

Abstract
Background
Ficolins were originally identified as proteins that bind to transforming growth factor-β1 (TGF-β1). They are capable of activating the complement system through lectin pathway for immune system protection. Ficolin-2 and 3 have been identified in patients with interstitial lung diseases (ILD) and their function in these diseases is currently being explored. In contrast, the functional role of ficolin-1 in pulmonary fibrosis is still elusive and remains to be elucidated.
Methods
The expression of ficolin-1 in the plasma of idiopathic pulmonary fibrosis (IPF) and connective tissue disease (CTD)-ILD patients was first determined. As the orthologue of human ficolin-1, ficolin-B knockout and ficolin-B overexpression were used to establish bleomycin (BLM)-induced pulmonary fibrosis mouse model. Co-immunoprecipitation, immunofluorescence and RNA sequencing were utilized to explore and expound on the expression and the functional mechanism of ficolin-1 in pulmonary fibrosis.
Results
Compared with healthy controls, plasma ficolin-1 was significantly decreased in patients with IPF and CTD-ILD. In the bleomycin (BLM)-induced mice model, ficolin-B deficiency aggravated lung injury and fibrosis. There was also observed increase in TGF-β1 levels and enhanced downstream signaling. However, the overexpression of ficolin-B showed preventative and therapeutic efficacy against lung fibrosis. Furthermore, coimmunoprecipitation studies revealed the direct interaction between ficolin-1 and TGF-β1 in human plasma, which was further confirmed by the colocalization of ficolin-1 and TGF-β1 in lung tissues.
Conclusions
Ficolin-1 inhibits pulmonary fibrosis by directly binding to the key profibrogenic factor TGF-β1, marking it as a potential target for therapy in the treatment of fibrotic lung diseases.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12967-024-05894-1.
Keywords: Pulmonary fibrosis, Bleomycin, Ficolin-1, Ficolin-B, TGF-β1
Background
Idiopathic pulmonary fibrosis (IPF) is a debilitating interstitial pneumonia with a rising incidence and a median survival of just 2–3 years from diagnosis. It is a chronic, progressive and irreversible fibrotic lung disease of unknown cause. It is marked by altered cellular composition and aberrant fibrotic remodeling of the distal lung tissue [1–3]. Despite variable underlying pathogenesis, it shares common pathophysiological pathways with other interstitial lung diseases (ILD), such as connective tissue disease (CTD)-related ILDs (CTD-ILD) and present similar clinical signs of interstitial fibrosis [4, 5]. The current available therapeutic drugs such as antifibrotic drugs, pirfenidone and nintedanib, cause detrimental side effects involving the decline of lung function over time while not halting the disease progression [6–8]. Therefore, it is imperative that we better understand the molecular mechanisms of pulmonary fibrosis for the development of new, essential and improved antifibrotic strategies.
Ficolins were originally documented as transforming growth factor (TGF)-β1 binding proteins, a crucial profibrotic factor in the fibrosis progress [9]. Three types of human ficolins have been identified to date, namely, ficolin-1 (M-ficolin), ficolin-2 (L-ficolin) and ficolin-3 (H-ficolin). Ficolin-1 is thought to function locally following synthesis and secretion from monocytes and granulocytes, with relatively low serum concentrations [10, 11]. On the other hand, ficolin-2 and ficolin-3 are mainly expressed in the liver and present in the circulation as serum lectins [12]. Though three ficolins have been discovered in humans, only two have been described in mice, which include ficolin-A and ficolin-B. A phylogenetic tree analysis has indicated that ficolin-A is regarded as the murine orthologue of human ficolin-2, while mouse ficolin-B is the orthologue of human ficolin-1 [13]. The mouse homologue of the ficolin-3 gene exists as a pseudo-gene. Ficolin-A is therefore functionally equivalent to ficolin-2, and just like in humans, it is mainly considered to be a serum protein derived from the liver and macrophages, whereas ficolin-B is stored and released from myeloid cells locally, and therefore considered functionally equivalent to ficolin-1 in humans [14].
Accumulating evidence has indicated that ficolins could form complexes with mannose-binding lectin-associated serine proteases (MASPs) to initiate the activation of the complement lectin pathway [15]. Several studies have linked pulmonary fibrosis to the complement system. In particular, complement factor 5 (C5) was shown to play an anti-inflammatory role during the acute phase, and a profibrotic role during the chronic stage in a murine model of bleomycin (BLM)-induced pulmonary fibrosis [16]. Another study revealed that C3 expression was higher in IPF patients than in healthy controls, and C3-deficient mice were protected from BLM-induced lung injury [17]. Hongmei et. reported a positive feedback crosstalk between TGF-β1 and complement activation, leading to epithelial injury in pulmonary fibrosis [18].
It is hypothesized that ficolins may be involved in the pulmonary fibrosis process. Lower ficolin-2 plasma levels were reported in IPF patients, which were inversely correlated with the forced vital capacity, whereas there was no observed difference in ficolin-3 [19]. Moreover, high ficolin-2 concentration was associated with better progression-free survival in IPF patients [20]. However, whether or not ficolin-1 plays a role in IPF remains to be confirmed.
To determine the functional role of ficolin-1 in pulmonary fibrosis, we initially had to establish the expression of ficolin-1 in the plasma of IPF and CTD-ILD patients. A mouse model was developed using ficolin-B knockout (Fcnb−/−) mice, the results from the model showed that ficolin-B deficiency exacerbated pulmonary fibrosis in BLM treated mice. In contrast, the overexpression of ficolin-B through delivery of adeno-associated virus (AAV)-FcnB remedied BLM-induced pulmonary fibrosis. Furthermore, co-immunoprecipitation assay was utilized to confirm the direct interaction between ficolin-1 and TGF-β1. Consistent with the findings from the present study, we can surmise that ficolin-1 may be a promising target for the development of improved therapeutic strategy for pulmonary fibrosis.
Materials and methods
Human samples
The diagnosis of IPF was done in accordance with the American Thoracic Society (ATS)/European Respiratory Society (ERS) consensus diagnostic criteria [21]. The classification criterias for CTD-ILD were conducted according to the American College of Rheumatology classification criteria for rheumatoid arthritis (RA), systemic sclerosis (SSc), Sjogren’s disease [22–24], and for other CTD we used current relevant criteria for diagnosis. Age- and sex-matched healthy people were included as control subjects. We recruited 20 healthy control subjects, 20 patients with IPF and 18 patients CTD-ILD. Blood samples from each subject, and lung explant material from IPF patient were collected. All subjects provided written informed consent for participation in the study prior to study onset, and the ethics committee of Tongji Hospital, Huazhong University of Science and Technology (Wuhan, China), reviewed and approved the study (IRB ID: 20150503). The clinical information was included in supplemental Table 1.
Murine model
All mice were housed in a specific-pathogen-free animal facility at the Tongji Hospital (Wuhan, China). The sample size of each group is 6–15. Mice were randomly assigned to different groups. The accessing process was conducted by an assessor blind to treatment allocation. All mice were eight-to-ten weeks male C57BL/6.
