

Abstract
Immune checkpoint blockade (ICB) enhances T cell responses against cancer, leading to long-term survival in a fraction of patients. CD8+ T cell differentiation in response to chronic antigen stimulation is highly complex and it remains unclear precisely which T cell differentiation states at which anatomic sites are critical for the response to ICB. We identified an intermediate-exhausted population in the white pulp of the spleen which underwent significant expansion in response to ICB and gave rise to the majority of tumor-infiltrating clonotypes. Increased systemic antigen perturbed differentiation of this population towards a most circulatory exhausted_KLR state, while a lack of cross-presented tumor-antigen blunted its differentiation in the spleen. An analogous population of exhausted_KLR CD8+ T cells in human blood samples exhibited diminished tumor-trafficking ability. Collectively, our data demonstrate the critical role of antigen density within the spleen for the differentiation and expansion of T cell clonotypes in response to ICB.
One-sentence summary:
Response to ICB depends on optimal splenic antigen levels, while increased antigen induces differentiation to circulatory T cells.
Introduction
Immune checkpoint blockade (ICB) using antagonist antibodies targeting CTLA-4 or PD-1/PD-L1 can provide durable responses against cancer for a subset of patients (1). Understanding the mechanisms of response to ICB could improve the selection of patients and the development of new therapies. Most evidence suggests that ICB enhances the functionality of tumor-reactive CD8+ T cells (2–4). An anti-tumor CD8+ T cell response is a sequential, multi-step process involving the coordination of a variety of cell types across multiple anatomic locations (5). This process initiates with the priming of naïve CD8+ T cells by dendritic cells (DCs) in the tumor-draining lymph node (TdLN) (6, 7). Tumor-reactive CD8+ T cells then undergo clonal expansion and differentiate into T cells with effector functions (8, 9), followed by migration to the tumor, where they eliminate tumor cells (10). Tumors use immune-suppressive mechanisms to avoid engagement from cytotoxic T cells (11), including chronic expression of tumor antigens, which induces exhaustion among tumor-reactive CD8+ T cells (12, 13). Exhausted CD8+ T cells express elevated levels of inhibitory checkpoint receptors that reduce T cell functionality (14). Blocking these receptors with ICB can increase T cell responsiveness to other costimulatory signals (14, 15), promote clonal expansion of functional CD8+ effector T cells (16), and enhance migration of CD8+ effector T cells to the tumor, reinvigorating the anti-tumor immune response.
Recent studies have demonstrated that T cell exhaustion represents a distinct trajectory of differentiation (17–21). An exhausted CD8+ T cell response comprises heterogenous differentiation states, including TCF-1+ “stem-like” progenitor-exhausted T cells (17–21) which exhibit an enhanced ability to respond to ICB (22, 23). TCF-1+ CD8+ T cells are critical for ICB responses, but the anatomic locations from which these ICB-responsive CD8+ T cells emerge is unclear (24–28). TdLNs and tumors have been proposed as sites that maintain ICB-responsive CD8+ T cell populations (24–28), but peripheral expansion also appears critical for generating reinvigorated tumor-infiltrating CD8+ T cells (29–31). Transcriptional analyses of LCMV-specific CD8+ T cells have highlighted additional transcriptional states associated with exhaustion, including intermediate-exhausted populations (32, 33). Nonetheless, the precise transcriptional states of T cells providing tumor control in response to ICB and the anatomic locations from which responses to ICB emerge remain unclear.
Using paired single-cell RNA and TCR sequencing, we profiled endogenous, tumor-reactive T cells isolated from tumors, TdLNs, and spleens of mice treated with ICB. We found that the spleen was a critical anatomic site for coordinating the differentiation of an intermediate-exhausted CD8+ T cell population into either a terminally-exhausted phenotype, which comprised the majority of tumor-infiltrating cells, or into an “exhausted_KLR” phenotype that was rarely found in the tumor. Increasing systemic antigen enhanced the differentiation of this splenic intermediate population towards the blood-resident exhausted_KLR phenotype, leading to reduced numbers of tumor-infiltrating T cells in untreated mice. The expansion and differentiation of splenic intermediate-exhausted T cells in response to ICB was dependent on cross-presenting DCs. This suggests that differentiation of CD8+ T cells is affected by both tissue site and antigen level. Using human data sets, we observed the exhausted_KLR transcriptional state in blood and determined that this differentiation state displayed limited infiltration into tumors, despite clonal expansion. These results demonstrate that antigen-specific restimulation in the spleen is required for the expansion of tumor-infiltrating clonotypes in response to ICB, and that increasing levels of systemic antigen perturbs the differentiation of these clonotypes to favor the exhausted_KLR state, thereby blunting tumor infiltration.
Results
Tumor-reactive CD8+ T cells accumulate in the spleen during ICB
To establish the impact of ICB on tumor growth, we inoculated mice subcutaneously with KP lung cancer cells and treated the mice with ICB (Figure 1A). Consistent with our previous report (9), ICB delayed the growth of subcutaneous KP tumors (Figure 1B). To track tumor-reactive CD8+ T cells we used a KP cell line engineered to express the model antigen SIY, which binds H2-Kb (KP.SIY). KP.SIY tumor growth was also delayed by ICB, with treated tumors displaying reduced size at day 14 post implantation (Supplemental Figure 1A–B, Figure 1C–D). At this timepoint, we observed an increase in the absolute number and frequency of SIY-reactive CD8+ T cells in the spleens of mice following ICB (Figure 1E–G, Supplemental Figure 1C). The specificity of the SIY-reactive tetramer was validated, and tetramer positive cells were CD44+ (Supplemental Figure 1D–G). To assess the generalizability of our findings, we also profiled SIY-reactive CD8+ T cells isolated from TdLNs, spleens, and tumors from two additional tumor models, MC38.SIY and LL/2.SIY. Similar to the KP.SIY model, nearly all SIY-reactive T cells were CD44+ (Supplemental Figure 2A–B), and we observed a strong accumulation of SIY-reactive T cells in the spleen following ICB (Supplemental Figure 2C–H).
Figure 1: Tumor-reactive CD8+ T Cells accumulate in the white pulp of the spleens of immune checkpoint blockade-treated mice.

A) Immune checkpoint blockade (ICB) treatment scheme of mice bearing subcutaneous KP tumors. B) KP flank tumor outgrowth, n=6, P-values calculated using 2-way ANOVA. C) ICB treatment scheme of mice bearing subcutaneous KP.SIY tumors. D) Weight of day 14 KP.SIY tumors, n=9, P-values calculated with a Mann-Whitney U test. E) Representative staining of SIY-reactive CD8+ T cells in TdLN, spleen, and tumor on day 14. F) Number of SIY-reactive CD8+ T cells in TdLN, spleen (white pulp), and tumor on day 14. Fold changes calculated using the median value from each condition, n=10, P-values calculated with one-way ANOVA. G) Percent of CD8+ T cells that are SIY-reactive in TdLN, spleen (white pulp), and tumor. Fold changes calculated using the median value from each condition, n=10, P-values calculated with a Mann-Whitney U test. H-I) Representative staining (H) and quantification (I) of the percentage of SIY-reactive CD8+ T cells in the white pulp (CD45-IV− fraction) of the spleen, n=6. P-values calculated with a Mann-Whitney U test.
We observed a similar expansion profile between splenic and blood-derived T cells (Supplemental Figure 2I, Figure 1G), which could reflect expanded splenic T cells reentering circulation or active expansion in the blood. To consider these possibilities, we distinguished CD8+ T cells in the white pulp and red pulp of the spleen by intravenously (IV) administered fluorescently-labeled anti-CD45 antibody 3 minutes before euthanasia (Supplemental Figure 2J–K). We observed a much greater expansion of SIY-reactive CD8+ T cells following ICB in the white pulp (CD45-IV−) (8.8x), compared to a more modest expansion in the red pulp (CD45-IV+) (4.4x) (Supplemental Figure 2L). Moreover, we found that 73.2% of splenic SIY-reactive CD8+ T cells in control mice were located in the white pulp, and this fraction increased to 82.6% following ICB treatment (Figure 1H–I). This observation suggests that SIY-reactive CD8+ T cells preferentially expand in the white pulp of the spleen.
Transcriptional states present among tumor-reactive CD8+ T cells comprise the full spectrum of T cell exhaustion
To determine the phenotypic and clonotypic features underlying the distinct expansion potential of SIY-reactive T cells in the white pulp of the spleen, we performed single-cell RNA and TCR sequencing of endogenous SIY-reactive T cells from the tumor, TdLN, and white pulp of five untreated and five ICB-treated KP.SIY tumor-bearing mice (Figure 2A) (34). After exclusion of naïve-like and low-quality single cells (Supplemental Figure 3A–F), we performed uniform manifold approximation and projection (UMAP) for dimensionality reduction and identified 10 distinct transcriptional states using unsupervised Louvain clustering (Figure 2B–C).
Figure 2: Single-cell, multi-tissue transcriptional atlas of SIY-reactive CD8+ T cells.
