

Abstract
Within the last decade, the scientific community has witnessed the importance of ferroptosis as a novel cascade of molecular events leading to cellular decisions of death distinct from apoptosis and other known forms of cell death. Notably, such non‐ apoptotic and iron‐dependent regulated cell death has been found to be intricately linked to several physiological processes as well as to the pathogenesis of various diseases. To this end, recent data support the notion that a potential molecular connection between ferroptosis and inherited bone marrow failure (IBMF) in individuals with ribosomopathies may exist. In this review, we suggest that in ribosome‐related IBMFs the identified mutations in ribosomal proteins lead to changes in the ribosome composition of the hematopoietic progenitors, changes that seem to affect ribosomal function, thus enhancing the expression of some mRNAs subgroups while reducing the expression of others. These events lead to an imbalance inside the cell as some molecular pathways are promoted while others are inhibited. This disturbance is accompanied by ROS production and lipid peroxidation, while an additional finding in most of them is iron accumulation. Once lipid peroxidation and iron accumulation are the two main characteristics of ferroptosis, it is possible that this mechanism plays a key role in the manifestation of IBMF in this type of disease. If this molecular mechanism is further confirmed, new pharmacological targets such as ferroptosis inhibitors that are already exploited for the treatment of other diseases, could be utilized to improve the treatment of ribosomopathies.
Keywords: anemia, bone marrow‐failure disorders, ferroptosis, hematopoiesis, iron metabolism, pharmacology, ribosomopathies
1. INTRODUCTION
Over the past few years, there has been a growing scientific interest in the mechanism of ferroptosis and the way it affects the physiology and etiology of various disorders keeping a promise for pharmacological exploitation. The term “ferroptosis,” was coined, for the first time, just more than a decade ago in 2012 and is referred to a unique form of a non‐apoptotic iron‐dependent regulated cell death that has captured the attention of researchers worldwide. 1 , 2 Ferroptosis, as an evolutionarily conserved process, serves a crucial role in developmental processes and pathophysiological conditions in many organisms, spanning across the plant and animal kingdom. 2 , 3 , 4 This intriguing type of programmed cell death occurs due to the cell membrane disruption because of excess iron accumulation and lipid peroxidation. To delve into the details, an excessive amount of intracellular iron, triggers a rise in lipid reactive oxygen species that surpasses the antioxidant activity of glutathione‐dependent peroxidase (GPX4), consequently disrupting the delicate cellular redox equilibrium. 5 This type of cell death is characterized by three main features: (1) the oxidation of membrane phospholipids containing polyunsaturated fatty acids (PUFAs), (2) the intracellular accumulation of active iron, and (3) the impairment of lipid reactive oxygen species detoxification capacity. 6 To date, through extensive research there has been demonstrated the role of ferroptosis as a mechanism in various human diseases, including many hematopoietic disorders. 7
It is important to highlight that ferroptosis distinguishes itself from other types of programmed cell death, including apoptosis. 8 Apoptosis, being an extensively researched form of regulated cell death, is primarily initiated by the activation of caspase family members. 8 On the other hand, ferroptosis can be considered as a cellular “sabotage” rather than a proactive “suicide” mechanism. 9 While “suicide” is actively triggered by specific molecular machinery dedicated to promoting cell death, the “sabotage” mechanism follows the deactivation or hyperactivation of physiological processes and leads to a fatal metabolic disruption. 10 During ferroptosis, cells are subjected to a form of cell death through their own ongoing metabolism. 11 , 12
Recent studies have shown a potential association between the ferroptosis pathway and ribosomal abnormalities. As is widely known, the ribosome is an intricate and finely controlled molecular machine responsible for protein synthesis within the cell. 13 Any deviations in the components of the ribosome or other cellular factors influencing its structure and function can result in a diverse group of diseases known as ribosomopathies. 13 , 14 These disorders may share the common characteristic of ribosomal dysfunction, but they vary greatly in their underlying mechanisms, clinical manifestations, and therapeutic prospects. However, this ribosomal dysfunction in most cases leads to bone marrow failure which is a very severe hematopoietic disorder and is responsible for lethal clinical symptoms of many ribosomopathies. 15 Nowadays, ribosome‐related inherited bone marrow failure (IBMF) has gained the interest of the researchers to unravel the molecular mechanisms and pathways connected generally with ribosomopathies and more especially with the ribosome‐related hematopoietic disorders. Even though much clinical data is available for these diseases, there is still much to uncover regarding the exact molecular mechanism and the “key player” molecular factors contributing to the pathophysiology and emergence of this type of diseases.
In this critical review, our objective is to comprehensively analyze and discuss the latest literature and data regarding the mechanism of ferroptosis in ribosomopathies. More specifically, we aim to examine the potential association of ferroptosis with bone marrow failure caused by ribosome dysfunctions, shedding light on underlying pathogenic mechanisms of pharmacological and therapeutic significance to the wide spectrum of ribosomopathies.
2. DISTINCTIVE FEATURES OF FERROPTOSIS
As already mentioned, ferroptosis stands out as a distinct type of regulated cell death, setting it apart from similar mechanisms, like apoptosis and necrosis, due to its unique morphological, biochemical, immune, and genetic characteristics. 16 , 17 , 18
2.1. Morphological features
According to the latest data, it is now widely accepted that ferroptotic cells often exhibit morphological features resembling necrosis. 18 These characteristics have been demonstrated in in vitro and in vivo models and are mainly concluded in the destruction of plasma membrane integrity, and cellular swelling (known as oncosis) including the cytoplasmic swelling as well as the swelling of cytoplasmic organelles, while moderate or no chromatin condensation is observed. 1 , 19 , 20 , 21 Additionally, in certain instances, ferroptosis is followed by cell detachment and rounding up, along with an increase in the presence of autophagosomes. 20 , 21 , 22 Furthermore, during ferroptosis, cells undergo a range of mitochondrial abnormalities. These include volume reduction, reduced or absent crista, disruption of the outer membrane, and increased membrane density. 10 , 18 , 19 , 20 , 21 , 22 These mitochondrial changes are crucial as they lead to mitochondrion‐mediated reactive oxygen species (ROS) production, which is essential for initiating lipid peroxidation and ultimately inducing ferroptosis. 10 It is important to highlight that unlike apoptosis, ferroptosis does not involve the formation of apoptotic bodies or cell shrinkage. Surprisingly, the cell membrane appears to maintain its integrity throughout the process. 10 , 18 , 23 It is worth noting that ferroptosis can also rapidly propagate from one cell to adjacent cells, showcasing its unique ability for intercellular spread. 18 , 23 , 24
2.2. Biochemical features
From the biochemical perspective, two are the main processes that trigger ferroptosis: excessive intracellular iron accumulation and lipid peroxidation. 16
2.2.1. Intracellular iron accumulation
Iron, a vital trace element, assumes a critical function in various subcellular organelles and pivotal cellular processes. The intricate regulation of intracellular iron levels encompasses a complex network involving absorption, storage, utilization, and outflow. 25 , 26 As anticipated, specific molecular regulators associated with iron homeostasis govern the susceptibility to ferroptosis. Excessive iron accumulation triggers an upsurge in ROS through the Fenton reaction, thereby intensifying oxidative stress. 27 Moreover, iron has been observed to promote the function of lipoxygenase (ALOX) enzymes, which are responsible for initiating lipid peroxidation. Unlike other metals, iron has a unique ability to activate downstream cascade reactions that ultimately lead to ferroptosis. 18 , 25 , 27 Recent studies have shown that transferrin (Tf) which is responsible for the iron uptake and its receptor (transferrin receptor, TfR) influences the intracellular iron levels and consequently the occurrence of ferroptosis. 16 Besides the existence of TfR1 and TfR2 it was shown that these receptors bind Tf through differing mechanisms. Moreover, TfR1 is mainly involved in the cellular iron uptake process, whereas TfR2 function is rather through the maintenance of iron levels within the body. 28 Also, the expression of TfR1 is ubiquitous in erythroblasts and shows dependency on iron loading and/or chelation, whereas the expression of TfR2 is high in hepatocytes and erythroid precursors and seems not to be substantially affected by iron loading and/or chelation. 29 Another noteworthy molecule, nuclear receptor activator 4 (NCOA4), assumes a significant role in ferritin degradation. 18 , 27 , 30 Furthermore, certain proteins, encompassing members of the Heat Shock Protein (HSP) family, manifest the ability to counteract ferroptosis. These proteins serve a dual purpose by facilitating ferritin export and reducing cytoskeleton‐mediated iron uptake. This intricate mechanism aids in maintaining iron homeostasis and safeguarding cells against the damaging effects of ferroptosis. 30
2.2.2. Lipid peroxidation
Lipid peroxidation refers to the process by which lipids undergo the loss of hydrogen atoms through their interaction with free radicals. 5 This oxidative mechanism leads to the oxidation, fragmentation, and shortened carbon chains of lipids and especially PUFAs, resulting in the synthesis of cytotoxic compounds such as lipid free radicals, lipid hydroperoxides, and active aldehydes (malonic aldehyde also known as MDA is the most studied biomarker of lipid peroxidation). 26 , 31 These detrimental substances play a significant role in causing cellular damage. 31 It has been observed that lipid peroxidation plays an important role in mediating ferroptosis. 25 , 31 , 32 Besides iron accumulation‐mediated lipid peroxidation, mitochondria, and NADPH oxidase (NOX) can also mediate ROS production and lead to the production of lipid‐free radicals. 1 , 31 , 33 , 34 , 35 It is worth noting that some enzyme families contribute to lipid peroxidation such as the isoforms of ALOX, such as ALOX5, ALOX12, ALOX15, ALOX15B, and ALOXE3 in humans 26 , 36 , 37 while the cytochrome P450 oxidoreductase system seems to induce ferroptosis without the implication of ALOX enzymes. 31 , 38
2.3. Genetic features
Ferroptosis is affected by a multitude of genes associated with iron metabolism, lipid synthesis, and oxidative stress pathways. These genes encompass TfR1, iron response element binding protein 2 (IREB2) for iron metabolism, and acyl‐CoA synthetase family member 2 (ACSF2) with acyl‐CoA synthetase long‐chain family member 4 (ACSL4) for lipid synthesis. ACSL4 is one of the most studied genes as a potential biomarker of ferroptosis. 39 , 40 , 41 , 42 This is due to the fact that upregulation of ACSL4 which is phosphorylated through the protein kinase CβΙΙ (PKCβΙΙ) results in an elevation of PUFAs. 41 These PUFAs are vulnerable to oxidation reactions, which can ultimately trigger the initiation of ferroptosis. 39 , 40 , 41 , 42 , 43 , 44 However, it is noteworthy that the ACSL4 presence is not always obligatory, as there are specific circumstances under which ACSL4‐depleted cells can still undergo ferroptosis. 44 Notably, prostaglandin‐endoperoxide synthase 2 (PTGS2/COX2), has also been recognized as a ferroptosis‐biomarker; however, its inhibitor does not seem to affect the procedure of ferroptosis. Additionally, genes associated with the detoxification mechanism of the cell, including the glutathione (GSH) pathway and coenzyme Q10 (CoQ10) pathway, as well as the nuclear factor, erythroid 2‐like 2 (NFE2L2), and membrane repair through pathways like the endosomal sorting complexes required for transport (ESCRT)‐III pathway, can confine membrane damage in ferroptosis. 45 , 46 , 47 Consequently, depending on the balance between damage and defense mechanisms, cells make the decision to either survive or succumb to ferroptotic stimuli. 18
2.4. Immune characteristics
Ferroptosis is an inflammatory type of cell death, thus it has been associated with the release of damage‐associated molecular patterns (DAMPs) and subsequent activation of DAMP receptors. More specifically, high mobility group box 1 (HMGB1), a DAMP located in nucleus, has been demonstrated to elicit immune responses in ferroptosis. 48 , 49 , 50 Subsequently, the receptor for HMGB1, known as advanced glycosylation end‐product specific receptor (AGER/RAGE), interacts with HMGB1 resulting in inflammatory responses when encountered by macrophages. 18 , 50 Ferroptotic cells may also release DNA or lipid oxidation products such as 4HNE, LTC4, and PGE2, activating many pathways including the NF‐kB pathway. 18 , 49 , 50 , 51
3. MAJOR SIGNALING PATHWAYS INVOLVED IN FERROPTOSIS
The onset of ferroptosis is primarily driven by two crucial biochemical changes: iron accumulation and lipid peroxidation. These processes initiate a cascade of reactions, subsequently activating a series of molecules that ultimately trigger ferroptosis. In this context, it is pertinent to elucidate the principal pathways intricately associated with this process. Ferroptosis is orchestrated by a complex network of cell signaling processes, as evidenced by recent scientific data. These data have highlighted the crucial role of various regulatory mechanisms in the evolution of ferroptosis, such as GSH metabolism, iron metabolism, lipid peroxidation, and the involvement of the ferroptosis‐suppressor protein 1 (FSP1)‐dihydroorotate dehydrogenase (DHODH) CoQ10 axis, GCH1‐BH4 axis MBOAT1/2‐MUFA axis, and SC5D‐7‐DHC axis. 1 , 40 , 43 , 51 , 52 , 53 , 54 , 55
3.1. Antioxidant defense pathways
3.1.1. SLC7A11‐GSH‐GPX4 axis
The antioxidant defense mechanism and especially GSH metabolism is strongly correlated to the regulation of ferroptosis. More specifically, GSH metabolism is connected with the system Xc−. This amino acid antiporter consists of two key subunits, the solute carrier family 3 member 2 (SLC3A2) and the solute carrier family 7 member 11 (SLC7A11). 51 , 56 The main function of system Xc− is the cell redox equilibrium by regulating the transportation of extracellular cysteine in exchange of intracellular glutamate. The imported cysteine will be converted to cysteine which is necessary for GSH synthesis. Subsequently, GPX4, exploits the antioxidant GSH to detoxify lipid‐ROS through the GPX4 detoxification reaction. 38 Inhibiting any of the major parts of this cascade can affect cell survival. 49 According to recent in vitro studies inhibiting system Xc− can decrease the cellular uptake of cysteine, leading to the reduction of GSH which inactivates GPX4 and triggers excessive lipid peroxidation and eventually ferroptosis. 38 , 57 In vivo studies have confirmed the core role of GPX4 in ferroptosis. 17 GSH constitutes a very important cofactor of GPX4, and its main role is to reduce lipid peroxides, having a ferroptosis‐resistance activity. 58 Many ferroptosis inducers like RAS‐selective‐lethal‐3 small molecule (RSL3) seem to interact and irreversibly inactivate GPX4, driving to ferroptosis. 58 According to Yan et al. 59 the nuclear transcription factor NFE2L2 also plays an essential role in the resistance to ferroptosis as it targets GPX4, glutathione synthetase (GSS), SLC7A11, and other genes of the system (Xc−)‐GSH (Figure 2).
FIGURE 2.