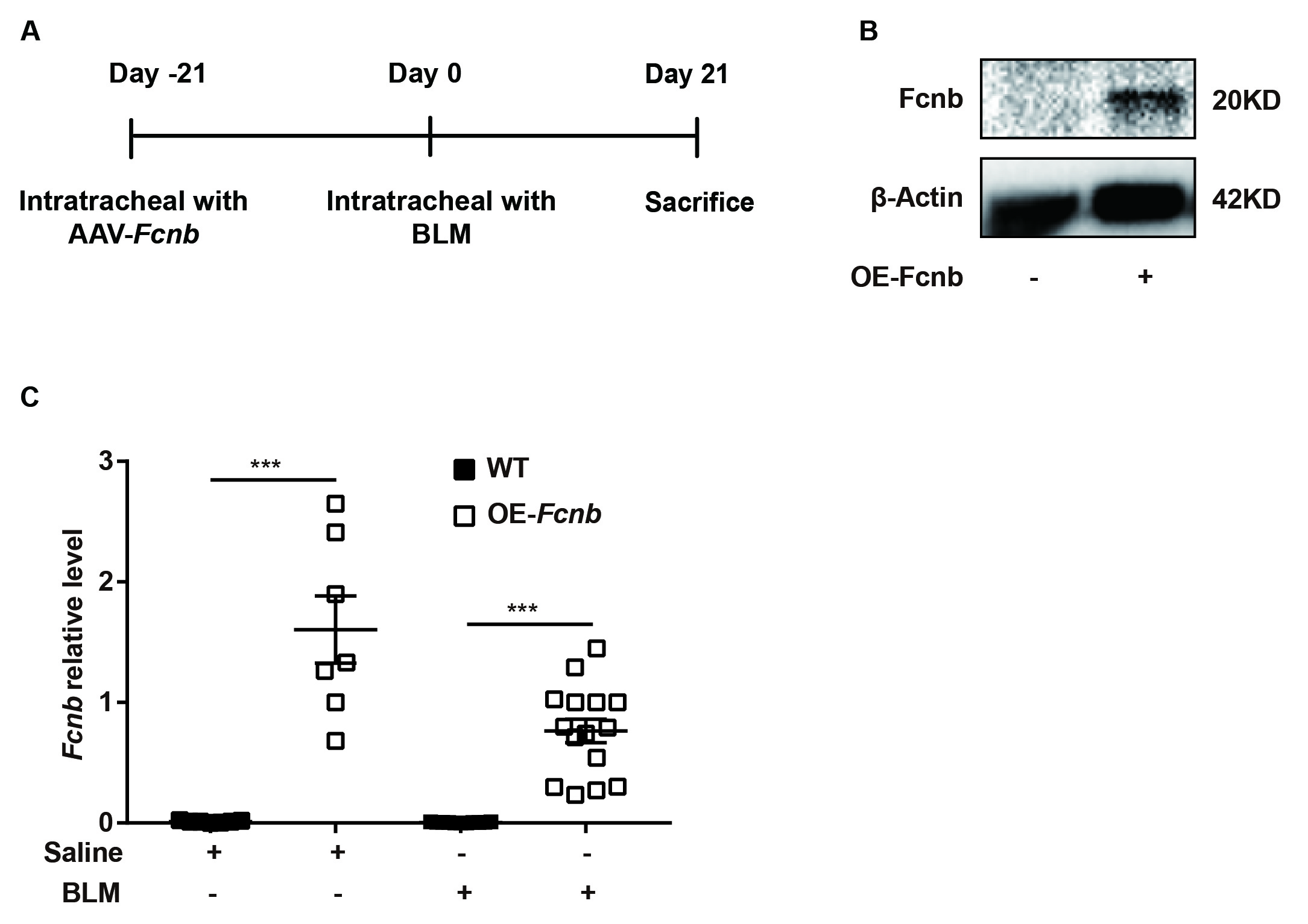
Ficolin-B knockout (Fcnb−/−) mice were generated by GemPharmatech (Nanjing, China), and the littermates were as the control. In this project we used CRISPR/Cas9 technology to modify Fcnb gene (exon2-exon9, Chr2), gRNA was target Fcnb DNA gene site. To generate mice with lung-specific transgenic expression of ficolin-B (OE-Fcnb), adeno-associated virus 6 (AAV6)-CMV-Fcnb-3xFlag-P2A-tWPA (OBIO, China) was delivered into mice via intratracheal instillation. The control mice were injected with AAV6-CMV-3xFlag-P2A-tWPA. The mice were all maintained in a specific pathogen-free animal center at the Tongji Hospital under a 12 h light-cycle environment with clean water and irradiated food. To avoid confounding factors, we randomly assigned mice to the experimental and control groups, and in order to observe mortality, the sample size of mice in the BLM group was 2–3 times that of the normal saline group. All the experiments were approved by the Animal Care and Use Committee of Tongji Hospital (TJH-202105013).
The mice were anesthetized by isoflurane inhalation and then treated with BLM or normal saline for the control group via intratracheal instillation. The bodyweight was recorded twice a day. At 21 days, the mice were anesthetized with 1% pentobarbital sodium. The lung function test was then performed using the FlexiVent system (SCIREQ, Canada). The BALF was collected from BLM-induced mice and the control group. The supernatant of BALF samples was stored at -80℃ and the total cells were counted. The right lungs were frozen in liquid nitrogen immediately and stored at -80℃ for subsequent measurements. The left lungs were soaked in 4% paraformaldehyde.
Regents and antibodies
Bleomycin (Catalog Number: HY-17565 A, dosage: 2 mg/kg) and human recombinant TGF-β1 (Catalog Number: HY-P73616, dosage:10ng/ml) were obtained from MedChemExpress. Primary antibodies were used as follows: Anti-human Ficolin-1 (Catalog Number: MBS2015772), mouse Ficolin-b (cat. MBS2028168) from MYBioSource, diluted 1:1000 for western blot and 1:50 for immunofluorescence. Anti-Fibronectin (Catalog Number: 15613-1-AP), β-Actin (Catalog Number: 20536-1-AP), Fsp1(Catalog Number: 16105-1-AP), Epcam (Catalog Number: 21050-1-AP) from Proteintech, diluted 1:2000 for western blot and 1:50 for immunofluorescence. Anti-TGF-β1(Catalog Number: sc-52893) from Santa cruz, diluted 1:500 for western blot and 1:50 for immunofluorescence. Anti-proSPC (Catalog Number: AB3786) from Millipore, diluted 1:200 for immunofluorescence. Anti-mouse/rabbit FITC and anti-mouse/rabbit Cy3 antibody from Boster, diluted 1:50 for immunofluorescence. ELISA kits were applied as follows: Human/mouse TGF-β1 ELISA (Catalog Number: DB100C) from R&D Systems. Human Ficolin-1 ELISA (Catalog Number: HK357) from Hycult Biotech. Hydroxyproline Colorimetric Assay Kit were from Biovison, Milpitas. RNA extraction kit and SYBR Premix Ex Taq were from Takara. Protein G Magnetic beads was from Cell Signaling Technology.
Histological analysis
The left lung was fixed and then subjected to Masson’s trichrome staining to detect pulmonary interstitial fibrosis of mice. Two pathologists who were blinded to the study assessed the degree of interstitial fibrosis using the Ashcroft scoring system [25]. Each group must contain at least six samples.
Hydroxyproline analysis
The content of hydroxyproline in the lung was used to quantify the collagen in whole mouse lungs. It was determined using a Hydroxyproline Colorimetric Assay Kit following the manufacturer’s instructions. Briefly, the vacuum-dried lung tissue was cracked with concentrated hydrochloric acid at 120℃. After adding the detection reagents to the supernatant, the absorbance was detected using a microplate reader at a wavelength of 560 nm.
Enzyme-linked immunosorbent assay
Human Ficolin-1 and active TGF-β1 were measured using ELISA kits according to the manufacturers’ instructions. The human serum samples were obtained and diluted at 1:50 while there was no dilution of bronchoalveolar lavage fluid (BALF) samples obtained from BLM-induced mice. The lower detection limit of the assays was 31.3 pg/ml for TGF-β1 and 3.1 ng/ml for Ficolin-1.
Western blot analysis
Western blotting was performed by first extracting protein from cultured cells or lung tissues from BLM-induced mice. The protein was then separated by electrophoresis and then transferred onto PVDF membranes. The samples were incubated with the appropriate primary antibodies and secondary antibodies, followed by visualization using the GelDoc XR + System from Bio-Rad, employing established methodologies [26].
Quantitative RT-PCR
Total RNA was extracted from mouse lung tissues or cell lysates using Trizol total RNA extraction kit and then the mRNA was reverse transcribed using reverse transcription kit. Quantitative RT-PCR was performed using SYBR Premix Ex Taq. The primers of the target genes are shown in Table 1 and 2. Relative mRNA expression was calculated using the 2−ΔΔCT method with β-actin as the endogenous control.
Table 1.
RT-PCR mouse primers of genes
| Fibronectin Forward | AATGGTGCCTTGTGCCACTTCC |
|---|---|
| Fibronectin Reverse | ATGTTGTCCCGCCTACCCTCA |
| Collagen I Forward | GCTCAGAGGCGAAGGCAACAG |
| Collagen I Reverse | GATGGGCAG GCGGGAGGTC |
| Tgfb1 Forward | CTCCCGTGGCTTCTAGTGC |
| Tgfb1 Reverse | GCCTTAGTTTGGACAGGATCTG |
| β-Actin Forward | GGCTGTATTCCCCTCCATCG |
| β-Actin Reverse | CCAGTTGGTAACAATGCCATG T |
| Fcnb Forward | CCCGAATTCCCAGCCATGGCCCTGGGATCT |
| Fcnb Reverse | CCCCTCGAGCTAGATGAGCCGCACCTTCAT |
Table 2.
