

Abstract
The Spectacled Fulvetta (Fulvetta ruficapilla) is the type species of Fulvetta, an evolutionarily distinct group whose species show a high degree of sympatry in distribution and phenotypic convergence. To pave the way for insights into their adaptive evolution and speciation, we have assembled the first high quality reference genome for F. ruficapilla using high-fidelity (HiFi) long-read and Hi-C sequencing technologies. The resulting assembly spans a total of ~1.21 Gb with a contig N50 of 18.8 Mb and scaffold N50 of 75.9 Mb, and has a BUSCO completeness of 97.0%. The quality assessment suggests a high standard in base accuracy, continuity, and completeness of the assembly, comparable or close to that of Vertebrate Genomes Project. On this basis, we have annotated 23,774 protein-coding genes, of which 18,832 are functionally identified. The availability of this high-quality genome provides a solid foundation for the future studies of evolution and local adaptation in birds.
Subject terms: Adaptive radiation, Conservation biology, Zoology, Genome, Conservation genomics
Background & Summary
Birds of the genus Fulvetta (Paradoxornithidae, Passeriformes) are mainly distributed in southwestern China, centred around the Hengduan Mountains and adjacent areas including the Himalayas, Indochina, and central to eastern China1–4. They were once grouped together in the genus Alcippe (Timaliidae, Passeriformes) due to the homogeneous morphology3. Recently, however, it has been shown that Fulvetta form an independent, well-supported phylogenetic cluster within the family Paradoxornithidae1,2,5, thus indicating their evolutionary independence and uniqueness. Consequently, how this speciose avian lineage has evolved from perspectives of genetic underpinnings deserves to be explored in depth. Interestingly, the species of Fulvetta show little sexual dimorphism in plumage and other morphological traits as well as a high degree of sympatry in distribution3,6. Therefore, morphological convergence and local adaptation may have played an important role in the evolutionary history of Fulvetta. All these suggest that the genus Fulvetta would be an ideal model for the study of avian evolution2. However, the lack of whole genomic data for the Fulvetta species has hindered in-depth exploration into their phylogeny, adaptive evolution, and genetic mechanisms under adaptive evolution and speciation.
Therefore, we choose the type species of the genus Fulvetta (Fulvetta ruficapilla), which is endemic to China, for genome sequencing and de novo assembling. We have assembled its reference genome in both high completeness and continuity, utilizing an integrated strategy of PacBio high-fidelity (HiFi) long-read combined with chromosome conformation capture (Hi-C) sequencing technologies. The resulting assembly spans a total of ~1.21 Gb with 580 contigs initially generated with an N50 of 18.8 Mb, which have been well-organized into 504 scaffolds by Hi-C data with a scaffold N50 of 75.9 Mb. We have further annotated 23,774 protein-coding genes, of which at least 18,832 have been identified with functions. In addition, we also identified repetitive and ncRNA elements in 17.82% and 0.06% of the assembly, respectively. Furthermore, our evaluation confirms an overall good quality of the final assembly in terms of base accuracy at QV 62.32 as well as BUSCO completeness of 97.0% for assembly and 98.3% for annotation, indicating a high standard even compared to some of the high-quality genomes of the Vertebrate Genomes Project (VGP)7 and other projects released recently. With this high-quality F. ruficapilla genome, we have briefly exemplified how it could contribute to evolutionary analyses. This assembly renders the first reference genome in high quality for this speciose avian lineage, which represents an exceptional model system to enhance our understanding about the genetic mechanism and avian evolution.
Methods
Ethics statement
All experiments and sample collection were for the scientific research and approved by the Institutional Animal Care and Use Committee of Kunming Institute of Zoology, Chinese Academy of Sciences (IACUC-OE-2023-08-004).