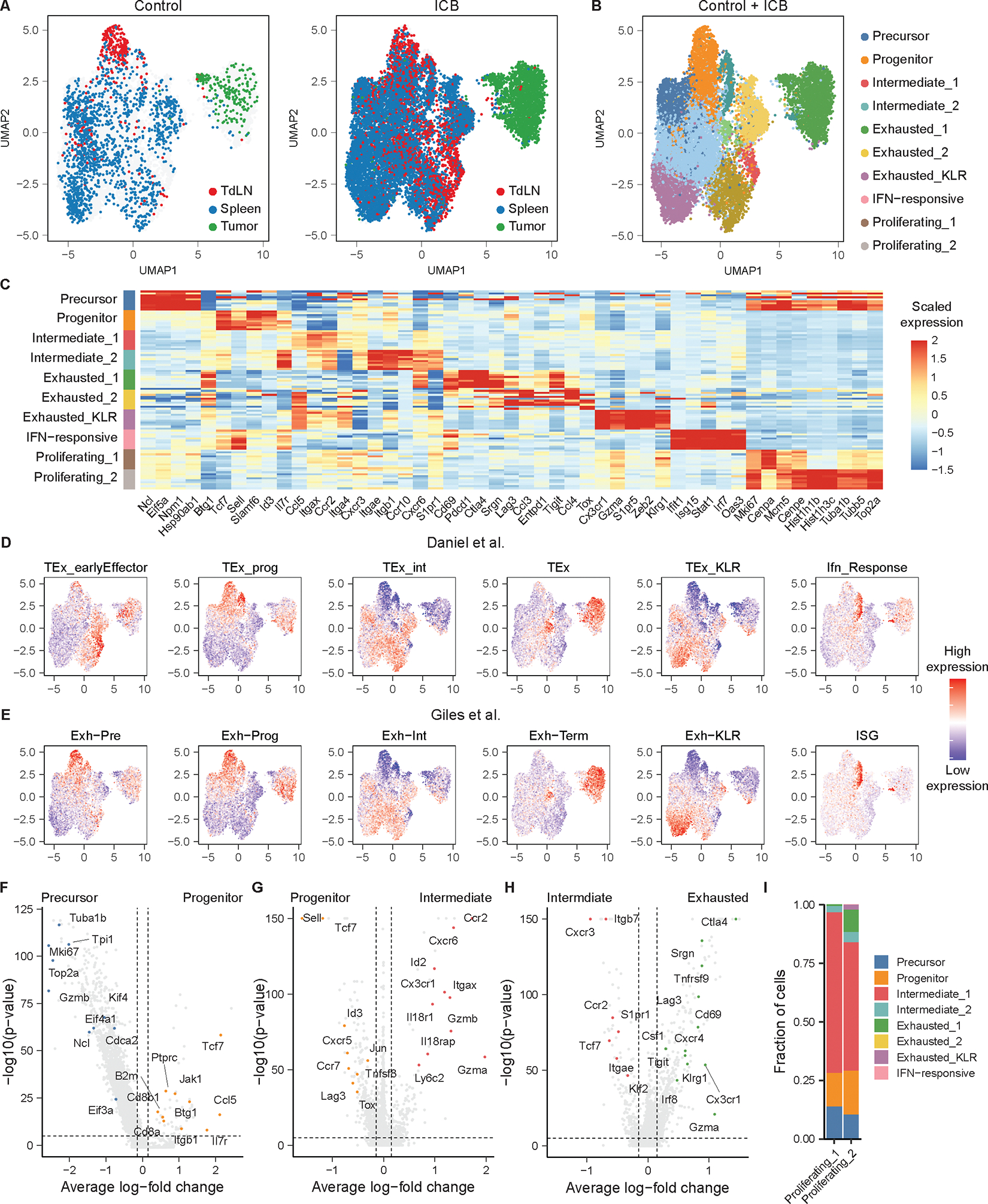
A) UMAP of SIY-reactive CD8+ T cells from untreated or ICB-treated KP tumor-bearing mice recovered colored by tissue (n = 5 untreated mice (hashed and pooled), n = 5 ICB-treated mice (hashed and pooled)). B) UMAP of SIY-reactive CD8+ T cells colored by phenotype. C) Heat map of scaled gene expression of marker genes in SIY-reactive CD8+ T cell phenotypes. Each row represents cells from one phenotype recovered from an individual mouse. D) UMAP of gene expression signatures defined among splenic gp33-reactive CD8+ T cells recovered from day 21 of clone 13 LCMV infection by Daniel et al. (33) E) UMAP of gene expression signatures defined among splenic gp33-reactive CD8+ T cells recovered from days 15 and 30 of clone 13 LCMV infection by Giles et al. (32, 61) F) Volcano plot of transcripts differentially expressed between precursor-exhausted T cells and progenitor-exhausted T cells. G) Volcano plot of transcripts differentially expressed between intermediate-exhausted T cells (Intermediate_1 and Intermediate_2) and progenitor-exhausted T cells. H) Volcano plot of transcripts differentially expressed between terminally-exhausted T cells (Exhausted_1, Exhausted_2, and Exhausted_KLR) and intermediate-exhausted T cells (Intermediate_1, Intermediate_2). I) Label transfer of non-proliferating T cell phenotypes onto proliferating phenotypes. P-values for volcano plots are calculated using a two-sided Wilcoxon rank-sum test and are adjusted using Bonferroni correction. Genes with an average log-fold change > 0.25 and an adjusted p-value < 1e-5 are considered significant. Genes of interest were manually curated and highlighted.
To identify how these transcriptional states related to the trajectory of T cell exhaustion, we generated gene expression signatures using two single-cell atlases of CD8+ T cell transcriptional phenotypes in chronic LCMV infection (32, 33) and overlaid the phenotypic signatures onto our data (Figure 2D–E). In our data, we identified: one cluster of precursor-exhausted T cells, one cluster of progenitor-exhausted T cells, two clusters of intermediate-exhausted T cells, two clusters of terminally-exhausted T cells, one cluster of exhausted_KLR T cells, one cluster that upregulated transcripts associated with interferon-response, and two clusters of proliferating T cells (Figure 2B–C). Consistent with these annotations, precursor- and progenitor-exhausted T cells expressed high levels of the transcript Slamf6 (Figure 2C). Precursor-exhausted T cells were highly proliferative, upregulated transcripts associated with translation and protein synthesis, and downregulated Btg1, while progenitor-exhausted T cells expressed higher levels of the transcription factor Tcf7 (Figure 2C, 2F). Intermediate-exhausted T cells expressed elevated levels of transcripts associated with tissue homing (Ccr2, Cxcr6, Itgax) and effector function (Gzma, Gzmb) and downregulated Sell and Tcf7 (Figure 2C, 2G). Among the two states of intermediate-exhausted T cells, intermediate_2 T cells upregulated transcripts such as Itgae and Ccr10, suggesting that they might comprise precursors of tissue-resident memory T cells (35, 36). Conversely, the intermediate_1 T cells upregulated transcripts such Itga4, Gzmk, and Eomes, consistent with tissue-homing effector T cells (Supplemental Figure 3G). Terminally-exhausted T cells upregulated transcripts such as Lag3, Tnfrsf9, and Cd69 (Figure 2C, 2H). Among terminally-exhausted T cells, exhausted_1 T cells expressed transcripts associated with cytokine signaling (Ifngr1, Il12rb2), while exhausted_KLR T cells upregulated transcripts encoding killer cell lectin-like receptors (KLRs) (Klrg1, Klrk1) as well as Cx3cr1 (Supplemental Figure 3H–J). Lastly, interferon-responsive T cells upregulated a variety of transcripts associated with interferon sensing (Supplemental Figure 3K).
To annotate the remaining two states of proliferating T cells, we performed label transfer of cluster identities from nonproliferating to proliferating T cells (Figure 2I). These results demonstrated that most proliferating T cells had transcriptional states consistent with either a Cxcr3+ intermediate_1 phenotype or a Slamf6+ progenitor-exhausted phenotype. In sum, these results demonstrate the transcriptional phenotypes present in this tumor model recapitulate the full spectrum of transcriptional phenotypes that have been observed in models of chronic viral infection in mice (32, 33).
States of T cell exhaustion vary in frequency between the tumor, lymph node, and spleen
We next examined the frequency of each transcriptional phenotype among the recovered SIY-reactive CD8+ T cells from the tumor, TdLN, and white pulp (Figure 3A, Supplemental Figure 4A). In control and ICB-treated mice, the TdLN was enriched in transcriptional states associated with early stages of T cell exhaustion, including precursor T cells and progenitor T cells. In contrast, the white pulp contained elevated frequencies of intermediate-exhausted T cell states, and the tumor comprised T cells with terminally-exhausted phenotypes. These data suggest that the TdLN, spleen and tumor are populated by increasingly differentiated phenotypes of tumor-reactive CD8+ T cells, respectively, highlighting a spatially-resolved trajectory of T cell differentiation.
Figure 3: Distribution of transcriptional states between TdLN, white pulp, and tumor.

A) Frequency of exhausted states in each tissue. B) Frequency of transcriptional states associated with exhaustion in control and ICB-treated mice. P-values are calculated using a two-sided Wilcoxon rank-sum test and are adjusted using Bonferroni correction. C) Heat map of normalized Cxcr3 and Cx3cr1 expression by progenitor, intermediate_1, and exhausted_KLR T cells. Each box is the average expression in one mouse. D) Representative flow cytometry plots of CXCR3 and CX3CR1 expression by SIY-reactive T cells in TdLN, white pulp (spleen), and tumor. E) Quantification of CXCR3+CX3CR1+ SIY-reactive CD8+ T cells in TdLN, white pulp (spleen), and tumor. F) Frequency of TCF-1 expression by CXCR3+CX3CR1+ SIY-reactive CD8+ T cells in the TdLN and spleen. G) Frequency of CXCR3+CX3CR1+ cells among TCF-1+ SIY-reactive CD8+ T cells. For E-G, n=6, P-values are calculated with one-way ANOVA.
We also examined the impact of ICB treatment on the frequency of specific transcriptional states across all three tissues (Figure 3B). ICB resulted in the expansion of intermediate_1 T cells, but not intermediate_2 T cells, in both the spleen and TdLN. Regardless of ICB treatment, T cells in tumors remained almost entirely within the exhausted_1 phenotype, while exhausted_KLR T cells were found predominantly in the white pulp and were largely absent from any other anatomic locations.
