

ABSTRACT
Reperfusion of ischemic skeletal muscle triggers oxidative stress and an immediate inflammatory reaction, leading to damage of distant organs such as the lungs. The inflammatory process implicates numerous mediators, including cytokines, chemokines, and arachidonic acid metabolites. In the orchestration of the inflammatory cascade, a critical role is played by the cluster of differentiation‐36 receptor (CD36), a scavenger receptor class B protein (SR‐B2) which is expressed on macrophages and functions as a Toll‐like receptor coreceptor. A mouse model of hind limb ischemia‐reperfusion has been used to investigate the interplay between CD36 signaling and remote inflammation: leukocyte recruitment, regulation of the nucleotide‐binding domain leucin‐rich repeat and pyrin‐containing receptor 3 (NLRP3) inflammasome, and release of nuclear factor‐kappa B (NF‐ĸB) and arachidonic acid metabolites. Levels of reactive oxygen species, inflammatory mediators, and gene expression were measured in blood and lung tissue samples collected from anesthetized mice on which unilateral hind limb ischemia was induced by rubber band constriction for 30 min followed by reperfusion for 3 h. The CD36 modulator EP 80317, a member of the growth hormone releasing peptide 6 family, was employed as a pharmacological agent to mitigate distant lung injury following skeletal limb ischemia‐reperfusion. Targeting CD36 on monocytes/macrophages, EP 80317 abated pro‐inflammatory signaling and transcriptional activity encompassing lipid and cytokine mediators. Targeting CD36 was shown to offer promise for curtailing tissue injury following hind limb ischemia‐reperfusion.
Keywords: CD36, hind limb, ischemia, remote inflammation, reperfusion
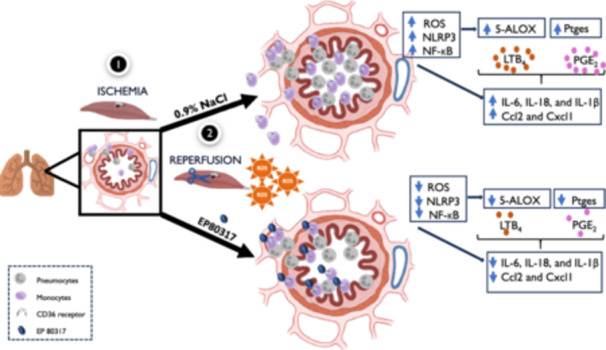
Proposed mechanism by which CD36 participates in generation of ROS and activation of leukocytes in the circulation and microvascular endothelium of lung tissue after reperfusion of an ischemic limb. Treatment with EP 80317, the CD36 modulator, decreased remote leukocyte recruitment, lipid mediators, cytokines, and lung tissue damage.
Abbreviations
- Alox
arachidonate lipoxygenase
- Alox5ap
arachidonate 5‐lipoxygenase activating protein
- ARDS
acute respiratory distress syndrome
- Atx
autotaxin
- BHT
butylated hydroxytoluene
- CCL2
chemokine C‐C motif ligand 2
- CD
cluster of differentiation
- Cox2
cyclooxygenase 2
- Cxcl1
chemokine C‐X‐C motif ligand 1
- EDTA
ethylenediaminetetraacetic acid
- HTAB
hexadecyltrimethylammonium bromide
- Igf‐1
insulin‐like growth factor 1
- IL
interleukin
- LTB4
leukotriene B4
- Ltb4r1
leukotriene B4 receptor 1
- Ltc4s
leukotriene C4 synthase
- Ly6G
lymphocyte antigen 6 complex locus G6D
- MDA
malondialdehyde
- MPO
myeloperoxidase
- NF‐κB
nuclear factor‐kappa B
- Nlrp3
nucleotide‐binding domain leucin‐rich repeat and pyrin‐containing receptor 3
- NOD
Nod‐like receptor
- Nox2
NADPH oxidase 2
- PGE2
prostaglandin E2
- PMNs
polymorphonuclear neutrophils
- Ptger2
prostaglandin E receptor 2 (subtype EP2)
- Ptger4
prostaglandin E receptor 4 (subtype EP4)
- Ptges
prostaglandin E synthase
- ROS
reactive oxygen species
- Siglecf
sialic acid binding Ig‐like lectin F
- SR‐B2
scavenger receptor B2
- TBARS
thiobarbituric acid reactive substance
- TLR
toll‐like receptor
- TMB
3,3′,5,5′‐tetramethylbenzidine
- TNF‐α
tumor necrosis factor alpha
1. Introduction
Reperfusion of skeletal ischemic tissue is crucial for functional recovery but leads paradoxically to remote organ injury [1]. For example, respiratory complications and lung damage have been correlated with the duration of ischemia preceding reperfusion [2]. Within the lungs, alveolar macrophages and monocyte‐derived lung macrophages play vital roles in innate defense mechanisms and respond to the sterile inflammatory response to ischemia and reperfusion [3]. In response to infection and injury, circulating polymorphonuclear neutrophils (PMNs) and monocytes are recruited to the lungs [2]. In the lungs, alveolar and interstitial macrophages play a crucial role in innate defense against infections and contribute to resolving sterile inflammation [4]. Reactive oxygen species (ROS) are generated and released in early events after hind limb ischemia and reperfusion and promote subsequent biosynthesis of arachidonic acid metabolites [5, 6]. Skeletal muscle tissue injury elicits release of inflammatory cytokines and systemic inflammation [7, 8, 9]. In rodent and rabbit models exposed to hind limb ischemic insult followed by reperfusion, lung damage has been shown to be due in part to activated PMNs, which are mainly trapped within lung capillaries [10, 11, 12, 13, 14]. In remote lung damage following hind limb ischemia and reperfusion, the causative and amplifying contributions remain incompletely characterized for PMNs, monocytes, and macrophages as sources of inflammatory mediators.
The cluster of differentiation 36 receptor [CD36, scavenger receptor B2 (SR‐B2)] is a Toll‐like receptor (TLR) coreceptor which regulates activation of the CD36/TLR heterodimer complex and consequent transcription of pro‐inflammatory cytokines, production of nitric oxide, and generation of ROS [15]. Situated on the cellular surfaces of monocytes and macrophages, the CD36/TLR heterodimer complex responds to endogenous ligands and plays pivotal roles in inflammation and innate immunity [16]. As coreceptor, CD36 is linked to the immune response and physio‐pathological conditions which share inflammation as a common feature, including atherosclerosis, atherothrombosis, diabetes, stroke, Alzheimer's disease, angiogenesis, and cancer [17]. In the lungs, CD36 is expressed on the surfaces of alveolar macrophages, type 2 pneumocytes, and interstitial macrophages, as well as blood‐derived monocytes, which are recruited during inflammation [18, 19]. The severity of tissue damage has been shown to be mediated by CD36 in brain ischemia [20] and myocardial ischemia‐reperfusion [21, 22, 23], as well as in acute lung injury induced by H2O2 [24] and lipopolysaccharide [25]. The latter is associated with M1 polarization and nuclear factor‐kappa B (NF‐κB) activation [25]. In isolated mononuclear phagocytes, CD36 plays a pro‐inflammatory role by activating NF‐κB and the Nod‐like receptors (NLR) family pyrin domain containing 3 (NLRP3) inflammasome [15].
Hypothesizing that activation of CD36 on monocytes and macrophages in the lungs could potentiate inflammation, the present study employs genetic and pharmacological methods to investigate the role of the coreceptor in regulating leukocyte recruitment to inflammatory sites in an acute inflammatory murine model of hind limb ischemia and reperfusion. The ligand EP 80317 has previously been shown to modulate inflammatory responses in a CD36‐dependent manner [21, 26]. Employing EP 80317 to modulate CD36 signaling, the relevance of the latter in exacerbating the inflammatory response has been indicated by measuring decreased production of cytokines, ROS, and arachidonic acid metabolites.
