

Abstract
Background
20(S)-Ginsenoside Rh2 (GRh2) has been extensively studied for multifaceted health benefits. However, the anti-melanoma effect of GRh2 remains poorly understood. Herein, the anti-melanoma effects and underlying mechanisms of GRh2 were investigated.
Methods
MTT assays, the EdU staining assay, flow cytometric analysis, the cellular thermal shift assay (CETSA), confocal microscope analysis, molecular docking, molecular dynamics (MD), immunoblotting, a B16F10 cell bearing mouse model were adopted to examine the anti-melanoma effect of mechanism of action of GRh2.
Results
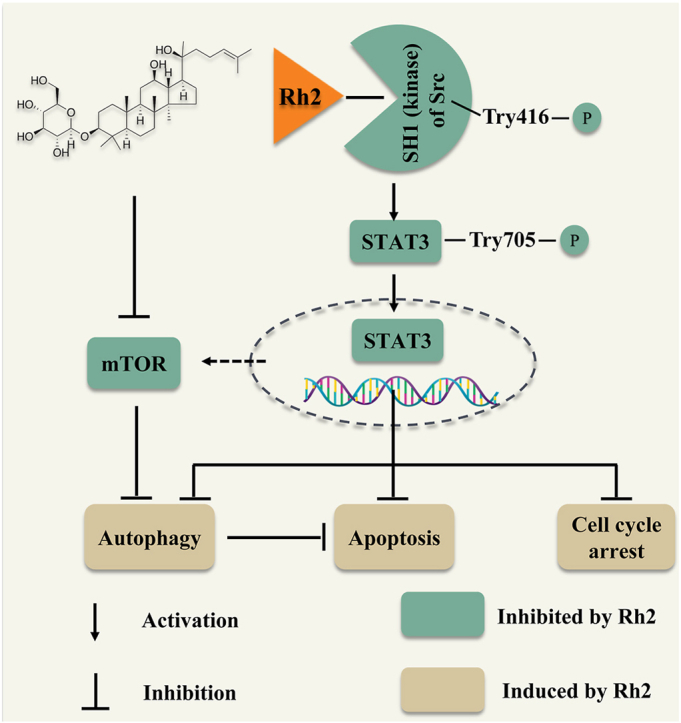
In melanoma cells, GRh2 was found to suppress cell proliferation, arrest cell cycle at G0/G1 phase and evoke apoptosis. GRh2 initiated autophagy and inhibited the activity of mTOR, the autophagy negative regulator, in melanoma cells. Repressing autophagy enhanced the anti-melanoma efficacy of GRh2. Molecular docking, MD and CETSA studies revealed that GRh2 stably bound to Src protein (one of the upstream kinases of STAT3). GRh2 suppressed Src and STAT3 activities, thereof prohibiting STAT3 nuclear translocation in melanoma cells. STAT3 over-activation attenuated the cytotoxic, apoptotic and autophagy inductive effects of GRh2. Additionally, GRh2 suppressed B16F10 tumor growth without inducing obvious toxicity in mice. It downregulated phospho-Src, phospho-STAT3, phospho-mTOR and Mcl-1 protein levels, while elevated cleaved-PARP and LC3B-II protein levels in B16F10 tumors.
Conclusion
GRh2 exerts anti-melanoma effects through suppressing Src/STAT3 signaling. This study advances our understanding on the anti-melanoma mechanism of GRh2 and indicates that the intake of GRh2 has the potential to retard melanoma progression.
Keywords: 20(S)-Ginsenoside Rh2, Melanoma, Apoptosis, Autophagy, Src/STAT3 signaling
Graphical abstract
1. Introduction
Melanoma is highly aggressive [1], which causes a formidable challenge to public health. The options for treating melanoma include surgery, radiotherapies, immunotherapies, targeted therapies and chemotherapies. Although these therapies could effectively inhibit melanoma progression, their clinical performance is not satisfactory. Because of the disadvantages of these therapeutic approaches, including severe side effects and drug resistance, the prognosis of patients with melanoma is still poor [2], which underscore an urgent need to search alternative strategies for melanoma treatment.
Ginseng (Panax ginseng) is commonly used as a functional food and medicine in Asia. Saponins derived from ginseng root are commonly used as herbal medicines and dietary supplements [3]. 20(S)-Ginsenoside Rh2 (GRh2), a bioactive triterpenoid saponin of ginseng, has potential cytotoxicity against various cancer cells, including breast, colorectal, cervical and pancreatic cancer cells [4]. Nevertheless, the anti-melanoma effects of GRh2 remain elusive.
