

Abstract
Advances in the molecular understanding of facioscapulohumeral muscular dystrophy (FSHD) have revealed that FSHD results from epigenetic de-repression of the DUX4 gene in skeletal muscle, which encodes a transcription factor that is active in early embryonic development but is normally silenced in almost all somatic tissues. These advances also led to the identification of targets for disease-altering therapies for FSHD, as well as an improved understanding of the molecular mechanism of the disease and factors that influence its progression. Together, these developments led the FSHD research community to shift its focus towards the development of disease-modifying treatments for FSHD. This Review presents advances in the molecular and clinical understanding of FSHD, discusses the potential targeted therapies that are currently being explored, some of which are already in clinical trials, and describes progress in the development of FSHD-specific outcome measures and assessment tools for use in future clinical trials.
Introduction
Facioscapulohumeral muscular dystrophy (FSHD) is one of the most common inherited muscle disorders of adulthood and, according to the most recent European epidemiological study of FSHD (published in 2014), has a prevalence of 5–12 affected individuals per 100,000 of the population1. After intensive research, consensus has been reached that FSHD results from epigenetic de-repression of the DUX4 gene in skeletal muscle. This gene encodes the transcription factor double homoebox 4 (DUX4), which is normally silenced in most somatic tissues2,3. These advances in understanding of the molecular mechanisms underlying FSHD led to new opportunities for the development of disease-altering therapies and prompted a substantial shift in focus for the FSHD research community towards the development of targeted treatments for FSHD. Academic groups and pharmaceutical companies now have active programmes developing targeted therapies for FSHD, and several clinical trials have already been initiated. In anticipation of additional upcoming trials, several challenges remain to be addressed owing to the rarity, clinical variability and slow progression of FSHD4.
This Review presents important advances in the molecular and clinical understanding of FSHD. In particular, we discuss the latest disease-altering therapies in various stages of research, ranging from proof-of-concept in vitro studies to clinical trials of a compound with proven DUX4-blocking effects. We propose a model for future clinical trials of disease-altering treatments for FSHD and discuss the outcome measures that are likely to be most clinically meaningful in the clinical trial setting. We also appraise the level of evidence for a proposed molecular biomarker of DUX4 activity in muscle and the utility of MRI neuroimaging biomarkers.
Clinical phenotype
FSHD is named after its distinctive phenotype, which consists of (often asymmetrical) weakness and atrophy of muscles of the face, shoulder girdle and upper arms5. Despite its name, the axial and leg muscles can also be affected. Muscle weakness is slowly progressive over the lifetime of a patient, although many individuals report long periods of disease stabilization interrupted by bursts of disease activity that are associated with rapid functional decline5,6. Although the pattern of muscle involvement is highly characteristic, the degree of muscle involvement is highly variable both between patients and between different muscles within the same patient. Eventually, approximately 20% of the patients with FSHD become wheelchair-dependent7.
The pattern of inheritance is most often autosomal dominant; however, in 10–30% of patients, FSHD is caused by de novo mutations, and individuals carrying FSHD mutations show a high frequency of somatic mosaicism8. Disease severity varies widely, even within affected families. Genetic studies reveal that up to 30% of family members carry FSHD mutations but have non-penetrant or minimally affected phenotypes that depend on their D4Z4 repeat length9–12. FSHD penetrance is unevenly distributed among the sexes. Non-penetrance is reported more often in women than in men9,13, and women with somatic mosaicism of DUX4-permissive, short D4Z4 repeat alleles were found to be non-affected carriers more often than were their male counterparts, even though the percentage of cells with DUX4-permissive, short D4Z4 repeat alleles was higher in mosaic women than in mosaic men14. Although some studies9,10,13,15 report higher clinical disease severity scores in male patients, other large cohort studies did not find a difference in disease severity between the sexes12,16,17. Contrary to the findings of earlier studies, a 2021 report of findings from a longitudinal study in a large FSHD1 cohort in the USA showed that, independent of genetics, female patients were more likely than male patients to progress to wheelchair use18. A protective effect of oestrogens in women with FSHD has been suggested on the basis of reports of female patients experiencing accelerated disease progression after receiving oestrogen-blocking therapy that triggered early menopause19. However, a clinical study of lifetime oestrogen exposure did not show any correlation between the (fairly small) variations in oestrogen exposure and FSHD severity20.
Age at symptom onset varies from infancy to late adulthood, but the disease mostly manifests between the age of 15 and 30 years. Disease onset below the age of 10 years is typically associated with a severe disease course21. A subgroup of patients with early-onset FSHD and a fairly consistent presentation defined by severe generalized muscle weakness and a high frequency of extramuscular disease manifestations is often referred to as having ‘infantile’ FSHD22,23. The extramuscular features of FSHD include sensorineural hearing loss, retinal vasculopathy that sometimes progresses to a pathology resembling Coats disease, an increased prevalence of (incomplete) right bundle branch block, restrictive lung disease and the possibility of cognitive impairment and epilepsy24. In patients with typical age at onset of FSHD, systemic features occur rarely and are mostly subclinical; moreover, involvement of the cardiac and respiratory muscles is rare and life expectancy is generally not reduced.
Currently, no curative or pharmacological treatments are available for FSHD. Treatment is focused on optimizing daily functioning, reducing the burden of pain and fatigue and surveillance for extramuscular complications25. Patients might benefit from the use of orthotic devices such as corsets for back support and leg braces for foot drop26. Patients with FSHD should be encouraged to engage in low-intensity aerobic exercise, which has a beneficial effect on chronic fatigue, physical activity and fitness27–29.
Disease mechanism
Identification of the FSHD-causing gene
In 1990, genetic linkage studies mapped an FSHD-associated locus to the distal end of chromosome 4q30,31. This work was soon followed by the discovery of de novo genetic rearrangements in the 4q35 locus in patients with FSHD32. These DNA rearrangements were defined as the partial deletion (shortening or contraction) of a tandem macrosatellite repeat called D4Z4 (ref. 33). Because the D4Z4 repeat itself was initially considered to be non-protein coding34, genes in close proximity to the D4Z4 repeat were among the first to be investigated as FSHD candidate genes35–38. Monosomy of distal 4q was not associated with FSHD, which excluded a loss-of-function mechanism for the observed mutations. Instead, a position-effect variegation model was considered, in which partial deletion of the D4Z4 repeat was thought to lead to altered cis-spreading of the 4q subtelomere chromatin structure resulting in the altered expression of proximal genes35,39–41. However, numerous follow-up genetic studies in FSHD families challenged this theory by failing to consistently demonstrate any transcriptional dysregulation of candidate proximal genes in FSHD muscle cells40,42–46. Indeed, some FSHD families have been identified in which the 4q subtelomere disease locus is physically separated from the so-called proximal genes by a 4;10 translocation, which casts further doubt on their involvement in FSHD pathogenesis47.
In healthy individuals, the D4Z4 macrosatellite repeat consists of between 8 and ~100 tandem repeat units of 3.3 kb each32 (Fig. 1). Each D4Z4 tandem repeat unit contains a retrogene that includes the full open reading frame of DUX430,31,34,48. The discovery of two equally common distal D4Z4 sequence variants, 4qA and 4qB (ref. 49), was followed by the observation that FSHD is exclusively associated with the 4qA variant50,51. The critical difference between these two genetic variants became apparent when a DUX4 polyadenylation signal (PAS) was found in the pLAM region33 flanking the most distal D4Z4 unit, which is specific to the 4qA genetic variant52,53. This PAS is used by DUX4 in FSHD muscle to stabilize its messenger RNAs (mRNAs) and hence facilitate protein translation. Of note, chromosome 10q also harbours a D4Z4 macrosatellite repeat that shows high sequence homology to the 4q D4Z4 macrosatellite repeat54,55, but size variations in the 10q D4Z4 repeat are typically not associated with FSHD because this chromosome lacks the DUX4 PAS56.
Fig. 1 |. The D4Z4 repeat array on chromosome 4q in healthy individuals and patients with facioscapulohumeral muscular dystrophy.
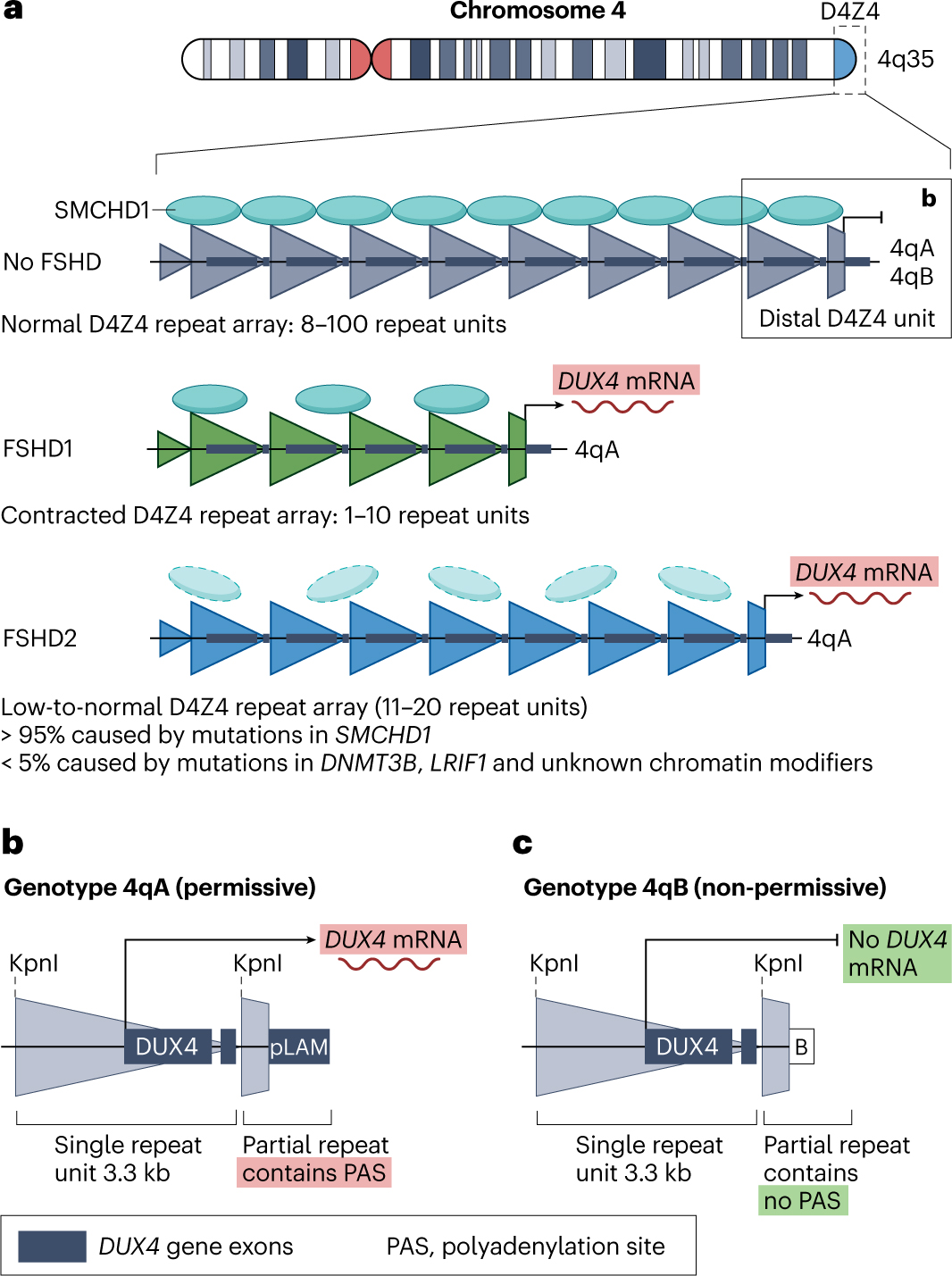
a, The D4Z4 macrosatellite tandem repeat array occurs in the subtelomeric region of the long arm of chromosome 4 (region 4q35). Each large triangle represents a single 3.3 kb D4Z4 unit. In healthy individuals, the D4Z4 array consists of 8–100 units and is epigenetically repressed, in part through structural maintenance of chromosomes flexible hinge domain containing protein 1 (SMCHD1) enrichment. In patients with facioscapulohumeral muscular dystrophy type 1 (FSHD1), the D4Z4 array is contracted on at least one allele to <10 repeats, leading to a more open chromatin structure and partial loss of SMCHD1 that ultimately enables DUX4 transcription from the most distal unit. In patients with FSHD2, a mutation either in SMCHD1 (>95% of patients) or in genes encoding other D4Z4 chromatin modifiers, such as DNMT3B and LRIF1 (≤5% of patients), de-represses DUX4 despite a low-to-intermediate normal-sized D4Z4 array. b, The most distal D4Z4 tandem repeat unit is flanked by an incomplete repeat (truncated triangle) followed by two equally prevalent genotypes, 4qA and 4qB, of which 4qA contains a 3ʹ-UTR DUX4 sequence. The pLAM region containing the PAS necessary to produce a stable DUX4 transcript is uniquely present in the 4qA genotype. Thus, transcription of the DUX4 retrogene can only occur from the most distal D4Z4 repeat unit in individuals with the 4qA genotype. KpnI, restriction enzyme cleavage site used for FSHD diagnostics; mRNA, messenger RNA; pLAM, DNA region flanking distal D4Z4 repeat unique to the 4qA genotype.
The presence of DUX4 mRNA and DUX4 protein in FSHD muscle cell cultures57 and their general absence (or occasionally very low abundance) in muscle cell cultures from symptom-free individuals57,58 confirmed the hypothesis that inappropriate expression of DUX4 in skeletal muscle causes FSHD2. This hypothesis is currently known as the unifying genetic model of FSHD.
Repression of the FSHD locus
The D4Z4 macrosatellite tandem repeat is epigenetically silenced in most somatic tissues of healthy individuals. This tandem repeat is CG-rich and has a higher than average frequency of CpG dinucleotides34, making it susceptible to transcriptional repression by DNA methylation59. The D4Z4 repeat is also characterized by other hallmarks of constitutive heterochromatin60, such as enrichment of the repressive modification trimethylation of histone H3 lysine 9 (H3K9me3) and the presence of repressive chromatin factors such as heterochromatin protein 1 (HP1) isoforms α and γ and cohesin61,62. Additionally, various chromatin modifiers have been identified that partake in D4Z4 repression, such as structural maintenance of chromosomes flexible hinge domain containing protein 1 (SMCHD1)3,63, nucleosome remodelling deacetylase complex, chromatin assembly factor 1 complex64, Polycomb repressive complex (PRC) 1 (ref. 65) and PRC2. PRC2 mediates the repressive histone mark trimethylation of histone H3 at lysine 27 (H3K27me3)62,66. The fact that several different transcriptional repressors are associated with D4Z4 suggests a degree of redundancy and highlights the importance of maintaining repression of this locus in somatic cells.
FSHD types 1 and 2
Two types of FSHD can be distinguished on the basis of the underlying epigenetic mechanism that causes D4Z4 chromatin relaxation and results in DUX4 de-repression in skeletal muscle. In autosomal dominant FSHD type 1 (FSHD1), which is the most common form of FSHD and affects 95% of all patients with this disease, de-repression is caused by partial deletion (also termed contraction) of the D4Z4 repeat on one 4qA allele32,33. Contraction reduces the number of repeat units to 1–10 (ref. 67), which leads to the partial loss of D4Z4 DNA methylation68,69 and a reduction in repressive H3K9me3 histone modifications on the affected allele61. Ultimately, these changes result in some degree of chromatin relaxation, which is sufficient to allow DUX4 transcription to occur in skeletal muscle.
In FSHD type 2 (FSHD2), which accounts for most of the remaining 5% of patients with FSHD, even the shortest D4Z4 allele typically still contains between 9 and 20 tandem repeat units67,70. Although this number of repeats is within the normal range for healthy individuals, in individuals with FSHD2, D4Z4 chromatin relaxation and DUX4 expression are both caused by a mutation in one of the chromatin repressors that normally participate in silencing of D4Z4. The most common cause of FSHD2 is a heterozygous mutation in the SMCHD1 gene, which accounts for >85% of the patients with FSHD2 (refs. 3,71). A rare cause of FSHD2 is a heterozygous mutation in DNMT3B, which encodes DNA methyltransferase 3β (ref. 72). Interestingly, mutations in these two genes can also cause two clinically unrelated conditions, bosma arhinia microphthalmia syndrome (BAMS) and immunodeficiency, centromeric instability and facial anomalies (ICF) syndrome, discussed subsequently. A homozygous mutation in LRIF1 (encoding ligand-dependent nuclear receptor-interacting factor 1) has also been identified as the cause of FSHD2 in one patient73. The roles of these genes in D4Z4 repression are discussed in more detail in the discussion of genotype–phenotype correlations and disease modifiers.
