

Abstract
Prostate cancer (PCa) is the most common cancer and second leading cause of cancer death in American men. Most patients with metastatic disease respond initially to androgen deprivation therapy (ADT), but almost inevitably progress to castration resistant prostate cancer (CRPC). Identification of markers and drivers of mCRPC that (a) represent a progenitor-type cancer cell population (b) persist in castration resistant disease (c) are actionable targets expressed on the cell surface, and (d) are induced by hypoxia, is required to facilitate the development of novel targeted therapies. We identified prostatic acid phosphatase (PAP), particularly the transmembrane form (TMPAP), as one such potential target. PAP is both a phosphatase and a 5’ectonucleotidase that generates adenosine. We herein demonstrate that PAP is expressed early on during fetal development and persists in castration-resistant disease. The VCaP and VCaP-enzalutamide-resistant PCa cell lines express secretory (sPAP) and TMPAP. Androgens downregulate while hypoxia upregulates PAP expression. In vivo, PAP persists in hypoxic areas of castration-resistant tumors. Knockdown of PAP decreases VCaP migration and colony formation. Finally, treatment of VCaP tumor-bearing mice with inhibitors of adenosine receptors reduces tumor growth. This data demonstrates that TMPAP is a novel therapeutic target in advanced prostate cancer.
Keywords: Prostate cancer, transmembrane prostatic acid phosphatase, progenitor/stem cell, metastatic, castration-resistant, hypoxia, tumor microenvironment, 5’ectonucleotidase, adenosine, adenosine receptors
Introduction
Prostate cancer (PCa) is the most common cancer and second leading cause of cancer death in American men [1]. The majority of patients with metastatic disease respond initially to androgen deprivation therapy (ADT), but almost inevitably progress to castration resistant prostate cancer (CRPC) [2,3]. Despite more potent novel anti-androgen therapies, metastatic CRPC (mCRPC) is incurable and contributing to patient lethality. Resistance to therapy may be intrinsic or may result from the adaptive responses to the pressure in the tumor microenvironment (TME). Identification of markers and drivers of mCRPC that (a) represent a progenitor-type cancer cell population (b) persist in castration resistant disease (c) are actionable targets expressed on the cell surface, and (d) are induced by hypoxia in the TME, is required to facilitate the development of novel targeted therapies to overcome resistance and lethal disease [4]. We identified prostatic acid phosphatase (PAP) as one such potential target.
PAP is a protein phosphatase and 5’ectonucleotidase expressed in normal and cancerous prostate epithelial cells that has served as a diagnostic marker for prostate cancer [5]. PAP expression persists in most mCRPC in bone metastases and may play a causal role in castrate-resistant osteoblastic metastasis and mineralization, possibly via effects on the RANK/RANKL/OPG system [6-10].
The prostate gland develops from epithelial buds arising from the urogenital sinus (urethra) immediately below the bladder [11]. Epstein et al. reported on a series of bladder cancers and tumors with mixed glandular and transitional features that expressed PAP, even those derived from female patients [12]. This data indicates that neither androgens nor AR signaling are needed for PAP expression and implies that PAP expressing cells represent a more progenitor/stem cell like phenotype. Elevated levels of PAP protein have been reported to be an independent predictor of tumor recurrence, suggesting that PAP expressing cells represent a more aggressive, PCa progenitor population [13].
Significantly, studies from our group and others indicate that PAP expression persists in bone metastases derived from patients with mCRPC [7-9]. More recent evidence suggests that in addition to secretory PAP, a membrane-bound splice variant, transmembrane PAP (TMPAP) is detected in mouse tissues (prostate lobes, salivary gland, thymus, lung, kidney, brain, spleen and thyroid) as well as a number of human cells [14]. Mechanistically, the ectonucleotidase function of TMPAP generates extracellular adenosine, which promotes cancer cell growth, migration, and invasion [15].