2. Materials and Methods
2.1. Animals
CD36‐deficient (CD36−/−) mice and wild‐type littermate controls on a C57Bl/6 J background were obtained from Jackson Laboratories (Bar Harbor, Maine, USA) and bred following established protocols. The mice were housed and cared for in local animal facilities, maintained on a 12:12‐h light/dark cycle, and provided with standard conditions, including ad libitum access to food and water. Thirty‐two wildtype (CD36+/+) and twelve CD36−/− male mice aged between 12 and 14 weeks were equally divided into two groups (control and EP 80317) and utilized for all experimental procedures. The Institutional Animal Ethics Committee approved all experimental protocols, ensuring compliance with guidelines outlined by the Canadian Council on Animal Care and the US National Institute of Health for the ethical treatment and use of laboratory animals.
2.2. Drug and Experimental Protocol
The drug EP 80317 (H‐Haic–D‐2MeTrp–d‐Lys–Trp–d‐Phe–Lys‐NH2) was obtained from Europeptides (Argenteuil, France) and reconstituted in sterile 0.9% NaCl (Baxter Corporation, Toronto, Ontario, Canada) before parenteral administration. The control group was treated with sterile 0.9% NaCl. For 14 days, CD36+/+ and CD36‐/‐ mice were subcutaneously injected daily with either EP 80317 (300 μg/kg) or 0.9% NaCl, excluding the day of experimentation. Following anesthesia with isoflurane, mice underwent a 30‐min ischemia of the right hind limb induced by applying a rubber band (black latex o‐ring, Miltex, York, Germany) above the greater trochanter using a McGivney hemorrhoidal ligator (Miltex, York, Germany). After 3 h of hind limb reperfusion, mice were euthanized via isoflurane overdose and exsanguination ( ~ 1000 µL in heparinized tubes) from the jugular vein. Lung tissues were collected and stored at –80°C until further analysis.
2.3. Histology
Lung tissues were fixed in formalin, sectioned into 5 µm‐thick slices, processed, and subsequently stained with hematoxylin and eosin by McGill University Health Centre's histopathology platform. Images were captured using a 40X objective using a NanoZoomer 2.0‐HT digital scanner and NDP view2 software (Hamamatsu Photonics, Shizuoka, Japan). Cells were counted manually using Adobe Photoshop CS3 software (San José, CA, United States) by two individuals who were blinded from knowledge of prior treatments.
2.4. Myeloperoxidase Assay in Lung Homogenates
A standard curve was constructed to determine the number of PMNs in tissue. PMNs were harvested from the mice peritoneal cavity 16 h after an intraperitoneal injection of 2 mL of 5% casein (Sigma‐Aldrich, St Louis, MO, USA), euthanasia was performed, and samples were collected and purified by positive selection (> 98% PMN) using an anti‐lymphocyte antigen 6 complex locus G6D (anti‐Ly6G) magnetic microbead kit (Miltenyi Biotec, Auburn, CA), according to the manufacturer's instructions. Hypotonic lysis of red blood cells was performed by resuspension of the cells in 0.2% NaCl for 20 s, followed by the addition of an equal volume of 1.6% NaCl. Aliquots of neutrophils (106 cells/mL) were stored at –80°C in acetate buffer (100 mM), pH 6, supplemented with 1% hexadecyltrimethylammonium bromide (HTAB) (Sigma‐Aldrich, St Louis, MO, USA) and 20 mM ethylenediaminetetraacetic acid (EDTA) (Sigma‐Aldrich, St Louis, MO, USA). Thawed aliquots were homogenized, incubated at 65°C for 2 h, centrifuged at 2000 g for 10 min, and the supernatants were used to construct the MPO standard curve. Lung tissue MPO was assayed as previously described, with slight modifications [27]. The lung tissue was homogenized in 1 mL PBS (Gibco Life Technologies, Grand Island, NY, USA), followed by centrifugation. The resulting pellets were suspended in 1 mL acetate buffer with 1% HTAB and 20 mM EDTA before undergoing another round of homogenization. Lung homogenates were heated to 65°C for 2 h, subjected to 3 freeze–thaw cycles, and then centrifuged at 2000 g for 10 min. Lung tissue MPO was assayed by incubating supernatants with 3.2 mM of 3,3′,5,5′‐tetramethylbenzidine (TMB, Sigma‐Aldrich, St Louis, MO, USA) and 1.0 mM of H2O2 (Sigma‐Aldrich, St Louis, MO, USA) for 5 min at 37°C. The reaction was stopped by the addition of 100 µL of 0.2 M sodium acetate (pH 3). Equivalent numbers of PMNs per tissue homogenate were calculated from the standard curve and normalized per g.
2.5. Real Time‐Quantitative Polymerase Chain Reaction (RT‐qPCR) in Lung Tissue
Total mRNA was extracted from lung tissue with the Ribozol RNA Extraction Reagent (VWR International, Radnor, PA, USA) with the PureLinkTM RNA Micro‐Kit (Invitrogen, Waltham, MA, USA), as described previously [28]. Relative mRNA expression levels of genes were determined using the comparative CT (2‐ΔΔCt) method, and results were normalized to the mean of 5 internal controls, βactin, Gapdh, Hprt, Rpl13a, and Ywhaz. The murine primer sequences are detailed in Table 1.
Table 1.
qPCR murine primer sequences.
