

Abstract
Fibroblast growth factor 21 (FGF21) is a promising target for treatment of obesity‐associated diseases including metabolic dysfunction‐associated steatohepatitis (MASH) and atherosclerosis. We evaluated the effects of the bispecific anti‐FGF21‐β klotho (KLB) agonist antibody bFKB1 in a preclinical model of MASH and atherosclerosis. Low‐density lipoprotein receptor knockout (Ldlr−/−).Leiden mice received a high‐fat diet for 20 weeks, followed by treatment with an isotype control antibody or bFKB1 for 12 weeks. Effects on plasma risk markers and (histo)pathology of liver, adipose tissue, and heart were evaluated alongside hepatic transcriptomics analysis. bFKB1 lowered body weight (−21%) and adipose tissue mass (−22%) without reducing food intake. The treatment also improved plasma insulin (−80%), cholesterol (−48%), triglycerides (−76%), alanine transaminase (ALT: −79%), and liver weight (−43%). Hepatic steatosis and inflammation were strongly reduced (macrovesicular steatosis −34%; microvesicular steatosis −100%; inflammation −74%) and while the total amount of fibrosis was not affected, bFKB1 did decrease new collagen formation (−49%). Correspondingly, hepatic transcriptomics and pathway analysis revealed the mechanistic background underlying these histological improvements, demonstrating broad inactivation of inflammatory and profibrotic transcriptional programs by bFKB1. In epididymal white adipose tissue, bFKB1 reduced adipocyte size (−16%) and inflammation (−52%) and induced browning, signified by increased uncoupling protein‐1 (UCP1) protein expression (8.5‐fold increase). In the vasculature, bFKB1 had anti‐atherogenic effects, lowering total atherosclerotic lesion area (−38%). bFKB1 has strong beneficial metabolic effects associated with a reduction in hepatic steatosis, inflammation, and atherosclerosis. Analysis of new collagen formation and profibrotic transcriptional programs indicate that bFKB1 treatment may have antifibrotic potential in a longer treatment duration as well.
Keywords: atherosclerosis, FGF21, MASH, metabolism, obesity
Overview of the effects of the FGF21 mimetic bFKB1 on the liver, white adipose tissue, and the vasculature. HFD, high‐fat diet; IFN, interferon; IL, interleukin; MCP, monocyte chemoattractant protein; SMAD, suppressor of mothers against decapentaplegic; TGF, transforming growth factor; TNF, tumor necrosis factor; UCP, uncoupling protein; VEGF, vascular endothelial growth factor; YAP/TAZ, yes‐associated protein/transcriptional coactivator with PDZ‐binding motif.
Abbreviations
- (V)LDL
(Very) low density lipoprotein
- ALT
Alanine transaminase
- AST
Aspartate transaminase
- bFKB1
Bispecific anti‐FGF21‐KLB agonist antibody
- DEG
Differentially expressed gene
- eWAT
Epididymal white adipose tissue
- FGF
Fibroblast growth factor
- FGFR
Fibroblast growth factor receptor
- H&E
Hematoxylin and eosin
- HFD
High fat diet
- HPS
Hematoxylin‐phloxine‐saffron
- IFN
Interferon
- IL
Interleukin
- KLB
β‐klotho
- LDLr
Low density lipoprotein receptor
- MASH
Metabolic dysfunction‐associated steatohepatitis
- MASLD
Metabolic dysfunction‐associated steatotic liver disease
- PCA
Principal component analysis
- SEM
Standard error of the mean
- SR
Sirius Red
- TGF
Transforming growth factor
- UCP1
Uncoupling protein‐1
1. INTRODUCTION
Chronic excessive food consumption combined with a sedentary lifestyle drives the current obesity pandemic. This lifestyle leads to metabolic complications that can induce development of metabolic dysfunction‐associated steatotic liver disease (MASLD), which is characterized by hepatic steatosis. MASLD can progress into metabolic dysfunction‐associated steatohepatitis (MASH), where hepatic steatosis is accompanied by inflammation and ultimately fibrosis. 1 When lifestyle changes cannot be sustained, pharmacological interventions are required to manage progression of MASLD‐MASH and accompanying cardiovascular disease, the primary cause of mortality in this patient population. 2 The high MASLD‐MASH prevalence and lack of approved pharmacological therapy represents a great unmet clinical need.
Fibroblast growth factors (FGFs) are a family of signaling proteins with a variety of effects on metabolism, making them promising candidates for treatment of obesity‐related disorders. FGF21, a hepatokine that signals through three main FGF receptor (FGFR) isoforms, namely 1c, 2c, and 3c, bound by the obligatory coreceptor β‐klotho (KLB), 3 is of particular interest. However, while FGF21 has beneficial effects on obesity, blood glucose and lipid levels, its efficacy is limited by rapid clearance from the circulation. 4 Several approaches have been implemented to stimulate signaling through FGFR1 and KLB, including native mature FGF21, short‐acting FGF21 analogues and more recently, long‐acting FGF21 analogues. 5 In various preclinical and clinical studies, these approaches have been shown to reduce obesity and improve energy and lipid metabolism. 6 , 7 Given their ameliorative effects on metabolic parameters, FGF21 interventions have therefore been suggested as therapeutic option for treatment of MASLD‐MASH as well.
The bispecific anti‐FGFR1‐KLB agonist antibody bFKB1 was designed to activate the FGFR1/KLB complex similarly to recombinant human FGF21 yet without interfering with endogenous FGF binding. 8 bFKB1 distinguishes itself from other FGF21 analogues by specifically targeting FGFR1c/KLB, mainly present in adipose tissue, pancreas, and brain, 8 , 9 which is thought to provide beneficial metabolic effects without the adverse effects associated with long‐term FGF21 administration. 8 On top of this receptor selectivity, bFKB1 does not induce cross‐reactive antibody development against endogenous FGF21. 8 Previous preclinical studies have demonstrated that bFKB1 treatment increases energy expenditure and ameliorates obesity and hyperlipidaemia. 6 , 7 , 8 However, its effects on MASLD‐MASH and atherosclerosis have not been studied to date. In this study, we used Ldlr−/−.Leiden mice, that develop MASH and atherosclerosis in the context of an obese phenotype with insulin resistance, dyslipidemia, and hypertriglyceridemia 10 as is typical for many patients with MASH. 11 , 12 , 13 The hyperlipidemic phenotype of low‐density lipoprotein receptor knockout (Ldlr−/−).Leiden mice (with increased triglyceride and cholesterol levels, confined mainly to the atherogenic (very) low‐density lipoprotein ((V)LDL) particles) allows investigation of efficacy of novel therapeutics for MASH and atherosclerosis simultaneously. 11 , 14 , 15 Ldlr−/−.Leiden mice also reflect underlying disease processes observed in man, shown by comparisons of transcriptomics and metabolomics profiles with those of patients. 11 , 12 , 16 , 17 Using this model, we demonstrate the beneficial effects of bFKB1 and mechanistic background of the changes on metabolic parameters, MASLD‐MASH, adipose tissue, and atherosclerosis in an intervention study.
2. MATERIALS AND METHODS
2.1. Animals and experimental design
This study was approved by The Netherlands Central Authority for Scientific Procedures on Animals (AVD5010020172064) and animal welfare body of TNO (IVD TNO; TNO‐430). The study was performed in compliance with European guidelines for the care and use of laboratory animals (European Directive 2010/63/EU). Mice were bred and housed at the AAALAC‐accredited SPF animal facility at TNO (TNO Metabolic Health Research, Leiden, the Netherlands). We specifically selected male mice because they are more sensitive to diet‐induced obesity and dysmetabolism than females. 18
Mice were group‐housed (two to five animals per cage) in a temperature‐controlled room (21 ± 2°C), at 50%–60% humidity and on a 12 h light–dark cycle. All mice had free access to food and heat‐sterilized tap water. Thirty mice (14–18 weeks old) were started on high‐fat diet (HFD; D12451, Research Diets, New Brunswick, NJ, USA) containing 45 kCal% fat from lard, 35 kCal% from carbohydrates, and 20 kCal% from casein. After a 20‐week induction period, mice were matched into two groups of 15 mice each, based on body weight, plasma cholesterol, plasma triglycerides, and blood glucose concentrations. Group numbers were calculated a priori using GPower3.1, based on a minimal effect size of 50%, using a one‐sided test with a 95% confidence interval and power of 0.80. One group received weekly intraperitoneal injections of an isotype control IgG antibody (5 mg/kg) (control group). The other group received weekly intraperitoneal injections of the monoclonal bispecific FGFR1/KLB agonist antibody bFKB1 (5 mg/kg) as described previously in detail. 8 Two mice from the bFKB1‐treated group were removed from the study in week 11, based on humane end‐point criteria due to body weight loss and cachexia and these mice were excluded from all analyses. Blood was sampled at t = 0, t = 6, and t = 12 weeks from the tail vein into EDTA‐coated tubes (Sarstedt, Nümbrecht, Germany) after a 5 h fasting period. After 12 weeks of treatment, 5 h‐fasted mice were sacrificed by gradual‐fill CO2 asphyxiation. Hearts with aortic roots, livers, and epididymal white adipose tissue were isolated and weighed and subsequently formalin‐fixed and paraffin‐embedded for histological analysis or snap‐frozen in liquid nitrogen and stored at −80°C.