FSHD types 1 and 2 can co-occur in the same individual when an FSHD1 allele size in the upper end of the range (9–10 D4Z4 tandem repeats) is inherited together with an FSHD2-causing mutation, typically in SMCHD1 (ref. 74). These patients are described as having FSHD1 + 2, which is often characterized by an earlier onset of disease and more rapid progression when compared with patients who carry only one FSHD1 allele of an equivalent D4Z4 tandem repeat size67. We therefore propose that, instead of considering FSHD1 and FSHD2 as separate disease entities, they rather form a disease continuum in which the D4Z4 tandem repeat copy number mainly dictates the disease characteristics in the lower end of the range (1–6 tandem repeat units), whereas the function of chromatin modifiers such as SMCHD1 takes an increasingly prominent role with increasing D4Z4 tandem repeat copy number67.
Rare forms and unexplained FSHD
Rare cases of FSHD have been attributed to unusual DNA alterations that result in D4Z4 chromatin relaxation and permit DUX4 expression47,75,76. These alterations include deep intronic mutations in SMCHD1 that affect mRNA splicing77, small duplications (expansions) of the D4Z4 macrosatellite repeat with or without co-segregation of an SMCHD1 mutation76,78 and partial deletions of the D4Z4 macrosatellite repeat region that extend proximally into flanking non-D4Z4 sequences79,80.
For a small number of patients with FSHD who have a normal-sized but transcription-permissive D4Z4 macrosatellite repeat on a 4qA background, the causative mutation has not been identified. Therefore, other FSHD-related genes might exist that perhaps represent other D4Z4 chromatin modifiers81.
Role of DUX4 in health and disease
To understand the deleterious effects of DUX4 expression in skeletal muscle, it is important to consider the role of this transcription factor in normal development. DUX4 function has been comprehensively reviewed elsewhere82; thus, this section summarizes some of the most important features of DUX4 in healthy individuals and patients with FSHD.
DUX4 is involved in embryonic genome activation, which occurs during early embryonic development at the cleavage stage and acts as a molecular switch that is required to traverse the stages of normal embryonic development83–85. DUX4 expression peaks at the two-cell stage in mouse embryos and at the four-cell stage in human embryos, and this peak is closely followed by expression of other genes and repetitive elements that are activated by direct binding of DUX4 to their promoters, in a process known as embryonic genome activation or maternal-to-zygotic transition83,84. DUX4 is capable of altering the chromatin structural landscape to orchestrate the activation of transcription in otherwise silenced regions through recruitment of p300 and CREB binding protein histone acetyltransferases, which induce global reorganization of H3K27 acetylation86. DUX4 also induces the expression and incorporation of histone variants H3.X and H3.Y throughout the bodies of DUX4 target genes, which results in increased relaxation of the chromatin structure at those loci and enhanced target gene perdurance87.
DUX4 expression is suppressed in most, but not all, adult somatic tissues. Some evidence supports DUX4 expression in human thymus and skin60,88, which suggests that DUX4 might have roles in these adult somatic tissues, although these studies have not been replicated. DUX4 is also expressed in testes, where its expression originates from both the 4q and 10q loci. An alternative (further downstream) PAS is present on both of these chromosomes, in addition to the one that is active in FSHD skeletal muscle. This downstream PAS results in a unique (germline-specific) splice form of DUX4 (ref. 58). The exact DUX4 gene structure and PAS used during the zygotic cleavage stage are not known.
Experimental induction of DUX4 expression, whether overexpressed or inducibly expressed, is cytotoxic in various types of somatic cells, although it is unclear which cell death pathway is the main contributor to DUX4-induced cytotoxicity. Evidence suggests that both p53-dependent89–92 and p53-independent93,94 apoptotic pathways can be effectors of DUX4-induced cell death. Conversely, in induced pluripotent stem cells derived from patients with FSHD, DUX4 is activated via a primate-specific subtelomeric region that contains a p53 binding site95. Therefore, p53 is not only induced by DUX4 but also has been identified as a direct activator of DUX4, which creates a DUX4-inducing feedforward loop. Accordingly, p53 is a potential therapeutic target in FSHD95. Moreover, p53 is a well-known tumour suppressor and is also the driver of apoptosis in the DNA damage response (DDR) pathway96. Further research to elucidate the mechanisms leading to cell death following DUX4 expression could prove beneficial for additional target discovery.
Myoblasts from patients with FSHD are highly susceptible to oxidative stress97. Indeed, in a patient-derived induced pluripotent stem cell model, oxidative stress induced by hydrogen peroxide led to DUX4 expression as a result of serine-protein kinase ataxia-telangiectasia mutated-dependent activation of the DDR pathway and subsequent activation of p53 (ref. 98). Of note, in this study, inhibition of mitogen-activated protein kinase (MAPK) family members — among which p38 MAPK can be activated directly by oxidative stress — did not result in decreased DUX4 activation, indicating that DUX4 activation is not a direct effect of oxidative stress itself, but rather is reliant on other factors, such as DDR98. Thus, oxidative stress is not only a consequence of DUX4 expression97,99 but also acts as an inducer of DUX4 expression, possibly creating another p53-mediated feedforward loop. Indeed, given that p53 is the main inducer of apoptosis, it partakes in any cascade that results in DDR activation95.
Direct detection of DUX4 protein in FSHD myonuclei is challenging, because this protein is only transiently present and occurs in very few myonuclei. In many studies, therefore, transcriptional upregulation of DUX4 targets in skeletal muscle is considered indirect evidence of the presence of DUX4 protein. Concurrent measures of RNA and protein levels in DUX4+ cells showed that expression of DUX4 target genes was highly induced at both the mRNA and protein levels100. Indeed, a pilot study has provided direct evidence of DUX4 protein in a muscle biopsy sample from a patient with FSHD using a proximity ligation assay101. DUX4 protein abundance in cultured FSHD myocytes increases with myogenic differentiation, but DUX4 positivity remains sporadic, ranging from 1 in 200 myonuclei102 to 1 in 1,000 (ref. 58) or 1 in 2,000 (ref. 57) myonuclei. DUX4 protein was also detected in differentiated myocytes from some unaffected family members of FSHD patients, albeit with at a much lower abundance, corresponding to a frequency for DUX4 positivity of approximately 1 in 11,000 myonuclei57.
In tissue culture, the typical expression pattern comprises a highly DUX4+ nucleus flanked by nuclei with somewhat reduced DUX4 positivity, which suggests that DUX4 positivity spreads to neighbouring nuclei after a transcriptional burst102,103. Expression of DUX4 target genes follows this same pattern of spread and either occurs coincidentally with DUX4 expression or persists after transient DUX4 activation, where it acts as an indicator of an extinguished DUX4 transcriptional peak in the affected cells64.
DUX4 has many downstream target loci that are most probably activated in a dose-dependent manner104. This circumstance would explain why healthy relatives of patients with FSHD can sometimes show low levels of DUX4 expression that do not result in either transcriptional activation of DUX4 target genes or cytotoxicity57. Among the genes most robustly upregulated by DUX4, LEUTX, KHDC1L, TRIM43, PRAMEF2, ZSCAN4 and MBD3L2 expression represent a DUX4 signature of potential utility as a molecular biomarker64,84,104,105. Of note, methyl-CpG-binding domain protein 3-like 2 (encoded by MBD3L2) is involved in a positive feedforward loop that amplifies DUX4 expression and potentially facilitates the spread of DUX4 to neighbouring nuclei within individual myofibres64.
Single-cell RNA sequencing (RNA-seq) of primary differentiated mononuclear myocytes identified a small subset of cells with an FSHD-specific transcriptional profile, including expression of DUX4 and/or DUX4 target genes. In combination with pseudotime trajectory modelling, early and late DUX4-responding genes were identified. Identification of these genes enabled the reconstruction of an FSHD cellular progression model in which bursts of DUX4 expression leave a long-lasting ‘scar’ of transcriptionally upregulated DUX4 target genes that remain activated long after the peak in DUX4 expression that triggered this transcriptional deregulation has passed106. Single-nucleus RNA-seq of differentiating primary FSHD2 myoblasts during a time course of 6 days captured native DUX4 expression and identified a subpopulation of cells that express DUX4 signature genes, including the DUX4 paralogue DUXA. DUXA is implicated in the maintenance of DUX4 target gene expression in late stages of myocyte differentiation, suggesting that DUXA can take over the role of DUX4 and create self-sustaining expression of DUX4 signature genes long after DUX4 itself has disappeared from the nuclei107.
Correlates and modifiers of disease
D4Z4 repeat size
Variations in the age at onset of FSHD, disease severity and penetrance can be partly explained by genetic and epigenetic factors that act as disease modifiers. In FSHD1, disease severity roughly and inversely correlates to the number of D4Z4 repeat units on the contracted allele, especially in patients carrying short repeats (1–6 D4Z4 units). Patients carrying repeats of 1–3 units have a higher risk of belonging to the ‘early onset’ FSHD group, are more severely affected and have faster disease progression than those with longer repeats21. However, a few reports have described individuals with unusual non-penetrant repeats of 1–3 D4Z4 units, suggesting that other genetic elements might also play a part in disease presentation108. Patients with repeats of 7–10 D4Z4 units are more likely than those with shorter repeats to present with increased clinical variability, lower CpG methylation than expected for their D4Z4 repeat size (given that D4Z4 methylation is dependent on repeat size) and generally also have milder disease severity109,110. Also, repeats of 7–10 D4Z4 units are more often observed in non-penetrant than in penetrant carriers12. Consistent with their reduced penetrance, D4Z4 repeats of 7–10 units can be observed in the general European population at a frequency of 1–2%111,112.
DNA methylation affects D4Z4 regulation
DNA methylation is an important epigenetic disease modifier and regulator of D4Z4. Although one study113 and a review114 questioned the predictive value and diagnostic relevance of DNA methylation in patients with FSHD, possibly because of technical challenges113,114, substantial evidence supports D4Z4 hypomethylation as an epigenetic hallmark of FSHD59,67,68,110,115–123. Unlike in FSHD1, in which hypomethylation is restricted to the contracted D4Z4 allele and is considered a consequence of the contraction, in FSHD2 all D4Z4 repeats are hypomethylated116,117. Therefore, considerable reductions in pan-D4Z4 methylation are observed, especially in patients with FSHD2 (refs. 72,73,124). Moreover, specific CpG sites within D4Z4 are differentially methylated in patients with FSHD versus control individuals125. The methylation status of diagnostic region 1, which lies 1 kb upstream of the DUX4 open reading frame, can differentiate muscle samples from patients with FSHD from those of healthy control individuals and could also distinguish between muscle samples from patients with FSHD1 and those from patients with FSHD2 (refs. 59,120–123,125). In addition, CpG methylation analysis of the pLAM region can also be used to detect FSHD-associated hypomethylation, with the advantage that this method offers increased specificity for the 4qA disease allele by preventing co-amplification of the 4qB allele (in 4qA/4qB heterozygous carriers) and also by preventing co-amplification of 10q alleles69,122. D4Z4 CpG methylation status has been associated with variations in clinical disease severity123: among individuals carrying a D4Z4 repeat of 7–10 units, a greater reduction in CpG methylation was found (corrected for D4Z4 repeat size) only in symptomatic individuals with FSHD1 and not in non-penetrant mutation carriers126. Moreover, asymptomatic carriers of a shortened D4Z4 repeat were shown to have methylation levels similar to those of healthy control indivduals118.
SMCHD1 acts as an FSHD modifier
SMCHD1 is a chromatin modifier that has been shown to bind to the D4Z4 repeat, and thereby to repress DUX4, in multiple cell lines and in cultured muscle cells from healthy individuals3,127,128. Heterozygous nonsense or truncating mutations in SMCHD1 and/or SMCHD1 haploinsufficiency lead to decreased SMCHD1 protein levels in fibroblasts from patients with FSHD3 and a decrease in D4Z4-bound SMCHD13,128, and both these changes are associated with pan-D4Z4 hypomethylation and DUX4 de-repression in patients with FSHD2 (refs. 3,127,128). Likewise, heterozygous missense mutations in SMCHD1 result in pan-D4Z4 hypomethylation, DUX4 expression in skeletal muscle and manifestation of FSHD2 in individuals with a permissive (4qA) genetic background3. SMCHD1 is not only a causative gene for FSHD2 but has also been shown to function as a disease modifier in FSHD in general. Patients whose FSHD1 allele included a D4Z4 repeat size in the upper end of the range were shown to be more severely affected when they also had a heterozygous SMCHD1 mutation compared with their less severely affected family members who carried the same D4Z4 disease allele but lacked the SMCHD1 mutation74,119.
Knockdown of SMCHD1 expression in myotubes from patients with FSHD1 increases DUX4 expression74,127. Moreover, moderate overexpression of SMCHD1 in myotubes from patients with either FSHD1 or FSHD2 resulted in reduced DUX4 expression127, as did restoring near-normal SMCHD1 levels by clustered regularly interspaced short palindromic repeat (CRISPR)–Cas9-mediated excision of an intronic cryptic splice site in SMCHD1 in muscle cells from a patient with FSHD2 (ref. 77). These findings not only mark SMCHD1 as an interesting therapeutic target but also demonstrate that DUX4 de-repression in myotubes can be rescued and its effects potentially reversed.
Heterozygous SMCHD1 missense mutations are a cause not only of FSHD2 but also of the rare developmental disorder BAMS. BAMS is characterized by severe craniofacial malformations such as incomplete development of nose and eyes129,130. Indeed, BAMS can be considered to lie at the severe end of a clinical spectrum that also includes congenital nasal hypoplasia, anosmia, arrhinia and isolated hypogonadotropic hypogonadism, all of which can be associated with SMCHD1 mutations129–131. Contrary to FSHD2, in which a broad spectrum of SMCHD1 mutations can be found spanning the entire locus, mutations that cause the BAMS phenotype are exclusively of the missense type and are concentrated in an area encoding the extended ATPase domain of SMCHD1110,132. The nature of these mutations suggest that some, but not all, BAMS mutations are hypermorphic, based on observations of increased ATP hydrolysis by recombinant mutant SMCHD1 in in vitro quantification assays of ATPase function133,134. Interestingly, two mutations are associated with both FSHD2 and BAMS130,132. One of these shared mutations has been shown to result in increased ATPase activity of the recombinant protein in vitro133.
Despite this overlap in the genetic causes of FSHD2 and BAMS, patients with FSHD2 seem to be unaffected by any of the dysmorphic features characteristic of BAMS135. Conversely, one patient with BAMS who presented with FSHD-like symptoms was found to have a moderately sized DUX4-permissive allele, which suggests that the two disorders can coexist within the same individual130. A study that investigated the overlap between BAMS and FSHD phenotypes reported that 3 of 11 patients with BAMS caused by a mutation in SMCHD1 also met epigenetic and genetic criteria for FSHD2: DUX4 expression was found in dermal fibroblasts from these three patients with BAMS after epigenetic de-repression136. DUX4 expression was also found in myotubes derived from two of these three patients, who were therefore considered at risk of developing FSHD2. However, DUX4 mRNA levels in the fibroblasts and myotubes of these three patients were much lower than those of patients with FSHD2, and the three patients with BAMS showed no evidence of skeletal muscle involvement on muscle ultrasound or muscle MRI136. These data suggest that patients with BAMS can have an (epi)genetic background compatible with FSHD2 yet show no manifestations of this disease and, furthermore, that the BAMS and FSHD2 phenotypes are unrelated136. These findings are also in concordance with the incomplete penetrance of FSHD in FSHD families, in which low levels of DUX4 expression in asymptomatic carriers of pathogenic SMCHD1 mutations have also been reported16,57. However, a molecular explanation for the disparate clinical outcomes of SMCHD1 mutations in BAMS and FSHD remains largely elusive.