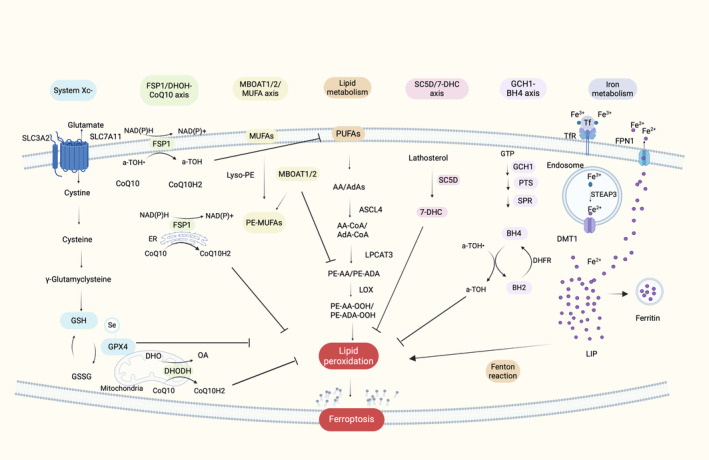
Schematic presentation of molecular mechanisms of ferroptosis. The major pathways involved in ferroptosis can be attributed to the interplay between the antioxidant defense system, represented by system Xc−, the FSP1‐CoQ10 and DHODH‐CoQ10 axis, MBOAT1/2‐MUFA axis, SC5D‐7‐DHC axis, GCH1‐BH4 axis, and the oxidative stress pathways, encompassing iron metabolism and lipid metabolism. First, system Xc− facilitates the uptake of cysteine and its conversion into cysteine, which subsequently contributes to the synthesis of GSH. GSH interacts with GPX4 to neutralize lipid reactive oxygen species. Second, FSP1 and DHODH convert CoQ10 enzyme into its reduced form, CoQ10H2, which aids in the detoxification of lipid reactive oxygen species. Moreover, MBOAT1/2, increase the intracellular level of PE‐MUFAs while decreasing the level of PE‐PUFAs, which are the main substrate for lipid peroxidation. Another antioxidant pathway is SC5D‐7‐DHC axis, in which 7‐DHC acts as a free radical trapping agent and protect cells from lipid peroxidation, while in the GCH1‐BH4 axis, BH4 alone and synergistically with a‐TOH also act as free radical trapping agents. Furthermore, lipid metabolism, PUFAs present in the plasma membrane interact with ACSL4, LPCAT3, and ALOXs, resulting in the generation of lipid reactive oxygen species. Additionally, Iron metabolism plays an important role in ferroptosis. Through the TfR1, Fe3+ is internalized and subsequently reduced to Fe2+. Subsequently, Fe2+ contributes to the formation of the LIP and reacts with hydrogen peroxide (H2O2) via the Fenton reaction, leading to the production of lipid reactive oxygen species. Excess intracellular iron is sequestered within ferritin, while the remaining iron is exported through FPN (this image was created with BioRender.com).
3.1.2. The FSP1‐CoQ10 axis and DHODH‐CoQ10 axis
Ferroptosis suppressor protein 1 (FSP1) was primarily called apoptosis inducing factor mitochondria associated 2 (AIFM2). However, it was renamed due to the back‐to‐back studies of Doll et al. (2019) and Bersuker et al. (2019) who identified FSP1 as a ferroptosis suppressor in a GPX4 independent manner 45 , 46 (Figure 2). FSP1 is located mainly in the plasma membrane but also in subcellular organelles such as the endoplasmic reticulum, 45 , 46 and consumes NADH/NADPH to reduce CoQ10 enzyme, which is also known as ubiquinone, to CoQ10H2 enzyme, also known as ubiquinol. Ubiquinol acts as an antioxidant that captures lipid reactive oxygen species and thus prevents lipid peroxidation. 5 , 60 , 61 Indirectly, CoQ10H2 can also contribute to α‐tocopherol (a‐TOH) synthesis, which is another important antioxidant to fortify the antioxidant mechanism of the cell. 61 , 62 , 63 Therefore, FSP1‐CoQ10‐NAD(P)H axis acts as a GPX4‐independent redox system, cooperating with GPX4 and GSH/GSSG to limit lipid peroxidation. 5 , 60
Similar axis is activated through DHODH in mitochondria. DHODH, a flavine‐dependent enzyme situated in the inner mitochondrial membrane, plays a pivotal role in the fourth step of pyrimidine nucleotide synthesis. 52 It facilitates the oxidation of dihydroorotate (DHO) to orotate (OA) while concurrently reducing ubiquinone to ubiquinol. This process, wherein ubiquinol scavenges free radicals, appears to protect the mitochondria against lipid peroxidation. Recent findings suggest that this axis further safeguards the cell against mitochondrial ferroptosis by modulating CoQH2 production within the inner mitochondrial membrane. 64
3.1.3. GCH1‐BH4 axis
In a 2020 research study conducted by Kraft et al., CRISPR/Cas9 screening was utilized to illustrate the role of the GTP cyclic hydrolase 1 (GCH1)‐tetrahydrobiopterin/dihydrobiopterin (BH4/BH2) system in the suppression of ferroptosis. 53 GCH1 catalyzes the hydrolysis of GTP in conjunction with 6‐pyruvoyltetrahydropterin synthase (PTS) and sepiapterin reductase (SPR), leading to the production of BH4 65 (Figure 2). BH4 is an antioxidant which plays a critical role in the metabolism of nitric oxide (NO), neurotransmitters, and aromatic amino acids, such as l‐arginine. 31 Additionally, both BH4 and its oxidized form, BH2, participate in a redox cycle that scavenges intracellular free radicals, thereby safeguarding lipid membranes against peroxidation. 53 , 66 This redox cycle is observed alone and synergistically with a‐TOH. BH4 can be regenerated from BH2, a process which is strictly regulated by dihydrofolate reductase (DHFR), with NADP+/NADPH serving as the cofactor. 66 Consequently, the GCH1‐BH4 axis plays a crucial role in inhibiting ferroptosis by acting as an antioxidant that shields cell membrane phospholipids from oxidation, thereby impeding lipid peroxidation. Notably, this system operates independently of the cysteine/GSH/GPX4 axis.
3.1.4. MBOAT1/2‐MUFA axis
Recently, Liang et al. (2023) have unveiled a novel pathway associated with the inhibition of ferroptosis. Specifically, membrane‐bound O‐acyltransferase domain‐containing 2 (MBOAT2), identified as a lysophospholipid acyltransferase (LPLAT), emerged as a suppressor of ferroptosis through CRISPR technology. 54 MBOAT2 exhibits a selective ability to transfer monounsaturated fatty acids (MUFAs) into lysophosphatidylethanolamine (lyso‐PE), leading to an increase in intracellular PE‐MUFA levels while concomitantly reducing cellular PE‐PUFA levels, which serve as the primary substrates for lipid peroxidation in ferroptosis 54 , 67 (Figure 2). Thus, MBOAT2 seem to act as a ferroptosis suppressor. Notably, MBOAT2 appears to impede ferroptosis independently of GPX4 and FSP1. Another noteworthy ferroptosis suppressor to be mentioned is MBOAT1, a member also of the LPLAT family, which exhibits a similar suppression pattern as MBOAT2. MBOAT2 and MBOAT1 are upregulated by the androgen receptor (AR) and estrogen receptor (ER), respectively. Hence, sex hormones appear to influence ferroptosis through the MBOAT1/2‐MUFA system, although the exact mechanism of action remains unknown. 67
3.1.5. SC5D‐7‐DHC axis
A newly bread axis with potential antiferroptotic properties is the SC5D‐7‐DHC axis, which is associated with the cholesterol biosynthesis pathway. Specifically, this pathway involves the conversion of lathosterol to 7‐DHC through the action of lathosterol oxidase (SC5D) and the subsequent reduction of 7‐DHC to cholesterol by DHCR7 55 , 68 (Figure 2). In a study conducted by Liu et al. (2024) and Yamada et al. (2024), it was observed that 7‐DHC, among other functions, robustly protects cell membrane phospholipids from lipid peroxidation by acting as a free radical trapping agent and getting oxidized instead of phospholipids thus preventing ferroptosis. 55 , 68 , 69
3.2. Iron metabolism
Iron homeostasis is very important for the physiological activity of the cell and especially the hematopoietic cells. Excess iron levels lead to ferroptosis through the induction of lipid peroxidation. It is thus considered essential, before describing the pathway of ferroptosis, to provide a brief description of the regulatory mechanisms involved in the highly orchestrated accumulation process of iron within the organism (Figure 1).
FIGURE 1.

Orchestrated accumulation process of iron within the organism. Iron absorbed by food has two forms: Non‐heme iron and heme iron. Non‐heme iron (Fe3+) is reduced to Fe2+ (ferrous) via DCYTB and further transported in the cell through DMT1 to join the LIP. In parallel, heme iron is transferred within the cell via the HCP1 and ferrous iron is released in the cell via HOX1 to join the LIP. Consequently, PCBP1 transfers ferrous iron to ferritin and vice versa, while it also delivers Fe2+ to FPN1 to export it in the cardiovascular system. Fe2+ is converted to Fe3+ (ferric) through hephaestin to interact with transferrin. This complex binds to TfR1 on the surface of the targeted cells. Furthermore, ferrous iron may enter into mitochondria for heme biosynthesis and generation of Fe‐S clusters. Subsequently, heme exports from mitochondria and the cell through the FLVCR. The exported heme molecules interact with hemopexin leading to the synthesis of heme‐hemopexin complex. This complex can be absorbed from hepatocytes and macrophages through the CD91 (this image was created with BioRender.com).
3.2.1. Iron absorption
On a daily basis, human organism absorbs ~1–2 mg of iron, which is only a small portion of the iron consumed. 70 , 71 Iron absorption primarily occurs in the duodenum and upper jejunum of the intestine and is regulated by two distinct pathways for non‐heme and heme iron. 63 Heme iron, found in animal products like meat, fish, and poultry, is absorbed more efficiently than non‐heme iron which is respectively found in fruits and vegetables. 71 The exact mechanism of heme‐iron absorption is yet to be fully understood, but it is believed that begins with the transfer of heme‐iron in enterocytes through the heme carrier protein 1 (HCP1) receptor, which is also associated with folate transport. 72 The heme oxygenase (HOX‐1) factor converts heme iron into divalent iron (ferrous), which is transported to the labile iron pool (LIP). 72 Similarly, non‐heme iron is in its trivalent (ferric) form and needs to be reduced to its ferrous form to enter the enterocyte. This reduction occurs in the presence of the duodenal cytochrome b transporter (DCYTB) and the six transmembrane epithelial antigen of the prostate 2 (Steap2). 73 , 74 The divalent metal transporter (DMT1), located in the membrane of duodenal enterocytes, facilitates the transport of iron across the enterocytes to join the LIP. 74 Consequently, ferrous is transported from the cytoplasm to ferritin via the poly(rC)‐binding protein 1 (PCBP1). 74 Within the cell, iron is stored in the form of ferritin and can be released through the pores, autophagy, and lysosomal degradation of ferritin. 3 Ferrous is then transported out of enterocytes by ferroportin (FP1) and converted to its ferric form in the presence of ferroxidases, namely hephaestin (Heph) and ceruloplasmin (Cp). 70 The trivalent iron combines with transferrin (Tf), a plasma protein, and is distributed through circulation to specific cells, where transferrin‐iron complex is bound and endocytosed by the TfR1. 70 , 71 It is important to note that a small amount of ferrous iron within enterocytes is transported to the mitochondria to form heme and iron–sulfur clusters (Fe‐S), which bind with mitochondrial enzymes. Heme‐iron is subsequently removed from the cell through the feline leukemia virus C receptor (FLVCR) and breast cancer resistance protein (Bcrp)/ATP‐binding cassette subfamily G member 2 (Abcg2), although the exact mechanism is once more, not fully understood. The exported free heme forms a complex with hemopexin, resulting in a heme–hemopexin complex. 73 This complex can be further absorbed by cluster of differentiation (CD91) receptors on hepatocytes and macrophages. 71 Hepatocytes serve as iron storage and also produce hepcidin, a hormone that is secreted into the plasma and is responsible for the regulation of iron levels in the plasma. Additionally, splenic, and hepatic macrophages scavenge heme‐iron from enterocytes and senescent erythrocytes, releasing iron from hemoglobin and making it available for another hemoglobin cycle, contributing significantly to the plasma iron pool. 75 , 76 Importantly, the intestinal iron absorption and macrophage iron recycling is blocked by high hepcidin levels, since hepcidin is essential for iron homeostasis in the body. 77 Reduced iron absorption, which can be affected by multiple factors such as increased hepcidin levels, can lead to iron deficiency, and consequently to iron deficiency anemia (IDA), as thoroughly described by. 78 , 79
3.2.2. Intracellular iron metabolism
After the endocytosis of Tf–Fe3+ complex through TfR1 of the target cell, Fe3+ is transported to the endosome and reduced to Fe2+ via the six‐transmembrane epithelial antigen of prostate 3 (STEAP3) metalloreductase. 80 , 81 , 82 Subsequently, Fe2+ ions transported in the cytoplasm through DMT1, form the LIP and interact with H2O2 through Fenton reaction leading to the formation of hydroxyl radicals. 5 These free radicals are very active and can cause oxidative damage in the macromolecules of the cell promoting among others lipid peroxidation and eventually cell death. The redundant cytoplasmic iron is stored in ferritin. 75 In ferroptotic cells the level of free intracellular iron is increased through the activated expression of TfR1 and the decrease of ferritin subunits mRNA levels. 57 , 83 Finally, ferroportin (FPN1) is an important molecule in ferroptosis as it is the only protein responsible for the efflux of excess iron from the cell 80 , 84 (Figure 2).