2.2. Biochemical analyses in plasma and liver
Fasted blood glucose levels were measured using a hand‐held glucometer (Freestyle Light, Abbott, Chicago, IL, USA) at the time of blood sampling. Enzyme‐linked immunosorbent assay (ELISA) was performed to determine plasma insulin levels (#90080; Chrystal Chem Inc., Elk Grove Village, IL, USA). Total cholesterol and triglycerides in plasma were measured using enzymatic assays (CHOD‐PAP and GPO‐PAP, respectively; Roche Diagnostics, Almere, the Netherlands). Total cholesterol exposure was expressed as cholesterol concentration × study duration in weeks. Lipoprotein profiles for cholesterol were determined in plasma samples pooled per group and by means of fast protein liquid chromatography (FPLC) in accordance with previously described protocols. 19 Plasma alanine transaminase (ALT) and aspartate transaminase (AST) levels were determined by spectrophotometric activity assay using a Reflotron® Plus analyzer (Hoffman‐La Roche, Mannheim, Germany).
For analysis of hepatic lipid content, liver tissue of six mice per group was homogenized in phosphate‐buffered saline (PBS) and lipids were extracted as described previously in detail. 20 Representative samples were selected based on MASH histopathology to reflect group average and variation. In short, lipids were separated by high‐performance thin‐layer chromatography (HPTLC) on silica gel plates and subsequently stained and analyzed with the ChemiDoc Touch Imaging System (Bio‐Rad Laboratories, Veenendaal, the Netherlands). Hepatic triglyceride, cholesteryl ester, and free cholesterol content was quantified using Image‐lab version 5.2.1. software (Bio‐Rad Laboratories) and expressed per mg liver protein.
Intrahepatic concentrations of cytokines and chemokines were determined in homogenized liver tissue for 15 isotype control antibody‐treated mice and 13 bFKB1‐treated mice. A multiplex immunoassay panel (#K0082207, V‐PLEX Proinflammatory Panel 1 Mouse Kit, Mesoscale Discovery (MSD, MD, USA)) was carried out in accordance with the manufacturer's protocol on a MESO QuickPlex SQ 120 reader (MSD). Total protein concentrations were determined with a BCA Protein Assay Kit (ThermoFisher Scientific, Waltham, MA, USA) in the same liver homogenates and cytokine and chemokine concentrations are expressed per mg protein.
2.3. De novo collagen synthesis
In the final 2 weeks of the study, de novo collagen formation was assessed by using stable isotope labelling and liquid chromatography mass spectrometry (LC–MS)‐based mass isotopomer analysis. 12 , 21 Mice received a bolus intraperitoneal injection of 100% 2H2O (17.5 mL/kg body weight) to bring total body water enrichment to approximately 2.5%. Mice received 4% 2H2O in the drinking water to maintain this level of body water enrichment until the study endpoint. Deuterated water incorporation into hydroxyproline was analyzed as described previously. 22 Mass spectrometry and analysis were performed by Metabolic Solutions (Nashua, NH, USA). New hydroxyproline content was expressed as μg per mg dry liver lobe.
2.4. Histological analysis of liver, adipose tissue, and heart
Formalin‐fixed and paraffin‐embedded tissue of the medial liver lobe was sectioned (3 μm) and stained with hematoxylin and eosin (H&E). A board‐certified pathologist scored sections blindly for MASH by examining two H&E‐stained sections per mouse using an adapted grading system of human MASH. 23 , 24 Hepatic macrovesicular and microvesicular steatosis were determined at 50–100× magnification and expressed as percentage relative to the total liver area. During the progression of MASLD into MASH, macrovesicular and microvesicular steatosis develop simultaneously. The former is characterized by a single large lipid droplet filling the hepatocyte cytoplasm while the latter is characterized by multiple small lipid droplets filling the hepatocyte cytoplasm. Macrovesicular steatosis is mainly associated with lobular inflammation 25 while microvesicular steatosis is thought to be associated with mitochondrial dysfunction and oxidative stress. 26 For scoring of hepatic inflammation, the number of inflammatory foci was determined at 100× magnification in five non‐overlapping fields (view size 4.2 mm2) and expressed as number of foci per mm2. For analysis of fibrosis, liver cross‐sections were stained with Sirius Red (SR). Hepatic fibrosis area and stage were assessed in two SR‐stained sections per mouse by the board‐certified pathologist using the scoring system described by Tiniakos et al., 27 where F0: absence of fibrosis, F1: fibrosis in perisinusoidal or periportal area, F2: fibrosis within both perisinusoidal and periportal areas, F3: bridging fibrosis and F4: cirrhosis.
Formalin‐fixed and paraffin‐embedded epididymal white adipose tissue (eWAT) was sectioned (5 μm), stained with hematoxylin‐phloxine‐saffron (HPS) and scanned for digital analysis (Aperio AT2, Leica Biosystems, Amsterdam, the Netherlands). Adipocyte size was analyzed using the Adiposoft 28 plugin in ImageJ (version 1.53; NIH) at 20× magnification in three non‐overlapping fields (1.56 mm2) as described previously. 12 , 18 , 29 Adipose tissue inflammation was quantified in the same fields used to quantify adipocyte size by determining the number of crown‐like structures as described previously. 12 , 18 , 29 Adipose tissue inflammation was expressed as number per 1000 adipocytes. 29
Atherosclerosis severity was determined histologically in line with previously described protocols. 30 , 31 In short, aortic roots were paraffin‐embedded and cross‐sectioned (5 μm) at 50 μm intervals and stained with HPS. Four sections per mouse were assessed for atherosclerotic lesion load and severity using the classification system of the American Heart Association, 31 where type I: early fatty streak; type II: regular fatty streak; type III: mild plaque; type IV: moderate plaque; and type V: severe plaque.
2.5. Protein expression in adipose tissue
Snap‐frozen eWAT was homogenized in lysis buffer containing Tris–HCL (pH 7.4), 150 mmol/L NaCl, 5 mmol/L CaCl2 (v/v), 1% Triton X‐100 and cOmplete™ mini protease inhibitor cocktail (Roche Diagnostics). Samples were centrifuged for 15 min at 4°C, 16 200 g, supernatant was collected, and this centrifuge step was repeated twice to remove excess fat. Protein concentrations were determined using the BCA Protein Assay Kit (ThermoFisher Scientific). Proteins (50 μg) in 2× SDS Laemmli Sample Buffer (1:1 v/v; Sigma‐Aldrich, St Louis, MO, USA) were boiled for 6 min at 95°C, separated on a 4%–20% (w/v) SDS‐page gel (mini‐Protean TGX stain‐free precast gel; Bio‐Rad) and subsequently transferred onto Trans‐blot Turbo mini PVDF blotting membranes (Bio‐Rad) using the MIXED MW program on the Trans‐blot Turbo Bio‐Rad machine. Membranes were blocked in 5% (w/v) milk powder in tris‐buffered saline with 0.1% Tween‐20 (TBST) for 1 h at room temperature. Membranes were incubated overnight at 4°C with primary antibody heat shock protein 90 (HSP90; #4874‐1:1000 v/v; Cell Signaling, Leiden, the Netherlands) or uncoupling protein 1 (UCP1; ab23841‐1:1000 v/v; Abcam, Cambridge, UK) in 5% milk in TBST. Secondary antibody was added in 5% milk in TBST (anti‐rabbit HRP conjugate #7074S‐1:2000 v/v; Cell Signaling) and visualized using SuperSignal West Femto (ThermoFisher Scientific). Blots were analyzed with ChemiDoc Touch Imaging system (Bio‐Rad) and UCP1 band intensities were normalized to HSP90.