Sample collection and sequencing
For this study, we trapped a sub-adult, healthy individual of F. ruficapilla (WMS3MU01) with a mist net in Kunming, China (10 August, 2022; Fig. 1), for which the gender could not be morphologically recognized and was further confirmed by the following assembled genome. We collected its brain, heart, kidney, liver, lung, muscle, spleen, and blood for the subsequent whole-genome, Hi-C, and RNA sequencing (Fig. 1). We first extracted total genomic DNA from the blood sample with the CTAB (cetyl trimethyl ammonium bromide) method (Grandomics Genomic kit). We next sheared the DNA using the Megaruptor 3 syste and screened for the target fragments after end repair and adapter ligation using SMRTbell prep kit 3.0 to prepare the sequencing library. We sequenced this library for the HiFi long reads of a ≥ Q20 single-molecule read accuracy by circular consensus sequencing (CCS) on the PacBio Sequel II system, with passes ≥ 3 and RQ ≥ 0.99 in CCS software (https://github.com/PacificBiosciences/ccs), which generated a total of 38.26 Gb (an estimated coverage of ~32×) of CCS reads with a read N50 of 18.23 Kb and the longest read of 55.33 Kb (Supplementary Fig. S1a, Table 1) after quality control by FastQC v0.12.1 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). For Hi-C sequencing, we fixed the muscle of the same individual with 1% formaldehyde and then collected the precipitated cells after treatment with Proteinase K. For the extracted, qualified DNA, we prepared a Hi-C library with the treatment of endonuclease DpnII and sequenced the library for 150-bp paired-end reads with a 350-bp insert size on the DNBSEQ-T7 platform of MGI Tech (Beijing Biomarker Technologies), which resulted in a total of 127.62 Gb (~105 × in estimated coverage) of raw Hi-C reads. After filtering with fastp (v0.23.4)8, we retained 126.54 Gb of Hi-C clean reads for assembling (Table 1). For RNA sequencing, we extracted RNA from the brain, muscle, heart, liver, spleen, lung, kidney, and testis separately. The tissues were carefully removed from RNAlater. We then isolated RNA following the instructions of HiPure Universal RNA Mini Kit (Magen, China). We mixed the RNA in equal amount of each tissue. The integrity of RNA was confirmed on 1% agarose gels. After steps including mRNA enrichment by magnetic oligo-dT beads, fragmentation, reverse transcription to cDNA, end repair and poly-A tail addition, adaptor ligation, and PCR enrichment for cDNA library, we sequenced it for 150-bp paired-end reads on the DNBSEQ-T7 platform (Beijing Biomarker Technologies), and obtained a total of 21.36 Gb of high-quality RNA reads (Table 1) after trimming by fastp (v0.23.4)8. All the above steps regarding library construction and sequencing, otherwise stated, were in accordance with official instructions, standard protocols, or default settings.
Fig. 1.

Flow chart of the pipeline for genome assembly and annotation of Fulvetta ruficapilla.
Table 1.
Summary of the sequencing strategy.
| Sequencing Strategy | Sequencing platform | Library size | Total clean data (Gb) | GC content (%) | Sequencing coverage (×) |
|---|---|---|---|---|---|
| PacBio HiFi | PacBio Sequel II | >10 Kb | 38.26 | 42.11 | ~32 |
| Hi-C | DNBSEQ-T7 PE150 | 350 bp | 126.54 | 43.31 | ~105 |
| RNA-seq | DNBSEQ-T7 PE150 | 350 bp | 21.36 | 48.71 | — |
| Total | — | — | 186.16 | — | >137 |
Genome assembly of F. ruficapilla
We first constructed a contig-level draft assembly for the F. ruficapilla genome based on the PacBio HiFi long reads and Hi-C clean data with hifiasm (v0.19.8)9 (Fig. 1). The resulting primary assembly consisted of 580 contigs with a contig N50 of 18.8 Mb, the longest contig of 70.2 Mb, the shortest contig of 16,685 bp, and a total size of 1.21 Gb (Table 2). To join the contigs, we aligned Hi-C reads to the contig-level assembly by BWA (v0.7.17)10. Then, we utilized YaHS (v1.1)11 to construct scaffolds of the genome assembly (Fig. 1). The resulting final assembly consisted of 504 scaffolds with an N50 of 75.9 Mb (Fig. 2a,b, & Supplementary Fig. S1b, Table 2). The GC content of the final assembly was generally consistent across most of the scaffolds with an average of 43.0% (Fig. 2c & Table 2).
Table 2.
