

Abstract
Perovskite solar cells (PSCs) offer an efficient, inexpensive alternative to current photovoltaic technologies, with the potential for manufacture via high-throughput coating methods. However, challenges for commercial-scale solution-processing of metal-halide perovskites include the use of harmful solvents, the expense of maintaining controlled atmospheric conditions, and the inherent instabilities of PSCs under operation. Here, we address these challenges by introducing a high volatility, low toxicity, biorenewable solvent system to fabricate a range of 2D perovskites, which we use as highly effective precursor phases for subsequent transformation to α-formamidinium lead triiodide (α-FAPbI3), fully processed under ambient conditions. PSCs utilising our α-FAPbI3 reproducibly show remarkable stability under illumination and elevated temperature (ISOS-L-2) and “damp heat” (ISOS-D-3) stressing, surpassing other state-of-the-art perovskite compositions. We determine that this enhancement is a consequence of the 2D precursor phase crystallisation route, which simultaneously avoids retention of residual low-volatility solvents (such as DMF and DMSO) and reduces the rate of degradation of FA+ in the material. Our findings highlight both the critical role of the initial crystallisation process in determining the operational stability of perovskite materials, and that neat FA+-based perovskites can be competitively stable despite the inherent metastability of the α-phase.
Subject terms: Solar cells, Energy, Devices for energy harvesting
The use of harmful solvents to fabricate stable devices hampers the commercialization of perovskite solar cells. Here, the authors introduce a biorenewable solvent system and precursor-phase engineering to realize stable formamidinium lead triiodide-based solar cells.
Introduction
Scalable, reproducible and controlled crystallisation of highly stable metal halide perovskite thin films is crucial to the commercial implementation of this promising class of semiconductor materials in optoelectronic applications. Ideally, solution-processed perovskite deposition should employ inks based on non-toxic and volatile solvents and should result in the controlled formation of a well-mixed and low defect perovskite phase that – crucially – possesses long term operational stability under external stimuli such as heat, light and moisture1–3.
Significant attention has been paid to the relationship between halide perovskite composition (ABX3 – where A is an organic or alkali-metal cation, B is a lead or tin cation, and X are halide anions) and operational stability. Exchanging the archetypical A-site methylammonium (MA+) cation for formamidinium (FA+) significantly increases thermal and photochemical stability but introduces a phase instability. The photoactive α-FAPbI3 phase is only metastable at room temperature, and the transition to the yellow non-perovskite δ-phase is accelerated by the presence of moisture4. This instability has been circumvented by alloying FA+ with caesium cations (Cs+)2. However, A-site inhomogeneity has been linked to both poor optoelectronic performance5 and other long-term degradation pathways, such as the emergence of Cs+-rich impurity phases6,7. Further, of all the elements employed in halide perovskites, the scarcity of Cs poses the greatest challenge in availability for TW scale PV production8 and thus routes realising stable neat-FA+ perovskites are greatly advantageous.
However, despite the emphasis on ABX3 composition as the key factor controlling perovskite operational stability, properties such as the types9 and density10 of defects, impurity phases11, compositional inhomogeneity6, residual solvents12–14 and residual strain15 have also been shown to influence long-term stability. All such properties are imparted into the perovskite thin layer during crystallisation of the ABX3 structure, which can proceed via a range of intermediates or precursor phases, including solvate16–18 and polytype7,19 phases and phases incorporating sacrificial ions20. Given the origin of these properties, it is imperative that the relationship between crystallisation pathway, precursor phase formation and resulting ABX3 perovskite stability be investigated and understood more widely.
Despite substantial research over the past decade21,22, highly toxic N,N-dimethylformamide (DMF) remains ubiquitous in halide perovskite inks, while operational instability of perovskites-based optoelectronics remains a key barrier to their wider application1. Here we simultaneously combat the challenges of perovskite ink toxicity, processing-induced instability and compositional heterogeneity. By developing a precursor ink that utilises a highly volatile, low toxicity solvent mixture, we replace conventional precursor phases with a solid-state 2D perovskite material. By exchanging the organic cations in the 2D precursor phase with FA+, we enable the subsequent crystallisation of α-FAPbI3 perovskite. We not only find that our α-FAPbI3 performs effectively in PSCs, but that 2D-precursor phase growth produces α-FAPbI3 with substantially improved ambient, thermal and photostability in comparison to any other perovskite composition or processing route that we fabricate herein2,3. We are able to fabricate neat FAPbI3 thin films that are stable for >3000 hours under harsh heat, light and moisture conditions, and achieve a promising champion lifetime to 80% of initial performance (t80) of 800 hours and no degradation (t100) for more than 1930 hours under ISOS-L-2 (85 °C, 1-sun equivalent) and ISOS-D-3 (85 °C, 85% relative humidity), respectively, when integrated into PSCs. Our work enables us to distinguish how different processing variables impact perovskite stability and demonstrate why conventional solution processing routes are not only problematic for toxicity but are fundamentally linked to halide perovskite instability.
Results and Discussion
A biorenewable and low toxicity solvent system
As solution processing at industrial scale dictates that the majority of solvents employed must be volatilised into the atmosphere23, the hazard of employing toxic solvents in PSC production at increasingly large scale are substantial. In light of this, several DMF-free solvent systems have been established for perovskite solution processing, notably based on N,N-dimethylacetamide (DMAc)24, γ-butyrolactone (GBL)25, 2-methoxyethanol (2-ME)26 and acetonitrile (ACN) in combination with methylamine gas27. However, as Fig. 1a shows, DMAc, 2-ME and ACN do not provide the desired reduction in toxicity, while GBL is an illegal narcotic on account of its metabolite, γ-hydroxybutyrate (GHB)28. Furthermore, the use of MA gas as a co-solvent, poses further problems of volatility, where escape of the MA gas can render the solution unstable and pose a toxicology risk itself. Moreover, while dimethyl sulfoxide (DMSO) – the typical companion to DMF in perovskite solution processing – is non-toxic as a neat liquid, in the context of lead salt solvation the proficiency of DMSO as a percutaneous absorption (skin-penetration) enhancer29,30 is of acute concern for at-scale processing (Fig. 1b).
Fig. 1. Scalable processing compatibility.
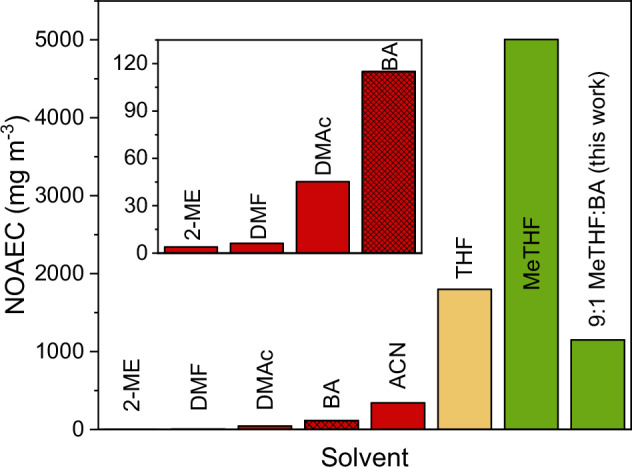
Preferential properties of 2-methyltetrahydrofuran (MeTHF) and THF as solvents in industrial processes where vapour phase exposure is unavoidable. Solvents shown in green are produced industrially from biorenewable sources, in yellow may be produced biorenewably but typically are not, and in red cannot be produced from renewable sources. No observed adverse effect concentrations (NOAECs) are taken from the European Chemicals Agency (ECHA) database and are calculated for workplace (except n-butylamine, BA, where the value for general population is used) exposure via inhalation of solvent vapours.
Here we investigate and develop a solvent system consisting of biorenewable cyclic ethers and alkylamine liquids, in which the latter serves both as a coordinating co-solvent and subsequently as a component within the 2D perovskite precursor phase. This system can be considered part of a family of solvent mixtures all comprising poorly Pb2+-coordinating solvents as the majority component alongside strongly Pb2+-coordinating solvents26. Here cyclic ethers take on the role of the poorly coordinating solvent while butylamine (BA), which is liquid at room temperature, acts as the strongly coordinating solvent. We find that both tetrahydrofuran (THF) and 2-methyltetrahydrofuran (MeTHF) are suitable poorly-coordinating solvents, however we focus on the latter on account of its biorenewable production, significantly lower toxicity and favourable vapour pressure for rapid at-scale solution processing. Vidal, et al23., recently highlighted that THF’s high vapour pressure renders its lifetime environmental impact relatively large compared with less volatile perovskite solvents due to additional greenhouse gas emissions resulting from energy-intensive solvent recapture. However, a lifecycle analysis of MeTHF conducted by Slater, et al31., demonstrated a 97 % reduction in lifetime emissions associated with the use of MeTHF as compared to THF, due mostly to its biorenewable production and significantly lower vapour pressure. Moreover, MeTHF has recently been recommended by both the European Medicines Agency (EMA) and the Food and Drug Administration (FDA) as a Class 3 solvent (“solvents with low toxic potential”) with a permitted daily exposure limit of 50 mg day−1 28. This exposure limit is much higher than that of THF (7.2 mg day−1), which Vidal, et al23., demonstrate already possesses significant toxicological advantages over other common perovskite solvents. Furthermore, as solution processing demands that the majority of solvents employed are volatilised, the other safety-critical parameters for at-scale processing are the no observed adverse effect concentration (NOAEC) via inhalation and the lower flammability limit, rather than more conventional measures of toxicity, such as the lethal dose (LD50) or concentration (LC50). Based on the European Chemicals Agency’s (ECHA) NOAECs (exposure via inhalation) for MeTHF and BA we calculate a NOAEC for our mixed solvent system (9:1 MeTHF:BA) of 1150 mg m−3, a 185-fold increase in comparison to DMF (6.2 mg m−3) (Fig. 1a). The lower flammability limit of all solvents commonly used for perovskite processing are comparable. Full discussion of these parameters is offered in Supplementary Note 2. Besides these, the critical advantage of both THF and MeTHF over the previously reported ACN systems is that the use of highly volatile MA gas is precluded. Use of a liquid – rather than gas phase – amine minimises the complexity of precursor preparation and handling, improving scalability and ensuring reproducible solution composition.
2D precursor phase-growth of 3D perovskites
Notwithstanding the volatility of MA, amines hold their own critical advantage against other Lewis basic Pb2+-coordinating solvents; they also behave as Brønsted-Lowry bases in precursor solutions. In our precursor ink the inclusion of BA alongside methylammonium iodide (MAI) leads to a Brønsted-Lowry H+ exchange in solution and the in-situ production of n-butylammonium (BA+). We dissolve MAI and lead (II) iodide (PbI2) in our MeTHF:BA solvent system and cast a thin film by spin-coating and drying at 70 °C for ten minutes. Incorporation of both BA+ and methylammonium (MA+) within the perovskite structure leads to highly crystalline 2D Ruddlesden-Popper phase perovskites (RPPs) of the (BA+)2(MA+)n-1PbnI3n+1 family, where BA+ serves as the large A-site cation (A’) and MA+ as the small A-site cation (A)32. The n described by this empirical formula is often used to differentiate RPP phases by the number (n) of lead-halide octahedra ([PbI6]4-) layers interspersed between layers of the large A’ cations. In steady-state photoluminescence (PL), X-ray diffraction (XRD) and absorbance measurements (Supplementary Figs. 2–4) we observe multiple peaks consistent with the presence of multiple RPP phases. By varying BA:MAPbI3 precursor stoichiometry (RBA-MA+) we are able to control the average RPP phase composition, denoted <n > (Supplementary Note 3).