2.6. Gene expression analysis of liver tissue
Transcriptomics analysis of liver tissue was performed for n = 12 samples per group in line with previously described protocols. 12 Briefly, RNA was isolated from snap‐frozen tissue from the left liver lobe using the RNA‐Bee total‐RNA isolation kit (Bio‐Connect, Huissen, the Netherlands). RNA quality was evaluated using a 2100 Bioanalyzer (Agilent Technologies, Amstelveen, The Netherlands). Total RNA was processed into tagged random sequence libraries (NEBNext Ultra II Directional RNA Library Prep Kit for Illumina, NEB #E7760S/L, Biolabs) and sample quality was checked for proper size distribution (300–500 bp peak, Fragment Analyzer). The mixed (multiplex) sample libraries were sequenced on an Illumina NovaSeq6000 sequencer with a paired‐read 150‐cycle sequencing protocol at GenomeScan BV (Leiden, the Netherlands), resulting in ~20–40 million read counts per sample. Clustering and DNA sequencing using the NovaSeq6000 was performed according to manufacturer's protocols. A concentration of 1.1 nM of DNA was used yielding paired end reads (2 × 150 bp). NovaSeq control software NCS v1.7 was used. Image analysis, base calling, and quality check was performed with the Illumina data analysis pipeline RTA3.4.4 and Bcl2fastq v2.20. One sample (in the isotype control antibody group) was excluded from analysis due to a technical error and a Principal Component Analysis (PCA) was carried out. Trimmed Fastq files were merged (in case of Paired‐end reads) and aligned to the reference genome (Mus_musculus.GRCm38.gencode.vM19) using the STAR 2.5 algorithm with default settings (https://github.com/alexdobin/STAR). Based on the mapped read locations and the gene annotation, Htseq‐count 0.6.1p1 was used to count the read mapping frequency/gene (transcript region) resulting in read mapping frequency per gene, that subsequently served as input for the differentially expressed genes (DEGs) analysis using the DESeq2‐method. 32 These were used as input for pathway analysis through Ingenuity Pathway Analysis (IPA, Ingenuity Systems Inc., Redwood City, CA, USA, accessed December 2020). IPA uses gene expression data of known downstream target genes to predict the activation state or deactivation state of upstream regulators (e.g., transcription factors, signaling proteins, and metabolites). Accordingly, positive z‐scores (z > 2) indicate enhanced activity and negative z‐scores (z < −2) indicate reduced activity of an upstream regulator. 12 The datasets obtained in this study are accessible at the NCBI Gene Expression Omnibus (GEO‐ID: GSE255321).
2.7. Statistical analysis
All values were evaluated for normality using Shapiro–Wilk normality tests and for equality of variances using Levene's test of homogeneity of variances. For normally distributed parameters, differences between groups were analyzed by a Student's t‐test in case of equal variances and a t‐test with Welch correction in case of unequal variances. For non‐normally distributed parameters, Mann–Whitney U testing was performed. For histopathological analyses, the null hypothesis that bFKB1 did not reduce development of MASH and atherosclerosis relative to the isotype control antibody‐treated group was tested using one‐sided tests. For all other data, the null hypothesis that these did not differ between groups was tested using two‐sided tests. All data are presented as mean ± standard error of the mean (SEM) and a p‐value<.05 was considered statistically significant with the exception for the upstream regulator analysis, where a p‐value<.01 was considered statistically significant. Statistical analyses were evaluated in SPSS (version 25, IBM Corp., Armonk, NY, USA).
3. RESULTS
3.1. The FGF21 mimetic agonist antibody bFKB1 improves metabolic parameters in obese Ldlr−/−.Leiden mice
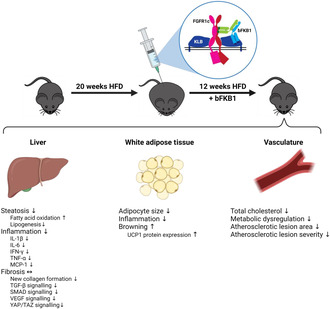
Ldlr−/−.Leiden mice with a mean starting body weight of 29.7 ± 2.4 g were fed an energy‐dense HFD for 20 weeks, which induced pronounced obesity as was evident from the 77% body weight increase (mean body weight at t = 0: 52.5 ± 3.4). Hereafter, mice were treated with either bFKB1 or an isotype control antibody for 12 weeks while continuing on the HFD. During this period, bFKB1‐treated mice lost weight relative to isotype controls (−21% at t = 12) (Figure 1A) while food intake was significantly higher at several timepoints throughout the study (Figure 1B), resulting in a 17% higher cumulative food intake during the 12‐week treatment period. Fasted blood glucose levels declined somewhat in the isotype control group toward the end of the study, a decline that was less pronounced in bFKB1‐treated mice, resulting in significantly higher levels in this group (+12% at t = 12) (Figure 1C). Plasma insulin levels on the other hand decreased significantly over time in bFKB1‐treated mice relative to isotype controls (−80% at t = 12) (Figure 1D). Similarly, plasma cholesterol concentrations were significantly reduced by bFKB1 (−48% at t = 12) (Figure 1E) and further analysis of lipoprotein profiles revealed that in isotype control mice, plasma cholesterol was mainly contained in the pro‐atherogenic VLDL and LDL particles (Figure 1F). The observed reduction in plasma cholesterol in bFKB1‐treated mice was attributable to reductions in these atherogenic lipoproteins, without an effect on HDL particles. In addition to plasma cholesterol levels, the bFKB1 antibody significantly reduced plasma triglycerides as well (−76% at t = 12) (Figure 1G). Liver weight was significantly lower in bFKB1‐treated mice (−43%) (Figure 1H). Consistently, plasma concentrations of the hepatic function markers ALT and AST were significantly reduced at the study endpoint in bFKB1‐treated mice compared with isotype controls (ALT: −79%; AST: −21%) (Figure 1I). These findings demonstrate that 33 weeks of HFD feeding induced severe obesity and metabolic dysfunction and that bFKB1 treatment during the last 12 weeks counteracted the observed metabolic complications despite increased caloric intake.
FIGURE 1.
The bispecific anti‐FGFR1/KLB agonist antibody bFKB1 improved metabolic parameters in Ldlr−/−.Leiden mice. Body weight (A), food intake (B), fasted blood glucose (C), plasma insulin (D), plasma cholesterol (E), lipoprotein profile for plasma cholesterol (F), plasma triglycerides (G), liver weight (H), and plasma alanine transaminase (ALT) and aspartate transaminase (AST) (I) were determined at several timepoints throughout the study or at the study endpoint. Values are presented as mean ± SEM for n = 15 mice on HFD treated with isotype control antibody and n = 13 mice on HFD treated with bFKB1. *p < .05, **p < .01, ***p < .001 bFKB1 versus isotype control antibody.
3.2. Treatment with bFKB1 reduces adipocyte size, adipose tissue inflammation, and induces browning of white adipose tissue
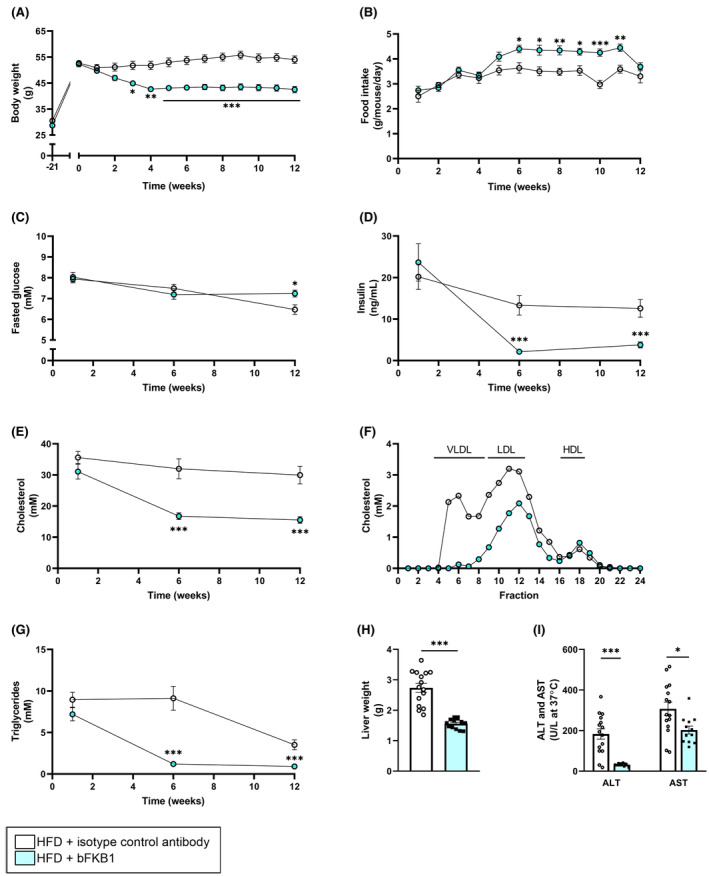
Since expression of FGFR1c, one of the two main target receptors of bFKB1, is minimal in the liver, its beneficial metabolic effects are likely exerted via different organs. FGFR1c and KLB are highly expressed in adipose tissue, which is therefore presumed to play a large role in mediating the metabolic effects of FGF receptor agonists. 33 , 34 Consequently, we determined the effects of bFKB1 on adipose tissue parameters. At sacrifice, epididymal white adipose tissue (eWAT) weight was significantly reduced in bFKB1‐treated mice relative to the isotype antibody controls (−22%) (Figure 2B). Morphological analysis of eWAT (Figure 2A) revealed a 16% reduction in adipocyte size (Figure 2C). Closer inspection of the distribution of cell size revealed that this reduction could predominantly be attributed to a significantly decreased number of large hypertrophic adipocytes (>8000 μm2) (−52%) (Figure 2D). Larger adipocytes are known to release more pro‐inflammatory mediators than smaller adipocytes. 35 This link between adipocyte hypertrophy and inflammation was reflected here, indicated by a significant reduction in the number of crown‐like structures in bFKB1‐treated mice (−70%) (Figure 2E). Further Western blot analysis of eWAT homogenates revealed a strong significant increase in UCP1 expression in bFKB1‐treated mice relative to isotype‐treated controls (8.5‐fold increase) (Figure 2F,G), indicative of white adipose tissue browning.