Summary of the genome assembly and annotation for Fulvetta ruficapilla.
| Genome characteristics | value | |
|---|---|---|
| Draft genome assembly | ||
| Total size (bp) | 1,208,292,158 | |
| No. of contigs | 580 | |
| N50 (bp)/L50 | 18,795,193/14 | |
| N90 (bp)/L50 | 2,218,513/82 | |
| Longest contig (bp) | 70,212,158 | |
| Shortest contig (bp) | 16,685 | |
| BUSCO completeness (aves_odb10) | C:8081 (96.9%) [S:7966 (95.5%), D:115 (1.4%)], F:42 (0.5%), M:215 (2.6%), n:8338* | |
| Scaffolded genome assembly | ||
| Total size (bp) | 1,208,308,458 | |
| GC content | 43.04% | |
| No. of scaffolds | 504 | |
| N50 (bp)/L50 | 75,915,673/5 | |
| N90 (bp)/L50 | 4,512,364/28 | |
| Longest scaffold (bp) | 172,205,748 | |
| QV (base quality value)** | 62.32 | |
| BUSCO completeness (aves_odb10) | C:8085 (97.0%) [S:7970 (95.6%), D:115 (1.4%)], F:41 (0.5%), M:212 (2.5%), n:8338 | |
| Protein-coding gene annotation | ||
| No. of predicted protein-coding genes | 23,774 | |
| No. of genes with some UTR† | 20,794 | |
| Average gene length (Kb) | 18.2 | |
| Average transcript length (Kb) | 21.9 | |
| Average CDS length (bp)†† | 164.0 | |
| Average number of CDS per gene | 20.0 | |
| Average number of CDS per transcript | 10.7 | |
| No. of functionally annotated protein-coding genes | 18,832 | |
| BUSCO completeness (aves_odb10) | C:8199 (98.3%) [S:8060 (96.7%), D:139 (1.7%)], F:57 (0.7%), M:82 (1.0%), n:8338 | |
| Repetitive sequences# | Length (Mb) | Proportion |
| SINEs | 0.5 | 0.3% |
| LINEs | 72.7 | 33.8% |
| LTR | 118.8 | 55.2% |
| DNA | 3.8 | 1.8% |
| Others [Simple repeat, microsatellite, RNA, Penelope, Rolling Circle] & Unclassified | 21.2 | 9.8% |
| Total (Non-redundant) | 215.3 | 17.82% (of assembly) |
| Non-coding RNA elements | Length (bp) | Count |
| rRNA | 584,756 | 307 |
| snRNA | 34,025 | 279 |
| tRNA | 20,392 | 275 |
| miRNA | 18,679 | 236 |
| lncRNA | 4,778 | 24 |
| ribozyme | 1,346 | 6 |
| IRES | 398 | 2 |
| frameshift_element | 115 | 2 |
| sRNA | 77 | 1 |
| Others | 10,946 | 125 |
| Total (Non-redundant) | 675,512 | 1,257 |
*C, complete; S, single-copy; D, duplicated; F, fragmented; M, missing; n, total orthologs.
**QV, base quality value assessed by Merqury, indicating base accuracy in the form of negative log-transformed number of base errors per 1 Mb of the assembly.
†UTR, untranslated region.
††CDS, coding region sequence.
#SINE, short interspersed nuclear element; LINE, long interspersed nuclear element; LTR, long terminal repeat.
Fig. 2.

Overview of the genome assembly for Fulvetta ruficapilla. (a) Snail plot of summary statistics of the genome assembly on total length, scaffold length, BUSCO completeness in assembly, and base composition. (b) Cumulative length distribution versus cumulative count of the assembled scaffolds. (c) Distributions of GC content and coverage depth of HiFi reads across the scaffolds. The larger of the circle indicates the longer of the scaffold. All plots were generated by BlobTools2 in the BlobToolKit (v4.3.5)50. The BUSCO results were generated by BUSCO v5.5.0 and fed into the BlobTools2 plotting.
Genome annotation of genes and repetitive sequences
To annotate the F. ruficapilla genome (Fig. 1), we first identified the repetitive elements with the Earl Grey (v4.1.0)12. The Earl Grey is a fully automated pipeline that leverages several of the most widely-used tools, including RepeatMasker v4.1.5 (http://www.repeatmasker.org) with Dfam (v3.7)13, RepeatModeler v2.0.5 (http://www.repeatmasker.org/RepeatModeler.html), RepeatScout (v1.0.6)14, Tandem Repeat Finder (v4.09)15, RECON16, and LTR_FINDER17 to identify transposable elements in an improved accuracy and efficiency12. As a result, we annotated 215.3 Mb of repetitive sequences, accounting for 17.82% of the newly assembled genome (Fig. 3 & Table 2). We also identified repetitive elements for the latest reference genomes of zebra finch and chicken, using both of the RepeatMasker and the same Earl Grey pipelines. Therefore, we could briefly test the efficiency of the repeat annotation by Earl Grey (Table 3).
