

RESUMEN
Fundamentos:
El Síndrome de Phelan-McDermid es una enfermedad poco frecuente de origen genético causada por la deleción del extremo terminal del cromosoma 22 región q13.3 o por mutaciones puntuales que afectan al gen SHANK3. Los objetivos de este trabajo fueron determinar la prevalencia de la enfermedad en la población española, establecer la distribución geográfica del síndrome entre las distintas comunidades autónomas, dilucidar el rango de edad en el que existen más pacientes y estudiar la relación enfermedad-sexo así como la edad media al diagnóstico.
Métodos:
Para la investigación se reclutaron pacientes diagnósticados con la enfermedad durante doce años en todo el territorio español. La información clínica de los pacientes se obtuvo de los médicos de referencia mediante dos cuestionarios estandarizados completados con datos de los informes médicos y la entrevista a los padres. El diagnóstico molecular de la enfermedad se realizó utilizando diferentes formatos de microarrays. Los datos se trataron utilizando Microsoft Excel y Statgraphics Centurion XVII.
Resultados:
Actualmente en España existen 201 personas diagnosticadas con la enfermedad siendo su prevalencia de 4x10-4/10.000 habitantes. La comunidad con más pacientes diagnosticados fue Madrid y no hubo diferencias significativas en cuanto al sexo y la enfermedad, la edad media al diagnóstico se sitúa en torno a los 6,67 años.
Conclusiones:
La prevalencia de la enfermedad en España es muy baja pudiéndose afirmar que es muy probable que en la población existan más personas con este síndrome.
Palabras clave: Síndrome Phelan-McDermid, Prevalencia, Sexo, Edad, Diagnóstico
ABSTRACT
Background:
Phelan-McDermid syndrome is a rare genetic condition caused by a deletion of the terminal end of chromosome 22 in the 13.3 region, as well as, by point mutations within SHANK3 gene. The aims of this research were to determine the prevalence of the disease in the Spanish population, to establish the geographical distribution of the syndrome among the different autonomous communities, to elucidate the age range that affects more patients, to study the disease-sex relationship, as well as the age at diagnosis.
Methods:
For the research, patients diagnosed with the disease for twelve years were recruited throughout the Spanish territory. The clinical patient information was obtained from the referral doctors using two standardized questionnaires completed with data from the medical reports and the interview with the parents. The molecular diagnosis of the disease was carried out using different formats of microarrays. Data were processed using Microsoft Excel and Statgraphics Centurion XVII.
Results:
Currently in Spain there are 201 people diagnosed with the disease. Currently in Spain there are 201 people diagnosed with the disease, its prevalence being 4x10-4/10,000 inhabitants. The community with the most diagnosed patients was Madrid and there were no significant differences in terms of sex and disease, the mean age at diagnosis was around 6.67 years.
Conclusions:
The prevalence of the disease in Spain is very low, and it can be stated that it is very likely that there are more people with this syndrome in the population.
Key words: Phelan-McDermid Syndrome; Preva-lence; Sex,Age; Diagnosis
INTRODUCCIÓN
El Síndrome de Phelan-McDermid (SPMD) es una condición genética causada por la deleción del extremo terminal del cromosoma 22 región q13 o por mutación puntual en el gen SHANK3 1 .
Por lo general, el extremo terminal que deleciona incluye desde 22q13.2 hasta 22qter 2 . En la mayoría de los casos, la alteración genética es el resultado de una mutación espontánea o de novo, pero existe también algunas formas hereditarias debida a translocaciones cromosómicas familiares y reordenamientos en forma de anillo involucrando el cromosoma 22 (el cromosoma paterno en el 75% de los casos). El tamaño de la deleción puede variar considerablemente entre 100 Kb-9,2 Mb, pero el principal responsable de la enfermedad es el gen SHANK3 3 , 4 .