Adenosine signaling via the adenosine receptor 2B (A2BR) promotes tumor progression, angiogenesis, metastasis and escape from immune attack in diverse cancers including prostate cancer [16-20]. Prostate cancer cell lines also express the A3 receptor and A3R inhibitors reduce tumor growth [21]. Extracellular adenosine promotes cell migration and invasion of glioblastoma via the A3 receptor [22]. Processes defining the landscape of the TME such as hypoxia and ischemia, can increase adenosine 1000-fold. Studies utilizing a glioblastoma (GBM) stem-like human cell line, U87MG, as well as primary cultures from GBM patients, demonstrated that (1) extracellular adenosine production was higher under hypoxia than normoxia mainly due to (2) hypoxia-inducible factor-2 alpha (HIF-2a) induction of expression of PAP driving adenosine production from AMP and resulting in (3) maintenance of the cancer stem cell phenotype and enhanced cell migration, invasion and EMT [18,19,22].
In the present study, we demonstrate that TMPAP is a marker of more progenitor-like prostate cancer cells that persist in CRPC. Mechanistic profiling demonstrates that TMPAP generates adenosine promoting cancer cell invasion and this activity is enhanced by hypoxia. Therefore, TMPAP is a novel target for the treatment of advanced prostate cancer.
Materials and methods
Tissue specimens
Archival specimens were obtained from male fetuses at the time of pregnancy interruption and prostate cancer bone metastases (from clinical specimens) under the guidelines of the Mount Sinai Medical Center Institutional Review Board as previously reported [7,23].
Immunohistochemistry
Immunohistochemical reagents were obtained from Vector Laboratories (Burlingame, CA). Formalin-fixed tissues were embedded in paraffin. Staining was carried out using an avidin-biotin complex method as previously described [23].
Reagents, cell culture, and Western blotting
VCaP, LNCaP and 22Rv1 (ATCC# CRL-2876, CRL-1740, CRL-2505) have been cultured as suggested by ATCC. LNCaP cell lines used in this study were maintained in RPMI with 10% fetal calf serum, while VCaP and 22Rv1 cell lines were maintained in DMEM with 10% fetal calf serum, at 37°C in 5% CO2, as per recommendation. For DHT studies, the cell lines are maintained in serum-free growth media supplemented with charcoal stripped serum. DHT treatments were maintained at the indicated concentration with addition of DHT every 24 hours. The following antibodies were used for Western Analysis: PAP, 1:2000 dilution (abcam: ab109004) or 1:1000 (Invitrogen: PA5-22322); AR, 1:1000 dilution (abcam: ab133273); PSA 1:1000 (Invitrogen: PA1-38514); PSMA 1:1000 (Proteintech: #66678-1-IG); GAPDH 1:1000 (Proteintech: #60004-1); and Vinculin 1:1000 dilution (Cell signaling #13901). AlexaFlour secondary antibodies were used at 1:10000 (Invitrogen: A21037, A21039, A21076, A21057). Hybridized blots were imaged and analyzed on Licor Odyssey FC 2800.
Soft agar assay and migration assay
Assays were performed essentially as described [24]. Briefly, 5% (w/v) agar (DIFCO, Detroit, MI, USA) was prepared in distilled water, autoclaved, then diluted to 0.5% final concentration with complete media and kept at 45°C in a water bath. A bottom layer of 0.5% agar was plated in each well of six-well plates to which cells were subsequently plated in a top layer of 0.37% agar cooled at 37°C. VCaP cells were treated with control and PAP siRNA for 48 hrs prior to plating on the soft agar. Samples were placed in humidified cell culture incubators at 37°C for 3 weeks after which colonies were stained using 1 ml solution of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (2 mg/ml in H2O). Colony numbers were quantified using ImageJ. Results represent Average ± SD of colony number per well from three wells. Migration assay was done using kit (abcam: CBA-101) per the manufacturer’s protocol. Assays were performed with VCaP cells treated for 48 hrs with control and PAP siRNA prior to plating on the transwell plate and observed for migration within a 24 hr-window.
Xenograft tumor model
10e6 VCaP cells were collected and resuspended in half volume of Matrigel. 100 ul of this cell suspension was injected into the flank of SCID mice (Charles River, Wilmington, MA). Surgical castration or treatments were initiated once tumors became palpable, followed by measurements with calipers weekly. The animals were euthanized when the tumor volume exceeded 500 mm3. The volume of the tumors was calculated according to the following formula: W2 * L/2 (W = shorter diameter, L = longer diameter of the tumor). The adenosine receptor inhibitor treatment groups were as follows: (1) Control (PBS injection), (2) the A2BR antagonist PSB-603 (Cat # 3198, Tocris Biosciences) 0.05 mg/kg and (3) the A3R antagonist MRS1220 (Cat # 1217, Tocris Biosciences) 0.05 mg/kg, all given intraperitoneally three times weekly for 4 weeks.
