

Abstract
Deciphering the specificity of T-cell receptor (TCR) repertoires is crucial for monitoring adaptive immune responses and developing targeted immunotherapies and vaccines. To elucidate the specificity of previously unseen TCRs, many methods employ the BLOSUM62 matrix to find TCRs with similar amino acid (AA) sequences. However, while BLOSUM62 reflects the AA substitutions within conserved regions of proteins with similar functions, the remarkable diversity of TCRs means that both TCRs with similar and dissimilar sequences can bind the same epitope. Therefore, reliance on BLOSUM62 may bias detection towards epitope-specific TCRs with similar biochemical properties, overlooking those with more diverse AA compositions. In this study, we introduce tcrBLOSUMa and tcrBLOSUMb, specialized AA substitution matrices for CDR3 alpha and CDR3 beta TCR chains, respectively. The matrices reflect AA frequencies and variations occurring within TCRs that bind the same epitope, revealing that both CDR3 alpha and CDR3 beta display tolerance to a wide range of AA substitutions and differ noticeably from the standard BLOSUM62. By accurately aligning distant TCRs employing tcrBLOSUMb, we were able to improve clustering performance and capture a large number of epitope-specific TCRs with diverse AA compositions and physicochemical profiles overlooked by BLOSUM62. Utilizing both the general BLOSUM62 and specialized tcrBLOSUM matrices in existing computational tools will broaden the range of TCRs that can be associated with their cognate epitopes, thereby enhancing TCR repertoire analysis.
Keywords: T-cell receptor, epitope-specific TCRs, TCR sequence similarity, TCR clustering, specialized amino acid substitution matrix, BLOSUM62
Graphical Abstract
Graphical Abstract.

Introduction
T cells orchestrate precise adaptive immune responses against pathogens and cancer by recognizing peptides (antigens, epitopes) presented by major histocompatibility complexes (MHCs). Each T cell expresses a single, distinct T-cell receptor (TCR) on its surface. With an estimated TCR diversity of more than 1011 different possible TCRs in every person [1], this ensures both comprehensive coverage and remarkable specificity in detecting foreign antigens. The uniqueness and specificity are largely governed by the amino acid (AA) sequence of the complementarity-determining region 3 (CDR3), the most diverse TCR region, which directly interacts with the presented epitope. Deciphering the antigen specificity of T cells through analysis of TCR repertoire sequences is crucial for unravelling adaptive immune responses and advancing targeted immunotherapies and vaccine development.
TCR-epitope prediction tools can be broadly categorized into three main groups: structure-based tools that rely on 3D structures of the TCR-pMHC complex, distance-based tools that use a single distance metric to calculate similarity between TCRs with known and unknown specificity, and feature-based tools that seek to identify common patterns within epitope-specific TCR sets. In the last two years, structure-based predictions of TCR-pMHC interactions began to develop and gain more attention [2–4]. With the growing availability of structural data [5] and the increasing reliability of structure predictions [6, 7], these approaches hold significant potential. The most active area of research focuses on developing feature-based machine learning (ML) tools. However, these methods face challenges with reproducibility across different datasets [8]. According to a benchmarking study published in 2023, their performance is still comparable to that of distance-based approaches [9]. This study also revealed that incorporating both alpha and beta CDR3s improves overall performance. Nevertheless, until sufficient paired-chain TCR sequencing data is accumulated, most tools remain devoted to optimizing results based solely on CDR3 beta [10]. A comprehensive review of the methods and challenges they face was published by Hudson et al. in 2023 [10].
To elucidate the specificity of TCRs that have not been previously characterized (unseen TCRs), both distance- and feature-based approaches often group TCRs based on their sequence similarity. This strategy leverages the observation that similar TCRs often recognize the same epitope [11]. However, it relies on a definition of ‘similarity’ between AA residues within CDR3s. For this reason, many tools, such as TCRdist [12], GLIPH [13], TITAN [14], TCRGP [15], epiTCR [16], GIANA [17], NetTCR 2.2 [18], etc., make use of the BLOCKS substitution matrix 62 (BLOSUM62) as the basis for quantifying AA similarity in the annotation process to infer epitope specificity of TCRs.
BLOSUM62 matrix reflects the evolutionary conservation of AA substitutions within closely related protein families [19] and is broadly used to determine sequence homology and functional similarity [20]. In the case of TCRs, that translates into whether TCR sequences are similar enough to bind the same epitope. However, the exceptionally diverse TCR repertoires do not fully adhere to the assumptions of traditional protein family evolution. Unlike standard proteins that have evolved through gene duplication, divergence, and mutation, every TCR is independently generated in a process known as V(D) J recombination, which involves quasi-random rearrangement of V(D)J gene segments during the development of every T cell [21]. Consequently, while traditional protein families often exhibit conserved regions across functionally related members, somatic recombination results in a highly diverse pool of sequences where even dissimilar TCRs can recognize the same epitope [22]. As a result, we hypothesize that BLOSUM62 does not optimally guide TCR data analysis approaches to the full spectrum of epitope-specific TCR repertoires.
Ideally, the most suitable substitution matrix for TCR-epitope annotation should reflect AA similarity and CDR3-epitope interaction patterns. AA substitution scores in BLOSUM62 approximate the similarity between the biochemical properties of AAs, reflecting the underlying evolutionary constraints within the data used for matrix construction. However, general-purpose matrices such as BLOSUM62 have demonstrated limitations in accurately aligning sequences of narrow protein classes, particularly those with distinct compositional biases [23, 24] or relative mutability of AAs [25]. Accordingly, various alternative substitution matrices have been developed over the years, including those based on structural alignments [26], peptide:MHC class I binding [27, 28], substitutions within protein families with non-standard compositions [29–32], and others. Similarly, BLOSUM62 may not fully capture the unique characteristics of TCR sequences, suggesting that TCR analysis may benefit from a specialized AA substitution matrix tailored to the substitutions within the CDR3 class. Such a matrix could offer new insights into the similarity of AAs and biological associations between them in the CDR3-epitope space, potentially enhancing TCR repertoire analysis.
In this study, we re-evaluate the use of BLOSUM62 for TCR analysis and present an alternative AA substitution matrix, tcrBLOSUM, which is consistent with AA frequencies and changes occurring within CDR3 sequences. Evaluation of tcrBLOSUM on a new set of TCRs with unseen epitopes showed that it enables the capture of a large number of epitope-specific TCRs with more diverse AA composition and physicochemical profiles, which were overlooked by BLOSUM62. By accurately aligning distant TCRs with tcrBLOSUM, we seek to expand current epitope annotation strategies, which rely on BLOSUM62, to include dissimilar CDR3s, thus enhancing TCR repertoire analysis.
Methods
Assembly of the datasets
Human CDR3 sequences with known epitope specificity used in this study were collected from VDJdb [33] (access date: 13.06.2023), McPAS [34] (access date: 10.09.2022), and ImmuneACCESS [35] (access date: 26.05.2020). After filtering to remove duplicates, CDR3 sequences (CDR3s) with spurious symbols, CDR3s that do not start with conserved cysteine (C), and CDR3s that do not end with phenylalanine (F) (in alpha & beta chains) or tryptophan (W) (in alpha chain), 85,053 CDR3 alpha/beta sequences specific to 1441 epitopes were retained. Identical CDR3 sequences differing in their V/J genes and/or cognate epitopes were kept since they have been generated independently and thus reflect the diversity and specificity patterns of epitope-specific TCR repertoires. In order to exclude the bias that could arise from conserved flanking AAs in CDR3s, all the sequences were processed. Specifically, flanking AA positions where 99% of the sequences had at most two different AAs were trimmed. In the case of CDR3 alpha, this was the first (position 1) and the last AA (position −1). In the case of CDR3 beta, this was two first (positions 1, 2) and the last AA (position −1).