| Gene | Primer | Product length (bp) | NCBI gGene ID |
|---|---|---|---|
| Βactin | Forward CAGCAAGCAGGAGTACGATGA | 93 | 11461 |
| Reverse GAAAGGGTGTAAAACGCAGCTC | |||
| Alox5 | Forward GTCCTGAGGGATGGACGTGCAAAAT | 89 | 11689 |
| Reverse TGCCGTGCCTCCAGTTCTTTACG | |||
| Alox5ap | Forward GGACTCTTGCCTTTGAGCGGGT | 139 | 11690 |
| Reverse TGCCGAAGATGTAGCCAGGGGT | |||
| Alox12 | Forward AGTGCGTTTGTGGCTGGTTGGG | 90 | 11684 |
| Reverse AAGTCAAACTCCTCCTCCTTGCCCC | |||
| Alox15 | Forward TGGGGCAACTGGAAGGATGGCA | 138 | 11687 |
| Reverse AACGGTGTCCATTGTCCCCAGAAC | |||
| Atx | Forward GACCCTAAAGCCATTATTGCTAA | 81 | 18606 |
| Reverse GGGAAGGTGCTGTTTCATGT | |||
| Ccl2 | Forward TCATGCTTCTGGGCCTGCTGTTCA | 101 | 20296 |
| Reverse GAATGAGTAGCAGCAGGTGAGTGGG | |||
| Cd11c | Forward CAGAGCCAGAACTTCCCAACTGCAC | 87 | 16411 |
| Reverse GATGCTACCCGAGCCATCAATCAGG | |||
| Cox2 | Forward GGACTGGGCCATGGAGTGGACTTAAA | 124 | 19225 |
| Reverse GGGGATACACCTCTCCACCAATGACC | |||
| Cxcl1 | Forward CACCCAAACCGAAGTCATAGCCACA | 125 | 14825 |
| Reverse TCTTTCTCCGTTACTTGGGGACACCT | |||
| Ptger2 | Forward TTGCCATACTTAGGCCACCG | 153 | 19217 |
| Reverse CGCATCCTCACAACTGTCCA | |||
| Ptger3 | Forward ATTGCAGTTCGCCTGGCTTCGC | 127 | 19218 |
| Reverse AGGTGGAGCTGGAAGCATAGTTGGT | |||
| Ptger4 | Forward GTGCTCATCTGCTCCATTCCGCT | 108 | 19219 |
| Reverse CCTGATGGCCTGCAAATCTGGGTT | |||
| Gapdh | Forward TCGGTGTGAACGGATTTGGCCG | 147 | 14433 |
| Reverse TGCCGTGAGTGGAGTCATACTGGA | |||
| Hprt | Forward TCCTCCTCAGACCGCTTTTTGCC | 80 | 15452 |
| Reverse CATCGCTAATCACGACGCTGGGA | |||
| Igf1 | Forward GCTTTTACTTCAACAAGCCCACAGGC | 150 | 16000 |
| Reverse AGCGGGCTGCTTTTGTAGGCT | |||
| Il1β | Forward GGACCCCAAAAGATGAAGGGCTGC | 146 | 16176 |
| Reverse TGCCACAGCTTCTCCACAGCCA | |||
| Il6 | Forward CTCTGGAGCCCACCAAGAACGA | 84 | 16193 |
| Reverse AAGGCAACTGGATGGAAGTCTCTTGC | |||
| Il18 | Forward TGCCAGTGAACCCCAGACCAGA | 86 | 16173 |
| Reverse CCTTCACAGAGAGGGTCACAGCCA | |||
| Ltb4r1 | Forward TGGCTGTGTTGCTCACTGCTCC Reverse ACAGGCGGCAACCCATCTCTCT | 84 | 16995 |
| Ltc4s | Forward CCCTGTGCGGACTGTTCTACCTGT | 93 | 17001 |
| Reverse GCAGGAGCATCTGGAGCCATCTGA | |||
| Nfκb1 | Forward GGATTTGCTGAGGGTTGGGGCT | 131 | 18033 |
| Reverse GGGGCGCTGCTTTTCTGCTCTT | |||
| Nfκb2 | Forward AGCAGGAGGCCAAGGAGCTGAA | 114 | 18034 |
| Reverse TCACAGGCTTCAGGGGCAAGGA | |||
| Nlrp3 | Forward AAGGACCAGCCAGAGTGGAATGACA | 108 | 216799 |
| Reverse GCGGGAGAGATATCCCAGCAAACCC | |||
| Nox2 | Forward TGCTGGAGACCCAGATGCAGGAA | 102 | 13058 |
| Reverse GCACAGCAAAGTGATTGGCCTGAGAT | |||
| Ptges | Forward CAAGATGTACGCGGTGGCTGTCA | 90 | 64292 |
| Reverse CCTCCACGTTTCAGCGCATCCT | |||
| RelA | Forward ATCGAACAGCCGAAGCAACGGG | 112 | 19697 |
| Reverse TGGTGGGGTGTGTCTTGGTGGT | |||
| RelB | Forward GGTTCCAGTGACCTCTCTTCCCTGT | 90 | 19698 |
| Reverse CAGGCCAAAGCCGTTCTCCTTAATGT | |||
| Rpl13a | Forward GAAGCAGATCTTGAGGTTACGGA | 131 | 22121 |
| Reverse GCAGGCATGAGGCAAACAGT | |||
| Siglecf | Forward CCAAAGGTCTCACAGGCAGGCAA | 100 | 233186 |
| Reverse AGAGGCAGACAGCAAGCAAGCC | |||
| Tnf | Forward TGTAGCCCACGTCGTAGCAAACCA | 149 | 21926 |
| Reverse CCTGGGAGTAGACAAGGTACAACCCA | |||
| Ywhaz | Forward TCCCCAATGCTTCGCAACCAGAA | 126 | 22631 |
| Reverse CTTGCTGTGACTGGTCCACAATTCCT |
2.6. Cytokine and Chemokine Levels in Lung Homogenates
The lung homogenates underwent analysis using commercial ELISA kits (eBioscience, Waltham, MA, USA) specific for IL‐1β (#88‐7013), following the manufacturer's guidelines.
2.7. PGE2 and LTB4 Levels in Lung Homogenates
Commercial ELISA kits (R&D System Inc., Minneapolis, USA) were used to measure PGE2 (# KGE004B) and LTB4 (# KGE006B) levels in lung homogenates using a competitive binding assay according to the manufacturer's instructions.
2.8. Malondialdehyde Plasma Levels
Malondialdehyde (MDA) which was generated from the breakdown of primary and secondary lipid peroxidation products, was quantified using the thiobarbituric acid reactive substance (TBARS) assay (Sigma‐Aldrich, St Louis, MO, USA). Plasma samples (25 μL) were mixed with PBS (475 μL) and incubated with 30 μL of butylated hydroxytoluene (BHT) and 1 mL of TBARS reagents for 60 min in glass tubes heated to 95°C in a heating block. After cooling, the samples were centrifuged at 1100 g for 10 min at 4°C, and the absorbances were measured at 532 nm. MDA concentrations were determined using the equation: MDA = Absorbance/1.56 × 105 (mole/L) as the molar absorbance coefficient (Kheradmand, Alirezaei, Asadian, Rafiei Alavi, & Joorabi, 2009).
2.9. Statistical Analysis
Data were analyzed using GraphPad Prism version 8.4.3 (San Diego, CA, USA) and expressed as mean ± SEM. To determine significant differences among groups in CD36+/+ and CD36‐/‐ mice, individual comparisons were made between groups using an unpaired t test with Welch's correction or the Mann–Whitney test. Statistical significance was considered at a p value < 0.05.
3. Results
3.1. EP 80317 Attenuates Plasma and Lung ROS and Inflammatory Mediators Following Hind Limb Ischemia and Reperfusion in a CD36‐dependent Manner
Wildtype (CD36+/+) and CD36‐deficient (CD36‐/‐) mice were pretreated daily with EP 80317 for 14 days before being subjected to a 30 min unilateral hind limb ischemia followed by 180 min reperfusion (I30/R180) (Figure 1A). EP 80317, identified previously as a nongrowth hormone secretagogue [29], did not alter the relative expression of Igf1 mRNA levels in lung tissue of CD36+/+ and CD36‐/‐ mice (Figure 1B,F). Mice treated with EP 80317 showed reduced ROS levels both systemically (Figure 1C,G) and remotely in lungs (Figure 1D,H) in a CD36‐dependent manner following hind limb ischemia and reperfusion. Using the MPO assay, total leukocyte recruitment into the lungs was assessed and shown to be reduced by 39% (p < 0.001) after pretreatment with EP 80317 compared to 0.9% NaCl vehicle in CD36+/+ but not CD36‐/‐ mice (Figure 1E,I). Manual microscopic analysis revealed a 28% decrease of mononuclear phagocyte cells (p < 0.05) (Figure 1J) without significant effect on granulocytes (Figure 1K), validating the effect of EP 80317 on CD36+/+ mice. Alveolar macrophage counts were unaltered in EP 80317‐treated versus vehicle‐treated mice (449 ± 32 vs. 495 ± 17 cells per mm2). Preserved alveolar structure, less vascular congestion, and diminished leukocyte accumulation, were observed in the photomicrographs of right lung sections from EP 80317‐treated CD36+/+ mice compared to those from vehicle‐treated CD36+/+ and CD36‐/‐ counterparts (Figure 1L).
Figure 1.