3.3. Lipid metabolism
Once lipid peroxidation is one of the main characteristics of ferroptosis, this type of cell death is strongly correlated with lipid metabolism. PUFAs are the main substrate of lipid peroxidation during ferroptosis 32 , 60 , 85 (Figure 2). Thus, the content of PUFAs crucially influences the trajectory of this process. More significantly, ACSL4, an important ferroptosis biomarker, drives the free arachidonic acid (AA) and adrenic acid (AdA, all‐cis‐7,10,13,16‐docosatetraenoic acid) to bind to coenzyme A (CoA) to form AA/AdA‐CoA. 32 , 40 , 57 , 86 , 87 Subsequently, lysophosphatidylcholine acyltransferase‐3 (LPCAT3) catalyzes the synthesis of phosphatidylethanolamines (AA/AdA Pes, PE‐PUFAs) under the esterification reaction of AA/AdA‐CoA. PEs are then oxidized into lipid hydroperoxides by ALOX15, inducing ferroptosis. 87 , 88 Therefore, ACSL4 and LPCAT3 as well as ALOX15, are three enzymes that influence ferroptosis sensitivity through the lipid metabolism pathway. Various studies confirm that decrease of ACSL4 and LPCAT3 or ALOXs affect lipid peroxidation and reduces ferroptosis sensitivity. 88 , 89 , 90 , 91 , 92 , 93 , 94 , 95 , 96 Experimental data confirmed that cells treated with arachidonic acid are sensitized to ferroptosis. 57
4. RIBOSOME BIOGENESIS AND “SPECIALIZED RIBOSOMES”
4.1. Ribosome biogenesis and protein synthesis
The discovery of ribosomes in 1950 by George Palade and Albert Claude marked a significant milestone in understanding their crucial role in protein synthesis. 97 , 98 In humans, ribosomes are composed of two subunits, namely the small subunit (40S) and the large subunit (60S), which combine to form the active ribosome (80S). 99 These subunits consist of ribosomal RNAs (rRNAs) and ribosomal proteins (RPs). Specifically, the small subunit (SSU) comprises 18S rRNA and 33 ribosomal proteins (RPSs), while the large subunit (LSU) contains 5S, 5.8S, and 28S rRNAs, along with 47 ribosomal proteins (RPLs). 99 , 100 , 101
Ribosome biogenesis is a dynamic and major energy‐consuming process that undergoes meticulous regulation. 99 , 100 , 101 To delve into the details, ribosome assembly begins with the RPs' transcription by RNA polymerase II (RNA pol II) and their subsequent synthesis in the cytoplasm. 101 , 102 , 103 The majority of these proteins return to the nucleus, where they interact with the ribosome binding factors (RBFs) and the precursor rRNA molecules to become part of the precursor ribosome assembly. 101 Concurrently, the transcription of rRNA molecules takes place within the nucleolus and the nucleoplasm. 104 , 105 , 106 Specifically, the ribosomal DNA sequences (rDNAs) encoding 5.8S, 18S, and 28S rRNAs are transcribed by RNA polymerase I (RNA pol I) in the nucleolus and the 5S by RNA polymerase III (RNA pol III) in the nucleoplasm. 106 , 107 , 108 , 109 , 110 The promoter site of the rDNA region consists of two distinct domains: AUS/UCE and CE. 94 , 95 , 96 These domains are recognized by various transcriptional factors, including the upstream binding factor (UBF) and the so‐called selective factor 1 (SL1) which form the preinitiation complex, with the RNA polymerase I‐associated transcription factor (RRN3) and allow the recruitment of the RNA pol I. 97 During this transcription process, a polycistronic precursor molecule called 47S pre‐rRNA is formed and interacts with the RBFs to form an intermediate 90S complex. The 47S pre‐rRNA contains external and internal transcribed regions (ETS and ITS) located between the rRNA sequences. These regions are consequently cleaved to generate the individual 18S, 5.8S, and 28S rRNAs 101 and result in the formation of 40S and 60S precursor subunits. While the 5S rRNA is transcribed separately. The maturation of rRNA precursor molecules involves a series of endonuclear and exonuclear events, facilitated by small nucleolar ribonucleoproteins (snoRNPs). 93 Notably, snoRNPs drive modifications such as 2′‐O‐ribose methylation (by box C/D) and pseudouridylation (by box H/ACA) in rRNAs. 106 Through these modifications, the interaction between rRNA molecules and RPs is enhanced, ultimately leading to the formation of mature ribosome subunits. 99 While a significant portion of ribosome assembly and maturation takes place in the nucleus, the 40S and 60S subunits undergo further maturation processes upon export to the cytosol. These final steps of ribosomal maturation involve indicative rRNA trimming, as well as incorporation of remaining RPs. 97
The process of protein synthesis begins with the recognition of the Kozak consensus sequence, which serves as the initiation site for translation in the majority of eukaryotic mRNAs. 98 , 102 During this process, an aminoacylated tRNA interacts with the translation start codon on the mRNA at the peptidyl‐site (P‐site), situated at the interface of the SSU and LSU, respectively. The initial amino acid forms a peptide bond with the subsequent amino acid, which is delivered to the aminoacylation site (A‐site) by tRNA. This newly formed peptide then moves to the P‐site, allowing the A‐site to become available for the next amino acid. The deacylated tRNA subsequently exits the ribosome through the exit site (E‐site). 98 , 99 This entire process ensures the accurate reading and translation of the information encoded by the mRNA molecule, codon by codon.
4.2. “Specialized ribosomes”
Translation is responsible for the protein synthesis, thus plays a key role in the protein level profile within the cell. Almost a few years back research community believed that translation is regulated through an interaction between mRNA structures and the cis‐ and trans‐regulators, which involve among others initiation (eIF), elongation (eEF) and termination eukaryotic factors (eTF), micro RNAs, lncRNAs, RNA binding proteins (RBPs), and internal ribosome entry site (IRES). 111 , 112 , 113 Ribosomes were believed to be the translation mediators with no direct regulatory activity on the translational process. However, according to the latest data ribosomes seem to have an essential role in the regulation of protein synthesis. 113 , 114 , 115 More specifically, the last few years research data reveal that ribosomes can exhibit heterogeneity at multiple levels, including RPs, RP paralogs, ribosome‐associated factors, rRNA, and rRNA modifications due to changes in the status, environment, developmental stage, or pathological conditions of the cell 116 , 117 , 118 , 119 , 120 , 121 , 122 , 123 , 124 , 125 , 126 , 127 , 128 , 129 (Figure 3). Those observations enhanced the theory of “specialized ribosomes,” which proposes that ribosome composition can drastically affect the translational activity of this ribosome and were first introduced as a definition by Xue and Barna. 115 These alterations in ribosome composition may lead to pathological phenotypes, leading to a group of conditions known as ribosomopathies. 130 , 131 , 132
FIGURE 3.
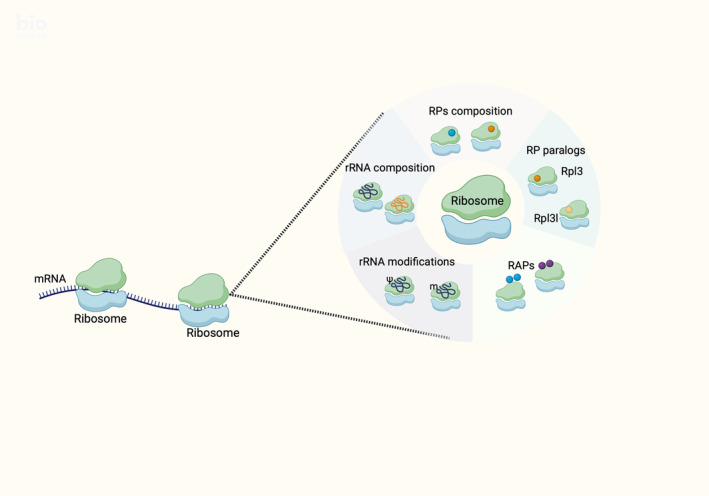
Diagrammatic depiction of “specialized ribosomes.” Ribosome heterogeneity can occur through differentiated levels of RPs or/and rRNAs composition, RP paralogs, ribosome‐associated factors (RAPs) and rRNA modifications (this image was created with BioRender.com).
5. RIBOSOMAL ABNORMALITIES AND FERROPTOSIS
Ribosomopathies are a group of diseases connected with the structure and function of ribosomes and have a great phenotypic heterogeneity. However, most of them are characterized by bone marrow failure and in some cases are accompanied by a severe anemic phenotype. To date, it is well‐established that the main mechanism leading to cell death and particularly in ribosome‐related IBMF syndromes (ribosome‐related IBMFs), is apoptosis. The primary mechanism associated with apoptosis in these conditions involves the activation of the p53 pathway, leading to cell cycle arrest and ultimately cell death through apoptosis. 15 , 131 , 132 , 133 Interestingly, recent data 134 suggest a susceptibility of hematopoietic stem cells (HSCs) to ferroptosis in various IBMFs. Since bone marrow failure is observed in all the ribosomopathies described below and iron levels play a particularly important role in the maturation and differentiation of hematopoietic cells, 134 the question arises whether, besides apoptosis, there is a connection between ribosomopathies and ferroptosis, particularly in the hematopoietic cell lineages. It is also important to be noted that recent reviews indicate a potential synergistic interaction between apoptosis and ferroptosis, thus an interaction between those two mechanisms might be connected with ribosome‐related IBMFs. 135 , 136 , 137
5.1. Diamond–Blackfan anemia, 5q− syndrome, and ferroptosis
Diamond–Blackfan anemia (DBA) stands as one of the most well‐known ribosomopathy. Its initial documentation can be traced back to Josephs' comprehensive review on anemia in infancy and childhood in 1936. 136 , 137 , 138 Subsequently, Diamond and Blackfan classified it as a congenital hypoplastic anemia in 1938. 139 The primary clinical manifestation associated with DBA is red cell aplasia. 139 Typically, DBA is diagnosed shortly after birth, presenting as a severe form of macrocytic or normocytic anemia accompanied by reticulocytopenia. Notably, individuals with DBA exhibit normal platelet and neutrophil counts, along with a deficiency of red blood cell precursors in the bone marrow. Physical malformations, primarily affecting the craniofacial region, upper limbs, and growth, are observed in ~35%–50% of cases. 138 , 139 To date, the most known ribosomal proteins involved in DBA are RPS7, RPS10, RPS15A, RPS17, RPS19, RPS24, RPS26, RPS27, RPS28, RPS29, RPL5, RPL11, RPL15, RPL18, RPL26, RPL27, RPL31, RPL35, RPL35A, and TSR2 encoding RPSs and RPLs. 137 , 139 , 140 , 141 , 142 , 143 , 144 Mutations in RPS 19 are observed in 25% of DBA patients. It is notable that these mutations in RPS19 have been related to decreased translation of FLVCR1, an erythroblastic heme exporter that plays a key role in erythroid differentiation. 145 , 146 , 147 , 148 , 149
Another known ribosomopathy is the 5q− syndrome, a subtype of myelodysplastic syndrome (MDS). It was initially described in 1974 by van Den Berghe in three patients who exhibited refractory anemia and a deletion of the long arm of chromosome 5. 150 Individuals with 5q− syndrome typically present severe macrocytic anemia, normal or increased platelet level, and hypolobulated micro‐megakaryocytes. 150 The underlying genetic abnormality associated with this disorder is a deletion or decreased expression of the RPS14 gene. 151 The reduced expression of RPS14 leads to impaired erythropoiesis, while the other lineages are relatively preserved. Interestingly, overexpression of RPS14 in samples from patients with 5q− syndrome has been shown to rescue erythropoiesis. 150 , 151 Additionally, RPS14 deficiency has been found to cause a block in the processing of the 18S rRNA and the formation of the 40S ribosomal subunit. 150 Inherited inactivation of RPS14 in the 5q− syndrome has the same manifestation as the haploinsufficiency in RPS19 and other ribosomal genes observed in DBA. 151
In both DBA and 5q− syndrome, a notable clinical feature is the presence of extensive red cell anemia. The process of erythropoiesis, which involves the production of red blood cells, is strictly regulated, whereas it relies significantly on the involvement of heme biosynthesis and iron metabolism. 149 , 150 Previous studies conducted by Doty et al. 152 , 153 , 154 and Yang et al. 155 have extensively elucidated the sequential progression of cells from the forming unit‐erythroid stage (BFU‐E, the earliest erythroid progenitors) to the colony forming unit‐erythroid (CFU‐E)/proerythroblast stage. During this transition, the expression of TfR1 is upregulated in response to erythropoietin. 153 , 154 , 155 , 156 Subsequently, the GATA binding factor 1 (GATA1) factor is upregulated, leading to the expression of δ‐aminolevulinate synthase 2 (ALAS2), which, in conjunction with the availability of iron, contributes to heme synthesis. The presence of heme induces the expression of globulin α and β by inhibiting the BTB domain and CNC homolog 1 factor (BACH1) 153 , 154 , 155 , 156 , 157 , 158 , 159 and eIF2a. 160 At this stage, the cellular iron levels are sufficient, heme levels are elevated, while globulin levels remain low. To prevent the accumulation of toxic free heme, the cell increases the expression of the FLVCR‐1 receptor, responsible for removing excess heme from the cell. 161 , 162 , 163 , 164 , 165 It is crucial to maintain a delicate balance between heme and iron levels at this stage, as an imbalance can drive the cell toward ferroptotic cell death. 134
In 2019, Doty et al. conducted studies on bone marrow samples from patients with DBA and MDS with 5q− syndrome to investigate the underlying mechanisms of red blood cell anemia. 154 Their findings revealed an imbalance between heme and globin, which likely contributes to the early termination of red blood cells differentiation. 155 , 156 , 157 According to their model, heme synthesis progresses normally in erythroid progenitors (burst/colony forming unit erythroid, BFU‐E/CFU‐E) and proerythroblasts stage, while globin expression is delayed due to ribosomal abnormalities. This delay in globin translation is suspected to be caused by the degradation of globin mRNA, which fails to bind to ribosomes due to their limited number or availability. 155 Consequently, there is an excess of free heme compared with globin in these cells. The accumulation of excessive free heme leads to oxidative stress and lipid peroxidation. 155 , 156 , 157 , 158
Furthermore, as already mentioned, it has been observed that HSCs of DBA patients with RPS19 mutations exhibit decreased translation of FLVCR‐1. 149 Recent data on ferroptosis also provide a connection between previous studies implicating P53 activation in the pathogenesis of DBA and 5q− syndrome anemia and the observation of excess heme. 155 , 158 , 159 , 160 , 161 , 162 , 163 , 164 Specifically, P53 transcriptionally represses SLC7A11, which inhibits cysteine uptake and reduces glutathione synthesis, thereby sensitizing cells to ferroptosis 156 , 165 , 166 , 167 Since SLC7A11 is a transcriptional target of BACH1 and uniquely activated by BACH1 159 , 160 , 161 the excess heme not only generates ROS, 167 but also further decreases SLC7A11. 165 Based on the aforementioned data, it can be concluded that in the case of DBA and 5q− syndrome, ferroptosis mechanism may be activated through the following events and synergistically with apoptosis contribute to erythroid progenitors' cell death: The mutations in ribosomal proteins, such as mutated RPS19, seem to modify the ribosome composition and subsequently affect ribosome function that impacts the mRNA expression patterns. The expression of globins seems to be reduced. Parallelly, the cell has already accumulated iron and heme to promote the synthesis of hemoglobin and to enhance erythroid differentiation process. For this reason, FLVCR1 has diminished promoting heme accumulation. The reduced levels of globins lead to an imbalanced synthesis of hemoglobin which enhance intracellular iron and heme accumulation. High levels of iron lead to increased free radicals through Fenton reaction while heme accumulation enhances oxidative stress and lipid peroxidation. Furthermore, high levels of heme decreases the expression of SLC7A11 receptor which is associated with the GSH synthesis pathway. The molecular events described above seem to lead to lipid peroxidation and iron accumulation which are the two main characteristics of ferroptosis (Figure 4). According to these data it is possible that ferroptosis synergistically with apoptosis promote cell death of erythroid progenitors in DBA and 5q− syndrome (Table 1).