Although 2D RPPs have been utilised as photo-absorbers in efficient PSCs, these typically display non-ideal bandgaps for use in single-junction photovoltaics33. Instead, we propose an alternative application of 2D mixed-phase RPPs; as solid-state precursor phases, or intermediates, in a solution-processed sequential deposition of 3D ABX3 perovskites34,35. This strategy allows fabrication of α-FAPbI3 by our solvent system but avoids the reported reactivity of amines with FA+ in solution36,37.
To convert the 2D precursor phase into α-FAPbI3, we dispense a solution of formamidinium iodide (FAI) in n-butanol (“conversion solution”) on top of the substrate and allow conversion via cation exchange to occur over a controlled time before spin-coating. In Fig. 2a we present the PL spectra of the precursor RPP layer as a function of conversion time. Supplementary Fig. 8 tracks the corresponding evolution of absorbance during conversion. While FA+ cations intercalate into the 2D material, we observe a gradual reduction in the PL intensity at the original emission wavelengths and the emergence of longer wavelength emission features. We interpret this to be due to FA+ exchanging with MA+ and BA+ and at first increasing the <n > -value in the now “triple cation” RPP perovskite, before the resultant film becomes predominantly FAPbI3, or a mixed FAxMA1-xPbI3 3D perovskite, as judged by the emission peak at 780 nm. After spin-coating to remove excess conversion solution, subsequent thermal annealing at low (70 °C, 10 minutes) and then high (180 °C, 30 minutes) temperature completes conversion to the 3D perovskite, with the high temperature step selected with the intention of driving out remnant BA+ and MA+. The final emission peak of the annealed perovskite film is centred at 821 nm. Figure 2b shows the same process tracked by in-situ GIWAXS, highlighting the evolution of crystalline domains during conversion and subsequent annealing. After solution conversion, we find that only a small quantity of the 2D precursor phase is retained, with the majority having been converted directly to 3D perovskite. Reflections consistent with a very small quantity of the preferred RT phase (2H-FAPbI3) are observed, however, these rapidly disappear along with remnant 2D intermediate upon high temperature curing. The direct conversion from 2D to 3D perovskite indicated by these combined in-situ GIWAXS and PL results indicate that the extended corner-sharing [PbI6] octahedral network present in the precursor phase is retained into the 3D perovskite.
Fig. 2. Conversion of 2D precursor phase to 3D α-FAPbI3 perovskite.

a Tracking photoluminescence (normalised) of optimised 2D precursor phase (RBA-MA+ = 1.5) converting to 3D α-FAPbI3 during soaking in “conversion solution” (FAI solution in n-butanol). Unconverted 2D precursor phase shown at t = 0 s. Insets: images of 2D precursor phase (red) and 3D (black) perovskite thin films. b In-situ grazing-incidence wide-angle X-ray scattering (GIWAXS) tracking evolution of crystalline phases present as 2D perovskite is converted to α-FAPbI3. 1H solution nuclear magnetic resonance (NMR) spectra of (c) 2D precursor phase (RBA-MA+ = 1.5) thin films (inset shows expanded region of spectrum corresponding to methyl and methylene signals, highlighted in dashed box) and (d) α-FAPbI3 thin films dissolved into DMSO-d6.
During in-situ GIWAXS measurement of the first five minutes of high temperature annealing, reflections corresponding to 6R-PbI2 gradually appear, but the intensity and position of these peaks stabilises – along with those of α-FAPbI3 – consistent with complete volatilisation of small quantities of remnant MA+ from the 3D perovskite phase. As MA+ is present in the precursor phase, it is important to identify if any residual MA+ is left in the final annealed film, since if it were this could contribute to the ABX3 phase stabilisation. Furthermore, previous work has indicated the phase stability of α-FAPbI3 is increased with the incorporation of some 2D phases38, and hence determining if any residual BA+ is present is also important. Ex-situ XRD characterisation of the fully-annealed 3D perovskite shows a reflection at 2θ = 13.92° (Supplementary Fig. 15a), consistent with that predicted for the (100) reflection of α-FAPbI3 at 298 K (CCDC: 2243718; 2θ = 13.924°)39. We also perform 1H solution NMR spectroscopy measurements of the as-annealed 3D perovskite films dissolved in DMSO-d6 (Fig. 2d, e), which confirm the absence of residual MA+. The clear resolution of 13C satellite signals (1J1H-13C coupling) indicates sensitivity to trace MA+ or BA+ as low as ~0.1 mol% (Supplementary Fig. 11), placing an upper limit residual MA+ or BA+ in our material. Further, in Supplementary Fig. 12 we show scanning electron diffraction (SED) measurements of the final annealed precursor phase-engineered 3D perovskite films. SED measurements allow detection and structural indexing of nanoscale crystalline domains that are too small for detection by bulk diffraction methods40. In these images we cannot find any diffraction spots which correspond to the unit cell dimensions expected from 2D perovskite phases. Thus, although this does not prove that there are no 2D domains present, we see no evidence that there are. From here on, we refer to our annealed 3D perovskite as α-FAPbI3.
To further investigate the mechanism of 2D precursor phase-conversion we assess the impact of precursor composition on the resulting 3D perovskite. Scanning electron microscopy (SEM) images of α-FAPbI3 processed via 2D precursor films of varying <n> reveal a wide variety of microstructures. Use of a high RBA-MA+ (BA:MA+ precursor solution stoichiometry) 2D precursor phase (low <n > ) results in a mesoporous structure in the 3D perovskite film (Fig. 3a-b), and even macroscopic cracking of the thin-films (Fig. 3a, inset). XRD analysis of α-FAPbI3 processed from intermediate RBA-MA+ ratios reveals how gradual reduction of RBA-MA+ (toward higher <n > ) leads to a pronounced increase in lead (II) iodide (6R-PbI2) content in the as annealed films (Fig. 3i, Supplementary Fig. 14). We observe a consistent trend with the appearance of hexagonal 6R-PbI2 platelets, which appear brighter in the SEM images (Fig. 3c–g)41. Conversion from high-n 2D precursor phase films also leads to mesoscopic porosity visible via SEM (Fig. 3h). However, in contrast to low-n precursor phases, XRD analysis of high-n-converted 3D materials demonstrates significant 6R-PbI2 content (Fig. 3i). We rationalise these results by considering the careful balance of factors that control optimum <n> for 2D precursor phase conversion and identify a ‘Goldilocks region’ in which optimal composition and morphology are achieved. We discuss our full mechanistic rationalisation of the conversion process in detail in Supplementary Note 4. Here we summarise our findings.
Fig. 3. Optimal 2D intermediate.

Scanning Electron Microscopy (SEM) images showing the microstructure of 2D-intermediate α-FAPbI3 layers when a series of 2D intermediate processed from THF with varying RBA-MA+ are converted sequentially. RBA-MA+ = (a) 3.00 (inset: photograph showing macroscopic cracking of 3D perovskite layers converted from RBA-MA+ = 3.00 intermediates due to extreme volume contraction), (b) 2.50, (c) 2.00, (d) 1.75, (e) 1.50, (f) 1.25, (g) 1.00, (h) 0.75. Scale bars: 5 μm. Schematic diagrams depicting the conversion of (i) BA2PbI4 (n = 1) (j) BA2MAPb2I7 (n = 2), and (k) BA2MA3Pb4I13 (n = 4) into 3D α-FAPbI3. FA+ highlighted in green has replaced BA+ in the organic channels of the 2D intermediate, while FA+ highlighted in yellow has replaced MA+ in the inorganic layers. Double-headed arrows display layer thickness. l Plot showing lead iodide (PbI2) content as a phase fraction in comparison to 2D-intermediate α-FAPbI3 extracted from XRD diffraction patterns (Supplementary Fig. 15) via peak integration of 6R-PbI2 (001) and α-FAPbI3 (100) scattering peaks. Inset shows an expanded view for low-PbI2 region. m 127I nuclear quadrupole resonance (NQR) spectra of 2D-intermediate FAPbI3 and FAPbI3 (processed from DMF:DMSO) thin films, which have been mechanically exfoliated and powdered for measurement.
For effective conversion of the precursor phase, we hypothesised that two processes should be optimised. (1) Facile intercalation of FA+ cations into, and expulsion of MA+ and BA+ from the precursor phase must occur. (2) Volume contraction and structural rearrangement required upon conversion from the 2D perovskite to α-FAPbI3 must be minimised. Both these processes are, in turn, controlled by two properties of the 2D RPP precursor phase; spacer molecule channel density, and the thickness of the corner-sharing haloplumbate, [PbI6]n, layers in the 2D precursor phase. Both these properties are determined by the <n> value.
The mesoscopic cracking observed in films converted from both high <n> and low <n > (low and high RBA-MA+, respectively; Fig. 3a–h) is consistent with the release of local strain in the films during thermal curing. High RBA-MA+ precursor phases possess high spacer channel density, which is expected to facilitate rapid cation exchange during conversion. However, 2D precursor films rich in the n = 1 phase possess low lead halide density (density of [PbI6] octahedra), and thus require significant volume contraction upon conversion to form a continuous network of 3D corner-sharing [PbI6] octahedra. By this mechanism, volume contraction leads to local strain in the converted film, and thus mesoscopic cracking upon curing. We show this process schematically in Fig. 3j. Conversion of low RBA-MA+ precursor phases to a 3D perovskite phase requires substantially less volume contraction. However, as shown in Fig. 3i, XRD analysis of converted RBA-MA+ = 0.75 films show that X-ray scattering intensity of the 6R-PbI2 is dominant over the 3D perovskite. XRD of the same series of 2D perovskites (Supplementary Fig. 2) revealed that the RBA-MA+ = 0.75 precursor phase is the only one to contain majority n ≥ 4 RPP phases. The lead halide density of PbI2 is even greater than of 3D perovskites and so conversion of a low RBA-MA+ precursor phase to PbI2 is also expected to result in substantial local strain, and thus mesoscopic cracking of the film, as observed in Fig. 3a.