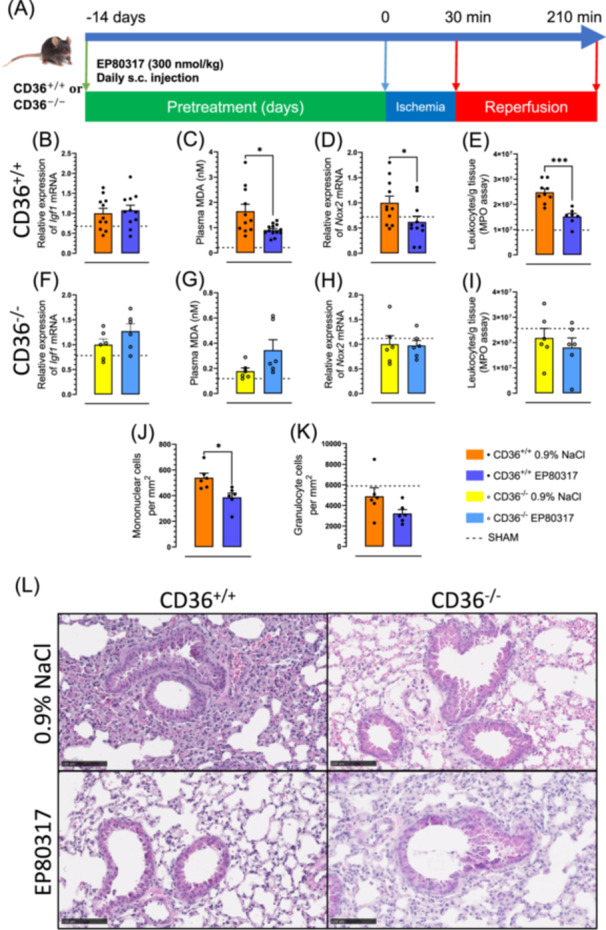
EP 80317 reduces systemic and lung homogenate ROS and inflammatory mediators. (A) Study design. (B) Bar graphs and dot plots represent the relative expression of Igf1 mRNA in lung tissue of CD36+/+ and (F) CD36‐/‐ mice. (C) Mean plasma MDA levels of CD36+/+ and (G) CD36‐/‐ mice, expressed as bar graphs and dot plots. (D) Bar graphs and dot plots represent the relative expression of Nox2 mRNA in lung tissue of CD36+/+ and (H) CD36‐/‐ mice. (E) Bar graphs and dot plots of the total leucocytes recruitment in lung tissue by MPO assay of CD36+/+ and (I) CD36‐/‐ mice. (J) Bar graphs and dot plots represent the mononuclear cells count per mm2 of photomicrographs of lung tissue of CD36+/+ and (K) CD36‐/‐. (L) Representative photomicrographs of lungs after staining with hematoxylin‐eosin (scale bar: 100 μm). Data are mean ± SEM. *p < 0.05 and ***p < 0.001, as assessed by an unpaired t test. n = 6–11 per group for CD36+/+ and n = 6 per group for CD36‐/‐.
Examination of mRNA extracted from the lungs of treated mice demonstrated that EP 80317 reduced Nlrp3, Il1β, Il18 gene expression and IL‐1β protein levels in a CD36‐dependent manner (Figure 2A–D). Moreover, EP 80317 diminished mRNA levels of nuclear factor‐kappa B (Nfκb) family members (Figure 2E–H), primarily the noncanonical RelA and RelB in a CD36‐dependent manner. Concurrent with reduced NF‐κB signaling, EP 80317 lowered gene expression of pro‐inflammatory cytokines and chemokines: e.g., tumor necrosis factor (Tnf) (p < 0.05) (Figure 2I), Il6 (p < 0.05) (Figure 2J), chemokine C‐C motif ligand 2 (Ccl2) (p < 0.05) (Figure 2K), and autotaxin (Atx) (p < 0.05) (Figure 2L). Furthermore, EP 80317 caused a reduction in mRNA expression for the chemokine (C‐X‐C motif) ligand 1 (Cxcl1) (Figure 2M) and for markers of granulocyte presence such as sialic acid binding Ig‐like lectin F (Siglecf) and Cd11c (Figure 2N–O). In contrast, mRNA levels of these biomarkers were unchanged in EP 80317‐treated CD36‐/‐ mice and vehicle treated CD36+/+ mice (Figure 2A–O).
Figure 2.
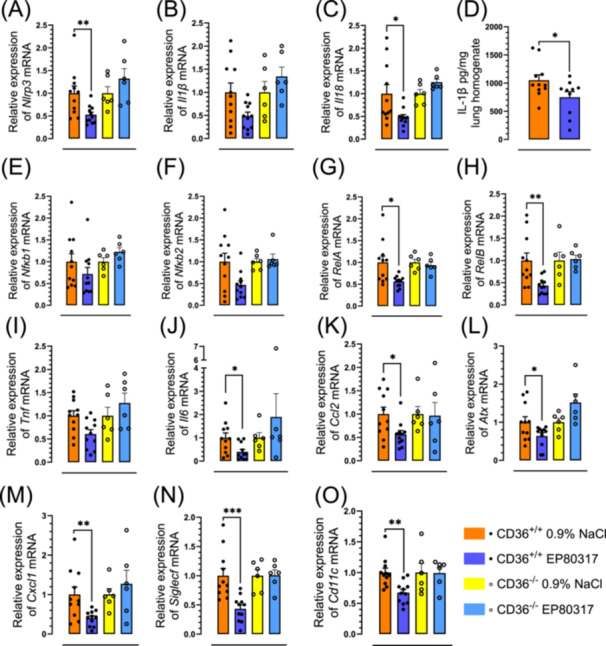
EP 80317 reduces lung NLRP3 inflammasome and pro‐inflammatory cytokines and chemokines. Bar graphs and dot plots represent the relative expression of (A) Nlrp3, (B) Il1β, and (C) Il18 mRNA in lung tissue of CD36+/+ and CD36‐/‐ mice. (D) Mean IL‐1β levels in lung homogenates of CD36+/+ mice, expressed as bar graph and dot plot. Bar graphs and dot plots represent the relative expression of (E) Nfκb1, (F) Nfκb2, (G) RelA, (H) RelB, (I) Tnf, (J) Il6, (K) Ccl2, (L) Atx, (M) Cxcl1, (N) Siglecf, and (O) Cd11c mRNA in lung tissue of CD36+/+ and CD36‐/‐. Data are mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001, as assessed by an unpaired t test. n = 11 for CD36+/+ and n = 6 for CD36‐/‐.
3.2. EP 80317 Decreases Arachidonic Acid Metabolites in Hind Limb Ischemia and Reperfusion
In lung homogenates of EP 80317‐treated CD36+/+ mice subjected to ischemia and reperfusion, the levels of PGE2 were decreased by 53% (p < 0.05) from 6.1 ± 1.0 × 104 to 2.9 ± 0.6 × 104 pg/mL compared to vehicle‐treated mice (Figure 3A). Pretreatment with EP 80317 caused no change on PGE2 levels in CD36‐/‐ mice (Figure 3B). After EP 80317 treatment, prostaglandin E synthase (Ptges) mRNA levels were reduced in CD36+/+ but not in CD36‐/‐ mice compared to vehicle (Figure 3C,D). No significant change was observed in cyclooxygenase 2 (Cox2) mRNA levels between groups (Figure 3E). The expression levels of the prostaglandin E receptors Ptger2 (Figure 3F) and Ptger4 (Figure 3G) but not Ptger3 (Figure 3H) were reduced by treatment of CD36+/+ mice with EP 80317, which had no effect on CD36‐/‐ mice (Figure 3I,J).
Figure 3.