DNMT3B and LRIF1: more FSHD modifiers
DNMT3B has also been identified as an FSHD disease-modifying gene. Mutations in this gene resulted in manifest FSHD in carriers of an FSHD1-sized D4Z4 repeat as well as in carriers of a normal-sized D4Z4 repeat72. Biallelic DNMT3B mutations also cause autosomal recessive ICF type 1 (ICF1)137,138. Monoallelic DNMT3B mutations, by contrast, are a cause of FSHD2: in individuals with a monoallelic DNMT3B mutation inherited along with a short (9 repeat units) permissive D4Z4 allele, de-repression of DUX4 has been observed in muscle cell cultures72. DNA methyltransferase 3B (DNMT3B) is co-responsible (along with DNMT3A and DNMT3L, which DNMT3B co-regulates) for de novo CpG methylation and plays a crucial part in the establishment of CpG methylation on the inactive X chromosome (among other loci) during early embryonic development137,139,140. One of the main features of ICF1 is hypomethylation of CpG islands in pericentromeric satellite repeats and other repeat regions in the genome, including D4Z4141,142. Therefore, DNMT3B acts as a D4Z4 repressor. As in a subset of patients with BAMS, low levels of expression of DUX4 and/or its target genes were observed in dermal fibroblasts from two patients with ICF1 carrying a DUX4 permissive allele, and low levels of DUX4 protein were detected in myotubes derived from a third patient with ICF1 (ref. 72). However, these three patients with ICF1 did not develop any muscle phenotype, possibly owing to their young age and reduced life expectancy72. Interestingly, ICF1 mutation carriers (that is, the parents of patients with ICF1, who each carry a monoallelic DNMT3B mutation) are generally unaffected by FSHD2, which suggests that, as is the case for SMCHD1 mutations causing either FSHD2 or BAMS, genotype–phenotype relationships are only partly understood for DNMT3B mutation carriers.
A homozygous mutation in LRIF1 (the gene encoding ligand-dependent nuclear-receptor-interacting factor 1) that abolishes the long isoform of this protein is associated with FSHD2 (ref. 73). LRIF1 is known to interact with SMCHD1 and is responsible for recruitment of SMCHD1 to the inactive X chromosome in a manner dependent on all subtypes143 (that is, α, β and γ144) of HP1. In patient-derived immortalized fibroblasts forced to form myocytes by ectopic expression of myostatin D, lack of the long isoform of LRIF1 not only resulted in a reduction in total LRIF1 binding but also in reduced SMCHD1 binding to D4Z4. As a result, these cells exhibited D4Z4 hypomethylation and DUX4 expression, marking LRIF1 as a D4Z4 chromatin modifier and LRIF1 as a potential FSHD2 gene73.
Other factors linked to DUX4 expression
Chromatin remodelling proteins.
Several proteins orchestrate the chromatin structure of D4Z4 and contribute to D4Z4 repeat silencing in somatic cells. In non-affected individuals, the D4Z4 repeat carries transcription-repressive H3K9me3 and H3K27me3 histone marks associated with heterochromatin as well as transcription-permissive H3K4me2 and H3 acetylation marks associated with euchromatin. In patients with FSHD, the ratio between repressive and permissive histone marks favours an open chromatin structure owing to a deficit of SUV39H1, a histone methyltransferase that trimethylates H3K9 (refs. 44,61). In turn, SUV39H1-mediated loss of H3K9me3 leads to the failure to recruit HP1γ and cohesin to the D4Z4 repeat array61. The chromatin compaction score, a ratio obtained by dividing the abundance of repressive H3K9me3 by that of permissive H3K4me2 in the D4Z4 repeat region, shows a trend towards correlation with age-corrected clinical symptom severity scores in patients with FSHD1 and FSHD2145.
PRC2, a multiprotein complex found at D4Z4, is a chromatin repressor responsible for deposition of the repressive histone modification H3K27me3 (ref. 62). In patients with FSHD2, reduced SMCHD1 activity coincides with increased recruitment of PRC2 to D4Z4, which results in increased levels of H3K27me3. However, this compensatory mechanism is insufficient to maintain D4Z4 repression in somatic cells127. D4Z4-binding element transcript, a long non-coding RNA that is expressed from the D4Z4 locus exclusively in patients with FSHD, recruits histone-lysine N-methyltransferase ASH1L to D4Z4. ASH1L, in turn, is responsible for deposition of the open-chromatin mark H3K36me2, thereby actively counteracting PRC2-mediated transcriptional repression146,147.
Several other proteins and complexes mediate the repressive chromatin environment at D4Z4, namely, zinc finger transcription factor YY1 (ref. 40), the nucleosome remodelling deacetylase (NuRD) complex and chromatin assembly factor 1 (ref. 64). Considering the organizational complexity of the D4Z4 locus, other as-yet unknown D4Z4 modifiers probably remain to be uncovered, some of which could lead to new therapeutic avenues.
Environmental factors.
Environmental factors can influence the course of FSHD, although the molecular mechanisms that underlie environmental interactions with disease are rarely investigated and poorly understood. Aerobic exercise has been proven beneficial in multiple studies, as evaluated by clinical outcomes such as fatigue and exercise performance27,148. Beneficial effects of aerobic exercise therapy were also detectable on quantitative muscle MRI as a reduction in the rate of progression of fatty infiltration of the muscle tissue, indicating slower disease progression149.
Disease modifiers are of interest as potential therapeutic targets in FSHD, especially those proven to influence D4Z4 chromatin structure and DUX4 expression. Not all variability in the clinical presentation of FSHD is explained by the currently known disease modifiers16. Modern research methods such as whole-genome CRISPR–Cas9 screens and next-generation sequencing can aid in identifying as-yet unknown disease modifiers, close the knowledge gap in the observed disease variability in FSHD and uncover novel candidates for targeted therapy.
An integrated model of FSHD progression
We propose an integrated model of FSHD progression on the basis of clinical features, muscle imaging data and molecular findings. According to this model, fatty infiltration of the muscle tissue of single muscles progresses in a nonlinear fashion with phases of muscle inflammation preceding episodes of rapid fatty replacement, consistent with discrete bursts of DUX4 expression17,150–152. At any given time, different muscles experience different disease stages. This model reflects a slow linear progression of whole body muscle weakness overall, within which individual muscles show a relapsing course.
Even in patients without any clinical symptoms or abnormalities on muscle MRI, mild-to-moderate histological changes in muscle biopsy samples as well as subtle structural changes on muscle ultrasonography can be detected in the leg muscles of patients with FSHD150,153,154. To date, no studies have been performed to directly link these ultrasonographic and histological changes. The abnormalities seen on muscle ultrasonography might be due to early histological changes, such as intramuscular fibrosis, that cannot be adequately detected with current MRI techniques.
FSHD muscle biopsy samples with no evidence of DUX4 target gene expression still show mild-to-moderate histological changes and elevated expression of genes encoding extracellular matrix components and inflammatory mediators or genes involved in immune responses150,155. On the basis of these findings, we hypothesize that expression of DUX4 and DUX4 target genes might still occur at levels below the detection threshold before disease progression in an individual muscle. This situation could occur either because DUX4 expression is transient or because DUX4 is expressed only in a very small number of nuclei at any one time, as has been shown in tissue culture58. The abnormal histological findings and gene expression patterns detected in FSHD muscles even in the absence of detectable DUX4 mRNA or DUX4 target gene expression might reflect the accumulated damage from intermittent DUX4 expression in nuclei that are too few or are too briefly DUX4+ to detect by RNA-seq. These episodes of transient and/or low-level expression of DUX4 might, nonetheless, be sufficient to activate a primary or secondary immune response.
Further muscle deterioration is characterized by progressive fatty infiltration of the muscle tissue, which can be detected and longitudinally tracked by muscle MRI and is associated with advancing muscle weakness17,149,156–158. Evidence is accumulating that this fatty infiltration is often preceded by a phase of muscle inflammation, which can be visualized on muscle MRI as signal hyperintensities on short-tau inversion recovery (STIR) scans (a T2-weighted sequence with nulling of the fat signal)159,160. Although some muscles initially show inflammation that disappears over time without leaving any macroscopic consequences on muscle MRI, a more common scenario is that muscle inflammation precedes rapid progression of fatty infiltration of the muscle tissue17,151,161–163 (Fig. 2).
Fig. 2 |. Progression of facioscapulohumeral muscular dystrophy can be measured with MRI.
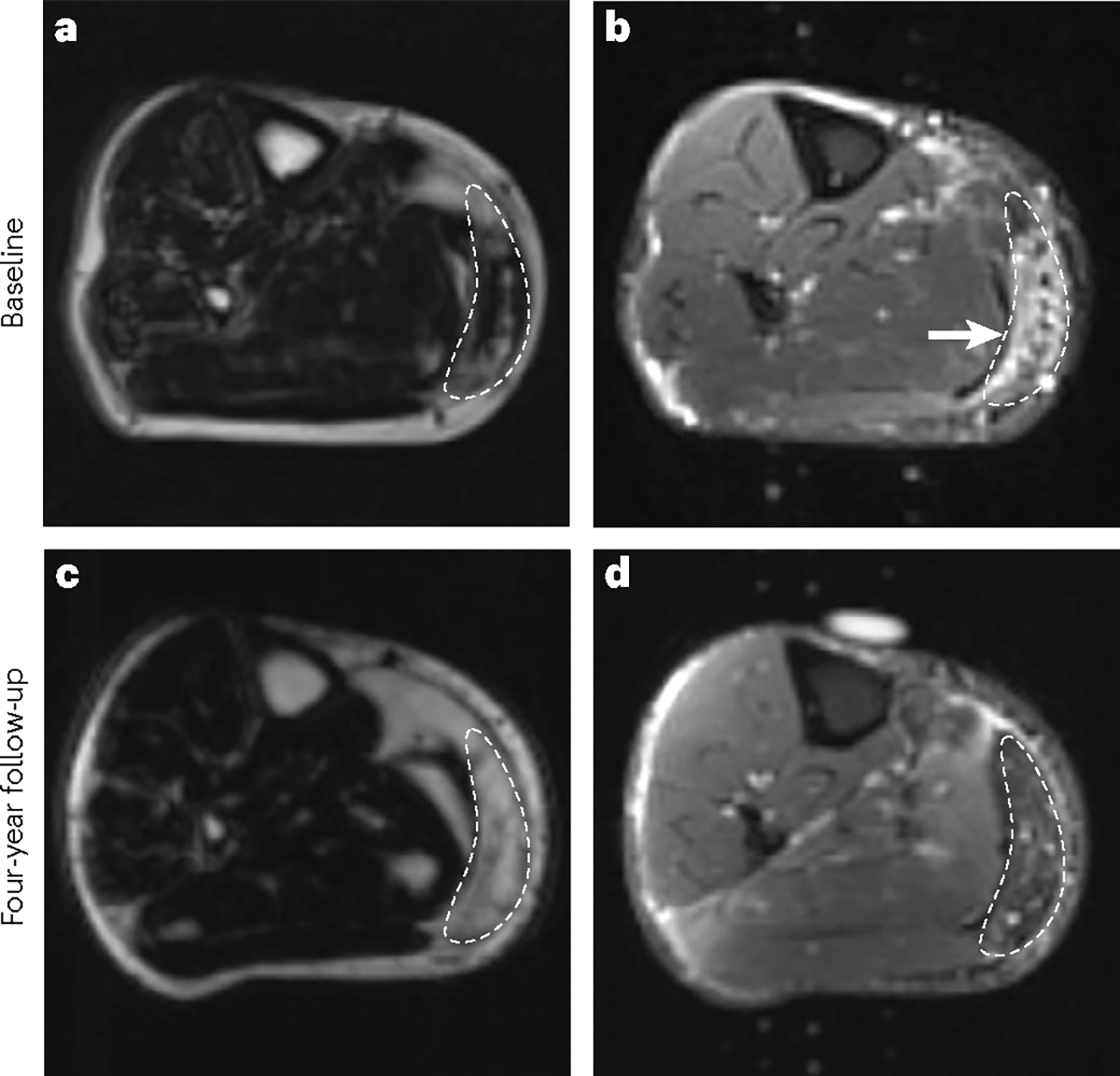
a, An MRI scan (Dixon sequence) showing mild-to-moderate fatty infiltration in a region of interest (outlined) of the right medial gastrocnemius muscle. b, A corresponding short tau inversion recovery image of the same patient shows a hyperintense signal (arrowhead) indicative of muscle inflammation. c, Follow-up MRI scan (Dixon sequence) of the right medial gastrocnemius muscle 4 years later of the same patient, showing facioscapulohumeral muscular dystrophy progression to near-complete fatty infiltration of the muscle tissue. d, A corresponding short tau inversion recovery image at the same time point shows that the hyperintense signal (muscle inflammation) has disappeared.
A study has investigated the correlation between two MRI characteristics: turbo inversion recovery magnitude (TIRM) hyperintensity (equivalent to the STIR sequence) and quantitative fat fraction; and two FSHD-associated molecular signatures — DUX4 target gene expression signature and PAX7 score — in muscle biopsy samples. TIRM hyperintensities correlated with active disease and were associated with increased expression of the DUX4 target gene signature, whereas increased expression of the DUX4 target gene signature and reduced PAX7 score were both found in muscles with a high fat fraction, which is characteristic of late stages of disease152. Molecular biomarkers of FSHD are described in more detail subsequently.
At a group level, disease progression is associated with increased expression of DUX4 and DUX4 target genes as well as increased expression of many extracellular matrix, inflammation and immune response genes in muscle biopsy samples, although in individual patients, biopsy samples might show fluctuating (either increased or decreased) expression of these genes over time104,150,155. In particular, the presence of STIR hyperintense areas seems to be strongly associated with aberrant DUX4 target gene expression, as well as with an active myopathic process marked by ongoing muscle fibre injury and repair in the presence or absence of associated inflammatory changes. Accelerated replacement of muscle by fat has also been associated with an intermediate fat content in muscles156,162,164. Graphs that arrange muscles in order of their fat content display an hourglass pattern, in which only a few muscles carry an intermediate fat content164. This fact suggests that once a muscle reaches an intermediate fat content, progression accelerates towards complete fatty replacement.
Once a muscle becomes nearly or completely replaced with fatty infiltrate, it reaches end-stage pathology and inflammation disappears151. At this point, the muscle has no contractile properties left and muscle damage is assumed to be irreversible.
Therapy development
Now that consensus has been reached on the underlying cause of FSHD — namely, the presence of DUX4 in skeletal muscle — the focus of research has shifted to development of disease-altering therapies (Fig. 3). In the past, several non-targeted therapies have been tested on a small scale (reviewed elsewhere165). In this Review, we focus on targeted therapies that are currently being developed and/or investigated in preclinical and clinical studies.
Fig. 3 |. DUX4-mediated pathways and possible methods of therapeutic inhibition.