Next, we consider the origin of PbI2 in the final FAPbI3 materials. For n > 2 precursor phases, complete extraction of MA+ from, and intercalation of FA+ cations into the intact haloplumbate structure requires cation migration across multiple perovskite A-sites, penetrating through unbroken layers of [PbI6] octahedra – which are not dissolved by the conversion solvent, n-butanol – and that act as an energetic barrier to ion intercalation. Any MA+ not extracted from the precursor phase during conversion is expected to volatilise during subsequent thermal curing at 180 °C, via mechanisms discussed in Supplementary Note 8. As Fig. 3l illustrates, such a process yields Pb2+ and I- -rich regions in the forming 3D perovskite, and ultimately regions of crystalline PbI2 upon grain ripening during thermal curing. The occurrence of such ripening is evidenced in the substantial evolution in morphology observed during high temperature curing (Supplementary Fig. 13g–j). This proposed mechanism is consistent with the gradual increase in PbI2 content observed in converted 1.75 ≥ RBA-MA+ ≥ 0.75 precursor phase films. To further demonstrate this effect, and the integral role spacer molecule channels play in conversion, we attempt the conversion of neat MAPbI3 (representing (BA+)2(MA+)n-1PbnI3n+1 with n = ∞; RBA-MA+ = 0) with the same solution conversion protocol. Absorbance spectra of this ‘MAPbI3 precursor phase’ and the material resulting from its attempted conversion show the formation of PbI2 in the converted layer, and a blue-shifted absorption onset compared to α-FAPbI3, confirming that complete conversion of the MAPbI3 precursor phase cannot been achieved (Supplementary Fig. 16a). Cross-sectional SEM images of the MAPbI3 precursor phase film and the converted film (Supplementary Fig. 16b, c) reveal the formation of significant voids at the bottom of the perovskite layer during conversion, similar to those observed in films converted from both high and low <n > 2D precursor phases (Fig. 3a–h).
In order to estimate the strain induced or released when going from the precursor phases to the 3D perovskite, we first employ thin film profilometry and simple structural considerations. Profilometer measurements indicate that the thickness of our optimum 2D precursor phase film (RBA-MA+ = 1.5; predominantly n = 2) contracts from 1,050 nm to 725 nm upon conversion to α-FAPbI3; a 32% volume contraction. Calculations using the reported unit cell volume of BA2MAPb2I7 (3118.7 Å3)42, which contains eight PbI64- octahedra, and that of FAPbI3 (256.4 Å3)43, indicate a theoretical volume contraction of approximately 34% should be observed. These data suggest that the majority of volume contraction upon conversion occurs in the vertical plane rather than laterally. This almost entirely accounts for the expected volume contraction due to the increased density of PbI6 octahedra in the 3D perovskites. Thus, almost all the strain is released in this conversion process. To further confirm the absence of microstrain in our α-FAPbI3 thin films, we employ 127I nuclear quadrupole resonance (NQR) spectroscopy39,40. As discussed in Supplementary Note 4, 127I NQR is highly sensitive to local symmetry distortions in the α-FAPbI3 structure induced by residual strain, A-site mixing (e.g. incorporation of MA+) or X-site mixing, which lead to a broadening of the NQR transition39,40. In comparison to α-FAPbI3 thin films made by a conventional DMF:DMSO solution processing route (in the absence of any additives), 2D precursor phase-engineered α-FAPbI3 thin films show a markedly narrower 127I NQR transition (Fig. 3m). This is compelling evidence that none of the phenomena noted above occur in our material, notably the absence of residual strain and further confirms that our α-FAPbI3 is MA+-free.
The mechanics of 2D precursor phase-engineered conversion and growth require significant further investigation. However, overall it is clear that, unlike intermediates featuring coordinating solvents or other lower-dimensionality materials reported to-date34,35,44, the <n > > 1 RPPs reported here hold two critical advantages as sequential deposition precursor phases for 3D perovskites: an extended corner-sharing haloplumbate structure already present in the precursor phase ready to serve as a scaffold for 3D perovskite growth, as evidenced by the direct conversion of 2D perovskite into α, not δ, phase FAPbI3; and built-in organic channels within the lead-halide scaffold that can facilitate rapid intercalation of a conversion solution containing A-site cations.
We note that very recently Sidhik, et al. reported a related route to fabricate α-phase FAPbI3, using DMF:DMSO solvents, and also observed very high quality, α-phase stable FAPbI345. Comparing our α-FAPbI3 to that published by Sidhik, et al., we determine a (cubic) lattice parameter of 6.341 Å by applying a Pawley fit (Supplementary Fig. S14), as compared to 6.369 Å reported by Sidhik, et al. The cubic lattice parameter of unstrained α-FAPbI3 single crystals has been reported as 6.355 Å39. The slightly larger lattice parameter of Sidhik, et al. may originate from the small fraction of retained BA+ reported in their material. Here, we find no evidence of any BA+ retained in our α-FAPbI3 material.
To investigate if our α-FAPbI3 films have useful optoelectronic properties, we compare them with α-FAPbI3 perovskites fabricated via two conventional solution processing approaches utilising DMF:DMSO solvent mixtures; with and without methylammonium chloride (MACl) additive46. MACl has been shown to improve perovskite material quality, stability and optoelectronic performance46–48. Time-resolved PL measurements reveal an order of magnitude improvement in PL lifetime for 2D precursor phase-engineered α-FAPbI3 compared with MACl-free FAPbI3, and a factor of three enhancement compared to MACl-additive FAPbI3 (Fig. 4a and Supplementary Fig. 19b). By fitting fluence-dependence of PL decays from films of the three materials (Supplementary Fig. 17), we determine a reduction in the monomolecular trap-assisted recombination rate constant, implying suppression of non-radiative recombination centres. Notably, for the conventional MACl-free FAPbI3, much higher excitation fluences and charge-carrier densities are required for recombination dynamics to transition from a “trap-mediated” monomolecular decay regime to intrinsic bimolecular radiative recombination49–51. To probe the charge carrier mobility of these materials, we perform optical-pump-terahertz-probe (OPTP) photoconductivity spectroscopy (Supplementary Fig. 18) measurements. We determine that the effective sum of mobilities (electron and hole) and the bimolecular recombination rate of charge carriers, are largely unaffected by differences in film fabrication (Supplementary Fig. 19a). All the FAPbI3 films studied have a very high mobility of approximately 60 cm2 V-1 s-1, which are some of the highest such values reported for thin film FAPbI3 and attest to the generally high quality of the fabricated layers52. However, as a result of the highly effective suppression of trap-mediated charge-carrier recombination, for FAPbI3 fabricated via the 2D precursor phase-engineered approach we calculate a significantly improved charge-carrier diffusion length under solar illumination conditions of 4.6 ± 0.2 μm, significantly higher than that for either FAPbI3 material processed from DMF:DMSO (1.3 ± 0.1 μm and 3.0 ± 0.2 μm for neat and MACl additive-processed FAPbI3) (Supplementary Fig. 19b).
Fig. 4. PSC Operational Performance.

a Time-resolved photoluminescence (PL) transients of FAPbI3 materials made using different fabrication methods. PL was detected at 810 nm following pulsed-laser excitations (398 nm at 2.5 MHz repetition rate) at an excitation fluence of 81 nJ cm-2. PL lifetimes, τPL, determined by stretched exponential fits are shown on the figure labelling. Solid lines are stretched-exponential fits to the measured data. b Cross-sectional scanning electron microscopy image of PSC fabricated utilising 2D precursor phase-engineered α-FAPbI3. J-V characteristics and corresponding maximum power point tracked efficiency (ηmpp) and current density (Jmpp) measured at maximum power point for champion devices fabricated from 2D precursor phase-engineered α-FAPbI3 with illuminated area 0.25 cm2 (c, e) and 1.00 cm2 (d, f). Light J-V and ηmpp measurements were performed under simulated AM1.5 100.7 mWcm-2 irradiance, accounting for the spectral mismatch factor. Jsc = short-circuit current density, Voc = open-circuit voltage, FF = fill factor, Vmpp = maximum power point voltage.
To determine if the improved optoelectronic properties of the 2D precursor phase-engineered α-FAPbI3 films translate into improved PSCs we fabricate n-i-p structured devices composed of FTO / SnO2 / α-FAPbI3 / PEAI passivation / spiro-OMeTAD / Au, which we show a cross-sectional SEM image of in Fig. 4b. In Fig. 4c-f we show the current-voltage curves and maximum power point tracked efficiency (ηmpp) for champion devices of both 0.25 cm2 and 1 cm2 respectively, and determine a ηmpp of 20.1 % and 18.4 %, respectively. In Supplementary Fig. 20a we present the distribution of optimised 0.25 cm2 and 1.00 cm2 PSCs. For comparison, in Supplementary Fig. 20b-d we present PSCs fabricated similarly but employing FAPbI3 with MACl additive (40 mol%), which achieve a maximum ηmpp of 18.8 %.
Investigating α-FAPbI3 stability
Despite the favourable bandgap of α-FAPbI3 for single-junction and for use as the middle junction in triple-junction PSCs, the well-known phase instability of this material is a drawback. At room temperature, the hexagonal non-perovskite 2H (δ) phase of FAPbI3 is thermodynamically preferred53. Although kinetic entrapment of the α-phase is possible, phase transformation is known to be accelerated by a range of external influences, including ambient humidity4, and materials properties, such as iodide-defects9. In Supplementary Figs. 21–23 we show the evolution in absorbance, microstructure (by means of SEM and visible light microscopy) and phase composition (by XRD) for the same three FAPbI3 materials investigated above, over 500 hours of storage under ambient conditions (18-22 °C, 20-55% relative humidity). We find that the composition of 2D precursor phase-engineered α-FAPbI3 remains almost entirely unaltered over this period, while α-FAPbI3 fabricated conventionally from DMF:DMSO solvents both with and without MACl additive show substantial secondary phase formation and reduction in optical density (absorbance), particularly at the band edge, over the first 300 hours of ambient storage.
In Supplementary Fig. 25 we show the unencapsulated “shelf stability” (stored in dry air, <10% relative humidity, at room temperature in darkness; adapted ISOS-D−1) of our n-i-p PSCs based on 2D precursor phase-engineered α-FAPbI3, where we observe only minimal drop in performance over 18,000 hours ( ~ 2 years). Generally, such estimates of stability are of only limited usefulness in assessing long-term PSC stability1. However, in this instance, since α-FAPbI3 is known to possess only kinetic metastability, such sustained performance confirms the unexpectedly promising phase-stability of our material.