La enfermedad se caracteriza por una variedad de síntomas clínicos con diferentes grados de severidad e incidencia y que afectan distintos sistemas orgánicos, siendo el más afectado el sistema nervioso. Algunas de las alteraciones más frecuentes son retraso del desarrollo y del lenguaje, hipotonía, trastorno del espectro autista, dismorfías faciales y corporales, afecciones en el sistema gastrointestinal, así como perturbaciones en el comportamiento que incluyen reacciones negativas a los cambios de rutina y variaciones en el ciclo sueño/vigilia 5 , 6 , 7 , 8 .
El SPMD es un síndrome infradiagnosticado debido a:
i) La dificultad de su diagnóstico, que implica el uso de diferentes técnicas genómicas y citogenéticas.
ii) Los datos clínicos no son específicos del síndrome, no presentando rasgos clínicos patognomónicos.
iii) La necesidad en la mejora de la práctica clínica, como consecuencia de ser una enfermedad poco frecuente y no existir guías clínicas que faciliten su manejo 9 .
Actualmente a nivel mundial existen diagnosticadas unas 2.000 personas. Los objetivos que se plantearon en esta investigación fueron:
i) Determinar la prevalencia de la enfermedad en la población española, y eventualmente su incidencia anual.
ii) Establecer la distribución geográfica del síndrome entre las distintas comunidades autónomas.
iii) Dilucidar el rango de edad en el que existen más pacientes y estudiar la relación de ésta en función del sexo.
iv) Establecer la edad media al diagnóstico.
MATERIALES Y MÉTODOS
Población. La población Phelan McDermid española estudiada, fue reclutada durante los últimos doce años por el INGEMM (Instituto de Genética Médica y Molecular) junto con la Asociación Española del síndrome Phelan-McDermid. Para la recogida y tratamiento de los datos, previamente se firmó sendos convenios de colaboración entre la Asociación Española del síndrome Phelan-McDermid y la Universidad Católica de Valencia San Vicente Mártir y la Asociación y INGEMM-FIBHULP.
Los familiares de los pacientes diagnosticados fueron informados del estudio y firmaron un documento de consentimiento informado. La investigación fue desarrollada de acuerdo con la Declaración de Helsinki y fue aprobada por el Comité de Ética del Hospital Universitario la Paz (PI: 2735 HULP, Madrid. España).
La información clínica de los pacientes se obtuvo de los médicos de referencia mediante dos cuestionarios estandarizados completados con datos de los informes médicos y la entrevista a los padres.
Datos genéticos. El diagnóstico molecular de la enfermedad se realizó utilizando diferentes formatos de microarrays:
- Personalizados (KaryoArray® 60K-arrayCGH para rutina clínica; INGEMM; un 60K aCGH específico para la región 22q13.3; INGEMM, no publicado; KaryoNIM 180K autismo; NIM Genetics; KaryoNIM 60K autismo; NIM Genetics; Q-chip®Post 60k, Q-Genomics).
- Comerciales (ISCA 60K, Agilent Tech.; Agilent 44K; Agilent 180K; Illumina Infinium CytoSNP-850k BeadChip; Affymetrix. Ther-moFisher Scientific 750K-chip).
Análisis de los datos. Los datos se analizaron utilizando los software Microsoft Excel y Statgraphics Centurion XVII (Stat Point, Inc., Herndon, 187 Virginia, USA).
Las variables estudiadas fueron número de pacientes, edad, sexo, comunidad autónoma y edad al diagnóstico. El análisis de los datos se realizó a principios del 2020.
Para analizar el número de pacientes, se registró la frecuencia absoluta anual (2007-2019) y se comparó el número de pacientes nuevos registrados durante un periodo de doce meses.
Para la determinación de la distribución geográfica de la enfermedad se analizaron las frecuencias absolutas en cada una de las comunidades autónomas de los pacientes reclutados al final del 2019. Para establecer el porcentaje en cuanto a prevalencia se refiere, se contrastaron los datos obtenidos con los habitantes de cada área según las cifras publicadas por el Instituto Nacional de Estadística (INE).
Los datos de sexo, edad y edad al diagnóstico de cada uno de los pacientes se recopilaron a través de una encuesta cumplimentada por sus familiares. Para el estudio de la relación enfermedad-sexo se codificó la variable sexo (1: mujer / 2: hombre) y para la edad se agruparon los años en intervalos de 5 años (hasta los 25 años).
