

Abstract
ERBIN and phosphoglucomutase 3 (PGM3) mutations both lead to rare primary atopic disorders characterized by allergic disease and connective tissue abnormalities, though each disorder has its own rather unique pattern of multisystem presentations. Pathway studies show how ERBIN mutations allow for enhanced TGFb signaling, and prevent STAT3 from negative-regulating TGFb signaling. This likely explains many elements of clinical overlap between disorders of STAT3 and TGFb signaling. The excessive TGFb signaling leading to increased IL-4 receptor expression also provides the rationale for precision-based therapy blocking the IL-4 receptor to treat the atopic disease. The mechanism by which PGM3 deficiency leads to atopic phenotypes is not well understood, nor is the broad variability in disease penetrance and expressivity, though preliminary studies suggest an overlap with IL-6 receptor signaling defects.
Introduction
Primary atopic disorders — monogenic causes of symptoms associated with the effector mechanisms of type-II immunity and allergy [1,2] — provide examples of how complex pathways normally prevent the development of allergic disease, and how disruption can cause it. Two examples, autosomal-dominant deficiency of ERBIN (Erbb2 interacting protein) and autosomal-recessive phosphoglucomutase 3 (PGM3) deficiency, lead to complex phenotypes, reflecting the known and unknown complex roles these genes, and the pathways they impact, serve. They also serve as excellent examples of the value in studying primary atopic disorders. By drawing our attention to the clinical overlap with other primary atopic disorders, detailed study of the overlap in cellular and biochemical immunopathogenic pathways helps clarify how each contributes to atopic disease and other clinical phenotypes.
ERBIN mutation
Normal Transforming Growth Factor Beta (TGF-β) signaling appears critical to control human allergic diseases such as eosinophilic esophagitis and asthma, as well as mouse models of atopy [3–7]. Increased TGFβ signaling is seen in certain atopic patients with connective tissue abnormalities [8,9]. Biallelic loss-of-function (LOF) mutations in TGFB1 lead to loss of immune tolerance in the form of inflammatory bowel disease — in addition to immune deficiency and CNS abnormalities [10], while gain-of-function (GOF) mutations lead to developmental defects and connective tissue abnormalities [11]. Neither is associated with a type 2 T helper (Th2) diathesis or atopic disease among the small number of affected patients. In contrast, substantial allergic disease has been observed in Loeys–Dietz syndrome (LDS) [12,13] — which is associated with multiple connective tissue phenotypes observed in patients with loss of Signal transducer and activator of transcription 3 (STAT3) function [14–24]. LDS is caused by TGF beta receptor 1 (TGFBR1) and TGFBR2 missense mutations, and while enhanced Small Mothers Against Decapentaplegic (SMAD) 2/3 phosphorylation can be seen in patient tissue, models of acute proximal mutant receptor-mediated signaling lead to loss of function [25]. In a mouse model of LDS, loss of TGFβ signaling in esophageal epithelial cells leads to a tissue-intrinsic predisposition to developing eosinophilic esophagitis [26], while TGFβ signaling LDS lymphocytes has not yet been carefully measured, LDS patient-derived naive T cells skew toward a Th2 bias in the presence of TGFβ in a cell-intrinsic fashion [12]. A well-characterized polymorphism that increases TGFb1 transcription leads to a direct effect on epithelial cells, which increases mucosal permeability that is associated with allergic inflammation [7]. The conflicting observations regarding mutant TGFBR1 signaling in LDS mirror conflicting observations regarding the link between TGFβ and allergy risk derived from tissue studies and patients with monogenic disease.
Of note, patients with loss of function of the IL-11 receptor and gp130 (a co-receptor for IL-11R, among others) — both of which signal through STAT3 — develop skeletal and dental abnormalities that overlap significantly with that seen in patients with dominant negative STAT3 mutations (STAT3DN) mutations. Patients with certain recessive or dominant IL6ST loss-of-function mutations have significant allergic inflammation and elevated immunoglobulin E (IgE) as well [27–30]. The IL-11R/gp130/STAT3 signaling pathway — and how it might interface with TGFβ signaling — is therefore of great interest to study in understanding the overlap between congenital connective tissue abnormalities and allergic disease.
The commonality between patients with TGFB mutations and STAT3 loss of function may in part be explained by insight found in a family with a unique missense mutation in erbb2- interacting protein, encoding ERBIN. The patients presented with connective tissue abnormalities, including joint hypermobility and aneurysm formation, bacterial infections, significant eosinophilic gastrointestinal disease (EGID), IgE elevation, and allergen-specific reactivity [31]. Common genomic variation in ERBIN is also associated with scoliosis [32], a major connective tissue abnormality seen in LDS and STAT3DN patients.