Several data subsets were then derived from the complete assembled dataset: a dataset only with CD8+ TCR sequences, only with CD4+ TCR sequences, only with alpha chain, and only with beta chain. Additionally, alpha and beta chain datasets were split into non-overlapping source and validation datasets. Source alpha/beta chain data subsets (SC2neg dataset) did not include alpha/beta TCRs specific to any of SARS-CoV-2 epitopes, while validation alpha/beta chain data subsets (SC2only dataset) contained only alpha/beta TCRs specific to SARS-CoV-2 epitopes. While no CDR3-epitope pairs occurred in both source and validation datasets, 181 alpha and 463 beta CDR3s were specific to epitopes from both datasets. A summary of all the datasets can be found in Table 1.
Table 1.
Summary statistics of all TCR datasets used in the study.
| alpha chain dataset | beta chain dataset | CD4+ dataset | CD8+ dataset | SC2neg alpha chain dataset | SC2neg beta chain dataset | SC2only alpha chain dataset | SC2only beta chain dataset | |
|---|---|---|---|---|---|---|---|---|
| N CDR3s | 17,669 | 63,445 | 4174 | 50,434 | 14,966 | 33,266 | 2703 | 30,179 |
| N unique CDR3s | 14,493 | 56,281 | 3571 | 42,136 | 12,014 | 27,194 | 2660 | 29,550 |
| N unique epitopes | 370 | 637 | 62 | 528 | 226 | 309 | 144 | 328 |
| N blocks | 1042 | 2388 | 309 | 2380 | 727 | 1220 | 315 | 1168 |
Construction of alternative AA substitution matrices
tcrBLOSUM
Traditional BLOSUM-series matrices were built from sequence blocks of the BLOCKS database, where one block referred to a multiple sequence alignment of protein regions that are conserved across different members of the protein family [19]. To design a BLOSUM-style matrix (tcrBLOSUM) reflective of TCR compositional bias, we utilized exclusively CDR3 sequences of TCRs.
Specifically, each of the assembled datasets was processed to define blocks of CDR3 sequences that would be conceptually similar to blocks of the BLOCKS database. One TCR block was defined as a group of at least two different CDR3 sequences that (i) recognize the same epitope, (ii) originate from the same chain (alpha/beta), and (iii) have the same length.
Similarly to BLOSUM62, tcrBLOSUM score for a particular AA substitution (Eq.1) is a log-odds score that provides a measure of the biological (observed) probability of a substitution in TCRs relative to the chance (expected) probability of the substitution.
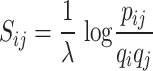 |
(1) |
pij - the probability of two AAs replacing each other in CDR3s recognizing the same epitope (observed frequency of a pair AAi – AAj in TCR blocks)
qi, qj - background probabilities of finding the AAs in all CDR3s (expected frequency of a pair AAi – AAj based on individual frequency of each AA in TCR blocks)
1/λ - scaling factor
As an initial step, the frequency of each AA in the entire dataset of TCR blocks was computed (qi, qj). Next, we counted the number of AA pairs (substitutions) in every column of all pairwise sequence combinations in every TCR block. Dividing these AA pair counts by the total number of all AA substitutions in the data, we obtained frequencies of the AA pairs (pij). Frequencies of AA pairs (pij) that have not been observed in the data of TCR blocks were substituted with the smallest frequency of all pairs in the data divided by 100. Finally, following the methodology proposed by Henikoff & Henikoff [19], a log-odds score of an AA substitution was calculated according to Equation 1. To ensure comparability with the original BLOSUM62 matrix, the scaling factor of two was applied, and computed substitution scores were rounded to the nearest integer. Scripts utilized for all the calculations were produced in-house and can be found under src/calculate_tcrBLOSUMs.py in the GitHub repository https://github.com/apostovskaya/tcrBLOSUM.
Physicochemical similarity matrix
The Physicochemical Similarity Matrix (PhysChemSim) is an AA substitution matrix designed to quantify the similarity between two AAs based on their physicochemical properties. Initially, each AA was encoded as a vector of z-descriptors of physicochemical properties, a common technique in quantitative structure–activity relationship modelling of biological activities of small peptides [36].
Next, the Euclidean distance was computed between all pairs of AAs, resulting in mostly negative substitution scores. However, certain AA pairs yielded positive scores, while self-substitution received a score of zero (representing the Euclidean distance to itself). Since any AA is most similar to itself in terms of biochemical properties, the self-substitution scores were adjusted to have the highest positive score.
Topological similarity matrix
The Topological Similarity Matrix (TopoSim) is an AA substitution matrix designed to quantify the similarity in atomic composition and atomic neighborhoods between pairs of AAs. Initially, each AA was encoded as a binary vector using extended-connectivity fingerprints with radius two (ECFP4) [37] from the RDKit (v. 2023.9.5) Python library [38]. ECFPs are circular topological fingerprints describing an atom’s circular chemical environment. They are commonly used for various applications in drug discovery, such as small molecule characterization, similarity searching, structure–activity modelling, and evaluation of high-throughput screening results [39]. Subsequently, Tanimoto similarity (Eq.2) was calculated to reflect the similarity between the fingerprints of two AAs [40], resulting in a range of positive substitution scores, with the self-substitution scores having the highest value.
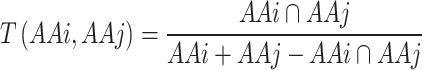 |
(2) |
AAi, AAj – ECFP4 binary vectors of two AAs for which similarity score is being calculated.
To ensure compatibility with other matrices, the similarity scores were rescaled to fit within the range of 1–10.
Comparison of matrices
To establish the relationship between all constructed matrices and BLOSUM62, Pearson correlation between the AA substitution score vectors of all the matrices was performed. Additionally, to evaluate the similarity of tcrBLOSUM matrices constructed using the full dataset and data subsets, we calculated the Euclidean distance (Frobenius norm) between them. Hierarchical clustering was then conducted based on the computed Pearson correlation values or Euclidean distances, respectively. The analysis was performed using SciPy (v. 1.10.1) Python library [41].
Evaluation of the tcrBLOSUM
TCRdist distance metric with subsequent clustering
To evaluate the effectiveness of tcrBLOSUM and BLOSUM62 in accurately grouping TCR sequences based on their epitope specificity, we employed TCRdist distance metric which uses BLOSUM62 by default. Firstly, we transformed the tcrBLOSUM similarity matrix into a distance matrix according to the rules of TCRdist metric [12]. Next, we aligned sequences using the Needleman-Wunsch algorithm from Parasail (v. 1.3.4) Python library [42] and computed the TCRdist distance adapted from the original TCRdist [12] and TCRdist3 [43] publications. TCRs with known epitope-specificity were then clustered with HDBSCAN algorithm from scikit-learn (v. 1.3.0) Python library [44] based on their TCRdist distance to each other.