To investigate the stability of our α-FAPbI3 material under operational conditions and explore the relationship between perovskite processing route and resultant stability we perform in-situ XRD tracking the accelerated thermal degradation (130 °C, ~30% RH) of 2D precursor phase-engineered α-FAPbI3, (Fig. 5a) which we discuss in detail in Supplementary Note 7. We find that α-FAPbI3, MAPbI3 and FA0.83Cs0.17Pb(I0.9Br0.1)3 thin films fabricated via DMF:DMSO solvent systems all degrade to PbI2 much more rapidly than our α-FAPbI3 processed from highly volatile MeTHF, BA and BuOH solvents (Fig. 5b–d). Use of higher curing temperatures (180 °C) during processing reduces the rate of thermal degradation, but compromises perovskite crystallinity and phase purity (Fig. 5e-f). We also show that, besides 2D precursor phase-engineered α-FAPbI3, remarkably the most thermally stable perovskite composition investigated is MAPbI3 processed from highly volatile ACN, which is much more thermally stable than MAPbI3 via DMF:DMSO (Fig. 5g). From these results, we hypothesised that poor thermal stability may be due to retention of residual processing solvents in thin films processed from low volatility DMF and DMSO. To confirm that such residual processing solvents are indeed released during thermal degradation, we employ thermal desorption-gas chromatography-mass spectrometry (TD-GCMS), whereby volatile organic compounds (VOCs) are desorbed from perovskite thin films and individually characterised. These may be VOCs retained during processing or degradation products of the perovskite material. TD-GCMS is highly sensitive; we calculate the detection limit of DMSO retained within a 25 cm2 thin film to be 0.002 wt.%54 (Supplementary Note 7). We confirm that DMSO is released from α-FAPbI3, MAPbI3 and FA0.83Cs0.17Pb(I0.9Br0.1)3 films fabricated from DMF:DMSO. No solvents are released from either MAPbI3 or our α-FAPbI3 processed from ACN and MeTHF, respectively, confirming a direct link between reduced perovskite thermal stability and release of residual processing solvents. Equally, we detect no low volatility residual solvents in α-FAPbI3 and FA0.83Cs0.17Pb(I0.9Br0.1)3 processed via DMF:DMSO and cured at 180 °C. However, we do detect release of sym-triazine from these materials, as is the case in the degradation of every FA+-containing perovskite investigated excluding 2D precursor phase-engineered α-FAPbI3 (15 different materials, Supplementary Fig. 28). A total of seven individual precursor phase-engineered α-FAPbI3 samples were analysed, with no sym-triazine detected. Sym-triazine has been reported as a thermal degradation product of FA+ 55–57. By isolating a range of processing parameters, we identify that the 2D precursor phase-engineering crystallisation process itself is most likely responsible for the suppression of FA+ volatilisation in our α-FAPbI3 (see discussion in Supplementary Note 7). We summarise these findings in Fig. 5h.
Fig. 5. Enhanced perovskite stability.

a–g Contour maps depicting in-situ x-ray diffraction (XRD) analysis of perovskite materials thermally degrading whilst held at 130 °C for 9 hours. Labels indicate perovskite composition and fabrication conditions (solvent, thermal curing). Full processing conditions described in Supplementary Note 6. Molecular structures shown correspond to the volatile species detected during thermal desorption-gas chromatography-mass spectrometry (TD-GCMS) analysis of each perovskite. h Schematic highlighting advantages to perovskite material properties of solution processing via MeTHF solvent system. Visible light microscopy images of encapsulated state-of-the-art perovskite thin films after aging for 3600 hours under 85 °C, 1-sun equivalent illumination (ISOS-L-2, i-j) and 3000 hours under 85 °C, 85 % relative humidity (ISOS-D-3, k, l) conditions. Evolution under ISOS-L-2 (m) and ISOS-D-3 (n) stressing of maximum power tracked efficiency (ηmpp) measured periodically on p-i-n PSCs employing 2D precursor phase-engineered FAPbI3 (7 cells and 8 cells, respectively) and FA0.83Cs0.17Pb(I0.9Br0.1)3 (8 cells, under each condition, “FACs”) as the photoactive layer. Cells undergoing ISOS-L-2 are encapsulated with a 250 nm layer of MoO3 followed by an on-cell epoxy resin and cover slip. Cells undergoing ISOS-D-3 are encapsulated in glass-glass laminates employing an industry standard butyl-rubber edge sealant (described in Supplementary Note 10). Dashed lines show the median t80 ηmpp values for each dataset. The median, range and interquartile range of each dataset are shown, as well as each data point within them.
To compare the stability of our α-FAPbI3 to other highly stable perovskite compositions under conventional environmental stressing conditions we encapsulate perovskite films in glass-glass laminates employing an industry standard butyl-rubber edge seal, and subject the perovskite material to the conditions of two of the most demanding stability tests for perovskites; damp-heat (85 °C and 85 % relative humidity; ISOS-D-3) and light-soaking under elevated temperature (85 °C under a 1-sun equivalent light source; ISOS-L-2). Impressively, we observe very little change in the UV-vis absorption spectra for our α-FAPbI3 films when aged for over 3000 hours (3 times the IEC standard) under both stressing conditions. In Supplementary Fig. 32a, b, we show the optical density (OD) of the films, at wavelengths between 500 to 510 nm, as a function of stressing time. This gives a relatively sensitive indication of the evolution of “pin-holes” in the perovskite films, which have previously been observed to form during degradation3,58. The relatively high OD of our α-FAPbI3 films over the aging time, indicates that few pinholes are forming, which we confirm this with visible light microscopy, where in Fig. 5i–l we show images of the different films after >3000 hours stressing.
To assess if the improved material stability translates into improved device stability, we integrate our α-FAPbI3 into p-i-n structured PSCs. Although we have demonstrated >2 year “shelf stability” (Supplementary Fig. 25), n-i-p PSCs are known to suffer from a number of inherent instabilities under elevated moisture59, temperature60 and light61, principally due to the instability of the transport materials usually employed in this architecture. However, reports of pristine FAPbI3 in the more stable p-i-n configuration are unusual62. Furthermore, for 2D precursor phase-engineered α-FAPbI3 we find that selection of the underlying hole transport material is severely limited by a combination of solvent incompatibility, high temperature processing of the perovskite and low mechanical adhesion with the as-deposited 2D precursor phase-engineered α-FAPbI3 layer. After mitigating these limitations (discussed in the Methods), we achieve substantial photovoltaic performance (ηmpp = 18.8 %, Supplementary Fig. 33).
Exposure of these devices to the ISOS-D-2 (85 °C, dark, N2) aging procedure confirms the remarkable long-term thermal stability of our α-FAPbI3-based PSCs (Fig. 5a), retaining 95% of their initial ηmpp after 1900 hours of aging (median of 15 cells, Supplementary Fig. 34). Next, we isolate the effect of light-induced degradation in the absence of elevated heat (adapted ISOS-L−1). Under these conditions we again find that our α-FAPbI3 PSCs are remarkably stable (projected t80 = 3940 hours, 21 cells, encapsulated, Supplementary Fig. 35). In comparison, PSCs of the same architecture employing FA0.83Cs0.17Pb(I0.9Br0.1)3 show reduced photostability, with a t80 of just 850 hours (10 cells). Although our target material is α-FAPbI3, we opt to compare this to an FAxCs1-x perovskite since these materials have been established as having state-of-the-art stability in single-junction PSCs2,3. Next, we subject similar PSCs to the ISOS-L-2 test (85 °C in ambient air, full spectrum 0.76 sun illumination with cells held at open circuit) in an Atlas Suntest CPS-Plus xenon-lamp aging box, as previously for thin film materials (Fig. 5i, j and Supplementary Fig. 32a, b). We find that device degradation of PSCs based on our precursor phase-engineered α-FAPbI3 achieve a promising ISOS-L-2 t80 of 570 hours (8 cells) in comparison to FA0.83Cs0.17Pb(I0.9Br0.1)3-based PSCs which exhibit a t80 of just 40 hours (7 cells) under these conditions (Fig. 5m). We note that this is typical for a standard FAxCs1-x PSC aged under these conditions, without any stability-enhancing additives2,3. Our champion α-FAPbI3 cell (0.25 cm2) achieves a t80 of 800 hours. Considering the finding that our α-FAPbI3 thin films appear to be unchanged after 3600 hours of aging under ISOS-L-2 (Fig. 5i and Supplementary Fig. 32), we postulated that the 570-hour t80 measured for PSCs based on this perovskite is still limited by factors besides photoabsorber bulk stability. Careful removal of the encapsulation and n-type contact layers after 1450 hours of ISOS-L-2 aging seems to confirm this inference. In Supplementary Fig. 37 we present XRD patterns of the aged devices, showing minimal change in the composition of either perovskite. However, SEM images of the pristine and aged perovskite layers (Supplementary Fig. 38) reveal degradation-induced void formation, which is substantially more severe in the FA0.83Cs0.17Pb(I0.9Br0.1)3 PSCs.
Notably, these enhancements in PSC stability are achieved despite the presence of a small quantity of lead (II) iodide in the photoabsorber layer (Fig. 3i). 2H-PbI2 has been shown by us and others to generate defect-rich interfaces with α-FAPbI3 and to undergo photodegradation to metallic Pb0 and I2, producing iodide vacancies. As both iodide vacancies63 and interstitial iodide9 defects have been implicated in accelerating the rate of α-to-δ phase transformation, the stability of our material is surprising. We note, however, that because of our high temperature processing, the PbI2 phase present in our material is the 6 R polytype, which has recently been shown to form highly coherent, low-defect interfaces with α-FAPbI364. Moreover, as Fig. 3a–h clearly show, the majority of 6R-PbI2 in 2D-intermediate α-FAPbI3 is in isolated grains. Thus, minimal PbI2-FAPbI3 interface is present in the material and any iodide vacancies generated by I2 formation in-operando are isolated from the α-FAPbI3 and so can play no part in accelerating its phase transformation.
Finally, by developing a whole-cell encapsulation strategy (Supplementary Note 10) p-i-n PSCs employing 2D precursor phase-engineered α-FAPbI3 are able to pass the IEC61625-1 damp heat test (85 °C, 85% RH, ISOS-D-3), with >100% of initial median ηmpp retained after 1930 hours (t100 > 1930 hours, 8 cells, Fig. 5n). By contrast, comparable FA0.83Cs0.17Pb(I0.9Br0.1)3-based cells achieve a median t95 of just 315 hours (8 cells). The IEC pass criterion is t95 > 1000 hours. Notably, the survival of the 2D precursor phase-engineered α-FAPbI3 for nearly 2000 hours under ISOS-D-3 conditions, despite the material’s moisture-accelerated 2H-phase instability, suggest that this simple three-ion perovskite may prove to be the most stable perovskite composition, matching the expectations for real world PV module deployment. To put our damp heat results into context, for copper indium gallium selenide PV modules, 3000 hours 85 °C, 85% RH stressing has been shown to be comparable to 20 years deployment in Miami, USA65. We summarise all lifetimes of PSCs presented in this work in Supplementary Table 3.