ERBIN appears to be a key mediator of the cross-talk between STAT3 and TGFβ signaling, in that it is induced by STAT3 and complexes with it to impair SMAD2/3 nuclear localization and propagation of the TGFBR signal. While this pathway may have important consequences within connective tissue, which could explain the overlap of connective tissue symptoms in STAT3DN, TGFBR mutations, and ERBIN LOF mutation, another key outcome of the excess signaling appears to be increased interleukin-4 (IL-4) receptor alpha (IL4Rα) expression [33]. The excessive IL4Rα activation can promote allergic inflammation, switching to IgE and pruritus [34,35], and may explain the allergic inflammation and IgE elevation seen in TGFB/STAT3DN/ERBIN patients [36–38]. Notably — treatment of STAT3DN and ERBIN LOF patients with IL4Rα blockade (dupilumab) led to marked success in treating the otherwise refractory skin and gut inflammation seen in these patients [39–45].
ERBIN mutations may also contribute to the atopic diathesis in an epithelium-specific fashion. Patients with loss of function in the epithelial barrier protein desmoglein (DSG1) develop severe atopic dermatitis, allergic inflammation and hypersensitivity and metabolic wasting (SAM) syndrome, another primary atopic disorder characterized by severe atopic dermatitis, allergic inflammation and hypersensitivity, and metabolic wasting. Mechanistic study suggests that DSG1 appears to drive normal ERBIN localization in epithelial cells, and as such, ERBIN dysfunction may contribute to the atopic phenotypes in SAM syndrome and related entities [46].
Phosphoglucomutase 3 deficiency
Autosomal-recessive hypomorphic mutations in PGM3 can lead to a glycosylation disorder with a variety of clinical outcomes with variable penetrance and expressivity. Immune dysregulation, connective tissue abnormalities, and neurodevelopmental deficits have been described. In those with sufficient effector immune function (unlike those PGM3-deficient patients with severe combined immunodeficiency), the immune dys-regulation includes substantial allergic disease ranging from severe atopic dermatitis, to food allergy, immediate and delayed hypersensitivity to medications, EGID, asthma, seasonal allergy, allergic bronchopulmonary aspergillosis, allergic fungal mastoiditis, and non-IgE-mediated, specific food-induced enteropathy [47–51]. PGM3 is required for the pathway that produces uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) [50,52], which is essential for both N- and O-linked glycosylation, and complete absence of PGM3 is embryonically lethal. Hypomorphic PGM3 function leads to reduced cytosolic UDP-GlcNAc that then variably impacts critical proteins throughout the body. Naive T cells appear more sensitive to reduced UDP-GlcNAc compared with memory T cells, presumably due to the lack of compensatory metabolic states [50,52].
Notably, patients can present with immune deficiency or atopic disease alone, without infection or developmental abnormalities, and hematopoietic stem cell transplant can fully restore normal immune function [53,54]. While these rather critical observations highlight the tissue-specific variability in penetrance and expressivity of disease, they also suggest that PGM3 variation could contribute to common allergic disease. A small screen of l-PHA (phytohemagglutinin) binding in naive T cells derived from nonsyndromic patients with atopic dermatitis showed no difference from healthy controls, nor was there overlap with the lower range seen in PGM3 deficiency [48]. However, further studies of larger populations could help determine if lower l-PHA binding in naive cells correlates with atopic disease risk.
The relevance of the PGM3 pathway in atopy and immune disorders may be all the more relevant given that exogenous GlcNAc treatment of PGM3mut cells can improve the lower cytosolic UDP-GlcNAc levels observed in PGM3 mutant cells — suggesting a potential precision therapeutic for PGM3mut patients, and in theory, those with impairments of the pathway, which lead to clinically relevant disease of any sort [50].
How any of the focal glycosylation defects in different immune cells lead to allergic disease is not understood. Measured glycosylation moieties on patient-derived IgE are normal [55], arguing against major B-cell-intrinsic glycosylation roles. One theory put forth suggests that adequate N-glycosylation of gp130 is required for its surface expression [56]. Reduction in N-glycosylation in relevant cells could therefore impair gp130-dependent cytokine signaling, leading to the overlap seen between PGM3 mutation and gp130/IL6ST LOF, including bacterial infection, connective tissue abnormalities, high IgE and allergic hypersensitivity, and inflammation, keratitis, neurodevelopmental delay, and other phenotypes [27–30]. While complicated by the fact that recently activated and/or effector T cells — that are enriched in PGM3-deficient patients — have lower gp130 surface expression in general, it nonetheless highlights the need to identify the focal pathways impacted, as they will likely unlock fundamental understanding of allergic disease pathogenesis.
Conclusion
ERBIN mutation and PGM3 deficiency are examples of very rare diseases that are nonetheless highly instructive to the biology behind multisystem syndromes impacting the immune system and allergy. Both point to potential precision diagnostics and therapies for the specific disorders, as well as the potential for relevance of those with common immunologic disease whose pathophysiology might overlap with these rare disorders. In addition, preliminary studies are suggestive of interactions with the IL-6/STAT3 signaling pathway in both disorders (Figure 1), potentially explaining the overlap of atopic and connective tissue abnormalities seen in patients with pathogenic mutations in ERBIN, PGM3, and the IL-6/STAT3 pathway. Further study of these patients, and expansion of the cohorts of those with pathogenic mutants in either gene, will help better define the mechanisms and impact of these mutations in allergic and immune disorders.