FIGURE 2.
Adipocyte size and adipose tissue inflammation are reduced by bFKB1. Representative histological photomicrographs of HPS‐stained epididymal white adipose tissue (eWAT) (A), epididymal WAT weight (B), average adipocyte size (C), adipocyte size distribution as percentage of total adipocytes (D), number of crown‐like structures (CLS) per 1000 adipocytes (E), uncoupling protein 1 (UCP1) protein expression in epididymal WAT normalized to HSP90 expression (F), and representative images of the Western blot bands (G). Values are presented as mean ± SEM for n = 15 mice on HFD treated with isotype control antibody and n = 13 mice on HFD treated with bFKB1. *p < .05, **p < .01, ***p < .001 bFKB1 versus isotype control antibody.
3.3. bKFB1 improves hepatic gene expression in Ldlr−/−.Leiden mice on HFD
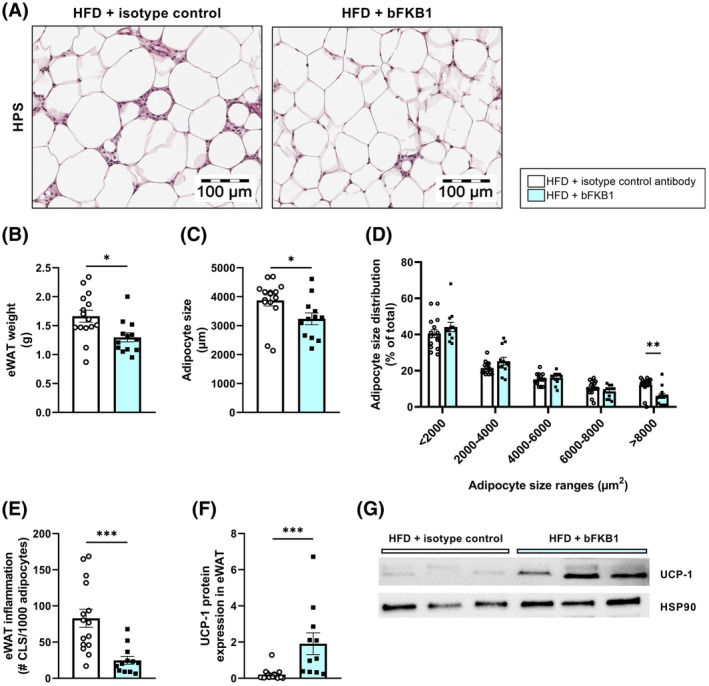
To investigate how treatment with bFKB1 affected the liver, next‐generation sequencing analysis of liver RNA was performed. A Principal Component Analysis (PCA) was carried out to visualize the entire transcriptome of each individual mouse (Figure 3A). The two component models explained 31% (PC1) and 10% (PC2) of the variation in the data. While there was an overlap between isotype control‐treated mice and bFKB1‐treated mice, both groups cluster clearly in their respective groups. bFKB1 treatment resulted in 3941 differentially expressed genes (DEGs) relative to isotype control‐treated mice. Next, we further investigated the nature of these DEGs and their involvement in specific biological pathways or processes was carried out, and showed that 97 pathways were significantly enriched, of which the pathways with Z‐scores ≤−2.0 and ≥2.0 (indicative of consistent inactivation or activation of a pathway) are shown in Figure 3B (the complete list is provided in Supplementary Figure S1). Among these, there were several pathways involved in lipid metabolism (e.g., bile acid and cholesterol biosynthesis and fatty acid oxidation pathways, all increased), inflammatory processes (e.g., leukocyte extravasation and IL‐8 signaling, both decreased) as well as fibrosis (e.g., integrin signaling, decreased). In bFKB1‐treated mice, the glucose‐6‐phosphate (GP6) signaling pathway was significantly inactivated as well. GP6 is the first intermediate of glucose metabolism and plays a pivotal role in several metabolic processes, including glycogen synthesis, de novo lipogenesis, and the pentose phosphate pathway. 36 This inactivation of the GP6 signaling pathway is reflected by the reduced insulin concentrations (Figure 1D), indicative of improved insulin sensitivity. A more elaborate breakdown of the hepatic transcriptomics data obtained in this study will be provided per pathological aspect of MASH.
FIGURE 3.
bFKB1 improves hepatic transcriptomics profile in Ldlr−/−.Leiden mice. Principal Component Analysis (PCA) in high‐fat diet (HFD)‐fed Ldlr−/−.Leiden mice treated with isotype control antibody or bFKB1 (A). Each dot represents the entire transcriptome of an individual mouse. The first principal component (PC1) accounts for as much variability in the data as possible and the second component (PC2) accounts for as much of the remaining variability. Gray and blue clusters represent the isotype control and bFKB1 groups, respectively. Heatmap showing significantly enriched pathways in bFKB1‐treated mice versus isotype control‐treated mice (B). Z‐scores indicate predicted activation (≥2.0, red) or inactivation (≤−2.0, green) and gray color indicates statistical significance (p < .01).
3.4. bFKB1 improves steatosis and lipid accumulation in the liver
To investigate the consequences of the beneficial metabolic changes induced by bFKB1, its effects in the liver were examined next. The HFD‐induced development of pronounced hepatic steatosis, which was reduced by bFKB1 (Figure 4A). Mice treated with the isotype control antibody developed both macrovesicular and microvesicular steatosis (Figure 4B,C). bFKB1 significantly lowered hepatic macrovesicular steatosis (−34%) and completely nullified hepatic microvesicular steatosis (−100%). Biochemical analysis of liver lipids revealed that bFKB1 significantly reduced hepatic triglycerides relative to isotype controls (−62%) (Figure 4D). Similar reductions were observed for hepatic cholesteryl esters (−56%) (Figure 4E) and hepatic‐free (unesterified) cholesterol (−34%) (Figure 4F) compared to isotype controls. To obtain more insight into how bFKB1 affected molecular processes in the liver, gene profiling analysis was performed. An upstream regulator analysis was carried out, which predicts the activation state of a protein, transcription factor or enzyme based on expression of downstream genes. Consistent with the observed effects on hepatic lipid accumulation, bFKB1 strongly activated ACOX1, indicative of increased fatty acid (beta) oxidation (Figure 4G). Moreover, bFKB1 modified regulators of lipogenesis including AGT and NR1I2, suggestive of reduced de novo lipogenesis.
FIGURE 4.
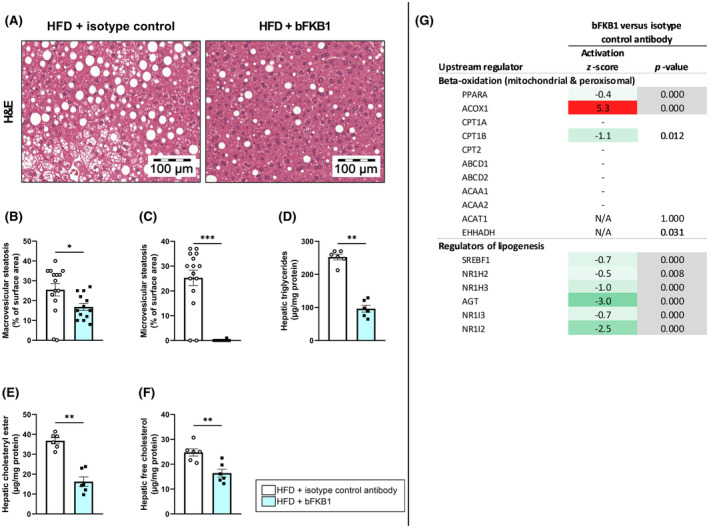
bFKB1 reduces hepatic steatosis and improves liver lipid concentrations. Representative histological photomicrographs of H&E‐stained liver cross‐sections (A), macrovesicular steatosis (B) and microvesicular steatosis (C) as percentage of surface area were determined at the study endpoint for n = 15 mice on HFD treated with isotype control antibody and n = 13 mice on HFD treated with bFKB1. Hepatic concentrations of triglycerides (D), cholesteryl ester (E), and free cholesterol (F) were determined at the study endpoint for n = 6 mice per group. Values are presented as mean ± SEM. *p < .05, **p < .01, ***p < .001 bFKB1 versus isotype control antibody. Activity of upstream regulators in homogenized liver tissue is expressed as z‐score that indicates the predicted activation state of a transcription factor or regulator. A z‐score <−2 indicates relevant inhibition (green) and z‐score >2 indicates relevant activation (red). The p‐value indicates significant enrichment of genes downstream of a regulator with p < .01 was considered statistically significant (gray). Hepatic expression of genes involved in beta‐oxidation and positive and negative regulators of lipogenesis (G).