Mouse primers of targeted knock-out genes
| KO Forward | CTACTTTGTGGCTGCCACATCC | 391 bp |
| KO Reverse | TCGACAGGGTTTCTCTGAATAGCC | |
| WT Forward | TCATTCCCAAGCAGTCCCACTC | 455 bp |
| WT Reverse | GTCAAGGCATCTGGTGTCACGG |
Immunofluorescence
Mouse and human lung tissue sections were fixed and paraffin-embedded then stained with primary antigens. The nuclei were counter-stained with DNA with 4′,6 diamidino-2-phenylindole (DAPI). All immunofluorescence images were captured using a Nikon inverted fluorescence microscope (Nikon Eclipse C1) and displayed through the professional analysis software (Nikon DS-U1). The quantification of recruited inflammatory cells was performed by visualizing sections under magnification 400×. Count the total number of cells and the number of cells that show double positivity in each image, then calculate the double positive cell percentage by dividing the number of double positive cells by the total number of cells and multiplying by 100.
RNA sequencing and bioinformatics
RNAs from WT and Fcnb−/− lung tissues were both extracted using an RNA isolation kit. RNA deep sequencing (RNA-seq) libraries were multiplexed and loaded into the Illumina HiSeq flow cell v3. All sequencing protocols were carried out as per the manufacturer’s instructions using the Illumina HiSeq 1000 system and HiSeq control software. In the DEG analysis, genes with a P-value of ≤ 0.05 and a fold change (FC) of ≥ 2 were considered significantly differentially expressed. To investigate genes of TGF-β1 signaling, 108 TGF-β1 signaling-related genes were extracted from the Gene Ontology and GO Annotations (https://www.ebi.ac.uk/QuickGO/), which provides detailed annotations of genes.
Co‑immunoprecipitation assay
Co-immunoprecipitation experiments were performed according to a standard protocol. Total plasma protein was extracted and pre-cleared with 20 µL Protein G Magnetic beads for 2 h. Then the pre-cleared supernatants were incubated with primary antibodies and shaken slowly overnight at 4 °C to form an immunocomplex. The immunocomplex solution was then transferred into a tube containing pre-washed magnetic beads and incubated rotated for 3–4 h at 4 °C. Finally, the immunocomplex was washed five times with 500 µL of 1×cell lysis buffer and analyzed by western blotting analysis.
Statistical analysis
There were no specific statistical tests done to predetermine the sample size. Comparisons among the different groups were performed using the GraphPad Prism 8 software (GraphPad, San Diego, CA). Data were expressed as the mean ± SEM values. Student’s t-test or ANOVA were used to compare normally distributed data. Mann-Whitney U-test was performed to compare the data that were not normally distributed. Pearson’s correlation was used to determine correlation analysis. Kaplan-Meier analysis was applied for the evaluation of survival analysis. A two-sided p-value of < 0.05 was considered statistically significant.
Results
Ficolin-1/ ficolin-B was downregulated in pulmonary fibrosis
Ficolins are capable of activating the complement system through the lectin pathway. In recent studies, the expression of the other two ficolins described in humans, ficolin-2 and 3 expressions have been explored in patients with ILD. However, the functional role of ficolin-1 in pulmonary fibrosis still remains to be studied. To examine and describe its role, the expression levels of ficolin-1 in the plasma of patients with ILD was examined and noted. It was discovered that IPF patients (median, 616.6 pg/ml; IQR, 430.7-745.8 pg/ml) and CTD-ILD patients (median, 666.1 pg/ml; IQR, 484.9-950.9 pg/ml) expressed significantly lower levels of ficolin-1 compared to the control group (median, 895.5 pg/ml; IQR, 724.4–1043.0 pg/ml) (Fig. 1A). On the other hand, the expression levels of TGF-β1 were increased in patients with IPF patients (median, 16527 pg/ml; IQR, 4770–31235 pg/ml) and CTD-ILD patients (median, 16436 pg/ml; IQR, 6981–25455 pg/ml), comparing with healthy control (median, 5695 pg/ml; IQR, 3041–7944 pg/ml) (Fig. 1B). The clinical information was included in supplemental Table 1.
Fig. 1.

Expression of Ficolin-1 and TGF-β1 in pulmonary fibrosis patients. (A-B) Elisa analysis of A) FCN1 and B) TGF-β1 expression in the serum of control subjects and pulmonary fibrosis patients (n = 18–20 per group). Data are presented as median ± IQR. *p < 0.05, **p < 0.01, ***p < 0.001 by Mann-Whitney’s unpaired non-parametric test (A-B). FCN1, ficolin-1; Fcnb, ficolin-B
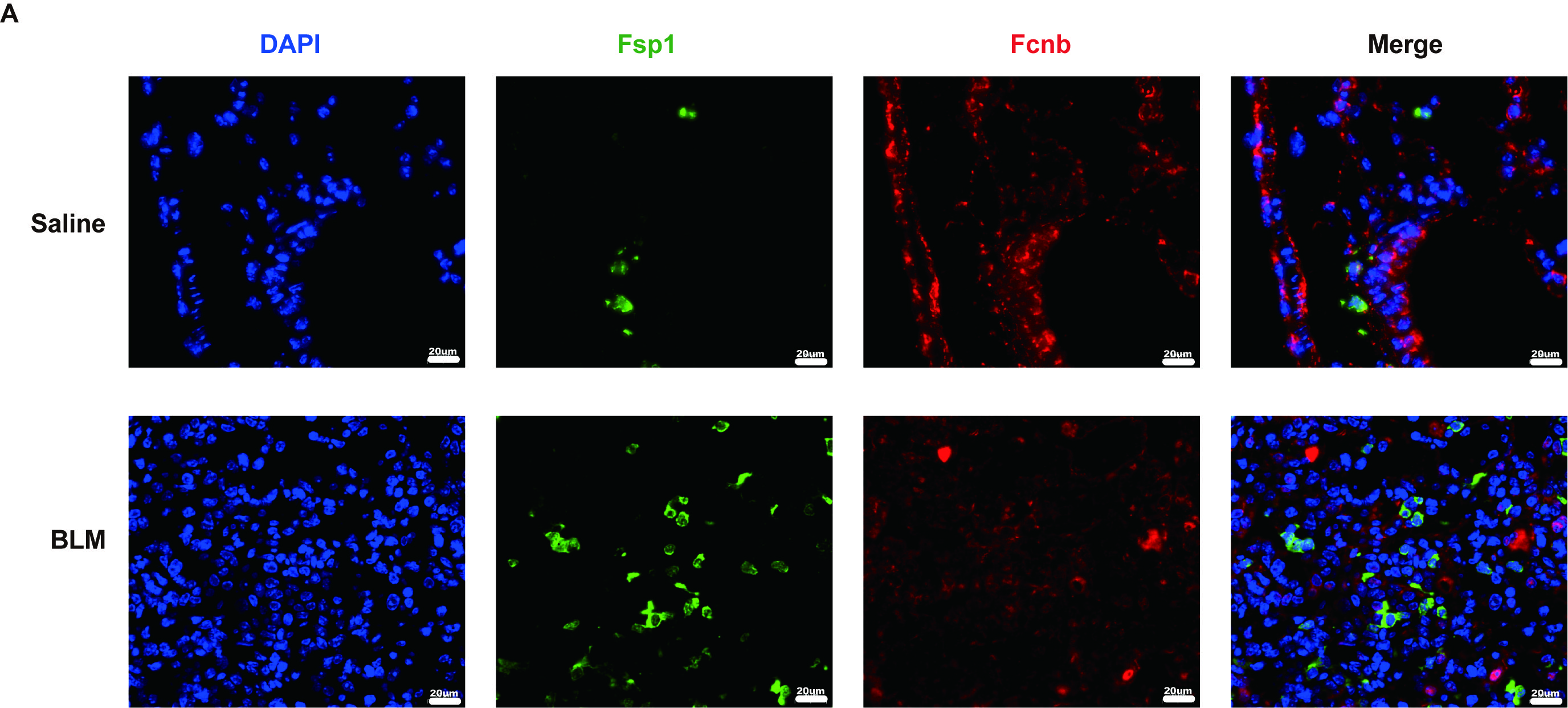
In the same way, the lungs obtained from BLM-induced mice also exhibited lower expression levels of ficolin-B than that of the control group, while fibronectin was upregulated in the fibrotic lungs (Fig. 2A and C). The expression of fibrosis-related genes (Acta2, Fn1 and Tgfβ1) revealed that they were negatively correlated to the expression of ficolin-B (Fig. 2D and F). Additionally, the co-immunostaining results further reinforced the conclusion that ficolin-B (red) was highly expressed in pro-Spc + alveolar epithelium (Fig. 2G) but not in Fsp1+ (green) myofibroblasts (Supplemental Fig. 1A). Taken together, our data indicate that pulmonary fibrosis is characterized by low ficolin-1/ficolin-B expression and high fibrosis markers.
