45:1238‐1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Xi H, Wang S, Wang B, et al. The role of interaction between autophagy and apoptosis in tumorigenesis (review). Oncol Rep. 2022;48:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang W, Li J, Tan J, et al. Endonuclease G promotes autophagy by suppressing mTOR signaling and activating the DNA damage response. Nat Commun. 2021;12:476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Roos WP, Kaina B. DNA damage‐induced cell death by apoptosis. Trends Mol Med. 2006;12:440‐450. [DOI] [PubMed] [Google Scholar]
- 61. Yeretssian G, Correa RG, Doiron K, et al. Non‐apoptotic role of BID in inflammation and innate immunity. Nature. 2011;474:96‐99. [DOI] [PubMed] [Google Scholar]
- 62. Masson F, Kupresanin F, Mount A, Strasser A, Belz GT. Bid and Bim collaborate during induction of T cell death in persistent infection. J Immunol. 2011;186:4059‐4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ward LD, Kellis M. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 2016;44:D877‐D881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Luo W, Li J, Zhang D, et al. Bid mediates anti‐apoptotic COX‐2 induction through the IKKbeta/NFkappaB pathway due to 5‐MCDE exposure. Curr Cancer Drug Targets. 2010;10:96‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang K, Yin XM, Chao DT, Milliman CL, Korsmeyer SJ. BID: a novel BH3 domain‐only death agonist. Genes Dev. 1996;10:2859‐2869. [DOI] [PubMed] [Google Scholar]
- 66. Zinkel SS, Ong CC, Ferguson DO, et al. Proapoptotic BID is required for myeloid homeostasis and tumor suppression. Genes Dev. 2003;17:229‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bertran‐Alamillo J, Gimenez‐Capitan A, Roman R, et al. BID expression determines the apoptotic fate of cancer cells after abrogation of the spindle assembly checkpoint by AURKB or TTK inhibitors. Mol Cancer. 2023;22:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491‐501. [DOI] [PubMed] [Google Scholar]
- 69. Lamparska‐Przybysz M, Gajkowska B, Motyl T. Cathepsins and BID are involved in the molecular switch between apoptosis and autophagy in breast cancer MCF‐7 cells exposed to camptothecin. J Physiol Pharmacol. 2005;56(Suppl 3):159‐179. [PubMed] [Google Scholar]
- 70. Sax JK, Fei P, Murphy ME, Bernhard E, Korsmeyer SJ, El‐Deiry WS. BID regulation by p53 contributes to chemosensitivity. Nat Cell Biol. 2002;4:842‐849. [DOI] [PubMed] [Google Scholar]
- 71. Xu X, Qian D, Liu H, et al. Genetic variants in the liver kinase B1‐AMP‐activated protein kinase pathway genes and pancreatic cancer risk. Mol Carcinog. 2019;58:1338‐1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chen F, Roberts NJ, Klein AP. Inherited pancreatic cancer. Chin Clin Oncol. 2017;6:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Katzke VA, Le Cornet C, Mahfouz R, et al. Are circulating immune cells a determinant of pancreatic cancer risk? A prospective study using epigenetic cell count measures. Cancer Epidemiol Biomarkers Prev. 2021;30:2179‐2187. [DOI] [PubMed] [Google Scholar]
- 74. Liu C, Cheng H, Luo G, et al. Circulating regulatory T cell subsets predict overall survival of patients with unresectable pancreatic cancer. Int J Oncol. 2017;51:686‐694. [DOI] [PubMed] [Google Scholar]
- 75. Cheng H, Luo G, Lu Y, et al. The combination of systemic inflammation‐based marker NLR and circulating regulatory T cells predicts the prognosis of resectable pancreatic cancer patients. Pancreatology. 2016;16:1080‐1084. [DOI] [PubMed] [Google Scholar]
- 76. Ito Y, Takeda T, Wakasa K, Tsujimoto M, Sakon M, Matsuura N. Expression of p73 and p63 proteins in pancreatic adenocarcinoma: p73 overexpression is inversely correlated with biological aggressiveness. Int J Mol Med. 2001;8:67‐71. [DOI] [PubMed] [Google Scholar]
- 77. Danilov AV, Neupane D, Nagaraja AS, et al. DeltaNp63alpha‐mediated induction of epidermal growth factor receptor promotes pancreatic cancer cell growth and chemoresistance. PLoS One. 2011;6:e26815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Flores ER, Sengupta S, Miller JB, et al. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell. 2005;7:363‐373. [DOI] [PubMed] [Google Scholar]