We established a lymphoid tissue-specific gating strategy for flow cytometry to distinguish between progenitor, intermediate_1, and exhausted_KLR T cells, which comprised the majority of SIY-reactive CD8+ T cells in the white pulp and TdLN. Cxcr3 and Cx3cr1 transcripts were differentially expressed among progenitor, intermediate_1, and exhausted_KLR T cell populations (Supplemental Figure 4B–C) (Figure 3C). Using flow cytometry, we confirmed the presence of a CXCR3+CX3CR1+ population, consistent with an intermediate_1 phenotype, and a CXCR3−CX3CR1+ population, consistent with an exhausted_KLR phenotype, in the white pulp of tumor-bearing mice (Figure 3D). To validate that these protein markers identify T cells with intermediate_1 and exhausted_KLR transcriptional phenotypes, we performed bulk RNA-sequencing of sorted splenic CXCR3+CX3CR1+ and CXCR3−CX3CR1+ SIY-reactive T cells. We observed a high degree of concordance among transcripts differentially expressed by these sorted populations and the intermediate_1 and exhausted_KLR populations in our single-cell data (Supplemental Figure 4D–F), suggesting that CXCR3 and CX3CR1 can be used to distinguish between these two transcriptional phenotypes.
We next assessed the frequency of CXCR3+CX3CR1− progenitor T cells, CXCR3+CX3CR1+ intermediate_1 T cells, and CXCR3−CX3CR1+ exhausted_KLR T cells in control and ICB-treated mice. Consistent with our scRNA-seq results, we observed an increase in the frequency and number of CXCR3+CX3CR1+ intermediate_1 T cells in both the TdLN and white pulp following ICB treatment (Figure 3E, Supplemental Figure 5A–B). Notably, the frequency of CXCR3+CX3CR1+ intermediate_1 T cells was highest in the white pulp, both in pre-treatment and post-ICB conditions (Figure 3E). These results were validated in MC38.SIY and LL/2.SIY tumor models (Supplemental Figure 5C). Analysis of T cells in the white pulp or the blood-accessible red pulp revealed that the expansion of intermediate_1 CXCR3+CX3CR1+ T cells occurred predominantly within the white pulp (Supplemental Figure 5D–F). This was further supported by the fact that the frequency of intermediate_1 T cells did not increase in the blood following ICB (Supplemental Figure 5E). Together, these data identify the white pulp as a critical anatomical site for the expansion of intermediate_1 CD8+ T cells upon ICB.
Several reports have demonstrated the importance of TCF-1 expression for CD8+ T cells to respond to ICB (2, 19, 22–24, 27). We examined the expression of TCF-1 among progenitor CXCR3+CX3CR1−, intermediate_1 CXCR3+CX3CR1+, and exhausted_KLR CXCR3−CX3CR1+ T cell populations. Progenitor T cells expressed the highest levels of TCF-1 in both the TdLN and white pulp (Supplemental Figure 6A–B). In the white pulp, progressive differentiation into intermediate_1 and exhausted_KLR T cells correlated with a gradual decrease in TCF-1 expression (Supplemental Figure 6A–B). Consistent with previous reports (19, 22, 23), the frequency of TCF-1+ T cells among all three of these T cell subsets decreased in response to ICB, suggesting enhanced T cell differentiation in response to ICB (Figure 3F, Supplemental Figure 6A–B). Additionally, in both the TdLN and white pulp, progenitor T cells comprised the majority of the TCF-1+ SIY-reactive T cells (Supplemental Figure 6C–D). Notably, the frequency of intermediate_1 T cells among TCF-1+ SIY-reactive T cells remained unchanged in the TdLN following ICB (Supplemental Figure 6C–D). In contrast, in the white pulp, the fraction of intermediate_1 T cells among TCF-1+ expressing SIY-reactive T cells increased following ICB, supporting that the expansion of this population constitutes a critical component of the T cell response elicited by ICB (Figure 3G, Supplemental 6C–D).
Additionally, we assessed the expression of GZMB, PD-1, and TIM-3 across these T cell populations. Intermediate_1 CXCR3+CX3CR1+ T cells expressed the highest level of GZMB, suggesting that these cells exhibit enhanced cytotoxic capacity (Supplemental Figure 7A–B). In the TdLN, progenitor CXCR3+CX3CR1− and intermediate_ 1 CXCR3+CX3CR1+ T cells displayed similar frequencies of TIM-3 and PD-1 expression (Supplemental Figure 7C–F). However, in the white pulp, the frequency of TIM-3 expression was highest among intermediate_1 CXCR3+CX3CR1+ T cells, while the frequency of PD-1 expression was reduced on exhausted_KLR CXCR3−CX3CR1+ T cells (Supplemental Figure 7C–F). Together, these results indicate that the frequency of distinct transcriptional states associated with the progression of T cell exhaustion varies between the tumor, TdLN, and spleen. Moreover, treatment with ICB selectively expands CD8+ T cells with an intermediate_1 phenotype that express elevated levels of effector molecules in both the white pulp and TdLN.
CXCR3+CX3CR1+ intermediate-exhausted T cells give rise to clonally distinct exhausted_1 or exhausted_KLR T cell phenotypes
To gain insights into the relationships among the expanded T cell clusters, we next examined the TCR repertoire of SIY-reactive T cells (Figure 4A). In untreated mice, we observed the largest levels of clonal expansion in the white pulp, while in mice treated with ICB, the magnitudes of clonal expansion observed were similar between the tumor, TdLN, and white pulp (Figure 4B). Consistent with this observation, TCR repertoire diversity decreased following ICB treatment, indicating that ICB promotes strong expansion of a subset of tumor-reactive clonotypes (Figure 4C). We also observed a greater clonal richness (i.e. total number of SIY-reactive clonotypes), demonstrating that ICB treatment simultaneously enables maintenance of a larger repertoire of tumor-reactive T cells (Figure 4D).
Figure 4: Clonal dynamics of SIY-reactive CD8+ T cells.

A) UMAP of SIY-reactive CD8+ T cells colored by clonal size. B) Stacked bar plots of clonal sizes among TdLN, white pulp (spleen), and tumor, of control and ICB-treated mice. Clonal sizes are computed separately in each tissue. C) Shannon diversity of TCR repertoire. D) Clonal richness of TCR repertoire. For C-D, n=5, P-values calculated using a two-sided Wilcoxon rank-sum test and are adjusted using Bonferroni correction. E) Heat map of phenotypes present within individual TCR clonotypes. A random sample of the top 150 most expanded clones is shown. Phenotypes present only once within a single clonotype are not shown. F) Transition matrix of transcriptional states. Boxes are shaded according to the geometric mean of normalized clonal frequencies between pairs of transcriptional states. G) Proposed model of T cell differentiation informed by clonal trajectories.
We next sought to define a hierarchy underlying the path of differentiation of tumor-reactive clonotypes. We considered a clonotype “representative” of a phenotype if at least two T cells from that clonotype were found in any given phenotype. Based on this definition, we constructed a heat map of the representative phenotypes present within individual TCR clonotypes (Figure 4E). We then computed a transition matrix using the geometric mean overlap of clonotypes between each pair of phenotypes (Figure 4F). We found that the repertoire of progenitor clonotypes shared substantial overlap with the repertoire of precursor clonotypes, consistent with precursor T cells undergoing differentiation to progenitor T cells. Likewise, intermediate_1 clonotypes exhibited the greatest clonal overlap with progenitor clonotypes, suggesting that intermediate_1 T cells differentiate from progenitor T cells. Both the exhausted_1 and exhausted_KLR clonotypes demonstrated the greatest clonal overlap to intermediate_1 clonotypes, suggesting that the majority of exhausted_1 and exhausted_KLR T cells differentiate from intermediate_1 T cells. Strikingly, the level of clonal overlap between exhausted_1 and exhausted_KLR phenotypes was minimal, suggesting that they represent distinct, non-interconvertible T cell states. Consistent with this analysis, upset plots depicting the patterns of phenotypic variation within clonotypes demonstrated that the most common pairs of phenotypes identified within a single clonotype were (progenitor, intermediate_1), (intermediate_1, exhausted_1), (progenitor, intermediate_2), (intermediate_1, exhausted_KLR), and (intermediate_1, intermediate_2) (Supplemental Figure 8A). Thus, we developed a model in which precursor T cells differentiate into progenitor T cells that in turn give rise to intermediate_1 and intermediate_2 T cells. Intermediate_1 T cells can bifurcate to either exhausted_1 T cells or exhausted_KLR T cells (Figure 4G). This model demonstrates high levels of concordance with recent studies of chronic viral infection of mice (32, 33), which include an intermediate phenotype preceding divergent exhausted phenotypes.