Figure 1.
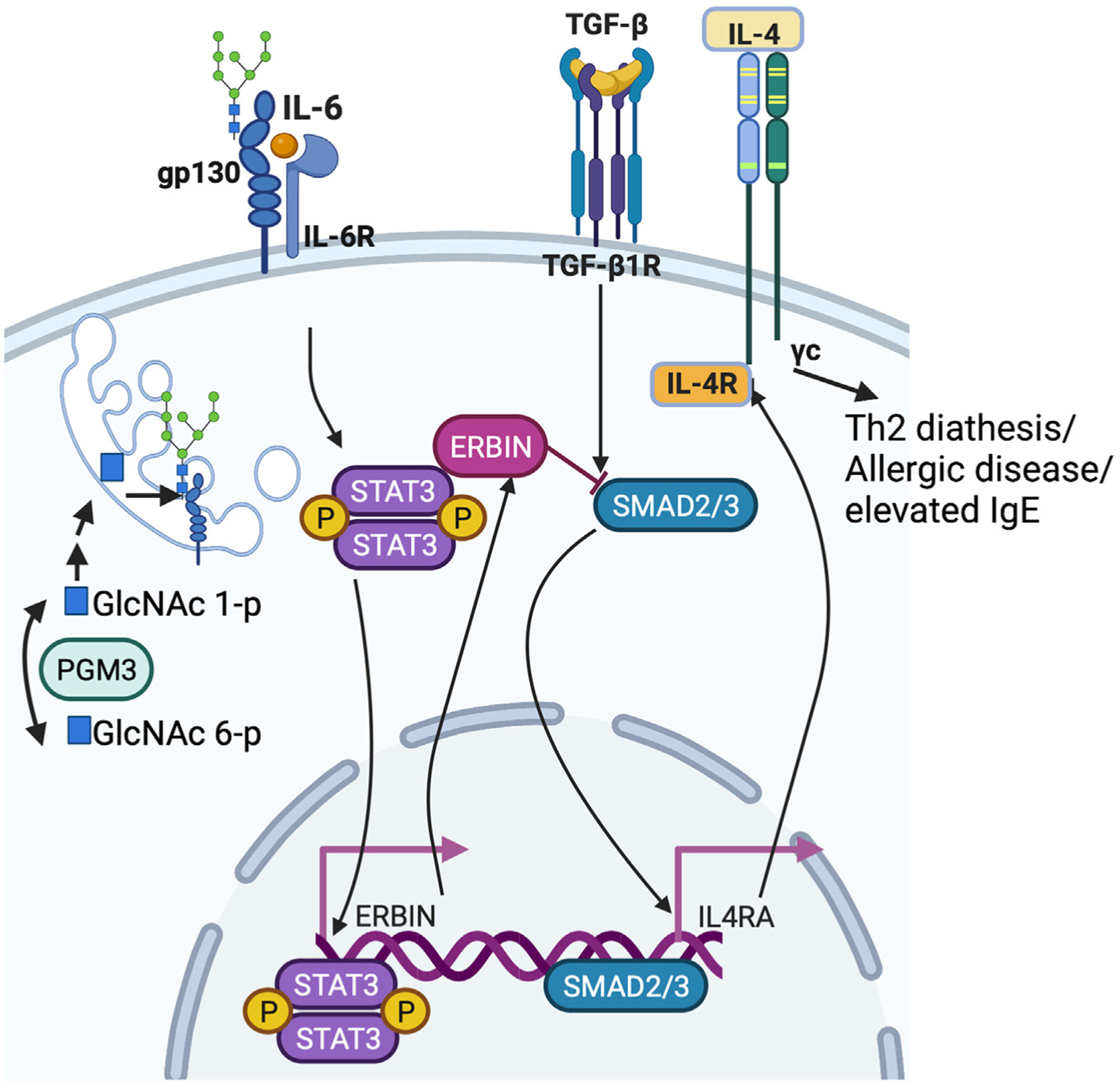
Il-6-mediated signaling via IL-6R and gp130 leads to activation of STAT3, which leads to transcription of ERBIN and physical complexing of STAT3/ERBIN/SMAD2/3, sequestering SMAD2/3 away from the nucleus, and normally preventing IL-4 receptor upregulation on the surface of lymphocytes, and therefore Th2-related phenotypes. PGM3 produces a precursor sugar amine required for N-linked glycosylation that provides stability for surface gp130 expression. Created with BioRender.com.
Funding
This work was supported by the Department of Pediatrics, CUIMC.