FIGURE 4.

Diagrammatic depiction of ferroptosis as a potential mechanism of Diamond–Blackfan anemia (DBA) and 5q− syndrome in HSCs, according to existing experimental data. In DBA, low expression of ribosomal proteins (especially the RPS19) and in 5q− syndrome, low expression most usually the RPS14, leads to reduced and impaired ribosomes. Ribosomes reduction appears to limit the globin genes expression and further hemoglobin levels. Consequently, the low levels of hemoglobin, combined with decreased levels of the heme exporter FLVCR1, result in heme accumulation and increased levels of intracellular lipid reactive oxygen species. Furthermore, both syndromes exhibit reduced expression of the subunit SLC7A11, one of the two subunits of the system Xc−. SLC7A11 reduction seems to decrease GSH synthesis, which is essential for the detoxification of lipid reactive oxygen species through the action of GPX4 (this image was created with BioRender.com).
TABLE 1.
Suggested ferroptosis connection with ribosome related bone marrow failure disorders.
| Type | Gene | Mode of action | Molecular events | Model |
|---|---|---|---|---|
| DBA and 5q− syndrome | ↓RPS19 and ↓RPS14 |
↑Heme ↓FLVCR1 ↑Fe2+ ↓SLC7A11 ↓ globins |
↑Heme accumulation ↑Iron accumulation ↑Lipid peroxidation ↓GSH synthesis |
DBA‐HSCs DBA bone marrow sample 5q− bone marrow sample |
| SDS | ↓SBDS |
↑NOX4 ↓GPX4 ↑ CYGB ↓FSP1 ↓RaRa ↑TfR2 |
↑Iron accumulation ↑Lipid peroxidation ↓Antioxidant defense |
SBDS silenced mammalian cells SBDS‐depleted mice Sdo1p‐silenced yeast cells |
| X‐linked DC | ↓DKC1 |
↑CBPB ↓FSP1 ↓TERC ↓GSTM1 ↓GSTM2 ↓GSH |
↑Lipid peroxidation ↓GSH synthesis |
DC‐HSCs DKC1‐silenced HeLa cells |
| CHH | ↓RMRP |
↓TERC ↓TERT |
Lipid peroxidation | CHH‐HSCs |
Abbreviations: CHH, cartilage hair hypoplasia; DBA, Diamond–Blackfan anemia; SDS, Shwachman–Diamond syndrome; X‐linked DC, X‐linked dyskeratosis congenita.
5.2. Shwachman–Diamond syndrome and ferroptosis
Shwachman–Diamond syndrome (SDS) is a ribosome disorder that was initially described in 1964 by Shwachman et al. 168 It is classified as IBMF and is characterized by various features, including neutropenia or cytopenia in multiple blood cell lineages, exocrine pancreatic insufficiency, metaphyseal chondrodysplasia, and deficient hematopoiesis. 169 The majority of SDS patients, around 90%, have present mutations in the Shwachman–Bodian–Diamond syndrome (SBDS) gene. 169 This gene encodes an essential protein involved in the maturation of 60S ribosomal subunits. Mutations in other genes such as EFL1, DNAJC21, and SRP54, which play a role in the final stages of 60S maturation, have also been associated with this syndrome. 170 SDS cells exhibit abnormal expression of several genes participating in ribosome synthesis, as well as rRNA and mRNA processing. Furthermore, they show limited expression of multiple ribosomal protein genes that are crucial for cell proliferation and survival, including RPS9, RPS20, RPL6, RPL15, RPL22, RPL23, and RPL29. 170 It is noteworthy to highlight that the SBDS protein, comprising a sequence of 250 amino acids, exhibits a high degree of conservation across various species. Furthermore, the SBDS mRNA demonstrates ubiquitous expression, underscoring the significance and essential role of this protein in biological processes. 139 , 170
In contrast to DBA and 5q− syndrome, SDS primarily manifests as neutropenia, while anemia occurs less frequently. However, when it occurs is usually connected with iron deficiency, due to dysregulation of hepcidin levels, leading to iron‐deficient erythropoiesis. 171 Previous research has indicated that the deficiency of the ribosome biogenesis gene SBDS, in hematopoietic stem and progenitor cells, leads to neutropenia in mice by impeding lineage progression in myelocytes. 171 , 172 , 173 , 174 In this context, Zampetti et al. 172 conducted a study on SBDS‐depleted mice to investigate the process of myelopoiesis and observed a decrease in the number of mature neutrophils. This decrease was attributed to the arrest of myelopoiesis in the myelocyte (MC)–metamyelocyte (MM) stage and an increased level of cell death, particularly among neutrophils. RNA sequencing of the FACS‐isolated c‐kit MC‐MM cells revealed a significant reduction in the levels of the retinoic acid receptor (RARα) gene. 175 RARα is known to be involved in the progression of myelopoiesis and cell cycle exit. 175 , 176 Notably, the RARs family has been associated with the prevention of ferroptosis by upregulating the expression of GPX4 and FSP1, which are key gatekeepers in eliminating lipid peroxidation. 177 Additionally, the RNA sequencing analysis showed a significant increase in p53 transcripts, and among the genes connected to the downstream cascade of the p53‐apoptosis axis, an increase in Bbc3 (PUMA) gene transcripts was observed. 176 PUMA has recently been linked to ferroptosis as a key regulatory gene in the interconnection between ferroptosis and other types of cell death. 5 The same study also reported a mild decrease in erythrocytes and other types of leukocytes. Another study conducted on CD34+ HSC/Ps cells isolated from SDS‐patient bone marrow and the human erythroleukemia cell line K562, after the induction of differentiation, revealed that the differentiation process in the red blood cells was not affected but cell death was induced. 177 Increased levels of ROS were observed in both cases compared with control cells. The study also highlighted an increase in NOX4 gene expression and a decrease in Eosinophil peroxidase (EPX), Cytoglobin (CYGB), and Forkhead Box M1 (FoxM1) genes. Interestingly, NOX4, along with other members of the NOX family, is known for inducing ROS production and especially lipid peroxidation this is why it has been associated with the induction of ferroptosis in various diseases such as heart failure and Alzheimer's. 178 , 179 Furthermore, the reduction of CYGB has been associated with increased sensitivity to ferroptosis in other cases. 180
It is essential to note that studies on SBDS silenced mammalian cells and Sdo1p silenced yeast cells, the orthologue gene of SBDS in yeast, have shown a significant increase in TfR2 expression, leading to enhanced iron uptake and increased sensitivity to oxidative stress. 179 , 180 , 181 , 182 , 183 , 184 This accumulation of iron, as suggested by previous studies, may impact the progression of SDS by exacerbating oxidative stress and potentially being connected to ferroptotic pathways. 184 These data tend to propose a connection between bone marrow failure in SDS and ferroptosis. In particular, in SDS, SBDS is downregulated affecting the maturation of 60S. Thus, it is possible that the ribosome composition is affected once again altering the ribosomal function and affecting onwards the mRNA expression patterns. The observed increase in NOX4 and CYGB, both associated with ferroptosis, along with the decrease in the RAR family, which has been furtherly associated with reduced levels of GPX4 and FSP1, may contribute to susceptibility to lipid peroxidation and reduced antioxidant defense, making cells more vulnerable to ferroptosis. To this end, these pathways seem to cause an increase in lipid peroxidation while together with the increase in TfR2 expression which enhances iron uptake, may lead to lipid peroxidation and iron accumulation activating ferroptosis pathways synergistically with apoptosis, thus contributing to the manifestation of SDS‐IBMF (Figure 5; Table 1).
FIGURE 5.
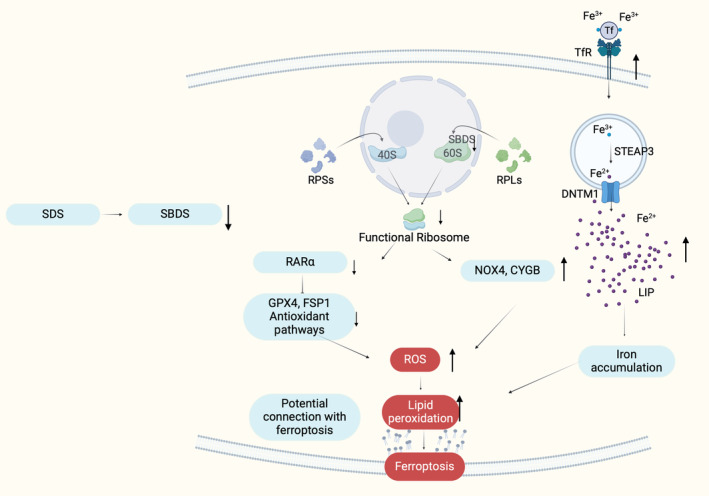
Illustration of the potential connection between SDS and ferroptosis in bone marrow cells, according to existing experimental data. The reduction in the expression of SBDS gene leads to impaired 60S formation, subsequently resulting in impaired ribosomes. This reduction in ribosomes seems to decrease the RARα expression. RARα has been associated with the upregulation of GPX4 and FSP1, which are antioxidant molecules crucial for the detoxification of intracellular ROS and for cell resistance to ferroptosis. Therefore, the downregulation of RARα in SDS may potentially affect the expression of these antioxidant molecules, compromising the cellular defense against ferroptosis. Moreover, increased NOX4, CYGB and TfR2, all associated with high levels of intracellular ROS and ferroptosis, have been observed in SBDS‐silenced cells. High levels of TfR2 may increase the intracellular iron and subsequently increase the production of lipid reactive oxygen species through the Fenton reaction (this image was created with BioRender.com).
5.3. X‐linked dyskeratosis congenita and ferroptosis
X‐linked dyskeratosis congenita (X‐linked DC) initially documented in 1910, represents a ribosomopathy that is also classified as telomeropathy. This condition is characterized by distinct manifestations, including abnormal skin pigmentation, nail dystrophy, and leukoplakia of the oral mucosa. 185 In the case of X‐linked DC, mutations in the DKC1 gene, responsible for encoding the dyskerin protein, have been identified. Dyskerin plays a crucial role in pseudouridylation during the processing of rRNA. Specifically, dyskerin functions as a constituent of H/ACA ribonuclear protein complexes, which are essential for modifying not only rRNAs but also various other types of RNAs, such as spliceosomal RNAs and the RNA component of telomerase. 185 , 186 It has been demonstrated that dysfunctional dyskerin can disrupt the translation of mRNAs containing IRES. 187 , 188 Furthermore, dyskerin plays a key role to the telomerase activity, which consists of the Telomerase RNA component (TERC) and the Telomerase Reverse Transcriptase (TERT), as it binds directly to TERC to confer the structural rigidity and stability necessary for its accumulation and function. 188 , 189
Limited knowledge exists regarding the underlying mechanism of how this disease induces anemia and in many cases pancytopenia. However, it has been established that the disease impacts the p53 pathway by sustaining heightened levels, resulting in cellular demise due to increased levels of ROS. 190 To date, there are no data relating this disease with the mechanism of ferroptosis, although certain findings suggest a potential association with this specific cellular death pathway.
According to Carrascoso‐Rubio et al., 191 suppression of the DKC1 gene in normal, healthy hematopoietic stem and progenitor cells (HSPCs) has been linked to a notable decrease in the expression of TERC and consequently telomerase activity. It is worth noting that the reduction of TERC has been observed to promote ferroptosis in lung cancer cells. 192 Additionally, this study confirmed that interference with DKC1 led to the manifestation of markers associated with DNA damage, cellular senescence, and cell death. 191 Moreover, another study conducted on CD34+ cells has pointed out that initial telomerase insufficiency and short telomeres primarily lead to a quantitative alteration in HSCs' numbers, rather than a qualitative change in their differentiation capacity. 193 RNA sequencing analysis performed on DC‐HSCs has further revealed a consistent reduction in the expression of CCAAT/Enhancer Binding Protein (C/EBP, CBPB). 193 CBPB is a transcription factor and one of its target genes is the FSP1 gene, which, as mentioned above, plays a significant role in conferring resistance against ferroptosis. 194 Notably, it has been observed that an increase in CBPB levels leads to enhanced FSP1 expression, thereby contributing to the resistance against ferroptosis. 194 Furthermore, in analyses performed on DC lymphocytes, increased levels of FOS, a transcription factor that suppresses the NRF‐2 antioxidant pathway, were observed compared with control samples. 195 Similarly, a decrease in the expression of the glutathione S‐transferase (GST), glutathione S‐transferase mu‐1 (GSTM1), and glutathione S‐transferase mu‐2 (GSTM2) genes was observed. 195 GSTM1 and GSTM2 are NRF2 transcriptional targets, and they have been previously implicated in the acquisition of aplastic anemia. 194 , 195 The pathways in which these genes are involved are related to the synthesis and transport of GSH, the main pathway that affects lipid peroxidation and could potentially be linked to ferroptosis. 179 Similar studies in DKC1 silenced fibroblasts confirmed these results. 180
It is important to highlight that in HeLa cells, a model cell line for many pathophysiological states, where the DKC1 gene was silenced, a reduction in the expression of glutathione‐related genes, such as glutathione synthase, glutamate‐cysteine ligase catalytic subunit (GCLC), and glutamate‐cysteine ligase regulatory subunit (GCLM), was observed. 147 Additionally, the levels of free glutathione were diminished compared with the control cells. 181 In the same context, the same scientific team observed high level of oxidative stress, as well as, GSSG/GSH ratio in DKC1 silenced HeLa cells. 196 Finally, it is noteworthy that since 1972 a similar pathophysiology to x‐linked DC has been observed in Fanconi anemia, which is also classified as telomeropathy. 197 , 198 , 199 In the case of Fanconi anemia, ferroptosis has been linked as a mechanism of cell death in HSCs. 134 The data described above may connect x‐linked DC‐IBMF with the pathway of ferroptosis. In particular, downregulation of DKC1 may affect the rRNA pseudouridylation and consequently the final ribosome subunits assembly. This alteration in coordination with the reduction of TERC which has been observed in DC‐HSCs may induce the expression of CBPB, reducing the translation of FSP1 and making cells more susceptible to lipid peroxidation and furtherly to ferroptosis. Parallelly, genes connected with antioxidant pathways and especially implicated in the prevention against lipid peroxidation are downregulated such as NRF‐2 pathway molecules and genes implicated in GSH synthesis and transport pathways. These data suggest a possible connection of ferroptosis with x‐linked DC IBMF synergistically with apoptosis, however the literature is still limited, and more research is needed to unravel the exact mechanism of action focusing indicatively on the intracellular iron levels (Table 1).
