

Abstract
Background
Diffusion-weighted magnetic resonance imaging (DWI) is essential for diagnosing Creutzfeldt–Jakob disease (CJD). Thalamic lesions are rarely detected by DWI in sporadic CJD (sCJD) cases with methionine homozygosity at polymorphic codon 129 (129MM) of the prion protein (PrP) gene. Here, we describe an unusual sCJD case, characterized by prolonged isolated thalamic diffusion hyperintensities and atypical brain pathology, in combination with the 129MM genotype.
Case presentation
A 72-year-old Japanese man developed a mild unsteady gait that had persisted for 1 year. DWI revealed isolated thalamic diffusion hyperintensities. Over the following 4 years, his condition progressed to include ataxia and cognitive decline. Repeated cerebrospinal fluid tests were negative for 14-3-3 protein, total tau protein, and real-time quaking-induced conversion assay. Electroencephalography did not show periodic sharp wave complexes or generalized periodic discharges. Despite these findings, thalamic DWI abnormalities persisted and evolved to include cortical lesions in the later stage of the disease. Genetic testing confirmed a 129MM genotype with no pathogenic PrP gene variants. Brain autopsy identified type 2 pathogenic PrP and the absence of the M2-thalamic prion strain, suggesting an MM2-cortical (MM2C)-subtype of sCJD. Histopathology revealed small vacuoles (sv) and patchy-perivacuolar PrP deposits without large vacuoles (lv). Patchy-perivacuolar deposits are a characteristic feature of the MM2C (lv) subtype and indicate MM2C (lv) pathology. Thus, this case was classified as a rare MM2C (sv + lv) subtype. No PrP protein staining was observed in the thalamus, despite spongiform changes with small vacuoles.
Conclusions
This case underscores the diagnostic challenges of atypical CJD with isolated thalamic abnormalities on DWI. Despite negative cerebrospinal fluid findings and clinical diagnostic criteria, persistent DWI abnormalities and evolving clinical symptoms continued to raise suspicion of CJD. A definitive diagnosis, being the MM2C (sv + lv) subtype of sCJD, was confirmed upon pathological examination. Even when atypical findings, such as isolated thalamic abnormalities, are observed and various tests are negative, if suspicion of CJD cannot be ruled out, it is important to confirm the diagnosis and pathological subtypes via postmortem analysis.
Keywords: Creutzfeldt–Jakob disease, Diffusion-weighted imaging, Thalamus, Vacuolation, MM2-cortical-type
Background
Diffusion-weighted magnetic resonance imaging (DWI) is a type of brain magnetic resonance imaging (MRI) that can be used as an informative diagnostic tool for Creutzfeldt–Jakob disease (CJD). Thalamic lesions on DWI are characteristic of variant CJD [1], genetic CJD [2], and iatrogenic CJD [3]. In sporadic CJD (sCJD), thalamic lesions are frequently reported in cases with type 2 pathogenic prion protein (PrPSc) and a polymorphic codon 129 Met/Val (MV) or Val/Val (VV) genotype of the prion protein gene (PRNP). Thalamic lesions are rare with the Met/Met (MM) genotype [4, 5]. In addition, thalamic lesions usually appear alongside other lesions, such as those in the basal ganglia or cerebral cortices.
Here, we describe a case of sCJD with the 129MM genotype, who presented with isolated thalamic DWI hyperintense signals for > 1 year and atypical MM2-cortical (MM2C) brain pathology. This case suggests that persistent isolated thalamic DWI hyperintensities may be a potential indicator of atypical MM2C pathology in sCJD.
Case presentation
A 72-year-old Japanese man visited a local physician complaining of a mild unsteady gait for over 1 year. The patient had no history of head surgery (including dura mater transplantation), hormone injections, or travel to Europe, including the United Kingdom. Brain MRI revealed symmetric restricted diffusion abnormalities in the thalamus (Fig. 1A). At the time of transfer to our hospital, neurological examination did not reveal any significant ataxia, signs of pyramidal or extrapyramidal dysfunction, sensory disturbance, or cognitive impairment. Single-photon emission computed tomography (SPECT) with technetium-99 m (99mTc)-ethylcysteinate dimer (ECD) did not detect any notable hypoperfusion lesions, including in the thalamus (Fig. 1F).
Fig. 1.