Abbreviations
- PIDD
primary immunodeficiency disease
- TCR
T cell receptor
- pMHC
peptide-major histocompatibility complex
- Th1
type 1 T helper
- Th17
type 17 T helper
- Th2
type 2 T helper
- SCID
severe combined immunodeficiency
- mTORC1
mammalian target of rapamycin complex 1
- Treg
regulatory T cell deaminase
- STAT3DN
dominant negative STAT3 mutations
- STAT5BGOF
gain-of-function
- LDS
Loeys-Dietz syndrome
- EGID
eosinophilic gastrointestinal disease
- JAK1GOF
gain-of-function JAK1 mutations
- UDP-GlcNAc
uridine diphosphate N-acetylglucosamine
- ABPA
allergic bronchopulmonary aspergillosis
- FPIES
food-protein induced enteropathy syndrome
- SAM
severe atopic dermatitis, allergic inflammation and hypersensitivity and metabolic wasting
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data Availability
No data were used for the research described in the article.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Lyons JJ, Milner JD: Primary atopic disorders. J Exp Med 2018, 215:1009–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Milner JD: Primary atopic disorders. Annu Rev Immunol 2020, 38:785–808. [DOI] [PubMed] [Google Scholar]
- 3.Rawson R, Yang T, Newbury RO, Aquino M, Doshi A, Bell B, Broide DH, Dohil R, Kurten R, Aceves SS: TGF-beta1-induced PAI-1 contributes to a profibrotic network in patients with eosinophilic esophagitis. J Allergy Clin Immunol 2016, 138:791–800 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balzar S, Chu HW, Silkoff P, Cundall M, Trudeau JB, Strand M, Wenzel S: Increased TGF-beta2 in severe asthma with eosinophilia. J Allergy Clin Immunol 2005, 115:110–117. [DOI] [PubMed] [Google Scholar]
- 5.Turner JA, Stephen-Victor E, Wang S, Rivas MN, Abdel-Gadir A, Harb H, Cui Y, Fanny M, Charbonnier LM, Hung Fong JJ, Benamar M, Wang L, Burton OT, Bansal K, Bry L, Zhu C, Li QZ, Clement RL, Oettgen HC, Crestani E, Rachid R, Sage PT, Chatila TA: Regulatory T cell-derived TGF-beta1 controls multiple checkpoints governing allergy and autoimmunity. Immunity 2020, 53:1331–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]; •This study highlights the positive role for TGFb1 in protecting against allergy via its production in regulatory T-cells.
- 6.Weissler KA, Frischmeyer-Guerrerio PA: Genetic evidence for the role of transforming growth factor-beta in atopic phenotypes. Curr Opin Immunol 2019, 60:54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duong LD, Rawson R, Bezryadina A, Manresa MC, Newbury RO, Dohil R, Liu Z, Barrett K, Kurten R, Aceves SS: TGFbeta1 single-nucleotide polymorphism C-509T alters mucosal cell function in pediatric eosinophilic esophagitis. Mucosal Immunol 2020, 13:110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]; •The authors provide an example of how a common germline variant which increases TGFb production mechanistically causes the worse atopic phenotype– in this case Eosinophilic esophagitis.
- 8.Abonia JP, Wen T, Stucke EM, Grotjan T, Griffith MS, Kemme KA, Collins MH, Putnam PE, Franciosi JP, von Tiehl KF, Tinkle BT, Marsolo KA, Martin LJ, Ware SM, Rothenberg ME: High prevalence of eosinophilic esophagitis in patients with inherited connective tissue disorders. J Allergy Clin Immunol 2013, 132:378–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morgan AW, Pearson SB, Davies S, Gooi HC, Bird HA: Asthma and airways collapse in two heritable disorders of connective tissue. Ann Rheum Dis 2007, 66:1369–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kotlarz D, Marquardt B, Baroy T, Lee WS, Konnikova L, Hollizeck S, Magg T, Lehle AS, Walz C, Borggraefe I, Hauck F, Bufler P, Conca R, Wall SM, Schumacher EM, Misceo D, Frengen E, Bentsen BS, Uhlig HH, Hopfner KP, Muise AM, Snapper SB, Stromme P, Klein C: Human TGF-beta1 deficiency causes severe inflammatory bowel disease and encephalopathy. Nat Genet 2018, 50:344–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kinoshita A, Saito T, Tomita H, Makita Y, Yoshida K, Ghadami M, Yamada K, Kondo S, Ikegawa S, Nishimura G, Fukushima Y, Nakagomi T, Saito H, Sugimoto T, Kamegaya M, Hisa K, Murray JC, Taniguchi N, Niikawa N, Yoshiura K: Domain-specific mutations in TGFB1 result in Camurati-Engelmann disease. Nat Genet 2000, 26:19–20. [DOI] [PubMed] [Google Scholar]