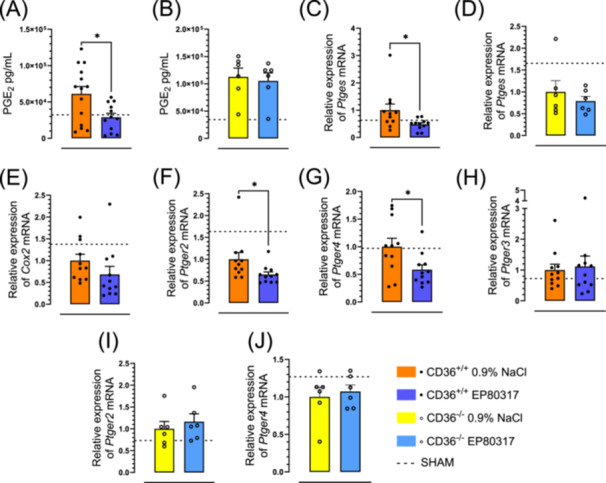
EP 80317 decreases arachidonic acid metabolites‐PGE2. PGE2 levels in lung homogenates of (A) CD36+/+ and (B) CD36‐/‐ mice, represented as bar graphs. Bar graphs and dot plots represent the relative expression of Ptges mRNA in lung tissue of (C) CD36+/+ and (D) CD36‐/‐ mice. (E) Bar graph and dot plot represent the relative expression of Cox2 mRNA in lung tissue of CD36+/+. Relative expression of (F) Ptger2, (G) Ptger4, and (H) Ptger3 mRNA in lung tissue of CD36+/+ mice, expressed as bar graphs and dot plots. Bar graphs and dot plots represent the relative expression of (I) Ptger2 and (J) Ptger4 mRNA in lung tissue of CD36‐/‐ mice. Data are mean ± SEM. *p < 0.05, as assessed by an unpaired t test. n = 14 for CD36+/+ and n = 6 for CD36‐/‐.
LTB4 levels in lung homogenates were increased by 2.7‐fold in CD36+/+ mice subjected to ischemia and reperfusion (Figure 4A) and compared to vehicle‐treated mice, were reduced by 22% (p < 0.05) from 2.7 ± 0.1 × 104 to 2.1 ± 0.3 × 104 pg/mL upon treatment with EP 80317, which did not affect LTB4 levels in CD36‐/‐ mice (Figure 4B). Arachidonate 5‐lipoxygenase (Alox5) mRNA levels were reduced by 1.6‐fold in EP 80317‐treated CD36+/+ mice, back to baseline levels of sham‐operated mice (Figure 4C), but no effect of the ligand was observed in CD36‐/‐ mice (Figure 4D). In contrast, EP 80317 pretreatment had no effect on the mRNA levels of Alox12 (Figure 4E), Alox15 (Figure 4F), arachidonate 5‐lipoxygenase activating protein (Alox5ap) (Figure 4G), nor leukotriene C4 synthase (Ltc4s) (Figure 4H). Finally, leukotriene B4 receptor 1 (Ltb4r1) mRNA expression was reduced 3.3‐fold in CD36+/+ mice pretreated with EP 80317 and subjected to ischemia and reperfusion (Figure 4I), but no effect was observed in the CD36‐/‐ counterpart (Figure 4J).
Figure 4.
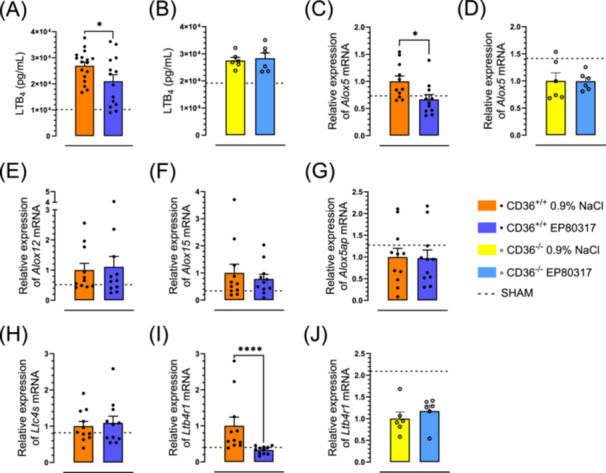
EP 80317 decreases arachidonic acid metabolites‐LTB4. (A) Bar graph and dot plot represent the LTB4 levels in lung homogenates of CD36+/+ and (B) CD36‐/‐ mice. (C) Relative expression of Alox5 mRNA in lung tissue of CD36+/+ and (D) CD36‐/‐ mice. Bar graph and dot plot represent the relative expression of (E) Alox12, (F) Alox15 and (G) Alox5ap mRNA in lung tissue of CD36+/+ mice. (H) Bar graph and dot plot of the relative expression of Ltc4s mRNA in lung tissue of CD36+/+. (I) Bar graph and dot plot represent the relative expression of Ltb4r1 mRNA in lung tissue of CD36+/+ and (J) CD36‐/‐ mice. Data are mean ± SEM. *p < 0.05 and ***p < 0.001, as assessed by an unpaired t test. n = 14 for CD36+/+ and n = 6 for CD36‐/‐.
A mechanism is proposed by which CD36 participates in the generation of ROS and activation of leukocytes in the circulation and microvascular endothelium of lung tissue after reperfusion of an ischemic limb. Treatment with EP 80317 attenuates the cascade of cytokines, chemokines, and arachidonic acid metabolites by mitigating CD36 signaling. Consequently, the CD36 modulator decreased remote leukocyte recruitment, lipid mediators, cytokines, and lung tissue damage.
4. Discussion
Remote injury following the reperfusion of an ischemic limb is known to affect well‐vascularized organs, such as the lungs, liver, and intestines. Consequences include organ injury and acute respiratory distress syndrome (ARDS). The principal finding of this study is that the scavenger receptor CD36 plays a critical role in these events by amplifying expression of key inflammatory mediators and recruiting leukocytes to vascularized tissue.
In skeletal muscle reperfusion, ROS play a well‐documented role in the release of pro‐inflammatory mediators, such as prostaglandins, thromboxane A2, and leukotriene B4, which mutually prime formation and activate leukocytes within the pulmonary circulation [10, 30, 31, 32]. Accordingly, pulmonary Nox2 expression and plasma MDA levels were decreased in EP 80317‐treated mice in a CD36‐dependent manner. Expression of Nox2 and p40phox were previously shown to decrease in atherosclerotic vascular tissue of apolipoprotein E (apoE)‐null mice treated with EP 80317 [33]. Moreover, a decrease in oxidative stress was observed in the left ventricle of mice that were treated with a selective azapeptide CD36 modulator before transient myocardial ischemia [23]. The systemic inflammatory response triggered by the reperfusion of the ischemic limb leads to remote organ injury, with greatest severity to the lungs [14, 34]. Activated vascular leukocytes are suggested to contribute to organ damage by releasing TNF‐α and IL‐1β, which upregulate cell adhesion molecules [7, 8, 35]. Injury of the lungs after limb reperfusion is associated with extensive infiltration of mononuclear cells as well as recruitment of PMNs, which mediate inflammatory responses [36, 37]. In the present study, lung tissue MPO and microscopic analysis of leukocyte counts indicated mononuclear phagocyte accumulation, which was curtailed by the CD36 modulator EP 80317.