To assess the accuracy of the clustering, we employed several metrics which have been introduced elsewhere [11]. Briefly, retention indicated the fraction of TCR sequences assigned to any cluster; purity measured the fraction of TCR sequences within a single cluster recognizing the same epitope; consistency represented the fraction of TCR sequences recognizing the same epitope and assigned to one cluster. These three metrics were computed for clustering performed on the TCRdist distance matrix computed with either tcrBLOSUM or BLOSUM62, providing test statistics. Since both related and unrelated TCRs could end up in the same cluster by chance, we have also calculated clustering metrics for randomly shuffled TCR-epitope pairs as baseline statistics. Test-to-baseline statistics ratios were then compared between the two approaches.
Evaluation of the similarity of the identified epitope-specific CDR3s
To explore the benefits of tcrBLOSUMb for epitope annotation of TCRs, an alignment-based approach was employed to identify epitope-specific CDR3s. The SC2only dataset was utilized to score against a query CDR3 (qCDR3) to identify hit CDR3s. Each CDR3 in the dataset served iteratively as a query, and every CDR3b in the SC2only dataset was aligned against all other CDR3s in the dataset using the Needleman-Wunsch (NW) algorithm. The alignment was performed using BLOSUM62, PhysChemSim, TopoSim, or tcrBLOSUMb, with the latter constructed on the SC2neg data subset to maintain independence between source and validation datasets. The NW alignment score provided a measure of similarity between the qCDR3 and other CDR3s, with higher scores indicating higher similarity and potential shared epitope specificity.
The top k-1 CDR3 hits with the highest NW scores (the most similar to qCDR3) that are known to bind the same epitope as qCDR3 were considered true positive (TP) CDR3 hits, with k as the number of TCRs with the given epitope specificity in the data. Subsequently, unique TP hits, identified exclusively with either of the compared substitution matrices, were further examined. To assess the (dis)similarity of unique TP CDR3 hits to their respective qCDR3s, we measured their differences in length, AA composition, and physicochemical properties. Two metrics were computed to estimate differences in AA composition: edit (Levenshtein) distance and TCRdist distance. NW alignment score calculated with PhysChemSim matrix represented similarity in physicochemical properties. Finally, the four obtained (dis) similarity metrics were compared between BLOSUM62 and tcrBLOSUMb to evaluate the overall (dis)similarity of unique TP CDR3 hits relative to the competitor matrix.
Results
Different BLOSUM-style AA substitution matrices are suitable for CDR3s of alpha and beta chains of CD8+ T cells
It has been noted that standard AA substitution matrices, such as BLOSUM62, may not be well-suited for sequence alignments within certain classes of proteins, primarily due to their non-standard composition, since AA frequencies are an important component in the calculation of substitution matrices. Comparing AA frequencies in TCR CDR3s with those in functional proteins from UniProt [45] reveals a unique AA distribution in CDR3 sequences. CDR3s were found to be enriched in AAs with polar side chains and depleted in positively charged AAs and the majority of non-polar hydrophobic AAs (Fig. 1, Fig. S1), therefore supporting the creation of a specialized BLOSUM-like matrix for TCRs (tcrBLOSUM).
Figure 1.

(a) Bar plot representing the frequency of amino acids in CDR3 sequences relative to the frequency in proteins from UniProt. CDR3s are enriched in AAs with polar side chains (amide, in white, and small hydroxy, in green) and depleted in positively charged (basic, in blue) and the majority of non-polar hydrophobic AAs (aliphatic, in grey). (b) Hierarchical clustering dendrogram depicting relationships between evaluated AA substitution matrices. While tcrBLOSUM matrices formed a distinct cluster from BLOSUM62 and the two descriptor matrices (PhysChemSim, TopoSim), the distance between tcrBLOSUMa and tcrBLOSUMb was larger than between any of the three matrices in the other cluster. AA: Amino acid.
First, we evaluated whether a single matrix for all T cells and TCR chains would suffice or if further specialization might be necessary. To this end, AA frequencies in different TCR data subsets were compared, including CD4+, CD8+, CDR3 alpha (CDR3a), and CDR3 beta (CDR3b). Due to the limited size of the CD4+ data subset (Table 1), which was 4–13 times smaller than other data subsets, further specification to both CD4+/CD8+ lineage and alpha/beta chains was not feasible. Notable differences were observed in the frequencies of CDR3a (Fig. S1), while the frequencies in CD4+ and CD8+ T-cell subsets were similar to those in CDR3b. Consequently, out of the four created BLOSUM-like matrices (Fig. S2), we focused on two chain-specific matrices, namely tcrBLOSUMa (Fig. 2, Table S1) and tcrBLOSUMb (Fig. 2, Table S2).
Figure 2.

Constructed BLOSUM-style AA substitution matrices for CDR3 alpha (tcrBLOSUMa, left) and CDR3 beta (tcrBLOSUMb, centre) and the original BLOSUM62 matrix (right). In contrast to BLOSUM62, constructed tcrBLOSUMa and tcrBLOSUMb matrices mostly feature neutral substitution scores of zero.
To provide additional reference points, two matrices reflecting the similarity between the physicochemical properties of AAs (PhysChemSim, Table S3) and their topological similarity (TopoSim, Table S4) were derived using z-descriptors and extended connectivity fingerprints (ECFPs), respectively (Fig. S2). Our analysis revealed distinct clustering patterns among the constructed matrices. As can be seen in Fig. 1-b, the two most similar matrices were BLOSUM62 and PhysChemSim matrix. Moreover, when AAs were grouped based on their substitution scores in a matrix, AAs with similar biochemical profiles formed clusters in BLOSUM62 and PhysChemSim but not in tcrBLOSUMs (Fig. S3). This is consistent with expectations, as both BLOSUM62 and PhysChemSim matrices were designed to reflect the biochemical similarity of AAs. Notably, tcrBLOSUM matrices formed a distinct cluster from BLOSUM62 and the two descriptor matrices (Fig. 1-b), indicating their unique characteristics. In agreement with the observed unique AA frequency profiles of alpha and beta chain CDR3s, the distance between tcrBLOSUMa and tcrBLOSUMb was larger than between any of the three matrices in the BLOSUM62 cluster (Fig. 1-b). Accordingly, we concluded that employing separate AA substitution matrices for each CDR3 chain would be appropriate.
CDR3s display tolerance to a wide range of AA substitutions
In tcrBLOSUM, as in BLOSUM62, the score indicates whether a particular AA pair (substitution) occurs more, less, or as frequently as expected and therefore can be considered favorable, unfavorable, or neutral, respectively. While most scores are negative (unfavorable substitutions) in BLOSUM62, both tcrBLOSUMa and tcrBLOSUMb predominantly feature neutral (equal to zero) scores (Fig. 2, Fig. S4). A unique characteristic of tcrBLOSUMb is the self-substitution scores of 0 for three aliphatic/hydrophobic AAs (A, G, V) and one small hydroxy AA (T). Intriguingly, despite being among the most unique and conserved AAs in BLOSUM62 (Fig. S3), all substitutions for tryptophan (W), except with cysteine (C), were neutral in tcrBLOSUMb (Fig. 2).