Fig. 2.

Expression of Fcnb in pulmonary fibrosis mice with bleomycin (BLM) induction. (A-B) Western blot analysis of Fcnb and fibronectin expression in the lungs of mice after BLM induction (n = 4 per group). (C) RT-PCR analysis of Fcnb in the lungs of mice after BLM treatment (n = 6–12 per group). (D-F) RT-PCR analysis of the correlation between Fcnb and D) Acta2, F) Fn1, F) Tgfb1 expression after BLM induction (n = 12 per group). (G) Representative results for co-immunostaining of Fcnb and pro-Spc (alveolar epithelium marker) in lung sections from BLM-induction mice. The nuclei were stained blue using 4′,6-diamidino-2-phenylindole (DAPI), and the images were taken under original magnification ×400. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by student’s unpaired t-test (B and C). Fcnb, ficolin-B
Ficolin-B deficiency exacerbated pulmonary fibrosis in BLM-treated mice
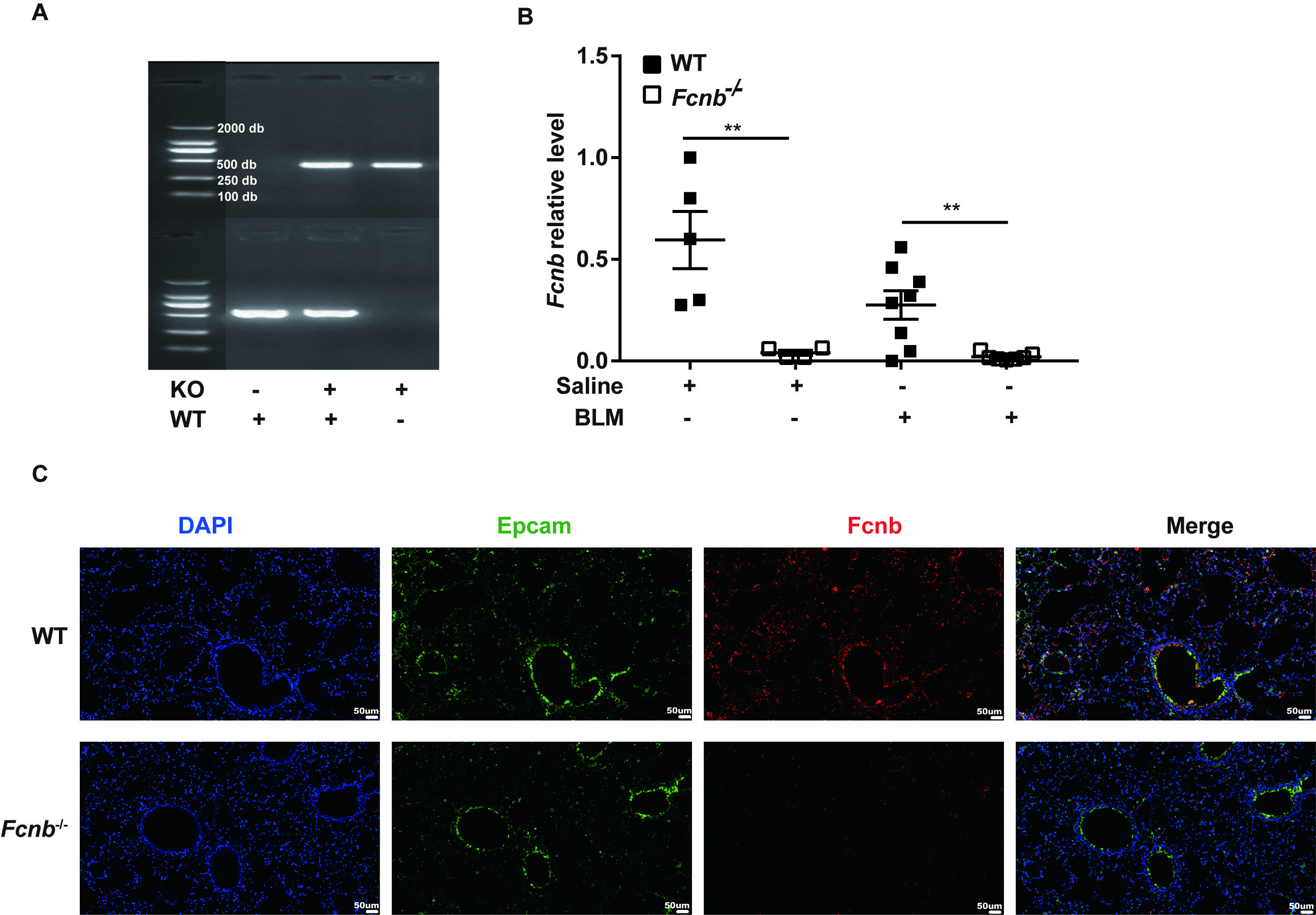
To further investigate the role of ficolin-B in pulmonary fibrosis, we generated ficolin-B knockout (Fcnb−/−) mice, and their wild-type (WT) littermates were used as the control group. The PCR analysis of Fcnb−/− mice DNA isolated from the lung showed that Fcnb was knocked out in mice (Supplemental Fig. 2A). The same results were obtained in RNA transcription level (Supplementary Fig. 2B). These results were further confirmed by co-immunostaining of lung sections (Supplemental Fig. 2C).
Considering frailty as an independent risk factor for pulmonary fibrosis, we first assessed the changes in body weight of the mice. At 21 days following the BLM treatment, Fcnb−/− mice presented with weight loss (Fig. 3A) indicating the debilitating effect of ficolin-B depletion in pulmonary fibrosis. The significantly decreased levels of ficolin-B greatly curbed mouse survival (Fig. 3B). Masson staining which is representative of tissue fibrosis showed immensely aggravated lung tissue injury and fibrosis in Fcnb−/− mice compared to the control group (Fig. 3C). Phenotypically, the Ashcroft score was much higher in Fcnb−/− mice compared with the WT group (Fig. 3D). Moreover, the parameters of lung compliance, lung elastance, and inspiratory capacity were significantly decreased in BLM-treated mice. In accordance with these observations, ficolin-B deficiency further exacerbated BLM-induced lung function loss in mice (Fig. 4A and E). Furthermore, WT mice showed lower hydroxyproline content in comparison with Fcnb−/− mice (Fig. 5A). The number of inflammatory cells (maingly macrophage) in the BALF and the amount of Fibronectin and Collagen I in lung tissue of BLM-treated Fcnb−/− mice were much higher than that in the WT mice (Fig. 5B and D). Collectively, these data support that the deletion of ficolin-B exacerbated lung injury and fibrosis.
Fig. 3.

Fcnb deficiency exacerbated pulmonary fibrosis in BLM-treated mice. (A) Work flow of murine models. (B) The body weight change during BLM treatment was monitored (n = 6–12 per group). (C) Survival studies were conducted in the BLM lung fibrosis model (n = 6–12 per group). (D) The lungs from WT and Fcnb−/− mice were stained with Masson’s trichrome (n = 6–10 per group). (E) Histological analysis of the severity of lung fibrosis in mice after BLM induction (n = 6–10 per group). Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA (D), two-way ANOVA (A) or Kaplan-Meier (B). Fcnb, ficolin-B
Fig. 4.

Lung function following prophylactic deletion of Fcnb in BLM-treated mice. (A-E) The pressure-volume-loops (PV-loops), inspiratory capacity, respiratory system elasticity (Ers), static compliance (Cst), and respiratory system compliance (Crs) of mice were measured (n = 6–10 per group). Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA (B-E) or two-way ANOVA (A). Fcnb, ficolin-B
Fig. 5.