Animal studies have highlighted the role of CD36 in cardiovascular diseases, particularly atherosclerosis [38] and atherothrombosis [39]. Previously, CD36‐selective ligands have reduced macrophage accumulation in aortic lesions, diminished foam cell formation, and mitigated atherosclerosis progression in apoE‐null mice [33]. Consistent with its role in chronic inflammatory diseases such as atherosclerosis, CD36 modulation was associated with reduced systemic inflammation and increased differentiation of vascular macrophages towards an M2 anti‐inflammatory phenotype [28, 29, 40]. The findings from a systematic review and meta‐analysis in humans demonstrated a significant association between the CD36 rs1761667 polymorphism and cardiometabolic risk factors, including circulating triglycerides, HDL cholesterol, and fasting blood glucose levels [41]. In a model of subretinal inflammation elicited by photo‐oxidative stress, the modulatory activity of the CD36 ligand azapeptide MPE‐298 reduced mononuclear phagocyte accumulation and production of inflammatory mediators with a change in cellular energy metabolism towards oxidative phosphorylation [15] and reduced mitochondrial stress in retinal pigment epithelium [42]. Azapeptide MPE‐298 has exhibited vascular protective effects and contributed to lesion regression in models of atherosclerosis [40]. In chronic obstructive pulmonary disease (COPD), CD36 has been shown to play a role in the progression of emphysema, particularly in the context of elastase‐induced disease advancement [43]. The role of CD36 expression in microvascular endothelial cells and circulating monocytes has now been studied in an acute inflammation model featuring hind limb ischemia and reperfusion.
The NLRP3 inflammasome is activated by CD36 signaling in response to TLR‐2 activation by lipopeptide and ROS [15]. Reduced levels of IL‐1β and decreased expression of Nlrp3 and Il18 mRNA were consistently found in lung homogenates from animals treated with EP 80317 before reperfusion compared to vehicle‐treated and CD36‐null counterparts. In addition to inhibiting the inflammasome NLRP3, EP 80317 caused a reduction in the expression of cytokines, chemokines, and inflammatory markers: Tnf, Il6, Ccl2, Cxcl1, Atx, Siglecf, and CD11c. The recruitment and localization of leukocytes in the lungs are significantly impacted by CD36 and likely involve both mononuclear phagocytes and polymorphonuclear leukocytes.
In mice subjected to acute inflammation caused by scorpion envenomation, treatment with EP 80317 effectively decreased leukocyte accumulation, prostaglandin E2, and IL‐1β levels remotely in bronchoalveolar fluid [44]. The latter activity of EP 80317 was attributed to effects that decreased NF‐κB phosphorylation and, in part, caused reductions of AMPc and PKA activation elicited by LTB4 and consequently inhibited NF‐κB activation. In mice subjected to myocardial ischemia‐reperfusion, the CD36 ligand azapeptide CP‐3(iv), increased circulating adiponectin levels, epididymal fat adiponectin gene expression, and transcriptional regulators (Pparg, Cebpb, Sirt1) after 6 h of reperfusion. Additionally, azapeptide CP‐3(iv) reduced myocardial oxidative stress and apoptosis [23]. In the context of lung inflammation and disease [24, 25, 43], a CD36 ligand reduced the cytokine storm elicited in experimental SARS‐Cov‐2 infected mice [45]. To our knowledge, no study has reported a link between remotely induced lung injury and CD36 expressed by alveolar or incoming mononuclear cells.
Our findings align with previous observations indicating what caused a decrease in the expression of Nfκb1 and Nfκb2 in lung tissue at the mRNA level. In the context of skeletal muscle limb ischemia and reperfusion, COX2 inhibition has been shown to prevent remote pulmonary dysfunction and increase permeability [10, 46]. In the current study, levels of PGE2 were reduced in lung homogenates from EP 80317‐treated mice, but not in CD36‐null counterparts. This reduction was associated with decreased expression of PGE2 synthase and reduced mRNA levels for the Ptger2 and Ptger4 receptors. In contrast, EP 80317 had no significant effect on Cox2 nor Ptger3 mRNA levels. Among the EP receptors, EP2 and EP4 bind PGE2 with high affinity and promote inflammation, consequently causing cytokine release and immune cell recruitment [47]. A previous study showed that PGE2‐mediated increase in IL‐1β is dependent on EP2 and EP4 signaling using a murine bone marrow transplant model [47, 48].
In systemic inflammation caused by hind limb ischemia and reperfusion, PGE2 has anti‐inflammatory properties and decreases cytokine secretion, but has also been linked with detrimental outcomes in the lungs such as heightened vascular permeability and plasma protein extravasation [49]. The anti‐inflammatory impact of PGE2, which curbs cytokine secretion and lung macrophage invasion, is facilitated through Ptger2 and Ptger4 signaling, underscoring an intricate role of PGE2 in lung inflammation secondary to systemic inflammation, which warrants further investigation. Arachidonic acid metabolism following hind limb ischemia and reperfusion leads to activated endothelial cell and leukocyte production of the powerful chemoattractant LTB4, which is released both systemically and locally in lung tissue [8, 36, 50]. In the context of the hind limb ischemia and reperfusion model, the inflammatory effects of CD36 were alleviated by EP 80317 treatment, which decreased LTB4 and IL‐1β levels as well as reduced Alox5, Ltb4r1, and Nlrp3 inflammasome mRNA expression in lung tissue but caused no alterations in the mRNA levels of Alox12, Alox15, Alox5ap nor Ltc4s. Expression of the Ltb4r1 receptor leads to intracellular signaling cascades through G proteins, affecting various cellular responses such as chemotaxis of immune cells, cytokine production, and oxidative burst [51]. In the present study, the reduction of levels of LTB4 and Ltb4r1 leads to inhibition of the inflammatory response. An inflammatory CD36‐LTB4 pathway is suggested in this model. More translational studies are needed to confirm the role of CD36 in remote organ injury. In a sequence of events leading to remote injury upon reperfusion of the ischemic hind limb, the reintroduction of oxygen may lead to the generation of ROS, triggering an acute inflammatory response in vascular endothelial cells and circulating leukocytes. The latter accumulate in well‐perfused tissues, become trapped, and migrate into the lung, upregulating NF‐κB‐dependent cytokine gene expression and NLRP3 inflammasome activation. Expressed by pulmonary macrophages, CD36 is an innate immune receptor that regulates the expression of pro‐inflammatory genes, such as Ptges and Alox5, and receptors on leukocytes. Treatment with a CD36 modulating ligand reduces lung leukocytosis as well as systemic and lung inflammatory mediator levels. Moreover, CD36‐deficient mice exhibit a similar pattern of events. Future research will explore the initial signaling molecules that regulate the formation of ROS, activate the NLRP3 inflammasome, and trigger NF‐ĸB inflammatory pathways, dissecting the sequential and concomitant pathways occurring in the inflammatory response. Targeting CD36 in hind limb ischemia and reperfusion could serve as a promising upstream pharmacological target.
Author Contributions
Hanan Elimam: conceptualization, methodology, investigation, formal analysis, writing original draft, writing–review and editing. Jade Gauvin: formal analysis, writing original draft, writing–review and editing. David N. Huynh: methodology, investigation, formal analysis, writing–review and editing. Liliane Ménard: methodology, investigation, formal analysis, writing–review and editing. Marie‐Lynn Al‐Hawat: writing original draft, writing–review and editing. Diala Harb: writing–review and editing. William D. Lubell: resources, writing–review and editing. André C. Carpentier: funding acquisition, writing–review and editing. Huy Ong: funding acquisition, writing–review and editing. Sylvie Marleau: conceptualization, funding acquisition, writing original draft, writing–review and editing.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgments
This work was supported by the Canadian Institutes of Health Research (PJT ‐ 178227), the Heart and Stroke Foundation of Canada (G‐18‐0022167), an educational grant from Mperia Therapeutics Inc., Natural Sciences and Engineering Research Council of Canada Discovery Grants (#04079 and #06647), and the Fonds de Recherche du Québec ‐ Nature et Technologies from the Centre in Green Chemistry and Catalysis (FRQNT‐ 2020‐RS4‐265155‐CCVC). HE is a recipient of a grant from the Direction des affaires internationales, Université de Montréal, Canada.
Hanan Elimam and Sylvie Marleau should be considered joint senior authors
Contributor Information
Hanan Elimam, Email: Hanan.Elimam@fop.usc.edu.eg.