- 12.Frischmeyer-Guerrerio PA, Guerrerio AL, Oswald G, Chichester K, Myers L, Halushka MK, Oliva-Hemker M, Wood RA, Dietz HC: TGFbeta receptor mutations impose a strong predisposition for human allergic disease. Sci Transl Med 2013, 5:195ra94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, Pannu H, De Backer JF, Oswald GL, Symoens S, Manouvrier S, Roberts AE, Faravelli F, Greco MA, Pyeritz RE, Milewicz DM, Coucke PJ, Cameron DE, Braverman AC, Byers PH, De Paepe AM, Dietz HC: Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med 2006, 355:788–798. [DOI] [PubMed] [Google Scholar]
- 14.Davis SD, Schaller J, Wedgwood RJ: Job’s Syndrome. Recurrent, “cold”, staphylococcal abscesses. Lancet 1966, 1:1013–1015. [DOI] [PubMed] [Google Scholar]
- 15.Buckley RH, Wray BB, Belmaker EZ: Extreme hyperimmunoglobulinemia E and undue susceptibility to infection. Pediatrics 1972, 49:59–70. [PubMed] [Google Scholar]
- 16.Buckley RH: The hyper-IgE syndrome. Clin Rev Allergy Immunol 2001, 20:139–154. [DOI] [PubMed] [Google Scholar]
- 17.Borges WG, Hensley T, Carey JC, Petrak BA, Hill HR: The face of Job. J Pediatr 1998, 133:303–305. [DOI] [PubMed] [Google Scholar]
- 18.Grimbacher B, Holland SM, Gallin JI, Greenberg F, Hill SC, Malech HL, Miller JA, O’Connell AC, Puck JM: Hyper-IgE syndrome with recurrent infections–an autosomal dominant multisystem disorder. N Engl J Med 1999, 340:692–702. [DOI] [PubMed] [Google Scholar]
- 19.Falah O, Thwaites SE, Chalmers RT: Ruptured thoracoabdominal aneurysm in a 27-year-old with hyper IgE syndrome. J Vasc Surg 2012, 55:830–832. [DOI] [PubMed] [Google Scholar]
- 20.Kim Y, Nard JA, Saad A, Casselman J, Wessell KR, Toller-Artis E, Tcheurekdijan H, Hostoffer RW: Cerebral aneurysm in a 12-year-old boy with a STAT3 mutation (hyper-IgE syndrome). Ann Allergy Asthma Immunol 2015, 114:430–431. [DOI] [PubMed] [Google Scholar]
- 21.Ling JC, Freeman AF, Gharib AM, Arai AE, Lederman RJ, Rosing DR, Holland SM: Coronary artery aneurysms in patients with hyper IgE recurrent infection syndrome. Clin Immunol 2007, 122:255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharma A, Kumar S, Jagia P: Pulmonary artery pseudoaneurysm in hyper-IgE syndrome: rare complication with successful endovascular management. Vasc Endovasc Surg 2018, 52:375–377. [DOI] [PubMed] [Google Scholar]
- 23.Takeuchi S, Wada K, Otani N, Nawashiro H: Multiple intracranial aneurysms associated with hyper-IgE syndrome. Intern Med 2012, 51:515–516. [DOI] [PubMed] [Google Scholar]
- 24.Freeman AF, Avila EM, Shaw PA, Davis J, Hsu AP, Welch P, Matta JR, Hadigan C, Pettigrew RI, Holland SM, Gharib AM: Coronary artery abnormalities in Hyper-IgE syndrome. J Clin Immunol 2011, 31:338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dewan AK, Tomlinson RE, Mitchell S, Goh BC, Yung RM, Kumar S, Tan EW, Faugere MC, Dietz HC 3rd, Clemens TL, Sponseller PD: Dysregulated TGF-beta signaling alters bone microstructure in a mouse model of Loeys-Dietz syndrome. J Orthop Res 2015, 33:1447–1454. [DOI] [PubMed] [Google Scholar]
- 26.Laky K, Kinard JL, Li JM, Moore IN, Lack J, Fischer ER, Kabat J, Latanich R, Zachos NC, Limkar AR, Weissler KA, Thompson RW, Wynn TA, Dietz HC, Guerrerio AL, Frischmeyer-Guerrerio PA: Epithelial-intrinsic defects in TGFbetaR signaling drive local allergic inflammation manifesting as eosinophilic esophagitis. Sci Immunol 2023, 8:eabp9940. [DOI] [PMC free article] [PubMed] [Google Scholar]; ••This mouse model of TGFBR1 mutation shows that spontaneous eosinophilic esophagitis emerges due to epithelium-specific effects of altered TGF beta receptor signaling.
- 27.Beziat V, Tavernier SJ, Chen YH, Ma CS, Materna M, Laurence A, Staal J, Aschenbrenner D, Roels L, Worley L, Claes K, Gartner L, Kohn LA, De Bruyne M, Schmitz-Abe K, Charbonnier LM, Keles S, Nammour J, Vladikine N, Maglorius Renkilaraj MRL, Seeleuthner Y, Migaud M, Rosain J, Jeljeli M, Boisson B, Van Braeckel E, Rosenfeld JA, Dai H, Burrage LC, Murdock DR, Lambrecht BN, Avettand-Fenoel V, Vogel TP, Undiagnosed Diseases N, Esther CR, Haskologlu S, Dogu F, Ciznar P, Boutboul D, Ouachee-Chardin M, Amourette J, Lebras MN, Gauvain C, Tcherakian C, Ikinciogullari A, Beyaert R, Abel L, Milner JD, Grimbacher B, Couderc LJ, Butte MJ, Freeman AF, Catherinot E, Fieschi C, Chatila TA, Tangye SG, Uhlig HH, Haerynck F, Casanova JL, Puel A: Dominant-negative mutations in human IL6ST underlie hyper-IgE syndrome. J Exp Med 2020, 217:e20191804. [DOI] [PMC free article] [PubMed] [Google Scholar]; ••This study showed how dominant negative mutations in IL6ST could lead to a discrete phenotypic version of the biallelic version of IL6ST-associated disease– one which is characterized by allergic lung inflammation with variable onset and severity.