3.5. Treatment with bFKB1 reduces inflammation and cytokine expression in the liver
Inflammation is a driving factor in progressing MASLD into MASH. bFKB1 significantly reduced the number of inflammatory cell clusters in the liver (−74%) (Figure 5A,B). Consistently, bFKB1 reduced intrahepatic protein concentrations of the pro‐inflammatory cytokines interleukin (IL)‐1β, IL‐6, interferon (IFN)‐γ, monocyte chemoattractant protein (MCP)‐1, and tumor necrosis factor (TNF)‐α as well as the anti‐inflammatory cytokine IL‐10 (Figure 5C). Hepatic expression of the anti‐inflammatory cytokine IL‐27p28/IL‐30, on the other hand, was significantly increased in bFKB1‐treated mice. Other cytokines including IL‐2, IL‐4, IL‐17A/F, IL‐33, interferon‐γ‐induced protein (IP)‐10, and keratinocyte chemoattractant/human growth‐regulated oncogene (KC/GRO), were not significantly altered with the intervention. An upstream regulator analysis of pro‐ and anti‐inflammatory cytokines revealed ameliorative effects of bFKB1 intervention at the transcriptomics level as well (Figure 5D). For example, in line with protein expression data, the upstream regulator analysis revealed reduced activation of the pro‐inflammatory cytokines IL‐1β, IL‐6, IFN‐ɤ, and TNF. Consistently, members of the nuclear factor κ light‐chain‐enhancer of activated B cells (NF‐κB) family also tended to be predicted to be inactivated with z‐scores of 1.5–1.7.
FIGURE 5.
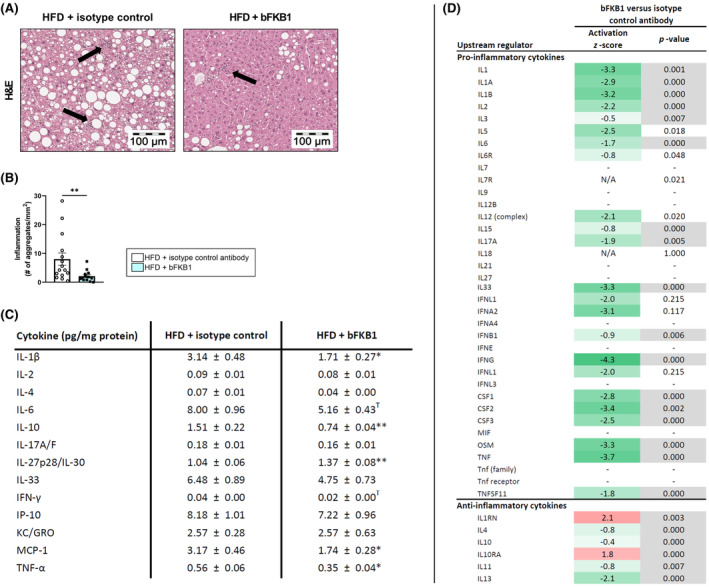
bFKB1 improves hepatic inflammation and cytokine expression. Representative histological photomicrographs of H&E‐stained liver cross‐sections with arrows indicating clusters of aggregated inflammatory cells (A). Inflammation expressed as the number of inflammatory aggregates pre mm2 (B). Concentrations of significantly modified cytokines measured in liver homogenates expressed as pg/mg protein (C). Values are presented as mean ± SEM for n = 15 mice on HFD treated with isotype control antibody and n = 13 mice on HFD treated with bFKB1. *p < .05, **p < .01, T: Tendency (p < .10), bFKB1 versus isotype control antibody. Activity of upstream regulators in homogenized liver tissue is expressed as z‐score that indicates the predicted activation state of a transcription factor or regulator. A z‐score <−2 indicates relevant inhibition (green) and z‐score >2 indicates relevant activation (red). The p‐value indicates significant enrichment of genes downstream of a regulator with p < .01 was considered statistically significant (gray). Hepatic expression of pro‐inflammatory and anti‐inflammatory cytokines (D). IFN, interferon; IL, interleukin; IP, interferon gamma‐induced protein; KC/GRO, keratinocyte chemoattractant/growth‐regulated oncogene; MCP, monocyte chemoattractant protein; TNF, tumor necrosis factor.
3.6. bFKB1 limits new collagen formation in the liver and improves hepatic expression of regulators of fibrosis
Hepatic fibrosis was assessed in Sirius Red‐stained liver sections (Figure 6A), revealing no significant difference in collagen deposition between mice treated with isotype control antibody or bFKB1 antibody (Figure 6B). Correspondingly, bFKB1 did not affect fibrosis stage: in both groups fibrosis was present within both perisinusoidal and periportal areas (stage F2 fibrosis) (Figure 6C). However, examination of de novo collagen formation in Ldlr−/−.Leiden mice revealed that bFKB1 had significant attenuating effects on the absolute amount of newly formed hydroxyproline in the liver during the final 2 weeks of the study (−49%) (Figure 6D). A reason for this apparent discrepancy is the presence of a relatively high amount of pre‐existent collagen at the start of the treatment period, which contributes significantly to the total amount of collagen at the endpoint. 10 At the transcriptomics level, bFKB1 had broad inactivating effects on transforming growth factor (TGF)‐β and suppressor of mothers against decapentaplegic (SMAD) signaling (downstream of TGFβ) (Figure 6E). One master regulator was significantly activated in bFKB1‐treated mice, namely SMAD7, an inhibitory SMAD that functions as TGFβ receptor antagonist. Other important regulators of profibrotic processes were significantly inactivated in bFKB1‐treated mice as well, including VEGF‐A and YAP/TAZ signaling (Figure 6E). Further investigation of collagen expression revealed the significant downregulation of several collagen sub‐types including COL1A1, COL3A1, and COL4A1 and of genes involved in collagen crosslinking, for example, LOX and LOXL2 (supplementary Figure S2). These expression patterns underline the inhibitory effects of bFKB1 on new collagen formation in the liver. Although total liver fibrosis at endpoint was not affected, bFKB1 reduced new collagen synthesis and had broad beneficial effects on genes and upstream regulators involved in fibrotic processes, suggesting that prolonged treatment with bFKB1 may be effective in reducing hepatic fibrosis.
FIGURE 6.
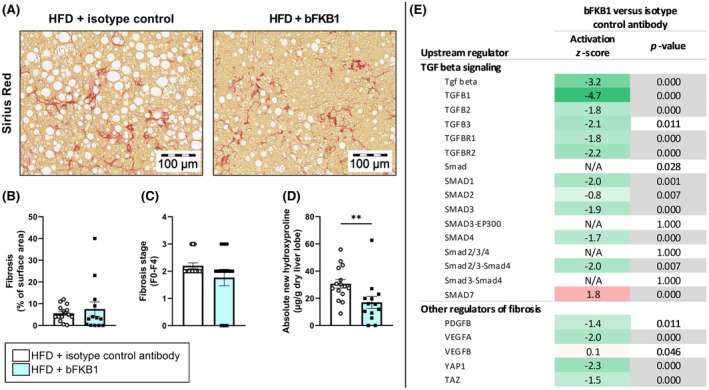
Treatment with bFKB1 reduces formation of new collagen in the liver and reduces hepatic expression of upstream regulators of fibrosis. Representative histological photomicrographs of Sirius Red‐stained liver cross‐sections (A), fibrosis as percentage of surface area (B), fibrosis stage (C), and absolute newly formed hydroxyproline (D). Values are presented as mean ± SEM for n = 15 mice on HFD treated with isotype control antibody and n = 13 mice on HFD treated with bFKB1. **p < .01 bFKB1 versus isotype control antibody. Activity of upstream regulators in homogenized liver tissue is expressed as z‐score that indicates the predicted activation state of a transcription factor or regulator. A z‐score <−2 indicates relevant inhibition (green) and z‐score >2 indicates relevant activation (red). The p‐value indicates significant enrichment of genes downstream of a regulator with p < .01 was considered statistically significant (gray). Hepatic expression of genes involved in transforming growth factor (TGF)‐beta signaling and other regulators of fibrosis (E).