Sylvie Marleau, Email: sylvie.marleau@umontreal.ca.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.
References
- 1. Bo H. and Feng X., “Post‐Treatment Curcumin Reduced Ischemia‐Reperfusion‐Induced Pulmonary Injury Via the Notch2/Hes‐1 Pathway,” Journal of International Medical Research 48, no. 4 (2020): 300060519892432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kalogeris T., Baines C. P., Krenz M., and Korthuis R. J., “Cell Biology of Ischemia/Reperfusion Injury,” International Review of Cell and Molecular Biology 298 (2012): 229–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chiu S. and Bharat A., “Role of Monocytes and Macrophages in Regulating Immune Response Following Lung Transplantation,” Current Opinion in Organ Transplantation 21, no. 3 (2016): 239–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hou F., Xiao K., Tang L., and Xie L., “Diversity of Macrophages in Lung Homeostasis and Diseases,” Frontiers in Immunology 12 (2021): 753940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nelson K., Herndon B., and Reisz G., “Pulmonary Effects of Ischemic Limb Reperfusion: Evidence for a Role for Oxygen‐Derived Radicals,” Critical Care Medicine 19, no. 3 (1991): 360–363. [DOI] [PubMed] [Google Scholar]
- 6. Koksel O., Ozdulger A., Aytacoglu B., et al., “The Influence of Iloprost on Acute Lung Injury Induced by Hind Limb Ischemia‐Reperfusion in Rats,” Pulmonary Pharmacology & Therapeutics 18, no. 4 (2005): 235–241. [DOI] [PubMed] [Google Scholar]
- 7. Seekamp A., Warren J. S., Remick D. G., Till G. O., and Ward P. A., “Requirements for Tumor Necrosis Factor‐Alpha and Interleukin‐1 in Limb Ischemia/Reperfusion Injury and Associated Lung Injury,” The American Journal of Pathology 143, no. 2 (1993): 453–463. [PMC free article] [PubMed] [Google Scholar]
- 8. Yassin M. M. I., Harkin D. W., Barros D'Sa A. A. B., Halliday M. I., and Rowlands B. J., “Lower Limb Ischemia‐Reperfusion Injury Triggers a Systemic Inflammatory Response and Multiple Organ Dysfunction,” World Journal of Surgery 26, no. 1 (2002): 115–121. [DOI] [PubMed] [Google Scholar]
- 9. Tu H. and Li Y. L., “Inflammation Balance in Skeletal Muscle Damage and Repair,” Frontiers in Immunology 14 (2023): 1133355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Punch J., Rees R., Cashmer B., Oldham K., Wilkins E., and Smith D. J., “Acute Lung Injury Following Reperfusion After Ischemia in the Hind Limbs of Rats,” The Journal of Trauma: Injury, Infection, and Critical Care 31, no. 6 (1991): 760–767. [DOI] [PubMed] [Google Scholar]
- 11. Kyriakides C., W. G. Austen, Jr. , Wang Y., Favuzza J., F. D. Moore, Jr. , and Hechtman H. B., “Neutrophil Mediated Remote Organ Injury After Lower Torso Ischemia and Reperfusion Is Selectin and Complement Dependent,” The Journal of Trauma: Injury, Infection, and Critical Care 48, no. 1 (2000): 32–38. [DOI] [PubMed] [Google Scholar]
- 12. Bianco‐Batlles M. D., Sosunov A., Polin R. A., and Ten V. S., “Systemic Inflammation Following Hind‐Limb Ischemia‐Reperfusion Affects Brain in Neonatal Mice,” Developmental Neuroscience 30, no. 6 (2008): 367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bitencourt C. S., Bessi V. L., Huynh D. N., et al., “Cooperative Role of Endogenous Leucotrienes and Platelet‐Activating Factor in Ischaemia‐Reperfusion‐Mediated Tissue Injury,” Journal of Cellular and Molecular Medicine 17, no. 12 (2013): 1554–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ibrahim M. A. A., Elwan W. M., and Elgendy H. A., “Role of Scutellarin in Ameliorating Lung Injury in a Rat Model of Bilateral Hind Limb Ischemia‐Reperfusion,” The Anatomical Record 302, no. 11 (2019): 2070–2081. [DOI] [PubMed] [Google Scholar]
- 15. Mellal K., Omri S., Mulumba M., et al., “Immunometabolic Modulation of Retinal Inflammation by CD36 Ligand,” Scientific Reports 9, no. 1 (2019): 12903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Febbraio M., Hajjar D. P., and Silverstein R. L., “CD36: a Class B Scavenger Receptor Involved in Angiogenesis, Atherosclerosis, Inflammation, and Lipid Metabolism,” Journal of Clinical Investigation 108, no. 6 (2001): 785–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Silverstein R. L. and Febbraio M., “CD36, a Scavenger Receptor Involved in Immunity, Metabolism, Angiogenesis, and Behavior,” Science Signaling 2, no. 72 (2009): re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kwak D., Bradley P. B., Subbotina N., et al., “CD36/Lyn Kinase Interactions Within Macrophages Promotes Pulmonary Fibrosis in Response to Oxidized Phospholipid,” Respiratory Research 24, no. 1 (2023): 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sharif O., Matt U., Saluzzo S., et al., “The Scavenger Receptor CD36 Downmodulates the Early Inflammatory Response While Enhancing Bacterial Phagocytosis During Pneumococcal Pneumonia,” The Journal of Immunology 190, no. 11 (2013): 5640–5648. [DOI] [PubMed] [Google Scholar]
- 20. Cho S., Park E. M., Febbraio M., et al., “The Class B Scavenger Receptor CD36 Mediates Free Radical Production and Tissue Injury in Cerebral Ischemia,” The Journal of Neuroscience 25, no. 10 (2005): 2504–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bessi V. L., Labbé S. M., Huynh D. N., et al., “EP 80317, a Selective CD36 Ligand, Shows Cardioprotective Effects Against Post‐Ischaemic Myocardial Damage in Mice,” Cardiovascular Research 96, no. 1 (2012): 99–108. [DOI] [PubMed] [Google Scholar]
- 22. Nagendran J., Pulinilkunnil T., Kienesberger P. C., et al., “Cardiomyocyte‐Specific Ablation of CD36 Improves Post‐Ischemic Functional Recovery,” Journal of Molecular and Cellular Cardiology 63 (2013): 180–188. [DOI] [PubMed] [Google Scholar]
- 23. Huynh D. N., Bessi V. L., Ménard L., et al., “Adiponectin has a Pivotal Role in the Cardioprotective Effect of CP‐3(iv), a Selective CD36 Azapeptide Ligand, After Transient Coronary Artery Occlusion in Mice,” The FASEB Journal 32, no. 2 (2018): 807–818. [DOI] [PubMed] [Google Scholar]
- 24. Suresh K., Servinsky L., Reyes J., et al., “CD36 Mediates H2O2‐Induced Calcium Influx in Lung Microvascular Endothelial Cells,” American Journal of Physiology‐Lung Cellular and Molecular Physiology 312, no. 1 (2017): L143–L153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sun S., Yao Y., Huang C., et al., “CD36 Regulates LPS‐Induced Acute Lung Injury by Promoting Macrophages M1 Polarization,” Cellular Immunology 372 (2022): 104475. [DOI] [PubMed] [Google Scholar]
- 26. Bujold K., Mellal K., Zoccal K. F., et al., “EP 80317, a CD36 Selective Ligand, Promotes Reverse Cholesterol Transport in Apolipoprotein E‐Deficient Mice,” Atherosclerosis 229, no. 2 (2013): 408–414. [DOI] [PubMed] [Google Scholar]