Tumor-specific T cell differentiation is associated with tissue-site trafficking
We next assessed the relationship between the differentiation of individual clonotypes and their tissue location. We found a robust correlation between the frequencies of individual clonotypes among the tumor, TdLN, and spleen, indicating that these tissues are populated by a common pool of SIY-reactive, tumor-specific clonotypes (Figure 5A). Using the constructed heatmap (Figure 4E), we identified the two most common phenotypes encountered within each clonotype, allowing us to classify clonotypes as either belonging to only a single phenotype or transitioning from one phenotype to another (Figure 4E). Clonotypes in which we could not unambiguously assign to one or two dominant phenotypes were excluded from this analysis. Comparing the clonal sizes and phenotypes present at each tissue site within transitioning clonotypes, we found that clonotypes differentiating from the progenitor state to intermediate_1 were strongly polarized towards the progenitor phenotype in the TdLN but were polarized towards the intermediate_1 phenotype in the white pulp, suggesting that transition between these two transcriptional states is strongly associated with the migration of T cells from the TdLN to the spleen (Figure 5B, Supplemental Figure 8B). Likewise, the transition between intermediate_1 and exhausted_1 phenotypes was strongly associated with trafficking from the white pulp to the tumor (Figure 5C). In contrast, while a small number of cells from clonotypes undergoing differentiation from intermediate_1 to exhausted_KLR states demonstrated an intermediate_1 phenotype in the TdLN, the majority of these clonotypes were absent from both the TdLN and the tumor but present in the white pulp, indicating that these transitions primarily occur within the spleen (Figure 5D, Supplemental Figure 8C). Overall, these results demonstrate that clonally related T cells possess distinct phenotypes in different tissues and that select phenotypic transitions, including progenitor to intermediate_1 and intermediate_1 to exhausted_1, are strongly associated with trafficking from one tissue to another, while others, such as intermediate_1 to exhausted_KLR, are not accompanied with a change in tissue site and instead take place primarily among cells resident in the spleen.
Figure 5. Clonal differentiation is accompanied by change in anatomic site.

A) Correlation between clonotype frequencies in the tumor, TdLN and white pulp (spleen). B) Tissue-site distribution of progenitor and intermediate_1 phenotypes and clonal sizes in progenitor->intermediate_1 clones. C) Tissue-site distribution of intermediate_1 and exhasuted_1 phenotypes and clonal sizes in intermediate_1 -> exhausted_1 clones. D) Tissue-site distribution of intermediate_1 and exhausted_KLR phenotypes and clonal sizes in intermediate_1 -> exhausted_KLR clones. For A-D, P-values are calculated with a paired, two-sided Wilcoxon rank-sum test and are adjusted with Bonferroni correction. E) Stacked bar chart of clonal behaviors assigned to single cells in control and ICB-treated mice. Only clonotypes consisting of more than two cells are shown. F) Absolute clonal sizes of transitioning clonotypes in control and ICB-treated mice. P-values are calculated with a two-sided Wilcoxon rank-sum test and are adjusted with Bonferroni correction. G) Clonal richness of transcriptional phenotypes in control and ICB-treated mice. P-values are calculated with a paired, two-sided Wilcoxon rank-sum test and are adjusted with Bonferroni correction.
The intermediate_1 to exhausted_1 transition limits clonal differentiation and is overcome by immune checkpoint blockade
To determine how ICB modulates the phenotypic transitions experienced by SIY-reactive clonotypes, we examined the distribution of clonal behaviors present in control and ICB-treated mice. Although the frequency of clonotypes classified as transitioning between intermediate_1 and exhausted_1 was similar (Supplemental Figure 8D), the fraction of T cells belonging to an intermediate_1 → exhausted_1 transitioning clonotype was substantially greater in mice treated with ICB (Figure 5E). We then calculated the absolute clonal sizes of individual clonotypes in control and ICB-treated mice undergoing the clonal transitions analyzed above. We also found that the clonal sizes of intermediate_1 → exhausted_1 clonotypes, but no other transitioning clonotypes, were significantly larger in mice treated with ICB, suggesting that ICB preferentially expands intermediate_1 clonotypes that are primed to undergo an intermediate_1 to exhausted_1 transition (Figure 5F).
We also computed the clonal richness of each phenotype encountered on our hypothesized differentiation trajectory (Figure 5G). We found that clonal richness peaked at the progenitor and intermediate_1 phenotype, suggesting that precursor T cells, the apparent direct precursors of progenitor T cells, are largely depleted by day 14 following tumor inoculation. In addition, this result demonstrates that there is an accumulation of clonotypes at the progenitor and intermediate_1 phenotype. These data suggests that while the transition from progenitor to intermediate_1 phenotype is efficient, occurring with high probability, the transition from intermediate_1 to an exhausted_1 phenotype is inefficient, and that the intermediate_1 phenotype is a rate-limiting step encountered during clonal differentiation. Thus, we concluded that one of the major results of ICB treatment is the expansion of splenic intermediate_1 clonotypes that are predisposed to undergo the intermediate_1 to exhausted_1 transition.
Splenic intermediate_1 T cells drive the expansion of tumor-reactive CD8+ T cells during immune checkpoint blockade
To confirm that the ability of tumor-reactive CD8+ T cells to expand in response to ICB was different between T cells from the TdLN and white pulp, we performed an adoptive transfer of TCR-transgenic in vivo activated 2C T cells into KP.SIY tumor-bearing mice (Figure 6A). In this system, expanded 2C T cells appear with comparable frequency and number in both the TdLNs and white pulp of mice within 72 hours after transfer (Figure 6B–C, Supplemental Figure 9A–B) and transcriptionally mirror endogenous SIY-reactive T cells (8, 9). Cell trace violet (CTV) dilution studies of labeled 2C T cells 72 hours post-transfer showed that 2C T cells undergo 4–6 cell divisions in the TdLN, while they undergo 8 or more divisions in the white pulp (Figure 6D–E), confirming that priming takes place in the TdLN, followed by splenic trafficking.
Figure 6: Splenic tumor-reactive CD8+ T cells drive the response to ICB.

A) Adoptive transfer experimental design. B-C) Frequency (B) and number (C) of 2C T cells in the TdLN and spleen 3 days after transfer into KP.SIY flank tumor-bearing mice, n = 10 (B) and n = 5 (C). P-values calculated with a Mann-Whitney U test. D) CTV dilution of 2C T cells in the TdLN and white pulp (spleen)3 days after transfer into KP.SIY flank tumor-bearing mice. E) Quantification of the fraction of 2C T cells in each cell division, n=4, P-values calculated with one-way ANOVA. F) Phenotype of 2C T cells 3 days after transfer to KP.SIY flank tumor-bearing mice. G) Quantification of the phenotype of 2C T cells 3 days after transfer to KP.SIY flank tumor-bearing mice, n = 10. P-value calculated with a Mann-Whitney U test. H) Experimental design of 2C T cell transfer into secondary recipients. I) Accumulation of transferred 2C T cells in secondary recipients. TdLN Control n = 13, TdLN ICB n=13, Spleen control n=14, Spleen ICB n=15. P-values calculated with one-way ANOVA.
We next investigated the phenotypes of transferred 2C T cells in TdLNs and white pulps of recipient mice. Consistent with our previous data, more 2C T cells in the white pulp displayed an intermediate_1 CXCR3+CX3CR1+ phenotype compared to 2C T cells in the TdLN (Figure 6F–G, Supplemental Figure 9C). This observation supports that differentiation from a progenitor to intermediate_1 T cell state is associated with trafficking to the spleen. Further, progenitor CXCR3+CX3CR1− and intermediate_1 CXCR3+CX3CR1+ T cells in the white pulp expressed similar levels of PD-1, while intermediate_1 T cells exhibited reduced SLAMF6 expression, a proxy for TCF-1(19), aligning with our prior findings on endogenous SIY-reactive CD8+ T cells (Supplemental Figures 9D–E, 6B, 7F). Overall, these data support a model in which transferred 2C T cells initially proliferate in the TdLN before trafficking to the white pulp of the spleen, where they undergo additional cycles of proliferation and acquire a more differentiated CXCR3+CX3CR1+ intermediate_1 T cell phenotype.
We next used a serial adoptive transfer approach to determine whether splenic 2C T cells exhibited enhanced responsiveness to ICB. We sorted 2C T cells from TdLNs and white pulp of primary recipient mice and transferred the sorted 2C T cells to secondary recipients with time-matched KP.SIY tumors (Figure 6H). Strikingly, only 2C T cells sorted from the white pulp of mice significantly expanded in response to ICB (Figure 6I, Supplemental Figure 10A–B). Further, GZMB expression by 2C T cells in the tumor was upregulated following ICB treatment (Supplemental Figure 10C). These data indicate that splenic T cells, which are enriched for the intermediate_1 phenotype, are the primary responders to ICB and can recirculate to both TdLNs and tumors.
Antigen density regulates intermediate_1 T cell differentiation and trafficking
We sought to further understand the regulators of tumor-reactive CD8+ T cell differentiation in the spleen. Chronic antigen stimulation is known to drive T cells into terminal exhaustion states (12, 13). Consistently, SIY-reactive CD8+ T cells infiltrating the tumor almost completely comprised an exhausted_1 phenotype. Progenitor-exhausted CD8+ T cells are also induced by high antigen levels in the lymph node (17), but how antigen affects differentiation of T cells into the intermediate_1 phenotype is unknown. To examine the extent of antigen trafficking to the spleen, we inoculated mice with KP cells that expressed the pH-stable fluorophore ZsGreen (KP.ZsGreen) and examined the levels of ZsGreen in CD45+ cells in TdLNs and spleens. TdLNs had a significantly higher frequency of ZsGreen+CD45+ cells compared to spleens, indicating that the spleen was a relatively antigen-low anatomic site (Supplemental Figure 11A–B). Using the 2C T cell adoptive transfer approach, we then determined whether antigen was required to drive tumor-reactive CD8+ T cell expansion in the spleen. We primed 2C T cells in vivo in primary KP.SIY tumor-bearing host mice before transferring 2C T cells isolated from the white pulp into secondary hosts bearing either KP.SIY or KP tumors. Secondary hosts were treated with ICB (Figure 7A). 2C T cells failed to accumulate in secondary hosts bearing KP tumors, indicating that antigen was required, even at low levels, for 2C T cell expansion in response to ICB (Figure 7B, Supplemental Figure 11C–D).
Figure 7: Antigen levels and cross-presentation by DC1 impact SIY-reactive T cell differentiation in the spleen and subsequent trafficking to the tumor.