Fig. 3.
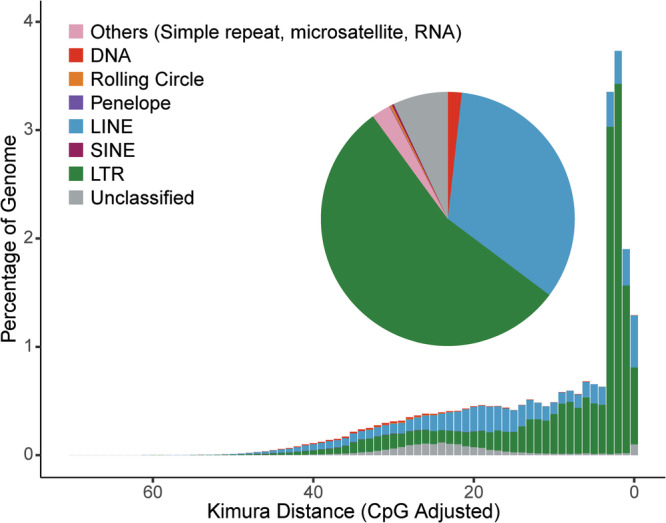
Repetitive sequences in the Fulvetta ruficapilla genome. Different categories of annotated repeats have been classified to show their proportions (pie chart) and percentage distribution of in the assembly (histogram).
Table 3.
Comparison of repeats annotation by Earl Grey and RepeatMasker.
| Species | Common name | Repeat rate by RepeatMasker | Repeat rate by Earl Grey |
|---|---|---|---|
| Fulvetta ruficapilla | Spectacled Fulvetta | 6.96% | 17.82% |
| Gallus gallus | Chicken | 13.36% | 15.35% |
| Taeniopygia guttata | Zebra Finch | 11.63% | 12.31% |
To further annotate gene models in the F. ruficapilla genome (Fig. 1), we integrated Braker3 (v3.0.8)18 with Gene Model Mapper (GeMoMa, v1.9)19. First, we prepared RNA-seq clean reads for the combined tissues of the F. ruficapilla (Methods) as well as 180 runs of downloaded RNA-seq data (~2979.69 Gb in total, ranging from 3.5 to 42.2 Gb for each run) of different tissues across representative clades of Passeriformes (Supplementary Table S1) with fastp v0.23.4, and mapped them onto the genome with hisat2 (v2.2.1)20 followed by sorting with SAMtools (v1.15.1)21. Next, we provided two sources of data in aid of the gene prediction by Braker3. One of them was a self-curated protein dataset, which included all the non-redundant vertebrate proteins from OrthoDB (v11)22 and all the Passeriformes protein sequences downloaded from NCBI Protein Database (accessed at 29 March, 2024), for annotation by homology. The another set of data was the RNA-seq data aligned onto the target genome for providing evidence of transcripts. After Braker3, we further used GeMoMa to improve the annotation, by feeding with the following three inputs, 1) the annotation output by Braker3, 2) all the mapped RNA-seq data in the bam format, and 3) four selected avian reference genomes with their associated annotations including pigeon (GCA_032206205.1)23, Anna’s hummingbird (GCF_003957555.1)7, zebra finch (GCF_003957565.2)7, and chicken (GCF_016699485.2)7. As a final result, we annotated 23,774 protein-coding genes with an average length of 18.2 Kb, of which 20,794 had at least one untranslated region (UTR) identified. The resulting gene models were generally comparable in quantity and length to that of several existing avian genomes (Table 4).
Table 4.