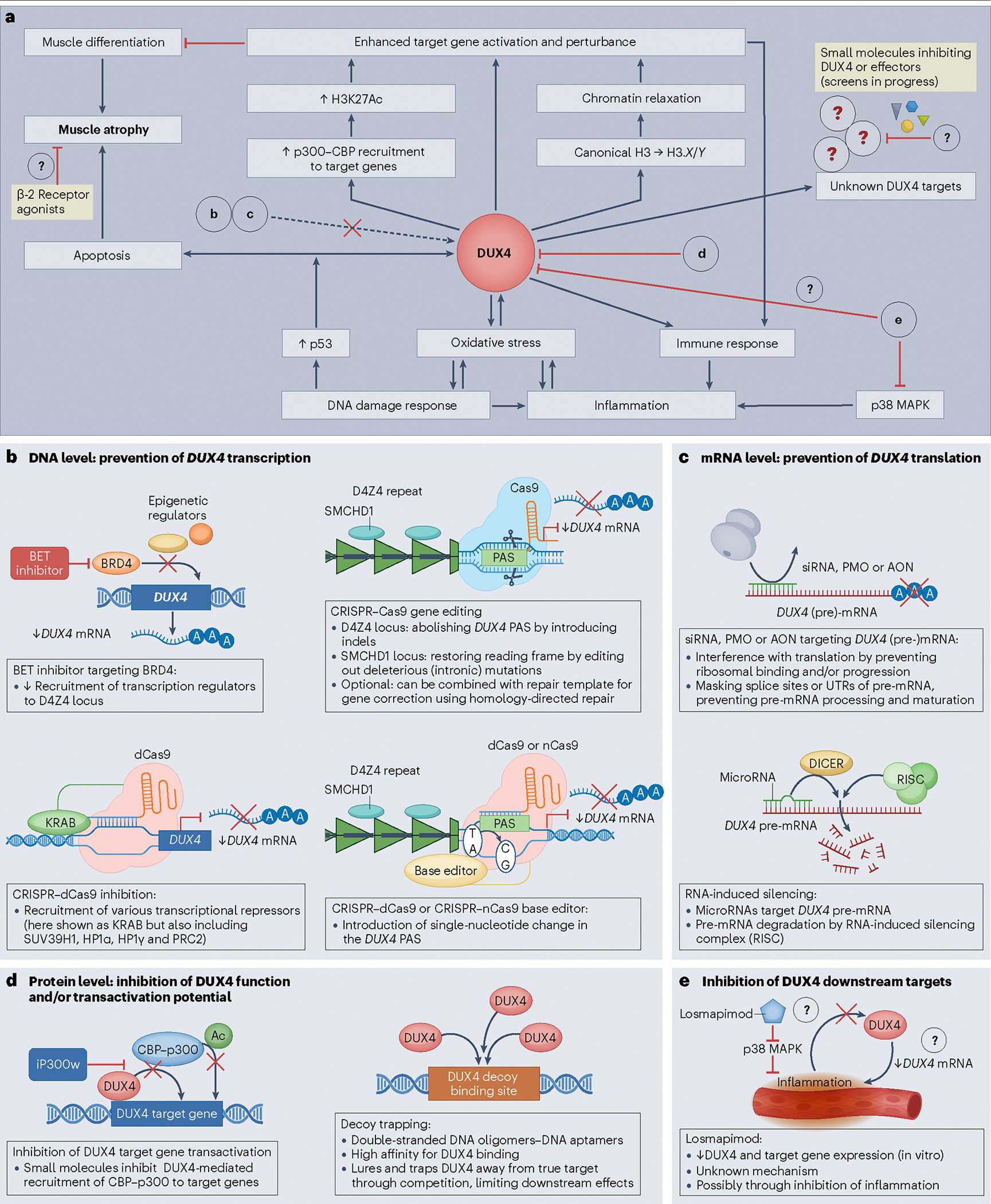
a, Pathways that are (partly) upregulated by and/or respond to DUX4 protein in skeletal muscle lead to transcriptional deregulation, inflammation and muscle atrophy. Possible interventions targeting either DUX4 function or DUX4 effectors are indicated with red inhibitory arrows. Bold letters refer to detailed figure panels showing the potential mechanisms that can attenuate the cytotoxic effects of DUX4. b, Top left. Bromodomain-containing protein 4 (BRD4) is an epigenetic regulator that activates DUX4 transcription by recruiting additional epigenetic regulators to the D4Z4 repeat. Bromodomain and extraterminal domain (BET) inhibitors block this BRD4-dependent recruitment and reduce DUX4 transcription. b, Bottom left. Clustered regularly interspaced short palindromic repeat (CRISPR)–dCas9 (‘dead’ caspase 9) inhibition recruits Krüppel-associated box (KRAB) zinc finger proteins and other transcriptional repressors to D4Z4, thereby reducing transcription of DUX4 and/or DUX4 target genes. b, right. CRISPR–Cas9 editing disrupts the polyadenylation signal (PAS), either through (top) inducing insertion–deletion mutations (indels) via double-stranded DNA breaks (traditional Cas9) or by (bottom) targeted conversion of a single DNA base pair using dCas9 or Cas9 nickase (nCas9) fused to a base editor protein. c, Phosphorodiamidate morpholino oligomers (PMOs), small interfering (si) RNAs or antisense oligonucleotides (AONs) bind to DUX4 transcripts and prevent maturation of pre-messenger RNA (mRNA) or ribosomal binding to mature mRNAs, thereby blocking translation. MicroRNAs that target DUX4 pre-mRNA can induce degradation of the RNA through the endoribonuclease DICER-activated RNA-induced silencing complex. d, At the protein level, DUX4 function can be blocked by the small molecule iP300w, which interferes with DUX4-dependent recruitment of the cyclic adenosine monophosphate response element binding protein (CREB)–p300 lysine acetyltransferase transcriptional activator complex, thereby inducing transcriptional deregulation. Alternatively, introducing competing high-affinity binding sites for DUX4 traps and prevents DUX4 from binding to its target genes. e, Although the exact mechanism of action of the p38 mitogen-activated protein kinase (MAPK) inhibitor losmapimod is unknown, this agent is proposed to reduce inflammation, possibly disrupting a feedforward loop that would otherwise increase DUX4 expression by increasing intracellular oxidative stress. SMCHD1, structural maintenance of chromosomes flexible hinge domain containing protein 1; UTR, untranslated region.
β2 Adrenergic agonists and p38 MAPK inhibitors
The results of two non-targeted trials of β2-adrenergic agonists in patients with FSHD showed limited beneficial effects on muscle mass and some measures of muscle strength in the short term166,167 (Fig. 3a). However, neither of these studies met their primary end point of a 52-week change in global strength as assessed by maximum voluntary isometric contraction testing166,167. More than a decade later, a targeted screen of a library enriched in pharmacological compounds with epigenetic activities revealed the DUX4-repressive action of β2 adrenergic agonists, which marked these compounds as potential DUX4 inhibitors168. Further investigations using small molecule inhibitors revealed that p38 MAPK is activated by β2 adrenergic signalling169. Interestingly, in this screen, the p38α and p38β MAPK isoforms were identified as potent inhibitors of DUX4, most probably independently of β2. This effect was confirmed in studies of the p38 MAPK inhibitor losmapimod in a mouse xenograft model, which provided evidence that DUX4-blocking effects can be achieved by targeting p38α and/or p38β MAPKs169,170. Oral losmapimod has also been tested in a randomized, double-blind phase 2b clinical trial involving 80 patients with FSHD171. Although no change in DUX4-driven gene expression was observed in losmapimod-treated patients, the researchers stated that losmapimod showed a clinically relevant benefit in terms of various functional and patient-reported outcome measures versus placebo. Patients in the treatment arm showed reduced progression of muscle fat infiltration on MRI over the course of the study in intermediately affected muscles when compared with the placebo group, but no difference in the progression of the muscle fat fraction and lean muscle volume. Given the anti-inflammatory properties of losmapimod169, it cannot be excluded that the benefits of losmapimod in this trial are (at least in part) due to its anti-inflammatory activity, independent of DUX4 reduction (Fig. 3a,e). The drug was well tolerated and no serious adverse effects were reported. These promising results still await peer-reviewed publication172,173.
BET-bromodomain inhibitors
The small molecule screen that identified the DUX4-repressive qualities of β2-adrenergic agonists also suggested that bromodomain and extra-terminal domain (BET) family members might attenuate DUX4 levels. BET inhibitors act by blocking the activity of bromodomain-containing protein 4 (BRD4), which in turn suppresses transcription of DUX4 mRNA and abrogates DUX4 activity168 (Fig. 3b, top left). BRD4 binds to acetylated open chromatin where it promotes transcription by recruiting transcription-regulatory complexes. BRD4 has been widely studied for its potential anticancer and anti-inflammatory effects174,175.
RNA interference
Small interfering RNAs (siRNAs), microRNAs (miRNAs or miRs) and antisense oligonucleotides (AONs) have therapeutic potential in FSHD owing to their capacity to block transcription and/or translation of DUX4 mRNA. Transcribed miRNA and siRNA molecules emerging from the D4Z4 repeat were identified early in the search for FSHD-causing genes53 and thought to be involved in RNA-mediated epigenetic silencing. Transfection with siRNAs bearing identical sequences to D4Z4-derived small siRNAs or miRNAs resulted in a strong reduction in DUX4 mRNA levels, increased H3K9me2 abundance at D4Z4 and increased recruitment of AGO2, an effector of RNA-mediated epigenetic silencing176 (Fig. 3c, bottom). Furthermore, inhibition of the DICER–AGO2 pathway led to de-repression of DUX4 expression, suggesting that this pathway provides a basal level of suppression176. Endogenous human miR-675 inhibits DUX4 mRNA in vitro and has muscle-protective effects when used as a gene therapy in an FSHD mouse model. Several small molecules have the potential to increase endogenous production of miR-675, including oestrogen and melatonin, marking miR-675 a candidate drug target177.
AONs comprise several different classes of molecules with unique chemical properties that can target RNA molecules in a sequence-specific manner. AONs can achieve knockdown of specific transcripts by preventing their translation and/or by inducing transcript degradation through the RNAse H1 pathway, depending on their specific chemistry178. Multiple studies have demonstrated the effectiveness of AONs for reducing levels of DUX4 protein, mature DUX4 mRNA levels and DUX4 target gene expression, both in vitro (in FSHD primary and immortalized myocytes) and in vivo (in mouse models)179–183. Systemic delivery of AONs targeting the DUX4 transcript in a transgenic mouse model proved to be successful in reducing both DUX4 mRNA transcript and DUX4 protein levels, as well as reducing DUX4 target gene expression. Compared with control mice that received a non-targeted AON, mice receiving the DUX4-targeted AON exhibited less skeletal muscle pathology and fibrosis, showed less inflammatory dysregulation and performed better on some functional outcome measures, specifically in relation to muscle strength182–185. AONs are therefore considered a feasible therapeutic strategy for FSHD (Fig. 3c, top).
Vector-based RNA interference offers the advantages of relatively easy in vivo delivery and a high cell transduction efficiency in post-mitotic cells. Adeno-associated virus (AAV) vectors have already been approved for use in human clinical trials, and trials of AAV-mediated therapies with many years of follow-up attest to their excellent safety profile186. Intramuscular administration of an AAV vector has been used to deliver miR-405 (which targets DUX4 mRNA) in mice co-transduced with AAV-DUX4. This approach successfully reduced DUX4 protein levels and DUX4-induced muscle pathology by directing the DUX4 mRNA transcript to the RNAi degradation pathway187,188.
Another class of oligonucleotides has demonstrated its potential for DUX4 inhibition. The targeting antisense sequence of the 5ʹ end of U7 small nuclear RNAs, a component of the small nuclear ribonucleoprotein complex involved in 3ʹ end processing of histone pre-mRNAs, was altered to specifically target DUX4 pre-mRNA, thereby interfering with its maturation. Treatment with this agent led to a substantial reduction in DUX4 transcript levels in FSHD muscle cells. The added benefit of this system compared with traditional AONs and RNAi-based therapies is the longevity of the small nuclear ribonucleoprotein complex that is redirected to target DUX4. This complex protects the included small nuclear RNA from degradation and enables intermittent administration of the therapeutic vector (in contrast to traditional AONs, which require continuous dosing)189. The necessity for lifelong administration (either continuous or intermittent) of RNAi-based therapies can be considered both a disadvantage (compared with single-dose gene-editing therapies, discussed subsequently) and a strength. RNAi-based treatment is generally non-permanent and self-inactivating, which enables dose correction or treatment discontinuation if adverse effects are observed.
DUX4 protein inhibition
DUX4 recruits the histone acetyltransferases p300 and CREB binding protein to its target loci, which then undergo local H3K27 acetylation that in turn leads to target gene expression86. Blocking the transactivation capacity of DUX4 could, therefore, be a viable method of interfering with the DUX4 cascade. Treatment with the p300-specific inhibitor iP300w is effective in blocking DUX4 target gene induction, inhibiting DUX4 cytotoxicity and reversing the DUX4-induced global accumulation of acetylated histone H3 (ref. 190) (Fig. 3d, left). Small molecule inhibitors have the added advantage of systemic administration, possibly even as drugs that can be taken orally.
Another method of blocking DUX4 transactivation has been tested in vitro and in vivo using ‘decoy’ DUX4 binding sites, which sequester endogenous DUX4 and prevent it from binding to and activating its target genes191 (Fig. 3d, right). DNA aptamers with optimized DUX4-binding affinity and specificity might further improve this decoy trapping strategy by increasing trapping efficiency192.
Gene editing
The CRISPR–Cas9 gene-editing system offers a versatile genetic toolbox for the development of DUX4-targeted therapy193–196. The first use of the CRISPR system in FSHD involved targeting the KRAB transcriptional inhibitor, fused to catalytically inactive, ‘dead’ Cas9 (dCas9), to the DUX4 promoter in FSHD myoblasts (Fig. 3b, bottom left). DUX4 and its target genes were successfully repressed. An increase of known repressive proteins on D4Z4 was also observed, although levels of H3K9me3 and H3K27me3 were not substantially altered197.
In FSHD2, deep intronic mutations in SMCHD1 that create an aberrant splice site have been targeted using the CRISPR–Cas9 system (Fig. 3b, top right). Excision of the mutation abolished the cryptic splice site and restored the wild-type SMCHD1 transcript, which was followed by a reduction in expression of DUX4 and DUX4 target genes. However, DNA methylation on the D4Z4 locus was not restored. Nonetheless, this study provided a proof-of-concept for the feasibility of this approach77.
Subsequent gene-editing strategies have mainly focused on abolishing the DUX4 exon 3 PAS. Targeting of this DUX4 PAS in FSHD myoblasts using CRISPR and TALEN (transcription activator-like effector nuclease) systems successfully eliminated the targeted DUX4 PAS, even though the editing efficiency was low198. However, this approach did not completely abolish DUX4 transcript levels, even though all three measured DUX4 target genes (MDB3L2, TRIM43 and LEUTX) assessed were downregulated, confirming the specificity of this approach. In fact, the researchers discovered that an alternative PAS, upstream of the targeted PAS, was responsible for the observed residual DUX4 transcript levels198. The observed redirection to an alternative PAS was identical to that described in earlier AON approaches that targeted DUX4 pre-mRNA179. Genetic and epigenetic targeting strategies were combined by targeting the DUX4 PAS directly and using the dCas9-KRAB inhibitor to restore the heterochromatin state at the D4Z4 locus. These two approaches led to reductions in DUX4 transcript levels and in DUX4 target gene expression, achieved by successfully deleting the PAS and restoration of the repressive H3K9me3 histone modification on the targeted locus, respectively199. In a separate study, the adenine base editor system200,201 was used along with Cas9-nickase202 to induce an AT to CG conversion (Fig. 3b, bottom right) in the DUX4 PAS in FSHD1 and FSHD2 patient-derived immortalized myoblasts, which effectively introduced mutations into the PAS that prevented DUX4 expression203.
Loci within repeats are challenging and risky to target. Owing to the repetitive nature of these sequences, the presence of multiple CRISPR–Cas9 binding sites can potentially lead to multiple DNA breaks along the locus, which not only creates unwanted off-target effects but also creates multiple danger signals to the cell and undermines editing efficiency at the on-target site by diluting the effect over a large area. Moreover, delivery of gene-editing systems such as CRISPR–Cas9 to muscle cells remains a considerable challenge that needs to be solved before this technique can develop beyond a promising proof-of-concept. One group tackled the delivery problem by using a dCas9 orthologue derived from Staphylococcus aureus (which is compact enough to be packaged into an AAV vector delivery system) rather than the more widely used Streptococcus pyogenes Cas9204. This strategy proved effective in restoring epigenetic repression of DUX4 without notable effects on the D4Z4 repeats adjacent to the targeted site or at the predicted and closest matching off-target genes. Moreover, this approach achieved repression of DUX4 and DUX4 target genes in vivo in an FSHD transgenic mouse model, albeit at a modest level of effect204. Altogether, the different CRISPR-based genetic and epigenetic strategies used so far have risen above a proof-of-principle stage and are viable methods that are worth pursuing towards therapeutic development.
Various approaches have been used with the aim of developing FSHD-specific, targeted therapies for clinical use (Fig. 3b and Table 1). The primary target of these therapies is DUX4, and these agents act by preventing or disrupting DUX4 transcription, preventing translation or inhibiting protein function (Fig. 3). All these potential therapies are in the preclinical stage, with the exception of losmapimod, which is progressing towards a phase 3 clinical trial. Each strategy presents challenges with respect to delivery, efficacy and safety and the need to address the sporadic nature of DUX4 expression. Moreover, the level of DUX4 that can be tolerated in the muscles of patients with FSHD remains unknown. Because DUX4 leaves a transcriptional memory in affected muscle cells, it is also unknown whether abolishing DUX4 expression can fully reverse or stabilize the damage already accumulated in affected muscles.
Table 1 |.