Fibrotic gene expression following prophylactic deletion of Fcnb in BLM-treated mice. (A)The content of hydroxyproline in the right middle lung was measured ((n = 6 per group). (B) The BALF total cells were counted (n = 6–10 per group). (C-D) RT-PCR analysis of Fibronectin (Fn1), Collagen I (Col1a2) (n = 4–9 per group). Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA (A-D). Fcnb, ficolin-B
Depletion of ficolin-B amplified TGF-β1 signaling
To outline more detailed mechanisms by which ficolin-B deficiency drives pulmonary fibrosis, RNA-seq was employed to compare the expression patterns of genes (Supplemental Fig. 3A). By comparing gene expression profiles between BLM-treated Fcnb−/− mice and BLM-treated WT mice, a total of 1684 differentially expressed genes (DEGs) were identified. Among these genes, 875 of them were upregulated and 809 were downregulated (Supplemental Fig. 3A, Supplemental Table 2). Given that TGF-β1 was associated with ficolin-1/ficolin-B, we embarked on confirming the expression levels of TGF-β1 downstream signaling molecules. As expected, the expression levels of the downstream molecules were changed sharply after the depletion ficolin-B (Fig. 6A, Supplemental Table 3), as evidenced by the ELISA analysis of TGF-β1 in BALF. Our data concomitantly suggested that the depletion of ficolin-B amplified TGF-β1 signaling.
Fig. 6.

Depletion of Fcnb amplified TGF-β1 signaling. (A) Heatmap of TGF-β1 signaling genes after BLM treated. (B) Elisa analysis of TGF-β1 expression in the BALF of WT and Fcnb−/− mice (n = 6–10 per group). Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA (B). Fcnb, ficolin-B
Lung-specific transgenic expression of ficolin-B protected mice against pulmonary fibrosis
Subsequently, to further validate the above findings, we generated mice with lung-specific transgenic expression of ficolin-B via intratracheal instillation of AAV6-Fcnb (Supplemental Fig. 4A). Western blot and RT-PCR analysis showed that ficolin-B was overexpressed in the Fcnb-overexpressed lung (Supplemental Fig. 4B and 4 C). The bodyweight of the mice was significantly improved by the overexpression of ficolin-B (Fig. 7A) as well as significantly improved BLM-induced pulmonary injury and fibrosis, as shown by histological analysis (Fig. 7B) and the Ashcroft score (Fig. 7C). Mice with lung-specific overexpression of ficolin-B also displayed reduced inflammatory cell infiltration (Fig. 8A) and decreased expression of fibrosis markers (Fig. 8B and C). Moreover, TGF-β1 transcriptional levels were also downregulated following prophylactic overexpression of ficolin-B in BLM-treated mice (Fig. 8D). These findings signify that ficolin-B overexpression in the lung might alleviate pulmonary fibrosis and its symptoms and therefore should be targeted as treatment option for pulmonary fibrosis.
Fig. 7.

Lung-specific transgenic expression of Fcnb protected mice against lung fibrosis. (A) Work flow of murine models. (B) The body weight change during BLM treatment was monitored (n = 6–15 per group). (C) The lungs from WT and OE-Fcnb mice were stained with Masson’s trichrome (n = 6 per group). (D) Histological analysis of the severity of lung fibrosis in mice after BLM induction (n = 6–15 per group). Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA (C) or two-way ANOVA (A). Fcnb, ficolin-B
Fig. 8.

Fibrotic gene expression following prophylactic overexpression of Fcnb in BLM-treated mice. (A) The BALF total cells were counted (n = 6–15 per group). (B-D) RT-PCR analysis of Fibronectin (Fn1), Collagen I (Col1a2) and Tgfb1 (n = 5–9 per group). Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA (A-D). Fcnb, ficolin-B
Ficolin-1 interacted with TGF-β1 in pulmonary fibrosis
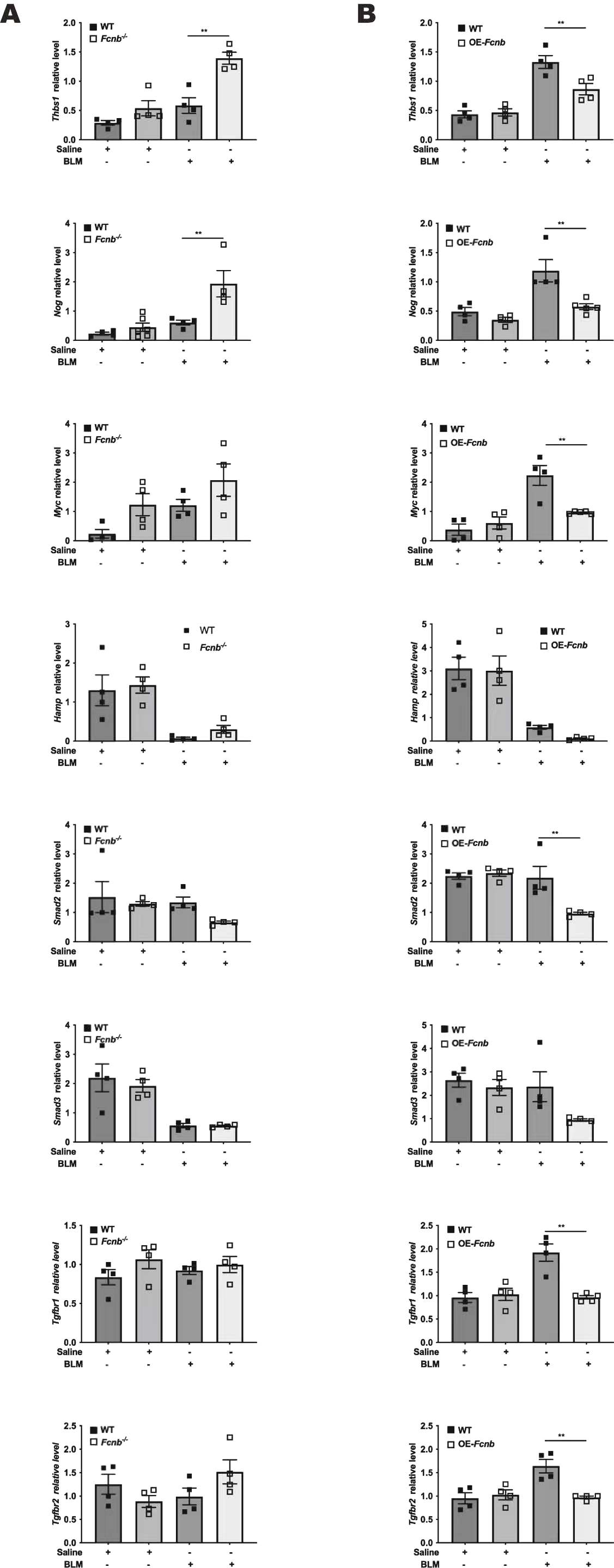
Studies have shown that ficolins were originally described to bind TGF-β1 in porcine uterus [9], but very little research has been done to confirm its interaction with TGF-β1 in humans. Our study has confirmed for the first time that this combination also existed in human by co-immunoprecipitation (Fig. 9A). Immunofluorescence analysis confirmed the co-localization of TGF-β1 and ficolin-1 in the lung sections of a patient with IPF (Fig. 9B). Importantly, the expression of ficolin-1 was decreased in a dose-dependent manner upon TGF-β1 stimulation (Fig. 9C and D). From the above findings, we can confirm the regulatory role of ficolin-B in vivo. The expression of TGF-β1 showed a negative correlation with the expression of ficolin-B (Fig. 2F). We found that the level of TGF-β1 was markedly increased in the lungs of BLM-treated mice, and further elevated in Fcnb−/− mice (Fig. 6B). Further, the expression of TGF-β1 signaling core genes (Thbs1, Nog, Myc, Hamp, Smad2, Smad3, Tgfbr1, Tgfbr2) were significantly changed in Fcnb−/− mice (Supplementary Fig. 5A), while this change was reversed in the ficolin-B-overexpressed mice (Supplementary Fig. 5B). Taken together, Ficolin-1 directly interacted with TGF-β1, thus leading to amplified TGF-β1 signaling in pulmonary fibrosis (See Fig. 10).
Fig. 9.