Signal transducer and activator of transcription 3 (STAT3), a transcriptional factor, is abnormally activated in melanoma tissues. It acts a vital role in melanoma development [5]. STAT3 undergoes activation/phosphorylation at Tyr705 through the activation of non-receptor tyrosine kinases (like Src). STAT3 phosphorylation at Tyr705 causes its nuclear translocation and genes transcription [6]. STAT3 also plays a pivotal role in manipulating autophagy in various cancers [7]. Autophagy is initially activated to maintain cell survival and establish cellular adaptation under stressful conditions by destroying harmful cellular components. However, the role of autophagy in cancers, including melanoma, is complicated, which may be related to the different cancer stages [8]. Growing evidence reveals the double edge consequences of autophagy regulation in melanoma treatments [8], highlighting the necessity of clarifying the role of autophagy in developing novel anti-melanoma agents. Overall, inhibiting STAT3 activation and manipulating autophagy have been proposed as attractive strategies for melanoma treatment.
In this study, we found that GRh2 shows anti-melanoma effect and suppressing Src/STAT3 pathway is partially involved in this effect.
2. Materials and methods
2.1. Reagents and chemicals
Dacarbazine (DTIC) was bought from MedChenExpress company (NJ, USA). Dimethyl sulfoxide (DMSO), rapamycin (Rapa), bafilomycin A1 (Baf-A1), and Chloroquine (CQ) were bought from Sigma-Aldrich (MO, USA). The Cell-Light™ EdU Apollo®567 kit was bought from Ribobio company (Guangzhou, China) GRh2 (Purity≥98 % as determined by HPLC analysis, Fig. 5A) was obtained from Chengdu Alfa Bio-Technology Co. Ltd (Chengdu, China).
Fig. 5.

GRh2 exerts anti-melanoma effects via suppressing Src/STAT3 signaling. (A) GRh2's chemical structure. (B) Three-dimensional binding mode of GRh2 (green) in complex with Src. (C) Surface crystal structure of Src-GRh2 complex at 0 ns (left panels) and 100 ns (right panels). (D) RMSD of heavy atoms of unbound Src (blue) and the Rh2-Src complex (red). (E) CETSA for the binding of GRh2 to Src protein in melanoma cell lysate. ##P < 0.01 vs. DMSO treatment. (F) GRh2 prohibits Src/STAT3 activation in melanoma cells. (G) GRh2 lowers nuclear STAT3 protein level in melanoma cells. (H) Immunofluorescence assay was performed to analyze the effects of GRh2 on STAT3 nuclear translocation in B16F10 and A375 cells. Cells were incubated with GRh2 for 24 h, then immunofluorescence of STAT3 was labeled with green, and the nuclei was labeled with DAPI. Scale bar, 25 μm. (I) STAT3 over-activation attenuated the inhibitory effects of GRh2 on A375 cell viability. (J) Effects of GRh2 on LC3B and PARP protein expression in empty vector and STAT3C transfected A375 cells. ∗∗P < 0.01 vs. control group. #P < 0.05, ##P < 0.01.
2.2. Cell culture
A375, B16F10 and L929 cells bought from ATCC were grown in DMEM containing 10 % FBS and 1 % penicillin/streptomycin at 37 °C with 5 % CO2.
2.3. Cell viability assays
Considering that murine B16F10 cells produce melanin that could disturb the absorbance detection, crystal violet staining assay was employed to detect the cytotoxic effects of GRh2 on B16F10 cells. The cytotoxic effect of GRh2 against A375 cells were examined using the MTT assays. A375 (5 × 103) seeded in 96-well plate and B16F10 cells (2 × 105 cells/well) seeded in 6-well plate were treated with GRh2 (0–60 μM) for 24 or 48 h [9]. A375 cells were then incubated with 5 mg/ml of MTT for 2 h and then dissolved with DMSO. The absorbance was detected using a microplate reader. B16F10 cells were fixed. Then cells were stained with 0.5 % crystal violet before taking photograph.
2.4. Colony formation and 5-ethynyl-2′-deoxyuridine (EdU) staining assays
For EdU staining assay, 4 × 104 melanoma cells seeded in 4-well plates were exposed to GRh2 for 24 h. Melanoma cell proliferation was detected by an EdU assay kit following the manufacturer's instruction. For colony formation assay, 400 melanoma cells seeded in 6-well plate were treated with GRh2 for 24 h. After replacing the medium, cells were allowed to grow for another 7 days [10].
2.5. Flow cytometric analysis
Melanoma cells were exposed to GRh2 (0–40 μM) for 24 h before harvested. For cell cycle analysis, the cells were fixed using cold 70 % ethanol. On the following day, cells were incubated with propidium iodide (PI)/RNase A staining buffer (Thermofisher, MA, USA). For apoptotic analysis, an Apoptosis Detection Kit (BD, USA) was employed to stain the cells following the manufactory's instruction. Flow cytometry (BD, USA) was employed for data acquisition and further analysis.