A) Experimental scheme of 2C T cell adoptive transfer into KP.SIY or KP flank tumor-bearing secondary recipients. B) Numbers of recovered 2C T cells in secondary recipients with either KP.SIY or KP flank tumors, n=5 (or 4 for KP tumor). P-values calculated with Mann-Whitney U test. C) Experimental scheme of 2C T cell adoptive transfer into KP.SIY flank tumor-bearing WT or Batf3−/− secondary recipients. D) Numbers of recovered 2C T cells in KP.SIY flank tumor-bearing WT or Batf3−/− secondary recipients, n=5. P-values calculated with Mann-Whitney U test. E) Experimental Scheme of SIY-pulsed splenocyte transfer. F) Example flow cytometry plots of SIY-reactive T cell expression of CXCR3 and CX3CR1 in the spleen G) Percentage of splenic SIY-reactive CD8+ T cells that are CXCR3+CX3CR1+. H) Percentage of splenic SIY-reactive CD8+ T cells that are CXCR3−CX3CR1+. I) CXCR3 MFI of splenic SIY-reactive CXCR3+CX3CR1+ CD8+ T cells. J) Relative CX3CR1 MFI of splenic SIY-reactive CXCR3+CX3CR1+ CD8+ T cell population. K) Number of SIY-reactive CD8+ T cells per gram of KP.SIY tumor in control mice with or without SIY-pulsed splenocyte transfer. L) Number of SIY-reactive CD8+ T cells per gram of KP.SIY tumor in ICB-treated mice with or without SIY-pulsed splenocyte transfer. For G-L, n=6, P-values calculated with one-way ANOVA (G-J) and Mann-Whitney U test (K-L).
Based on these results, we aimed to determine whether T cell expansion in the spleen was dependent on DC-mediated antigen presentation. Previous work has shown that cross-presenting dendritic cells (DC1), driven by the transcription factor Batf3, are the dominant DC subset that prime CD8+ T cells in TdLN (8, 37). Further, we observed that splenic DC1 are exclusively present in the white pulp (Supplemental Figure 12A–C). Thus, we compared the expansion of adoptively transferred 2C T cells in Batf3−/− hosts to wildtype hosts following ICB therapy (Figure 7C). Consistent with prior studies, we observed poor infiltration into the tumor and a reduced expansion of 2C T cells in the TdLN of Batf3−/− hosts (Figure 7D, Supplemental Figure 12D–E) (38–40). Strikingly, we also observed a 5-fold reduction in the expansion of SIY-reactive T cells in the spleen of Batf3−/− mice (Figure 7D, Supplemental Figure 12D–E). Further, Batf3−/− mice had a reduction in the proportion of 2C T cells with an intermediate_1 CXCR3+CX3CR1+ phenotype in the white pulp, while the less differentiated progenitor CXCR3+CX3CR1− T cell state was enriched (Supplemental Figure 12F–G).
Next, we aimed to specifically address whether levels of systemic antigen affect the expansion of antigen-reactive intermediate_1 T cells in the spleen. To directly test this, we isolated splenocytes from naïve mice, pulsed them with SIY, and transferred them into KP.SIY tumor-bearing mice, which were subsequently treated with ICB (Figure 7E). This approach systemically increases cell-associated antigen and enhances cross-presentation, including in the spleen (41, 42). On day 14 post-tumor inoculation, we observed accelerated differentiation of tumor-reactive T cells, as evidenced by the decrease in progenitor CXCR3+CX3CR1− T cells in the white pulp, and to a lesser degree in the TdLN, of mice that received SIY-pulsed splenocytes (Supplemental Figure 12H–I). Elevated levels of systemic antigen increased the proportion of intermediate_1 CXCR3+CX3CR1+ T cells in untreated mice but not in ICB treated animals and abrogated the previously observed expansion of intermediate_1 T cells in mice that received ICB (Figure 7F–G). Additionally, and independently of ICB therapy, systemic antigen resulted in an increased frequency of exhausted_KLR CXCR3−CX3CR1+ T cells in the white pulp (Figure 7F–H) accompanied by lower expression of CXCR3 and higher expression of CX3CR1 on intermediate_1 T cells (Figure 7I–J). Together, this suggests that intermediate_1 T cells in the white pulp undergo accelerated differentiation to the exhausted_KLR phenotype in response to increased levels of systemic antigen. This biased T cell differentiation toward exhausted_KLR T cells in the white pulp in response to SIY-pulsed splenocytes was associated with a significant reduction of tumor-infiltrating SIY-reactive CD8+ T cells in untreated mice but not mice treated with ICB therapy (Figure 7K, 7L). Increasing systemic antigen did not influence the viability of tumor-infiltrating SIY-reactive CD8+ T cells (Supplemental Figure 12J–K). This is consistent with our previous observation that exhausted_KLR T cells reside primarily in the spleen and do not traffic to tumors (Figure 3B). The observation that ICB therapy was able rescue T cell infiltration into the tumor suggests that the remaining fraction of intermediate_1 T cells in SIY-pulsed mice can differentiate into sufficient numbers of tumor-trafficking, exhausted_1 T cells upon ICB, resulting in some degree of tumor control (Supplemental Figure 12L). In sum, these results suggest that low but detectable levels of cross-presented antigen in the white pulp are critical for the accumulation and retention of splenic T cells in the intermediate_1 state, and that perturbation of splenic intermediate_1 T cells can affect migration of T cells to the tumor.
Exhausted_KLR T cells in human patients exhibit reduced migration to tumors relative to other tumor-homing clonotypes
To assess the extent to which the transcriptional phenotypes and clonotypic trends observed among SIY-reactive T cells in our mouse model are present in human patients, we conducted a reanalysis of a pan-cancer atlas of tumor-infiltrating lymphocytes recovered from 316 cancer patients (43). We focused this analysis on a subset of the larger atlas, comprising four types of solid tumors (colorectal carcinoma, hepatocellular carcinoma, non-small cell lung cancer, and cholangiocarcinoma) for which single-cell RNA and TCR sequencing data from matched tumor tissue and peripheral blood were available. Based on transcriptional expression, the authors annotated the majority of CD8+ T cells as either: naïve (Tn), memory (Tm), resident memory (Trm), effector memory (Tem), exhausted (Tex), terminally differentiated memory or effector cells (Temra), NK-like (Tk), or ISG-positive (ISG).
We first examined the concordance between SIY-reactive transcriptional phenotypes identified in our mouse model and the transcriptional phenotypes present among tumor-infiltrating lymphocytes (TIL) from human patients. We found strong agreement between the signatures identified in our mouse model and the phenotypes annotated by Zheng et al: specifically, Tn expressed strong levels of our progenitor signature, Tm, Trm, and Tem upregulated our intermediate_1 signature, with a subset of Trm also upregulating our intermediate_2 signature, Tex upregulated our exhausted_1 and exhausted_2 signatures, and Temra upregulated our exhausted_KLR signature (Figure 8A–C, Supplemental Figure 13). Thus, the transcriptional states present in our mouse model can differentiate phenotypes of T cells present in the tumor and peripheral blood of human patients, with Temra being the analogous counterpart of the exhausted_KLR cells. Next, we analyzed T cells in the peripheral blood that were clonally related to tumor-infiltrating clonotypes. Temra cells in the peripheral blood were the only population increased in frequency among tumor-trafficking clonotypes relative to non-tumor-trafficking clonotypes, suggesting that this population in peripheral blood is enriched for tumor-specific CD8+ T cells (Figure 8D). The tumor-infiltrating clonal relatives of Temra cells from the peripheral blood were also enriched for a Temra phenotype, relative to other tumor-infiltrating clonotypes found in peripheral blood, suggesting that this phenotype is conserved upon entry into the tumor (Figure 8E).
Figure 8. Exhausted_KLR cells in human patients exhibit decreased migration from peripheral blood to tumor.

A-C) Expression of signatures for intermediate_1, exhausted_1, and exhausted_KLR phenotypes on clusters defined by Zheng et al. (43) D) Frequency of phenotypes present in peripheral blood among tumor-trafficking and non-tumor-trafficking clonotypes. P-values are calculated with a two-sided Wilcoxon rank-sum test and are adjusted with Bonferroni correction. E) Frequency of phenotypes present among tumor-infiltrating T cells related to Temra clonotypes from peripheral blood and all other tumor-infiltrating clonotypes. P-values are calculated with a two-sided Wilcoxon rank-sum test and are adjusted with Bonferroni correction. F) Frequency of phenotypes present among tumor-trafficking CD8+ T cells in blood and tumor. P-values are calculated with a two-sided Wilcoxon rank-sum test and are adjusted with Bonferroni correction. G) Ratio of clonal frequency in tissue to clonal frequency among Tm, Trm, Tem, Temra, and all other clonotypes. P-values are calculated by a Kruskal-Wallis rank-sum test followed by Dunn’s post-test. H) Frequency of endogenous CXCR3−CX3CR1+ exhausted_KLR SIY-reactive CD8+ T cells from the white pulp, red pulp, and blood of day 14 KP.SIY tumor-bearing mice, n=11, P-values calculated with one-way ANOVA.