Another pronounced difference between both tcrBLOSUMs and BLOSUM62 lies in the strongly negative scores for cysteine (C) in tcrBLOSUMs, as this AA is the rarest in CDR3s (excluding the first conserved position). Notably, in both tcrBLOSUMa and tcrBLOSUMb, three additional AAs displayed a high degree of conservation, with at least 50% of their non–self-substitution scores (≥10/19) being negative (Fig. S4): one basic AA (K) and two aliphatic/hydrophobic AAs (A, L) in tcrBLOSUMa; one amide AA (Q) and two aromatic AAs (Y, F) in tcrBLOSUMb. In this instance, it appears unrelated to the abundance of these AAs in the dataset, as all of them had frequencies higher than the median (Fig. S1).
Among favorable substitutions, several pairs in tcrBLOSUMa stood out because these substitutions occurred between AAs from different classes: C-R and T-I. Interestingly, some favorable substitutions were observed between AAs with opposite charges of their side chains across all three matrices: K-E in BLOSUM62 and tcrBLOSUMb, H-E in tcrBLOSUMa. Additionally, several substitutions between AAs with polar charged and uncharged chains received positive scores in all three matrices: N-D in BLOSUM62, K-N and K-Q in tcrBLOSUMa, T-E and N-D in tcrBLOSUMb.
tcrBLOSUM matrices are stable towards new epitopes
Given the tendency for similar TCRs to recognize the same epitopes, we considered the possibility that the constructed tcrBLOSUMs might not generalize well to new TCRs recognizing epitopes not present in the current source TCR-epitope data. If the constructed tcrBLOSUMa/b matrices do not represent the entire TCR space, the inclusion of new epitope-specific TCRs could significantly alter the scores in the matrices. To assess the impact of the size and (TCR-epitope) diversity of the source TCR-epitope data, we leveraged the abundance of TCR sequences recognizing SARS-CoV-2 epitopes generated during the COVID-19 pandemic.
To this end, we split the full dataset into two non-overlapping subsets, each lacking TCR-epitope pairs present in the other. The first data subset (SC2neg dataset) contained CDR3s specific to all epitopes except SARS-CoV-2 epitopes. Conversely, the second data subset (SC2only dataset) exclusively contained SARS-CoV-2-specific CDR3s. The two resulting beta subsets were comparable in size, number of unique epitopes and CDR3s, as well as the number of CDR3 blocks (Table 1). In contrast, there was a noticeable discrepancy in the size of the two alpha subsets, with the SC2only alpha subset having five and two times fewer unique epitopes and CDR3s, respectively, compared to the SC2neg alpha subset (Table 1). tcrBLOSUMa and tcrBLOSUMb matrices were constructed for each subset, resulting in four new matrices (Fig. S5), which were then compared to each other and the respective matrices derived from the full dataset.
Among three matrices (full, SC2neg, SC2only) within the same class (alpha/beta), a slight variation in scores was observed (Fig. S5). Furthermore, while two beta matrices built on data subsets were equidistant from the full tcrBLOSUMb (Fig. S6), SC2only tcrBLOSUMa resembled full tcrBLOSUMa less than SC2neg tcrBLOSUMa (Fig. S6), highlighting the importance of source dataset diversity in epitopes and CDR3s to obtain a representative AA substitution matrix. Despite minor differences, the alpha chain matrices and beta chain matrices formed two distinct clusters when compared to BLOSUM62 (Fig. 3). The predominant similarity between the matrices of the same class indicates that the most distinct features of AA substitutions in alpha and beta CDR3s were captured in the respective tcrBLOSUM matrices constructed from the full dataset, thus demonstrating their stability.
Figure 3.

Hierarchical clustering dendrogram depicting relationships between alpha/beta tcrBLOSUM matrices constructed using the full dataset or either of the two data subsets (SC2neg and SC2only). The matrices formed two distinct chain-specific clusters, alpha chain tcrBLOSUMs (green) and beta chain tcrBLOSUMs (orange), illustrating the predominant similarity between the matrices of the same class.
tcrBLOSUM results in higher cluster purity and consistency
After producing the tcrBLOSUM matrix, our subsequent objective was to assess whether this matrix enhances the identification of TCRs sharing the same cognate epitope. A common approach to annotating TCRs involves clustering them based on sequence similarity, as TCRs within the same cluster are likely to bind the same epitope. To quantify the similarity between two TCRs, we employed the TCRdist distance metric, which is the most widely used, performant, and interpretable method for TCR repertoire analysis.
To rigorously evaluate the performance of tcrBLOSUM on TCR data unseen during matrix construction, we made use of the two datasets described in the previous section. The first dataset named SC2neg was used as a source dataset to construct tcrBLOSUMb matrix, while the computation of the TCRdist distance metric and the subsequent TCR clustering were conducted on the second dataset named SC2only, which therefore served as a validation dataset. This approach enabled us to estimate the tcrBLOSUM performance on unseen TCR-epitope pairs not utilized in the matrix construction, ensuring a robust evaluation of its efficacy. The evaluation of tcrBLOSUM was conducted only for the beta chain matrix, as alpha chain subsets demonstrated noticeable variation, therefore compromising the accuracy of the test.
Our analysis demonstrated that using the TCRdist distance metric with tcrBLOSUMb improved the clustering accuracy of beta chain CDR3s compared to BLOSUM62. Notably, the ratio of test to baseline statistics consistently showed higher values for both purity and consistency of clusters when using tcrBLOSUMb (Table 2). This improvement is noteworthy because there is typically a trade-off between these two metrics, where higher purity often comes at the expense of lower consistency, and vice versa. An example of clusters specific to the same epitope, generated with either tcrBLOSUMb or BLOSUM62, is provided in Table S5. Upon further examination, clusters of size 2 were found to constitute 50% of all clusters in both cases. Among these, 72 epitopes were shared between the two methods, with each method also identifying clusters for 20 unique epitopes that were not detected by the other. In the clusters specific to these 20 unique epitopes, the CDR3 sequences in the tcrBLOSUMb clusters exhibited a higher median TCRdist distance and lower median physicochemical similarity than the CDR3s in the BLOSUM62 clusters (Table S6).
Table 2.
Clustering performance on the validation (SC2only) dataset.
| SC2neg tcrBLOSUM | BLOSUM62 | |||||
|---|---|---|---|---|---|---|
| metrics | test | baseline | test/baseline ratio | test | baseline | test/baseline ratio |
| retention | 0.703 | 0.703 | 1.000 | 0.726 | 0.726 | 1.000 |
| purity | 0.743 | 0.287 | 2.588 | 0.729 | 0.294 | 2.482 |
| consistency | 0.095 | 0.021 | 4.546 | 0.094 | 0.021 | 4.408 |
The values have been rounded to three decimal places.
To illustrate how a similar approach could be beneficial for TCR repertoire analysis, we applied this workflow to two previously published TCR repertoires [46]: one from a critically ill COVID-19 patient and one from a non-critical COVID-19 patient (Supplementary material 1). When standard database matching was used to identify SARS-CoV-2-specific TCRs, both patients exhibited similarly low repertoire breadth devoted to the virus (Table S7). However, after combining TCRs from SARS-CoV-2-specific clusters uniquely identified with BLOSUM62 and tcrBLOSUM, the repertoire breadths clearly aligned with the original publication's findings derived from repertoire depth [46]. Specifically, the non-critical patient displayed markedly higher proportion of unique SARS-CoV-2-specific TCR sequences than the critical patient (Table S7). Expanding the number of identified virus-specific TCRs enabled clearer differentiation between the repertoire breadths of these individuals without considering their repertoire depth.
tcrBLOSUM identifies ‘unseen’ epitope-specific TCRs with more diverse AA composition and physicochemical profile than BLOSUM62
To further explore the advantages provided by utilizing tcrBLOSUMb for epitope annotation of TCRs, we aimed to investigate epitope-specific CDR3s that could be identified using either tcrBLOSUMb or BLOSUM62. We opted for an alignment-based approach that would not require subsequent clustering, thereby eliminating the potential effects of the interplay between distance metric and clustering method on the performance.