Confocal microscopy
VCaP cells were grown on collagen coated 35-mm glass-bottom culture dishes (MatTek #P35GCOL-1.0-14-C), fixed with 3.7% formaldehyde, permeabilized with methanol and blocked with 5% BSA in PBS. PAP protein is labeled with 1:100 dilution of PAP primary antibody (abcam: ab109004) followed by 1:1000 anti-rabbit Alexafluor 488 conjugated secondary antibody (Invitrogen: A32731). Hoechst 33342 (ThermoFisher: 62249) was used as a nuclear stain. E-Cadherin is labeled 1:200 (Invitrogen: MA1-10192) followed by 1:1000 anti-mouse Alexafluor 555 conjugated secondary antibody (Invitrogen: A21422). Confocal images are captured using Leica SP8 confocal microscopy housed at Leica Microsystems Center of Excellence at Icahn School of Medicine at Mount Sinai.
Sphere formation assay
Cells were seeded in six-well ultralow attachment plates (TPP; Thermo Fisher Scientific) at 1000 cells/ml of culture medium. The cells have the capability to grow and form spheres in serum-free DMEM/F12 media (Lonza, USA) supplemented with 0.2 ng/ml of epidermal growth factor and 25 µg/ml of bovine pituitary extract. In addition, 827 S0x (Thermo Fisher Scientific), 20 ng/mL epidermal growth factors (Thermo Fisher Scientific), and 20 ng/ml basic fibroblast growth factors (Thermo Fisher Scientific) were added to the cell culture media.
Statistical methods
Data Analysis and visualization was performed on Graphpad Prism 5.0. All data was analyzed with Two-Way ANOVA with Bonferroni’s multiple comparisons test, significance as indicated by * is P < 0.05.
Results
Immunohistochemical profiling of human fetal prostates demonstrates PAP expression in the transitional cells of the prostatic urethra and prostate epithelial cells in the solid ducts of the prostatic buds as early as 11.5 weeks with continued expression in canalized ducts at 16 weeks gestation (Figure 1A-D). In CRPC bone metastasis, there is limited expression of PSA, modest expression of AR and robust immunoreactivity for PAP (Figure 1E-H).
Figure 1.

Immunohistochemical expression of PAP in fetal prostate and bone metastases. (A, B) Low and High magnification of 11.5 week (C, D) and 16 week human fetus, stained for PAP; σ: Transitional cells of the prostatic urethra; ɛ: Ductal budding into primitive mesenchyme; (E-H) mCRPC in bone demonstrating minimal PSA expression (F), heterogeneous AR (G) and persistent strong expression of PAP (H).
We subsequently examined PAP protein expression in human PCa cell lines. Neither the LNCaP nor the 22Rv1 cell lines expressed measurable levels of PAP by Western blotting (Figure 2A). However, the castration-resistant cell line VCaP derived from a vertebral bone metastases expressed PAP protein, both the monomer of the secretory form and the transmembrane TMPAP (Figure 2A). Confocal imaging of VCaP cells demonstrated primarily cytoplasmic PAP (Figure 2B); although subcellular fractionation revealed that there was both cytosolic and TMPAP expression in VCaP cells (Figure 2C).
Figure 2.

PAP expression in PCa cell lines. A. Immunoblot representing PAP expression in various PCa cell lines. B. Confocal image representing intracellular localization of PAP in VCaP cell line. C. Subcellular fractionation representing the localization of a pool of PAP in the Plasma Membrane (Top band).