3.7. The anti‐FGFR1/KLB agonist antibody bFKB1 improves HFD‐induced atherosclerosis
Atherosclerosis development in the aortic root was determined next. Prolonged HFD feeding resulted in development of severe atherosclerotic lesions (Figure 7A). Cholesterol exposure (concentration × weeks) throughout the whole study, including the 20‐week run‐in on HFD was significantly lower in bFKB1‐treated mice compared to untreated controls (−22%) (Figure 7B). Accordingly, bFKB1 significantly reduced the total atherosclerotic lesion area compared to isotype control antibody mice (−38%) (Figure 7C). Atherosclerotic lesions in the isotype control antibody group were predominantly severe type V lesions accompanied by some type IV lesions (Figure 7D). The lesion‐lowering effect of bFKB1 could be attributed specifically to a significant reduction in severe type V lesions compared to controls (−35%). These data indicate that bFKB1 attenuates the progression of atherosclerosis to more severe lesions, which is mainly caused by the marked cholesterol‐lowering effect of bFKB1.
FIGURE 7.
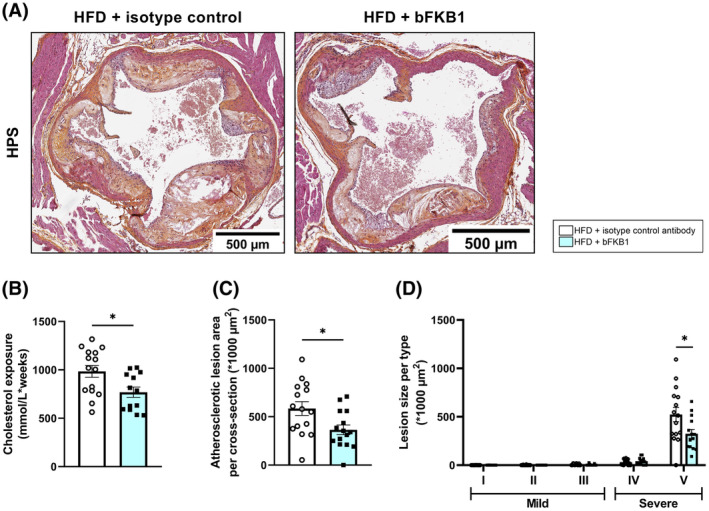
bFKB1 reduces atherosclerosis parameters in Ldlr−/−.Leiden mice. Representative images of HPS‐stained liver cross‐sections (A), total cholesterol exposure calculated as concentration*weeks (B), atherosclerotic lesion area per cross‐section (C), and lesion size per type (D) were determined at the study endpoint. Values are presented as mean ± SEM for n = 15 mice on HFD treated with isotype control antibody and n = 13 mice on HFD treated with bFKB1. *p < .05 bFKB1 versus isotype control antibody.
4. DISCUSSION
The current obesity pandemic and accompanying rise in metabolic complicatio™ns necessitates development of therapeutic interventions and FGF21 forms an interesting target due to its beneficial metabolic effects. Previous preclinical studies have demonstrated that the FGF21 mimetic bFKB1 increases energy expenditure and may ameliorate obesity and hyperlipidaemia. 6 , 7 , 8 Using a model that shows high translatability to humans with obesity and associated comorbidities, we have demonstrated the beneficial effects of bFKB1 on metabolic parameters, liver steatosis, liver inflammation, adipose inflammation, and atherosclerosis. Furthermore, analysis of new collagen synthesis and gene expression profiles in the liver indicated a slow‐down of fibrosis‐related processes.
In this study, while treatment with bFKB1 increased food intake at several timepoints, body weight was significantly reduced throughout the treatment period. This is indicative of increased energy expenditure, consistent with reported effects of this and other FGF21 analogues in preclinical studies. 6 , 7 bFKB1 significantly and strongly reduced plasma insulin concentrations as well, indicative of improved insulin sensitivity compared to untreated controls, where insulin remained high throughout the study. We demonstrate that in white adipose tissue, bFKB1 increases protein expression of the thermogenic protein UCP1, which enables conversion of energy to heat upon stimulation by cold exposure. 37 This is indicative of white adipose tissue browning and thereby increased energy expenditure, which may in turn explain the increased food intake observed in these mice. Consistently, FGF21 has been shown to play a role in thermogenic recruitment of white adipose tissues and browning thereof, and FGF21‐deficient mice were demonstrated to have impaired ability to adapt to cold exposure. 38 In addition, Kolumam et al. reported that bFKB1 significantly increased energy expenditure without changing the relative rate of fat and carbohydrate utilization. 8
Hepatic expression of the FGF receptor isoform 1c is low and the hepatic effects of bFKB1 observed here are therefore likely indirect, for example by signaling through other organs such as adipose tissue or brain. Lan et al. demonstrated that KLB expression in neuronal tissue is particularly indispensable for FGF21 signaling, resulting in weight loss and improved glycemia. 7 The improved insulin resistance induced by FGF21 analogues contributes to lowering of free fatty acid levels in the circulation. 39 The resulting reduction in flux of free fatty acids from adipose tissue to the liver ultimately improves liver health as well, therefore making the liver a tertiary target of FGF21 signaling. In our study, bFKB1 treatment significantly reduced inflammation in the epididymal white adipose tissue depot, which is likely to have directly contributed to alleviating hepatic inflammation. Previously, it was demonstrated that surgical removal of inflamed epididymal white adipose tissue attenuated MASH development, signifying its causal involvement in MASH pathogenesis. 40 In addition, bFKB1 indeed improved hepatic steatosis and triglyceride, cholesteryl ester, and free cholesterol concentrations. The latter data are in line with clinical data, with bFKB1 showing substantial and dose‐dependent reduction of liver fat in a phase 1b study. 41
Hepatic inflammation is a driving factor in progressing MASLD into MASH and was shown here to be drastically improved by bFKB1, as demonstrated by the reduced number of aggregated inflammatory cells. Moreover, bFKB1 reduced gene and protein expression of pro‐inflammatory cytokines including IL‐1β, IL‐6, and TNF, that are (in part) regulated through the NF‐κB signaling pathway. In an in vitro study in macrophages, FGF21 was shown to have anti‐inflammatory effects mediated by the nuclear transcription factor‐E2‐related factor 2 (Nrf2), 42 which inhibits both NF‐κB activation and nuclear translocation. 43 In the aforementioned in vitro studies, the authors demonstrate that FGF21 suppressed NF‐κB signaling and consequently reduced secretion of downstream effector pro‐inflammatory cytokines. 42 Our findings illustrate that the beneficial effects of the FGF21 mimetic bFKB1 on metabolic parameters and adipose tissue inflammation translate into reduced hepatic steatosis and inflammation.
In the current study, mice were fed an HFD for 32 weeks in total, with intervention in the final 12 weeks. This exposure to the HFD resulted in development of substantial hepatic fibrosis in both perisinusoidal and periportal areas. Treatment with bFKB1 did not affect total hepatic collagen deposition at the study endpoint. However, de novo collagen synthesis during the final 2 weeks of the study and expression of collagen genes at endpoint were significantly decreased. These data demonstrate that while pre‐existing collagen is unaffected by the treatment, intervention with bFKB1 does reduce the deposition of newly formed collagen, thereby preventing further progression of hepatic fibrosis. FGF21 inhibits the activation of stellate cells via downregulation of TGFβ, 32 which we confirmed here, with bFKB1 intervention resulting in broad inactivation of TGFβ signaling, with concomitantly reduced downstream activation of SMAD signaling. Additionally, our upstream regulator analysis revealed a significant inactivation of YAP/TAZ signaling in bFKB1‐treated mice. TAZ is the downstream activator in the Hippo pathway that plays a critical role in progression of MASLD into MASH. 32 In a clinical study, it was demonstrated that compared to patients with simple steatosis, livers of patients with MASH exhibited increased TAZ expression. 44 Moreover, elevated hepatic‐free cholesterol concentrations are known to stabilize TAZ, ultimately promoting collagen synthesis, secretion, and fibrogenesis. 45 Here, bFKB1 reduced hepatic‐free cholesterol concentrations and since changes detected at the transcriptomics level have been shown to precede changes in histologically measured fibrosis, 16 the effects of bFKB1 on YAP/TAZ signaling may ultimately translate into reduced hepatic fibrosis. Other preclinical studies have demonstrated the attenuating effects of FGF21 analogues on hepatic fibrosis, either in preventive 46 or treatment settings. 47 Similarly, in a clinical study in patients with MASH with stage F2/F3 fibrosis, treatment with the long‐acting pegylated FGF21 analogue pegozafermin resulted in improved fibrosis and MASH resolution. 48 In a different clinical phase 2b study, it was shown that after 24 weeks, intervention with the bivalent Fc‐FGF21 analog efruxifermin improved liver fibrosis and resolved NASH, 49 leading to further evaluation in a phase 3 clinical study. Considering the substantial effects of bFKB1 on processes involved in fibrogenesis observed here, a longer treatment duration may eventually result in improvement in histologically measured fibrosis as well.