- 28.Schwerd T, Twigg SRF, Aschenbrenner D, Manrique S, Miller KA, Taylor IB, Capitani M, McGowan SJ, Sweeney E, Weber A, Chen L, Bowness P, Riordan A, Cant A, Freeman AF, Milner JD, Holland SM, Frede N, Muller M, Schmidt-Arras D, Grimbacher B, Wall SA, Jones EY, Wilkie AOM, Uhlig HH: A biallelic mutation in IL6ST encoding the GP130 co-receptor causes immunodeficiency and craniosynostosis. J Exp Med 2017, 214:2547–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen YH, Zastrow DB, Metcalfe RD, Gartner L, Krause F, Morton CJ, Marwaha S, Fresard L, Huang Y, Zhao C, McCormack C, Bick D, Worthey EA, Eng CM, Gold J, Undiagnosed Diseases N, Montgomery SB, Fisher PG, Ashley EA, Wheeler MT, Parker MW, Shanmugasundaram V, Putoczki TL, Schmidt-Arras D, Laurence A, Bernstein JA, Griffin MDW, Uhlig HH: Functional and structural analysis of cytokine-selective IL6ST defects that cause recessive hyper-IgE syndrome. J Allergy Clin Immunol 2021, 148:585–598. [DOI] [PubMed] [Google Scholar]
- 30.Shahin T, Aschenbrenner D, Cagdas D, Bal SK, Conde CD, Garncarz W, Medgyesi D, Schwerd T, Karaatmaca B, Cetinkaya PG, Esenboga S, Twigg SRF, Cant A, Wilkie AOM, Tezcan I, Uhlig HH, Boztug K: Selective loss of function variants in IL6ST cause Hyper-IgE syndrome with distinct impairments of T-cell phenotype and function. Haematologica 2019, 104:609–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lyons JJ, Liu Y, Ma CA, Yu X, O’Connell MP, Lawrence MG, Zhang Y, Karpe K, Zhao M, Siegel AM, Stone KD, Nelson C, Jones N, DiMaggio T, Darnell DN, Mendoza-Caamal E, Orozco L, Hughes JD, McElwee J, Hohman RJ, Frischmeyer-Guerrerio PA, Rothenberg ME, Freeman AF, Holland SM, Milner JD: ERBIN deficiency links STAT3 and TGF-beta pathway defects with atopy in humans. J Exp Med 2017, 214:669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu J, Zhou Y, Liu S, Song X, Yang XZ, Fan Y, Chen W, Akdemir ZC, Yan Z, Zuo Y, Du R, Liu Z, Yuan B, Zhao S, Liu G, Chen Y, Zhao Y, Lin M, Zhu Q, Niu Y, Liu P, Ikegawa S, Song YQ, Posey JE, Qiu G, Study D, Zhang F, Wu Z, Lupski JR, Wu N: The coexistence of copy number variations (CNVs) and single nucleotide polymorphisms (SNPs) at a locus can result in distorted calculations of the significance in associating SNPs to disease. Hum Genet 2018, 137:553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen C, Akiyama K, Wang D, Xu X, Li B, Moshaverinia A, Brombacher F, Sun L, Shi S: mTOR inhibition rescues osteopenia in mice with systemic sclerosis. J Exp Med 2015, 212:73–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hershey GK, Friedrich MF, Esswein LA, Thomas ML, Chatila TA: The association of atopy with a gain-of-function mutation in the alpha subunit of the interleukin-4 receptor. N Engl J Med 1997, 337:1720–1725. [DOI] [PubMed] [Google Scholar]
- 35. Oetjen LK,Mack MR, Feng J, Whelan TM, Niu H, Guo CJ, Chen S, Trier AM, Xu AZ, Tripathi SV, Luo J, Gao X, Yang L, Hamilton SL, Wang PL, Brestoff JR, Council ML, Brasington R, Schaffer A, Brombacher F, Hsieh CS, Gereau RWt, Miller MJ, Chen ZF, Hu H, Davidson S, Liu Q, Kim BS: Sensory neurons co-opt classical immune signaling pathways to mediate chronic itch. Cell 2017, 171:217–28 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, Freeman AF, Demidowich A, Davis J, Turner ML, Anderson VL, Darnell DN, Welch PA, Kuhns DB, Frucht DM, Malech HL, Gallin JI, Kobayashi SD, Whitney AR, Voyich JM, Musser JM, Woellner C, Schaffer AA, Puck JM, Grimbacher B: STAT3 mutations in the hyper-IgE syndrome. N Engl J Med 2007, 357:1608–1619. [DOI] [PubMed] [Google Scholar]
- 37.Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, Kawamura N, Ariga T, Pasic S, Stojkovic O, Metin A, Karasuyama H: Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 2007, 448:1058–1062. [DOI] [PubMed] [Google Scholar]