RESULTADOS
Prevalencia de la enfermedad en España. Actualmente en España existen 201 personas diagnosticadas con el síndrome de Phelan-McDermid. Teniendo en cuenta que la población española es de 46.818.763 (2019) habitantes, la prevalencia de la enfermedad actualmente en el país es de un 4x10-4/10.000 habitantes.
La incidencia anual del SPMD, se estableció en 3-4 casos/año durante los primeros años, incrementándose a 10-15 casos/año en periodos intermedios del reclutamiento y a 25-35 casos/año en los últimos años. La mejora del reclutamiento de los pacientes fue debida a los convenios de colaboración establecidos, los avances tecnológicos y la mejora diagnostica desde el punto de vista genético (figura 1).
Figura 1. Evolución de pacientes reclutados con SMPD.

Distribución geográfica. La figura 2 muestra cómo se distribuyen los pacientes en el territorio español.
Figura 2. Distribución geográfica de los pacientes con SPMD diagnosticados en España.

Madrid en porcentaje es la comunidad autónoma que más enfermos presenta (29.85%), seguido por Cataluña (15,42%), Andalucía (14,42%), Comunidad Valenciana (8,95%), Murcia (7,46%), Baleares (6,50%), Castilla la Mancha (3,98%), País Vasco, Galicia (2,48%), Canarias (1,98%), Asturias (1,49%), Castilla León, Extremadura (1,49%), Aragón y Navarra (0,99%). En Cantabria, La Rioja, Ceuta y Melilla, actualmente no existen personas diagnosticadas con el síndrome (figura 3).
Figura 3. Distribución geográfica en porcentaje de los casos diagnosticados con la enfermedad en España.

Si se considera la población de cada una de las comunidades autónomas, la prevalencia de la enfermedad en cada una de ellas, los datos son muy bajos (tabla 1).
Tabla 1. Prevalencia de SPMD en las distintas comunidades autónomas en relación con la población establecida por el INE en 2019.

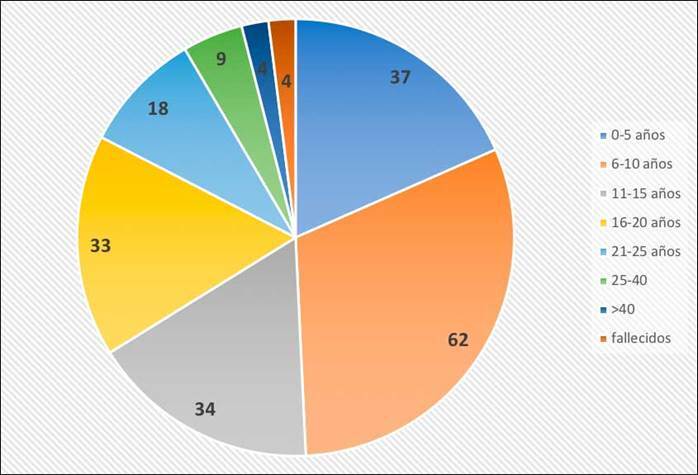
Relación enfermedad-edad. Actualmente en España, el rango de edad de pacientes diagnosticados con SPMP varía desde el nacimiento hasta los 54 años con una media de 13,06 y una mediana de 10,80 años, respectivamente, siendo entre los 6 y 10 años dónde más pacientes existen diagnosticados. No obstante, en este estudio el número de pacientes en edad juvenil-adolescente/mayoría de edad, se situó en un número similar al grupo etario anterior. Adicionalmente, adultos (por encima de 20 años) se han diagnosticado en menor número (figura 4).
Figura 4. Distribución en función de la edad de pacientes españoles con el síndrome de Phelan-McDermid.
Prevalencia por sexo. Analizando la prevalencia por sexo en España, se registra que existe un número similar de mujeres y hombres afectados por este síndrome (102 hombres por 99 mujeres), no habiendo diferencias estadísticamente significativas entre ambos sexos.