Comparison of genome annotation of protein-coding genes for Fulvetta ruficapilla with other avian assemblies.
| Common name | Fulvetta ruficapilla | Gallus gallus | Taeniopygia guttata | Calypte anna | Columba livia |
|---|---|---|---|---|---|
| Spectacled Fulvetta | Chicken | Zebra finch | Anna’s Hummingbird | Pigeon | |
| Genome accession | GWHETLV00000000.1* | GCF_016699485.2 | GCF_003957565.2 | GCF_003957555.1 | GCA_032206205.1 |
| Annotation source | This study** | Ensembl 111 | NCBI 101 | NCBI 106 | NCBI |
| No. Gene | 23,774 | 17,007 | 15,620 | 14,711 | 16,853 |
| No. Transcript | 44,387 | 44,876 | 41,214 | 29,214 | 18,431 |
| No. CDS | 475,115 | 527,464 | 577,950 | 353,175 | 201,099 |
| No. mRNA/Gene | 1.87 | 2.64 | 2.64 | 1.99 | 1.09 |
| No. CDS/Gene | 20.00 | 31.01 | 37.00 | 24.00 | 11.93 |
| No. CDS/mRNA | 10.70 | 11.75 | 14.02 | 12.09 | 10.92 |
| Avg. Gene (Kb) | 18.2 | 34.5 | 34.9 | 34.5 | 33.2 |
| Avg. mRNA (Kb) | 21.9 | 43.5 | 58.1 | 41.9 | 35.6 |
| Avg. CDS (bp) | 164.0 | 163.0 | 161.7 | 161.2 | 164.1 |
*The final scaffolded genome assembly has been deposited in the GWH database under the accession GWHETLV00000000.1 (publicly accessible at https://ngdc.cncb.ac.cn/gwh/Assembly/85202/show) and NCBI GenBank under the accession JBGGOM00000000046.
**The associated annotation files have been deposited in Science Data Bank47 and Figshare48.
We next annotated functions of these protein-coding genes with three approaches (Fig. 1). First, we blasted the protein sequences of the gene models to the UniProtKB/Swiss-Prot database (accessed at 17 April, 2024) with diamond (v2.1.9)24, and extracted the genes whose names were annotated in the database. Then, we used eggNOG-mapper (v2.1.12)25 and the online Pannzer2 (accessed at 11 June, 2024)26 to annotate the proteins translated from the gene models. For a transcript if conflicting gene names were annotated by different methods, we manually determined the name in a priority order as 1) identical in at least two methods, 2) in eggNOG-mapper or Pannzer2, 3) in eggNOG-mapper. Consequently, of the 23,774 protein-coding gene models, a total of ≥ 18,832 were successfully annotated with functions or gene names by at least one approach, more than the protein-coding genes that were identified in several representatives of avian genomes by NCBI or Ensembl (Tables 2 & 4).
We also annotated the non-coding RNA (ncRNA) in the newly assembled genome with Infernal (v1.1.5)27 (Fig. 1). We downloaded the latest Rfam database (v14.10)28, and then run an Infernal search for RNA homology with cmscan program. As a result, we annotated 1,257 non-redundant ncRNA elements spanning 675.5 Kb (~0.06% of the assembly), of which the majority (87.3% in quantity and 97.4% in length) comprised rRNA, snRNA, tRNA, and miRNA (Table 2).
Assessment of the genome assembly
To reasonably determine the gender of the collected sample (WMS3MU01), we first blasted the female-specific EE0.6 sequence (GenBank accession: D85617.1) and CHDW gene (GenBank accessions: XP_058718501.1, XP_050842274.1, and NP_001071646.1) on the avian W chromosome to the F. ruficapilla assembly (identity > 85%, E-value < 1.0e-10) with BLAST (v2.15.0)29. For the general quality metrics of the assembly (Fig. 1), we first evaluated the completeness and quality with a k-mer-based approach by Merqury (v1.3)30, which could show the QV (consensus base quality) and the proportion of the k-mers in the HiFi reads that the resulting assemblies covered (Supplementary Fig. S2). We also compared the synteny between the F. ruficapilla assembly and the latest reference genomes of zebra finch7 and chicken7 using JCVI (v1.4.15)31 to check for how the scaffolds we assembled would be close to these high-quality reference genomes (Fig. 4). We further used BUSCO (v5.5.0)32 with the aves_odb10 database (8338 avian single-copy genes) to assess the completeness of the final assembly and its gene annotation in genome and protein modes. We also performed a BUSCO assessment for several selected avian genomes23,30,33–36 that were deemed a good quality, aiming at comparing the completeness with these high-quality references (Supplementary Fig. S3). We further briefly assessed how the F. fulvetta genome could contribute to evolutionary analyses37–41 using PSMC (v0.6.5-r67)42 and PAML (v4.10.7)43 (Supplementary Figs. S4 & S5, Tables S2 & S3).