FCN1 interacted with TGF-β1 in pulmonary fibrosis. (A) Co‑immunoprecipitation analysis of TGF-β1 and FCN1 in serum of IPF patients. (B) Representative results for co-immunostaining of FCN1 and TGF-β1 in lung sections derived from lung explant material from an IPF patient. The nuclei were stained DAPI, and the images were taken under original magnification ×400. (C-D) Western blot analysis of FCN1 expression in A549 cells after TGF-β1 induction (n = 3 per group). Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA (D). FCN1, ficolin-1; Fcnb, ficolin-B
Fig. 10.

Diagram of the mechanisms underlying ficolin-1 regulation of pulmonary fibrosis
Discussion
Pulmonary fibrosis is an endpoint pathological change that occurs in the lungs in many chronic respiratory diseases due to chronic lung injury. It is associated with an atypical immune response which tends to lead to aberrant wound healing and the continuous deposition of extracellular matrix [27]. Fundamentally, our study revealed that plasma ficolin-1 was significantly decreased in patients with IPF and CTD-ILD when compared with healthy controls. Correlatively, the expression levels of ficolin-B were also observed to be downregulated in bleomycin-induced pulmonary fibrosis mouse model. Furthermore, Fcnb−/− mice presented with aggravated pulmonary fibrosis along with an elevated expression of TGF-β1 levels and enhanced downstream signaling, this finding reveals a regulatory relationship between ficolin-1/ficolin-B and TGF-β1, a key profibrogenic molecule. It points to the fact that the downregulation of ficolin-1/ficolin-B in pulmonary fibrosis leads to the elevated expression of TGF-β1, thereby making TGF-β1 a driver of pulmonary fibrosis and ficolin-1/ficolin-B a preventer or limiter of the onset or progression of pulmonary fibrosis. This relationship has been previously been described in porcine uterus and our results from the colocalization of ficolin-1 and TGF-β1 in lung tissues have confirmed the direct interaction between ficolin-1 and TGF-β1 in human plasma. This observation is noteworthy and is further confirmed in bleomycin-induced pulmonary fibrosis mouse model, where the overexpression of ficolin-B improved pulmonary compliance, and reduced pulmonary fibrosis related genes, including Collagen I and fibronectin.
It is commonly accepted that ficolins are vital triggers of the complement lectin pathway. They are mainly involved in protective roles against various bacterial, fungal, and viral pathogens [28–35]. However, other studies such as those conducted by Jarlhelt L et al. and Wu X et al. showed that the levels of both ficolin-A and B were significantly increased in LPS-induced acute lung injury (ALI) model. The extent of lung injury and local inflammation in the acute phase were aggravated [36, 37], and this was also accompanied by attenuated complement activation observed only in FcnA−/− but not FcnB−/− mice [37]. The same group also reported that ficolin-A exacerbated severe H1N1 influenza virus infection-induced ALI via excessive complement activation, whereas ficolin-B deficiency showed no effect in either the lung injury or complement activation [38]. One particular observation to note was that ficolin-A and B showed different expressions in the lungs of the two ALI mouse models mentioned above [37, 38], indicating that these two mouse ficolins may have distinct physiological functions. Even more interestingly, it was observed that ficolin-B deficiency led to the delayed repair of LPS-induced local lung injury [37]. In view of all these findings pertaining to the key role played by ficolins in chronic lung injury in pulmonary fibrosis, it may be worth noting that the repair process dysregulation in FcnB−/− mice may partly contribute to exacerbated pulmonary fibrosis, which was further confirmed in our study.
Although TGF-β1 has long been considered a key profibrotic mediator in organ fibrosis, few studies exploring the role of ficolins, initially found as TGF-β1 binding proteins, in pulmonary fibrosis have been reported. Lower ficolin-2 plasma levels were observed in IPF patients [19], whereas higher ficolin-2 levels were associated with interstitial lung involvement in patients with systemic sclerosis [39, 40]. It is presumed that ficolin-2 expression was reversely affected in the pulmonary fibrosis process shared by IPF and SSc-ILD, as well as the pathogenesis of SSc itself. In terms of ficolin-3, despite of no difference between SSc patients and healthy controls, SSc patients with decreased ficolin-3 levels tended to have a higher prevalence of ILD. Besides, serum ficolin-3 levels inversely correlated with ground-glass opacity score on chest computed tomography in SSc-ILD patients [41]. Curiously, there was no difference in ficolin-3 expression between IPF and healthy subjects [19]. Unlike ficolin-2 and 3, we firstly reported that plasma ficolin-1 concentrations were decreased both in IPF and CTD-ILD patients, accompanied with increased TGF-β1 levels, which can be partly explained by the direct interaction between ficolin-1 and TGF-β1 in human plasma, suggesting that ficolin-1 may play a protective role against pulmonary fibrosis. Besides, ficolin-1 decreased with TGF-β1 stimulation in type II alveolar epithelial cells (AECII) in a time-dependent manner, as another explanation for the low of ficolin-1 expression in patients with lung fibrosis.
Ficolin-B is considered the ortholog of human ficolin-1 in mice, and consistently with ficolin-1, it was significantly downregulated in BLM-treated mice, just as in patients with pulmonary fibrosis. The location of ficolin-B remains unclear, it has nonetheless been reported to be primarily stored and released locally from macrophages and neutrophils with very low serum concentrations, which is just as similar as ficolin-1 in human [14]. In our study, ficolin-B was further confirmed to be detected in the lungs, especially in AECIIs and fibroblasts, playing vital roles in the initiation and development of IPF. We further observed and reported that pulmonary fibrosis appeared to be aggravated in BLM-treated FcnB−/− mice, and progressed alongside elevated levels of TGF-β1 levels and augmented downstream signaling.
In contrast with our results, Xu and colleagues reported elevated ficolin-1 levels in ILD patients, and profibrotic role of ficolin-B in BLM-induced lung injury model [42]. It should be emphasized that ILD consists of a group of pulmonary disorders, and different types of ILD have distinct pathophysiology, clinical manifestations, and prognoses. Given the high heterogeneity of ILD, a simple comparison of ficolin-1 levels between ILD patients and healthy controls may not be appropriate, especially with relatively small sample size [42]. In their animal experiments, 10 mice were divided into each group, and the survival rate of BLM-induced WT mice was < 50% [42]. As ficolin-B treatment was reported to exacerbate bleomycin-induced lung injury [42], which may lead to lower survival rate, whether survived mice were sufficient for statistical analysis was an important concern.
Under the intervention of Fcnb, the most significantly changed genes in the TGF-β1 signaling pathway are Thbs1 and Nog. Synthesized by various cellular sources such as endothelial cells, smooth muscle cells, and fibroblasts, Thbs1 contributes to the cell-matrix interactions, blood vessel formation, and tissue repair processes [43, 44]. In our previous study, we identified Thbs1 as an aging-associated core gene in the development of pulmonary fibrosis [45]. But Micheal ect. demonstrated that Thbs1 deficiency did not protect mice from systemic bleomycin challenge [46]. The relationship of Thbs1 and aged pulmonary fibrosis was worth deep-investigation. The Nog gene encodes the Noggin protein, which plays an important role in the development of many body tissues, including neural tissues, muscles, and bones [47]. Noggin may affect the repair and fibrotic processes of lung tissue by regulating the BMP signaling pathway in pulmonary fibrosis [48, 49]. However, the specific role and mechanism of Nog in pulmonary fibrosis still require further research to elucidate.
The causal relationship between decreased ficolin-1 and the development of pulmonary fibrosis is not yet completely determined. However, the in vitro experimental results of this study show that TGF-β1 intervention inhibits the expression of ficolin-1 in a time-dependent manner, we can therefore speculate that the occurrence of pulmonary fibrosis is more likely to act as a driving factor for the downregulation of ficolin-1 expression. The reduced expression of ficolin-1 leads to a reduction in its competitive binding with TGF-β1, which consequently causes excessive activation of the downstream signaling pathway of TGF-β1, forming a feedback loop which promotes the progression of pulmonary fibrosis.
In conclusion, we established the association between decreased ficolin-1 expression and the lung fibrosis process. Using ficolin-B, the ortholog of human ficolin-1 in vivo, we were able to determine that ficolin-B deficiency exacerbated BLM-induced pulmonary fibrosis. The suppressed levels of ficolin-B were accompanied by elevated TGF-β1 expression and signaling. When overexpressed, ficolin-B exerted an antifibrotic effect and alleviated pulmonary fibrosis. From the results obtained in patients with IPF and the mouse model, we can draw that ficolin-1 competitively binds to TGF-β1, and when its expression is repressed just as in pulmonary fibrosis, the expression of TGF-β1 becomes elevated and pulmonary fibrosis becomes exacerbated.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We would like to thank GemPharmatech Co., Ltd. for Fcnb−/− mice construction.