To determine whether trafficking of T cells into the tumor was associated with any specific T cell state, we assessed the fraction of T cells within each state between tumor and peripheral tissues. Strikingly, among tumor-trafficking clonotypes, the Temra phenotype was substantially decreased in frequency in the tumor relative to peripheral blood, suggesting poor tumor infiltration (Figure 8F). In contrast, Trm, Tem, and ISG cells exhibited an increase in frequency in the tumor, suggesting that these populations undergo efficient recruitment to the tumor. To assess whether these trends in tumor trafficking potential were apparent at the level of individual clonotypes, we computed the ratio between the frequency of each clonotype in the tumor and the blood. Relative to other clonotypes in the peripheral blood, Temra clonotypes were substantially more expanded in peripheral blood compared to the tumor, indicating that individual Temra clonotypes exhibit a reduced ability to enter the tumor, relative to other phenotypes in the peripheral blood (Figure 8G). Considering this observation in human tumors, we revisited our analysis of exhausted_KLR T cells in the blood and observed an increase in frequency of exhausted_KLR T cells in the blood of both control and ICB-treated mice, with only a modest expansion in the white and red pulp of the spleen following ICB (Figure 8H). These data are consistent with the finding that the human analog of the exhausted_KLR T cell population resides primarily in the blood but not cancerous tissue (Figure 8H). Overall, these data support our model in which intermediate_1-like T cells (Tm, Trm, and Tem) undergo efficient entry into the tumor from lymphoid tissues, while exhausted_KLR-like clonotypes (Temra) exhibit a decreased potential to migrate to the tumor, especially considering their relative level of clonal expansion within peripheral blood.
Discussion
Here, we analyzed tumor-reactive CD8+ T cells from the tumor, TdLN, and white pulp of ICB-treated and untreated mice with scRNA and TCR sequencing. Our findings corroborate previous reports on the step-wise differentiation along distinct transcriptional phenotypes (32, 33) and demonstrate that these transcriptional states appear largely restricted to individual anatomic sites. We observed that the intermediate_1 T cell population is enriched in the white pulp of the spleen and gives rise to two terminally differentiated populations of CD8+ T cells: exhausted_1, which is largely restricted to the tumor, and exhausted_KLR which is enriched in the blood of mice and humans. While increasing systemic antigen promoted the differentiation of intermediate_1 to the exhausted_KLR T cell state, the absence of antigen or cross-presenting DCs reduced differentiation of intermediate_1 T cells in the white pulp, suggesting that some level of antigen on DCs is necessary to sustain T cells in the intermediate_1 phenotype. Together, our results demonstrate that microenvironmental factors present in individual anatomic sites play a role in guiding exhausted T cell differentiation and are critical in determining the outcome of ICB.
The spleen is frequently analyzed in models of chronic viral infection including LCMV, but in these models, the spleen is also a primary site of infection. In contrast, the spleen is rarely studied in the context of cancer (19, 22–24, 26, 27). We found that splenic intermediate_1 T cells exhibit the greatest expansion in response to ICB. Consistent with this result, a recent study proposed that intermediate-exhausted T cells are most expanded by anti-PD-L1 treatment in murine LCMV infection (32). We found that a subset of intermediate_1 T cells retains TCF-1 expression, consistent with previous reports identifying TCF-1+ CD8+ T cells as the mediators of T cell self-renewal and expansion in response to ICB (2, 19, 22–24, 27). Moreover, the proportion of intermediate_1 T cells within the TCF-1+ splenic SIY-reactive T cell population strongly increased after ICB, reinforcing the idea that the expansion of this subset plays a crucial role in the T cell response induced by ICB.
Intermediate_1 T cells also bear some resemblance to a population of “transitory” proliferating cells reported to express CX3CR1 (22, 44). These studies have suggested that these transitory T cells are derived directly from CXCR5+ progenitor T cells; our data suggests most ICB-triggered expansion can be attributed to cells undergoing transitions from intermediate_1 to exhausted_1. A reported population of CXCR5+ CD8+ progenitor exhausted T cells, which proliferated after PD-L1 blockade, also express CXCR3 and bear strong similarities to intermediate_1 T cells (23). It may be that in cancer, the intermediate_1 population combines aspects of progenitor and transitory T cells found in chronic LCMV infection. Intermediate_1 T cells therefore likely have a greater role in the response to ICB than previously appreciated, especially in cancer. We propose that these patterns of tissue-restricted antigen presence may encourage important yet transient antigen encounter in infected or tumor tissue and their draining lymph nodes, followed by low density antigen encounter in the spleen. Our work highlights the crucial role of cross-presentation by DC1 in facilitating optimal T cell differentiation, even in scenarios of low antigen density. This pattern of antigen encounter may represent a key component of the anti-tumor immune response that is not well-modeled by chronic LCMV infection, which results in high antigen levels in the spleen.
Similar to previous studies (32, 33), we found that the intermediate_1 population served as a hub for differentiation into multiple phenotypic states, including exhausted_1 and exhausted_KLR T cells. Of these phenotypes, only exhausted_1 cells exhibited efficient trafficking to the tumor, suggesting that state decisions made at this bottleneck substantially impact the effectiveness of an anti-tumor T cell response. These two T cell states also exhibited minimal clonal overlap, suggesting that they represent divergent fates, raising the possibility that TCR signaling characteristics may influence fate decisions. Indeed, prior studies have demonstrated that higher TCR affinity promotes differentiation into a terminally exhausted phenotype, rather than a phenotype resembling exhausted_KLR (33). Future studies could seek to understand factors that regulate the fate commitment of intermediate_1 T cells, as well as assess to what extent these transitions may be reversible. Understanding how to effectively target and manipulate the intermediate_1 to exhausted_1 fate decision may offer new strategies to increase the tumor-infiltrating abilities of T cells and enhance the efficacy of ICB.
By examining the relationships between anatomic sites and transcriptional phenotypes within individual clonotypes, we observed that some phenotypic transitions, including progenitor intermediate_1 and intermediate_1 exhausted_1, are strongly associated with transit between different tissues. We are unable to conclude whether entry into different tissues is a key factor promoting differentiation, or whether differentiation itself promotes transit to different tissue sites. One previous study found, however, that full effector differentiation of progenitor T cells requires entry into the tumor, suggesting that T cell state transitions can result from trafficking to new anatomic locations (45).
In contrast to intermediate_1 T cells, our results indicate that mouse and human CX3CR1hi CD8+ T cells, including exhausted_KLR and Temra T cells, respectively, do not traffic efficiently to the tumor. This result is consistent with previous findings where CX3CR1hi memory CD8+ T cells were preferentially found in blood, while CXCR3+CX3CR1int memory T cells, which phenotypically resemble intermediate_1 T cells, were resident in non-lymphoid tissues (46). These differential homing patterns may be explained by the expression of CXCR3. Intermediate_1 T cells express significantly more CXCR3 than exhausted_KLR cells, and CXCR3 expression is critical for the homing of CD8+ T cells into tumors (47). Consistent with our finding of inefficient tumor homing by exhausted_KLR T cells, two previous studies using subcutaneous mouse models of cancer found that CX3CR1hi CD8+ T cells contribute little to anti-tumor immunity (48, 49). One could imagine, however, that a population of circulating or spleen-patrolling tumor-reactive CD8+ T cells could surveil for disseminated tumor cells and protect against metastasis (50). Thus, future studies could focus on the contributions of exhausted_KLR CD8+ T cells to anti-tumor immunity, including whether these cells can eventually reach the tumor and undergo differentiation to a tumor-homing exhausted_1 phenotype. These populations could also potentially provide a blood-based biomarker for anti-tumor immunity or response to ICB (51).
One limitation of our study is that there remains insufficient available data from tumor-reactive CD8+ T cells recovered from the TdLN and spleen of human cancer patients to assess to what extent the transcriptional states and phenotypic transitions that we identified in our mouse models reflect patterns of differentiation experienced by anti-tumor CD8+ T cells in human cancer patients. However, a recent clinical study using adjuvant mRNA vaccination in patients with pancreatic cancer identified an enrichment of non-responders in patients that had their spleen removed during debulking surgery (52). Like us, the authors speculated that “optimal” antigen restimulation in the spleen might be associated with better therapeutic responses. Nonetheless, it remains unknown whether similar patterns of differentiation also occur in other lymphoid tissues, such as non-draining, distal lymph nodes, that may provide low but non-zero levels of antigen that support the differentiation of intermediate clonotypes. These studies might be particularly interesting in models of metastasis.
Together, our data highlight a previously unappreciated role for the spleen in coordinating the differentiation of tumor-reactive CD8+ T cells as they respond to ICB. The splenic intermediate_1 phenotype that we identified comprised the majority of tumor-reactive CD8+ T cells that expanded upon ICB treatment and gave rise to a majority of tumor-infiltrating clonotypes, suggesting that is a crucial part of the anti-tumor immune response. This study provides mechanistic insight into ICB responses and will inform future studies of anti-tumor immune responses.
Materials and Methods
Study design
Here, we used paired single-cell RNA and TCR sequencing to profile the endogenous, tumor-reactive T cells isolated from the tumors, TdLNs, and spleens of mice treated with ICB to identify the transcriptional T cell states that maximally respond to ICB, as well as the anatomic location they reside in. We functionally validated our findings using adoptive T cell transfer approaches and multiparameter flowcytometry.
ICB treatment
Mice were injected intraperitoneally with anti-CTLA-4 (clone UC10–4F10–11, Bio X Cell) and anti-PD-L1 (clone 10F.9G2, Bio X Cell) antibodies on days 7, 10, 13, and 16 post-tumor inoculation for tumor outgrowth studies, or on days 7 and 10 post-tumor inoculation for day 14 analyses. Each mouse received 100 μg of each antibody per treatment.