2.6. Subcellular fractionation
Melanoma cells grown in 90-mm dishes were treated with GRh2 for 24 h before collecting. A NE-PER™ Nuclear and Cytoplasmic Extraction Reagents Kit (Thermofisher, MA, USA) was adopted to separate the cytoplasmic and nuclear protein following the manufacturer's instructions.
2.7. Molecular docking and molecular dynamics (MD)
To explore the binding mechanism of Src (PDB ID: 1YOL) and GRh2 (Pubchem CID: 119307), molecular docking was conducted using Autodocktools 1.5.6. MD simulations were performed using YASARA as previously described [11].
2.8. Cellular thermal shift assay (CETSA)
B16F10 and A375 cells were lysed using radioimmunoprecipitation (RIPA) containing phenylmethylsulfonyl fluoride (PMSF), phosphatase and protease inhibitors before centrifugation. The supernatants were obtained and then exposed to GRh2 (80 μM) or DMSO for half an hour at 25 °C. Thereafter, lysates were heated at 37, 41, 45, 49, 53 and 57 °C for 5 min. These samples were centrifuged after cooling down and then subjected to immunoblotting [11].
2.9. Immunoblotting analysis
Proteins from cultured cells and tumors were extracted using RIPA buffer. Equal amount of proteins were employed to perform Western blotting as previously report [12]. The primary antibodies used in this study include LC3B (Cell Signaling Technology (CST), #2775), CDK6 (CST, #13331), Mcl-1 (CST, #4572), Bcl-xL (CST, #2764), Cyclin D1 (CST, #2978), Src (CST, #2108), phospho-Src (Tyr416) (CST, #6943), STAT3 (CST, #9139), phospho-STAT3 (Tyr705) (CST, #9145), Cyclin A2 (CST, #91500), CDK4 (CST, #12790), Poly (ADP-Ribose) Polymerase (PARP) (CST, #9542), CDK2 (CST, #2546), phospho-mTOR (Ser2448) (CST, #5536), mTOR (CST, #2983) and β-actin (Santa Cruz, CA, USA, 1:3000). The dilution ratio of antibodies obtained from Cell Signaling Technology was 1:1000.
2.10. LC3 puncta analysis
A375 and B16F10 cells were transfected with the Premo™ autophagy tandem sensor RFP-GFP-LC3B kit (Invitrogen, USA) for 24 h and then exposure to GRh2 for another 24 h. Thereafter, cell images were obtained using laser confocal microscope (Leica, Germany) [10].
2.11. Immunofluorescence analysis
A375 and B16F10 cells (2 × 104 cells/well) were seeded in 4-well plates and then treated with GRh2 (10, 20, and 40 μM) for 24 h. Thereafter, the cells come to fixation of 4 % PFA for 10 min at room temperature, followed by permeabilization (0.1 % Triton X-100) and blocking (5 % BSA). Subsequently, the cells were co-incubated with an anti-STAT3 antibody (diluted at 1:100) at 4 °C overnight. Then cells were incubated with a homologous secondary antibody marked with fluorescence (Alex 251 Fluor@488) for 1 h in the dark. Finally, cell nuclei were labeled with DAPI (diluted in PBS, 1:1000) for 5 min. Images were captured under an immunofluorescence microscopy (Nikon, Japan).
2.12. STAT3 overactivation in A375 cells
The establishment of STAT3 overactivated A375 cells were described in a previous study [9].
2.13. In vivo studies
B16F10 cells were suspended in PBS (1 × 106/100 μl). The cells were subcutaneously injected into the flank of eight-week-old C57BL/6 mice which were obtained from the Chinese University of Hong Kong. After injection, mice were randomly divided into five groups according to their body weight, namely vehicle control (i.p solvent), DTIC (i.p 50 mg/kg, positive group) or GRh2 (i.p 20, 40 or 60 mg/kg) every day for 15 days. The vehicle solvent includes PBS, 5 % Tween-80 and 5 % PEG-400. Mice tumor volumes and body weights were measured on day 1, 7, 10, 13 and 15. After last dosing, all mice were euthanized using excessive 5 % isoflurane. The removed tumors were photographed and weighed. All animal procedure was approved by the Department of Health, Hong Kong [No.: (22–147) in DH/HT&A/8/2/6 Pt.6].
2.14. Statistical analysis
All data were shown as means ± SD in triplicate. The results were analyzed using GraphPad Prism 9 software (USA). Statistical comparisons between two groups were examined by student's t-test. Multiple comparisons were examined using one-way ANOVA with Dunnett's test. P < 0.05 were considered as statistically significant.