Abbreviations
- IPF
idiopathic pulmonary fibrosis
- ILD
interstitial lung diseases
- CTD
connective tissue disease
- TGF
transforming growth factor
- MASP
mannose-binding lectin-associated serine protease
- C5
complement factor 5
- BLM
bleomycin
- AAV
adeno-associated virus
- RA
rheumatoid arthritis
- SSc
systemic sclerosis
- BALF
bronchoalveolar lavage fluid
- DAPI
DNA with 4′,6 diamidino-2-phenylindole
- MFI
mean fluorescence intensity
- WT
wild-type
- DEG
differentially expressed genes
- ALI
acute lung injury
- AECII
type II alveolar epithelial cells
Author contributions
Pengfei Gao, Yanjiao Lu, Jianping Zhao and Yimin Mao contributed to the design of this study. Pengfei Gao, Yanjiao Lu, Kun Tang and Wei Wang contributed to animal experiments and evaluations. Pengfei Gao and Yanjiao Lu analyzed the results and wrote the manuscript. Tongsheng Wang and Yingwei Zhu analyzed and interpreted the data, and thoroughly revised the manuscript. Jianping Zhao and Yimin Mao provided overall supervision and contributed to the overall revision of the manuscript. All authors read and approved the final manuscript.
Funding
The work in this study was supported by Henan Provincial Science and Technology Research Project (No. 222103810052, 222102310533), Natural Science Foundation of Henan Province (No. 232300421304), Henan Provincial Medical Science and Technology Research Project (No. LHGJ20220671), National Natural Science Foundation of China (No.82300094), and Young Elite Scientists Sponsorship Program by Luoyang Association for Science and Technology (No. 2022HLTJ18).
Data availability
Data will be made available upon reasonable request.
Declarations
Ethics approval and consent to participate
The use of clinical specimens was approved by the ethics committee of Tongji Hospital, Huazhong University of Science and Technology (IRB ID: 20150503). All the animal experiments were approved by the Animal Care and Use Committee of Tongji Hospital (TJH-202105013).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Pengfei Gao and Yanjiao Lu contributed equally to this work.
Contributor Information
Pengfei Gao, Email: gpf_working@163.com.
Jianping Zhao, Email: Zhaojp88@126.com.
Yimin Mao, Email: yimin6107@sina.com.
References
- 1.Lederer DJ, Martinez FJ: Idiopathic Pulmonary Fibrosis. N Engl J Med 2018, 378:1811–1823. [DOI] [PubMed]
- 2.Olson AL, Gifford AH, Inase N, Fernandez Perez ER, Suda T: The epidemiology of idiopathic pulmonary fibrosis and interstitial lung diseases at risk of a progressive-fibrosing phenotype. Eur Respir Rev 2018, 27. [DOI] [PMC free article] [PubMed]
- 3.Ley B, Collard HR, King TE, Jr.: Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011, 183:431–440. [DOI] [PubMed]
- 4.Mathai SK, Newton CA, Schwartz DA, Garcia CK: Pulmonary fibrosis in the era of stratified medicine. Thorax 2016, 71:1154–1160. [DOI] [PMC free article] [PubMed]
- 5.Goodwin AT, Jenkins G: Molecular Endotyping of Pulmonary Fibrosis. Chest 2016, 149:228–237. [DOI] [PubMed]
- 6.Behr J, Prasse A, Kreuter M, Johow J, Rabe KF, Bonella F, Bonnet R, Grohe C, Held M, Wilkens H, et al: Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): a double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir Med 2021, 9:476–486. [DOI] [PubMed]
- 7.King TE, Jr., Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, Gorina E, Hopkins PM, Kardatzke D, Lancaster L, et al: A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014, 370:2083–2092. [DOI] [PubMed]
- 8.Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, Cottin V, Flaherty KR, Hansell DM, Inoue Y, et al: Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014, 370:2071–2082. [DOI] [PubMed]
- 9.Ichijo H, Hellman U, Wernstedt C, Gonez LJ, Claesson-Welsh L, Heldin CH, Miyazono K: Molecular cloning and characterization of ficolin, a multimeric protein with fibrinogen- and collagen-like domains. J Biol Chem 1993, 268:14505–14513. [PubMed]
- 10.Wittenborn T, Thiel S, Jensen L, Nielsen HJ, Jensenius JC: Characteristics and biological variations of M-ficolin, a pattern recognition molecule, in plasma. J Innate Immun 2010, 2:167–180. [DOI] [PubMed]
- 11.Garred P, Genster N, Pilely K, Bayarri-Olmos R, Rosbjerg A, Ma YJ, Skjoedt MO: A journey through the lectin pathway of complement-MBL and beyond. Immunol Rev 2016, 274:74–97. [DOI] [PubMed]
- 12.Endo Y, Matsushita M, Fujita T: New insights into the role of ficolins in the lectin pathway of innate immunity. Int Rev Cell Mol Biol 2015, 316:49–110. [DOI] [PubMed]
- 13.Garred P, Honore C, Ma YJ, Rorvig S, Cowland J, Borregaard N, Hummelshoj T: The genetics of ficolins. J Innate Immun 2010, 2:3–16. [DOI] [PubMed]
- 14.Liu Y, Endo Y, Homma S, Kanno K, Yaginuma H, Fujita T: Ficolin A and ficolin B are expressed in distinct ontogenic patterns and cell types in the mouse. Mol Immunol 2005, 42:1265–1273. [DOI] [PubMed]
- 15.Howard M, Farrar CA, Sacks SH: Structural and functional diversity of collectins and ficolins and their relationship to disease. Semin Immunopathol 2018, 40:75–85. [DOI] [PMC free article] [PubMed]
- 16.Addis-Lieser E, Kohl J, Chiaramonte MG: Opposing regulatory roles of complement factor 5 in the development of bleomycin-induced pulmonary fibrosis. J Immunol 2005, 175:1894–1902. [DOI] [PubMed]
- 17.Okamoto T, Mathai SK, Hennessy CE, Hancock LA, Walts AD, Stefanski AL, Brown KK, Lynch DA, Cosgrove GP, Groshong SD, et al: The relationship between complement C3 expression and the MUC5B genotype in pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 2018, 315:L1-L10. [DOI] [PMC free article] [PubMed]
- 18.Gu H, Mickler EA, Cummings OW, Sandusky GE, Weber DJ, Gracon A, Woodruff T, Wilkes DS, Vittal R: Crosstalk between TGF-beta1 and complement activation augments epithelial injury in pulmonary fibrosis. FASEB J 2014, 28:4223–4234. [DOI] [PMC free article] [PubMed]
- 19.Vogt S, Trendelenburg M, Tamm M, Stolz D, Hostettler KE, Osthoff M: Local and Systemic Concentrations of Pattern Recognition Receptors of the Lectin Pathway of Complement in a Cohort of Patients With Interstitial Lung Diseases. Front Immunol 2020, 11:562564. [DOI] [PMC free article] [PubMed]
- 20.Ashley SL, Xia M, Murray S, O’Dwyer DN, Grant E, White ES, Flaherty KR, Martinez FJ, Moore BB: Six-SOMAmer Index Relating to Immune, Protease and Angiogenic Functions Predicts Progression in IPF. PLoS One 2016, 11:e0159878. [DOI] [PMC free article] [PubMed]
- 21.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, et al: An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011, 183:788–824. [DOI] [PMC free article] [PubMed]
- 22.Shiboski CH, Shiboski SC, Seror R, Criswell LA, Labetoulle M, Lietman TM, Rasmussen A, Scofield H, Vitali C, Bowman SJ, Mariette X: 2016 American College of Rheumatology/European League Against Rheumatism classification criteria for primary Sjögren’s syndrome: A consensus and data-driven methodology involving three international patient cohorts. Ann Rheum Dis 2017, 76:9–16. [DOI] [PubMed]