Cardiovascular complications are the main cause of death in patients with MASLD‐MASH. 17 Consequently, therapeutics for MASLD‐MASH should preferably also have beneficial effects on cardiovascular risk factors and disease end points. Therefore, we studied the effects of bFKB1 on progression of pre‐existent atherosclerosis. Despite the 20‐week induction period that elevated plasma cholesterol levels substantially, 12 weeks of bFKB1 intervention reduced cholesterol levels to such an extent that total cholesterol exposure was significantly reduced relative to controls. The ameliorative effects of bFKB1 on plasma cholesterol and triglycerides are consistent with other preclinical 6 , 8 and two clinical studies 41 , 50 that used the same intervention. Consequently, progression of pre‐existent atherosclerosis 11 , 14 was significantly reduced, which could primarily be attributed to a reduction in severe type V lesions. Using a preventive study design, similar effects were reported in another study that used recombinant human FGF21 in non‐obese mice, showing reduced lesion severity and improved lesion stability. 51 Apart from preclinical data, no clinical studies have been carried out that studied the application of FGF21 interventions in cardiovascular disease. However, some clinical studies report improvements in lipid profiles with bFKB1 and other FGF21 analogues, 52 , 53 which in the long run may be expected to result in improved cardiovascular outcomes. We add to these data by demonstrating the significant beneficial effects of bFKB1 on atherosclerosis in a model with established obesity and insulin resistance.
In conclusion, we have demonstrated the beneficial effects of the FGF21 mimetic bFKB1 in a preclinical model showing high translatability to humans with obesity and associated comorbidities (See Graphical abstract). Treatment with bFKB1 reduced plasma lipids, hepatic steatosis, hepatic inflammation, and newly formed collagen in the liver. Additional anti‐atherosclerotic effects alongside reduced adipose tissue inflammation demonstrate that bFKB1 has potential for the treatment of metabolic complications as a result of obesity.
AUTHOR CONTRIBUTIONS
José A. Inia, Hans M. G. Princen, and Martine C. Morrison contributed to the conception and design of the research. José A. Inia, Joline Attema, Christa de Ruiter, Aswin L. Menke, Martien P. M. Caspers, and Lars Verschuren performed the experiments. José A. Inia and Martine C. Morrison analyzed the data. José A. Inia, Hans M. G. Princen, and Martine C. Morrison interpreted the results of the experiments. José A. Inia prepared the figures. José A. Inia, Hans M. G. Princen, and Martine C. Morrison drafted the manuscript. José A. Inia, Joline Attema, Christa de Ruiter, Aswin L. Menke, Martien P. M. Caspers, Lars Verschuren, Maria Wilson, Alexander Arlantico, Hans D. Brightbill, J. Wouter Jukema, Anita M. van den Hoek, Hans M.G. Princen, Mark Z. Chen, and Martine C. Morrison approved the final version of the manuscript.
FUNDING INFORMATION
Genentech Inc. provided a part of the funding for this study. The work was further supported by the TNO research programs “Lifestyle‐Related Disease Models” and “Functional Biomarkers.” The funders had no role in data collection and raw data analysis, decision to publish, or preparation of the manuscript.
DISCLOSURES
M.W., H.D.B., and A.A. are employees of Genentech Inc., South San Francisco, USA. M.C. is a former employee of Genentech Inc. and present employee of Takeda Pharmaceuticals, San Diego, USA.
Supporting information
Supplementary Figure S1.
ACKNOWLEDGMENTS
The authors would like to thank Jessica Snabel, Nicole Worms, Anita van Straalen‐van Nieuwkoop, Nanda Keijzer, Wim van Duyvenvoorde, and Nikki van Trigt for their excellent technical assistance.
Inia JA, Attema J, de Ruiter C, et al. Therapeutic effects of FGF21 mimetic bFKB1 on MASH and atherosclerosis in Ldlr−/−.Leiden mice. The FASEB Journal. 2024;38:e70087. doi: 10.1096/fj.202401397R
Mark Z. Chen and Martine C. Morrison contributed equally to this study.
DATA AVAILABILITY STATEMENT
The transcriptomics data are available on Gene Expression Omnibus (GEO), data set GSE255321.
REFERENCES
- 1. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Friedman SL, Neuschwander‐Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24(7):908‐922. doi: 10.1038/S41591-018-0104-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Goetz R, Beenken A, Ibrahimi OA, et al. Molecular insights into the klotho‐dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol Cell Biol. 2007;27(9):3417‐3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dunshee DR, Bainbridge TW, Kljavin NM, et al. Fibroblast activation protein cleaves and inactivates fibroblast growth factor 21. J Biol Chem. 2016;291(11):5986‐5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jin L, Yang R, Geng L, Xu A. Fibroblast growth factor‐based pharmacotherapies for the treatment of obesity‐related metabolic complications. Annu Rev Pharmacol Toxicol. 2023;63:359‐382. [DOI] [PubMed] [Google Scholar]
- 6. Chen MZ, Chang JC, Zavala‐Solorio J, et al. FGF21 mimetic antibody stimulates UCP1‐independent brown fat thermogenesis via FGFR1/βKlotho complex in non‐adipocytes. Mol Metab. 2017;6(11):1454‐1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lan T, Morgan DA, Rahmouni K, et al. FGF19, FGF21, and an FGFR1/β‐klotho‐activating antibody act on the nervous system to regulate body weight and glycemia. Cell Metab. 2017;26(5):709‐718.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kolumam G, Chen MZ, Tong R, et al. Sustained Brown fat stimulation and insulin sensitization by a humanized bispecific antibody agonist for fibroblast growth factor receptor 1/βKlotho complex. EBioMedicine. 2015;2(7):730‐743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tillman EJ, Rolph T. FGF21: an emerging therapeutic target for non‐alcoholic steatohepatitis and related metabolic diseases. Front Endocrinol (Lausanne). 2020;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gart E, Van Duyvenvoorde W, Snabel JM, et al. Translational characterization of the temporal dynamics of metabolic dysfunctions in liver, adipose tissue and the gut during diet‐induced NASH development in Ldlr−/−.Leiden mice. Heliyon. 2023;9(3):e13985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Van den Hoek AM, Verschuren L, Worms N, et al. A translational mouse model for NASH with advanced fibrosis and atherosclerosis expressing key pathways of human pathology. Cells. 2020;9(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morrison MC, Verschuren L, Salic K, et al. Obeticholic acid modulates serum metabolites and gene signatures characteristic of human NASH and attenuates inflammation and fibrosis progression in Ldlr−/−.Leiden mice. Hepatol Commun. 2018;2(12):1513‐1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gutiérrez‐Cuevas J, Santos A, Armendariz‐Borunda J. Pathophysiological molecular mechanisms of obesity: a link between MAFLD and NASH with cardiovascular diseases. Int J Mol Sci. 2021;22(21). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Inia JA, Stokman G, Morrison MC, et al. Semaglutide has beneficial effects on non‐alcoholic steatohepatitis in Ldlr−/−.Leiden mice. Int J Mol Sci. 2023;24(10):8494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Inia JA, De Jong JCBC, Keijzer N, et al. Effects of repeated weight cycling on non‐alcoholic steatohepatitis in diet‐induced obese mice. FASEB J. 2024;38(7):e23579. [DOI] [PubMed] [Google Scholar]
- 16. Van Koppen A, Verschuren L, Van den Hoek AM, et al. Uncovering a predictive molecular signature for the onset of NASH‐related fibrosis in a translational NASH mouse model. Cell Mol Gastroenterol Hepatol. 2018;5(1):83‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martínez‐Arranz I, Bruzzone C, Noureddin M, et al. Metabolic subtypes of patients with NAFLD exhibit distinctive cardiovascular risk profiles. Hepatology. 2022;76(4):1121‐1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jacobs SAH, Gart E, Vreeken D, et al. Sex‐specific differences in fat storage, development of non‐alcoholic fatty liver disease and brain structure in juvenile HFD‐induced obese Ldlr−/−.Leiden mice. Nutrients. 2019;11(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Westerterp M, Van der Hoogt CC, De Haan W, et al. Cholesteryl ester transfer protein decreases high‐density lipoprotein and severely aggravates atherosclerosis in APOE*3‐Leiden mice. Arterioscler Thromb Vasc Biol. 2006;26(11):2552‐2559. [DOI] [PubMed] [Google Scholar]