As a TCR repertoire can be scanned to find TCRs similar to a TCR with known epitope-specificity, the SC2only data subset (search space) was scored against a query CDR3 (qCDR3) to find the most similar CDR3s as detailed in Fig. 4. To this end, every CDR3 beta in SC2only dataset was aligned against all other CDR3s in the dataset using the Needleman-Wunsch (NW) algorithm with one of the AA substitution matrices (Fig. 4, steps 1–2). In the case of tcrBLOSUMb, the matrix constructed on the SC2neg data subset was used, adhering to the aforementioned approach of having independent source and validation datasets. The NW alignment score provided a measure of similarity between a qCDR3 with known specificity and ‘unlabelled’ CDR3s, where the epitope-specificities were masked. Higher NW scores indicated higher similarity between TCRs, which should be a proxy for shared epitope specificity. CDR3s which were most similar to a qCDR3 and were identified exclusively with either of the compared substitution matrices (step 5), termed unique true positive (TP) CDR3 hits, were further examined. This section aimed to investigate the distinctions between TP CDR3 hits derived from BLOSUM62 and tcrBLOSUM matrices, rather than assessing method performance, given that the identification of epitope-specific TCRs typically involves more advanced approaches than simple sequence alignment.
Figure 4.

Workflow used to identify and evaluate epitope-specific TCRs uniquely identified by aligning CDR3s from search space to query CDR3 (qCDR3, purple) using Needleman-Wunsch (NW) algorithm with either BLOSUM62 (blue) or tcrBLOSUMb (orange). SC2only dataset was used for evaluation, while tcrBLOSUMb was constructed on a SC2neg dataset. (1) qCDR3 and search space are selected from the dataset. (2) CDR3s from search space are aligned against qCDR3 using NW-algorithm with either BLOSUM62 (blue) or tcrBLOSUMb (orange). (3) aligned CDR3s with the highest NW-scores are retained. (4) CDR3s that recognize different epitope than qCDR3 are removed from retained top-scoring CDR3s, therefore only true positive (TP) CDR3s are labelled as hits. (5) to compare TP CDR3 hits identified exclusively with BLOSUM62 or tcrBLOSUMb (unique TP CDR3 hits), overlapping TP CDR3 hits are filtered out. (6) similarity metrics are computed between qCDR3 and unique TP CDR3 hits derived with BLOSUM62/tcrBLOSUMb. (7) similarity metrics of BLOSUM62-derived unique TP CDR3 hits and tcrBLOSUMb-derived unique TP CDR3 hits are compared.
TP CDR3 hits uniquely identified with BLOSUM62 or tcrBLOSUMb were found for 85% (25656) and 84% (25314) of qCDR3s, respectively. On average, for every qCDR3, slightly more TP CDR3 hits were identified exclusively with BLOSUM62 (median = 68 (4.5%), range = [1–1321] ([0.1–45]%)) than with tcrBLOSUMb (median = 57 (3.8%), range = [1–1389] ([0.1–50]%)). When the alignment query used the PhysChemSim or TopoSim matrices, the number of unique TP CDR3 hits decreased by at least two-fold compared to those with BLOSUM62, with a median value being no greater than 30 unique TP CDR3 hits per query for any of the three matrices. These findings illustrate that with BLOSUM62 predominantly reflecting biochemical similarities between AAs, a higher degree of overlap occurs between TP CDR3 hits identified with BLOSUM62 versus PhysChemSim or TopoSim matrices. Conversely, tcrBLOSUMb specifically captures AA usage patterns unique to CDR3b, leading to the identification of a larger number of non-overlapping epitope-specific TCRs.
To further evaluate the resemblance of unique TP CDR3 hits identified with BLOSUM62 versus tcrBLOSUMb to their respective qCDR3s, we measured their closeness in length, AA composition, and physicochemical properties (step 6). We observed that although BLOSUM62 worked with a wider range of CDR3 lengths than tcrBLOSUMb, the most common length difference between qCDR3 and TP hits was −1 and 0, respectively (Fig. S7). As can be seen in Fig. 5-a, −b, TP CDR3s discovered using tcrBLOSUMb exhibited greater edit (Levenshtein) distance (median = 8, max = 18) and TCRdist distance (median = 72, max = 198) to the qCDR3 compared to those identified with BLOSUM62 (edit distance: median = 6, max = 16; TCRdist distance: median = 56, max = 170). Similarly, physicochemical similarity to qCDR3 (Fig. 5-c) was higher for CDR3s identified with BLOSUM62 (median = 22, max = 77) than tcrBLOSUMb (median = 4, max = 57). An example of the query and detected TP CDR3s can be found in Fig. 5-d. These findings indicate the ability of tcrBLOSUMb to capture previously overlooked epitope-specific CDR3s characterized by more diverse AA composition and physicochemical profiles, which makes them more challenging to detect with traditional methods.
Figure 5.

(a-c) Distributions of the metrics used to evaluate the resemblance of unique TP CDR3 hits, identified either with BLOSUM62 (blue) or with tcrBLOSUM (orange), to their respective qCDR3s. (a) Edit (Levenshtein) distance; (b) TCRdist distance; and (c) physicochemical similarity demonstrated that tcrBLOSUM-derived unique TP CDR3 hits were characterized by more diverse AA composition and physicochemical profiles. (d) Examples of unique TP CDR3 hits identified with BLOSUM62 versus tcrBLOSUM, which demonstrate a smaller sequence identity in the case of tcrBLOSUM. Amino acids found in the query CDR3 and in both groups of unique TP CDR3 hits are in orange, while AAs found in the query CDR3 and only in BLOSUM62-derived unique TP CDR3 hits are in blue.
Discussion
One of the challenges on the way to solving the code of TCR-pMHC interactions is the immense diversity of TCRs, where even TCRs with dissimilar sequences can have the same cognate epitope [22]. The solution to this problem entails considering four distinct groups of CDR3 sequences: CDR3s with similar and dissimilar AA compositions and physicochemical profiles, either of similar or dissimilar lengths. Existing methods excel at identifying CDR3s with similar lengths and AA compositions, leveraging metrics like edit (Levenshtein) distance [47–52] and the TCRdist distance [43, 49, 53–55]. However, these methods struggle with CDR3s of dissimilar lengths, AA composition, and physicochemical profiles.
In this study, we utilized tcrBLOSUMb and BLOSUM62 with TCRdist distance followed by clustering or with the classical Needleman-Wunsch alignment algorithm to find CDR3 sequences similar in epitope-recognition space. Substituting the general-purpose BLOSUM62 matrix with the CDR3-specific tcrBLOSUMb matrix, we correctly identified CDR3s with identical epitope specificity but with dissimilar AA compositions and physicochemical profiles. We believe the grouping of distant epitope-specific TCRs became possible because tcrBLOSUM captures the actual AA variability observed across epitope-specific TCRs, thus avoiding the restrictions imposed by the physicochemical similarity encoded in BLOSUM62.