In conclusion, we have developed a volatile, low toxicity solvent system for α-FAPbI3, one of the most desirable PV materials today, which enables fabrication of efficient and highly stable PSCs. This approach has allowed us to present an unconventional crystallisation pathway for a 3D perovskite in which the pre-existing corner-sharing [PbI6]4- network present in a solid state 2D RPP perovskite is retained to direct the growth of α-FAPbI3. Competitive PSC device efficiencies validate the 2D precursor phase-engineered crystallisation route, particularly considering the absence of MA+ in the final 3D perovskite material and the use of a green solvent system. We find that α-FAPbI3 fabricated by our processing approach possesses substantially improved thermal and phase-stability, as compared to a broad range of state-of-the-art perovskite compositions and processing routes. We demonstrate that this improved stability is the product of several factors. Critical among these is the 2D precursor phase crystallisation route itself, which both avoids the use of low volatility solvents – which we show are often retained in perovskite thin films, leading to long-term instability – and leads to a material in which FA+ degradation to sym-triazine under thermal stressing is supressed. Integration of our 2D precursor phase-engineered α-FAPbI3 into p-i-n PSCs confirm that the enhancement in bulk perovskite stability translates to impressive device stability. In particular, the photostability under elevated temperature (ISOS-L-2) of our α-FAPbI3 gives a champion t80 lifetime of 800 hours in comparison to just 100 hours for FA0.83Cs0.17Pb(I0.9Br0.1)3-based PSCs. Further, we determine t100 lifetimes of >1900 hours under damp heat stressing (ISOS-D-3). These damp heat results are particularly surprising since α-FAPbI3 is considered to be metastable under ambient operational conditions. FAPbX3 perovskites inherently bypass instability issues associated with A-site cation segregation and inhomogeneity6, the possibility of creating Cs-rich impurity phases7 and the hygroscopic nature of alkali-metal halide salts. Furthermore, by avoiding caesium, future material scarcity issues are avoided, representing a highly sustainable perovskite composition for terawatt scale PV deployment.
Methods
Materials
Fluorine-doped tin oxide-coated glass substrates (8 or 15 Ω cm-2, AMG), tin (IV) oxide (15 wt. % in H2O colloidal dispersion, Alfa Aesar), MeO-2PACz ([2-(3,6-Dimethoxy-9H-carbazol-9-yl)ethyl]phosphonic acid, >98.0%, Tokyo Chemical Industries), lead(II) iodide (99.99 %, trace metal basis, Tokyo Chemical Industries), methylammonium iodide ( > 99.99 %, Greatcell Solar Materials), formamidinium iodide ( > 99.99 %, Greatcell Solar Materials), phenethylammonium iodide ( > 99 %, Greatcell Solar Materials), ethylenediammonium diiodide (Sigma Aldrich), PC60BM ([6,6]-Phenyl-C61-butyric acid methyl ester), bathocuproine (BCP, >95.0%, Tokyo Chemical Industries), spiro-OMeTAD (2,2’,7,7’-Tetrakis(N,N-di-p-methoxyphenylamino)-9,9’-spirobifluoren, >99.5 %, Luminescence Technology Corp.), FK209 Co(III) TFSI salt (tris(2-(1H-pyrazol-1-yl)-4-tert-butylpyridine)cobalt(III) tri[bis(trifluoromethane)sulfonimide], 98 %, Sigma Aldrich), bis(trifluoromethylsulfonyl)amine lithium salt (99.95 %, Sigma Aldrich), gold pellets (99.999 %, Kurt J. Lesker Company). Ethanol (200 proof, anhydrous, >99.5%, Sigma Aldrich), n-butylamine (99.5 %, Sigma Aldrich), 2-methyltetrahydrofuran (Biorenewable, anhydrous, >99.0 %, contains 250 ppm BHT as inhibitor, Sigma Aldrich), tetrahydrofuran (anhydrous, >99.9 %, contains 250 ppm BHT as inhibitor, Sigma Aldrich), n-butanol (anhydrous, 99.8 %, Sigma Aldrich), aluminium oxide nanoparticles (20 wt.% in 2-propanol, Sigma Aldrich), 2-propanol (anhydrous, 99.5 %, Sigma Aldrich), chlorobenzene (anhydrous, 99.8 %, Sigma Aldrich), acetonitrile (anhydrous, 99.8 %, Sigma Aldrich), 4-tert-butylpyridine (98 %, Sigma Aldrich), toluene (anhydrous, 99.8%, Sigma Aldrich), chlorobenzene (anhydrous, 99.8%, Sigma Aldrich), 1,2-dichlorobenzene (anhydrous, 99%, Sigma Aldrich). Prior to use, all FTO-coated substrates were scrubbed with an aqueous 2 vol % Decon 90 solution, rinsed with deionised water, sonicated in acetone, and subsequently sonicated in 2-propanol. Besides aluminium oxide nanoparticles, all other non-aqueous chemicals were stored in a N2-filled glovebox before use and protected from exposure to light.
n-i-p configuration perovskite solar cells
A colloidal suspension of tin oxide nanoparticles (400 μL) was diluted with ultrapure water (2600 μL). In an atmosphere containing minimum moisture ( < 5 % relative humidity), 200 μL of this solution was placed statically on a UV-ozone treated (15 minutes) substrate coated with a fluorine-doped tin oxide layer (15 Ω cm-2), then spun at 4000 rpm (1000 rpm s-1) for 30 seconds before being immediately annealed at 150 °C for 30 minutes in the same environment. The substrates were allowed to cool, then immediately subjected to a further 15 minutes of UV-ozone treatment before being immediately used in the following processing step.
2-methyltetrahydrofuran (1500 μL) was added to lead iodide (1.266 mmol, 583.5 mg) and methylammonium iodide (1.139 mmol, 181.1 mg), followed by n-butylamine (1.899 mmol, 187.6 μL), and agitated until all solids were fully dissolved. This corresponds to an RBA-MA+ value of 1.67. 50 μL of this solution was spin-coated dynamically on top of the SnO2 layer at 2500 rpm for 45 seconds in an environment at between 20–22 % relative humidity and <22 °C. The substrates were immediately annealed at 70 °C for 10 minutes in the same environment. After cooling, the substrates were coated with 350 μL of a 0.1 M solution of formamidinium iodide (0.500 mmol, 86.0 mg) dissolved in n-butanol (5000 μL). After 45 seconds of static soaking, the substrates were spun at 4000 rpm (1000 rpm s-1) for 45 seconds, then immediately annealed for 10 minutes at 70 °C, followed by 30 minutes at 180 °C. We summarise this process schematically in Supplementary Fig. 1. Note: The full fabrication procedures for all other perovskite compositions utilised in this work are described in Supplementary Note 6.
The substrates were allowed to cool, then a 20 mM solution of phenethylammonium iodide (0.200 mmol, 49.8 mg) in 2-propanol (10,000 μL) was spin-coated dynamically on top at 5000 rpm for 45 seconds, in a N2-containing glovebox.
A solution of spiro-OMeTAD (0.070 mmol, 85.8 mg) dissolved in chlorobenzene (1000 μL) and doped with 8.4 μL of a 0.250 M solution of tris(2-(1H-pyrazol-1-yl)-4-tert-butylpyridine)cobalt(III) tri[bis(trifluoromethane)sulfonimide] in acetonitrile, 19.4 μL of a 1.800 M solution of bis(trifluoromethylsulfonyl)amine lithium salt in acetonitrile, and tert-butyl pyridine (0.231 mmol, 38.0 μL). This solution was spin-coated on the PEAI-passivated substrates dynamically at 2500 rpm for 30 seconds, in a N2-containing glovebox.
Finally, 75 nm of gold was evaporated on top of the substrates at an initial rate of 0.1 A s-1 (to 5 nm, then ramped gradually to 1.0 A s-1) at a pressure <2 × 10-6 torr.
p-i-n configuration perovskite solar cells
In a N2 glovebox, a filtered (0.22 μm PTFE syringe filter) 0.33 mg mL-1 solution of MeO-2PACz ([2-(3,6-Dimethoxy-9H-carbazol-9-yl)ethyl]phosphonic acid) in anhydrous ethanol (350 μL) was prepared and placed statically on a UV-ozone treated (30 minutes) substrate coated with a fluorine-doped tin oxide layer (15 Ω cm-2), allowed to spread for 10 seconds, then spun at 3000 rpm (600 rpm s-1) for 30 seconds before being immediately annealed at 100 °C for 10 minutes in the same environment.
Separately, a colloidal suspension of alumina nanoparticles (50 μL) was diluted with 2-propanol (7500 μL) and ultrasonicated for at least 30 minutes (“1:150 Al2O3 NP:IPA”). This solution was filtered immediately before use (0.22 μm PTFE syringe filter). Preparation of this solution was carried out in ambient air, but all spin-coating was carried out in a N2 glovebox.
The MeO-2PACz-coated substrates were allowed to cool, then in the same environment the solution of alumina nanoparticles (80 μL) was dispensed dynamically whilst the substrate was spun at 5000 rpm (5000 rpm s-1) for 20 seconds. The substrate was then annealed at 100 °C for 5 minutes in the same environment. We have included this layer in the p-i-n device architecture in response to the poor wettability of perovskite solutions on carbazole-based SAMs, as has been reported elsewhere66. The use of Al2O3 nanoparticles in this way forms a thin mesoporous layer on top of the SAM, improving wettability67.
2-methyltetrahydrofuran (1500 μL) and n-butylamine (1.688 mmol, 166.8 μL) were added to lead iodide (1.125 mmol, 518.6 mg) and methylammonium iodide (1.013 mmol, 161.0 mg) and agitated until all solids were fully dissolved. This corresponds to an RBA-MA+ value of 1.67. 50 μL of this solution was spin-coated dynamically on top of the MeO-2PACz layer at 2500 rpm for 40 seconds in an environment at between 20–22 % relative humidity and <22 °C. The substrates were immediately annealed at 70 °C for 10 minutes in the same environment. After cooling, the substrates were coated with 350 μL of a 0.1 M solution of formamidinium iodide (0.500 mmol, 86.0 mg) dissolved in n-butanol (5000 μL). After 45 seconds of static soaking, the substrates were spun at 4000 rpm (1000 rpm s-1) for 45 seconds, then immediately annealed for 10 minutes at 70 °C, followed by 30 minutes at 180–185 °C.
In ambient air, a colloidal suspension of alumina nanoparticles (50 μL) was diluted with 2-propanol (5000 μL) and ultrasonicated for at least 30 minutes (“1:100 Al2O3 NP:IPA”)67. In a N2 glovebox, a 0.5 mg mL-1 solution of ethylenediammonium diiodide (EDAI2) in a 1:1 mixture of anhydrous 2-propanol and toluene was prepared and heated for between 2 and 4 hours68. Also, a 30 mg mL-1 solution of PC60BM ([6,6]-Phenyl-C61-butyric acid methyl ester) in a 3:1 mixture of chlorobenzene:dichlorobenzene, and a 0.5 mg mL-1 solution of bathocuproine (BCP) in anhydrous 2-propanol. All these solutions were filtered immediately before use (0.22 μm PTFE syringe filter).
In a N2 glovebox, the solution of EDAI2 (325 μL) was deposited on the on the previously deposited perovskite layer, and the substrate immediately spun at 3000 rpm (1333 rpm s-1) to remove excess solution. The substrate was immediately annealed for 10 minutes at 100 °C in the same environment. EDAI2 surface treatment has previously been reported as a highly effective strategy of improving both the Voc and operational stability of PSCs69.