- 23.van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, Matucci-Cerinic M, Naden RP, Medsger TA, Jr., Carreira PE, et al: 2013 classification criteria for systemic sclerosis: an American college of rheumatology/European league against rheumatism collaborative initiative. Ann Rheum Dis 2013, 72:1747–1755. [DOI] [PubMed]
- 24.Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO, 3rd, Birnbaum NS, Burmester GR, Bykerk VP, Cohen MD, et al: 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis 2010, 69:1580–1588. [DOI] [PubMed]
- 25.Hubner RH, Gitter W, El Mokhtari NE, Mathiak M, Both M, Bolte H, Freitag-Wolf S, Bewig B: Standardized quantification of pulmonary fibrosis in histological samples. Biotechniques 2008, 44:507–511, 514 − 507. [DOI] [PubMed]
- 26.Lu Y, Tang K, Wang S, Tian Z, Fan Y, Li B, Wang M, Zhao J, Xie J: Dach1 Deficiency Drives Alveolar Epithelium Apoptosis in Pulmonary Fibrosis via Modulating C-Jun/Bim Activity. Transl Res 2023. [DOI] [PubMed]
- 27.Kolb M, Vasakova M: The natural history of progressive fibrosing interstitial lung diseases. Respir Res 2019, 20:57. [DOI] [PMC free article] [PubMed]
- 28.Genster N, Praestekjaer Cramer E, Rosbjerg A, Pilely K, Cowland JB, Garred P: Ficolins Promote Fungal Clearance in vivo and Modulate the Inflammatory Cytokine Response in Host Defense against Aspergillus fumigatus. J Innate Immun 2016, 8:579–588. [DOI] [PMC free article] [PubMed]
- 29.Pan Q, Chen H, Wang F, Jeza VT, Hou W, Zhao Y, Xiang T, Zhu Y, Endo Y, Fujita T, Zhang XL: L-ficolin binds to the glycoproteins hemagglutinin and neuraminidase and inhibits influenza A virus infection both in vitro and in vivo. J Innate Immun 2012, 4:312–324. [DOI] [PMC free article] [PubMed]
- 30.Endo Y, Takahashi M, Iwaki D, Ishida Y, Nakazawa N, Kodama T, Matsuzaka T, Kanno K, Liu Y, Tsuchiya K, et al: Mice deficient in ficolin, a lectin complement pathway recognition molecule, are susceptible to Streptococcus pneumoniae infection. J Immunol 2012, 189:5860–5866. [DOI] [PubMed]
- 31.Bidula S, Kenawy H, Ali YM, Sexton D, Schwaeble WJ, Schelenz S: Role of ficolin-A and lectin complement pathway in the innate defense against pathogenic Aspergillus species. Infect Immun 2013, 81:1730–1740. [DOI] [PMC free article] [PubMed]
- 32.Jensen K, Lund KP, Christensen KB, Holm AT, Dubey LK, Moeller JB, Jepsen CS, Schlosser A, Galgoczy L, Thiel S, et al: M-ficolin is present in Aspergillus fumigatus infected lung and modulates epithelial cell immune responses elicited by fungal cell wall polysaccharides. Virulence 2017, 8:1870–1879. [DOI] [PMC free article] [PubMed]
- 33.Kobayashi T, Kuronuma K, Saito A, Ikeda K, Ariki S, Saitou A, Otsuka M, Chiba H, Takahashi S, Takahashi M, Takahashi H: Insufficient serum L-ficolin is associated with disease presence and extent of pulmonary Mycobacterium avium complex disease. Respir Res 2019, 20:224. [DOI] [PMC free article] [PubMed]
- 34.Luo F, Chen T, Liu J, Shen X, Zhao Y, Yang R, Zhang X: Ficolin-2 binds to HIV-1 gp120 and blocks viral infection. Virol Sin 2016, 31:406–414. [DOI] [PMC free article] [PubMed]
- 35.Zhao Y, Ren Y, Zhang X, Zhao P, Tao W, Zhong J, Li Q, Zhang XL: Ficolin-2 inhibits hepatitis C virus infection, whereas apolipoprotein E3 mediates viral immune escape. J Immunol 2014, 193:783–796. [DOI] [PubMed]
- 36.Jarlhelt I, Genster N, Kirketerp-Moller N, Skjoedt MO, Garred P: The ficolin response to LPS challenge in mice. Mol Immunol 2019, 108:121–127. [DOI] [PubMed]
- 37.Wu X, Yao D, Bao L, Liu D, Xu X, An Y, Zhang X, Cao B: Ficolin A derived from local macrophages and neutrophils protects against LPS-induced acute lung injury by activating complement. Immunol Cell Biol 2020. [DOI] [PubMed]
- 38.Wu X, Bao L, Hu Z, Yao D, Li F, Li H, Xu X, An Y, Wang X, Cao B, Zhang X: Ficolin A exacerbates severe H1N1 influenza virus infection-induced acute lung immunopathological injury via excessive complement activation. Cell Mol Immunol 2021. [DOI] [PMC free article] [PubMed]
- 39.Osthoff M, Jaeger VK, Heijnen I, Trendelenburg M, Jordan S, Distler O, Walker UA: Role of lectin pathway complement proteins and genetic variants in organ damage and disease severity of systemic sclerosis: a cross-sectional study. Arthritis Res Ther 2019, 21:76. [DOI] [PMC free article] [PubMed]
- 40.Osthoff M, Ngian GS, Dean MM, Nikpour M, Stevens W, Proudman S, Eisen DP, Sahhar J: Potential role of the lectin pathway of complement in the pathogenesis and disease manifestations of systemic sclerosis: a case-control and cohort study. Arthritis Res Ther 2014, 16:480. [DOI] [PMC free article] [PubMed]
- 41.Miyagawa T, Asano Y, de Mestier Y, Saigusa R, Taniguchi T, Yamashita T, Nakamura K, Hirabayashi M, Miura S, Ichimura Y, et al: Serum H-ficolin levels: Clinical association with interstitial lung disease in patients with systemic sclerosis. J Dermatol 2017, 44:1168–1171. [DOI] [PubMed]
- 42.Wu X, Jiang Y, Li R, Xia Y, Li F, Zhao M, Li G, Tan X: Ficolin B secreted by alveolar macrophage exosomes exacerbates bleomycin-induced lung injury via ferroptosis through the cGAS-STING signaling pathway. Cell Death Dis 2023, 14:577. [DOI] [PMC free article] [PubMed]
- 43.Kaiser R, Frantz C, Bals R, Wilkens H: The role of circulating thrombospondin-1 in patients with precapillary pulmonary hypertension. Respir Res 2016, 17:96. [DOI] [PMC free article] [PubMed]
- 44.Rogers NM, Sharifi-Sanjani M, Csányi G, Pagano PJ, Isenberg JS: Thrombospondin-1 and CD47 regulation of cardiac, pulmonary and vascular responses in health and disease. Matrix Biol 2014, 37:92–101. [DOI] [PMC free article] [PubMed]
- 45.Lu Y, Chen J, Wang S, Tian Z, Fan Y, Wang M, Zhao J, Tang K, Xie J: Identification of Genetic Signature Associated With Aging in Pulmonary Fibrosis. Front Med (Lausanne) 2021, 8:744239. [DOI] [PMC free article] [PubMed]
- 46.Ezzie ME, Piper MG, Montague C, Newland CA, Opalek JM, Baran C, Ali N, Brigstock D, Lawler J, Marsh CB: Thrombospondin-1-deficient mice are not protected from bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol 2011, 44:556–561. [DOI] [PMC free article] [PubMed]
- 47.Karunaraj P, Tidswell O, Duncan EJ, Lovegrove MR, Jefferies G, Johnson TK, Beck CW, Dearden PK: Noggin proteins are multifunctional extracellular regulators of cell signaling. Genetics 2022, 221. [DOI] [PMC free article] [PubMed]
- 48.De Langhe E, Cailotto F, De Vooght V, Aznar-Lopez C, Vanoirbeek JA, Luyten FP, Lories RJ: Enhanced endogenous bone morphogenetic protein signaling protects against bleomycin induced pulmonary fibrosis. Respir Res 2015, 16:38. [DOI] [PMC free article] [PubMed]
- 49.Murphy N, Gaynor KU, Rowan SC, Walsh SM, Fabre A, Boylan J, Keane MP, McLoughlin P: Altered Expression of Bone Morphogenetic Protein Accessory Proteins in Murine and Human Pulmonary Fibrosis. Am J Pathol 2016, 186:600–615. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available upon reasonable request.