5.4. Cartilage‐hair hypoplasia and ferroptosis
Cartilage‐hair hypoplasia (CHH) is a type of skeletal dysplasia. This disorder was first reported in 1965 by McKusick et al. mainly in Amish families. 200 CHH includes hair hypoplasia, bone marrow failure, and immunodeficiency. Other usual symptoms are macrocytic anemia and lymphopenia. CHH is associated with biallelic mutations in RMRP. RMRP encodes the non‐coding RNA subunit to form the ribonuclease MRP complex which is classified as a snoRNA and is located in the nucleolus. 201 , 202 This complex has an emerging role in the synthesis of ribosomal RNA and specifically in the maturation of the 5′ end of the 5.8S rRNA. 203 , 204
Until now, there is limited information available regarding the potential association between bone marrow failure in individuals with CHH and ferroptosis. However, it is noteworthy to mention that RNA component of mitochondrial RNA processing endoribonuclease (RMPR) can form a ribonucleoprotein complex with TERT, thereby negatively regulating the expression of RMRP. 205 TERT, as already mentioned, is a catalytic reverse transcriptase, and is intricately connected to the non‐coding RNA component TERC creating the Telomerase complex. 205 This complex is responsible for the maintenance of genomic integrity, by elongating the telomeres. Interestingly, in patients with CHH, it has been observed that telomeres are shorter in peripheral blood cells, as well as in granulocytes and lymphocytes. 206 , 207 The shortening of telomeres can trigger the activation of DNA damage response and lipid peroxidation and furtherly ROS production, which may explain the increased apoptosis of lymphocytes in CHH. 208 Recent studies have suggested a potential role of TERC 209 , 210 and TERT 210 as suppressors of ferroptosis, however the exact mechanism remains unknown. Therefore, it is plausible that in CHH, downregulation of RMRP may affect TERC and TERT making cells more susceptible to ferroptosis and leading to reduced telomerase activity which is connected with shorter telomeres and furtherly ROS production and lipid peroxidation (Figure 6). In addition to apoptosis, ferroptosis may also play a role in definitive hematopoiesis in individuals with CHH, however more experiments on this field focusing once again on the intracellular iron levels may clarify the exact molecular mechanisms connected with deficient hematopoiesis in CHH (Table 1).
FIGURE 6.
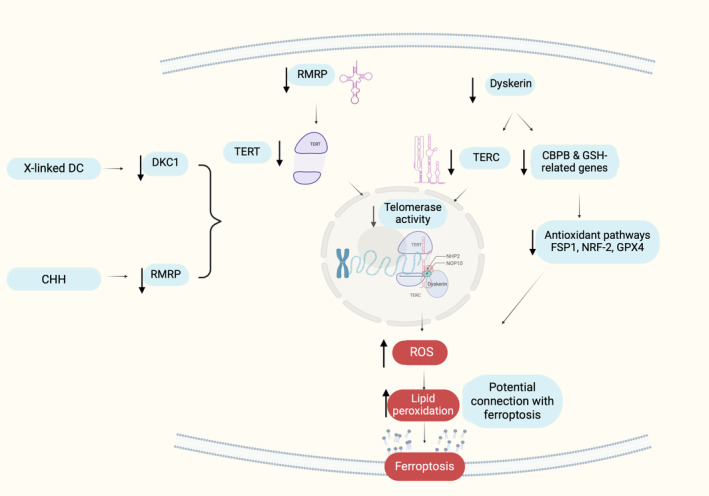
Depiction of the potential pathways connecting X‐linked dyskeratosis congenita (DC) and cartilage hair hypoplasia (CHH) with ferroptosis. Existing data suggests that reduced expression of RMRP and DKC1 results in low levels of TERT and TERC, respectively. TERT and TERC play crucial roles in telomerase activity, and their diminished expression leads to increased levels of intracellular ROS. Previous studies have already established the association between TERC, TERT, and ferroptosis. Furthermore, the decreased expression of DKC1 leads to reduced levels of CBPB and genes related to GSH metabolism. These genes are involved in various antioxidant pathways, including FSP1, NRF‐2, and GPX4 pathways. Low levels of CBPB and GSH‐related genes may disrupt these pathways, resulting in intracellular ROS accumulation and rendering cells more susceptible to ferroptosis (this image was created with BioRender.com).
6. PHARMACOLOGICAL EXPLOITATION OF FERROPTOSIS AND FUTURE PERSPECTIVES
Once ferroptosis may play a key role to ribosome related IBMFs it is important to present the recent pharmacological landscape of ferroptosis inhibitors. The last decade, it has been experimentally confirmed that many diseases are connected with ferroptosis. More specifically, in neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease as well as ischemia/reperfusion (I/R) injury, the observed increased cell death has been connected with ferroptosis. For this reason, even though the exact molecular mechanism in many cases is not totally clarified, several ferroptosis targets have been pharmacologically exploited to improve the existing therapeutic strategies, as thoroughly described by Ge et al., Wang et al., and Scarpellini et al. 211 , 212 , 213 In Table 2 some of the most well studied ferroptosis inhibitors are presented with regard to the molecular mechanism of action and the respective study models in each case. Most of the anti‐ferroptotic agents target regulators of lipid metabolism, iron metabolism and cellular antioxidant system, to prevent iron accumulation and lipid peroxidation. The initial ferroptosis inhibitor was first introduced in 2012 by Stockwell laboratory as Ferostatin‐1 (Fer‐1), an arylalkylamine compound which inhibits lipid peroxidation by scavenging the alkoxyl radicals produced by ferrous iron from lipid hydroperoxides, while it also creates a complex with Fe2+ reducing the LIP. At the same time Fer‐1 binds to ALOX15 thus inhibiting the synthesis of lipid hydroperoxides and thus lipid peroxidation. 1 , 2 , 214 , 223 , 224 , 225 , 226 Beyond Fer‐1, other known anti‐ferroptotic molecules targeting lipid metabolism are liproxstatin‐1 (Lip‐1) as well as deferiprone (DFP). These molecules inhibit ACSL4 and ALOX15 decreasing the free lipid hydroperoxides content of the cell and inhibiting lipid peroxidation. 197 , 215 , 223 , 224 Interestingly, DFP is also an iron chelator, as well as deferoxamine (DFO) and dexrazoxane (DXZ) targeting iron metabolism and especially reducing free Fe2+. 216 , 217 DFP is undergoing phase II clinical trial for Parkinson's disease and Friedreich's ataxia. 197 , 223 , 224 The same mechanism is followed by another broad category of antioxidant inducers in Alzheimer's disease are thiazolidinediones (TZDs) as well as, substances like N‐acetylcysteine (NAC), which function as ACSL4 inhibitors, again inhibiting free lipid radicals. 211 , 221 Additionally, certain vitamins such as a‐TOH and coQ10 serve as natural antioxidants by targeting scavenging lipid hydroperoxides thus preventing lipid peroxidation and are utilized against ferroptosis. 18 , 218 , 221 , 227 , 228 , 229 Finally, it is noteworthy to mention, that a promising anti‐ferroptotic clinical candidate is copper(II)‐diacetyl‐bis(N4‐methylthiosemicarbazone) (CuATSM) which acts as a potent radical trapping agent and is currently undergoing phase I clinical trials for early idiopathic Parkinson's disease, as well as phase II and III for amyotrophic lateral sclerosis/motor neuron disease. 211 , 216 , 217 , 218 , 219 Finally, Inducers of system Xc−, such as β‐mercaptoethanol, also belong to the broad category of ferroptosis inhibitors. 211 , 212 , 213 , 220 , 222 , 225 , 230 , 231 , 232
TABLE 2.
Most well‐studied drugs or chemicals used as ferroptosis inhibitors with their pharmacological targets and mode of action.
| Drug name | Targets | Mode of action | Molecular pathway | Model | References |
|---|---|---|---|---|---|
| Ferrostatin‐1 | ALOX15, Alkoxyl radicals |
↓ LPO, ↓ Fe2+ |
Lipid metabolism |
AKI mice | [214] |
| Liproxstatin‐1 | ASCL4/ALOX15 | ↑GPX4, ↓ LPO |
Lipid metabolism |
MAFLD mice | [215] |
| CuASTM | PE AA/ADA OOH |
↓ LPO ↑GPX4 |
Lipid metabolism |
Embryonic fibroblasts Hippocampal cells |
[216, 217, 218, 219] |
| CoQ10 | PE AA/ADA OOH | ↓ LPO ↑GPX4 | Lipid metabolism | MF‐dCas9‐MS2 cells | [220] |
| TZDs | ASCL4/LPCAT3 |
↓ LPO, ↑GPX4 |
Lipid metabolism |
AD mice | [221] |
| NAC | ALOX5 |
↓ LPO, ↑GPX4 |
Lipid metabolism |
ICH mice and rats | [211] |
| α‐tocopherol (Vitamin E) | ALOX15 | ↓ LPO ↑GPX4 | Lipid metabolism | SD rats | [202, 203, 204] |
| β‐mercaptoethanol | Cysteine | ↑GSH, ↑GPX4 | GSH synthesis | L1210 cell line | [18, 219, 222] |
| DFP | ASCL4/ALOX15 | ↓ Fe2+, ↓ LPO | Lipid metabolism | MAFLD mice | [196, 223, 224] |
| DFO, DXZ | Fe2+ | ↓ Fe2+, ↑GSH | Iron metabolism | SD rats | [216, 217] |
Abbreviations: AD mice, Alzheimer's disease mice model; AKI mice, acute kidney injury mice model; CuATSM, copper(II)‐diacetyl‐bis(N4‐methylthiosemicarbazone); DFO, deferoxamine; DFP, deferiprone; DXZ, dexrazoxane; GSH, glutathione; ICH mice and rats, intracerebral hemorrhage mice and rats model; L1210 cell line, murine lymphocytic leukemia cell line; LPO, lipid peroxidation; MAFLD mice, metabolic associated fatty liver disease mice model; MF‐dCas9‐MS2 cells, dCas9‐MS2 (SAM system) murine fibroblasts; SD rats, Sprague–Dawley rats; TZDs, thiazolidinediones.
Finally, it is important to note that among the 15 active clinical trials in Clincaltrials.gov associated with ferroptosis, 1 of them is dealing with MDS. 233 More specifically, this trial involves 80 participants separated in Splicing Factor 3B Subunit 1A (SF3B1)‐mutant MDS and normal bone marrow control individuals and aims to demonstrate the activation of ferroptosis pathways in MDS patients compared with the control ones (NCT05924074). From the data mentioned above, it seems that ferroptosis inhibitors is a promising group of drugs for the treatment of many diseases. If the role of ferroptosis is clarified in blood disorders and especially in ribosome related IBMFs then a promising therapeutic avenue is emerged in the clinical setting.
7. CONCLUSIONS
Although, the term “ferroptosis” was coined about a decade ago, many data support its involvement in various physiological and pathophysiological processes. Based on the knowledge accumulated thus far, we attempted in this review to present evidence supporting potential molecular links between the ribosome‐related IBMF syndromes with ferroptosis. Within this frame, deficient hematopoiesis is associated with molecular pathways that lead to cell death in bone marrow cells. We suggest that in the hematopoietic progenitors of each of the described ribosomopathies, mutations in RPs lead to changes in ribosome composition which seems to affect ribosomal function enhancing the expression of some mRNAs subgroups while reducing the expression of others facilitating an intracellular imbalance. In each case, this disturbance is accompanied by ROS production and lipid peroxidation, while an additional finding in most of them is iron accumulation. Once lipid peroxidation and iron accumulation are the two main characteristics of ferroptosis, it is possible that this mechanism play a key role in the manifestation of IBMF in this type of disease. Despite the recent scientific advancements in the area of ferroptosis, the development and use of specific inhibitors of ferroptosis still presents a challenge for research to develop new therapeutics to combat diseases. Nowadays, ferroptosis inhibitors are under evaluation in trials running in clinical practice, with this fact reinforcing the promise for healthcare to broaden its pharmacological repertoire, identifying new druggable molecular targets and establishing innovative therapeutic strategies of ribosome‐related IBMF syndromes. Importantly, however, and in any case, further research is needed in the years to come, to uncover the exact pathophysiological role of ferroptosis in human disorders.