The standard BLOSUM62 matrix has produced distinct sets of epitope-specific CDR3s compared to substitution matrices based on physicochemical properties, such as PhysChemSim. With these findings, we would like to highlight the complementarity of tcrBLOSUM and BLOSUM62 matrices, as well as the importance of selecting the appropriate substitution matrix based on the research objective. tcrBLOSUM, designed to capture observed AA substitutions in CDR3s, is particularly effective at identifying epitope-specific TCRs that share little sequence identity. In contrast, BLOSUM62 is more suited for finding biochemically similar CDR3s. Thus, combining the results from these two complementary matrices will enable identification of both similar and dissimilar epitope-specific TCRs. This dual approach can increase the proportion of disease-specific TCRs extracted during TCR repertoire analysis. The increased sensitivity of in silico TCR-epitope annotations would enhance the resolution with which disease-specific TCR clones can be monitored, particularly with respect to their breadth and depth. In turn, this would improve tracking of responding clones during [56] and after an infection [57], and after vaccination [58]. Finally, knowing epitope specificity of a TCR from its sequence is essential for designing long-lasting vaccines [59] and engineering TCRs for personalized T-cell therapy [60].
Another advantage of the specialized AA substitution matrix is that it offers some insight into general rules of TCR-epitope interaction which become reflected in the substitution scores. Surprisingly, most of the substitutions in tcrBLOSUMs are neutral. Although this could be reflective of true interaction patterns, we lean towards a different explanation: non-participation of most CDR3 AAs in epitope binding. Since for most CDR3-epitope pairs, it is not known which AAs in a CDR3 form a bond with which AAs in a cognate epitope, these interaction patterns were not considered when the matrix was constructed. Observed AA pairs between non-interacting AAs add noise, resulting in more neutral tcrBLOSUM matrices.
Despite predominantly neutral scores, AAs that have a significant proportion of non-zero, particularly negative, scores could point towards the relative importance of specific residues for epitope binding. Remarkably, AAs identified as conserved in tcrBLOSUMb are often located within 4 Å of the epitope in crystal structures of TCR-pMHC complexes [5], suggesting their crucial role in successful interactions. Furthermore, out of three highly conserved AAs we identified in tcrBLOSUMb (Y, Q, F), two (Y, Q) were among the 20% of residues most frequently involved in TCR-pMHC binding, as reported in a study published in July 2024 [4]. Additionally, certain scores hint that, in some interactions, size might be more important than biochemical similarity. For example, although threonine (T) and glutamine (Q) both have polar uncharged side chains, their size difference may have contributed to an unfavorable substitution score. Conversely, histidine (H), phenylalanine (F), and tyrosine (Y) have positive substitution scores, possibly due to their shared bulky ring structure, despite being from different biochemical classes.
The alignment of our findings with crystal structures indicates that examining the locations of conserved AAs in CDR3s, along with their neighboring residues, could provide some insight into potential epitope-interaction sites and epitope-binding solutions. This could ultimately enhance our understanding of the key principles underlying productive epitope recognition by diverse TCRs.
The study exhibits several limitations that warrant consideration. Given that epitope-specific TCRs tend to have inherent biases, the data employed in this study may not adequately represent the entire TCR-epitope landscape. This is particularly due to the fact that most of the publicly available TCR-epitope data is concentrated around the 3–6 most common MHC alleles [33], which could introduce bias into the resulting tcrBLOSUMs. The absence of a threshold for CDR3 sequence identity within TCR blocks may also affect AA substitution scores in tcrBLOSUM matrices due to the inclusion of highly (dis)similar sequences. Currently, CD4 and CD8 TCRs were combined into a single matrix, as the AA frequencies were highly similar. However, prior studies have noted slight AA preferences between T-cell lineages [61].
Finally, the accuracy and representativeness of the matrices could be improved if ground truth data were available regarding the specific positions and AAs that actively participate in epitope binding. Such data would also allow the evaluation of the contribution of the two TCR chains to the interaction with an epitope, accounting for potential synergistic effects. Presented limitations could reduce the application potential of the matrices, highlighting the need for reliable and extensive ground truth data as well as continued refinement and validation of methodologies in TCR repertoire analysis.
In conclusion, we presented tcrBLOSUMa and tcrBLOSUMb, specialized AA substitution matrices tailored to AA frequencies and variations occurring within epitope-specific TCRs. Analysis of these matrices revealed a notable tolerance for any AA substitution in the majority of the AAs of alpha and beta CDR3s. Distinct conservation patterns observed for aliphatic and aromatic AAs within CDR3a and CDR3b, respectively, suggest their higher relative importance for epitope binding. Lastly, tcrBLOSUMs enable accurate alignment of CDR3 sequences with dissimilar AA compositions and physicochemical properties. As a result, by accounting for distant CDR3s, tcrBLOSUMs offer the potential to enhance TCR repertoire analysis and epitope-annotation of TCRs in various therapeutic applications.
Key Points
We present tcrBLOSUMa and tcrBLOSUMb, specialized BLOSUM-style AA substitution matrices consistent with the AA frequencies and variations occurring within CDR3 alpha and CDR3 beta sequences of epitope-specific TCRs, respectively.
tcrBLOSUMs differ noticeably from the standard BLOSUM62, revealing that both CDR3 alpha and CDR3 beta are tolerant to a wide range of AA substitutions.
Constructed tcrBLOSUMs proved to be stable towards new epitopes.
Application of tcrBLOSUMb instead of BLOSUM62 improved clustering performance and enabled the identification of epitope-specific TCRs with more diverse AA compositions and physicochemical profiles.
Utilizing both general BLOSUM62 and specialized tcrBLOSUMs in computational tools that infer the epitope-specificity of TCRs will expand the number and diversity of detected TCRs that bind the same epitope, thereby enhancing TCR repertoire analysis.
Supplementary Material
Contributor Information
Anna Postovskaya, Adrem Data Lab, Department of Computer Science, University of Antwerp, Antwerp, Belgium; Antwerp Unit for Data Analysis and Computation in Immunology and Sequencing (AUDACIS), University of Antwerp, Antwerp, Belgium; Clinical Virology Unit, Department of Clinical Sciences, Institute of Tropical Medicine, Antwerp, Belgium.
Koen Vercauteren, Clinical Virology Unit, Department of Clinical Sciences, Institute of Tropical Medicine, Antwerp, Belgium.
Pieter Meysman, Adrem Data Lab, Department of Computer Science, University of Antwerp, Antwerp, Belgium; Antwerp Unit for Data Analysis and Computation in Immunology and Sequencing (AUDACIS), University of Antwerp, Antwerp, Belgium.
Kris Laukens, Adrem Data Lab, Department of Computer Science, University of Antwerp, Antwerp, Belgium; Antwerp Unit for Data Analysis and Computation in Immunology and Sequencing (AUDACIS), University of Antwerp, Antwerp, Belgium; Biomedical Informatics Research Network Antwerp (BIOMINA), University of Antwerp, Antwerp, Belgium.
Conflict of interest: KL and PM are co-founders, board directors, and shareholders of ImmuneWatch, an immunoinformatics company. ImmuneWatch had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Funding
This work was supported by the Research Foundation Flanders (FWO) [1S38723N to A.P.]; the Interuniversity Special Research Fund (iBOF) [‘Modulating Immunity and the Microbiome for Effective CRC Immunotherapy’ (MIMICRY) Project]; and the Flemish Government [‘Onderzoeksprogramma Artificiële Intelligentie (AI) Vlaanderen’ Program].