Targeted therapies for facioscapulohumeral muscular dystrophy
| Agent | Mechanism | Preclinical and clinical findings | Refs. |
|---|---|---|---|
| Small molecules | |||
| β2 Adrenergic receptor agonists | Presumed to inhibit DUX4 transcription by promoting muscle hypertrophy, in part through modulating cAMP levels | ↓DUX4 mRNA, ↓DUX4 target gene expression and ↑intracellular cAMP in FSHD patient-derived muscle cells RCTs in 90 and 65 patients with FSHD: ↑lean muscle mass, ↑grip strength; no change or limited improvement in maximum voluntary isometric strength in some muscles |
166–168 |
| BET protein BRD4 inhibitor | Inhibition of DUX4 transcription blocks BRD4- dependent recruitment of transcription regulators to D4Z4 | ↓DUX4 mRNA and ↓ DUX4 target gene expression in FSHD patient-derived muscle cells | 168 |
| p38 MAPK inhibitors (losmapimod) | Inhibition of p38 MAPK signalling has anti-inflammatory effects. Decreased DUX4 expression observed through unknown mechanism | ↓DUX4 mRNA and ↓DUX4 target gene expression in FSHD patient-derived muscle cells; ↓DUX4 and DUX4 target gene expression in a mouse FSHD xenograft model Phase 2b RCT in 80 patients with FSHD (awaits peer review): improvement in some patient-reported outcomes albeit without DUX4 attenuation (possibly through anti-inflammatory properties); phase 3 trial planned |
169–173 |
| P300 histone acetyltransferase inhibitor (iP300w) | Inhibits DUX4-mediated recruitment of p300–CBP to DUX4 target genes by blocking p300 | ↓DUX4 target gene expression; ↓global acetylated histone H3 abundance (accessible chromatin marker) in FSHD myotubes and in a transgenic FSHD mouse model | 190 |
| Oligotherapeutics | |||
| PMO and/or AON targeting 3’-UTR of DUX4 pre-mRNA (PAS and/or cleavage sites) | Prevents correct pre-mRNA processing (intron splicing or polyadenylation), inhibiting mRNA stability and promoting transcript degradation | ↓DUX4 mRNA and ↓DUX4 target gene expression; cleavage site redirection that rescued DUX4 observed with one PMO; ↓DUX4 protein and ↓atrophic myotubes; ↑muscle mass and strength; ↓myofibre central nucleation and muscle fibrosis; in FSHD patient-derived muscle cells, FSHD patient-derived primary muscle cells and in FSHD xenograft and/or transgenic FSHD mouse models | 179–184 |
| AON targeting exon 1 of DUX4 transcript | Inhibits DUX4 mRNA translation, promoting transcript degradation | ↓DUX4 mRNA, DUX4 protein and DUX4 target gene expression; ↓skeletal muscle pathology; improved treadmill test results; no improvement in strength test results in a transgenic FSHD mouse model | 185 |
| siRNAs bearing sequence homology to DUX4 | Exogenous siRNA targeting the 5’-UTR of DUX4 pre-mRNA | ↓DUX4 mRNA; ↑H3K9me2 and recruitment of AGO2 (indicating RNA-mediated epigenetic silencing) in FSHD patient-derived muscle cells | 176 |
| miR-405 | RNA interference through artificial miRNA targeting human DUX4 mRNA open reading frame, inhibiting transcription | ↓DUX4 mRNA in luciferase assay screen; ↓skeletal muscle pathology but no notable improvement in grip strength with AAV-miDUX4.405 overexpression in the AAV-DUX4-transgenic mouse model | 187,188 |
| miR-675 | RNA interference through naturally occurring human miR-675 directly targeting both the 3’-UTR and open reading frame of DUX4 mRNA | ↓DUX4 mRNA, ↓DUX4 protein, ↓DUX4 target gene expression, ↓DUX4-induced cell death; ↓transactivation of DUX4-responsive reporter in HEK293T cells transfected with DUX4 and in FSHD patient-derived muscle cells; ↓skeletal muscle pathology in transgenic (AAV-DUX4 induced) FSHD mouse model | 177 |
| U7 small nuclear (sn) RNA antisense expression cassettes | Alters target specificity of sn ribonucleoprotein complex to target DUX4 pre-mRNA, inhibiting its maturation | ↓DUX4-induced cell death; ↓DUX4 mRNA; ↓DUX4 protein in transfected HEK293 cells overexpressing DUX4; ↓DUX4 mRNA and ↓DUX4 target gene expression in FSHD patient-derived muscle cells | 189 |
| Double-stranded (ds) DNA oligonucleotides encoding DUX4 binding sites | Inhibits DUX4 transactivational activity through competition of decoy binding sites with real DUX4 target gene-binding sites | ↓DUX4 target gene expression on direct electrotransfection of dsDNA in FSHD patient-derived muscle cells and on systemic delivery of AAV-dsDNA decoy in mice overexpressing DUX4 from transfected plasmid vector | 191 |
| DNA aptamers | Bind DUX4 protein with increased affinity and specificity, inhibiting DUX4 target binding and activation | Aptamers enhance affinity and specificity of decoy binding site sequences for DUX4 using recombinant DUX4 (SELEX-derived aptamers have not yet been tested in DUX4-expressing cells) | 192 |
| Gene editing | |||
| CRISPR–Cas9 | Restores SMCHD1 reading frame by removing an intronic mutation causing a cryptic splice site | ↑Wild-type SMCHD1; ↓DUX4 mRNA; ↓DUX4 target gene expression in FSHD2 patient-derived muscle cells; no effect on D4Z4 DNA methylation | 77 |
| CRISPR–Cas9 | CRISPR–Cas9 and TALEN-based elimination of DUX4 PAS (exon 3) | ↓DUX4 mRNA and ↓DUX4 target gene expression in double-knockout HCT116 (human colorectal cancer cell) model; use of alternative PAS upstream of targeted PAS in FSHD patient-derived muscle cells | 198 |
| CRISPR–Cas9 | CRISPR–Cas9 elimination of DUX4 PAS (exon 3) | ↓DUX4 mRNA and ↓DUX4 target gene expression in double-knockout HCT116 human colorectal cancer cell model and in FSHD patient-derived muscle cells | 199 |
| CRISPR–dCas9 adenine base editor | Induces mutation (AT → CG conversion) in DUX4 PAS | ↓DUX4 mRNA and ↓DUX4 target gene expression in FSHD patient-derived muscle cells | 203 |
| CRISPR–dCas9 inhibition | |||
| CRISPR–dCas9 transcriptional repression | CRISPR–dCas9 orthologue fused to transcriptional repressors of DUX4 | ↓DUX4 mRNA and ↓DUX4 target gene expression in FSHD primary myocytes; ↓DUX4 and DUX4 target gene expression in an FSHD transgenic mouse model (more modest repression) | 204 |
| CRISPR–dCas9–KRAB transcriptional repression | KRAB-repressor targeting the DUX4 gene promoter | ↓DUX4 mRNA and ↓DUX4 target gene expression in FSHD primary myocytes; no effect on expression of other genes in the D4Z4 locus (FRG1 and FRG2) | 197 |
| CRISPR–dCas9–KRAB transcriptional repression | KRAB-repressor targeting DUX4 transcription activators | ↓DUX4 mRNA; ↑H3K9me3 (evidence of altered chromatin) at the targeted D4Z4 locus in FSHD primary myocytes | 147 |
| CRISPR–dCas9–KRAB transcriptional repression | KRAB-repressor targeting the DUX4 PAS | ↓DUX4 mRNA and DUX4 target gene expression; (partial) restoration of repressive H3K9me3 in FSHD patient-derived muscle cells | 199 |
AAV, adeno-associated virus; AGO2, argonaute RISC catalytic component 2; AON, antisense oligonucleotide; BET, bromodomain and extra terminal domain; BRD4, bromodomain-containing protein 4; cAMP, cyclic adenosine monophosphate; Cas9, caspase 9; CBP, cyclic adenosine monophosphate response element binding protein (CREB) binding protein; CRISPR, clustered regularly interspaced short palindromic repeat; dCas9, ‘dead’ (DNA cleavage-inactivated) Cas9; DUX4, double homeobox protein 4; FSHD, facioscapulohumeral muscular dystrophy; KRAB, Krüppel-associated box; MAPK, mitogen-activated protein kinase; PAS, polyadenylation signal; PMO, phosphorodiamidate morpholino oligomer; RCT, randomized controlled trial; SELEX, systematic evolution of ligands by exponential enrichment; siRNA, small interfering RNA; TALEN, transcription activator-like effector nucleases; UTR, untranslated region of pre-mRNA; mRNA, messenger RNA; miR, microRNA; SMCHD1, structural maintenance of chromosomes flexible hinge domain containing protein 1.
Clinical trial readiness
Important challenges also remain in the development of highly reliable and responsive biomarkers and clinical outcome measures for use in future clinical trials4,205. As variability between patients can be large and FSHD progression is generally slow, clinical trials are likely to require high numbers of individuals with this rare disease and a long follow-up time. For example, a natural history study in the 1990s, in which 50 patients with FSHD were followed up for 1 year, calculated that a two-armed clinical trial that used quantitative muscle strength as the primary outcome would require 160 patients per group and 1 year of follow-up to provide 80% power to detect a complete halt in disease progression206. To tackle these challenges, improved clinical trial access, sensitive biomarkers and relevant clinical outcome measures are required.
Clinical trial access
Access to clinical trials is facilitated by the use of patient registries in 14 different countries207,208. In addition to being an effective means of informing large numbers of patients about new trials, patients who fulfil the (strict) eligibility criteria can be approached selectively through the registry if clinical and genetic data are collected.
Biomarkers
Reliable biomarkers are also required, especially for early (phase 1–2) clinical trials, to accelerate the drug development process. Of most interest are pharmacodynamic biomarkers that can both serve as (secondary) clinical trial end points and provide a ‘proof of mechanism’ that confirms target engagement, thereby improving the chances of a novel therapy moving forward to later-phase and confirmatory clinical trials.
Although DUX4 protein levels might seem at first glance to be an appropriate molecular biomarker for disease activity in patients with FSHD, DUX4 itself is not ideal because this protein can be difficult to detect owing to its low levels and sporadic DUX4 gene expression in skeletal muscle57,58,209. Instead, the expression of several DUX4 target genes that comprise a DUX4 signature can be measured. This signature is useful for drug screening in patient-derived cells and inducible animal models (that is, models in which DUX4 expression can be switched on specifically in muscle cells) and is also measurable in patient-derived muscle biopsy samples104,105,150,210,211. Knockdown of DUX4 expression substantially decreases the expression of DUX4 target genes104,180. Various studies64,77,128,198 use combinations of 2–4 DUX4-upregulated target genes, among which LEUTX, KHDC1L, PRAMEF2, MDB3L2, ZSCAN4 and TRIM43 comprise a DUX4 signature panel. One study showed that a set of four genes (LEUTX, PRAMEF2, TRIM43 and KHDC1L) showed slightly superior performance in discriminating muscle biopsy samples from patients with FSHD from those of healthy control individuals104. However, the muscle biopsy samples from healthy control individuals could not be accurately distinguished from those of mildly affected patients with FSHD (mimicking early stage of disease) on the basis of expression of the signature genes alone155. An RNA-seq study in 36 patients with FSHD suggests that an expanded set of 52 genes, which comprises DUX4 target genes as well as other genes that are differentially expressed in FSHD muscle, encoding complement proteins, inflammatory mediators, lymphocyte markers, immunoglobulins and cell cycle regulators, might have increased discriminative power compared with just the above four DUX4-upregulated target genes for identifying early changes in FSHD muscle155.
Another candidate biomarker is the PAX7 score212. PAX7 is produced in satellite cells and is a marker for these quiescent myogenic progenitors. PAX7 and DUX4 exhibit high sequence similarity in their DNA binding domains and compete for DNA binding to their respective target loci212,213. DUX4 expression, therefore, competitively inhibits PAX7 target genes in differentiating myogenic cells, which results in an altered ratio between PAX7-induced and PAX7-repressed genes. This ratio is known as the PAX7 score212. The PAX7 score correlates with histological and muscle imaging measures of FSHD disease activity in a manner independent of, but partially overlapping with, DUX4 target gene expression152,214,215. Like DUX4, PAX7 also undergoes sporadic expression, a characteristic that similarly limits its suitability for use as a singular molecular biomarker. In areas where the DUX4 signature was increased in TIRM+ muscle biopsy samples, combining both signatures enabled detection of the later stages of disease, which are characterized by fat infiltration. These data suggest that although the PAX7 score and DUX4 signature overlap, they represent different stages of disease152. A study of FSHD muscle biopsy samples obtained from 36 participants in a different longitudinal study155 showed an increase in PAX7 target gene repression over 1 year of follow-up, suggesting that this biomarker is associated with FSHD disease progression215. Further validation of these potential muscle tissue biomarkers in independent patient cohorts is required, in addition to determining their test–retest reliability, variability between different samples within the same muscle and how large a change would be necessary to see a clinical effect.
Multiple exploratory studies have identified several potential serum biomarkers for FSHD216–223, the most promising of which are creatine kinase-myocardial band isoforms216,217, innate immunity mediator protein S100-A8 (ref. 220), IL-6 (ref. 221), complement components222, miR-206 (ref. 223) and a panel of 19 miRNAs that are upregulated in patients with FSHD219. Confirmatory studies need to be conducted to validate these biomarkers.
Several muscle imaging biomarkers are promising and could be especially useful to support the transition from early phase clinical trials to phase 3 trials. Both quantitative muscle MRI and quantitative ultrasonography are able to discriminate between-patient differences in the severity of muscle involvement and, on a cross-sectional level, both correlate strongly with clinical outcome measures153,158,164,224–230. MRI is better than ultrasonography for detecting the late stages of fatty infiltration, but ultrasonography can detect changes in muscles that appear normal on MRI153. In the past few years, muscle MRI has been used in two multicentre clinical trials: to assess muscle volume in a phase Ib–II study of recombinant human histidyl-tRNA synthetase and to assess quantitative fat fraction in a phase I and II study of losmapimod. In future clinical trials that include patients in the early disease stages of FSHD, ultrasonography biomarkers might be of particular interest.
As mentioned previously, muscle MRI can identify the muscles with active disease that are at highest risk of accelerated disease progression17,162,163. MRI-based targeting of such muscles (which are also likely to show high levels of DUX4 target gene expression) for muscle biopsy could be advantageous for assessing early responses of patients to treatment with a DUX4-targeted therapy. As such, the presence of STIR+ areas on muscle MRI might serve as an inclusion criterion for clinical trials. This use of imaging biomarkers is especially relevant because finding an appropriate time window for treatment administration might be challenging owing to the variability in disease stages between different muscles within one patient, the variability in disease course between patients and the high likelihood of only transient DUX4 expression. Longitudinal data show that both MRI and ultrasonography can detect progression of fatty infiltration in muscle tissue17,149,156,162,230. Whether the changes in imaging metrics correlate with clinical changes is yet to be established, because in most of these studies no changes occurred in clinical parameters over the course of the study, either because of small cohort sizes (up to 45 patients) or short follow-up periods (a maximum of 2 years). Additional longitudinal imaging studies are also required to define the increase in muscle fatty infiltration that represents a clinically relevant change.
Another MRI metric that could be used to measure disease progression or response to therapy is muscle volume. Although some early studies used muscle computed tomography or dual-energy X-ray absorptiometry to measure changes in muscle SMCHD1 mass, the clinical relevance of this parameter remains questionable because an increase in muscle mass in several clinical trials did not result in increased muscle strength166,167,231.
Clinical trial outcome measures
Clinical outcome measures that reflect how a patient ‘feels, functions or survives’ are essential for late phase clinical trials and for obtaining treatment approval. Regulatory agencies such as the FDA and EMA favour the use of clinical outcome measures in phase 3 clinical trials232,233. FSHD-specific outcomes are preferred, because outcome measures designed to assess function in patients with other neuromuscular disorders are often not fully suited to assess change in patients with FSHD, because the rate of progression and distribution of weakness across the body vary across different neuromuscular disorders234.
In the past few years, multiple FSHD-specific measurement instruments have been developed and are currently undergoing further validation. These include functional outcome measures, most importantly the FSHD Composite Outcome Measure (FSHD-COM)235 and Reachable Workspace (RWS)236–238, and two FSHD-specific patient-reported outcomes, namely, the FSHD Health Index (FSHD-HI)239 and FSHD-Rasch-built Overall Disability Scale (FSHD-RODS)4,207. A detailed description of these instruments is beyond the scope of this Review. All these outcome measures are currently being tested in longitudinal studies. The results of these studies will be important to determine the sensitivity to change, sample size requirements and minimal clinically relevant changes for each tool. Until these data are available, these outcome measures should be used with caution in clinical trials.
Conclusions
After decades of studying the disease mechanism of FSHD and uncovering many potential therapeutic targets, consensus has been reached that aberrant DUX4 expression is the root cause of FSHD and, as a result, the field is entering a new era focusing on the development of potentially disease-modifying therapy. Many promising molecular pathways and (epi)genetic tools have successfully demonstrated proof-of-concept applications in the treatment of FSHD, and many more are rapidly being uncovered by emerging single-cell and single-molecule approaches, as well as CRISPR–Cas9 genome editing tools. The first DUX4-targeted therapies are either already being investigated or moving towards in vivo studies and making their way towards clinical approval. Of these, losmapimod, a p38 MAPK inhibitor, has been tested in a recent phase 2 trial.