- 79. Rouschop KM, van den Beucken T, Dubois L, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest. 2010;120:127‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zhang X, Lao M, Yang H, et al. Targeting cancer‐associated fibroblast autophagy renders pancreatic cancer eradicable with immunochemotherapy by inhibiting adaptive immune resistance. Autophagy. 2024;20:1314‐1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Omar N, Yan B, Salto‐Tellez M. HER2: an emerging biomarker in non‐breast and non‐gastric cancers. Pathogenesis. 2015;2:1‐9. [Google Scholar]
- 82. Meyers N, Gerard C, Lemaigre FP, Jacquemin P. Differential impact of the ERBB receptors EGFR and ERBB2 on the initiation of precursor lesions of pancreatic ductal adenocarcinoma. Sci Rep. 2020;10:5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ortega MA, Pekarek L, Fraile‐Martinez O, et al. Implication of ERBB2 as a predictive tool for survival in patients with pancreatic cancer in histological studies. Curr Oncol. 2022;29:2442‐2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Li Z, Shao C, Liu X, et al. Oncogenic ERBB2 aberrations and KRAS mutations cooperate to promote pancreatic ductal adenocarcinoma progression. Carcinogenesis. 2020;41:44‐55. [DOI] [PubMed] [Google Scholar]
- 85. Shibata W, Kinoshita H, Hikiba Y, et al. Overexpression of HER2 in the pancreas promotes development of intraductal papillary mucinous neoplasms in mice. Sci Rep. 2018;8:6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Gore J, Imasuen‐Williams IE, Conteh AM, Craven KE, Cheng M, Korc M. Combined targeting of TGF‐beta, EGFR and HER2 suppresses lymphangiogenesis and metastasis in a pancreatic cancer model. Cancer Lett. 2016;379:143‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Stoecklein NH, Luebke AM, Erbersdobler A, et al. Copy number of chromosome 17 but not HER2 amplification predicts clinical outcome of patients with pancreatic ductal adenocarcinoma. J Clin Oncol. 2004;22:4737‐4745. [DOI] [PubMed] [Google Scholar]
- 88. Aumayr K, Soleiman A, Sahora K, et al. HER2 gene amplification and protein expression in pancreatic ductal adenocarcinomas. Appl Immunohistochem Mol Morphol. 2014;22:146‐152. [DOI] [PubMed] [Google Scholar]
- 89. Lin Y, Nakatochi M, Hosono Y, et al. Genome‐wide association meta‐analysis identifies GP2 gene risk variants for pancreatic cancer. Nat Commun. 2020;11:3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Campa D, Rizzato C, Capurso G, et al. Genetic susceptibility to pancreatic cancer and its functional characterisation: the PANcreatic Disease ReseArch (PANDoRA) consortium. Dig Liver Dis. 2013;45:95‐99. [DOI] [PubMed] [Google Scholar]
- 91. Campa D, Gentiluomo M, Stein A, et al. The PANcreatic Disease ReseArch (PANDoRA) consortium: ten years' experience of association studies to understand the genetic architecture of pancreatic cancer. Crit Rev Oncol Hematol. 2023;186:104020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting Information.
Table S1. List of selected genes.
Table S2. List of autophagy‐related SNPs.
Data Availability Statement
The genotype data used in the present study come from three pancreatic cancer GWAS datasets downloaded from the NCBI database of genotypes and phenotypes (dbGaP; study accession numbers phs000206.v5.p3 and phs000648. v1.p1; project reference #12644), namely PanScan I, PanScan II and PanC4. Genotyping data of the genetic markers analyzed in the PANDORA cohort are available on request from the corresponding author. Functional data used in this project have been meticulously catalogued and archived in the BBMRI‐NL data infrastructure (https://hfgp.bbmri.nl/) using the MOLGENIS open‐source platform for scientific data. This allows flexible data querying and download, including sufficiently rich metadata and interfaces for machine processing (R statistics, REST API) and using FAIR principles to optimize Findability, Accessibility, Interoperability and Reusability. Further information is available from the corresponding author upon request.