Papadimitriou‐Tsantarliotou A, Avgeros C, Konstantinidou M, Vizirianakis IS. Analyzing the role of ferroptosis in ribosome‐related bone marrow failure disorders: From pathophysiology to potential pharmacological exploitation. IUBMB Life. 2024;76(12):1011–1034. 10.1002/iub.2897
REFERENCES
- 1. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron‐dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stockwell BR. Ferroptosis turns 10: emerging mechanisms, physiological functions, and therapeutic applications. Cell. 2022;185:2401–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Distéfano AM, Martin MV, Córdoba JP, Bellido AM, D'Ippólito S, Colman SL, et al. Heat stress induces ferroptosis‐like cell death in plants. J Cell Biol. 2017;216:463–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, et al. Selenium utilization by GPX4 is required to prevent hydroperoxide‐induced ferroptosis. Cell. 2018;172:409–422. [DOI] [PubMed] [Google Scholar]
- 5. Chen X, Li J, Kang R, Klionsky DJ, Tang D. Ferroptosis: machinery and regulation. Autophagy. 2020;17:2054–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dixon SJ, Stockwell BR. The hallmarks of ferroptosis. Annu Rev Cancer Biol. 2019;3:35–54. [Google Scholar]
- 7. Lv Q, Niu H, Yue L, Liu J, Yang L, Liu C, et al. Abnormal ferroptosis in myelodysplastic syndrome. Front Oncol. 2020;10:1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gong L, Huang D, Shi Y, Liang Z, Bu H. Regulated cell death in cancer: from pathogenesis to treatment. Chin Med J (Engl). 2022;136:653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Green DR, Victor B. The pantheon of the fallen: why are there so many forms of cell death? Trends Cell Biol. 2012;22:555–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Battaglia AM, Chirillo R, Aversa I, Sacco A, Costanzo F, Biamonte F. Ferroptosis and cancer: mitochondria meet the “iron maiden” cell death. Cells. 2020;9:1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zou Y, Schreiber SL. Progress in understanding ferroptosis and challenges in its targeting for therapeutic benefit. Cell Chem Biol. 2020;27:463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miller SC, MacDonald CC, Kellogg MK, Karamysheva ZN, Karamyshev AL. Specialized ribosomes in health and disease. Int J Mol Sci. 2023;24:6334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fan Y, Cheng Y, Li Y, Chen B, Wang Z, Wei T, et al. Phosphoproteomic analysis of neonatal regenerative myocardium revealed important roles of checkpoint kinase 1 via activating mammalian target of rapamycin C1/ribosomal protein S6 kinase B‐1 pathway. Circulation. 2020;141:1554–1569. [DOI] [PubMed] [Google Scholar]
- 14. Domitrovic T, Moreira MH, Carneiro RL, Ribeiro‐Alves M, Palhano FL. Natural variation of the cardiac transcriptome in humans. RNA Biol. 2020;18:1374–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nakhoul H, Ke J, Zhou X, Liao W, Zeng SX, Lu H. Ribosomopathies: mechanisms of disease. Clin Med Insights Blood Disord. 2014;7:7‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lyu T, Li X, Song Y. Ferroptosis in acute leukemia. Chin Med J (Engl). 2023;136:886–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Berghe TV, Linkermann A, Jouan‐Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non‐apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–147. [DOI] [PubMed] [Google Scholar]
- 18. Seiler A, Schneider M, Förster H, Roth S, Wirth EK, Culmsee C, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15‐lipoxygenase dependent‐ and AIF‐mediated cell death. Cell Metab. 2008;8:237–248. [DOI] [PubMed] [Google Scholar]
- 19. Conrad M, Angeli JP, Vandenabeele P, Stockwell BR. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2016;15:348–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype‐selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3:285–296. [DOI] [PubMed] [Google Scholar]
- 21. Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. Ras–Raf–Mek‐dependent oxidative cell death involving voltage‐dependent anion channels. Nature. 2007;447:865–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator GPX4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li J, Cao F, Yin H, Huang Z, Lin Z, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis. 2020;11:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Riegman M, Sagie L, Galed C, Levin T, Steinberg N, Dixon SJ, et al. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat Cell Biol. 2020;22:1042–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Katikaneni A, Jelcic M, Gerlach GF, Ma Y, Overholtzer M, Niethammer P. Lipid peroxidation regulates long‐range wound detection through 5‐lipoxygenase in zebrafish. Nat Cell Biol. 2020;22:1049–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen X, Comish PB, Tang D, Kang R. Characteristics and biomarkers of ferroptosis. Front Cell Dev Biol. 2021;9:637162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ouyang J, Zhou L, Wang Q. Spotlight on iron and ferroptosis: research progress in diabetic retinopathy. Front Endocrinol. 2023;14:1234824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kleven MD, Jue S, Enns CA. Transferrin receptors tfr1 and TFR2 bind transferrin through differing mechanisms. Biochemistry. 2018;57:1552–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kawabata H. Transferrin and transferrin receptors update. Free Radic Biol Med. 2019;133:46–54. [DOI] [PubMed] [Google Scholar]
- 30. Chen X, Yu C, Kang R, Tang D. Iron metabolism in ferroptosis. Front Cell Dev Biol. 2020;8:590226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kuang F, Liu J, Tang D, Kang R. Oxidative damage and antioxidant defense in ferroptosis. Front Cell Dev Biol. 2020;8:586578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci. 2016;113:E4966–E4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59:298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 2017;20:1692–1704. [DOI] [PubMed] [Google Scholar]
- 35. Yang W‐H, Ding C‐KC, Sun T, Rupprecht G, Lin C‐C, Hsu D, et al. The hippo pathway effector Taz regulates ferroptosis in renal cell carcinoma. Cell Rep. 2019;28:2501‐2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, et al. ALOX12 is required for p53‐mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. 2019;21:579–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li C, Zhang Y, Liu J, Kang R, Klionsky DJ, Tang D. Mitochondrial DNA stress triggers autophagy‐dependent ferroptotic death. Autophagy. 2020;17:948–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, et al. Cytochrome P450 oxidoreductase contributes tophospholipid peroxidation in ferroptosis. Nat Chem Biol. 2020;16:302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Feng H, Stockwell BR. Unsolved mysteries: how does lipid peroxidation cause ferroptosis? PLoS Biol. 2018;16:e2006203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2016;13:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang H‐L, Hu B‐X, Li Z‐L, Du T, Shan J‐L, Ye Z‐P, et al. PKCΒII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat Cell Biol. 2022;24:88–98. [DOI] [PubMed] [Google Scholar]
- 42. Yuan H, Li X, Zhang X, Kang R, Tang D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun. 2016;478:1338–1343. [DOI] [PubMed] [Google Scholar]
- 43. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2016;13:91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The COQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione‐independent ferroptosis suppressor. Nature. 2019;575:693–698. [DOI] [PubMed] [Google Scholar]
- 47. Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62‐keap1‐nrf2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2015;63:173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jang EJ, Kim DH, Lee B, Lee EK, Chung KW, Moon KM, et al. Activation of proinflammatory signaling by 4‐hydroxynonenal‐src adducts in aged kidneys. Oncotarget. 2016;7:50864–50874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao L, et al. HMGB1 in health and disease. Mol Aspects Med. 2014;40:1–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wen Q, Liu J, Kang R, Zhou B, Tang D. The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun. 2019;510:278–283. [DOI] [PubMed] [Google Scholar]
- 51. Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26:165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. Dhodh‐mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593:586–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kraft VA, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, et al. GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. 2020;6:41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liang D, Feng Y, Zandkarimi F, Wang H, Zhang Z, Kim J, et al. Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones. Cell. 2023;186:2748–2764.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Freitas FP, Alborzinia H, dos Santos AF, Nepachalovich P, Pedrera L, Zilka O, et al. 7‐dehydrocholesterol is an endogenous suppressor of ferroptosis. Nature. 2024;626:401–410. [DOI] [PubMed] [Google Scholar]
- 56. Yan H‐F, Tuo Q‐Z, Yin Q‐Z, Lei P. The pathological role of ferroptosis in ischemia/reperfusion‐related injury. Zool Res. 2020;41:220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang S, Hu R, Geng Y, Chen K, Wang L, Imam MU. The regulatory effects and the signaling pathways of natural bioactive compounds on ferroptosis. Foods. 2021;10:2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dixon SJ. Ferroptosis: bug or feature? Immunol Rev. 2017;277:150–157. [DOI] [PubMed] [Google Scholar]
- 59. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yan R, Lin B, Jin W, Tang L, Hu S, Cai R. NRF2, a superstar of ferroptosis. Antioxidants. 2023;12:1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li W, Liang L, Liu S, Yi H, Zhou Y. FSP1: a key regulator of ferroptosis. Trends Mol Med. 2023;29:753–764. [DOI] [PubMed] [Google Scholar]
- 62. Chen Y, Yi X, Huo B, He Y, Guo X, Zhang Z, et al. BRD4770 functions as a novel ferroptosis inhibitor to protect against aortic dissection. Pharmacol Res. 2022;177:106122. [DOI] [PubMed] [Google Scholar]
- 63. Zeng F, Chen X, Deng G. The anti‐ferroptotic role of FSP1: current molecular mechanism and therapeutic approach. Mol Biomed. 2022;3:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Liu M, Kong X‐Y, Yao Y, Wang X‐A, Yang W, Wu H, et al. The critical role and molecular mechanisms of ferroptosis in antioxidant systems: a narrative review. Ann Transl Med. 2022;10:368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol. 2020;16:1351–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Liu Y, Lu S, Wu L, Yang L, Yang L, Wang J. The diversified role of mitochondria in ferroptosis in cancer. Cell Death Dis. 2023;14:519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Belavgeni A, Tonnus W, Linkermann A. Cancer cells evade ferroptosis: sex hormone‐driven membrane‐bound O‐acyltransferase domain‐containing 1 and 2 (MBOAT1/2) expression. Signal Transduct Target Ther. 2023;8:336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li Y, Ran Q, Duan Q, Jin J, Wang Y, Yu L, et al. 7‐Dehydrocholesterol dictates ferroptosis sensitivity. Nature. 2024;626:411–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yamada N, Karasawa T, Ito J, Yamamuro D, Morimoto K, Nakamura T, et al. Inhibition of 7‐dehydrocholesterol reductase prevents hepatic ferroptosis under an active state of sterol synthesis. Nat Commun. 2024;15(1):2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ravingerová T, Kindernay L, Barteková M, Ferko M, Adameová A, Zohdi V, et al. The molecular mechanisms of iron metabolism and its role in cardiac dysfunction and cardioprotection. Int J Mol Sci. 2020;21:7889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Piskin E, Cianciosi D, Gulec S, Tomas M, Capanoglu E. Iron absorption: factors, limitations, and improvement methods. ACS Omega. 2022;7:20441–20456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Johnson‐Wimbley TD, Graham DY. Diagnosis and management of iron deficiency anemia in the 21st century. Therap Adv Gastroenterol. 2011;4:177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Seyoum Y, Baye K, Humblot C. Iron homeostasis in host and gut bacteria – a complex interrelationship. Gut Microbes. 2021;13:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Le Blanc S, Garrick MD, Arredondo M. Heme carrier protein 1 transports heme and is involved in heme‐FE metabolism. Am J Physiol Cell Phys Ther. 2012;302:C1780–C1785. [DOI] [PubMed] [Google Scholar]
- 75. Canonne‐Hergaux F, Gruenheid S, Ponka P, Gros P. Cellular and subcellular localization of the NRAMP2 iron transporter in the intestinal brush border and regulation by dietary iron. Blood. 1999;93:4406–4417. [PubMed] [Google Scholar]
- 76. Silva B, Faustino P. An overview of molecular basis of iron metabolism regulation and the associated pathologies. Biochim Biophys Acta (BBA) – Mol Basis Dis. 2015;1852:1347–1359. [DOI] [PubMed] [Google Scholar]
- 77. Krishnamurthy P, Xie T, Schuetz J. The role of transporters in cellular heme and porphyrin homeostasis. Pharmacol Ther. 2007;114:345–358. [DOI] [PubMed] [Google Scholar]
- 78. Yang J, Tang Q, Zeng Y. Melatonin: potential avenue for treating iron overload disorders. Ageing Res Rev. 2022;81:101717. [DOI] [PubMed] [Google Scholar]
- 79. Pagani A, Nai A, Silvestri L, Camaschella C. Hepcidin and anemia: a tight relationship. Front Physiol. 2019;10:1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kumar A, Sharma E, Marley A, Samaan MA, Brookes MJ. Iron deficiency anaemia: pathophysiology, assessment, practical management. BMJ Open Gastroenterol. 2022;9:e000759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Yang J, Li Q, Feng Y, Zeng Y. Iron deficiency and iron deficiency anemia: potential risk factors in bone loss. Int J Mol Sci. 2023;24:6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Clifford W‐F. Coenzyme Q 10 and vitamin E synergy, electron transfer, antioxidation in cell membranes, and interaction with cholesterol. Adelaide: Eigenenergy; 2023. [Google Scholar]
- 83. Han M, Xu R, Wang S, Yang N, Ni S, Zhang Q, et al. Six‐transmembrane epithelial antigen of prostate 3 predicts poor prognosis and promotes glioblastoma growth and invasion. Neoplasia. 2018;20:543–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Gao G, Li J, Zhang Y, Chang Y‐Z. Cellular iron metabolism and regulation. In: Chang YZ, editor. Brain iron metabolism and CNS diseases. Singapore: Springer; 2019. p. 21–32. [Google Scholar]