- 27. Bélanger C., Elimam H., Lefebvre J., Borgeat P., and Marleau S., “Involvement of Endogenous Leukotriene B4 and Platelet‐Activating Factor in Polymorphonuclear Leucocyte Recruitment to Dermal Inflammatory Sites in Rats,” Immunology 124, no. 3 (2008): 295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Frégeau G., Sarduy R., Elimam H., et al., “Atheroprotective and Atheroregressive Potential of Azapeptide Derivatives of GHRP‐6 as Selective CD36 Ligands in Apolipoprotein E‐Deficient Mice,” Atherosclerosis 307 (2020): 52–62. [DOI] [PubMed] [Google Scholar]
- 29. Marleau S., Harb D., Bujold K., et al., “EP 80317, a Ligand of the CD36 Scavenger Receptor, Protects Apolipoprotein E‐Deficient Mice From Developing Atherosclerotic Lesions,” The FASEB Journal 19, no. 13 (2005): 1869–1871. [DOI] [PubMed] [Google Scholar]
- 30. Klausner J. M., Anner H., Paterson I. S., et al., “Lower Torso Ischemia‐Induced Lung Injury Is Leukocyte Dependent,” Annals of Surgery 208, no. 6 (1988): 761–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Klausner J. M., Paterson I. S., Goldman G., et al., “Thromboxane A2 Mediates Increased Pulmonary Microvascular Permeability Following Limb Ischemia,” Circulation Research 64, no. 6 (1989): 1178–1189. [DOI] [PubMed] [Google Scholar]
- 32. Klausner J. M., Paterson I. S., Kobzik L., Valeri C. R., Shepro D., and Hechtman H. B., “Leukotrienes but not Complement Mediate Limb Ischemia‐Induced Lung Injury,” Annals of Surgery 209, no. 4 (1989): 462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Harb D., Bujold K., Febbraio M., Sirois M. G., Ong H., and Marleau S., “The Role of the Scavenger Receptor CD36 in Regulating Mononuclear Phagocyte Trafficking to Atherosclerotic Lesions and Vascular Inflammation,” Cardiovascular Research 83, no. 1 (2009): 42–51. [DOI] [PubMed] [Google Scholar]
- 34. Hsu K. Y., Tsai P. S., Lee J. J., Wang T. Y., and Huang C. J., “Platonin Mitigates Acute Lung Injury Induced by Bilateral Lower Limb Ischemia‐Reperfusion in Rats,” Journal of Surgical Research 167, no. 2 (2011): e255–e262. [DOI] [PubMed] [Google Scholar]
- 35. Atahan E., Ergun Y., Belge Kurutas E., Cetinus E., and Guney Ergun U., “Ischemia‐Reperfusion Injury in Rat Skeletal Muscle Is Attenuated by Zinc Aspartate,” Journal of Surgical Research 137, no. 1 (2007): 109–116. [DOI] [PubMed] [Google Scholar]
- 36. Bengisun U., Köksoy C., Bengisun J. S., Bayraktaroğlu G., Çamur A., and Aras N., “Ischemia and Reperfusion Injury: Prevention of Pulmonary Hypertension and Leukosequestration Following Lower Limb Ischemia,” Prostaglandins, Leukotrienes and Essential Fatty Acids 56, no. 2 (1997): 117–120. [DOI] [PubMed] [Google Scholar]
- 37. Kassab A. A., Aboregela A. M., and Shalaby A. M., “Edaravone Attenuates Lung Injury in a Hind Limb Ischemia‐Reperfusion Rat Model: A Histological, Immunohistochemical and Biochemical Study,” Annals of Anatomy ‐ Anatomischer Anzeiger 228 (2020): 151433. [DOI] [PubMed] [Google Scholar]
- 38. Febbraio M., Podrez E. A., Smith J. D., et al., “Targeted Disruption of the Class B Scavenger Receptor CD36 Protects Against Atherosclerotic Lesion Development in Mice,” Journal of Clinical Investigation 105, no. 8 (2000): 1049–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang M., Cooley B. C., Li W., et al., “Platelet CD36 Promotes Thrombosis by Activating Redox Sensor ERK5 in Hyperlipidemic Conditions,” Blood 129, no. 21 (2017): 2917–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gauvin J., Frégeau G., Elimam H., et al., “A Cyclic Azapeptide Ligand of the Scavenger Receptor CD36/SR‐B2 Reduces the Atherosclerotic Lesion Progression and Enhances Plaque Stability in Apolipoprotein E‐Deficient Mice,” Frontiers in Pharmacology 14 (2023): 1204905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yazdanpanah Z., Mozaffari‐Khosravi H., Mirzaei M., Sheikhha M. H., and Salehi‐Abargouei A., “A Systematic Review and Meta‐Analysis on the Association Between CD36 rs1761667 Polymorphism and Cardiometabolic Risk Factors in Adults,” Scientific Reports 12, no. 1 (2022): 5916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dorion M.‐F., Mulumba M., Kasai S., Itoh K., Lubell W. D., and Ong H., “The CD36 Ligand‐Promoted Autophagy Protects Retinal Pigment Epithelial Cells From Oxidative Stress,” Oxidative Medicine and Cellular Longevity 2021 (2021): 6691402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang J., Wang R., Li Y., et al., “Lipolysis Engages CD36 to Promote ZBP1‐mediated Necroptosis‐Impairing Lung Regeneration in COPD,” Cell Reports Medicine 5, no. 9 (2024): 101732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zoccal K. F., Gardinassi L. G., Bordon K. C. F., et al., “EP80317 Restrains Inflammation and Mortality Caused by Scorpion Envenomation in Mice,” Frontiers in Pharmacology 10 (2019): 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gauvin J., Huynh D. N., Dubuc I., et al., “Pharmacological Targeting of the Hyper‐Inflammatory Response to SARS‐CoV‐2‐infected K18‐hACE2 Mice Using a Cluster of Differentiation 36 Receptor Modulator,” Frontiers in Pharmacology 15 (2024): 1303342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ye Y., Shan Y., Bao C., Hu Y., and Wang L., “Ginsenoside Rg1 Protects against Hind‐Limb Ischemia Reperfusion Induced Lung Injury Via NF‐κB/COX‐2 Signaling Pathway,” International Immunopharmacology 60 (2018): 96–103. [DOI] [PubMed] [Google Scholar]
- 47. Sugimoto Y. and Narumiya S., “Prostaglandin E Receptors,” Journal of Biological Chemistry 282, no. 16 (2007): 11613–11617. [DOI] [PubMed] [Google Scholar]
- 48. Martinez‐Colon G. J., Taylor Q. M., Wilke C. A., Podsiad A. B., and Moore B. B., “Elevated Prostaglandin E(2) Post‐Bone Marrow Transplant Mediates Interleukin‐1Beta‐Related Lung Injury,” Mucosal Immunology 11, no. 2 (2018): 319–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cebulla D., van Geffen C., and Kolahian S., “The Role of PGE2 and EP Receptors on Lung's Immune and Structural Cells; Possibilities for Future Asthma Therapy,” Pharmacology & Therapeutics 241 (2023): 108313. [DOI] [PubMed] [Google Scholar]
- 50. Goldman G., Welbourn R., Klausner J. M., Valeri C. R., Shepro D., and Hechtman H. B., “Oxygen Free Radicals Are Required for Ischemia‐Induced Leukotriene B4 Synthesis and Diapedesis,” Surgery 111, no. 3 (1992): 287–293. [PubMed] [Google Scholar]
- 51. Li P., Oh D. Y., Bandyopadhyay G., et al., “LTB4 Promotes Insulin Resistance in Obese Mice by Acting on Macrophages, Hepatocytes and Myocytes,” Nature Medicine 21, no. 3 (2015): 239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.