3. Results
3.1. GRh2 inhibits melanoma cell proliferation
MTT and crystal violet results in Fig. 1A and B demonstrate that GRh2 reduced A375 and B16F10 cell viabilities, respectively. We used normal mouse fibroblast L929 cell line to evaluate the cytotoxic effect of GRh2 in normal cells. MTT results showed that the cytotoxic effect of GRh2 against L929 cells were lower than that in melanoma cells (Fig. S1). To determine whether GRh2 suppresses melanoma cell proliferation, the EdU staining and colony formation assays were performed. We detected that GRh2 evidently reduced the ratio of EdU positive cells (Fig. 1C) and the number of colonies in melanoma cells (Fig. 1D).
Fig. 1.

GRh2 inhibits melanoma cell proliferation. GRh2 reduces the viabilities of (A) A375 and (B) B16F10 cells. (C) GRh2 decreases the proportion of EdU positive melanoma cells. (D) GRh2 reduces melanoma cell colony number. ∗P < 0.05, ∗∗P < 0.01 vs. control group.
3.2. GRh2 arrests melanoma cell cycle at G0/G1 phase
In comparison to control group, the proportion of G0/G1 phase was significantly raised in GRh2 treated-B16F10 and -A375 cells. Meanwhile, ratio of cells distributed in S phase was decreased (Fig. 2A). Immunoblotting results demonstrated that GRh2 downregulated levels of G0/G1 cell cycle regulated proteins, including CDK2, CDK4, CDK6, Cyclin A2 and Cyclin D1 (Fig. 2B).
Fig. 2.

GRh2 arrests melanoma cell cycle at G0/G1 phase. (A) Effects of GRh2 on cell cycle progression in melanoma cells. (B) Western blot analysis for G0/G1 phase cell cycle arrest related protein expression. ∗∗P < 0.01 vs. control group.
3.3. GRh2 evokes apoptosis in melanoma cells
As shown in Fig. 3A, GRh2 markedly elevated the proportion of apoptotic cells in B16F10 and A375 cells. PARP cleavage is considered to be a prominent marker of apoptosis. Additionally, the pro-apoptotic proteins (e.g. Bax) and anti-apoptotic proteins (e.g. Bcl-XL and Mcl-1) have also been identified to play a vital role in apoptosis. We found that GRh2 significantly elevated cleaved-PARP and Bax levels, whereas downregulated the antiapoptotic regulators Bcl-XL and Mcl-1 protein levels, in melanoma cells (Fig. 3B). However, GRh2 did not induce the cleavage of PARP in L929 cells (Fig. S2), highlighting that GRh2 did not trigger apoptosis in normal cells.
Fig. 3.

GRh2 evokes apoptosis in melanoma cells. (A) GRh2 increases the proportion of apoptotic cells. (B) GRh2 alters PARP, Bcl-XL, BAX and Mcl-1 protein expression in melanoma cells. ∗P < 0.05, ∗∗P < 0.01 vs. control group.
3.4. GRh2 initiates autophagy in melanoma cells
To examine whether GRh2 affects autophagy in melanoma cells, LC3B protein level was examined using Western blot analysis. In comparison to DMSO treated cells, a significant increment of LC3B-II protein expression was observed in GRh2 treated B16F10 and A375 cells, indicating that GRh2 initiates autophagosome formation in melanoma cells (Fig. 4A). Apart from autophagosome formation, autophagic flux blockage also results in the increase of LC3B-II protein level [13]. We, therefore, explored whether GRh2 initiates or suppresses autophagic flux in melanoma cells. In comparison to GRh2 treatment alone, autophagy inhibitors CQ (Fig. 4B) and Baf-A1 (Fig. 4C) pretreatment further upregulated LC3B-II protein expression in GRh2-treated B16F10 and A375 cells, implying that GRh2 induces autophagic flux in melanoma cells.
Fig. 4.

GRh2 initiates cytoprotective autophagy in melanoma cells. (A) Effect of GRh2 on LC3B-II protein expression in melanoma cells. (B and C) Immunoblotting analysis of LC3B protein expression in melanoma cells following GRh2 treatment with or without (B) CQ (25 μM) and (C) Baf-A1 (25 nM) pretreatment. (D) GRh2 promotes autophagic flux in B16F10 and A375 cells. scale bar = 25 μm. (E) GRh2 lowered phospho-mTOR protein level in melanoma cells. Viabilities of GRh2-plus-CQ or -Baf-A1-treated B16F10 (F) and A375 (G) cells. ∗∗P < 0.01 vs. control group. #P < 0.05, ##P < 0.01.
A tandem RFP-GFP-LC3B plasmid was transiently transfected into melanoma cells to confirm whether GRh2 promotes autophagic flux. Results in Fig. 4D show that the average red fluorescence puncta in GRh2-treated B16F10 and A375 cells were markedly increased, compared with that in DMSO-treated group, highlighting that GRh2 induces autophagy in melanoma cells. Meanwhile, Rapa (positive control), an autophagy inducer, also caused to a significant rise in red fluorescent spots number in melanoma cells (Fig. 4D). mTOR is a canonically negative regulator of autophagy. In this study, we found that GRh2 lowered p-mTOR protein level in melanoma cells (Fig. 4E).