DHT addition to VCaP cells resulted in downregulation of PAP mRNA in VCaP cells (Figure 3A). Figure 3B demonstrates the differential protein expression of monomeric PAP, TMPAP, AR, ARv7, PSMA, PSA and synaptophysin in parental VCaP cells vs. enzalutamide resistant VCaP (VCaP-Enz). Of note, the protein expression of all forms of PAP as well as AR, PSMA and synaptophysin are increased in VCaP-Enz whereas PSA is decreased compared to parental VCaP (Figure 3B). DHT decreases and enzalutamide restores AR, ARv7, TMPAP, monomeric sPAP and synaptophysin in both VCaP and VCaP-Enz cells. In contrast, there is no significant effect of DHT or enzalutamide on PSMA but a clear increase in PSA with DHT that is decreased with the addition of enzalutamide to DHT. VCaP tumors grown in vivo ± castration express AR and PAP, but castration decreases AR expression while increasing PAP expression (Figure 3C). These data indicate that all forms of PAP are downregulated by DHT and that PAP is increased in Enz-resistant and castrated tumors.
Figure 3.

Regulation of PAP expression by androgenic signaling. (A) PAP transcription is downregulated by DHT in VCaP cells (B) monomeric and TMPAP protein is downregulated by DHT and restored by Enzalutamide treatment in VCaP and VCaP-Enzalutamide Resistant cell lines. (C) AR and PAP staining in VCaP mouse xenograft models with and without castration.
DHT addition to VCaP cells grown as spheroids resulted in more viable cancer cells with less areas of necrosis. In non-DHT treated spheroids, AR was only expressed at the periphery whereas PAP was demonstrable both at the periphery and in the central necrotic core (Figure 4). These findings imply that AR is expressed in more differentiated PCa cells whereas PAP expression persists in hypoxic cells.
Figure 4.

PAP and AR localization in VCaP cells grown as spheroids: PCa cell lines were seeded in Matrigel and grown in standard culture conditions to form 3D spheroids, treated without (A and B) or with DHT (C and D).
Hypoxia is a key factor that enriches cancer progenitor cell populations. VCaP cells grown under hypoxic conditions had increased TMPAP and dimeric sPAP with downregulation of monomeric sPAP, AR and PSA (Figure 5A). In vivo, hypoxic areas of tumors away from blood vessels continued to express PAP but lost AR expression (Figure 5B).
Figure 5.

PAP expression in response to hypoxia. A. Western blot demonstrating PAP upregulation in VCaP cells cultured under hypoxic conditions either with 5% FCS (Fetal Calf Serum) or 5% CSS (Charcoal Stripped Serum); downregulation of AR in hypoxia. B. VCaP xenograft stained for AR (red) and PAP (black) showing a progression of decreasing AR and increasing PAP towards the hypoxic, necrotic areas (yellow arrow) away from blood supply (*) (limited vascularity).
To investigate the effect of PAP on VCaP cell migration and anchorage-independent growth, PAP siRNA knockdown was performed. Figure 6 demonstrates the successful dose and time-related knockdown of PAP expression under both normoxic and hypoxic conditions with PAP siRNA (Figure 6A-D). Reduction in PAP protein expression resulted in decreased VCaP cell migration and anchorage-independent growth, indicating that PAP promotes PCa invasiveness. We next investigated the effects of adenosine receptor inhibitors on VCaP growth in vivo, as hypoxia-induced TMPAP expression has been demonstrated to increase adenosine levels via its 5’ectonucleotidase activity [15] and promote cell migration/invasion of glioblastoma cells via the A3R [25].
Figure 6.

siRNA knockdown of PAP decreases migration and anchorage independent growth. (A, B) Immunoblot showing dose and time dependent effects of PAP siRNA on PAP protein expression in VCaP cells. (C, D) The effect of hypoxia (18 hrs) on PAP expression in VCaP cells with/without PAP siRNA demonstrating that PAP levels increase with hypoxia and silenced with siRNA under both normoxic and hypoxic conditions siRNA knockdown of PAP decreases migration (E) and colony formation (F).
It was reported that both A2BR and A3R are expressed in human prostate cancer specimens [21] and cell lines [18]. We confirmed A2BR and A3R receptor expression in VCaP cells (data not shown). Figure 7 demonstrates that treatment of in vivo VCaP tumors with either the A2BR antagonist PSB (Figure 7A) or the A3R antagonist MRS1220 (Figure 7B) inhibited tumor growth compared to control.