- 38.Arora M, Bagi P, Strongin A, Heimall J, Zhao X, Lawrence MG, Trivedi A, Henderson C, Hsu A, Quezado M, Kleiner DE, Venkatesan AM, Holland SM, Freeman AF, Heller T: Gastrointestinal manifestations of STAT3-deficient hyper-IgE syndrome. J Clin Immunol 2017, 37:695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Droghini HR, Abonia JP, Collins MH, Milner JD, Lyons JJ, Freeman AF, Mukkada VA, Risma KA, Rothenberg ME, Schwartz JT: Targeted IL-4Ralpha blockade ameliorates refractory allergic eosinophilic inflammation in a patient with dysregulated TGF-beta signaling due to ERBIN deficiency. J Allergy Clin Immunol Pract 2022, 10:1903–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]; ••This study provides a prime example of precision therapy– whereby a monogenic disorder leading to refractory to treatment allergic inflammation can be ameliorated via inhibition of a pathway abnormally activated by the underlying mutation. In this case excessive IL-4 receptor expression caused by poor ERBIN function could be inhibited by antibodies targeting the IL-4 receptor.
- 40.James AE, West L, Schloss K, Nataraj P, Urban A, Hirsch A, Krausz M, Kumar S, Raasch J, Risma K, Church JA, Grimbacher B, Bergerson JRE, Chong H, Freeman AF: Treatment of STAT3-deficient hyper-immunoglobulin E syndrome with monoclonal antibodies targeting allergic inflammation. J Allergy Clin Immunol Pract 2022, 10:1367–1370 e1. [DOI] [PubMed] [Google Scholar]; ••This study provides a prime example of precision therapy– whereby a monogenic disorder leading to refractory to treatment allergic inflammation can be ameliorated via inhibition of a pathway abnormally activated by the underlying mutation. In this case excessive IL-4 receptor expression caused by poor STAT3 function could be inhibited by antibodies targeting the IL-4 receptor. It also contrasts with the efficacy of other biologics which impact other atopy causing pathways.
- 41.Lu CW, Lee WI, Chung WH: Dupilumab for STAT3-hyper-IgE syndrome with refractory intestinal complication. Pediatrics 2021, 148:e2021050351. [DOI] [PubMed] [Google Scholar]
- 42.Matucci-Cerinic C, Viglizzo G, Pastorino C, Corcione A, Prigione I, Bocca P, Bustaffa M, Cecconi M, Gattorno M, Volpi S: Remission of eczema and recovery of Th1 polarization following treatment with Dupilumab in STAT3 hyper IgE syndrome. Pediatr Allergy Immunol 2022, 33:e13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sogkas G, Hirsch S, Jablonka A, Witte T, Schmidt RE, Atschekzei F: Dupilumab to treat severe atopic dermatitis in autosomal dominant hyper-IgE syndrome. Clin Immunol 2020, 215:108452. [DOI] [PubMed] [Google Scholar]
- 44.Staudacher O, Kruger R, Kolsch U, Thee S, Gratopp A, Wahn V, Lau S, von Bernuth H: Relieving job: dupilumab in autosomal dominant STAT3 hyper-IgE syndrome. J Allergy Clin Immunol Pract 2022, 10:349–351 e1. [DOI] [PubMed] [Google Scholar]
- 45.Wang HJ, Yang TT, Lan CE: Dupilumab treatment of eczema in a child with STAT3 hyper-immunoglobulin E syndrome. J Eur Acad Dermatol Venereol 2022, 36:e367. [DOI] [PubMed] [Google Scholar]
- 46.Polivka L, Hadj-Rabia S, Bal E, Leclerc-Mercier S, Madrange M, Hamel Y, Bonnet D, Mallet S, Lepidi H, Ovaert C, Barbet P, Dupont C, Neven B, Munnich A, Godsel LM, Campeotto F, Weil R, Laplantine E, Marchetto S, Borg JP, Weis WI, Casanova JL, Puel A, Green KJ, Bodemer C, Smahi A: Epithelial barrier dysfunction in desmoglein-1 deficiency. J Allergy Clin Immunol 2018, 142:702–706 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bernth-Jensen JM, Holm M, Christiansen M: Neonatal-onset T(−)B (−)NK(+) severe combined immunodeficiency and neutropenia caused by mutated phosphoglucomutase 3. J Allergy Clin Immunol 2016, 137:321–324. [DOI] [PubMed] [Google Scholar]