We next studied the relationship between GRh2-induced autophagy and melanoma cell death. Our results showed that blocking autophagy further decreased viabilities of GRh2 treated B16F10 and A375 cells (Fig. 4F). Immunoblotting results showed that the GRh2 and CQ (Fig. 4B) or Baf-A1 (Fig. 4C) cotreatment markedly upregulated cleaved-PARP protein expression in melanoma cells, when compared with GRh2 treatment alone, indicating that GRh2 induces protective autophagy in melanoma cells (Fig. 4B and C). Blocking autophagy could further improve the anti-melanoma efficacy of GRh2.
3.5. Inhibiting Src/STAT3 signaling contributes to GRh2-induced cell death and autophagy in melanoma cells
STAT3 is a downstream survival cascades of Src in melanoma cells [14]. To determine whether Src is a molecular target of GRh2, molecular docking was determined to analyze the interplay between Src and GRh2. Our results showed that GRh2 fitted into the hydrophobic fissure provided by SER-347, GLU-312, PHE-309 and GLN-311 of Src protein, suggesting that GRh2 binds to the SH1 (acting as a kinase domain) domain of Src protein. The binding energy was −8.79 kcal/mol (Fig. 5A and B). MD simulation was employed to further confirm the results of molecular docking. The surface visualization models of the Src-GRh2 complex demonstrated that GRh2 stably remained at the center of Src binding site until the end of the 100 ns simulation (Fig. 5C). All atoms root-mean-square deviation (RMSD) track of Src mildly fluctuated at about 2.0–3.0 Å during 0–100 ns, and the RMSD track of unbound Src fluctuated at approximately 1.1 Å during MD simulation (Fig. 5D).
To verify the binding activity of GRh2 to Src protein in melanoma cells, CETSA was employed. GRh2 incubation resulted in an enhancement in the thermal polymerization temperature of Src from 41 °C to 49 °C in B16F10 cells, and from 49 °C to 57 °C in A375 cells, as compared to DMSO treatment, indicating that GRh2 enhanced the thermal stability of Src (Fig. 5E). Based on the above analyses, we can conclude that GRh2 directly and stably binds to Src in melanoma cells.
The SH1 domain plays a pivotal role in Src phosphorylation at Tyr416, which contributes to its active form [15]. To verify that GRh2 could suppress the kinase activity of Src, immunoblotting analyses were performed. Results in Fig. 5F showed that GRh2 prominently lowered phospho-Src (Tyr416) and phospho-STAT3 (Tyr705) protein levels in melanoma cells. Furthermore, in melanoma cells, GRh2 significantly lowered STAT3 nuclear protein level, while upregulated the cytoplasmic STAT3 protein level (Fig. 5G). Immunofluorescence analysis further confirmed that GRh2 inhibited the nuclear translocation of STAT3 in both A375 and B16F10 cells (Fig. 5H). To study the involvement of STAT3 suppression in GRh2-induced melanoma cell apoptosis and autophagy, A375 cells expressing overactivated STAT3 were used. MTT results showed that STAT3 overactivation markedly alleviated the cytotoxic effect of GRh2 against A375 cells (Fig. 5H). In comparison to GRh2-treated A375 cells transfected with empty vector, LC3B-II and cleaved-PARP protein expression was evidently lowered in GRh2-treated cells expressing overactivated STAT3, implying that suppressing STAT3 contributes to GRh2-induced apoptosis and autophagy (Fig. 5I).
3.6. GRh2 retards tumor growth in B16F10 cell-bearing mice
After 15 days of GRh2 treatment, a significant inhibitory effect on B16F10 tumor growth between GRh2 treated groups and control group was observed. GRh2 at 60 mg/kg demonstrated the strongest melanoma tumor growth suppressive effects among 20, 40 and 60 mg/kg. The average tumor weights of 20, 40 and 60 mg/kg of GRh2 groups were decreased by 56.5 %, 78.1 % and 88.3 %, respectively, when compared with vehicle control group (Fig. 6B). In comparison to vehicle control group, after dosing for 9 days, the average tumor volume in GRh2 groups were apparently lowered (Fig. 6C). On the last day of the experiment, the average tumor volume of 20, 40 and 60 mg/kg GRh2-treated groups were reduced to 49.1 %, 21.6 % and 19.8 % of that of control groups. Additionally, DTIC (50 mg/kg, positive control) significantly impeded B16F10 tumor growth, evidenced by markedly reduced tumor weight and tumor volume compared to control group. No obvious toxicity was seen in clinical signs and necropsy for pivotal organs. There was no marked difference in body weight between GRh2 or DTIC treated groups and control group (Fig. 6D). Immunoblotting results demonstrated that GRh2 significantly lowered phospho-Src, phospho-mTOR and phospho-STAT3 protein levels, while upregulated cleaved-PARP and LC3B-II protein levels in B16F10 allografts (Fig. 6E). Overall, these findings suggest that GRh2 retards melanoma tumor growth in vivo and inhibits Src/STAT3 signaling in tumor tissues.