Figure 7.
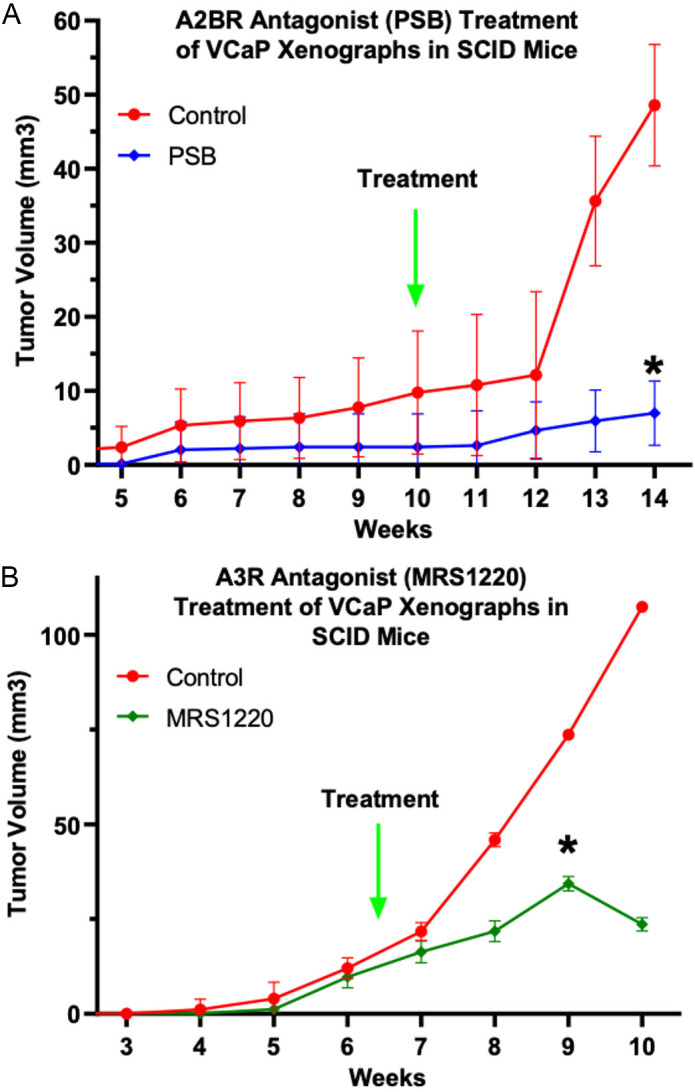
Adenosine receptor antagonist inhibited prostate xenografts in mice: VCaP cells were grown subcutaneously in SCID mice. Treatment with A2BR antagonist PSB (A) and A3R antagonist MRS1220 (B) inhibited tumor growth compared to untreated. Values represent Geometric Average ± SD, significance as indicated by * = P < 0.05 as measured by Bonferroni’s multiple comparisons test, Control vs. Treatment at each timepoint.
Discussion
Prostatic acid phosphatase (PAP) is the oldest serum tumor marker ever described [26]. In the landmark studies by Huggins and Hodges, serum PAP decreased in response to castration, leading to the widely accepted concept that PAP, like PSA, is purely an indicator of androgen action in well-differentiated prostate cancer cells [27]. In this study we report new results demonstrating that PAP is expressed in human fetal prostate epithelial cells in the solid ducts of the prostatic buds as early as 11.5 weeks, prior to the reported expression of AR or PSA [28]. Moreover, there is evidence that PAP persists in metastatic CRPC, when PSA expression is low, and that survival duration is significantly decreased in PAP+ groups [29]. Early expression of PAP in prostate development coupled with its persistence in castration-resistant, metastatic disease suggests that PAP is a marker of a more progenitor/stem cell.
PAP has two splice variants encoding a secretory form and a type I transmembrane (TM) protein [14]. We herein demonstrate that both the secretory and TMPAP are negatively regulated by androgens in the human PCa cell line VCaP, derived from a castration-resistant vertebral bone metastasis. Further, the enzalutamide-resistant subline of VCaP (VCaP-Enz) has higher basal expression levels of PAP, implying that TMPAP may serve as a therapeutic target in advanced, metastatic, castration-resistant disease. DHT addition decreases the expression of both PAP isoforms as well as AR and its splice variant ARv7. However, despite decreased AR levels, DHT increases PSA indicating that androgens have differential effects on PAP and PSA independent of AR levels.