In vivo studies also require reliable efficacy biomarkers. Although the expression of DUX4 signature genes and the PAX7 score seem to be viable biomarkers of active FSHD disease processes in cultured cells and muscle biopsy samples, the search for easily applicable (serum) biomarkers continues. Several muscle imaging modalities are currently being investigated to identify metrics that could be used as non-invasive clinical biomarkers, and these studies have also demonstrated the suitability of quantitative MRI and ultrasonography for measuring FSHD progression.
Various functional and patient-reported outcome measures are currently being tested in longitudinal studies. With more trials on the way, an increasingly important research focus is the further development of clinical outcome measures and investment in worldwide patient registries and trial readiness. At the same time, studies of the basic molecular underpinnings of this disease continue to fuel translational studies that lead to the development of new therapies. This exciting new era is expected to benefit patients and bear witness to the joint efforts of many research groups and industry in making the final leap from bench to bedside.
Key points.
Facioscapulohumeral muscular dystrophy (FSHD), a disorder for which there currently is no cure, is characterized by muscle weakness, predominantly affecting muscles in the face, shoulder girdle and upper arms.
FSHD is associated with epigenetic de-repression of the DUX4 gene, which leads to aberrant expression of the transcription factor DUX4 and cytotoxicity in skeletal muscle cells.
Clinical disease progression occurs in a nonlinear and muscle-by-muscle fashion with phases of muscle inflammation preceding rapid fatty replacement of muscle tissue and muscle wasting.
Consensus has been reached on the pathogenetic mechanism of FSHD, and the field is entering a new era of targeted therapy development.
Disease-altering therapies currently in development range from proof-of-principle gene-editing technologies focusing on reducing DUX4 expression to clinical trials of DUX4-blocking agents.
Clinical trials in FSHD require the development of meaningful patient outcome measures, identification of reliable biomarkers and accurate methods of measuring disease progression, such as MRI and ultrasonography.
Acknowledgements
The authors apologize to those whose work was not cited or insufficiently cited because of size constraints. Our work is supported by the Prinses Beatrix Spierfonds (W.OR19-06, W.OR21-04), public–private partnerships allowances made available by Health-Holland, Top Sector Life Sciences and Health, The Netherlands, the Medical Research Council UK, Stichting Spieren voor Spieren, the National Institute of Neurological Disorders and Stroke (Grant No. P01NS069539) and the National Institute of Arthritis and Musculoskeletal and Skin Diseases (Grant No. P50AR065139). Some of the authors are members of the Radboud University Medical Centre (UMC), Center of Expertise for Neuromuscular Disorders (Radboud-NMD), Netherlands Neuromuscular Center (NL-NMD) and the European Reference Network for Rare Neuromuscular Diseases [ERN EURO-NMD].
Glossary
- cis-spreading
Spreading of epigenetic chromatin modifications from a locus to neighbouring loci on the same chromatin strand
- FSHD Composite Outcome Measure
(FSHD-COM) An evaluator-administered instrument composed of individually validated functional motor tasks that assess leg, shoulder and arm, trunk and hand function and balance
- FSHD Health Index
(FSHD-HI) A patient-reported outcome measures to assess total FSHD health-related quality of life and 14 separate subdomains, each identified by FSHD patients as having the greatest importance to their specific population
- FSHD-Rasch-built Overall Disability Scale
(FSHD-RODS) A linear-weighted patient-reported outcome measure for detecting activity and participation restrictions in patients with FSHD
- PAX7 score
The ratio of genes induced to genes repressed indicated by the satellite cell marker PAX7, which is closely related to DUX4
- Perdurance
Lasting target gene expression followed after a short DUX4 transcriptional burst
- pLAM
A DNA region flanking the most distal D4Z4 unit that contains the polyadenylation signal (PAS) of the DUX4 gene, named after the cosmid clone used for its isolation
- Proximity ligation assay
An antibody assay based on rolling-circle amplification of a DNA loop formed from ligation of oligonucleotide-labelled probes bound to distinct primary antibodies, used for detection of proteins at single-molecule resolution in unmodified cells and tissues
- Pseudotime trajectory modelling
A computational method used in single-cell transcriptomics to establish the pattern of a dynamic cellular process; followed by ordering of cells based on their progression through the process
- Reachable Workspace
(RWS) A depth-ranging sensor (Kinect)-based upper extremity motion analysis system that is applied to determine the spectrum of reachable workspace encountered with the arms and shoulders
- Retrogene
A segment of DNA reverse-transcribed from mRNA and randomly inserted into a genome
Footnotes
Competing interests
K.M. declares that she has acted as a consultant for Avidity Biosciences. S.J.T. declares that he has acted as a consultant for Avidity Biosciences and is a Board member for Renogenyx. R.T. declares that he has acted as a consultant and/or is a member of the advisory board for Arrowhead Pharma, Avidity Biosciences, Dyne Therapeutics, Fulcrum Therapeutics, Mitsubishi Tanabe Pharma, miRecule Biotech and Roche. J.M.S. declares that he has acted as a consultant and/or is a member of the advisory board for Acceleron, Avidity Biosciences, Dyne Therapeutics, Fulcrum Therapeutics, Ionis, ML Bio Solutions, Mitsubishi Tanabe Pharma, Roche and Sarepta. S.M.v.d.M. declares that he has acted as consultant and/or is a member of the advisory board for Avidity Biosciences, Dyne Therapeutics and Fulcrum Therapeutics and is a Board member for Renogenyx. The other authors declare no competing interests.
Additional information
Peer review information Nature Reviews Neurology thanks the anonymous reviewers for their contribution to the peer review of this work.
References
- 1.Deenen JC et al. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology 83, 1056–1059 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lemmers RJ et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 329, 1650–1653 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lemmers RJ et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet. 44, 1370–1374 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tawil R et al. Clinical trial preparedness in facioscapulohumeral muscular dystrophy: clinical, tissue, and imaging outcome measures 29–30 May 2015, Rochester, New York. Neuromuscul. Disord. 26, 181–186 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Mul K et al. What’s in a name? The clinical features of facioscapulohumeral muscular dystrophy. Pract. Neurol. 16, 201–207 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Tawil R & Van Der Maarel SM Facioscapulohumeral muscular dystrophy. Muscle Nerve 34, 1–15 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Statland JM & Tawil R Risk of functional impairment in facioscapulohumeral muscular dystrophy. Muscle Nerve 49, 520–527 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Lemmers RJ et al. Somatic mosaicism in FSHD often goes undetected. Ann. Neurol. 55, 845–850 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Tonini MM et al. Asymptomatic carriers and gender differences in facioscapulohumeral muscular dystrophy (FSHD). Neuromuscul. Disord. 14, 33–38 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Ricci G et al. Large scale genotype–phenotype analyses indicate that novel prognostic tools are required for families with facioscapulohumeral muscular dystrophy. Brain 10.1093/brain/awt226 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salort-Campana E et al. Low penetrance in facioscapulohumeral muscular dystrophy type 1 with large pathological D4Z4 alleles: a cross-sectional multicenter study. Orphanet J. Rare Dis. 10, 2 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wohlgemuth M et al. A family-based study into penetrance in facioscapulohumeral muscular dystrophy type 1. Neurology 91, e444–e454 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zatz M et al. The facioscapulohumeral muscular dystrophy (FSHD1) gene affects males more severely and more frequently than females. Am. J. Med. Genet. 77, 155–161 (1998). [PubMed] [Google Scholar]
- 14.van der Maarel SM et al. De novo facioscapulohumeral muscular dystrophy: frequent somatic mosaicism, sex-dependent phenotype, and the role of mitotic transchromosomal repeat interaction between chromosomes 4 and 10. Am. J. Hum. Genet. 66, 26–35 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sakellariou P et al. Mutation spectrum and phenotypic manifestation in FSHD Greek patients. Neuromuscul. Disord. 22, 339–349 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Mul K et al. Phenotype–genotype relations in facioscapulohumeral muscular dystrophy type 1. Clin. Genet. 94, 521–527 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Monforte M et al. Tracking muscle wasting and disease activity in facioscapulohumeral muscular dystrophy by qualitative longitudinal imaging. J. Cachexia Sarcopenia Muscle 10, 1258–1265 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Katz NK et al. Predictors of functional outcomes in patients with facioscapulohumeral muscular dystrophy. Brain 144, 3451–3460 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teveroni E et al. Estrogens enhance myoblast differentiation in facioscapulohumeral muscular dystrophy by antagonizing DUX4 activity. J. Clin. Invest. 127, 1531–1545 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mul K, Horlings CGC, Voermans NC, Schreuder THA & van Engelen BGM Lifetime endogenous estrogen exposure and disease severity in female patients with facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 28, 508–511 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Goselink RJM et al. Early onset as a marker for disease severity in facioscapulohumeral muscular dystrophy. Neurology 92, e378–e385 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klinge L et al. Severe phenotype in infantile facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 16, 553–558 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Brouwer OF, Padberg GW, Wijmenga C & Frants RR Facioscapulohumeral muscular dystrophy in early childhood. Arch. Neurol. 51, 387–394 (1994). [DOI] [PubMed] [Google Scholar]
- 24.Goselink RJM et al. Early onset facioscapulohumeral dystrophy — a systematic review using individual patient data. Neuromuscul. Disord. 27, 1077–1083 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Tawil R et al. Evidence-based guideline summary: evaluation, diagnosis, and management of facioscapulohumeral muscular dystrophy: report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular and Electrodiagnostic Medicine. Neurology 85, 357–364 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aprile I et al. Balance and walking involvement in facioscapulohumeral dystrophy: a pilot study on the effects of custom lower limb orthoses. Eur. J. Phys. Rehab. Med. 49, 169–178 (2013). [PubMed] [Google Scholar]
- 27.Voet N et al. Both aerobic exercise and cognitive-behavioral therapy reduce chronic fatigue in FSHD: an RCT. Neurology 83, 1914–1922 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Andersen G, Prahm KP, Dahlqvist JR, Citirak G & Vissing J Aerobic training and postexercise protein in facioscapulohumeral muscular dystrophy: RCT study. Neurology 85, 396–403 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Andersen G, Heje K, Buch AE & Vissing J High-intensity interval training in facioscapulohumeral muscular dystrophy type 1: a randomized clinical trial. J. Neurol. 264, 1099–1106 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Wijmenga C et al. Location of facioscapulohumeral muscular dystrophy gene on chromosome 4. Lancet 336, 651–653 (1990). [DOI] [PubMed] [Google Scholar]
- 31.Wijmenga C et al. Mapping of facioscapulohumeral muscular dystrophy gene to chromosome 4q35-qter by multipoint linkage analysis and in situ hybridization. Genomics 9, 570–575 (1991). [DOI] [PubMed] [Google Scholar]
- 32.Wijmenga C et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat. Genet. 2, 26–30 (1992). [DOI] [PubMed] [Google Scholar]
- 33.van Deutekom JC et al. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum. Mol. Genet. 2, 2037–2042 (1993). [DOI] [PubMed] [Google Scholar]
- 34.Hewitt JE et al. Analysis of the tandem repeat locus D4Z4 associated with facioscapulohumeral muscular dystrophy. Hum. Mol. Genet. 3, 1287–1295 (1994). [DOI] [PubMed] [Google Scholar]
- 35.van Deutekom JC et al. Identification of the first gene (FRG1) from the FSHD region on human chromosome 4q35. Hum. Mol. Genet. 5, 581–590 (1996). [DOI] [PubMed] [Google Scholar]
- 36.van Geel M et al. The FSHD region on human chromosome 4q35 contains potential coding regions among pseudogenes and a high density of repeat elements. Genomics 61, 55–65 (1999). [DOI] [PubMed] [Google Scholar]
- 37.van Geel M et al. Identification of a novel β-tubulin subfamily with one member (TUBB4Q) located near the telomere of chromosome region 4q35. Cytogenet. Cell Genet. 88, 316–321 (2000). [DOI] [PubMed] [Google Scholar]
- 38.Rijkers T et al. FRG2, an FSHD candidate gene, is transcriptionally upregulated in differentiating primary myoblast cultures of FSHD patients. J. Med. Genet. 41, 826–836 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tupler R et al. Monosomy of distal 4q does not cause facioscapulohumeral muscular dystrophy. J. Med. Genet. 33, 366–370 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gabellini D, Green MR & Tupler R Inappropriate gene activation in FSHD: a repressor complex binds a chromosomal repeat deleted in dystrophic muscle. Cell 110, 339–348 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Bodega B et al. Remodeling of the chromatin structure of the facioscapulohumeral muscular dystrophy (FSHD) locus and upregulation of FSHD-related gene 1 (FRG1) expression during human myogenic differentiation. BMC Biol. 7, 41 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klooster R et al. Comprehensive expression analysis of FSHD candidate genes at the mRNA and protein level. Eur. J. Hum. Genet. 17, 1615–1624 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang G et al. Testing the position-effect variegation hypothesis for facioscapulohumeral muscular dystrophy by analysis of histone modification and gene expression in subtelomeric 4q. Hum. Mol. Genet. 12, 2909–2921 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Cheli S et al. Expression profiling of FSHD-1 and FSHD-2 cells during myogenic differentiation evidences common and distinctive gene dysregulation patterns. PLoS ONE 6, e20966 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thijssen PE et al. DUX4 promotes transcription of FRG2 by directly activating its promoter in facioscapulohumeral muscular dystrophy. Skelet. Muscle 4, 19 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferri G, Huichalaf CH, Caccia R & Gabellini D Direct interplay between two candidate genes in FSHD muscular dystrophy. Hum. Mol. Genet. 24, 1256–1266 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lemmers R et al. Chromosome 10q-linked FSHD identifies DUX4 as principal disease gene. J. Med. Genet. 59, 180–188 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gabriels J et al. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene 236, 25–32 (1999). [DOI] [PubMed] [Google Scholar]
- 49.van Geel M et al. Genomic analysis of human chromosome 10q and 4q telomeres suggests a common origin. Genomics 79, 210–217 (2002). [DOI] [PubMed] [Google Scholar]
- 50.Lemmers RJ et al. Facioscapulohumeral muscular dystrophy is uniquely associated with one of the two variants of the 4q subtelomere. Nat. Genet. 32, 235–236 (2002). [DOI] [PubMed] [Google Scholar]
- 51.Lemmers RJ et al. Contractions of D4Z4 on 4qB subtelomeres do not cause facioscapulohumeral muscular dystrophy. Am. J. Hum. Genet. 75, 1124–1130 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dixit M et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc. Natl Acad. Sci. USA 104, 18157–18162 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Snider L et al. RNA transcripts, miRNA-sized fragments and proteins produced from D4Z4 units: new candidates for the pathophysiology of facioscapulohumeral dystrophy. Hum. Mol. Genet. 18, 2414–2430 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bakker E et al. The FSHD-linked locus D4F104S1 (p13E-11) on 4q35 has a homologue on 10qter. Muscle Nerve. 2, S39–S44 (1995). [PubMed] [Google Scholar]
- 55.Deidda G et al. Physical mapping evidence for a duplicated region on chromosome 10qter showing high homology with the facioscapulohumeral muscular dystrophy locus on chromosome 4qter. Eur. J. Hum. Genet. 3, 155–167 (1995). [DOI] [PubMed] [Google Scholar]