Fig. 6.

GRh2 impedes tumor growth in mice. (A) GRh2 decreases average tumor weights. (B) Images of removed tumors. (C) GRh2 reduced tumor volumes in mice. (D) GRh2 did not affect the body weights of mice. From (A) to (D), n = 6. (E) Effects of GRh2 on protein expression of Src, phospho-Src, STAT3, phospho-STAT3, PARP and LC3B in B16F10 allografts. ∗P < 0.05, ∗∗P < 0.01 vs. the vehicle control group.
4. Discussion
Ginseng is one of the globally best-selling natural products due to its prominent tonifying effects [16]. Ginsenosides have great potential in drug development for cancer treatment, either as a stand-alone agent or as an adjuvant drug [17]. Among the ginsenosides, GRh2 has shown potent anticancer property in several cancer models [18]. However, the anti-melanoma effects of GRh2 are rarely studied. Here, we showed that GRh2 suppresses cell proliferation of, arrests cell cycle at G0/G1 phase, evokes apoptosis, and promotes autophagy in, melanoma cells. Moreover, GRh2 evidently restricted B16F10 tumor growth. In line with previous studies, no obvious toxicities were observed following GRh2 treatment [19], indicating that GRh2 is a safe and effective medication for treating melanoma.
Autophagy in cancer progression and treatment is critical and complicated, either cytoprotective or cytotoxic [20]. Therefore, the role of autophagy for cancer therapy development should be considered rigorously [21]. Here, we found that GRh2 induced autophagic flux in melanoma cells. mTOR negatively regulates autophagy [22]. We showed that GRh2 significantly lowered phospho-mTOR protein level in melanoma cells, corroborating the notion that GRh2 initiates autophagy in melanoma cells. Next, our findings show that inhibiting autophagy by autophagy inhibitors further decreased the viabilities of, while increased apoptosis in, GRh2-treated melanoma cells. These findings indicate that GRh2-initiated autophagy is cytoprotective in melanoma cells, thereof the combination of GRh2 and autophagy inhibitors may represent as a strategizing treatment modality for melanoma.
STAT3 activation, initiated by phosphorylation of Tyr705 residue, is related to cell survival, apoptosis resistance and cancer invasion and metastasis [23]. Moreover, persistent STAT3 activation occurs frequently in melanoma [24]. Therefore, STAT3 is commonly considered as an effective target for developing inhibitors for treating cancers [25]. The beneficial effects of STAT3 inhibitors have also been exploited. However, the severe side effects remain the greatest obstacle for the clinical application of STAT3 inhibitors [26]. Therefore, identification of safe and effective STAT3 inhibitors is of great significance. GRh2 restrains STAT3 activation in non-small-cell lung cancer [27] and liver cancer cells [28]. Results in our study show that GRh2 represses STAT3 activity in melanoma cells and allograft tumors. Moreover, GRh2 downregulated nuclear STAT3 protein level, as well as STAT3 targeted genes, including Bcl-xL and Mcl-1 (cell survival related genes [12]) and Cyclin D1 (G0/G1 phase cell cycle regulated gene), in melanoma cells. STAT3 overexpression attenuated the anti-melanoma effects of GRh2, implying the involvement of STAT3 suppression in the anti-melanoma effects of GRh2. Additionally, we revealed that GRh2 directly binds to Src, one of the upstream kinases of STAT3, with a strong binding affinity and suppresses its activation, suggesting that GRh2 suppresses STAT3 activation by directly binding to Src.
Shen et al., revealed that STAT3 inhibitors stimulate autophagy in cancer cells via disrupting STAT3-PKR interaction [29]. Wu et al., also indicated that STAT3 can negatively regulate autophagy via regulating Bcl2-Beclin 1 axis in cervical cancer cells [30]. The translocation of STAT3 into nucleus can transcriptionally regulate autophagy-related genes (PIK3C3, CTSB, Bcl-2, BECN1 and CTSL) expression [31]. In our study, we found that overactivation of STAT3 lowered LC3B-II protein expression in A375 cells.
However, our study has some limitations. Somatic mutations, such as BRAF, PTEN, NRAS and p53, are frequently detected in melanoma. Among them, BRAFV600E are the most recurrent mutation [32]. In addition to A375 (BRAFV600E), more melanoma cell lines with different genetic background, such as A2058 (BRAFV600E, p53V274F, PTENnull), Hs294T (BRAFwt, p53 wt), WM-115 (BRAFV600D), IGR1 (BRAFV600K) and SK-MEL-2 (NRASQ61R, p53S245G) will be employed in the future study to validate the anti-melanoma effects of GRh2. In addition, we detected that GRh2 markedly upregulated the protein level of LC3B-II in L929 cells (Fig. S2). Whether GRh2 induces autophagy in L929 cells will be investigated in future study. Additionally, the mechanisms underlying GRh2-mediated STAT3 inhibition and autophagy induction remains unclear which will be investigated in the future.