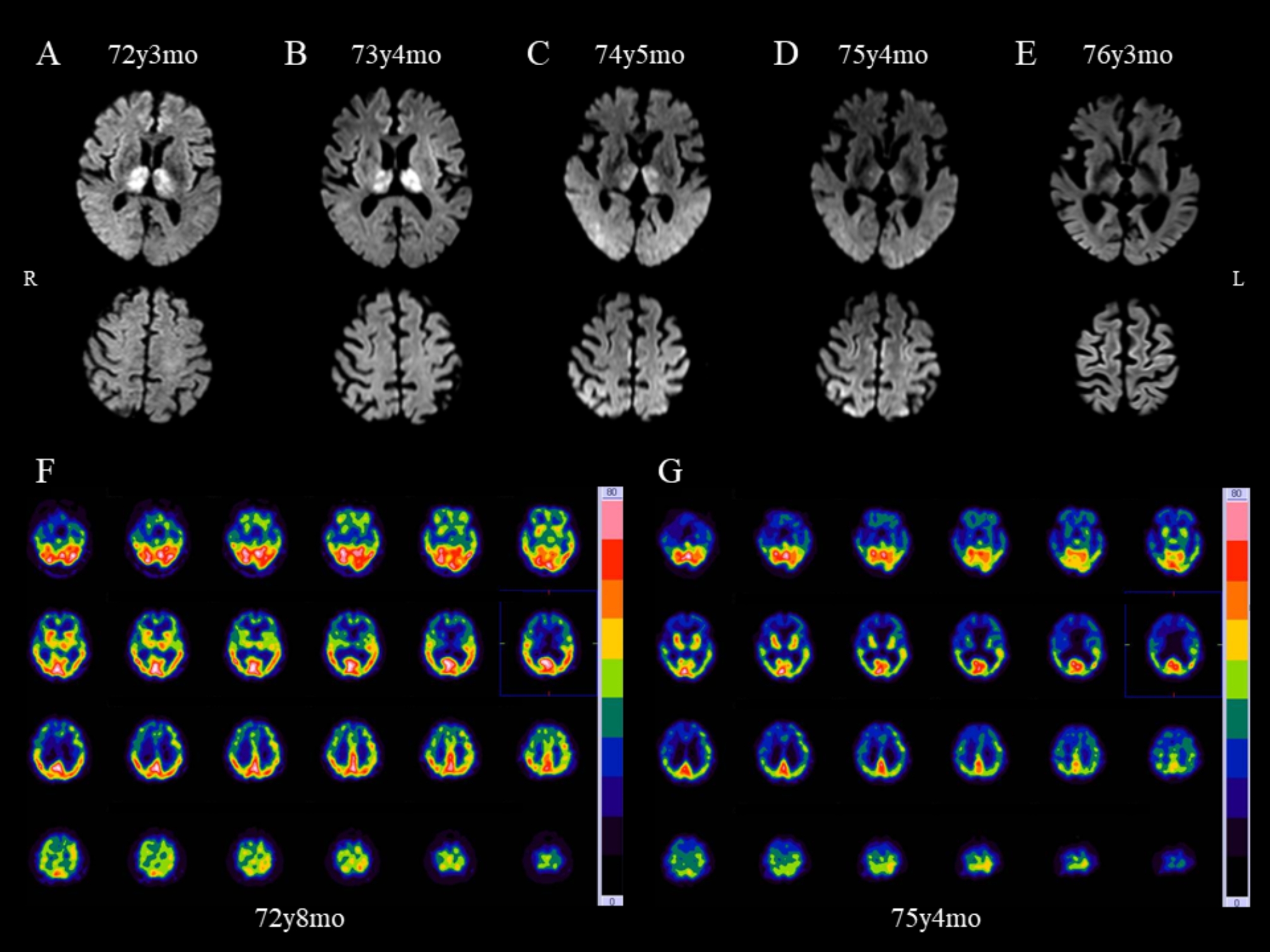
Brain imaging in an individual with Creutzfeldt–Jakob disease. Serial brain diffusion-weighted magnetic resonance imaging (DWI) scans (A–E) and technetium-99 m (99mTc)-ethylcysteinate dimer (ECD) single-photon emission computed tomography (SPECT) (F, G). Initial DWI performed 1 year after disease onset showed bilateral thalamic high signals with no cortical abnormalities (A). DWI conducted 2 years after disease onset exhibited findings similar to those in A (B). Three years after onset, DWI hyperintensities in the thalamus were less distinct and reduced in size (C upper); a few cortical lesions appeared (C lower). Four years (D) and 5 years (E) after onset, the thalamus showed atrophy and cortical hyperintensities became increasingly apparent over time. SPECT imaging in the early stage of the disease showed no thalamic hypoperfusion (F), whereas in the advanced stage, generalized hypoperfusion including in bilateral thalami was observed (G). The age of the individual is indicated on the images. y, years; mo, months
Approximately 2 years after the onset of gait disturbance, his tendency to fall progressively worsened, and the patient was admitted to our hospital at age 73. Neurological examination revealed apparent ataxic gait and dysarthria, with a Mini-Mental State Examination score of 27/30. DWI continued to show symmetrical hyperintense lesions limited to the thalamus, with no abnormalities in other areas (Fig. 1B). Electroencephalography (EEG) examination showed no periodic sharp wave complexes (PSWCs) or generalized periodic discharges (GPDs). Cerebrospinal fluid (CSF) was negative for 14-3-3 protein and total tau protein, and a second-generation real-time quaking-induced conversion (RT-QuIC) assay did not detect PrPSc. Genetic testing showed that the PRNP genotype at codons 129 and 219 were MM and Glu/Glu (EE), respectively, with no pathogenic variants identified.