Fig. 4.

Genome synteny analyses. Synteny comparison of the Fulvetta ruficapilla (Spectacled Fulvetta, blue) genome with the latest reference genomes of zebra finch (green) and chicken (orange). The horizontal bars represent scaffolds or chromosomes. The Z and W chromosomes of zebra finch and chicken are indicated along the corresponding bars.
Data Records
The raw data of PacBio HiFi, Hi-C and RNA sequencing reads are deposited in the Genome Sequence Archive (GSA)44 of National Genomics Data Center under the accession CRA017143 with runs of CRR1203752, CRR1203753–CRR1203754, and CRR1203755, respectively (publicly accessible at https://ngdc.cncb.ac.cn/gsa/browse/CRA017143). The final genome assembly is deposited in Genome Warehouse (GWH)45 under the accession GWHETLV00000000.1 (publicly accessible at https://ngdc.cncb.ac.cn/gwh/Assembly/85202/show) and NCBI GenBank under the accession JBGGOM00000000046. The genome annotations are deposited in the Science Data Bank47 and Figshare48 repositories.
Technical Validation
The final genome assembly for the F. ruficapilla has yielded a total length of 1.21 Gb with a scaffold N50 of 75.92 Mb. The Hi-C interacting heatmap shows generally well-organized compartments consistent with the assembled scaffolds (Supplementary Fig. S1b), which show a clearly recognizable synteny with the latest reference genomes of both zebra finch and chicken (Fig. 4). In addition, the female-specific EE0.6 sequence and CHDW gene on the avian W could be reliably blasted onto the scaffold 8 of the F. ruficapilla assembly (identity > 85%, E-value < 1.0e-10), which is generally consistent with the synteny pattern (Fig. 4), and therefore show the female status of the F. ruficapilla sample.
The BUSCO score indicates that 97.0% of the single-copy orthologs are complete and 2.5% missing for the assembly. In addition, BUSCO shows 98.3% and < 1% of the completeness and missing rate, respectively, for the annotated protein-coding sequences (Supplementary Fig. S3), of which at least 18,832 have been annotated with functions or names (Table 2). Although the duplicate BUSCO is slightly higher in the F. ruficapilla, it would not lay a significantly negative impact on the assembly as the assembler hifiasm has built-in the duplication purging algorithm and suggested not necessarily to remove duplicates additionally9. Notably, the annotated repetitive elements in the final F. ruficapilla genome show a slightly higher rate than that reported for many of the passerine birds49, which is perhaps due to the high completeness of our F. ruficapilla genome assembled with the latest long-read sequencing technologies (e.g., PacBio HiFi). In comparison, Earl Grey has actually produced higher repeat rates for the F. ruficapilla assembly and the reference genomes of chicken and zebra finch than RepeatMasker (Table 3). It thus confirms the high efficiency of the Earl Grey pipeline and validates our repeats annotation. The primary assembly covers 92.93% of the total sequenced k-mers, while if taken the alternate assembly into account, the k-mer completeness has reached 99.74% (Supplementary Fig. S2). Moreover, the mapping rate of the HiFi reads back onto the final assembly has reached 99.97%. These results confirmed the high level of completeness of the F. ruficapilla assembly from both the perspectives of genome assembly and gene annotation. Even among other avian genomes considered to be of good quality or most commonly used as references in evolutionary analyses, especially those recently released by VGP and other projects, our assembly still shows a comparable or close completeness and continuity (Supplementary Figs. S3 & S6). The QV (consensus base quality value) of our assembly has also achieved 62.32, which corresponds to <0.59 potentially erroneous bases in per 1 Mb. This indicates a surprisingly high base accuracy of our assembly with assembling base errors hardly detectable (Supplementary Fig. S2). It is comparable to the “finished” standard of VGP (QV > 60) as well as many of the recent VGP genomes7, and thus validates the high accuracy of the assembly for F. ruficapilla. At last, we briefly validated that the newly assembled genome of F. ruficapilla could be employed to investigate the demographic history and adaptive evolution of F. ruficapilla that it might have experienced (Supplementary Figs. S4 & S5, Tables S2 & S3).