5. Conclusions
In summary, GRh2 exerts anti-melanoma effects partially by inhibiting Src/STAT3 signaling. Blocking autophagy augments the anti-melanoma efficacy of GRh2. Findings of this work advances our understanding on the anti-melanoma mechanism of GRh2 and indicates that the intake of GRh2 has the potential to retard melanoma progression.
Declaration of competing interest
All authors declare that there are no conflicts of interest.
Acknowledgments
This work was supported by Natural Science Foundation of Guangdong Province (2023A1515011076, 2021A1515012093 and 2021A1515010657) and National Natural Science Foundation of China (81903868).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jgr.2024.07.002.
Contributor Information
Bin Liu, Email: xmhoolv@163.com.
Ken-Kin-Lam Yung, Email: kklyung@eduhk.hk.
Pei-Li Zhu, Email: pellyzhu@hkbu.edu.hk.
Appendix A. Supplementary data
The following is the Supplementary data to this article.
References
- 1.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., et al. Global cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Curti B.D., Faries M.B. Recent advances in the treatment of melanoma. N Engl J Med. 2021;384(23):2229–2240. doi: 10.1056/NEJMra2034861. [DOI] [PubMed] [Google Scholar]
- 3.Ratan Z.A., Haidere M.F., Hong Y.H., Park S.H., Lee J.-O., Lee J., et al. Pharmacological potential of ginseng and its major component ginsenosides. J Ginseng Res. 2021;45(2):199–210. doi: 10.1016/j.jgr.2020.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu Q.Q., Pan Y., Wu H.C., Dai Z.Z., Huang Q.X., Luo T., et al. The ways for ginsenoside Rh2 to fight against cancer: the molecular evidences in vitro and in vivo. J Ginseng Res. 2022;47(2):173–182. doi: 10.1016/j.jgr.2022.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fu X.Q., Liu B., Wang Y.-P., Li J.-K., Zhu P.-L., Li T., et al. Activation of STAT3 is a key event in TLR4 signaling-mediated melanoma progression. Cell Death Dis. 2020;11(4):246. doi: 10.1038/s41419-020-2440-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhattacharya S., Ray R.M., Johnson L.R. STAT3-mediated transcription of Bcl-2, Mcl-1 and c-IAP2 prevents apoptosis in polyamine-depleted cells. Biochem J. 2005;392(Pt 2):335–344. doi: 10.1042/BJ20050465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu J., Zhang J., Mao Q.-F., Wu J., Wang Y. The interaction between autophagy and JAK/STAT3 signaling pathway in tumors. Front Genet. 2022;13 doi: 10.3389/fgene.2022.880359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Debnath J., Gammoh N., Ryan K.M. Autophagy and autophagy-related pathways in cancer. Nat Rev Mol Cell Biol. 2023;24(8):560–575. doi: 10.1038/s41580-023-00585-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J.K., Zhu P.L., Wang Y., Jiang X.L., Zhang Z., Zhang Z., et al. Gracillin exerts anti-melanoma effects in vitro and in vivo: role of DNA damage, apoptosis and autophagy. Phytomedicine. 2023;108 doi: 10.1016/j.phymed.2022.154526. [DOI] [PubMed] [Google Scholar]
- 10.Li J., Zhu P., Chen Y., Zhang S., Zhang Z., Zhang Z., et al. Isoalantolactone induces cell cycle arrest, apoptosis and autophagy in colorectal cancer cells. Front Pharmacol. 2022;13 doi: 10.3389/fphar.2022.903599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cai M., Xu Y.C., Deng B., Chen J.B., Chen T.F., Zeng K.F., et al. Radix Glycyrrhizae extract and licochalcone a exert an anti-inflammatory action by direct suppression of toll like receptor 4. J Ethnopharmacol. 2023;302 doi: 10.1016/j.jep.2022.115869. [DOI] [PubMed] [Google Scholar]
- 12.Siveen K.S., Sikka S., Surana R., Dai X., Zhang J., Kumar A.P., et al. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochim Biophys Acta Rev Cancer. 2014;1845(2):136–154. doi: 10.1016/j.bbcan.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 13.Parzych K.R., Klionsky D.J. An overview of autophagy: morphology, mechanism, and regulation. Antioxidants Redox Signal. 2014;20(3):460–473. doi: 10.1089/ars.2013.5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y.X., Xu B.W., Niu X.D., Chen Y.J., Fu X.Q., Wang X.Q., et al. Inhibition of Src/STAT3 signaling-mediated angiogenesis is involved in the anti-melanoma effects of dioscin. Pharmacol Res. 2022;175 doi: 10.1016/j.phrs.2021.105983. [DOI] [PubMed] [Google Scholar]