The substrate was allowed to cool, then the 1:150 Al2O3 NP:IPA solution (80 μL) was spin coated dynamically at 5000 rpm (5000 rpm s-1), before being further annealed at 100 °C for 5 minutes in the same environment. Analogously to our use of Al2O3 nanoparticles to form a mesoporous underlayer beneath the perovskite, here we use the same strategy to combat the roughness of the perovskite surface. This roughness is associated with the large ( > 1500 nm) grains often observed in our FAPbI3 material and leads to very poor adhesion and atomic-level contact between the PCBM electron transport layer and the perovskite. Recently in our group we have a developed a strategy by which a mesoporous alumina layer is introduced, and the PCBM solution allowed to intercalate into this structure prior to spin-coating (see below) to improve the contact between the perovskite and the fullerene layer67. We have also found that this layer is beneficial for long-term operational stability of the PSC, and as the aim of our p-i-n devices was to achieve the highest possible stability for control and target devices, this strategy was particularly selected for this reason67.
The substrate was allowed to cool, then the PC60BM solution (200 μL) was deposited on the substrate and allowed to spread for 10 seconds67, before being spin coated at 2000 rpm (2000 rpm s-1). The substrate was then annealed at 100 °C for 5 minutes in the same environment. Finally, once the substrates had cooled, the BCP solution (100 μL) was spin coated dynamically at 5000 rpm (5000 rpm s-1), and the substrate annealed at 100 °C for 2 minutes in the same environment.
Using a chromium-coated tungsten bar, 3.5 nm of chromium was deposited on top of the substrate at a rate of 0.2 A s-1, followed immediately by 100 nm of gold, which was evaporated on top of the substrates at an initial rate of 0.1 A s-1 (to 5 nm, then ramped gradually to 1.2 A s-1), all at a pressure <2 × 10-6 torr. A layer of chromium was deposited prior to gold as this has previously been reported as reducing Au migration through to the perovskite layer thus increasing the long-term operational stability of the PV devices, which was our aim in fabricating these p-i-n configuration PSCs. We note, however, that use of this Cr layer typically reduces device PCE by ~1%.
Time-integrated photoluminescence
PL spectra were acquired using a time-correlated single photon counting (TCSPC) setup (FluoTime 300, PicoQuant GmbH). Samples were photoexcited using a 507 nm laser (LDH-P-C-510, Pico Quant GmbH) pulsed at a frequency of 40 MHz. The PL was dispersed using a monochromator, detected with hybrid photomultiplier detector assembly (PMA Hybrid 40, PicoQuant GmbH) and integrated over time.
Ultraviolet-visible absorption spectroscopy
Reflectance and transmittance spectra were recorded on a Varian Cary 1050 UV Vis spectrophotometer equipped with an integrating sphere. From these measurements, in combination with the photoactive layer thickness, absorption coefficients were calculated assuming a direct bandgap semi-conductor. Separately, absorbance spectra were measured with a Varian Cary 300 Bio UV-visible spectrophotometer with a 50×50 mm reflective neutral density filter with an optical density of 3.0 (made of UV fused silica).
Visible light microscopy
Optical microscope images were taken on a Nikon Eclipse LV100ND microscope with Nikon TU Plan Fluor lenses (10x/0.30 A, 20x/0.45 A, 50x/0.60 B, 100x/0.90 A). The images are taken with an attached Nikon Digital Camera D6.10.
Time-correlated single photon counting (TCSPC)
Time-resolved PL of the thin films on quartz substrates was measured using TCSPC following excitation by a 398 nm picosecond pulsed diode laser at a repetition rate of 2.5 MHz (PicoHarp, LDH-D-C-405M). The resultant PL was collected and coupled into a grating spectrometer (Princeton Instruments, SP-2558), which directed the spectrally dispersed PL onto a photon-counting detector (PDM series from MPD), whose timing was controlled with a PicoHarp300 TCSPC event timer. The PL transients were measured at 810 nm, corresponding to the peak PL intensity in the steady-state PL spectrum of this composition.
The excitation fluence of 81 nJ cm-2, corresponding to a charge-carrier density of the order of 1015 cm-3, was used to ensure that the dominant recombination pathway is through monomolecular recombination. To account for both the time dependence and the local distribution of k1 recombination rates, we fitted a stretched exponential function to the PL intensity to account for the local distribution of monoexponential decay rates, whose effective lifetime is given by . is the characteristic lifetime corresponding to the time taken for the PL intensity to drop to , and is the distribution coefficient that encompasses the case for monoexponential decays for the case , and the cases for a range of decay times (or potentially higher-order effects) as it decreases70,71. To account for the PL originating from electron-hole recombination we further double the effective fitted lifetime in order to extract the monomolecular charge-carrier recombination rate , such that , assuming at our excitation fluence of 81 nJ cm-2 50,51. The samples were all mounted in a vacuum cell under low pressure ( mbar). We note that the absence of oxygen prevents the possible passivation of the photo-generated defects in the film and could have resulted in an underestimation of the lifetimes extractable under ambient operating conditions.
This spectroscopic technique initially generated charge-carriers closer to the surface of the film through which excitation occurs, owing to most of the 398 nm excitation being absorbed in the first 100 nm. This also accounts for the fast decay at very early times; most of the photogenerated charge-carriers will be initially generated closer to the surface where the density of trap states is higher than in the bulk, resulting in a rapid drop in PL intensity at very early times before the charge-carriers diffuse away further from the surface. The diffusion along the thickness of the film towards the bulk and perpendicular diffusion away from the excitation spot results in the local distribution of mono-exponential recombination rates assumed for our use of the stretch exponential fits.
Optical-pump terahertz-probe (OPTP) spectroscopy
An amplified laser system (Spectra Physics, MaiTai – Ascend – Spitfire), with a 5 kHz repetition rate, centre wavelength of 800 nm and pulse duration of 35 fs is used to generate the THz radiation using a spintronic emitter. The THz probe is then focused onto the sample, overlaid with a 400 nm excitation pump that is generated using a Beta Barium Borate (BBO) crystal. The THz radiation transmitted through the sample is then detected via free-space electro-optical sampling in a ZnTe (110) crystal of thickness 200 μm72.
X-ray diffraction (XRD)
The 1D-XRD patterns were obtained with a Panalytical X’Pert Pro X-Ray diffractometer and In-situ 2D-XRD patterns (Fig. 5a–g, Supplementary Figs. 8 and 20) using a Rigaku SmartLab X-ray diffractometer and a HyPix-3000 2D hybrid pixel array detector, both with CuKα1 (1.54060 Å) source. A heating stage was employed in conjunction with the Rigaku SmartLab diffractometer for these measurements.
Grazing-incidence wide-angle X-ray scattering (GIWAXS) data (Supplementary Fig. 6) was acquired at the I07 beamline at Diamond Light Source. X-rays with energy 10 keV were incident on the samples at a grazing incidence angle αi = 0.5-3°, with scattering collected by a Pilatus 2 M (DECTRIS) hybrid photon-counting detector with a sample-to-detector distance (SDD) of 375 mm, with the geometry calibrated using LaB6. In-situ GIWAXS data (Fig. 2b) was measured using the same configuration, with spin-coated 2D intermediate samples aligned before FAI solutions were deposited on top using an in-situ blade coater in ambient conditions. The coating apparatus incorporates a syringe driver, motorised blade, integrated hotplate and an N2 outlet directed at the sample to remove excess solvent after conversion. Solutions were deposited via a microfluidic tube adjacent to the measured area, and coated perpendicular to the beam with the blade set with a shim height of 500 μm and coating speed of ~10 mm s-1, and measured with a grazing incidence angle of 2°.
Additional GIWAXS data (Supplementary Fig. 24) was acquired with a SmartLab (RIGAKU) diffractometer with a 3 kW Cu X-ray source (8.04 keV) in parallel beam configuration with 0.5° pinhole optics, a 1° in-plane parallel slit collimator and 0.3° long collimator attachments. Scattering was collected with a HyPix-3000 (RIGAKU) 2D detector with 65 mm SDD, also at αi = 2°. For each measurement, the detector goniometer arm was rotated through 2θ angles from 0° to 40° in 1° steps, with 15 min acquisition at each position, with detector images then remapped into Q-space and combined together. All data reduction was performed using scripts based on the PyFAI and pygix libraries73,74.
Scanning electron microscopy (SEM)
A FEI Quanta 600 FEG Environmental Scanning Electron Microscope (ESEM) was employed to investigate perovskite layer morphology. Accelerating voltages between 4-15 kV were employed for various analyses.
Nuclear magnetic resonance (NMR) spectroscopy
A two-channel Bruker Avance III HD Nanobay 400 MHz instrument running TOPSPIN 3 equipped with a 5 mm z-gradient broadband/fluorine observation probe is used. The signal from residual non-deuterated DMSO solvent is used for reference.
Nuclear quadrupole resonance (NQR) spectroscopy
For 127I NQR spectroscopy, thin films were mechanically exfoliated from FTO (2D-intermediate FAPbI3) or glass (FAPbI3, DMF:DMSO) substrates using a razor blade to produce powders. These thin film powders were packed into 2.5 mm zirconia rotors and compacted. 127I NQR spectra were recorded on a Bruker Avance III spectrometer equipped with a 2.5 mm CPMAS probe using 42 kHz RF field amplitude, a recycle delay of 0.05 s and with the probe placed outside of any external magnetic field. The sample rotor was static during the measurement.
Thermal desorption-gas chromatography-mass spectrometry (TD-GCMS)
Thin films ( < 1 μm) of perovskite material on fluorine-doped tin oxide-coated glass substrates were loaded into a thermal extractor unit (Micro-Chamber/Thermal Extractor M-CTE250, Markes International)and heated at 165 °C for 60 minutes under a flow of N2 gas (50 mL min-1). Volatile components released during extraction were collected onto sorbent-packed collection tubes. The sorbent tubes are then loaded into a thermal desorption unit and heated rapidly to desorb the volatiles concentrated in the tube, which are then passed via N2 carrier gas into the gas chromatography-mass spectrometry instrument (Agilent 5977B GC/MSD). Identification of the volatile components was done by comparison to the NIST 17 Mass Spectral Library. All reported species showed a > 85 % match with the database compound.
Optically-modulated surface photovoltage measurements (SPV)
Modulated SPV spectra were measured in the configuration of a parallel plate capacitor (quartz cylinder partially coated with the SnO2:F electrode, mica sheet as insulator), under ambient atmosphere. The SPV signal is defined as the change in the surface potential as a result of the illumination. In our case the Illumination was provided by a Halogen lamp, coupled to a quartz prism monochromator (SPM2), and modulated at a frequency of 8 Hz by using an optical chopper. The overall SPV amplitude (combined of the in-phase and out of phase SPV signals) were detected with a high-impedance buffer and a double phase lock-in amplifier (EG&G 5210).