- 48.Sassi A, Lazaroski S, Wu G, Haslam SM, Fliegauf M, Mellouli F, Patiroglu T, Unal E, Ozdemir MA, Jouhadi Z, Khadir K, Ben-Khemis L, Ben-Ali M, Ben-Mustapha I, Borchani L, Pfeifer D, Jakob T, Khemiri M, Asplund AC, Gustafsson MO, Lundin KE, Falk-Sorqvist E, Moens LN, Gungor HE, Engelhardt KR, Dziadzio M, Stauss H, Fleckenstein B, Meier R, Prayitno K, Maul-Pavicic A, Schaffer S, Rakhmanov M, Henneke P, Kraus H, Eibel H, Kolsch U, Nadifi S, Nilsson M, Bejaoui M, Schaffer AA, Smith CI, Dell A, Barbouche MR, Grimbacher B: Hypomorphic homozygous mutations in phosphoglucomutase 3 (PGM3) impair immunity and increase serum IgE levels. J Allergy Clin Immunol 2014, 133:1410–1419 9 e1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stray-Pedersen A, Backe PH, Sorte HS, Morkrid L, Chokshi NY, Erichsen HC, Gambin T, Elgstoen KB, Bjoras M, Wlodarski MW, Kruger M, Jhangiani SN, Muzny DM, Patel A, Raymond KM, Sasa GS, Krance RA, Martinez CA, Abraham SM, Speckmann C, Ehl S, Hall P, Forbes LR, Merckoll E, Westvik J, Nishimura G, Rustad CF, Abrahamsen TG, Ronnestad A, Osnes LT, Egeland T, Rodningen OK, Beck CR, Baylor-Johns Hopkins Center for Mendelian G, Boerwinkle EA, Gibbs RA, Lupski JR, Orange JS, Lausch E, Hanson IC: PGM3 mutations cause a congenital disorder of glycosylation with severe immunodeficiency and skeletal dysplasia. Am J Hum Genet 2014, 95:96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Y, Yu X, Ichikawa M, Lyons JJ, Datta S, Lamborn IT, Jing H, Kim ES, Biancalana M, Wolfe LA, DiMaggio T, Matthews HF, Kranick SM, Stone KD, Holland SM, Reich DS, Hughes JD, Mehmet H, McElwee J, Freeman AF, Freeze HH, Su HC, Milner JD: Autosomal recessive phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. J Allergy Clin Immunol 2014, 133:1400–1409 9 e1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ben-Khemis L, Mekki N, Ben-Mustapha I, Rouault K, Mellouli F, Khemiri M, Bejaoui M, Essaddam L, Ben-Becher S, Boughamoura L, Hassayoun S, Ben-Ali M, Barbouche MR: A founder mutation underlies a severe form of phosphoglutamase 3 (PGM3) deficiency in Tunisian patients. Mol Immunol 2017, 90:57–63. [DOI] [PubMed] [Google Scholar]
- 52.Carlson RJ, Bond MR, Hutchins S, Brown Y, Wolfe LA, Lam C, Nelson C, DiMaggio T, Jones N, Rosenzweig SD, Stone KD, Freeman AF, Holland SM, Hanover JA, Milner JD, Lyons JJ: Detection of phosphoglucomutase-3 deficiency by lectin-based flow cytometry. J Allergy Clin Immunol 2017, 140:291–294 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garcia-Garcia A, Buendia Arellano M, Deya-Martinez A, Lozano Blasco J, Serrano M, Van Den Rym A, Garcia-Solis B, Esteve-Sole A, Yiyi L, Vlagea A, Solanich X, Fisher MR, Lyons JJ, de Diego RP, Alsina L: Novel PGM3 compound heterozygous variants with IgE-related dermatitis, lymphopenia, without syndromic features. Pediatr Allergy Immunol 2021, 32:566–575. [DOI] [PubMed] [Google Scholar]; ••This report critically demonstrates that PGM3 mutation can lead to a clinical presentation which is more commonly encountered– severe atopic disease in the absence of other syndromic or infectious co-morbidities– another example of a Primary Atopic Disorder underlying common allergic disease.
- 54.Winslow A, Jalazo ER, Evans A, Winstead M, Moran T: A De Novo cause of PGM3 deficiency treated with hematopoietic stem cell transplantation. J Clin Immunol 2022, 42:691–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu G, Hitchen PG, Panico M, North SJ, Barbouche MR, Binet D, Morris HR, Dell A, Haslam SM: Glycoproteomic studies of IgE from a novel hyper IgE syndrome linked to PGM3 mutation. Glycoconj J 2016, 33:447–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ben-Ali M, Ben-Khemis L, Mekki N, Yaakoubi R, Ouni R, Benabdessalem C, Ben-Mustapha I, Barbouche MR: Defective glycosylation leads to defective gp130-dependent STAT3 signaling in PGM3-deficient patients. J Allergy Clin Immunol 2019, 143:1638–1640 e2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data were used for the research described in the article.