- 85. Hong M, Rong J, Tao X, Xu Y. The emerging role of ferroptosis in cardiovascular diseases. Front Pharmacol. 2022;13:822083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Chen Y, Li X, Wang S, Miao R, Zhong J. Targeting iron metabolism and ferroptosis as novel therapeutic approaches in cardiovascular diseases. Nutrients. 2023;15:591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Mou Y, Wang J, Wu J, He D, Zhang C, Duan C, et al. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol. 2019;12:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Fan X, Li A, Yan Z, Geng X, Lian L, Lv H, et al. From iron metabolism to ferroptosis: pathologic changes in coronary heart disease. Oxid Med Cell Longev. 2022;2022:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron‐dependent, nonapoptotic cell death in oncogenic‐ras‐harboring cancer cells. Chem Biol. 2008;15:234–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Di Paola A, Tortora C, Argenziano M, Marrapodi MM, Rossi F. Emerging roles of the iron chelators in inflammation. Int J Mol Sci. 2022;23:7977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Mortensen MS, Ruiz J, Watts JL. Polyunsaturated fatty acids drive lipid peroxidation during ferroptosis. Cells. 2023;12:804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Li D, Li Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct Target Ther. 2020;5:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Jia B, Li J, Song Y, Luo C. ACSL4‐mediated ferroptosis and its potential role in central nervous system diseases and injuries. Int J Mol Sci. 2023;24:10021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Yan N, Zhang J‐J. The emerging roles of ferroptosis in vascular cognitive impairment. Front Neurosci. 2019;13:811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Singh NK, Rao GN. Emerging role of 12/15‐lipoxygenase (ALOX15) in human pathologies. Prog Lipid Res. 2019;73:28–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. 2015;10:1604–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Palade GE. A small particulate component of the cytoplasm. J Cell Biol. 1955;1:59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Genuth NR, Barna M. The discovery of ribosome heterogeneity and its implications for gene regulation and organismal life. Mol Cell. 2018;71:364–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Boisvert F‐M, van Koningsbruggen S, Navascués J, Lamond AI. The multifunctional nucleolus. Nat Rev Mol Cell Biol. 2007;8:574–585. [DOI] [PubMed] [Google Scholar]
- 100. Jiao L, Liu Y, Yu X‐Y, Pan X, Zhang Y, Tu J, et al. Ribosome biogenesis in disease: new players and therapeutic targets. Signal Transduct Target Ther. 2023;8:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Baßler J, Hurt E. Eukaryotic ribosome assembly. Annu Rev Biochem. 2019;88:281–306. [DOI] [PubMed] [Google Scholar]
- 102. Dörner K, Ruggeri C, Zemp I, Kutay U. Ribosome biogenesis factors—from names to functions. EMBO J. 2023;42:e112699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Warner JR. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci. 1999;24:437–440. [DOI] [PubMed] [Google Scholar]
- 104. Islam RA, Rallis C. Ribosomal biogenesis and heterogeneity in development, disease, and aging. Epigenomes. 2023;7:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Turowski TW, Tollervey D. Cotranscriptional events in eukaryotic ribosome synthesis. WIREs RNA. 2015;6:129–139. [DOI] [PubMed] [Google Scholar]
- 106. Marintchev A, Wagner G. Translation initiation: structures, mechanisms and evolution. Q Rev Biophys. 2004;37:197–284. [DOI] [PubMed] [Google Scholar]
- 107. Russell J, Zomerdijk JCBM. The RNA polymerase I transcription machinery. Biochem Soc Symp. 2006;73:203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Arabi A, Wu S, Ridderstråle K, Bierhoff H, Shiue C, Fatyol K, et al. C‐MYC associates with ribosomal DNA and activates RNA polymerase I transcription. Nat Cell Biol. 2005;7:303–310. [DOI] [PubMed] [Google Scholar]
- 109. Grandori C, Gomez‐Roman N, Felton‐Edkins ZA, Ngouenet C, Galloway DA, Eisenman RN, et al. C‐MYC binds to human ribosomal DNA and stimulates transcription of rrna genes by RNA polymerase I. Nat Cell Biol. 2005;7:311–318. [DOI] [PubMed] [Google Scholar]
- 110. Knutson BA, Hahn S. TFIIB‐related factors in RNA polymerase I transcription. Biochim Biophys Acta (BBA) – Gene Regul Mech. 2013;1829:265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Liu S, Hua Y, Wang J, Li L, Yuan J, Zhang B, et al. RNA polymerase III is required for the repair of DNA double‐strand breaks by homologous recombination. Cell. 2021;184:1314–1329. [DOI] [PubMed] [Google Scholar]
- 112. Henderson AS, Warburton D, Atwood KC. Location of ribosomal DNA in the human chromosome complement. Proc Natl Acad Sci U S A. 1972;69:3394–3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Higa‐Nakamine S, Suzuki T, Uechi T, Chakraborty A, Nakajima Y, Nakamura M, et al. Loss of ribosomal RNA modification causes developmental defects in zebrafish. Nucleic Acids Res. 2011;40:391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Dalla Venezia N, Vincent A, Marcel V, Catez F, Diaz J‐J. Emerging role of eukaryote ribosomes in translational control. Int J Mol Sci. 2019;20:1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Xue S, Barna M. Specialized ribosomes: a new frontier in gene regulation and organismal biology. Nat Rev Mol Cell Biol. 2012;13:355–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Kondrashov N, Pusic A, Stumpf CR, Shimizu K, Hsieh AC, Xue S, et al. Ribosome‐mediated specificity in Hox mrna translation and vertebrate tissue patterning. Cell. 2011;145:383–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Crick FH, Barnett L, Brenner S, Watts‐Tobin RJ. General nature of the genetic code for proteins. Nature. 1961;192:1227–1232. [DOI] [PubMed] [Google Scholar]
- 118. Shi Z, Fujii K, Kovary KM, Genuth NR, Röst HL, Teruel MN, et al. Heterogeneous ribosomes preferentially translate distinct subpools of mrnas genome‐wide. Mol Cell. 2017;67:71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Shi Z, Barna M. Translating the genome in time and space: specialized ribosomes, RNA regulons, and RNA‐binding proteins. Annu Rev Cell Dev Biol. 2015;31:31–54. [DOI] [PubMed] [Google Scholar]
- 120. Guimaraes JC, Zavolan M. Patterns of ribosomal protein expression specify normal and malignant human cells. Genome Biol. 2016;17:236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Gupta V, Warner JR. Ribosome‐omics of the human ribosome. RNA. 2014;20:1004–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Kyritsis KA, Ouzounis CA, Angelis L, Vizirianakis IS. Sequence variation, common tissue expression patterns and learning models: a genome‐wide survey of vertebrate ribosomal proteins. NAR Genom Bioinform. 2020;2:lqaa088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Papagiannopoulos CI, Kyritsis KA, Psatha K, Mavridou D, Chatzopoulou F, Orfanoudaki G, et al. Invariable ribosome stoichiometry during murine erythroid differentiation: implications for understanding ribosomopathies. Front Mol Biosci. 2022;9:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Gay DM, Lund AH, Jansson MD. Translational control through ribosome heterogeneity and functional specialization. Trends Biochem Sci. 2022;47:66–81. [DOI] [PubMed] [Google Scholar]
- 125. Alkan F, Wilkins OG, Hernández‐Pérez S, Ramalho S, Silva J, Ule J, et al. Identifying ribosome heterogeneity using ribosome profiling. Nucleic Acids Res. 2022;50:e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Wong QW‐L, Li J, Ng SR, Lim SG, Yang H, Vardy LA. RPL39L is an example of a recently evolved ribosomal protein paralog that shows highly specific tissue expression patterns and is upregulated in escs and HCC tumors. RNA Biol. 2013;11:33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Zhang Y, Duc A‐CE, Rao S, Sun X‐L, Bilbee AN, Rhodes M, et al. Control of hematopoietic stem cell emergence by antagonistic functions of ribosomal protein paralogs. Dev Cell. 2013;24:411–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Little RD, Braaten DC. Genomic organization of human 5S rDNA and sequence of one tandem repeat. Genomics. 1989;4:376–383. [DOI] [PubMed] [Google Scholar]
- 129. Stults DM, Killen MW, Pierce HH, Pierce AJ. Genomic architecture and inheritance of human ribosomal RNA gene clusters. Genome Res. 2008;18:13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Hiregange D, Rivalta A, Yonath A, Zimmerman E, Bashan A, Yonath H. Mutations in rps19 may affect ribosome function and biogenesis in diamond blackfan anemia. FEBS Open Bio. 2022;12:1419–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Narla A, Ebert BL. Ribosomopathies: human disorders of ribosome dysfunction. Blood. 2010;115:3196–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Aspesi A, Ellis SR. Rare ribosomopathies: insights into mechanisms of cancer. Nat Rev Cancer. 2019;19:228–238. [DOI] [PubMed] [Google Scholar]
- 133. Kang J, Brajanovski N, Chan KT, Xuan J, Pearson RB, Sanij E. Ribosomal proteins and human diseases: molecular mechanisms and targeted therapy. Signal Transduct Target Ther. 2021;6:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Zhao J, Jia Y, Mahmut D, Deik AA, Jeanfavre S, Clish CB, et al. Human hematopoietic stem cell vulnerability to ferroptosis. Cell. 2023;186:732–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Wu P, Zhang X, Duan D, Zhao L. Organelle‐specific mechanisms in crosstalk between apoptosis and ferroptosis. Oxid Med Cell Longev. 2023;2023:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Zhou L, Dong C, Ding L, Feng W, Yu L, Cui X, et al. Targeting ferroptosis synergistically sensitizes apoptotic sonodynamic anti‐tumor nanotherapy. Nano Today. 2021;39:101212. [Google Scholar]
- 137. Hong SH, Lee D‐H, Lee Y‐S, Jo MJ, Jeong YA, Kwon WT, et al. Molecular crosstalk between ferroptosis and apoptosis: emerging role of ER stress‐induced p53‐independent Puma expression. Oncotarget. 2017;8:115164–115178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Josephs HW. Anaemia of infancy and early childhood. Medicine. 1936;15:307–451. [Google Scholar]
- 139. Diamond LK, Blackfan KD. Hypoplastic anemia. Am J Dis Child. 1938;56:464–467. [Google Scholar]
- 140. Vlachos A, Muir E. How I treat Diamond‐Blackfan anemia. Blood. 2010;116:3715–3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Vlachos A, Ball S, Dahl N, Alter BP, Sheth S, Ramenghi U, et al. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol. 2008;142:859–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Glader BE, Backer K. Elevated red cell adenosine deaminase activity: a marker of disordered erythropoiesis in diamond‐blackfan anaemia and other haematologic diseases. Br J Haematol. 1988;68:165–168. [DOI] [PubMed] [Google Scholar]
- 143. Da Costa L, Tchernia G, Gascard P, Lo A, Meerpohl J, Niemeyer C, et al. Nucleolar localization of RPS19 protein in normal cells and mislocalization due to mutations in the nucleolar localization signals in 2 Diamond‐Blackfan anemia patients: potential insights into pathophysiology. Blood. 2003;101:5039–5045. [DOI] [PubMed] [Google Scholar]
- 144. Gazda HT, Grabowska A, Merida‐Long LB, Latawiec E, Schneider HE, Lipton JM, et al. Ribosomal protein S24 gene is mutated in diamond‐blackfan anemia. Am J Hum Genet. 2006;79:1110–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Cmejla R, Cmejlova J, Handrkova H, Petrak J, Pospisilova D. Ribosomal protein S17 gene (RPS17) is mutated in diamond‐blackfan anemia. Hum Mutat. 2007;28:1178–1182. [DOI] [PubMed] [Google Scholar]
- 146. Farrar JE, Nater M, Caywood E, McDevitt MA, Kowalski J, Takemoto CM, et al. Abnormalities of the large ribosomal subunit protein, RPL35A, in Diamond‐Blackfan anemia. Blood. 2008;112:1582–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Gazda HT, Sheen MR, Vlachos A, Choesmel V, O'Donohue M‐F, Schneider H, et al. Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond‐Blackfan anemia patients. Am J Hum Genet. 2008;83:769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Orrù S, Aspesi A, Armiraglio M, Caterino M, Loreni F, Ruoppolo M, et al. Analysis of the ribosomal protein S19 interactome. Mol Cell Proteomics. 2007;6:382–393. [DOI] [PubMed] [Google Scholar]
- 149. Rey MA, Duffy SP, Brown JK, Kennedy JA, Dick JE, Dror Y, et al. Enhanced alternative splicing of the FLVCR1 gene in Diamond Blackfan anemia disrupts FLVCR1 expression and function that are critical for erythropoiesis. Haematologica. 2008;93:1617–1626. [DOI] [PubMed] [Google Scholar]
- 150. Van Den Berghe H, Cassiman J‐J, David G, Fryns J‐P, Michaux J‐L, Sokal G. Distinct haematological disorder with deletion of long arm of no. 5 chromosome. Nature. 1974;251:437–438. [DOI] [PubMed] [Google Scholar]
- 151. Ebert BL, Pretz J, Bosco J, Chang CY, Tamayo P, Galili N, et al. Identification of RPS14 as the 5Q‐syndrome gene by RNA interference screen. Blood. 2007;110:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Doty RT, Phelps SR, Shadle C, Sanchez‐Bonilla M, Keel SB, Abkowitz JL. Coordinate expression of heme and globin is essential for effective erythropoiesis. J Clin Investig. 2015;125:4681–4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Keel SB, Doty RT, Yang Z, Quigley JG, Chen J, Knoblaugh S, et al. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science. 2008;319:825–828. [DOI] [PubMed] [Google Scholar]
- 154. Doty RT, Yan X, Lausted C, Munday AD, Yang Z, Yi D, et al. Single‐cell analyses demonstrate that a heme–GATA1 feedback loop regulates red cell differentiation. Blood. 2019;133:457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Yang Z, Keel SB, Shimamura A, Liu L, Gerds AT, Li HY, et al. Delayed globin synthesis leads to excess heme and the macrocytic anemia of Diamond Blackfan anemia and DEL(5Q) myelodysplastic syndrome. Sci Transl Med. 2016;8:338ra67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Hu J, Liu J, Xue F, Halverson G, Reid M, Guo A, et al. Isolation and functional characterization of human erythroblasts at distinct stages: implications for understanding of normal and disordered erythropoiesis in vivo. Blood. 2013;121:3246–3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Chiabrando D, Vinchi F, Fiorito V, Mercurio S, Tolosano E. Heme in pathophysiology: a matter of scavenging, metabolism and trafficking across cell membranes. Front Pharmacol. 2014;5:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Li J, Hale J, Bhagia P, Xue F, Chen L, Jaffray J, et al. Isolation and transcriptome analyses of human erythroid progenitors: Bfu‐E and cfu‐e. Blood. 2014;124:3636–3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Suzuki H, Tashiro S, Hira S, Sun J, Yamazaki C, Zenke Y, et al. Heme regulates gene expression by triggering CRM1‐dependent nuclear export of bach1. EMBO J. 2004;23:2544–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Tahara T, Sun J, Nakanishi K, Yamamoto M, Mori H, Saito T, et al. Heme positively regulates the expression of β‐globin at the locus control region via the transcriptional factor bach1 in erythroid cells. J Biol Chem. 2004;279:5480–5487. [DOI] [PubMed] [Google Scholar]
- 161. Han A‐P. Heme‐regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J. 2001;20:6909–6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Dutt S, Narla A, Lin K, Mullally A, Abayasekara N, Megerdichian C, et al. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood. 2011;117:2567–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Jaako P, Debnath S, Olsson K, Zhang Y, Flygare J, Lindström MS, et al. Disruption of the 5s RNP–mdm2 interaction significantly improves the erythroid defect in a mouse model for Diamond‐Blackfan anemia. Leukemia. 2015;29:2221–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Barlow JL, Drynan LF, Hewett DR, Holmes LR, Lorenzo‐Abalde S, Lane AL, et al. A p53‐dependent mechanism underlies macrocytic anemia in a mouse model of human 5q– syndrome. Nat Med. 2009;16:59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Jiang L, Kon N, Li T, Wang S‐J, Su T, Hibshoosh H, et al. Ferroptosis as a p53‐mediated activity during tumour suppression. Nature. 2015;520:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Warnatz H‐J, Schmidt D, Manke T, Piccini I, Sultan M, Borodina T, et al. The BTB and CNC homology 1 (BACH1) target genes are involved in the oxidative stress response and in control of the cell cycle. J Biol Chem. 2011;286:23521–23532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Ghaffari S. Oxidative stress in the regulation of normal and neoplastic hematopoiesis. Antioxid Redox Signal. 2008;10:1923–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Shwachman H, Diamond LK, Oski FA, Khaw K‐T. The syndrome of pancreatic insufficiency and bone marrow dysfunction. J Pediatr. 1964;65:645–663. [DOI] [PubMed] [Google Scholar]