- 56.Lemmers RJ et al. Worldwide population analysis of the 4q and 10q subtelomeres identifies only four discrete interchromosomal sequence transfers in human evolution. Am. J. Hum. Genet. 86, 364–377 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jones TI et al. Facioscapulohumeral muscular dystrophy family studies of DUX4 expression: evidence for disease modifiers and a quantitative model of pathogenesis. Hum. Mol. Genet. 21, 4419–4430 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Snider L et al. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet. 6, e1001181 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huichalaf C, Micheloni S, Ferri G, Caccia R & Gabellini D DNA methylation analysis of the macrosatellite repeat associated with FSHD muscular dystrophy at single nucleotide level. PLoS ONE 9, e115278 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Das S & Chadwick BP Influence of repressive histone and DNA methylation upon D4Z4 transcription in non-myogenic cells. PLoS ONE 11, e0160022 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zeng W et al. Specific loss of histone H3 lysine 9 trimethylation and HP1γ/cohesin binding at D4Z4 repeats is associated with facioscapulohumeral dystrophy (FSHD). PLoS Genet 5, e1000559 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boros J, Arnoult N, Stroobant V, Collet JF & Decottignies A Polycomb repressive complex 2 and H3K27me3 cooperate with H3K9 methylation to maintain heterochromatin protein 1α at chromatin. Mol. Cell Biol. 34, 3662–3674 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen K et al. Genome-wide binding and mechanistic analyses of Smchd1-mediated epigenetic regulation. Proc. Natl Acad. Sci. USA 112, E3535–E3544 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Campbell AE et al. NuRD and CAF-1-mediated silencing of the D4Z4 array is modulated by DUX4-induced MBD3L proteins. eLife 7, e31023 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Casa V et al. Polycomb repressive complex 1 provides a molecular explanation for repeat copy number dependency in FSHD muscular dystrophy. Hum. Mol. Genet. 26, 753–767 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Haynes P, Bomsztyk K & Miller DG Sporadic DUX4 expression in FSHD myocytes is associated with incomplete repression by the PRC2 complex and gain of H3K9 acetylation on the contracted D4Z4 allele. Epigenet. Chromatin 11, 47 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sacconi S et al. FSHD1 and FSHD2 form a disease continuum. Neurology 92, e2273–e2285 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Overveld PG et al. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat. Genet. 35, 315–317 (2003). [DOI] [PubMed] [Google Scholar]
- 69.Calandra P et al. Allele-specific DNA hypomethylation characterises FSHD1 and FSHD2. J. Med. Genet. 53, 348–355 (2016). [DOI] [PubMed] [Google Scholar]
- 70.Rieken A, Bossler AD, Mathews KD & Moore SA CLIA Laboratory testing for facioscapulohumeral dystrophy: a retrospective analysis. Neurology 96, e1054–e1062 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lemmers RJ et al. Hemizygosity for SMCHD1 in facioscapulohumeral muscular dystrophy type 2: consequences for 18p deletion syndrome. Hum. Mutat. 36, 679–683 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.van den Boogaard ML et al. Mutations in DNMT3B modify epigenetic repression of the D4Z4 repeat and the penetrance of facioscapulohumeral dystrophy. Am. J. Hum. Genet. 98, 1020–1029 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hamanaka K et al. Homozygous nonsense variant in LRIF1 associated with facioscapulohumeral muscular dystrophy. Neurology 94, e2441–e2447 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sacconi S et al. The FSHD2 gene SMCHD1 is a modifier of disease severity in families affected by FSHD1. Am. J. Hum. Genet. 93, 744–751 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.van Deutekom JC et al. Evidence for subtelomeric exchange of 3.3 kb tandemly repeated units between chromosomes 4q35 and 10q26: implications for genetic counselling and etiology of FSHD1. Hum. Mol. Genet. 5, 1997–2003 (1996). [DOI] [PubMed] [Google Scholar]
- 76.Nguyen K et al. Deciphering the complexity of the 4q and 10q subtelomeres by molecular combing in healthy individuals and patients with facioscapulohumeral dystrophy. J. Med. Genet. 56, 590–601 (2019). [DOI] [PubMed] [Google Scholar]
- 77.Goossens R et al. Intronic SMCHD1 variants in FSHD: testing the potential for CRISPR-Cas9 genome editing. J. Med. Genet. 56, 828–837 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lemmers RJLF et al. Cis D4Z4 repeat duplications associated with facioscapulohumeral muscular dystrophy type 2. Hum. Mol. Genet. 27, 3488–3497 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lemmers R et al. High-resolution breakpoint junction mapping of proximally extended D4Z4 deletions in FSHD1 reveals evidence for a founder effect. Hum. Mol. Genet. 31, 748–760 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lemmers RJ et al. D4F104S1 deletion in facioscapulohumeral muscular dystrophy: phenotype, size, and detection. Neurology 61, 178–183 (2003). [DOI] [PubMed] [Google Scholar]
- 81.van den Boogaard ML et al. Double SMCHD1 variants in FSHD2: the synergistic effect of two SMCHD1 variants on D4Z4 hypomethylation and disease penetrance in FSHD2. Eur. J. Hum. Genet. 24, 78–85 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mocciaro E, Runfola V, Ghezzi P, Pannese M & Gabellini D DUX4 role in normal physiology and in FSHD muscular dystrophy. Cells 10.3390/cells10123322 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Geng LN et al. DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev. Cell 22, 38–51 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hendrickson PG et al. Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL/HERVL retrotransposons. Nat. Genet. 49, 925–934 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.De Iaco A et al. DUX-family transcription factors regulate zygotic genome activation in placental mammals. Nat. Genet. 49, 941–994 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Choi SH et al. DUX4 recruits p300/CBP through its C-terminus and induces global H3K27 acetylation changes. Nucleic Acids Res. 44, 5161–5173 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Resnick R et al. DUX4-induced histone variants H3.X and H3.Y mark DUX4 target genes for expression. Cell Rep. 29, 1812–1820.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gannon OM, Merida de Long L & Saunders NA DUX4 is derepressed in late-differentiating keratinocytes in conjunction with loss of H3K9me3 epigenetic repression. J. Invest. Dermatol. 136, 1299–1302 (2016). [DOI] [PubMed] [Google Scholar]
- 89.Kowaljow V et al. The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscul. Disord. 17, 611–623 (2007). [DOI] [PubMed] [Google Scholar]
- 90.Knopp P et al. DUX4 induces a transcriptome more characteristic of a less-differentiated cell state and inhibits myogenesis. J. Cell Sci. 129, 3816–3831 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wallace LM et al. DUX4, a candidate gene for facioscapulohumeral muscular dystrophy, causes p53-dependent myopathy in vivo. Ann. Neurol. 69, 540–552 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lek A et al. Applying genome-wide CRISPR–Cas9 screens for therapeutic discovery in facioscapulohumeral muscular dystrophy. Sci. Transl. Med. 10.1126/scitranslmed.aay0271 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bosnakovski D et al. p53-independent DUX4 pathology in cell and animal models of facioscapulohumeral muscular dystrophy. Dis. Model. Mech. 10, 1211–1216 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shadle SC et al. DUX4-induced dsRNA and MYC mRNA stabilization activate apoptotic pathways in human cell models of facioscapulohumeral dystrophy. PLoS Genet. 13, e1006658 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Grow EJ et al. p53 convergently activates Dux/DUX4 in embryonic stem cells and in facioscapulohumeral muscular dystrophy cell models. Nat. Genet. 53, 1207–1220 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vogelstein B, Lane D & Levine AJ Surfing the p53 network. Nature 408, 307–310 (2000). [DOI] [PubMed] [Google Scholar]
- 97.Winokur ST et al. Expression profiling of FSHD muscle supports a defect in specific stages of myogenic differentiation. Hum. Mol. Genet. 12, 2895–2907 (2003). [DOI] [PubMed] [Google Scholar]
- 98.Sasaki-Honda M et al. A patient-derived iPSC model revealed oxidative stress increases facioscapulohumeral muscular dystrophy-causative DUX4. Hum. Mol. Genet. 27, 4024–4035 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tsumagari K et al. Gene expression during normal and FSHD myogenesis. BMC Med. Genom. 4, 67 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jagannathan S, Ogata Y, Gafken PR, Tapscott SJ & Bradley RK Quantitative proteomics reveals key roles for post-transcriptional gene regulation in the molecular pathology of facioscapulohumeral muscular dystrophy. eLife 8, e41740 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Beermann ML, Homma S & Miller JB Proximity ligation assay to detect DUX4 protein in FSHD1 muscle: a pilot study. BMC Res. Notes 15, 163 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tassin A et al. DUX4 expression in FSHD muscle cells: how could such a rare protein cause a myopathy. J. Cell. Mol. Med. 17, 76–89 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rickard AM, Petek LM & Miller DG Endogenous DUX4 expression in FSHD myotubes is sufficient to cause cell death and disrupts RNA splicing and cell migration pathways. Hum. Mol. Genet. 24, 5901–5914 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yao Z et al. DUX4-induced gene expression is the major molecular signature in FSHD skeletal muscle. Hum. Mol. Genet. 23, 5342–5352 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jagannathan S et al. Model systems of DUX4 expression recapitulate the transcriptional profile of FSHD cells. Hum. Mol. Genet. 25, 4419–4431 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Heuvel AVD et al. Single-cell RNA-sequencing in facioscapulohumeral muscular dystrophy disease etiology and development. Hum. Mol. Genet. 28, 1064–1075 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jiang S et al. Single-nucleus RNA-seq identifies divergent populations of FSHD2 myotube nuclei. PLoS Genet 16, e1008754 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nikolic A et al. Clinical expression of facioscapulohumeral muscular dystrophy in carriers of 1–3 D4Z4 reduced alleles: experience of the FSHD Italian National Registry. BMJ Open 6, e007798 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Statland JM et al. Milder phenotype in facioscapulohumeral dystrophy with 7–10 residual D4Z4 repeats. Neurology 85, 2147–2150 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lemmers RJ et al. Inter-individual differences in CpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Hum. Mol. Genet. 24, 659–669 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Scionti I et al. Large-scale population analysis challenges the current criteria for the molecular diagnosis of fascioscapulohumeral muscular dystrophy. Am. J. Hum. Genet. 90, 628–635 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lemmers RJ et al. Specific sequence variations within the 4q35 region are associated with facioscapulohumeral muscular dystrophy. Am. J. Hum. Genet. 81, 884–894 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nikolic A et al. Interpretation of the epigenetic signature of facioscapulohumeral muscular dystrophy in light of genotype–phenotype studies. Int. J. Mol. Sci. 21, 2635 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Salsi V, Magdinier F & Tupler R Does DNA methylation matter in FSHD. Genes 11, 258 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.van Overveld PG et al. Variable hypomethylation of D4Z4 in facioscapulohumeral muscular dystrophy. Ann. Neurol. 58, 569–576 (2005). [DOI] [PubMed] [Google Scholar]
- 116.de Greef JC et al. Hypomethylation is restricted to the D4Z4 repeat array in phenotypic FSHD. Neurology 69, 1018–1026 (2007). [DOI] [PubMed] [Google Scholar]
- 117.de Greef JC et al. Common epigenetic changes of D4Z4 in contraction-dependent and contraction-independent FSHD. Hum. Mutat. 30, 1449–1459 (2009). [DOI] [PubMed] [Google Scholar]
- 118.Gaillard MC et al. Differential DNA methylation of the D4Z4 repeat in patients with FSHD and asymptomatic carriers. Neurology 83, 733–742 (2014). [DOI] [PubMed] [Google Scholar]
- 119.Larsen M et al. Diagnostic approach for FSHD revisited: SMCHD1 mutations cause FSHD2 and act as modifiers of disease severity in FSHD1. Eur. J. Hum. Genet. 23, 808–816 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jones TI et al. Individual epigenetic status of the pathogenic D4Z4 macrosatellite correlates with disease in facioscapulohumeral muscular dystrophy. Clin. Epigenet. 7, 37 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Roche S et al. Methylation hotspots evidenced by deep sequencing in patients with facioscapulohumeral dystrophy and mosaicism. Neurol. Genet. 5, e372–e372 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jones TI et al. Identifying diagnostic DNA methylation profiles for facioscapulohumeral muscular dystrophy in blood and saliva using bisulfite sequencing. Clin. Epigenet. 6, 23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Erdmann H et al. Methylation of the 4q35 D4Z4 repeat defines disease status in facioscapulohumeral muscular dystrophy. Brain 10.1093/brain/awac336 (2022). [DOI] [PubMed] [Google Scholar]
- 124.Mason AG et al. SMCHD1 regulates a limited set of gene clusters on autosomal chromosomes. Skelet. Muscle 7, 12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hartweck LM et al. A focal domain of extreme demethylation within D4Z4 in FSHD2. Neurology 80, 392–399 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lemmers RJLF et al. Inter-individual differences in CpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Hum. Mol. Genet. 24, 659–669 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Balog J et al. Increased DUX4 expression during muscle differentiation correlates with decreased SMCHD1 protein levels at D4Z4. Epigenetics 10, 1133–1142 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Balog J et al. Monosomy 18p is a risk factor for facioscapulohumeral dystrophy. J. Med. Genet. 55, 469–478 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gordon CT et al. De novo mutations in SMCHD1 cause bosma arhinia microphthalmia syndrome and abrogate nasal development. Nat. Genet. 49, 249–255 (2017). [DOI] [PubMed] [Google Scholar]
- 130.Shaw ND et al. SMCHD1 mutations associated with a rare muscular dystrophy can also cause isolated arhinia and bosma arhinia microphthalmia syndrome. Nat. Genet. 49, 238–248 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kinjo K et al. Rare variant of the epigenetic regulator SMCHD1 in a patient with pituitary hormone deficiency. Sci. Rep. 10, 10985 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lemmers RJLF et al. SMCHD1 mutation spectrum for facioscapulohumeral muscular dystrophy type 2 (FSHD2) and bosma arhinia microphthalmia syndrome (BAMS) reveals disease-specific localisation of variants in the ATPase domain. J. Med. Genet. 56, 693–700 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gurzau AD et al. FSHD2- and BAMS-associated mutations confer opposing effects on SMCHD1 function. J. Biol. Chem. 293, 9841–9853 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dion C et al. SMCHD1 is involved in de novo methylation of the DUX4-encoding D4Z4 macrosatellite. Nucleic Acids Res 47, 2822–2839 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mul K et al. FSHD type 2 and bosma arhinia microphthalmia syndrome: two faces of the same mutation. Neurology 91, e562–e570 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mohassel P et al. Cross-sectional neuromuscular phenotyping study of patients with arhinia with SMCHD1 variants. Neurology 98, e1384–e1396 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hansen RS et al. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc. Natl Acad. Sci. USA 96, 14412–14417 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Xu GL et al. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 402, 187–191 (1999). [DOI] [PubMed] [Google Scholar]
- 139.Gendrel AV et al. Smchd1-dependent and -independent pathways determine developmental dynamics of CpG island methylation on the inactive X chromosome. Dev. Cell 23, 265–279 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Thijssen PE et al. Mutations in CDCA7 and HELLS cause immunodeficiency-centromeric instability-facial anomalies syndrome. Nat. Commun. 6, 7870 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Jeanpierre M et al. An embryonic-like methylation pattern of classical satellite DNA is observed in ICF syndrome. Hum. Mol. Genet. 2, 731–735 (1993). [DOI] [PubMed] [Google Scholar]
- 142.Kondo T et al. Whole-genome methylation scan in ICF syndrome: hypomethylation of non-satellite DNA repeats D4Z4 and NBL2. Hum. Mol. Genet. 9, 597–604 (2000). [DOI] [PubMed] [Google Scholar]
- 143.Nozawa RS et al. Human inactive X chromosome is compacted through a PRC2-independent SMCHD1–HBiX1 pathway. Nat. Struct. Mol. Biol. 20, 566–573 (2013). [DOI] [PubMed] [Google Scholar]
- 144.Brideau NJ et al. Independent mechanisms target SMCHD1 to H3K9me3-modified chromatin and the inactive X chromosome. Mol. Cell Biol. 35, 4053–4068 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Balog J et al. Correlation analysis of clinical parameters with epigenetic modifications in the DUX4 promoter in FSHD. Epigenetics 7, 579–584 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Cabianca DS et al. A long ncRNA links copy number variation to a polycomb/trithorax epigenetic switch in FSHD muscular dystrophy. Cell 149, 819–831 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Himeda CL et al. Identification of epigenetic regulators of DUX4-fl for targeted therapy of facioscapulohumeral muscular dystrophy. Mol. Ther. 26, 1797–1807 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Olsen DB, Ørngreen MC & Vissing J Aerobic training improves exercise performance in facioscapulohumeral muscular dystrophy. Neurology 64, 1064–1066 (2005). [DOI] [PubMed] [Google Scholar]
- 149.Janssen B, Voet N, Geurts A, van Engelen B & Heerschap A Quantitative MRI reveals decelerated fatty infiltration in muscles of active FSHD patients. Neurology 86, 1700–1707 (2016). [DOI] [PubMed] [Google Scholar]
- 150.Wang LH et al. MRI-informed muscle biopsies correlate MRI with pathology and DUX4 target gene expression in FSHD. Hum. Mol. Genet. 28, 476–486 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dahlqvist JR et al. Evaluation of inflammatory lesions over 2 years in facioscapulohumeral muscular dystrophy. Neurology 95, e1211–e1221 (2020). [DOI] [PubMed] [Google Scholar]