Edad al diagnóstico. Uno de los principales problemas en las enfermedades poco frecuentes es el tiempo que se establece de media para sus diagnósticos. Este hecho es uno de los objetivos principales de Instituciones de Investigación en EERR, como el IRDIC o el CIBERER.
El tiempo medio de diagnósticos en España se sitúa en 80 meses de media (6,67 años) y una mediana de 48 meses (4 años).
DISCUSIONES
Este es el primer estudio que describe la prevalencia del síndrome Phelan-McDermid en España siendo ésta muy baja (4x10-4/10.000). Este dato se sitúa lejos de entre 1/10.000 y 1/20.000 habitantes previamente sugerido a nivel global. En este sentido, se puede afirmar que esta enfermedad está infradiagnosticada en España, por lo que es muy posible que en la población existan más personas con este síndrome.
Si se consultan diferentes organizaciones, a nivel mundial existen unos 2.000 casos diagnosticados (el 10,26% son españoles).
La Comunidad que mayor número de pacientes registra es Madrid. La enfermedad se da prácticamente igual en hombres que en mujeres. Estos resultados coinciden con otras investigaciones realizadas previamente en las que registraron que la prevalencia de la enfermedad en ambos sexos es la misma 8 , 9 .
El mayor número de pacientes diagnosticados son niños entre 6 y 10 años siendo los datos obtenidos similares a los que existen a nivel mundial, en los que establecen que la mayoría de pacientes se encuentran en edad pediátrica (0-15 años).
Actualmente se conoce poco sobre la esperanza de vida de enfermos con SPMD y existen pocos estudios sobre personas adultas. Sí que existe un caso reportado de una persona de 70 años 10 .
La edad al diagnóstico de la enfermedad está altamente condicionada por la complejidad de las tecnologías utilizadas para el diagnóstico genético. De hecho, los microarrays sólo están disponibles en la rutina asistencial desde 2008 (en España desde 2010, en adelante) y la NGS (Secuenciación de Segunda Generación) desde 2016. Pero los datos reportados por la AEGH (Asociación Española de Genética Humana), establecen que en 2017 sólo el 16% de los hospitales públicos en España hacía el uso diagnóstico establecido de los microarrays.
Cita sugerida: Gómez Taylor B, Moreno Sancho ML, Drehmer Rieger E, Carrera Julia S, Nevado J, Sempere Ferre F. Prevalencia del síndrome de Phelan-McDermid en España. Rev Esp Salud Pública. 2020; 94: 21 de diciembre e202012121.
BIBLIOGRAFÍA
- 1.. Kolevzon A, Angarita B, Bush L, Wang AT, Frank Y, Yang A, et al. Phelan-McDermid syndrome: a review of the literature and practice parameters for medical assessment and monitoring. J. Neurodev. Disord. 2014; 6: 39. [DOI] [PMC free article] [PubMed]; Kolevzon A, Angarita B, Bush L, Wang AT, Frank Y, Yang A. Phelan-McDermid syndrome a review of the literature and practice parameters for medical assessment and monitoring. . J. Neurodev Disord. 2014;6:39–39. doi: 10.1186/1866-1955-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.. Costales JL, Kolevzon A. Phelan-McDermid Syndrome and SHANK3: Implications for treatment. Neurotherapeutics. 2015; 12: 620-630. [DOI] [PMC free article] [PubMed]; Costales JL, Kolevzon A. Phelan-McDermid Syndrome and SHANK3 Implications for treatment. Neurotherapeutics. 2015;12:620–630. doi: 10.1007/s13311-015-0352-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.. Anderlid BM, Schoumans J, Anneren G, et al. FISH-mapping of a 100-kb terminal 22q13 deletion. Hum Genet 2002; 110: 439-443. [DOI] [PubMed]; Anderlid BM, Schoumans J, Anneren G. FISH-mapping of a 100-kb terminal 22q13 deletion. Hum Genet. 2002;110:439–443. doi: 10.1007/s00439-002-0713-7. [DOI] [PubMed] [Google Scholar]