- 20. Post SM, De Crom R, Van Haperen R, Van Tol A, Princen HMG. Increased fecal bile acid excretion in transgenic mice with elevated expression of human phospholipid transfer protein. Arterioscler Thromb Vasc Biol. 2003;23(5):892‐897. [DOI] [PubMed] [Google Scholar]
- 21. Blaauboer ME, Emson CL, Verschuren L, et al. Novel combination of collagen dynamics analysis and transcriptional profiling reveals fibrosis‐relevant genes and pathways. Matrix Biol J Int Soc Matrix Biol. 2013;32(7–8):424‐431. [DOI] [PubMed] [Google Scholar]
- 22. Xi Y, LaCanna R, Ma HY, et al. A WISP1 antibody inhibits MRTF signaling to prevent the progression of established liver fibrosis. Cell Metab. 2022;34(9):1377‐1393.e8. [DOI] [PubMed] [Google Scholar]
- 23. Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41(6):1313‐1321. [DOI] [PubMed] [Google Scholar]
- 24. Liang W, Menke AL, Driessen A, et al. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS ONE. 2014;9(10):1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mulder P, Liang W, Wielinga P, et al. Macrovesicular steatosis is associated with development of lobular inflammation and fibrosis in diet‐induced non‐alcoholic steatohepatitis (NASH). Inflamm Cell Signal. 2015;2:e804. [Google Scholar]
- 26. Fromenty B, Berson A, Pessayre D. Microvesicular steatosis and steatohepatitis: role of mitochondrial dysfunction and lipid peroxidation. J Hepatol. 1997;26(suppl 1):13‐22. [DOI] [PubMed] [Google Scholar]
- 27. Tiniakos DG, Vos MB, Brunt EM. Nonalcoholic fatty liver disease: pathology and pathogenesis. Annu Rev Pathol. 2010;5:145‐171. [DOI] [PubMed] [Google Scholar]
- 28. Galarraga M, Campión J, Munõz‐Barrutia A, et al. Adiposoft: automated software for the analysis of white adipose tissue cellularity in histological sections. J Lipid Res. 2012;53(12):2791‐2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tengeler AC, Gart E, Wiesmann M, et al. Propionic acid and not caproic acid, attenuates nonalcoholic steatohepatitis and improves (cerebro) vascular functions in obese Ldlr−/−.Leiden mice. FASEB J. 2020;34(7):9575‐9593. [DOI] [PubMed] [Google Scholar]
- 30. Kühnast S, Van der Tuin SJL, Van der Hoorn JWA, et al. Anacetrapib reduces progression of atherosclerosis, mainly by reducing non‐HDL‐cholesterol, improves lesion stability and adds to the beneficial effects of atorvastatin. Eur Heart J. 2015;36(1):39‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pouwer MG, Pieterman EJ, Worms N, et al. Alirocumab, evinacumab, and atorvastatin triple therapy regresses plaque lesions and improves lesion composition in mice. J Lipid Res. 2020;61(3):365‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xu P, Zhang Y, Liu Y, et al. Fibroblast growth factor 21 attenuates hepatic fibrogenesis through TGF‐β/smad2/3 and NF‐κB signaling pathways. Toxicol Appl Pharmacol. 2016;290:43‐53. [DOI] [PubMed] [Google Scholar]
- 33. Fon Tacer K, Bookout AL, Ding X, et al. Research resource: comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol Endocrinol. 2010;24(10):2050‐2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hultman K, Scarlett JM, Baquero AF, et al. The central fibroblast growth factor receptor/beta klotho system: comprehensive mapping in Mus musculus and comparisons to nonhuman primate and human samples using an automated in situ hybridization platform. J Comp Neurol. 2019;527(12):2069‐2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Skurk T, Alberti‐Huber C, Herder C, Hauner H. Relationship between adipocyte size and adipokine expression and secretion. J Clin Endocrinol Metab. 2007;92(3):1023‐1033. [DOI] [PubMed] [Google Scholar]
- 36. Rajas F, Gautier‐Stein A, Mithieux G. Glucose‐6 phosphate, a central hub for liver carbohydrate metabolism. Metabolites. 2019;9(12). doi: 10.3390/METABO9120282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84(1):277‐359. [DOI] [PubMed] [Google Scholar]
- 38. Fisher FF, Kleiner S, Douris N, et al. FGF21 regulates PGC‐1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 2012;26(3):271‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Boden G. 45Obesity, insulin resistance and free fatty acids. Curr Opin Endocrinol Diabetes Obes. 2011;18(2):139‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mulder P, Morrison MC, Wielinga PY, Van Duyvenvoorde W, Kooistra T, Kleemann R. Surgical removal of inflamed epididymal white adipose tissue attenuates the development of non‐alcoholic steatohepatitis in obesity. Int J Obes. 2016;40(4):675‐684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wong C, Dash A, Fredrickson J, et al. Fibroblast growth factor receptor 1/Klothoβ agonist BFKB8488A improves lipids and liver health markers in patients with diabetes or NAFLD: a phase 1b randomized trial. Hepatology. Published online. 2022;78(3):847‐862. [DOI] [PubMed] [Google Scholar]
- 42. Yu Y, He J, Li S, et al. Fibroblast growth factor 21 (FGF21) inhibits macrophage‐mediated inflammation by activating Nrf2 and suppressing the NF‐κB signaling pathway. Int Immunopharmacol. 2016;38:144‐152. [DOI] [PubMed] [Google Scholar]
- 43. Saha S, Buttari B, Panieri E, Profumo E, Saso L. An overview of Nrf2 signaling pathway and its role in inflammation. Molecules. 2020;25(22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang X, Zheng Z, Caviglia JM, et al. Hepatocyte TAZ/WWTR1 promotes inflammation and fibrosis in nonalcoholic steatohepatitis. Cell Metab. 2016;24(6):848‐862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang X, Cai B, Yang X, et al. Cholesterol stabilizes TAZ in hepatocytes to promote experimental non‐alcoholic steatohepatitis. Cell Metab. 2020;31(5):969‐986.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu C, Schönke M, Spoorenberg B, et al. FGF21 protects against hepatic lipotoxicity and macrophage activation to attenuate fibrogenesis in nonalcoholic steatohepatitis. elife. 2023;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bao L, Yin J, Gao W, Wang Q, Yao W, Gao X. A long‐acting FGF21 alleviates hepatic steatosis and inflammation in a mouse model of non‐alcoholic steatohepatitis partly through an FGF21‐adiponectin‐IL17A pathway. Br J Pharmacol. 2018;175(16):3379‐3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Loomba R, Sanyal AJ, Kowdley KV, et al. Randomized, controlled trial of the FGF21 analogue pegozafermin in NASH. N Engl J Med. 2023;389(11):998‐1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Harrison SA, Frias JP, Neff G, et al. Safety and efficacy of once‐weekly efruxifermin versus placebo in non‐alcoholic steatohepatitis (HARMONY): a multicentre, randomised, double‐blind, placebo‐controlled, phase 2b trial. Lancet Gastroenterol Hepatol. 2023;8(12):1080‐1093. [DOI] [PubMed] [Google Scholar]
- 50. Baruch A, Wong C, Chinn LW, et al. Antibody‐mediated activation of the FGFR1/Klothoβ complex corrects metabolic dysfunction and alters food preference in obese humans. Proc Natl Acad Sci USA. 2020;117(46):28992‐29000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu C, Schönke M, Zhou E, et al. Pharmacological treatment with FGF21 strongly improves plasma cholesterol metabolism to reduce atherosclerosis. Cardiovasc Res. 2022;118(2):489‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kim AM, Somayaji VR, Dong JQ, et al. Once‐weekly administration of a long‐acting fibroblast growth factor 21 analogue modulates lipids, bone turnover markers, blood pressure and body weight differently in obese people with hypertriglyceridaemia and in non‐human primates. Diabetes Obes Metab. 2017;19(12):1762‐1772. [DOI] [PubMed] [Google Scholar]
- 53. Rader DJ, Maratos‐Flier E, Nguyen A, et al. LLF580, an FGF21 analog, reduces triglycerides and hepatic fat in obese adults with modest hypertriglyceridemia. J Clin Endocrinol Metab. 2022;107(1):e57‐e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1.
Data Availability Statement
The transcriptomics data are available on Gene Expression Omnibus (GEO), data set GSE255321.