- 152.van den Heuvel A et al. Facioscapulohumeral dystrophy transcriptome signatures correlate with different stages of disease and are marked by different MRI biomarkers. Sci. Rep. 12, 1426 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Mul K et al. Quantitative muscle MRI and ultrasound for facioscapulohumeral muscular dystrophy: complementary imaging biomarkers. J. Neurol. 265, 2646–2655 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lassche S et al. Correlation between quantitative MRI and muscle histopathology in muscle biopsies from healthy controls and patients with IBM, FSHD and OPMD. J. Neuromuscul. Dis. 7, 495–504 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wong CJ et al. Longitudinal measures of RNA expression and disease activity in FSHD muscle biopsies. Hum. Mol. Genet. 29, 1030–1043 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Andersen G et al. MRI as outcome measure in facioscapulohumeral muscular dystrophy: 1-year follow-up of 45 patients. J. Neurol. 264, 438–447 (2017). [DOI] [PubMed] [Google Scholar]
- 157.Fatehi F et al. Long-term follow-up of MRI changes in thigh muscles of patients with facioscapulohumeral dystrophy: a quantitative study. PLoS One 12, e0183825 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mul K et al. Adding quantitative muscle MRI to the FSHD clinical trial toolbox. Neurology 89, 2057–2065 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Frisullo G et al. CD8+ T cells in facioscapulohumeral muscular dystrophy patients with inflammatory features at muscle MRI. J. Clin. Immunol. 31, 155–166 (2011). [DOI] [PubMed] [Google Scholar]
- 160.Tasca G et al. Different molecular signatures in magnetic resonance imaging-staged facioscapulohumeral muscular dystrophy muscles. PLoS ONE 7, e38779 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ferguson MR et al. MRI change metrics of facioscapulohumeral muscular dystrophy: STIR and T1. Muscle Nerve 57, 905–912 (2018). [DOI] [PubMed] [Google Scholar]
- 162.Dahlqvist JR et al. Relationship between muscle inflammation and fat replacement assessed by MRI in facioscapulohumeral muscular dystrophy. J. Neurol. 266, 1127–1135 (2019). [DOI] [PubMed] [Google Scholar]
- 163.Friedman SD et al. Longitudinal features of STIR bright signal in FSHD. Muscle Nerve 49, 257–260 (2014). [DOI] [PubMed] [Google Scholar]
- 164.Janssen BH et al. Distinct disease phases in muscles of facioscapulohumeral dystrophy patients identified by MR detected fat infiltration. PLoS ONE 9, e85416 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Cohen J, DeSimone A, Lek M & Lek A Therapeutic approaches in facioscapulohumeral muscular dystrophy. Trends Mol. Med. 27, 123–137 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kissel JT et al. Randomized, double-blind, placebo-controlled trial of albuterol in facioscapulohumeral dystrophy. Neurology 57, 1434–1440 (2001). [DOI] [PubMed] [Google Scholar]
- 167.van der Kooi EL et al. Strength training and albuterol in facioscapulohumeral muscular dystrophy. Neurology 63, 702–708 (2004). [DOI] [PubMed] [Google Scholar]
- 168.Campbell AE et al. BET bromodomain inhibitors and agonists of the β2 adrenergic receptor identified in screens for compounds that inhibit DUX4 expression in FSHD muscle cells. Skelet. Muscle 7, 16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Oliva J et al. Clinically advanced p38 inhibitors suppress DUX4 expression in cellular and animal models of facioscapulohumeral muscular dystrophy. J. Pharmacol. Exp. Ther. 370, 219–223 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Rojas LA et al. p38α regulates expression of DUX4 in a model of facioscapulohumeral muscular dystrophy. J. Pharmacol. Exp. Ther. 374, 489–498 (2020). [DOI] [PubMed] [Google Scholar]
- 171.Mellion ML et al. Phase 1 clinical trial of losmapimod in facioscapulohumeral dystrophy: safety, tolerability, pharmacokinetics, and target engagement. Br. J. Clin. Pharmacol. 87, 4658–4669 (2021). [DOI] [PubMed] [Google Scholar]
- 172.Tawil R & Wagner K Clinical research: O.5 A phase 2, randomized, double-blind, placebo-controlled, 48-week study of the efficacy and safety of losmapimod in subjects with FSHD: ReDUX4. Neuromuscul. Disord. 31, S48–S49 (2021). [Google Scholar]
- 173.Jagannathan S et al. Meeting report: the 2021 FSHD International Research Congress. Skelet. Muscle 12, 1 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wang N, Wu R, Tang D & Kang R The BET family in immunity and disease. Signal. Transduct. Target. Ther. 6, 23 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Yang H, Wei L, Xun Y, Yang A & You H BRD4: an emerging prospective therapeutic target in glioma. Mol. Ther. Oncolyt. 21, 1–14 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lim JW et al. DICER/AGO-dependent epigenetic silencing of D4Z4 repeats enhanced by exogenous siRNA suggests mechanisms and therapies for FSHD. Hum. Mol. Genet. 24, 4817–4828 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Saad NY et al. Human miRNA miR-675 inhibits DUX4 expression and may be exploited as a potential treatment for facioscapulohumeral muscular dystrophy. Nat. Commun. 12, 7128 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kuijper EC, Bergsma AJ, Pijnappel WWMP & Aartsma-Rus A Opportunities and challenges for antisense oligonucleotide therapies. J. Inherit. Metab. Dis. 44, 72–87 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Marsollier AC et al. Antisense targeting of 3ʹ end elements involved in DUX4 mRNA processing is an efficient therapeutic strategy for facioscapulohumeral dystrophy: a new gene-silencing approach. Hum. Mol. Genet. 25, 1468–1478 (2016). [DOI] [PubMed] [Google Scholar]
- 180.Chen JC et al. Morpholino-mediated knockdown of DUX4 toward facioscapulohumeral muscular dystrophy therapeutics. Mol. Ther. 24, 1405–1411 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ansseau E et al. Antisense oligonucleotides used to target the DUX4 mRNA as therapeutic approaches in faciosscapulohumeral muscular dystrophy (FSHD). Genes 8, 93 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Lu-Nguyen N, Dickson G, Malerba A & Popplewell L Long-term systemic treatment of a mouse model displaying chronic FSHD-like pathology with antisense therapeutics that inhibit DUX4 expression. Biomedicines 10, 1623 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Lu-Nguyen N, Malerba A, Antoni Pineda M, Dickson G & Popplewell L Improving molecular and histopathology in diaphragm muscle of the double transgenic ACTA1-MCM/FLExDUX4 mouse model of FSHD with systemic antisense therapy. Hum. Gene Ther. 33, 923–935 (2022). [DOI] [PubMed] [Google Scholar]
- 184.Lu-Nguyen N, Malerba A, Herath S, Dickson G & Popplewell L Systemic antisense therapeutics inhibiting DUX4 expression ameliorates FSHD-like pathology in an FSHD mouse model. Hum. Mol. Genet. 30, 1398–1412 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Bouwman LF et al. Systemic delivery of a DUX4-targeting antisense oligonucleotide to treat facioscapulohumeral muscular dystrophy. Mol. Ther. Nucleic Acids 26, 813–827 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.George LA et al. Long-term follow-up of the first in human intravascular delivery of AAV for gene transfer: AAV2-hFIX16 for severe hemophilia B. Mol. Ther. 28, 2073–2082 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wallace LM et al. RNA interference inhibits DUX4-induced muscle toxicity in vivo: implications for a targeted FSHD therapy. Mol. Ther. 20, 1417–1423 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wallace LM et al. Pre-clinical safety and off-target studies to support translation of AAV-mediated RNAi therapy for FSHD. Mol. Ther. Methods Clin. Dev. 8, 121–130 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Rashnonejad A, Amini-Chermahini G, Taylor NK, Wein N & Harper SQ Designed U7 snRNAs inhibit DUX4 expression and improve FSHD-associated outcomes in DUX4 overexpressing cells and FSHD patient myotubes. Mol. Ther. Nucleic Acids 23, 476–486 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Bosnakovski D et al. A novel P300 inhibitor reverses DUX4-mediated global histone H3 hyperacetylation, target gene expression, and cell death. Sci. Adv. 5, eaaw7781 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Mariot V et al. A deoxyribonucleic acid decoy trapping DUX4 for the treatment of facioscapulohumeral muscular dystrophy. Mol. Ther. Nucleic Acids 22, 1191–1199 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Klingler C et al. DNA aptamers against the DUX4 protein reveal novel therapeutic implications for FSHD. FASEB J. 10.1096/fj.201902696 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ran FA et al. Genome engineering using the CRISPR–Cas9 system. Nat. Protoc. 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Cong L et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Richardson CD, Ray GJ, DeWitt MA, Curie GL & Corn JE Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 34, 339–344 (2016). [DOI] [PubMed] [Google Scholar]
- 196.Larson MH et al. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 8, 2180–2196 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Himeda CL, Jones TI & Jones PL CRISPR/dCas9-mediated transcriptional inhibition ameliorates the epigenetic dysregulation at D4Z4 and represses DUX4-fl in FSH muscular dystrophy. Mol. Ther. 24, 527–535 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Joubert R, Mariot V, Charpentier M, Concordet JP & Dumonceaux J Gene editing targeting the DUX4 polyadenylation signal: a therapy for FSHD? J. Pers. Med. 10.3390/jpm11010007 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Das S & Chadwick BP CRISPR mediated targeting of DUX4 distal regulatory element represses DUX4 target genes dysregulated in facioscapulohumeral muscular dystrophy. Sci. Rep. 11, 12598 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Gaudelli NM et al. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Komor AC, Kim YB, Packer MS, Zuris JA & Liu DR Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ran FA et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380–1389 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Šikrová D et al. Adenine base editing of the DUX4 polyadenylation signal for targeted genetic therapy in facioscapulohumeral muscular dystrophy. Mol. Ther. Nucleic Acids 25, 342–354 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Himeda CL, Jones TI & Jones PL Targeted epigenetic repression by CRISPR/dSaCas9 suppresses pathogenic DUX4-fl expression in FSHD. Mol. Ther. Methods Clin. Dev. 20, 298–311 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Tawil R, Shaw DW, van der Maarel SM & Tapscott SJ Clinical trial preparedness in facioscapulohumeral dystrophy: outcome measures and patient access: 8–9 April 2013, Leiden, The Netherlands. Neuromuscul. Disord. 24, 79–85 (2014). [DOI] [PubMed] [Google Scholar]
- 206.The FSH-DY group. A prospective, quantitative study of the natural history of facioscapulohumeral muscular dystrophy (FSHD): implications for therapeutic trials. Neurology 48, 38–46 (1997). [DOI] [PubMed] [Google Scholar]
- 207.Kinoshita J, Magdinier F & Padberg GW 26th Annual Facioscapulohumeral Dystrophy International Research Congress Marseille, France, 19–20 June 2019. Neuromuscul. Disord. 29, 811–817 (2019). [DOI] [PubMed] [Google Scholar]
- 208.Mul K et al. 225th ENMC international workshop: a global FSHD registry framework, 18–20 November 2016, Heemskerk, The Netherlands. Neuromuscul. Disord. 27, 782–790 (2017). [DOI] [PubMed] [Google Scholar]
- 209.Tassin A et al. FSHD myotubes with different phenotypes exhibit distinct proteomes. PLoS ONE 7, e51865 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Jones TI et al. Transgenic mice expressing tunable levels of DUX4 develop characteristic facioscapulohumeral muscular dystrophy-like pathophysiology ranging in severity. Skelet. Muscle 10, 8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Jones T & Jones PL A cre-inducible DUX4 transgenic mouse model for investigating facioscapulohumeral muscular dystrophy. PLoS One 13, e0192657 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Banerji CRS et al. PAX7 target genes are globally repressed in facioscapulohumeral muscular dystrophy skeletal muscle. Nat. Commun. 8, 2152 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Bosnakovski D et al. An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. EMBO J. 27, 2766–2779 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Banerji CRS & Zammit PS PAX7 target gene repression is a superior FSHD biomarker than DUX4 target gene activation, associating with pathological severity and identifying FSHD at the single-cell level. Hum. Mol. Genet. 28, 2224–2236 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Banerji CRS PAX7 target gene repression associates with FSHD progression and pathology over one year. Hum. Mol. Genet. 29, 2124–2133 (2020). [DOI] [PubMed] [Google Scholar]
- 216.Petek LM et al.A cross sectional study of two independent cohorts identifies serum biomarkers for facioscapulohumeral muscular dystrophy (FSHD). Neuromuscul. Disord. 26, 405–413 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Statland J, Donlin-Smith CM, Tapscott SJ, van der Maarel S & Tawil R Multiplex screen of serum biomarkers in facioscapulohumeral muscular dystrophy. J. Neuromuscul. Dis. 1, 181–190 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Signorelli M et al. Evaluation of blood gene expression levels in facioscapulohumeral muscular dystrophy patients. Sci. Rep. 10, 17547 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Heier CR et al. Multi-omics identifies circulating miRNA and protein biomarkers for facioscapulohumeral dystrophy. J. Pers. Med. 10, 236 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Corasolla Carregari V et al. Proteomics of muscle microdialysates identifies potential circulating biomarkers in facioscapulohumeral muscular dystrophy. Int. J. Mol. Sci. 22, 290 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Gros M et al. Identification of serum interleukin 6 levels as a disease severity biomarker in facioscapulohumeral muscular dystrophy. J. Neuromuscul. Dis. 9, 83–93 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Wong CJ et al. Elevated plasma complement components in facioscapulohumeral dystrophy. Hum. Mol. Genet. 31, 1821–1829 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Nunes AM, Ramirez M, Jones TI & Jones PL Identification of candidate miRNA biomarkers for facioscapulohumeral muscular dystrophy using DUX4-based mouse models. Dis. Model. Mech. 14, dmm049016 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Tasca G et al. Magnetic resonance imaging in a large cohort of facioscapulohumeral muscular dystrophy patients: pattern refinement and implications for clinical trials. Ann. Neurol. 79, 854–864 (2016). [DOI] [PubMed] [Google Scholar]
- 225.Regula JU et al. Clinical muscle testing compared with whole-body magnetic resonance imaging in facio-scapulo-humeral muscular dystrophy. Clin. Neuroradiol. 26, 445–455 (2016). [DOI] [PubMed] [Google Scholar]
- 226.Olsen DB, Gideon P, Jeppesen TD & Vissing J Leg muscle involvement in facioscapulohumeral muscular dystrophy assessed by MRI. J. Neurol. 253, 1437–1441 (2006). [DOI] [PubMed] [Google Scholar]
- 227.Leung DG, Carrino JA, Wagner KR & Jacobs MA Whole-body magnetic resonance imaging evaluation of facioscapulohumeral muscular dystrophy. Muscle Nerve 52, 512–520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Lareau-Trudel E et al. Muscle quantitative MR imaging and clustering analysis in patients with facioscapulohumeral muscular dystrophy type 1. PLoS ONE 10, e0132717 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Dahlqvist JR, Vissing CR, Thomsen C & Vissing J Severe paraspinal muscle involvement in facioscapulohumeral muscular dystrophy. Neurology 83, 1178–1183 (2014). [DOI] [PubMed] [Google Scholar]
- 230.Goselink RJM et al. Muscle ultrasound is a responsive biomarker in facioscapulohumeral dystrophy. Neurology 94, e1488–e1494 (2020). [DOI] [PubMed] [Google Scholar]
- 231.Vera KA, McConville M, Kyba M & Keller-Ross ML Sarcopenic obesity in facioscapulohumeral muscular dystrophy. Front. Physiol. 11, 1008 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.U.S. Food and Drug Administration. Guidance document: patient-reported outcome measures: use in medical product development to support labeling claims. Guidance for Industry (docket number FDA-2006-D-0362). https://www.fda.gov/regulatory-information/search-fda-guidance-documents/patient-reported-outcome-measures-use-medical-product-development-support-labeling-claims (2009).
- 233.European Medicines Agency. Appendix 2 to the guideline on the evaluation of anticancer medicinal products in man. The use of patient-reported outcome (PRO) measures in oncology studies (EMA/CHMP/292464/2014). https://www.ema.europa.eu/en/documents/other/appendix-2-guideline-evaluation-anticancer-medicinal-products-man_en.pdf (2016).
- 234.Mul K, Horlings CGC, Faber CG, van Engelen BGM & Merkies ISJ Rasch analysis to evaluate the motor function measure for patients with facioscapulohumeral muscular dystrophy. Int. J. Rehabil. Res. 44, 38–44 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Eichinger K et al. Facioscapulohumeral muscular dystrophy functional composite outcome measure. Muscle Nerve 58, 72–78 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Han JJ et al. Reachable workspace reflects dynamometer-measured upper extremity strength in facioscapulohumeral muscular dystrophy. Muscle Nerve 52, 948–955 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Hatch MN et al. Longitudinal study of upper extremity reachable workspace in fascioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 29, 503–513 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Hatch MN, Kurillo G, Chan V & Han JJ Motion sensor-acquired reachable workspace correlates with patient-reported upper extremity activities of daily living (ADL) function in facioscapulohumeral dystrophy. Muscle Nerve 63, 250–257 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Hamel J et al. Patient-reported symptoms in facioscapulohumeral muscular dystrophy (PRISM-FSHD). Neurology 93, e1180–e1192 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]