Although it is well established that progenitor/stem cells re-side in hypoxic niches [30,31], our data provide the first evidence that hypoxia increases PAP protein while decreasing AR protein both in vitro and in vivo models of prostate cancer. This is consistent with the concept that PAP is a marker of cancer cell stemness. Loss of expression of PAP decreased tumor cell migration and invasion. The findings from glioblastoma cell lines, indicate that hypoxia induces TMPAP expression and increases adenosine via its 5’ectonucleotidase activity, driving tumor cell invasion and aggressivity [22]. Blockade of A2A and A3 adenosine receptors reduced PCa tumor growth in vivo, suggesting that TMPAP is working similarly via its 5’ectonucleotidase activity in PCa as was demonstrated in glioblastoma.
A growing body of evidence has implicated TMPAP as a tumor-suppressor gene in prostate cancer. In a PAP mouse knockout model, there was an increase in slow-growing, non-metastatic prostate adenocarcinoma [32]. In that same report and in a subsequent article, the authors demonstrated that stable overexpression of TMPAP in the LNCaP PCa cell line reduced cell growth via accumulation of cells in the G1 phase of the cell cycle [31]. LNCaP cells, although derived from a lymph node of a patient with castration resistant disease, still retain more androgen sensitivity than VCaP and its enzalutamide resistant subline, VCaP-Enz. The demonstration that in VCaP and VCaP-Enz there are high basal and hypoxia-induced TMPAP levels, decreased invasiveness with siRNA to PAP and tumor inhibition with adenosine receptor inhibitors, indicates that in more aggressive tumors, TMPAP is mechanistically involved in tumor progression.
Our findings that TMPAP is a characteristic feature of a more progenitor/stem cell phenotype, persists in CRPC, is upregulated by hypoxia and promotes tumor invasion via its 5’ectonucleotidase activity generating adenosine, identifies this cell surface protein as a novel therapeutic target in advanced prostate cancer.
Acknowledgements
Studies are supported by a grant from CDMRP DOD Award Number W81XWH-21-1-0330 and a grant from Prostate Action, Inc.
Disclosure of conflict of interest
None.
Abbreviations
- ADT
androgen deprivation therapy
- CRPC
castration resistant prostate cancer
- PCa
prostate cancer
- PAP
prostatic acid phosphatase
- SPAP
secretory prostatic acid phosphatase
- TMPAP
transmembrane prostatic acid phosphatase
- TME
tumor microenvironment
References
- 1.Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA Cancer J Clin. 2024;74:12–49. doi: 10.3322/caac.21820. [DOI] [PubMed] [Google Scholar]
- 2.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, Socci ND, Lash AE, Heguy A, Eastham JA, Scher HI, Reuter VE, Scardino PT, Sander C, Sawyers CL, Gerald WL. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kulasegaran T, Oliveira N. Metastatic castration-resistant prostate cancer: advances in treatment and symptom management. Curr Treat Options Oncol. 2024;25:914–31. doi: 10.1007/s11864-024-01215-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dong L, Zieren RC, Xue W, de Reijke TM, Pienta KJ. Metastatic prostate cancer remains incurable, why? Asian J Urol. 2019;6:26–41. doi: 10.1016/j.ajur.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gutman EB, Sproul EE, Gutman AB. Significance of increased phosphatase activity of bone at the site of osteoplastic metastases secondary to carcinoma of the Prostate Gland. Am J Cancer. 1936;28:485–95. [Google Scholar]