- 4.. Tabet A-C, Rolland T, Ducloy M, Lévy J, Buratti J, Mathieu A, et al. A framework to identify contributing genes in patients with Phelan-McDermid syndrome. NPJ Genom. Med. 2017;2(1). [DOI] [PMC free article] [PubMed]; Tabet A-C, Rolland T, Ducloy M, Lévy J, Buratti J, Mathieu A, et al. A framework to identify contributing genes in patients with Phelan-McDermid syndrome. NPJ Genom. Med. 2017;2(1) doi: 10.1038/s41525-017-0035-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.. Bonaglia MC, Giorda R, Borgatti R, Felisari G, Gagliardi C, Selicorni A, et al. Disruption of the ProSAP2 gene in at (12;22) (q24.1;q13.3) is associated with the 22q13.3 deletion syndrome. Am. J. Hum. Genet. 2011; 69: 261-268. [DOI] [PMC free article] [PubMed]; Bonaglia MC, Giorda R, Borgatti R, Felisari G, Gagliardi C, Selicorni A. Disruption of the ProSAP2 gene in at (12;22) (q24 1;q13.3) is associated with the 22q13.3 deletion syndrome . . Am. J. Hum Genet. 2011;69:261–268. doi: 10.1086/321293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.. Luciani JJ, de Mas P, Depetris D, Mignon-Ravix C, Bottani A, Prieur M, et al. Telomeric 22q13 deletions resulting from rings, simple deletions, and translocations: cytogenetic, molecular, and clinical analyses of 32 new observations. J. Med. Genet. 2003; 40: 690-696. [DOI] [PMC free article] [PubMed]; Luciani JJ, de Mas P, Depetris D, Mignon-Ravix C, Bottani A, Prieur M. Telomeric 22q13 deletions resulting from rings, simple deletions, and translocations cytogenetic, molecular, and clinical analyses of 32 new observations. . J. Med Genet. 2003;40:690–696. doi: 10.1136/jmg.40.9.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.. Wilson HL, Wong ACC, Shaw SR, Tse WY, Stapleton GA, et al. Molecular characterization of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/ PROSAP2 in the major neurological symptoms. J. Med. Genet. 2003; 40:575-584. [DOI] [PMC free article] [PubMed]; Wilson HL, Wong ACC, Shaw SR, Tse WY, Stapleton GA. Molecular characterization of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/ PROSAP2 in the major neurological symptoms . J. Med Genet. 2003;40:575–584. doi: 10.1136/jmg.40.8.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.. Phelan MC. Deletion 22q13.3 syndrome. Orphanet J. Rare Dis. 2008; 3: 14. [DOI] [PMC free article] [PubMed]; Phelan MC. Deletion 22q13 3 syndrome. . Orphanet J Rare Dis. 2008;3:14–14. doi: 10.1186/1750-1172-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.. Hernández-Gómez M, Meléndez-Hernández R, Ramírez-Arroyo E, Mayén-Molina DG. Síndrome de Phelan-McDermid: reporte de un caso y revisión de la literatura. Acta Pediatr. Mex. 2018; 39(1):42-51.ICAL; Hernández-Gómez M, Meléndez-Hernández R, Ramírez-Arroyo E, Mayén-Molina DG. Síndrome de Phelan-McDermid: reporte de un caso y revisión de la literatura. Acta Pediatr. Mex. . 2018;39(1):42–51. ICAL. [Google Scholar]
- 10.. Verhoeven WMA, Egger JI, Cohen-Snuijf R, Kant SG, de Leeuw N. (2013). Phelan-McDermid syndrome: clinical report of a 70-year-old woman. Am. J. Med. Genet. 2013; Part A 161A: 158-161. [DOI] [PubMed]; Verhoeven WMA, Egger JI, Cohen-Snuijf R, Kant SG, de Leeuw N. Phelan-McDermid syndrome: clinical report of a 70-year-old woman. Am. J. Med. Genet. . 2013;2013(Part A 161A):158–161. doi: 10.1002/ajmg.a.35597. [DOI] [PubMed] [Google Scholar]