- 169. Boussaid I, Le Goff S, Floquet C, Gautier E‐F, Raimbault A, Viailly P‐J, et al. Integrated analyses of translatome and proteome identify the rules of translation selectivity in RPS14‐deficient cells. Haematologica. 2020;106:746–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Boocock GRB, Morrison JA, Popovic M, Richards N, Ellis L, Durie PR, et al. Mutations in SBDS are associated with Shwachman‐Diamond syndrome. Nat Genet. 2002;33:97–101. [DOI] [PubMed] [Google Scholar]
- 171. Ganapathi KA, Austin KM, Lee C‐S, Dias A, Malsch MM, Reed R, et al. The human shwachman‐diamond syndrome protein, SBDS, associates with ribosomal RNA. Blood. 2007;110:1458–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Gallagher PG. Anemia in the pediatric patient. Blood. 2022;140:571–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Zambetti NA, Bindels EM, Van Strien PM, Valkhof MG, Adisty MN, Hoogenboezem RM, et al. Deficiency of the ribosome biogenesis gene Sbds in hematopoietic stem and progenitor cells causes neutropenia in mice by attenuating lineage progression in myelocytes. Haematologica. 2015;100:1285–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Bush TS, St Coeur M, Resendes KK, Rosmarin AG. Ga‐binding protein (GABP) and SP1 are required, along with retinoid receptors, to mediate retinoic acid responsiveness of CD18 (Β2 leukocyte integrin): a novel mechanism of transcriptional regulation in myeloid cells. Blood. 2003;101:311–317. [DOI] [PubMed] [Google Scholar]
- 175. Walkley CR, Purton LE, Snelling HJ, Yuan Y‐D, Nakajima H, Chambon P, et al. Identification of the molecular requirements for an RARΑ‐mediated cell cycle arrest during granulocytic differentiation. Blood. 2004;103:1286–1295. [DOI] [PubMed] [Google Scholar]
- 176. Tschuck J, Nair VP, Galhoz A, Ciceri G, Rothenaigner I, Tchieu J, et al. Suppression of ferroptosis by vitamin A or antioxidants is essential for neuronal development. bioRxiv. 2023:2023.04.05.535746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Sen S, Wang H, Nghiem CL, Zhou K, Yau J, Tailor CS, et al. The ribosome‐related protein, SBDS, is critical for normal erythropoiesis. Blood. 2011;118:6407–6417. [DOI] [PubMed] [Google Scholar]
- 178. Cheng Y, Song Y, Chen H, Li Q, Gao Y, Lu G, et al. Ferroptosis mediated by lipid reactive oxygen species: a possible causal link of neuroinflammation to neurological disorders. Oxid Med Cell Longev. 2021;2021:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Park MW, Cha HW, Kim J, Kim JH, Yang H, Yoon S, et al. NOX4 promotes ferroptosis of astrocytes by oxidative stress‐induced lipid peroxidation via the impairment of mitochondrial metabolism in Alzheimer's diseases. Redox Biol. 2021;41:101947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. De Backer J, Maric D, Zuhra K, Bogaerts A, Szabo C, Vanden Berghe W, et al. Cytoglobin silencing promotes melanoma malignancy but sensitizes for ferroptosis and pyroptosis therapy response. Antioxidants. 2022;11:1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Kanprasoet W, Jensen LT, Sriprach S, Thitiananpakorn K, Rattanapornsompong K, Jensen AN. Deletion of mitochondrial porin alleviates stress sensitivity in the yeast model of Shwachman‐Diamond syndrome. J Genet Genomics. 2015;42:671–684. [DOI] [PubMed] [Google Scholar]
- 182. Ambekar C, Das B, Yeger H, Dror Y. SBDS‐deficiency results in deregulation of reactive oxygen species leading to increased cell death and decreased cell growth. Pediatr Blood Cancer. 2010;55:1138–1144. [DOI] [PubMed] [Google Scholar]
- 183. Herbison CE, Thorstensen K, Chua AC, Graham RM, Leedman P, Olynyk JK, et al. The role of transferrin receptor 1 and 2 in transferrin‐bound iron uptake in human hepatoma cells. Am J Physiol Cell Physiol. 2009;297:C1567–C1575. [DOI] [PubMed] [Google Scholar]
- 184. Jain A, Nilatawong P, Mamak N, Jensen LT, Jensen AN. Disruption in iron homeostasis and impaired activity of iron‐sulfur cluster containing proteins in the yeast model of Shwachman‐Diamond syndrome. Cell Biosci. 2020;10:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Zinsser F. Atrophia cutis reticularis cum pigmentations, dystrophia unguium et leukoplakis oris (Poikioodermia atrophicans vascularis Jacobi). Ikonographia Dermatol. 1910;5:219–223. [Google Scholar]
- 186. Tollervey D, Kiss T. Function and synthesis of small nucleolar RNAS. Curr Opin Cell Biol. 1997;9:337–342. [DOI] [PubMed] [Google Scholar]
- 187. Filipowicz W, Pogačić V. Biogenesis of small nucleolar ribonucleoproteins. Curr Opin Cell Biol. 2002;14:319–327. [DOI] [PubMed] [Google Scholar]
- 188. Yoon A, Peng G, Brandenburg Y, Zollo O, Xu W, Rego E, et al. Impaired control of IRES‐mediated translation in X‐linked dyskeratosis congenita. Science. 2006;312:902–906. [DOI] [PubMed] [Google Scholar]
- 189. Richards LA, Kumari A, Knezevic K, Thoms JA, von Jonquieres G, Napier CE, et al. dkc1 is a transcriptional target of GATA1 and drives upregulation of telomerase activity in normal human erythroblasts. Haematologica. 2019;105:1517–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Pereboom TC, van Weele LJ, Bondt A, MacInnes AW. A zebrafish model of dyskeratosis congenita reveals hematopoietic stem cell formation failure resulting from ribosomal protein‐mediated p53 stabilization. Blood. 2011;118:5458–5465. [DOI] [PubMed] [Google Scholar]
- 191. Carrascoso‐Rubio C, Zittersteijn HA, Pintado‐Berninches L, Fernández‐Varas B, Lozano ML, Manguan‐Garcia C, et al. Generation of dyskeratosis congenita‐like hematopoietic stem cells through the stable inhibition of DKC1. Stem Cell Res Ther. 2021;12:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Zhou N, Liu Z, Huang X, Yu H, Huang Z, Chen Y, et al. Suppression of the telomerase RNA component induces autophagy and ferroptosis in lung cancer via regulation of AMP‐activated protein kinase. Durham, North Carolina: Research Square; 2022. [Google Scholar]
- 193. Goldman F. DNA damage and oxidative stress in dyskeratosis congenita: analysis of pathways and therapeutic strategies using CRISPR and iPSC model systems. Available from: https://apps.dtic.mil/sti/pdfs/AD1093071.pdf
- 194. Wang S, Chen J, Li P, Chen Y. Linc01133 can induce acquired ferroptosis resistance by enhancing the FSP1 mrna stability through forming the LINC01133‐fus‐FSP1 complex. Cell Death Dis. 2023;14:767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Westin E, Pereboeva L, Devadasan D, Townes TM, Goldman FD. Suppression of antioxidant responses in dyskeratosis congenita cells. Blood. 2015;126:2412. [Google Scholar]
- 196. Ibáñez‐Cabellos JS, Seco‐Cervera M, Picher‐Latorre C, Pérez‐Machado G, García‐Giménez JL, Pallardó FV. Acute depletion of telomerase components DKC1 and NOP10 induces oxidative stress and disrupts ribosomal biogenesis via NPM1 and activation of the P53 pathway. Biochim Biophys Acta (BBA) – Mol Cell Res. 2020;1867:118845. [DOI] [PubMed] [Google Scholar]
- 197. Ibáñez‐Cabellos JS, Pérez‐Machado G, Seco‐Cervera M, Berenguer‐Pascual E, García‐Giménez JL, Pallardó FV. Acute telomerase components depletion triggers oxidative stress as an early event previous to telomeric shortening. Redox Biol. 2018;14:398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Steier W, Van Voolen GA, Selmanowitz VJ. Dyskeratosis congenita: relationship to Fanconi's anemia. Blood. 1972;39:510–521. [PubMed] [Google Scholar]
- 199. Fiesco‐Roa M, García‐de Teresa B, Leal‐Anaya P, Vant Hek R, Wegman‐Ostrosky T, Frías S, et al. Fanconi anemia and dyskeratosis congenita/telomere biology disorders: two inherited bone marrow failure syndromes with genomic instability. Front Oncol. 2022;12:949435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. McKusick VA, Eldridge R, Hostetler JA, Ruangwit U, Egeland JA. Dwarfism in the Amish: II. Cartilage‐hair hypoplasia. Bull Johns Hopkins Hosp. 1965;116:285–326. [PubMed] [Google Scholar]
- 201. Welting TJ. Mutual interactions between subunits of the human RNase MRP ribonucleoprotein complex. Nucleic Acids Res. 2004;32:2138–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Vakkilainen S, Skoog T, Einarsdottir E, Middleton A, Pekkinen M, Öhman T, et al. The human long non‐coding RNA gene RMRP has pleiotropic effects and regulates cell‐cycle progression at G2. Sci Rep. 2019;9:13758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Schmitt ME, Clayton DA. Nuclear RNase MRP is required for correct processing of pre‐5.8s rrna in saccharomyces cerevisiae. Mol Cell Biol. 1993;13:7935–7941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Thiel CT, Horn D, Zabel B, Ekici AB, Salinas K, Gebhart E, et al. Severely incapacitating mutations in patients with extreme short stature identify RNA‐processing endoribonuclease RMRP as an essential cell growth regulator. Am J Hum Genet. 2005;77:795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Vakkilainen S, Taskinen M, Mäkitie O. Immunodeficiency in cartilage‐hair hypoplasia: pathogenesis, clinical course and management. Scand J Immunol. 2020;92:e12913. [DOI] [PubMed] [Google Scholar]
- 206. Kostjukovits S, Degerman S, Pekkinen M, Klemetti P, Landfors M, Roos G, et al. Decreased telomere length in children with cartilage‐hair hypoplasia. J Med Genet. 2016;54:365–370. [DOI] [PubMed] [Google Scholar]
- 207. Aubert G, Strauss KA, Lansdorp PM, Rider NL. Defects in lymphocyte telomere homeostasis contribute to cellular immune phenotype in patients with cartilage‐hair hypoplasia. J Allergy Clin Immunol. 2017;140:1120. [DOI] [PubMed] [Google Scholar]
- 208. Rosado‐Pérez J, Santiago‐Osorio E, Ortiz R, Mendoza‐Núñez VM. Tai chi diminishes oxidative stress in Mexican older adults. J Nutr Health Aging. 2012;16:642–646. [DOI] [PubMed] [Google Scholar]
- 209. de la Fuente MA, Recher M, Rider NL, Strauss KA, Morton DH, Adair M, et al. Reduced thymic output, cell cycle abnormalities, and increased apoptosis of T lymphocytes in patients with cartilage‐hair hypoplasia. J Allergy Clin Immunol. 2011;128:139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Alder JK, Hanumanthu VS, Strong MA, DeZern AE, Stanley SE, Takemoto CM, et al. Diagnostic utility of telomere length testing in a hospital‐based setting. Proc Natl Acad Sci. 2018;115:E2358–E2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Ge C, Zhang S, Mu H, Zheng S, Tan Z, Huang X, et al. Emerging mechanisms and disease implications of ferroptosis: potential applications of natural products. Front Cell Dev Biol. 2022;9:774957. 10.3389/fcell.2021.774957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Wang Y, Wu S, Li Q, Sun H, Wang H. Pharmacological inhibition of ferroptosis as a therapeutic target for neurodegenerative diseases and strokes. Adv Sci. 2023;10:e2300325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Scarpellini C, Klejborowska G, Lanthier C, Hassannia B, Vanden Berghe T, Augustyns K. Beyond ferrostatin‐1: a comprehensive review of ferroptosis inhibitors. Trends Pharmacol Sci. 2023;44:902–916. [DOI] [PubMed] [Google Scholar]
- 214. Anthonymuthu TS, Tyurina YY, Sun W‐Y, Mikulska‐Ruminska K, Shrivastava IH, Tyurin VA, et al. Resolving the paradox of ferroptotic cell death: ferrostatin‐1 binds to 15LOX/PEBP1 complex, suppresses generation of peroxidized ETE‐PE, and protects against ferroptosis. Redox Biol. 2021;38:101744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Feng Y, Madungwe NB, Aliagan AI, Tombo N, Bopassa JC. Liproxstatin‐1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem Biophys Res Commun. 2019;520:606–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Zhu R, Kang Y, Li Q, Peng K, Shi X, Yin Z, et al. Alpha‐tocopherol inhibits ferroptosis and promotes neural function recovery in rats with spinal cord injury via downregulating Alox15. Biomed Pharmacother. 2024;175:116734. [DOI] [PubMed] [Google Scholar]
- 217. Yang W, Liu X, Song C, Ji S, Yang J, Liu Y, et al. Structure‐activity relationship studies of phenothiazine derivatives as a new class of ferroptosis inhibitors together with the therapeutic effect in an ischemic stroke model. Eur J Med Chem. 2021;209:112842. [DOI] [PubMed] [Google Scholar]
- 218. Shah R, Shchepinov MS, Pratt DA. Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent Sci. 2018;4:387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Zilka O, Poon J‐F, Pratt DA. Radical‐trapping antioxidant activity of copper and nickel bis(thiosemicarbazone) complexes underlies their potency as inhibitors of ferroptotic cell death. J Am Chem Soc. 2021;143:19043–19057. [DOI] [PubMed] [Google Scholar]
- 220. Ishii T, Bannai S, Sugita Y. Mechanism of growth stimulation of L1210 cells by 2‐mercaptoethanol in vitro. Role of the mixed disulfide of 2‐mercaptoethanol and cysteine. J Biol Chem. 1981;256:12387–12392. [PubMed] [Google Scholar]
- 221. Southon A, Szostak K, Acevedo KM, Dent KA, Volitakis I, Belaidi AA, et al. Cuii(atsm) inhibits ferroptosis: implications for treatment of neurodegenerative disease. Br J Pharmacol. 2020;177:656–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Dennys CN, Roussel F, Rodrigo R, Zhang X, Sierra Delgado A, Hartlaub A, et al. CuATSM effectively ameliorates als patient astrocyte‐mediated motor neuron toxicity in human in vitro models of amyotrophic lateral sclerosis. Glia. 2022;71:350–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Van Coillie S, Van San E, Goetschalckx I, Wiernicki B, Mukhopadhyay B, Tonnus W, et al. Targeting ferroptosis protects against experimental (multi)organ dysfunction and death. Nat Commun. 2022;13:1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Van San E, Debruyne AC, Veeckmans G, Tyurina YY, Tyurin VA, Zheng H, et al. Ferroptosis contributes to multiple sclerosis and its pharmacological targeting suppresses experimental disease progression. Cell Death Differ. 2023;30:2092–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Zhang J‐J, Du J, Kong N, Zhang G‐Y, Liu M‐Z, Liu C. Mechanisms and pharmacological applications of ferroptosis: a narrative review. Ann Transl Med. 2021;9:1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Miotto G, Rossetto M, Roveri A, Venerando R, Vučković A‐M, Di Paolo ML, et al. Insight the mechanism of ferroptosis inhibition by ferrostatin‐1. Free Radic Biol Med. 2018;120:S120–S121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Matsuo T, Adachi‐Tominari K, Sano O, Kamei T, Nogami M, Ogi K, et al. Involvement of ferroptosis in human motor neuron cell death. Biochem Biophys Res Commun. 2021;566:24–29. [DOI] [PubMed] [Google Scholar]
- 228. Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12:497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Muthukumaran K, Kanwar A, Vegh C, Marginean A, Elliott A, Guilbeault N, et al. Ubisol‐Q10 (a nanomicellar water‐soluble formulation of coq10) treatment inhibits alzheimer‐type behavioral and pathological symptoms in a double transgenic mouse (TGAPESWE, PSEN1DE9) model of alzheimer's disease. J Alzheimers Dis. 2017;61:221–236. [DOI] [PubMed] [Google Scholar]
- 230. Lum JS, Brown ML, Farrawell NE, McAlary L, Ly D, Chisholm CG, et al. CUATSM improves motor function and extends survival but is not tolerated at a high dose in sod1g93a mice with a C57BL/6 background. Sci Rep. 2021;11:19392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Yang J, Shi X, Wang Y, Ma M, Liu H, Wang J, et al. Multi‐target neuroprotection of thiazolidinediones on alzheimer's disease via neuroinflammation and ferroptosis. J Alzheimers Dis. 2023;96:927–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Karuppagounder SS, Alin L, Chen Y, Brand D, Bourassa MW, Dietrich K, et al. N‐acetylcysteine targets 5 lipoxygenase‐derived, toxic lipids and can synergize with prostaglandin E2 to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice. Ann Neurol. 2018;84:854–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. U.S. National Library of Medicine Home – ClinicalTrials.gov. Available from: https://clinicaltrials.gov