2C T cell adoptive transfer
Spleens and inguinal lymph nodes of 2C RAG2−/−CD45.1+ mice were dissected and made into single cell suspensions as described above. Approximately 1 million cells were transferred to KP.SIY tumor-bearing mice seven days post tumor inoculation. Recipient CD45.2+ animals were euthanized and analyzed three days post 2C T cell transfer. For transfers into secondary recipients, 2C T cells were transferred into primary recipients as described above. Three days post adoptive transfer, the tumor-draining iLNs or spleens of recipient mice were isolated and CD8+ T cells were enriched with the Miltenyi CD8+ T cell isolation kit. Congenically marked CD45.1+ 2C T cells were then isolated from the CD8+ T cells using fluorescence activated cells sorting and 50,000 2C T cells were transferred intravenously into secondary recipient C57BL/6 mice bearing day 7 flank KP.SIY tumors. Additional serial transfer experiments were conducted by transferring isolated 50,000 2C T cells (as described above) into secondary C57BL/6 recipient mice with day 7 KP flank tumors and into secondary Batf3−/− recipient mice with day 7 KP.SIY flank tumors. Secondary recipients received ICB one and three days after adoptive transfer of in vivo primed 2C T cells. TdLNs, spleens and tumors from secondary recipients were analyzed seven days after adoptive transfer of in vivo primed 2C T cells.
In vivo antigen delivery
C57BL/6 splenocytes were isolated as described above, then ACK lysed, washed, and pulsed in complete medium for 1 hour at 37 degrees Celsius with 0.2 μM SIY peptide. 20 ×106 SIY-pulsed splenocytes were washed and injected intravenously into KP.SIY tumor-bearing mice on days 4, 6, and 9 post tumor inoculation. During this experiment, some mice were also treated with ICB on days 7 and 10 post-tumor inoculation. Mice were analyzed on day 14 post-tumor inoculation.
Single-cell RNA sequencing with Seq-Well
Sorted cells were then processed for scRNA-seq using the Seq-Well platform with second strand chemistry, as previously described (53, 54). Whole transcriptome libraries were barcoded and amplified using the Nextera XT kit (Illumina) and were sequenced on a Novaseq 6000 (Illumina). Hashtag oligo libraries were amplified as described previously and were sequenced on a Nextseq 550(34). Whole-transcriptome libraries were sequenced to a median depth of > 80,000 reads/cell recovered. Cell hashing libraries were sequenced to a medium depth of >3,000 reads/cell recovered.
Processing of cell hashing data
Cell hashing data was aligned to HTO barcodes using CITE-seq-Count v1.4.2. First, cells receiving fewer than five total HTO counts were classified as negatives. Downstream deconvolution of the hash-tag barcodes and analysis was conducted as previously described(55). Thresholds calculated for each sample were manually inspected and adjusted if necessary. Cells marked as “doublets” or “negatives” by this procedure were excluded from downstream analysis.
Processing of single-cell RNA sequencing data
Raw read processing of scRNA-seq reads was performed as previously described(56). Briefly, reads were aligned to the mm10 reference genome and collapsed by cell barcode and unique molecular identifier (UMI). Then, cells with less than 300 unique genes detected or with greater than 25% mitochondrial gene counts and genes detected in fewer than 5 cells were filtered out. Cell cycle scores for individual cells were computed using CellCycleScoring function in Seurat. Data was then integrated by batch using Seurat v4.1.1(56). The ScaleData function was used to regress out the number of RNA features in each cell, as well as S and G2/M cell cycle scores and fraction of mitochondrial gene expression. The number of principal components used for visualization was determined by assessing of the elbow plot, and two-dimensional embeddings were generated using uniform manifold approximation and projection (UMAP). Clusters were identified using Louvain clustering, as implemented in the FindClusters function in Seurat. DEG analysis was performed for each cluster and between indicated cell populations using the FindMarkers function. Data was iteratively reclustered to remove clusters with gene expression consistent with naïve T cells, monocytes, and NK cells. Label transfer of cluster labels onto proliferating cell populations was performed using the FindTransferAnchors and TransferData functions in Seurat (56).
Paired single-cell TCR sequencing and analysis
Paired TCR sequencing and read alignment was performed as described previously (57). In summary, TCR transcripts using biotinylated Tcrb and Tcra probes and magnetic streptavidin beads were enriched from the whole transcriptome amplification product from each single-cell library and further amplified using V-region primers and Nextera sequencing handles, and the resulting libraries were sequenced on an Illumina Novaseq 6000. Processing of raw sequencing reads was performed using the Immcantation software suite (58, 59). First, the FilterSeq.py function was used to remove reads with an average quality score less than 25. Subsequent steps in CDR3 mapping were previously described (55). In brief, reads were sorted by cell barcode and UMI, and UMI with less than 10 reads were discarded. Sets of sequences that comprised less than 30% of the sequences obtained for that UMI were discarded. BuildConsensus.py function and IgBlast (60) were used to determine consensus sequences and sequences with no consensus sequence were discarded. the remaining TCR sequences were mapped to single cell transcriptomes by matching cell barcodes. To define clonotypes of cells, we first segregated cells by mouse and unique Tcrb CDR3 junction nucleotide sequences. For each unique combination of mouse and CDR3β junction, we determined the most common TCRα sequence in cells with paired TCR recovery. We then imputed missing beta chains from cells with recovery of only alpha chain by matching to these combinations of mouse, beta chain, and alpha chain.
Statistical analysis
Statistical analyses were performed using GraphPad Prism (GraphPad) and R. All data are shown as means ± SEM. For flow cytometry and tumor outgrowth studies, statistical analyses were performed with Mann-Whitney U (MWU) test for comparisons of two groups, one-way analysis of variance (ANOVA) for comparisons between multiple groups, or two-way ANOVA for multiple comparisons over time (unless explicitly stated otherwise), with *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001. For one-way and two-way ANOVAs, Šídák’s or Tukey’s multiple comparisons tests were utilized as post-tests. For each experiment 2–4 independent repeats were performed and combined; n numbers are indicated in figure captions. Statistical tests accompanying scRNA and scTCR sequencing analyses have been described in detail in figure captions.
Supplementary Material
Acknowledgments
We would like to thank M. Duquette for mouse colony maintenance and P. Thompson and Judy Teixeira for administrative support. We thank the Koch Institute’s Robert A. Swanson (1969) Biotechnology Center for providing core services.
We would like to extend a special acknowledgement of the significant contributions of Brendan L. Horton to the field of tumor immunology during his research career, which was too short. Brendan was a born scientist with the desire to understand the unknowns and he pursued his desire in an unparalleled collegial and collaborative way. He will be forever missed as a scientist, colleague, and friend.
Funding:
This work was supported by NIH cancer core grant P30 CA014051–49, a postdoctoral fellowship from the Ludwig Center at MIT’s Koch Institute for Integrative Cancer Research, the Pew-Stewart Scholarship and the NCI funded 1R37CA273819–01A1.
Footnotes
Competing interests
J.C.L. is a co-founder and advisor to Honeycomb Biotechnologies. S.S. is a SAB member for Related Sciences, Arcus Biosciences, Ankyra Therapeutics, and Repertoire Immune Medicines. S.S. is a co-founder of Danger Bio. S.S. is a consultant for TAKEDA, Merck, and receives funding for unrelated projects from Leap Therapeutics and iTeos Therapeutics. J.C.L.’s and S.S.’s interests were reviewed and managed under MIT’s policies for potential conflicts of interest. All other authors declare no competing interests.
Data and materials availability:
Tabulated data underlying the figures are provided in Data file S1. Single-cell transcriptome data from untreated flank tumors was previously published in GSE184388. All remaining sequencing data is also available on GEO, accession number GSE270050. Codes used to perform preprocessing and analysis of single-cell data are available at https://github.com/duncanmorgan/KPSpleen and is archived on Zenodo (10.5281/zenodo.12786217). All other data needed to support the conclusions of the paper are present in the paper or the Supplementary Materials.