By the age of 74, the patient’s cognitive decline had progressed, and he required assistance with eating and walking, although simple conversation was still possible. DWI showed that the thalamic high signal lesions were gradually smaller and less distinct, with the appearance of a few cortical lesions (Fig. 1C).
At 75 years and 4 months, approximately 4 years after onset, he became bedridden but could still respond to his name and answer questions by nodding. Re-examination of CSF showed that 14-3-3 protein and total tau protein levels remained normal, and the RT-QuIC assay continued to be negative. EEG revealed diffuse slow wave activity on a background pattern, with no PSWCs or GPDs. Thalamic high signal lesions became less distinct. Additionally, the thalamus showed atrophy and spreading cortical lesions (Fig. 1D). 99mTc-ECD SPECT showed marked hypoperfusion throughout the brain, including bilateral thalami (Fig. 1G).
At age 76 years and 2 months, approximately 5 years after onset, the patient was readmitted to hospital owing to urethral injury and fever. He was nearly in an akinetic mutism state but could open his eyes in response to verbal stimuli. Thalamic high signal lesions were almost undetectable by DWI, whereas the cortical lesions had become more prominent (Fig. 1E). The urethral injury led to the development of an iliopsoas abscess, which worsened his overall condition and resulted in his death 1 month later. A brain autopsy was performed after obtaining informed consent from his family.
The autopsy was conducted 62 h after death. The brain weighed 1010 g, and diffuse cortical atrophy was observed. Western blot analysis of protease K-resistant PrP [6] using the frontal and parietal lobes revealed a 19 kDa unglycosylated band corresponding to type 2 PrPSc. This band was positive using a type 2-specific antibody but was not detected using a type 1-specific antibody (Fig. 2 shows only the results from the frontal lobe). Protein misfolding cyclic amplification (PMCA) detected no significant signal for the M2-thalamic (M2T) prion strain, which indicated that this case should be classified as MM2C rather than MM2T [7].
Fig. 2.