Supplementary information
Acknowledgements
This work was supported by grants from the National Key R&D Program of China (2022YFC2602500), the Yunnan Fundamental Research Projects (202301AW070012, 202401AV070007, and 202401CF070065), Yunnan Province (202305AH340006), and the “Yunnan Revitalization Talent Support Program: High-end Foreign Expert Project and Young Talent Project” (XDYC-QNRC-2022-0770). The National Natural Science Foundation of China, Talent Program of Chinese Academy of Sciences (CAS), and Animal Branch of the Germplasm Bank of Wild Species of CAS (the Large Research Infrastructure Funding) also supported this project.
Author contributions
M.-S.W. and F.W. conceived, designed, and supervised the research. S.S., Y.-T.Z., L.-M.L., F.W., and M.-S.W. collected and prepared samples. S.S. extracted DNA and RNA from samples. C.Y. and H.-M.C. performed bioinformatic analyses and prepared data, figures, and tables. C.Y. drafted the original manuscript. M.-S.W., F.W., C.Y., and H.-M.C. revised the manuscript. All authors have agreed on the submission of the manuscript for publication.
Code availability
Relevant software, programs, core options, and pipelines regarding the analyses of data filtering, genome assembly, assembly assessment, and annotation have been stated in Methods. The key parameters, code, and scripts for the pipeline are publicly accessible at https://github.com/YanCheer/Fruf_v1_assembly.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Chen Yan, Si Si.
Contributor Information
Fei Wu, Email: wufei@mail.kiz.ac.cn.
Ming-Shan Wang, Email: wangmingshan@mail.kiz.ac.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41597-024-04094-2.
References
- 1.Gill, F., Donsker, D. & Rasmussen, P. IOC World Bird List (v14.2)10.14344/IOC.ML.14.1 (2024).
- 2.Zheng, G. et al. A Checklist on the Classification and Distribution of the Birds of the World, Second Edition. (Science Press, Beijing, 2021). [Google Scholar]
- 3.Pasquet, E., Bourdon, E., Kalyakin, M. V. & Cibois, A. The fulvettas (Alcippe, Timaliidae, Aves): a polyphyletic group. Zool. Scr.35, 559–566 (2006). [Google Scholar]
- 4.Collar, N. & Robson, C. in Birds of the World. (eds. del Hoyo, J., A. Elliott, J. Sargatal, D.A. Christie & E. de Juana) (Cornell Lab of Ornithology, Ithaca, NY, USA, 2023).
- 5.Cai, T. et al. Near-complete phylogeny and taxonomic revision of the world’s babblers (Aves: Passeriformes). Mol. Phylogenet. Evol.130, 346–356 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Xia, J., Wu, F., Hu, W. Z., Fang, J. L. & Yang, X. J. The coexistence of seven sympatric fulvettas in Ailao Mountains, Ejia Town, Yunnan Province. Zool. Res.36, 18–28 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rhie, A. et al. Towards complete and error-free genome assemblies of all vertebrate species. Nature592, 737–746 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics34, i884–i890 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods18, 170–175 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou, C., McCarthy, S. A. & Durbin, R. YaHS: yet another Hi-C scaffolding tool. Bioinformatics39, btac808 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baril, T., Galbraith, J. & Hayward, A. Earl Grey: A fully automated user-friendly transposable element annotation and analysis pipeline. Mol. Biol. Evol.41, msae068 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Storer, J., Hubley, R., Rosen, J., Wheeler, T. J. & Smit, A. F. The Dfam community resource of transposable element families, sequence models, and genome annotations. Mobile DNA12, 2 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics21(Suppl 1), i351–358 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res27, 573–580 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bao, Z. & Eddy, S. R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res12, 1269–1276 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res35, W265–268 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gabriel, L. et al. BRAKER3: Fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS and TSEBRA. bioRxiv, 2023.2006.2010.544449 (2024). [DOI] [PMC free article] [PubMed]
- 19.Keilwagen, J., Hartung, F. & Grau, J. in Gene Prediction: Methods and Protocols. (ed. Kollmar, M.) 161-177 (Springer, New York, 2019).