Data availability
Constructed alternative substitution matrices, including tcrBLOSUMs, are provided in online supplementary materials. The matrices as well as the data and code underlying this article are available on GitHub at https://github.com/apostovskaya/tcrBLOSUM and can be openly accessed. The datasets were derived from sources in the public domain: VDJdb [33] (https://vdjdb.cdr3.net/), McPAS [34] (http://friedmanlab.weizmann.ac.il/McPAS-TCR/), and ImmuneACCESS [35] (https://clients.adaptivebiotech.com/immuneaccess).
References
- 1. Jenkins MK, Chu HH, McLachlan JB. et al. On the composition of the preimmune repertoire of T cells specific for peptide-major histocompatibility complex ligands. Annu Rev Immunol 2010;28:275–94. 10.1146/annurev-immunol-030409-101253. [DOI] [PubMed] [Google Scholar]
- 2. Bradley P. Structure-based prediction of T cell receptor:Peptide-MHC interactions. Elife 2023;12:12. 10.7554/eLife.82813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ji H, Wang XX, Zhang Q. et al. Predicting TCR sequences for unseen antigen epitopes using structural and sequence features. Brief Bioinform 2024;25:bbae210. 10.1093/bib/bbae210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Karnaukhov VK, Shcherbinin DS, Chugunov AO. et al. Structure-based prediction of T cell receptor recognition of unseen epitopes using TCRen. Nature Computational Science 2024;4:510–21. 10.1038/s43588-024-00653-0. [DOI] [PubMed] [Google Scholar]
- 5. Leem J, De Oliveira SHP, Krawczyk K. et al. STCRDab: The structural T-cell receptor database. Nucleic Acids Res 2018;46:D406–12. 10.1093/nar/gkx971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jensen KK, Rantos V, Jappe EC. et al. TCRpMHCmodels: Structural modelling of TCR-pMHC class I complexes. Sci Rep 2019;9:1–12. 10.1093/sw/swae047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yin R, Ribeiro-Filho HV, Lin V. et al. TCRmodel2: High-resolution modeling of T cell receptor recognition using deep learning. Nucleic Acids Res 2023;51:W569–76. 10.1093/nar/gkad356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grazioli F, Mösch A, Machart P. et al. On TCR binding predictors failing to generalize to unseen peptides. Front Immunol 2022;13:1014256. 10.3389/fimmu.2022.1014256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Meysman P, Barton J, Bravi B. et al. Benchmarking solutions to the T-cell receptor epitope prediction problem: IMMREP22 workshop report. ImmunoInformatics 2023;9:100024. 10.1016/j.immuno.2023.100024. [DOI] [Google Scholar]
- 10. Hudson D, Fernandes RA, Basham M. et al. Can we predict T cell specificity with digital biology and machine learning? Nat Rev Immunol 2023;23:511–21. 10.1093/jnci/djae276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Meysman P, De Neuter N, Gielis S. et al. On the viability of unsupervised T-cell receptor sequence clustering for epitope preference. Bioinformatics 2019;35:1461–8. 10.1093/bioinformatics/bty821. [DOI] [PubMed] [Google Scholar]
- 12. Dash P, Fiore-Gartland AJ, Hertz T. et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature 2017;547:89–93. 10.1038/nature22383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Glanville J, Huang H, Nau A. et al. Identifying specificity groups in the T cell receptor repertoire. Nature 2017;547:94–8. 10.1038/nature22976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Weber A, Born J, Rodriguez MM. TITAN: T-cell receptor specificity prediction with bimodal attention networks. Bioinformatics 2021;37:i237–44. 10.1093/bioinformatics/btab294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jokinen E, Huuhtanen J, Mustjoki S. et al. Predicting recognition between T cell receptors and epitopes with TCRGP. PLoS Comput Biol 2021;17:e1008814. 10.1371/journal.pcbi.1008814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pham MDN, Nguyen TN, Tran LS. et al. epiTCR: A highly sensitive predictor for TCR–peptide binding. Bioinformatics 2023;39:btad284. 10.1093/bioinformatics/btad284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang H, Zhan X, Li B. GIANA allows computationally-efficient TCR clustering and multi-disease repertoire classification by isometric transformation. Nat Commun 2021;12:1–11. 10.3389/fimmu.2022.1014256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jensen MF, Nielsen M. NetTCR 2.2 - improved TCR specificity predictions by combining pan- and peptide-specific training strategies, loss-scaling and integration of sequence similarity. Elife 2024;12:RP93934. 10.7554/eLife.93934.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Biochemistry 1992;89:10915–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Altschul SF, Gish W, Miller W. et al. Basic local alignment search tool. J Mol Biol 1990;215:403–10. 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 21. Davis MM, Bjorkman PJ. T-cell antigen receptor genes and T-cell recognition. Nature 1988;334:395–402. 10.1016/j.jvoice.2024.09.033. [DOI] [PubMed] [Google Scholar]
- 22. Mayer A, Callan CG. Measures of epitope binding degeneracy from T cell receptor repertoires. Proc Natl Acad Sci U S A 2023;120:e2213264120. 10.1073/pnas.2213264120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yu YK, Altschul SF. The construction of amino acid substitution matrices for the comparison of proteins with non-standard compositions. Bioinformatics 2005;21:902–11. 10.1093/bioinformatics/bti070. [DOI] [PubMed] [Google Scholar]
- 24. Yu YK, Wootton JC, Altschul SF. The compositional adjustment of amino acid substitution matrices. Proc Natl Acad Sci U S A 2003;100:15688–93. 10.1073/pnas.2533904100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Trivedi R, Nagarajaram HA. Substitution scoring matrices for proteins - an overview. Protein Sci 2020;29:2150. 10.1002/pro.3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Keul F, Hess M, Goesele M. et al. PFASUM: A substitution matrix from Pfam structural alignments. BMC Bioinformatics 2017;18:1–14. 10.1186/s12859-017-1703-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim Y, Sidney J, Pinilla C. et al. Derivation of an amino acid similarity matrix for peptide:MHC binding and its application as a Bayesian prior. BMC Bioinformatics 2009;10:1–11. 10.1186/1471-2105-10-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shen W-J, Wong H-S, Xiao Q-W. et al. Towards a Mathematical Foundation of Immunology and Amino Acid Chains. arXiv 2012, 1205.6031. 10.48550/arXiv.1205.6031. [DOI]
- 29. Rios S, Fernandez MF, Caltabiano G. et al. GPCRtm: An amino acid substitution matrix for the transmembrane region of class a G protein-coupled receptors. BMC Bioinformatics 2015;16:1–11. 10.1186/s12859-015-0639-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Trivedi R, Nagarajaram HA. Amino acid substitution scoring matrices specific to intrinsically disordered regions in proteins. Sci Rep 2019;9:1–12. 10.1021/acssynbio.4c00542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ng PC, Henikoff JG, Henikoff S. PHAT: A transmembrane-specific substitution matrix. Bioinformatics 2000;16:760–6. 10.1093/bioinformatics/16.9.760. [DOI] [PubMed] [Google Scholar]
- 32. Müller T, Rahmann S, Rehmsmeier M. Non-symmetric score matrices and the detection of homologous transmembrane proteins. Bioinformatics 2001;17:S182–9. 10.1093/bioinformatics/17.suppl_1.S182. [DOI] [PubMed] [Google Scholar]
- 33. Goncharov M, Bagaev D, Shcherbinin D. et al. VDJdb in the pandemic era: A compendium of T cell receptors specific for SARS-CoV-2. Nat Methods 2022;19:1017–9. 10.17116/neiro20248804122. [DOI] [PubMed] [Google Scholar]
- 34. Tickotsky N, Sagiv T, Prilusky J. et al. McPAS-TCR: A manually curated catalogue of pathology-associated T cell receptor sequences. Bioinformatics 2017;33:2924–9. 10.1093/bioinformatics/btx286. [DOI] [PubMed] [Google Scholar]
- 35. Nolan S, Vignali M, Klinger M. et al. A Large-Scale Database of T-Cell Receptor Beta (TCRβ) Sequences and Binding Associations from Natural and Synthetic Exposure to SARS-CoV-2. Res Sq, 2020, rs.3.rs-51964. 10.21203/rs.3.rs-51964/v1. [DOI] [Google Scholar]
- 36. Hellberg S, Sjostrom M, Skagerberg B. et al. Peptide quantitative structure-activity relationships, a multivariate approach. J Med Chem 1987;30:1126–35. 10.1021/jm00390a003. [DOI] [PubMed] [Google Scholar]
- 37. Rogers D, Hahn M. Extended-connectivity fingerprints. J Chem Inf Model 2010;50:742–54. 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- 38. RDKit: Open-source cheminformatics . http://www.rdkit.org/.