Characterisation of solar cells
Current-voltage (J–V) and maximum power point (MPP) measurements were measured (2400 series source meter, Keithley Instruments) in ambient air under both light (simulated AM 1.5 irradiance generated by a Wavelabs SINUS-220 simulator) and in the dark. The active area of the solar cell was masked with a black-anodised metal aperture to either 0.25 or 1.00 cm2, within a light-tight holder. The ‘forward’ J–V scans were measured from forward bias to short-circuit and the ‘backward’ scans were from short-circuit to forward bias, both at a scan rate of 245 mV s−1. Active MPP tracking measurements using a gradient descent algorithm were performed for at least 30 s to obtain the steady-state power conversion efficiency. Some cells exhibited improvement over multiple measurements, in which case the peak performance was reported. This typically took two to five J–V scan plus MPP tracking iterations for the highest efficiency cells, in a measurement time of around 2–5 minutes. The intensity of the solar simulator was set periodically such that the short-circuit current density from a KG3-filtered Si reference photodiode (Fraunhofer ISE) matched its 1-sun certified value. A local measurement of the intensity before each batch of solar cell measurements were performed, was made by integrating the spectrum obtained from the solar simulator’s internal spectrometer. By taking the ratio of this internal intensity measurement to one obtained at the time of calibration we determined the equivalent irradiance at the time of measurement. For the data presented in this publication, this gave values ranging from 0.985-1.005 suns equivalent, which have been applied to the calculation of power conversion efficiencies for each individual measurement. The spectral mismatch factor was estimated to be 1.022 according to a previously reported method75. This has also been applied to calculate power conversion efficiencies. We estimate the systematic error of this setup to be on the order of ± 5% (relative).
External quantum efficiency (EQE)
External quantum efficiency (EQE) measurements were performed using a custom setup. The solar cells were illuminated with a 250 W quartz-tungsten halogen lamp that was first passed through a monochromator (Princeton Instruments SP2150) with a filter wheel (Princeton Instruments FA2448), then chopped with an optical chopper (Thorlabs MC2000B) at 280 Hz, and finally focussed onto the sample with a smaller spot size than the solar cell area (as defined by the metallic top contact). The amplitude of the resulting AC current signal was measured with a lock-in amplifier (Stanford Research Systems SR830) as the voltage drop across a 50 Ohm resistor in series with the solar cell. A reference measurement of a calibrated Si photodiode of known EQE (Thorlabs FDS100-CAL) was used to correct the measured signal from the solar cell to absolute units. White light bias was applied using LEDs.
Operational stability accelerated-aging equipment
PSCs (p-i-n configuration) aged under ISOS-L-1 and ISOS-L-2 conditions were encapsulated using a cover glass and UV-curable epoxy adhesive (Everlight Eversolar AB341), which was spread across the full active area of the PSCs. As discussed in Supplementary Note 9, for ISOS-L-2 testing, in addition to the PSC fabrication described above, prior to on-cell encapsulation (where the encapsulation glue is spread across the full device area) a 250 nm layer of MoO3 was thermally evaporated76. For ISOS-D-3 aging, an industry standard edge-sealant was employed to encapsulate PSCs, as described in Supplementary Note 10.
For ISOS-D-2 aging, PSCs were not encapsulated and were placed in an oven controlled at 85 ˚C in a N2 filled glovebox.
ISOS-D-3 aging was carried out in a Weiss Technik LabEvent L C/64/40/3 environmental testing chamber at 85 ˚C and 85% relative humidity (RH).
For adapted ISOS-L-1 aging, a bespoke setup was used. RS Peltier Pro modules were employed to actively cool the PSCs to approximately 22 ˚C (measured by a photodiode encased in a black thermally conductive material to simulate the conditions in the photoactive layer). As shown in Supplementary Fig. 29b, an LED array positioned such as to provide 0.86 sun equivalents to the surface of the PSCs was employed. Aging was carried out at Voc under these conditions.
For ISOS-L-2 aging an Atlas SUNTEST CPS+ light-soaking chamber under simulated full spectrum AM1.5 sunlight was employed (no UV filter was applied during the aging process). Aging was carried out at Voc under these conditions. The temperature for the aging chamber was measured by a black standard temperature control unit.
To perform J-V characterisation and stabilised Voc, Jsc and active MPP tracking measurements, all substrates were removed from the aging setups and immediately tested before being returned to the relevant aging condition.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Acknowledgements
We acknowledge the contribution of Shuan Reeksting at the MC2 Institute, University of Bath, for performing and advising on our TD-GCMS measurements, and Kilian Lohmann for providing the evaporated FAPbI3 sample for TD-GCMS. Further, we acknowledge Diamond Light Source for time on beamline I07 under proposal SI33462-1 for the GIWAXS measurements and thank Jonathan Rawle for assistance with these measurements. This project was supported by the Plastic Electronics Centre for Doctoral Training, funded by EPSRC UK, the EPSRC grant number EP/V027131/1 (B.M.G.) and has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 764787 (P.H.). D.J.K. acknowledges the ERC-selected UKRI Horizon Europe guarantee funding (PhotoPeroNMR - grant agreement number EP/Y01376X/1). I.L. thanks the AiF project (ZIM-KK5085302DF0) for financial support.
Author contributions
B.M.G. conceived the experiments with support from P.H. and J.A.S. B.M.G. developed the ether-amine solvent systems, carried out characterisation of the 2D perovskites, investigation of the mechanism of 2D-intermediate conversion, fabrication of the perovskite solar cells, TD-GCMS analysis and all materials and device stability measurements besides those involving in-situ XRD measurements. B.M.G. also carried out the NQR measurements, supervised by D.J.K. S.C. and P.C. assisted with fabrication of p-i-n configuration solar cells. B.M.G., P.H. and J.A.S. carried out XRD analysis. K.A.E. carried out all OPTP and TCSPC measurements and associated calculations, supervised by L.M.H. J.A.S., B.M.G. and F.Y. conducted the GIWAXS characterisation and analysis. D.T.W.T, R.C.K. and A.K.A.Z. designed and built the GIWAXS setup. A.S. analysed the SED data, supervised by N.K.N. E.Y-H.H carried out Pawley fitting of XRD of optimised material. J.M.B. constructed the setup used for EQE measurements and developed the software used to measure and analyse all PSC device performance. M.G.C. designed and implemented the solar simulator and cell layout used for all device performance measurements. I.L. carried out and analysed the modulated SPV measurements. H.J.S supervised the project, developed the ideas and concepts with B.M.G, and raised the funds for the work. B.M.G. wrote the manuscript and P.H., J.A.S. and H.J.S. edited it. All authors contributed to proofing of the manuscript and writing/editing sections related to their contributions.
Peer review
Peer review information
Nature Communications thanks Gang Li, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Data availability
All relevant data are provided in the figures, tables and Supplementary Information. The raw data that support the findings of this study are available via the Zenodo repository at: 10.5281/zenodo.14008401.
Competing interests
H.J.S. is a co-founder and Chief Scientific Officer of Oxford PV Ltd., a company industrialising perovskite PV. The other authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-54113-4.
References
- 1.Zhu, H. et al. Long-term operating stability in perovskite photovoltaics. Nat. Rev. Mater.8, 569–586 (2023). [Google Scholar]
- 2.McMeekin, D. P. et al. A mixed-cation lead mixed-halide perovskite absorber for tandem solar cells. Science (1979)351, 151–155 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Lin, Y. H. et al. A piperidinium salt stabilizes efficient metal-halide perovskite solar cells. Science (1979)369, 96–102 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Yun, J. S. et al. Humidity-Induced Degradation via Grain Boundaries of HC(NH2)2PbI3 Planar Perovskite Solar Cells. Adv. Funct. Mater.28, 1705363 (2018).
- 5.Knight, A. J. et al. Halide segregation in mixed-halide perovskites: influence of a-site cations. ACS Energy Lett.6, 799–808 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mundt, L. E. et al. Mixing matters: nanoscale heterogeneity and stability in metal halide perovskite solar cells. ACS Energy Lett.7, 471–480 (2022). [Google Scholar]
- 7.Shen, X. et al. Chloride-Based Additive Engineering for Efficient and Stable Wide-Bandgap Perovskite Solar Cells. Adv. Mater.35, 2211742 (2023). [DOI] [PubMed]
- 8.Vidal, R., Alberola-Borràs, J. A. & Mora-Seró, I. Abiotic depletion and the potential risk to the supply of cesium. Resources Policy68, 101792 (2020).
- 9.Tan, S. et al. Shallow iodine defects accelerate the degradation of α-phase formamidinium perovskite. Joule4, 2426–2442 (2020). [Google Scholar]
- 10.Woo, Y. W., Li, Z., Jung, Y. K., Park, J. S. & Walsh, A. Inhomogeneous defect distribution in mixed-polytype metal halide perovskites. ACS Energy Lett.8, 356–360 (2023). [Google Scholar]
- 11.Macpherson, S. et al. Local nanoscale phase impurities are degradation sites in halide perovskites. Nature607, 294–300 (2022). [DOI] [PubMed] [Google Scholar]
- 12.Chen, S. et al. Stabilizing perovskite-substrate interfaces for high-performance perovskite modules. Science 373, 902–907 (2021). [DOI] [PubMed] [Google Scholar]
- 13.Chen, L., Yoo, J. W., Hu, M., Lee, S. U. & Seok, S. Il. Intrinsic phase stability and inherent bandgap of formamidinium lead triiodide perovskite single crystals. Angew. Chem. - Int. Ed.61, 1–7 (2022). [DOI] [PubMed]
- 14.McMeekin, D. P. et al. Intermediate-phase engineering via dimethylammonium cation additive for stable perovskite solar cells. Nat. Mater.22, 73–83 (2023). [DOI] [PubMed] [Google Scholar]
- 15.Du, T. et al. Additive-free, low-temperature crystallization of stable α-fapbi3 perovskite. Adv. Mater.34, 1–10 (2022). [DOI] [PubMed] [Google Scholar]
- 16.Bu, T. et al. Lead halide-templated crystallization of methylamine-free perovskite for efficient photovoltaic modules. Science 372, 1327–1332 (2021). [DOI] [PubMed] [Google Scholar]
- 17.Huang, X. et al. Solvent gaming chemistry to control the quality of halide perovskite thin films for photovoltaics. ACS Cent. Sci.8, 1008–1016 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao, J. et al. Identifying the molecular structures of intermediates for optimizing the fabrication of high-quality perovskite films. J. Am. Chem. Soc.138, 9919–9926 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Gratia, P. et al. The many faces of mixed ion perovskites: unraveling and understanding the crystallization process. ACS Energy Lett.2, 2686–2693 (2017). [Google Scholar]
- 20.Moore, D. T. et al. Crystallization kinetics of organic-inorganic trihalide perovskites and the role of the lead anion in crystal growth. J. Am. Chem. Soc.137, 2350–2358 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Park, J. et al. Controlled growth of perovskite layers with volatile alkylammonium chlorides. Nature616, 724–730 (2023). [DOI] [PubMed] [Google Scholar]
- 22.Jiang, Q. et al. Surface reaction for efficient and stable inverted perovskite solar cells. Nature611, 278–283 (2022). [DOI] [PubMed] [Google Scholar]
- 23.Vidal, R. et al. Assessing health and environmental impacts of solvents for producing perovskite solar cells. Nat. Sustain4, 277–285 (2021). [Google Scholar]
- 24.Zhou, Y. et al. Room-temperature crystallization of hybrid-perovskite thin films via solvent-solvent extraction for high-performance solar cells. J. Mater. Chem. A Mater.3, 8178–8184 (2015). [Google Scholar]
- 25.Jeon, N. J. et al. Solvent engineering for high-performance inorganic-organic hybrid perovskite solar cells. Nat. Mater.13, 897–903 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Deng, Y. et al. Tailoring solvent coordination for high-speed, room-temperature blading of perovskite photovoltaic films. Sci. Adv.5, 1–8 (2019). [DOI] [PMC free article] [PubMed]
- 27.Noel, N. K. et al. A low viscosity, low boiling point, clean solvent system for the rapid crystallisation of highly specular perovskite films. Energy Environ. Sci.10, 145–152 (2017). [Google Scholar]
- 28.International council for harmonisation of technical requirements for pharmaceuticals for human use. Guideline for Residual Solvents: Q3C(R8). ICH (2021).