- 20.Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol.37, 907–915 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuznetsov, D. et al. OrthoDB v11: annotation of orthologs in the widest sampling of organismal diversity. Nucleic Acids Res51, D445–D451 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holt, C. et al. Improved genome assembly and annotation for the rock pigeon (Columba livia). G3-Genes Genomes Genet8, 1391–1398 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods12, 59–60 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol.38, 5825–5829 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Törönen, P., Medlar, A. & Holm, L. PANNZER2: a rapid functional annotation web server. Nucleic Acids Res46, W84–W88 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics29, 2933–2935 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kalvari, I. et al. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res49, D192–D200 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics10, 421 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol21, 245 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang, H. et al. JCVI: A versatile toolkit for comparative genomics analysis. iMeta3, e211 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Manni, M., Berkeley, M. R., Seppey, M., Simao, F. A. & Zdobnov, E. M. BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol.38, 4647–4654 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.NCBI GenBankhttps://identifiers.org/ncbi/insdc.gca:GCA_020745825.3 (2023).
- 34.NCBI GenBankhttps://identifiers.org/ncbi/insdc.gca:GCA_013377495.2 (2022).
- 35.NCBI GenBankhttps://identifiers.org/ncbi/insdc.gca:GCA_028551555.1 (2023).
- 36.Black, A. N. et al. A highly contiguous and annotated genome assembly of the lesser prairie-chicken (Tympanuchus pallidicinctus). Genome Biol. Evol.15, evad043 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leroy, T. et al. Island songbirds as windows into evolution in small populations. Curr. Biol.31, 1303–1310 (2021). [DOI] [PubMed] [Google Scholar]
- 38.Hiller, A. E., Brumfield, R. T. & Faircloth, B. C. A reference genome for the nectar-robbing Black-throated Flowerpiercer (Diglossa brunneiventris). G3. Genes Genomes Genet11, jkab271 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robledo-Ruiz, D. A. et al. Chromosome-length genome assembly and linkage map of a critically endangered Australian bird: the helmeted honeyeater. Gigascience11, giac025 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.NCBI GenBankhttps://identifiers.org/ncbi/insdc.gca:GCA_022539395.1 (2022).
- 41.Peona, V. et al. An annotated chromosome-scale reference genome for Eastern black-eared wheatear (Oenanthe melanoleuca). G3-Genes Genomes Genet.13, jkad088 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li, H. & Durbin, R. Inference of human population history from individual whole-genome sequences. Nature475, 493–496 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol.24, 1586–1591 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Chen, T. et al. The Genome Sequence Archive Family: Toward explosive data growth and diverse data types. Genom. Proteom. Bioinf.19, 578–583 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen, M. et al. Genome Warehouse: A public repository housing genome-scale data. Genom. Proteom. Bioinf.19, 584–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.NCBI GenBankhttps://identifiers.org/ncbi/insdc.gca:GCA_042477295.1 (2024).
- 47.Yan, C. & Wang, M.-S. Genome annotation of the assembly for Fulvetta ruficapilla (Fruf_v1). Science Data Bank10.57760/sciencedb.09502 (2024). [Google Scholar]
- 48.Yan, C. & Wang, M.-S. Genome annotation of the assembly for Fulvetta ruficapilla (Fruf_v1). Figshare10.6084/m9.figshare.26531713.v1 (2024).
- 49.Feng, S. et al. Dense sampling of bird diversity increases power of comparative genomics. Nature587, 252–257 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Challis, R., Richards, E., Rajan, J., Cochrane, G. & Blaxter, M. BlobToolKit - Interactive quality assessment of genome assemblies. G3. Genes Genomes Genet10, 1361–1374 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Yan, C. & Wang, M.-S. Genome annotation of the assembly for Fulvetta ruficapilla (Fruf_v1). Figshare10.6084/m9.figshare.26531713.v1 (2024).
Supplementary Materials
Data Availability Statement
Relevant software, programs, core options, and pipelines regarding the analyses of data filtering, genome assembly, assembly assessment, and annotation have been stated in Methods. The key parameters, code, and scripts for the pipeline are publicly accessible at https://github.com/YanCheer/Fruf_v1_assembly.