- 39. Carracedo-Reboredo P, Liñares-Blanco J, Rodríguez-Fernández N. et al. A review on machine learning approaches and trends in drug discovery. Comput Struct Biotechnol J 2021;19:4538. 10.1016/j.csbj.2021.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bajusz D, Rácz A, Héberger K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J Chem 2015;7:1–13. 10.1186/s13321-015-0069-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Virtanen P, Gommers R, Oliphant TE. et al. SciPy 1.0: Fundamental algorithms for scientific computing in python. Nat Methods 2020;17:261–72. 10.1038/s41592-019-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Daily J. Parasail: SIMD C library for global, semi-global, and local pairwise sequence alignments. BMC Bioinformatics 2016;17:1–11. 10.1186/s12859-016-0930-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mayer-Blackwell K, Schattgen S, Cohen-Lavi L. et al. TCR meta-clonotypes for biomarker discovery with tcrdist3 enabled identification of public, HLA-restricted clusters of SARS-CoV-2 TCRs. Elife 2021;10:68605. 10.7554/eLife.68605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pedregosa F, Varoquaux G, Gramfort A. et al. Scikit-learn: Machine learning in python. The Journal of Machine Learning Research 2011;12:2825–30. 10.7554/eLife.68605. [DOI] [Google Scholar]
- 45. Bateman A, Martin MJ, Orchard S. et al. UniProt: The universal protein knowledgebase in 2023. Nucleic Acids Res 2023;51:D523–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Postovskaya A, Vujkovic A, Block T. et al. Leveraging T-cell receptor – Epitope recognition models to disentangle unique and cross-reactive T-cell response to SARS-CoV-2 during COVID-19 progression/resolution. Front Immunol 2023;14:1130876. 10.3389/fimmu.2023.1130876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Høye E, Dagenborg VJ, Torgunrud A. et al. T cell receptor repertoire sequencing reveals chemotherapy-driven clonal expansion in colorectal liver metastases. Gigascience 2022;12:1–11. 10.1093/gigascience/giad032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huisman W, Hageman L, Leboux DAT. et al. Public T-cell receptors (TCRs) revisited by analysis of the magnitude of identical and highly-similar TCRs in virus-specific T-cell repertoires of healthy individuals. Front Immunol 2022;13:851868. 10.3389/fimmu.2022.851868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Miho E, Yermanos A, Weber CR. et al. Computational strategies for dissecting the high-dimensional complexity of adaptive immune repertoires. Front Immunol 2018;9:330957. 10.3389/fimmu.2018.00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dahal-Koirala S, Risnes LF, Neumann RS. et al. Comprehensive analysis of CDR3 sequences in gluten-specific T-cell receptors reveals a dominant R-motif and several new minor motifs. Front Immunol 2021;12:639672. 10.3389/fimmu.2021.639672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Smith NP, Ruiter B, Virkud YV. et al. Identification of antigen-specific TCR sequences based on biological and statistical enrichment in unselected individuals. Insight 2021;6:6. 10.1172/jci.insight.140028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Madi A, Poran A, Shifrut E. et al. T cell receptor repertoires of mice and humans are clustered in similarity networks around conserved public CDR3 sequences. Elife 2017;6:6. 10.7554/eLife.22057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chiffelle J, Genolet R, Perez MA. et al. T-cell repertoire analysis and metrics of diversity and clonality. Curr Opin Biotechnol 2020;65:284–95. 10.1016/j.copbio.2020.07.010. [DOI] [PubMed] [Google Scholar]
- 54. Olson BJ, Schattgen SA, Thomas PG. et al. Comparing T cell receptor repertoires using optimal transport. PLoS Comput Biol 2022;18:e1010681. 10.1371/journal.pcbi.1010681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chronister WD, Crinklaw A, Mahajan S. et al. TCRMatch: Predicting T-cell receptor specificity based on sequence similarity to previously characterized receptors. Front Immunol 2021;12:640725. 10.3389/fimmu.2021.640725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. DeWitt WS, Emerson RO, Lindau P. et al. Dynamics of the cytotoxic T cell response to a model of acute viral infection. J Virol 2015;89:4517–26. 10.1128/JVI.03474-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Luo L, Liang W, Pang J. et al. Dynamics of TCR repertoire and T cell function in COVID-19 convalescent individuals. Cell Discovery 2021;7:1–17. 10.1038/s41598-024-78498-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pogorelyy MV, Minervina AA, Touzel MP. et al. Precise tracking of vaccine-responding T cell clones reveals convergent and personalized response in identical twins. Proc Natl Acad Sci U S A 2018;115:12704–9. 10.1073/pnas.1809642115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bravi B. Development and use of machine learning algorithms in vaccine target selection. npj Vaccines 2024;9:1–14. 10.3390/jcm13196016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Klebanoff CA, Chandran SS, Baker BM. et al. T cell receptor therapeutics: Immunological targeting of the intracellular cancer proteome. Nat Rev Drug Discov 2023;22:996–1017. 10.1038/s41573-023-00809-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li HM, Hiroi T, Zhang Y. et al. TCRβ repertoire of CD4+ and CD8+ T cells is distinct in richness, distribution, and CDR3 amino acid composition. J Leukoc Biol 2016;99:505–13. 10.1189/jlb.6A0215-071RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Constructed alternative substitution matrices, including tcrBLOSUMs, are provided in online supplementary materials. The matrices as well as the data and code underlying this article are available on GitHub at https://github.com/apostovskaya/tcrBLOSUM and can be openly accessed. The datasets were derived from sources in the public domain: VDJdb [33] (https://vdjdb.cdr3.net/), McPAS [34] (http://friedmanlab.weizmann.ac.il/McPAS-TCR/), and ImmuneACCESS [35] (https://clients.adaptivebiotech.com/immuneaccess).