- 29.Williams, A. C. & Barry, B. W. Penetration enhancers. Adv. Drug Deliv. Rev.56, 603–618 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Wang, Z., Richter, S. M., Gates, B. D. & Grieme, T. A. Safety concerns in a pharmaceutical manufacturing process using dimethyl sulfoxide (DMSO) as a solvent. Org. Process Res Dev.16, 1994–2000 (2012). [Google Scholar]
- 31.Slater, C. S., Savelski, M. J., Hitchcock, D. & Cavanagh, E. J. Environmental analysis of the life cycle emissions of 2-methyl tetrahydrofuran solvent manufactured from renewable resources. J. Environ. Sci. Health A Tox Hazard Subst. Environ. Eng.51, 487–494 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Zhang, F. et al. Advances in two-dimensional organic-inorganic hybrid perovskites. Energy. Environ. Sci.13, 1154–1186 (2020).
- 33.Zheng, X. et al. Managing grains and interfaces via ligand anchoring enables 22.3%-efficiency inverted perovskite solar cells. Nat Energy5, 131–140 (2020).
- 34.Miao, Y. et al. From 1D to 3D: Fabrication of CH3NH3PbI3 Perovskite Solar Cell Thin Films from (Pyrrolidinium)PbI3 via Organic Cation Exchange Approach. Energy Technol.2000148, 1–5 (2020). [Google Scholar]
- 35.Adjokatse, S., Fang, H. H., Duim, H. & Loi, M. A. Scalable fabrication of high-quality crystalline and stable FAPbI3 thin films by combining doctor-blade coating and the cation exchange reaction. Nanoscale11, 5989–5997 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Taddei, M. et al. Ethylenediamine addition improves performance and suppresses phase instabilities in mixed-halide perovskites. ACS Energy Lett.7, 4265–4273 (2022). [Google Scholar]
- 37.O’Kane, M. E. et al. Perovskites on ice: an additive-free approach to increase the shelf-life of triple-cation perovskite precursor solutions. ChemSusChem14, 2537–2546 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang, Z. et al. Efficient ambient-air-stable solar cells with 2D-3D heterostructured butylammonium-caesium-formamidinium lead halide perovskites. Nat. Energy2, 1–10 (2017). [Google Scholar]
- 39.Duijnstee, E. A. et al. Understanding the degradation of methylenediammonium and its role in phase-stabilizing formamidinium lead triiodide. J. Am. Chem. Soc.145, 10275–10284 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doherty, T. A. S. et al. Stabilized tilted-octahedra halide perovskites inhibit local formation of performance-limiting phases. Science 374, 1598–1605 (2021). [DOI] [PubMed] [Google Scholar]
- 41.Luo, D. et al. Enhanced photovoltage for inverted planar heterojunction perovskite solar cells. Science (1979)360, 1442–1446 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Stoumpos, C. C. et al. Ruddlesden-popper hybrid lead iodide perovskite 2d homologous semiconductors. Chem. Mater.28, 2852–2867 (2016). [Google Scholar]
- 43.Le Corre, V. M. et al. Revealing charge carrier mobility and defect densities in metal halide perovskites via space-charge-limited current measurements. ACS Energy Lett.6, 1087–1094 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang, Y. et al. From 2D to 3D: A facile and effective procedure for fabrication of planar CH3NH3PbI3 perovskite solar cells. J. Mater. Chem. A Mater.6, 17867–17873 (2018). [Google Scholar]
- 45.Sidhik, S. et al. Two-dimensional perovskite templates for durable, efficient formamidinium perovskite solar cells. Science384, 1227–1235 (2024). [DOI] [PubMed] [Google Scholar]
- 46.Kim, M. et al. Methylammonium chloride induces intermediate phase stabilization for efficient perovskite solar cells. Joule3, 2179–2192 (2019).
- 47.Ye, F. et al. Roles of MACl in Sequentially Deposited Bromine-Free Perovskite Absorbers for Efficient Solar Cells. Adv. Mater.2007126, 1–8 (2020). [DOI] [PubMed] [Google Scholar]
- 48.Xie, F. et al. Vertical recrystallization for highly efficient and stable formamidinium-based inverted-structure perovskite solar cells. Energy Environ. Sci.10, 1942–1949 (2017). [Google Scholar]
- 49.Stranks, S. D. et al. Recombination kinetics in organic-inorganic perovskites: excitons, free charge, and subgap states. Phys. Rev. Applied2, 034007 (2014).
- 50.Yamada, Y., Endo, M., Wakamiya, A. & Kanemitsu, Y. Spontaneous defect annihilation in CH3NH3PbI3 thin films at room temperature revealed by time-resolved photoluminescence spectroscopy. J. Phys. Chem. Lett.6, 482–486 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Yamada, Y., Nakamura, T., Endo, M., Wakamiya, A. & Kanemitsu, Y. Photocarrier recombination dynamics in perovskite CH3NH3PbI3 for solar cell applications. J. Am. Chem. Soc.136, 11610–11613 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Herz, L. M. Charge-carrier mobilities in metal halide perovskites: fundamental mechanisms and limits. ACS Energy Lett.2, 1539–1548 (2017). [Google Scholar]
- 53.Chen, T. et al. Entropy-driven structural transition and kinetic trapping in formamidinium lead iodide perovskite. Sci. Adv.2, 1–7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Asfaw, A. A., Wolfs, K., Van Schepdael, A. & Adams, E. Determination of residual dimethylsulphoxide in drug loaded gelatin using thermal desorber – gas chromatography. J. Pharm. Biomed. Anal.153, 193–198 (2018). [DOI] [PubMed] [Google Scholar]
- 55.Shi, L. et al. Gas chromatography-mass spectrometry analyses of encapsulated stable perovskite solar cells. Science368, 6497 (2020). [DOI] [PubMed]
- 56.Ma, L. et al. Temperature-dependent thermal decomposition pathway of organic-inorganic halide perovskite materials. Chem. Mater.31, 8515–8522 (2019). [Google Scholar]
- 57.Juarez-Perez, E. J., Ono, L. K. & Qi, Y. Thermal degradation of formamidinium based lead halide perovskites into: Sym -triazine and hydrogen cyanide observed by coupled thermogravimetry-mass spectrometry analysis. J. Mater. Chem. A Mater.7, 16912–16919 (2019). [Google Scholar]
- 58.Fu, F. et al. I2 vapor-induced degradation of formamidinium lead iodide based perovskite solar cells under heat-light soaking conditions. Energy Environ. Sci.12, 3074–3088 (2019). [Google Scholar]
- 59.Ren, G. et al. Strategies of modifying spiro-OMeTAD materials for perovskite solar cells: a review. J. Mater. Chem. A Mater.9, 4589–4625 (2021). [Google Scholar]
- 60.Thampy, S., Zhang, B., Hong, K. H., Cho, K. & Hsu, J. W. P. Altered stability and degradation pathway of CH3NH3PbI3 in contact with metal oxide. ACS Energy Lett.5, 1147–1152 (2020). [Google Scholar]
- 61.Wang, K. et al. Novel inorganic electron transport layers for planar perovskite solar cells: Progress and prospective. Nano Energy68, 104289 (2020). [Google Scholar]
- 62.Jacobsson, T. J. et al. An open-access database and analysis tool for perovskite solar cells based on the FAIR data principles. Nat. Energy7, 107–115 (2022). [Google Scholar]
- 63.Chen, T. et al. Inhibition of defect-induced α-to-δ phase transition for efficient and stable formamidinium perovskite solar cells. Nat. Commun.14, 1–9 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rothmann, M. U. et al. Atomistic Understanding of the Coherent Interface Between Lead Iodide Perovskite and Lead Iodide. Adv. Mater Interfaces10, 2300249 (2023).
- 65.Kempe, M. D., Panchagade, D., Reese, M. O. & Dameron, A. A. Modelling moisture ingress through polyisobutylene-based edge-seals. Prog. Photovol. Res. Appl.23, 570–581 (2014). [Google Scholar]
- 66.Al-Ashouri, A. et al. Wettability improvement of a carbazole-based hole-selective monolayer for reproducible perovskite solar cells. ACS Energy Lett.8, 898–900 (2023). [Google Scholar]
- 67.Jin, H. et al. Alumina nanoparticle interfacial buffer layer for low-bandgap lead-tin perovskite solar cells. Adv. Funct. Mater.33, 1–10 (2023). [Google Scholar]
- 68.Hu, S. et al. A universal surface treatment for p-i-n perovskite solar cells. ACS Appl Mater. Interfaces14, 56290–56297 (2022). [DOI] [PubMed] [Google Scholar]
- 69.Hu, S. et al. Optimized carrier extraction at interfaces for 23.6% efficient tin-lead perovskite solar cells. Energy Environ. Sci.15, 2096–2107 (2022).
- 70.Longo, G. et al. Understanding the Performance-Limiting Factors of Cs2AgBiBr6 Double-Perovskite Solar Cells. ACS Energy Lett.5, 2200–2207 (2020). [Google Scholar]
- 71.Savill, K. J. et al. Impact of tin fluoride additive on the properties of mixed tin-lead iodide perovskite semiconductors. Adv. Funct. Mater.30, 1–13 (2020). [Google Scholar]
- 72.Joyce, H. J., Boland, J. L., Davies, C. L., Baig, S. A. & Johnston, M. B. A review of the electrical properties of semiconductor nanowires: Insights gained from terahertz conductivity spectroscopy. Semicond. Sci. Technol.31, 103003 (2016).
- 73.Ashiotis, G. et al. The fast azimuthal integration Python library: PyFAI. J. Appl Crystallogr48, 510–519 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dane, T. G. The pygix library. https://github.com/tgdane/pygix.
- 75.Snaith, H. J. How should you measure your excitonic solar cells? Energy Environ. Sci.5, 6513–6520 (2012). [Google Scholar]
- 76.Belich, N. A. et al. How to stabilize standard perovskite solar cells to withstand operating conditions under an ambient environment for more than 1000 hours using simple and universal encapsulation. J. Energy Chem.78, 246–252 (2023). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data are provided in the figures, tables and Supplementary Information. The raw data that support the findings of this study are available via the Zenodo repository at: 10.5281/zenodo.14008401.