References
- 1.Karasarides M, Cogdill AP, Robbins PB, Bowden M, Burton EM, Butterfield LH, et al. Hallmarks of Resistance to Immune-Checkpoint Inhibitors. Cancer Immunol Res. 2022;10(4):372–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zehn D, Thimme R, Lugli E, de Almeida GP, Oxenius A. ‘Stem-like’ precursors are the fount to sustain persistent CD8(+) T cell responses. Nat Immunol. 2022;23(6):836–47. [DOI] [PubMed] [Google Scholar]
- 3.Chow A, Perica K, Klebanoff CA, Wolchok JD. Clinical implications of T cell exhaustion for cancer immunotherapy. Nat Rev Clin Oncol. 2022;19(12):775–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359(6382):1350–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10. [DOI] [PubMed] [Google Scholar]
- 6.Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8alpha+ dendritic cells. J Exp Med. 2011;208(10):2005–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208(10):1989–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zagorulya M, Yim L, Morgan DM, Edwards A, Torres-Mejia E, Momin N, et al. Tissue-specific abundance of interferon-gamma drives regulatory T cells to restrain DC1-mediated priming of cytotoxic T cells against lung cancer. Immunity. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horton BL, Morgan DM, Momin N, Zagorulya M, Torres-Mejia E, Bhandarkar V, et al. Lack of CD8(+) T cell effector differentiation during priming mediates checkpoint blockade resistance in non-small cell lung cancer. Sci Immunol. 2021;6(64):eabi8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uyttenhove C, Maryanski J, Boon T. Escape of mouse mastocytoma P815 after nearly complete rejection is due to antigen-loss variants rather than immunosuppression. J Exp Med. 1983;157(3):1040–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14(10):1014–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oliveira G, Stromhaug K, Klaeger S, Kula T, Frederick DT, Le PM, et al. Phenotype, specificity and avidity of antitumour CD8(+) T cells in melanoma. Nature. 2021;596(7870):119–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557(7706):575–9. [DOI] [PubMed] [Google Scholar]
- 14.Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science. 2017;355(6332):1423–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017;355(6332):1428–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fairfax BP, Taylor CA, Watson RA, Nassiri I, Danielli S, Fang H, et al. Peripheral CD8(+) T cell characteristics associated with durable responses to immune checkpoint blockade in patients with metastatic melanoma. Nat Med. 2020;26(2):193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Utzschneider DT, Gabriel SS, Chisanga D, Gloury R, Gubser PM, Vasanthakumar A, et al. Early precursor T cells establish and propagate T cell exhaustion in chronic infection. Nat Immunol. 2020;21(10):1256–66. [DOI] [PubMed] [Google Scholar]
- 18.Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, et al. Developmental Relationships of Four Exhausted CD8(+) T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity. 2020;52(5):825–41 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nature immunology. 2019;20(3):326–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Z, Ji Z, Ngiow SF, Manne S, Cai Z, Huang AC, et al. TCF-1-Centered Transcriptional Network Drives an Effector versus Exhausted CD8 T Cell-Fate Decision. Immunity. 2019;51(5):840–55 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science. 2012;338(6111):1220–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hudson WH, Gensheimer J, Hashimoto M, Wieland A, Valanparambil RM, Li P, et al. Proliferating Transitory T Cells with an Effector-like Transcriptional Signature Emerge from PD-1(+) Stem-like CD8(+) T Cells during Chronic Infection. Immunity. 2019;51(6):1043–58 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature. 2016;537(7620):417–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang Q, Wu X, Wang Z, Chen X, Wang L, Lu Y, et al. The primordial differentiation of tumor-specific memory CD8(+) T cells as bona fide responders to PD-1/PD-L1 blockade in draining lymph nodes. Cell. 2022;185(22):4049–66 e25. [DOI] [PubMed] [Google Scholar]
- 25.Francis DM, Manspeaker MP, Schudel A, Sestito LF, O’Melia MJ, Kissick HT, et al. Blockade of immune checkpoints in lymph nodes through locoregional delivery augments cancer immunotherapy. Sci Transl Med. 2020;12(563). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dammeijer F, van Gulijk M, Mulder EE, Lukkes M, Klaase L, van den Bosch T, et al. The PD-1/PD-L1-Checkpoint Restrains T cell Immunity in Tumor-Draining Lymph Nodes. Cancer Cell. 2020;38(5):685–700 e8. [DOI] [PubMed] [Google Scholar]
- 27.Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, et al. Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity. 2019;50(1):195–211 e10. [DOI] [PubMed] [Google Scholar]
- 28.Jansen CS, Prokhnevska N, Master VA, Sanda MG, Carlisle JW, Bilen MA, et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature. 2019;576(7787):465–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu TD, Madireddi S, de Almeida PE, Banchereau R, Chen YJ, Chitre AS, et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature. 2020;579(7798):274–8. [DOI] [PubMed] [Google Scholar]
- 30.Kamphorst AO, Pillai RN, Yang S, Nasti TH, Akondy RS, Wieland A, et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc Natl Acad Sci U S A. 2017;114(19):4993–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. 2017;545(7652):60–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giles JR, Ngiow SF, Manne S, Baxter AE, Khan O, Wang P, et al. Shared and distinct biological circuits in effector, memory and exhausted CD8(+) T cells revealed by temporal single-cell transcriptomics and epigenetics. Nat Immunol. 2022;23(11):1600–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daniel B, Yost KE, Hsiung S, Sandor K, Xia Y, Qi Y, et al. Divergent clonal differentiation trajectories of T cell exhaustion. Nat Immunol. 2022;23(11):1614–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stoeckius M, Zheng S, Houck-Loomis B, Hao S, Yeung BZ, Mauck WM, 3rd, et al. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 2018;19(1):224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xia M, Hu S, Fu Y, Jin W, Yi Q, Matsui Y, et al. CCR10 regulates balanced maintenance and function of resident regulatory and effector T cells to promote immune homeostasis in the skin. J Allergy Clin Immunol. 2014;134(3):634–44 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kok L, Dijkgraaf FE, Urbanus J, Bresser K, Vredevoogd DW, Cardoso RF, et al. A committed tissue-resident memory T cell precursor within the circulating CD8+ effector T cell pool. J Exp Med. 2020;217(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell. 2014;26(5):638–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spranger S, Dai D, Horton B, Gajewski TF. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell. 2017;31(5):711–23 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meiser P, Knolle MA, Hirschberger A, de Almeida GP, Bayerl F, Lacher S, et al. A distinct stimulatory cDC1 subpopulation amplifies CD8(+) T cell responses in tumors for protective anti-cancer immunity. Cancer Cell. 2023;41(8):1498–515 e10. [DOI] [PubMed] [Google Scholar]
- 40.Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, et al. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell. 2016;30(2):324–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen X, MacNabb BW, Flood B, Blazar BR, Kline J. Divergent fates of antigen-specific CD8(+) T cell clones in mice with acute leukemia. Cell Rep. 2021;37(6):109991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.MacNabb BW, Tumuluru S, Chen X, Godfrey J, Kasal DN, Yu J, et al. Dendritic cells can prime anti-tumor CD8(+) T cell responses through major histocompatibility complex cross-dressing. Immunity. 2022;55(6):982–97 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng L, Qin S, Si W, Wang A, Xing B, Gao R, et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science. 2021;374(6574):abe6474. [DOI] [PubMed] [Google Scholar]
- 44.Zander R, Schauder D, Xin G, Nguyen C, Wu X, Zajac A, et al. CD4(+) T Cell Help Is Required for the Formation of a Cytolytic CD8(+) T Cell Subset that Protects against Chronic Infection and Cancer. Immunity. 2019;51(6):1028–42 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prokhnevska N, Cardenas MA, Valanparambil RM, Sobierajska E, Barwick BG, Jansen C, et al. CD8(+) T cell activation in cancer comprises an initial activation phase in lymph nodes followed by effector differentiation within the tumor. Immunity. 2023;56(1):107–24 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gerlach C, Moseman EA, Loughhead SM, Alvarez D, Zwijnenburg AJ, Waanders L, et al. The Chemokine Receptor CX3CR1 Defines Three Antigen-Experienced CD8 T Cell Subsets with Distinct Roles in Immune Surveillance and Homeostasis. Immunity. 2016;45(6):1270–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mikucki ME, Fisher DT, Matsuzaki J, Skitzki JJ, Gaulin NB, Muhitch JB, et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nature communications. 2015;6:7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamauchi T, Hoki T, Oba T, Kajihara R, Attwood K, Cao X, et al. CD40 and CD80/86 signaling in cDC1s mediate effective neoantigen vaccination and generation of antigen-specific CX3CR1(+) CD8(+) T cells. Cancer Immunol Immunother. 2022;71(1):137–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamauchi T, Hoki T, Oba T, Saito H, Attwood K, Sabel MS, et al. CX3CR1-CD8+ T cells are critical in antitumor efficacy but functionally suppressed in the tumor microenvironment. JCI Insight. 2020;5(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burrack AL, Spartz EJ, Rollins MR, Miller EA, Firulyova M, Cruz E, et al. Cxcr3 constrains pancreatic cancer dissemination through instructing T cell fate. Cancer Immunol Immunother. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamauchi T, Hoki T, Oba T, Jain V, Chen H, Attwood K, et al. T-cell CX3CR1 expression as a dynamic blood-based biomarker of response to immune checkpoint inhibitors. Nat Commun. 2021;12(1):1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rojas LA, Sethna Z, Soares KC, Olcese C, Pang N, Patterson E, et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature. 2023;618(7963):144–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gierahn TM, Wadsworth MH 2nd, Hughes TK, Bryson BD, Butler A, Satija R, et al. Seq-Well: portable, low-cost RNA sequencing of single cells at high throughput. Nat Methods. 2017;14(4):395–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hughes TK, Wadsworth MH 2nd, Gierahn TM, Do T, Weiss D, Andrade PR, et al. Second-Strand Synthesis-Based Massively Parallel scRNA-Seq Reveals Cellular States and Molecular Features of Human Inflammatory Skin Pathologies. Immunity. 2020;53(4):878–94 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zagorulya M, Yim L, Morgan DM, Edwards A, Torres-Mejia E, Momin N, et al. Tissue-specific abundance of interferon-gamma drives regulatory T cells to restrain DC1-mediated priming of cytotoxic T cells against lung cancer. Immunity. 2023;56(2):386–405 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell. 2015;161(5):1202–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tu AA, Gierahn TM, Monian B, Morgan DM, Mehta NK, Ruiter B, et al. TCR sequencing paired with massively parallel 3’ RNA-seq reveals clonotypic T cell signatures. Nat Immunol. 2019;20(12):1692–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vander Heiden JA, Yaari G, Uduman M, Stern JN, O’Connor KC, Hafler DA, et al. pRESTO: a toolkit for processing high-throughput sequencing raw reads of lymphocyte receptor repertoires. Bioinformatics. 2014;30(13):1930–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gupta NT, Vander Heiden JA, Uduman M, Gadala-Maria D, Yaari G, Kleinstein SH. Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics. 2015;31(20):3356–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ye J, Ma N, Madden TL, Ostell JM. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res. 2013;41(Web Server issue):W34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Giles JR, Manne S, Freilich E, Oldridge DA, Baxter AE, George S, et al. Human epigenetic and transcriptional T cell differentiation atlas for identifying functional T cell-specific enhancers. Immunity. 2022;55(3):557–74 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Tabulated data underlying the figures are provided in Data file S1. Single-cell transcriptome data from untreated flank tumors was previously published in GSE184388. All remaining sequencing data is also available on GEO, accession number GSE270050. Codes used to perform preprocessing and analysis of single-cell data are available at https://github.com/duncanmorgan/KPSpleen and is archived on Zenodo (10.5281/zenodo.12786217). All other data needed to support the conclusions of the paper are present in the paper or the Supplementary Materials.