- 6.Ishibe M, Rosier RN, Puzas JE. Human prostatic acid phosphatase directly stimulates collagen synthesis and alkaline phosphatase content of isolated bone cells. J Clin Endocrinol Metab. 1991;73:785–92. doi: 10.1210/jcem-73-4-785. [DOI] [PubMed] [Google Scholar]
- 7.Kirschenbaum A, Liu XH, Yao S, Leiter A, Levine AC. Prostatic acid phosphatase is expressed in human prostate cancer bone metastases and promotes osteoblast differentiation. Ann N Y Acad Sci. 2011;1237:64–70. doi: 10.1111/j.1749-6632.2011.06198.x. [DOI] [PubMed] [Google Scholar]
- 8.Kirschenbaum A, Izadmehr S, Yao S, O’Connor-Chapman KL, Huang A, Gregoriades EM, Yakar S, Levine AC. Prostatic acid phosphatase alters the RANKL/OPG system and induces osteoblastic prostate cancer bone metastases. Endocrinology. 2016;157:4526–33. doi: 10.1210/en.2016-1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Larson SR, Chin J, Zhang X, Brown LG, Coleman IM, Lakely B, Tenniswood M, Corey E, Nelson PS, Vessella RL, Morrissey C. Prostate cancer derived prostatic acid phosphatase promotes an osteoblastic response in the bone microenvironment. Clin Exp Metastasis. 2014;31:247–56. doi: 10.1007/s10585-013-9625-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quiroz-Munoz M, Izadmehr S, Arumugam D, Wong B, Kirschenbaum A, Levine AC. Mechanisms of osteoblastic bone metastasis in prostate cancer: role of prostatic acid phosphatase. J Endocr Soc. 2019;3:655–64. doi: 10.1210/js.2018-00425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liaw A, Cunha GR, Shen J, Cao M, Liu G, Sinclair A, Baskin L. Development of the human bladder and ureterovesical junction. Differentiation. 2018;103:66–73. doi: 10.1016/j.diff.2018.08.004. [DOI] [PubMed] [Google Scholar]
- 12.Epstein JI, Kuhajda FP, Lieberman PH. Prostate-specific acid phosphatase immunoreactivity in adenocarcinomas of the urinary bladder. Hum Pathol. 1986;17:939–42. doi: 10.1016/s0046-8177(86)80645-1. [DOI] [PubMed] [Google Scholar]
- 13.Han M, Piantadosi S, Zahurak ML, Sokoll LJ, Chan DW, Epstein JI, Walsh PC, Partin AW. Serum acid phosphatase level and biochemical recurrence following radical prostatectomy for men with clinically localized prostate cancer. Urology. 2001;57:707–11. doi: 10.1016/s0090-4295(00)01073-6. [DOI] [PubMed] [Google Scholar]
- 14.Quintero IB, Araujo CL, Pulkka AE, Wirkkala RS, Herrala AM, Eskelinen EL, Jokitalo E, Hellström PA, Tuominen HJ, Hirvikoski PP, Vihko PT. Prostatic acid phosphatase is not a prostate specific target. Cancer Res. 2007;67:6549–54. doi: 10.1158/0008-5472.CAN-07-1651. [DOI] [PubMed] [Google Scholar]
- 15.Zimmermann H. Prostatic acid phosphatase, a neglected ectonucleotidase. Purinergic Signal. 2009;5:273–5. doi: 10.1007/s11302-009-9157-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sepúlveda C, Palomo I, Fuentes E. Role of adenosine A2b receptor overexpression in tumor progression. Life Sci. 2016;166:92–9. doi: 10.1016/j.lfs.2016.10.008. [DOI] [PubMed] [Google Scholar]
- 17.Ryzhov S, Novitskiy SV, Goldstein AE, Biktasova A, Blackburn MR, Biaggioni I, Dikov MM, Feoktistov I. Adenosinergic regulation of the expansion and immunosuppressive activity of CD11b+Gr1+ cells. J Immunol. 2011;187:6120–9. doi: 10.4049/jimmunol.1101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei Q, Costanzi S, Balasubramanian R, Gao ZG, Jacobson KA. A2B adenosine receptor blockade inhibits growth of prostate cancer cells. Purinergic Signal. 2013;9:271–80. doi: 10.1007/s11302-012-9350-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vecchio EA, Tan CY, Gregory KJ, Christopoulos A, White PJ, May LT. Ligand-independent adenosine A2B receptor constitutive activity as a promoter of prostate cancer cell proliferation. J Pharmacol Exp Ther. 2016;357:36–44. doi: 10.1124/jpet.115.230003. [DOI] [PubMed] [Google Scholar]