- 15.Boggon T.J., Eck M.J. Structure and regulation of Src family kinases. Oncogene. 2004;23(48):7918–7927. doi: 10.1038/sj.onc.1208081. [DOI] [PubMed] [Google Scholar]
- 16.Li Z., Wang Y., Xu Q., Ma J., Li X., Tian Y., et al. Ginseng and health outcomes: an umbrella review. Front Pharmacol. 2023;14 doi: 10.3389/fphar.2023.1069268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahuja A., Kim J.H., Kim J.H., Yi Y.-S., Cho J.Y. Functional role of ginseng-derived compounds in cancer. J Ginseng Res. 2018;42(3):248–254. doi: 10.1016/j.jgr.2017.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li X., Chu S.-F., Lin M., Gao Y., Liu Y., Yang S., et al. Anticancer property of ginsenoside Rh2 from ginseng. Eur J Med Chem. 2020;203 doi: 10.1016/j.ejmech.2020.112627. [DOI] [PubMed] [Google Scholar]
- 19.Zhang H., Yi J.K., Huang H., Park S., Kwon W., Kim E., et al. 20 (S)-ginsenoside Rh2 inhibits colorectal cancer cell growth by suppressing the Axl signaling pathway in vitro and in vivo. J Ginseng Res. 2022;46(3):396–407. doi: 10.1016/j.jgr.2021.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levy J.M.M., Towers C.G., Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17(9):528–542. doi: 10.1038/nrc.2017.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pangilinan C., Xu X., Herlyn M., Liang C. Autophagy Paradox: strategizing treatment modality in melanoma. Curr Treat Options Oncol. 2023;24(2):130–145. doi: 10.1007/s11864-023-01053-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan V.P., Miyamoto S. Nutrient-sensing mTORC1: Integration of metabolic and autophagic signals. J Mol Cell Cardiol. 2016;95:31–41. doi: 10.1016/j.yjmcc.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tolomeo M., Cascio A. The multifaced role of STAT3 in cancer and its Implication for anticancer therapy. Int J Mol Sci. 2021;22(2):603. doi: 10.3390/ijms22020603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu R., Zhang Y.G., Sun J. STAT3 activation in infection and infection-associated cancer. Mol Cell Endocrinol. 2017;451:80–87. doi: 10.1016/j.mce.2017.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong J., Cheng X.D., Zhang W.D., Qin J.J. Recent Update on development of small-molecule STAT3 inhibitors for cancer therapy: from phosphorylation inhibition to protein Degradation. J Med Chem. 2021;64(13):8884–8915. doi: 10.1021/acs.jmedchem.1c00629. [DOI] [PubMed] [Google Scholar]
- 26.Yang L., Lin S., Xu L., Lin J., Zhao C., Huang X. Novel activators and small-molecule inhibitors of STAT3 in cancer. Cytokine Growth Factor Rev. 2019;49:10–22. doi: 10.1016/j.cytogfr.2019.10.005. [DOI] [PubMed] [Google Scholar]
- 27.Sun X., Zhao P., Li H., Liu Y., Wang T., Cheng Y. Ginsenoside Rh2 inhibits Glycolysis through the STAT3/c-MYC Axis in non-small-cell lung cancer. JAMA Oncol. 2021;2021 doi: 10.1155/2021/9715154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y.S., Chen C., Zhang S.Y., Li Y., Jin Y.H. (20S) ginsenoside Rh2 inhibits STAT3/VEGF signaling by targeting Annexin A2. Int J Mol Sci. 2021;22(17):9289. doi: 10.3390/ijms22179289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shen S., Niso-Santano M., Adjemian S., Takehara T., Malik S.A., Minoux H., et al. Cytoplasmic STAT3 represses autophagy by inhibiting PKR activity. Mol Cell. 2012;48(5):667–680. doi: 10.1016/j.molcel.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 30.Wu L., Shen B., Li J., Zhang H., Zhang K., Yang Y., et al. STAT3 exerts pro-tumor and anti-autophagy roles in cervical cancer. Diagn Pathol. 2022;17(1):13. doi: 10.1186/s13000-021-01182-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y., Liao S., Bennett S., Tang H., Song D., Wood D., et al. STAT3 and its targeting inhibitors in osteosarcoma. Cell Prolif. 2021;54(2) doi: 10.1111/cpr.12974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Motwani J., Eccles M.R. Genetic and Genomic pathways of melanoma development, invasion and metastasis. Genes. 2021;12(10):1543. doi: 10.3390/genes12101543. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.