- 20.Mediero A, Cronstein BN. Adenosine and bone metabolism. Trends Endocrinol Metab. 2013;24:290–300. doi: 10.1016/j.tem.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mousavi S, Panjehpour M, Izadpanahi MH, Aghaei M. Expression of adenosine receptor subclasses in malignant and adjacent normal human prostate tissues. Prostate. 2015;75:735–47. doi: 10.1002/pros.22955. [DOI] [PubMed] [Google Scholar]
- 22.Torres Á, Erices JI, Sanchez F, Ehrenfeld P, Turchi L, Virolle T, Uribe D, Niechi I, Spichiger C, Rocha JD, Ramirez M, Salazar-Onfray F, San Martín R, Quezada C. Extracellular adenosine promotes cell migration/invasion of glioblastoma stem-like cells through A(3) adenosine receptor activation under hypoxia. Cancer Lett. 2019;446:112–22. doi: 10.1016/j.canlet.2019.01.004. [DOI] [PubMed] [Google Scholar]
- 23.Levine AC, Wang JP, Ren M, Eliashvili E, Russell DW, Kirschenbaum A. Immunohistochemical localization of steroid 5 alpha-reductase 2 in the human male fetal reproductive tract and adult prostate. J Clin Endocrinol Metab. 1996;81:384–9. doi: 10.1210/jcem.81.1.8550782. [DOI] [PubMed] [Google Scholar]
- 24.Clark GJ, Cox AD, Graham SM, Der CJ. Biological assays for Ras transformation. Methods Enzymol. 1995;255:395–412. doi: 10.1016/s0076-6879(95)55042-9. [DOI] [PubMed] [Google Scholar]
- 25.Torres A, Vargas Y, Uribe D, Jaramillo C, Gleisner A, Salazar-Onfray F, López MN, Melo R, Oyarzún C, San Martín R, Quezada C. Adenosine A3 receptor elicits chemoresistance mediated by multiple resistance-associated protein-1 in human glioblastoma stem-like cells. Oncotarget. 2016;7:67373–86. doi: 10.18632/oncotarget.12033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gutman AB, Gutman EB. An “ACID” phosphatase occurring in the serum of patients with metastasizing carcinoma of the prostate gland. J Clin Invest. 1938;17:473–8. doi: 10.1172/JCI100974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huggins C, Hodges CV. Studies on prostatic cancer. I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate*. Cancer Res. 1941;1:293–7. [Google Scholar]
- 28.Cunha GR, Cao M, Franco O, Baskin LS. A comparison of prostatic development in xenografts of human fetal prostate and human female fetal proximal urethra grown in dihydrotestosterone-treated hosts. Differentiation. 2020;115:37–52. doi: 10.1016/j.diff.2020.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu H, Wang F, Li H, Ji J, Cao Z, Lyu J, Shi X, Zhu Y, Zhang C, Guo F, Fang Z, Yang B, Sun Y. Prostatic acid phosphatase (PAP) predicts prostate cancer progress in a population-based study: the renewal of PAP? Dis Markers. 2019;2019:7090545. doi: 10.1155/2019/7090545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hira VVV, Breznik B, Vittori M, Loncq de Jong A, Mlakar J, Oostra RJ, Khurshed M, Molenaar RJ, Lah T, Van Noorden CJF. Similarities between stem cell niches in glioblastoma and bone marrow: rays of hope for novel treatment strategies. J Histochem Cytochem. 2020;68:33–57. doi: 10.1369/0022155419878416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rouault-Pierre K, Hamilton A, Bonnet D. Effect of hypoxia-inducible factors in normal and leukemic stem cell regulation and their potential therapeutic impact. Expert Opin Biol Ther. 2016;16:463–76. doi: 10.1517/14712598.2016.1133582. [DOI] [PubMed] [Google Scholar]
- 32.Araujo CL, Quintero IB, Ovaska K, Herrala AM, Hautaniemi S, Vihko PT. Transmembrane prostatic acid phosphatase (TMPAP) delays cells in G1 phase of the cell cycle. Prostate. 2016;76:151–62. doi: 10.1002/pros.23105. [DOI] [PubMed] [Google Scholar]