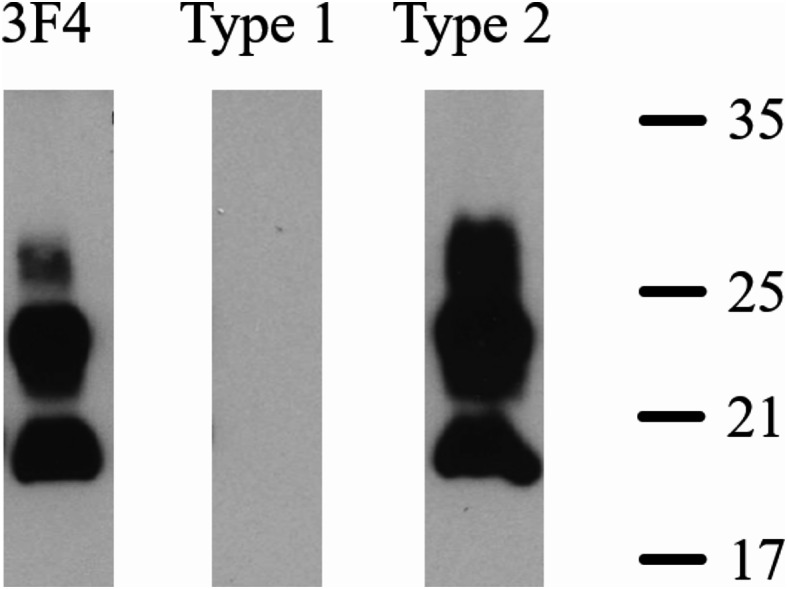
Western blotting for the prion protein subtype in frontal lobe tissue from an individual with Creutzfeldt–Jakob disease. Western blot analysis using the 3F4 antibody, T1 antibody (type 1 pathogenic prion protein [PrPSc]-specific), and T2 antibody (type 2 PrPSc-specific) [16]. Our case showed a 19 kDa unglycosylated band corresponding to type 2 PrPSc
Microscopic examination revealed widespread small vacuole (sv)-type spongiform changes (diameter ≤ 18 μm) [8] in the cerebral cortex with no large vacuoles (lv) (Fig. 3A). The degree of spongiform changes varied even within the same sections. The thalamus exhibited moderate spongiform changes (Fig. 3C), whereas the caudate nucleus showed mild gliosis (Fig. 3E). The cerebellum showed no pathological changes (Fig. 3G), and there was no neuronal loss in the inferior olivary nucleus (Fig. 3I). PrP immunostaining revealed diffuse synaptic-type PrP deposition and patchy-perivacuolar aggregates of PrP in the frontal lobe (Fig. 3B) and caudate nucleus (Fig. 3F), but not in the thalamus (Fig. 3D) or cerebellum (Fig. 3H). This case primarily corresponds to MM2C (sv), one of the pathological subtypes of MM2C sCJD, and is also considered to exhibit overlapping MM2C (lv) pathology owing to the presence of patchy-perivacuolar PrP deposition typically observed around large vacuoles [9, 10]. Therefore, the final pathological diagnosis of this case was MM2C (sv + lv).
Fig. 3.

Postmortem brain pathology in an individual with Creutzfeldt–Jakob disease. Microscopic findings of the frontal lobe (A, B), anterior thalamic nucleus (C, D), caudate nucleus (E, F), cerebellum (G, H), and inferior olivary nucleus (I) with hematoxylin and eosin staining (A, C, E and G), anti-prion protein (PrP) immunostaining using 3F4 antibody (B, D, F and H), and Klüver–Barrera staining (I)
Discussion and conclusions
Bilateral thalamic DWI hyperintensities can be observed in a range of disorders, including vascular diseases, metabolic disorders, inflammatory diseases, trauma, tumors, and infections [11]. In this case, based on various examinations and medical history, diseases other than CJD were deemed unlikely. However, a definitive diagnosis could not be made until pathological analysis because of the absence of myoclonus, lack of PSWCs or GPDs on EEG, and negative results for 14-3-3 protein, total tau protein, and the RT-QuIC test of the CSF.
In sCJD, partial thalamic DWI hyperintensities in the pulvinar, mediodorsal nuclei, or anterolateral nuclei are often observed in cases with V2 prion strains, i.e., MV2 or VV2 [5]. Although less pronounced than in cases with V2 strains, hyperintensities in the mediodorsal thalamic nuclei have been reported in cases of MM1 + 2 C and MM2C + T types of sCJD [12]. However, these cases typically present with abnormal signals confined to parts of the thalamus, accompanied by hyperintensities in the basal ganglia or cortex. Our case exhibited diffuse thalamic hyperintensities without additional cortical abnormalities until the late stage of the disease, which is atypical for the expected DWI findings for the MM2C subtype of sCJD. Moreover, the patient did not meet the recently proposed diagnostic criteria for this subtype, which include DWI findings confined to the cerebral cortex [13]. However, our case exhibited characteristics of the MM2C subtype, such as the absence of abnormalities in CSF and EEG tests [13]. Additionally, although the prolonged disease course aligned with features typical for the MM2C subtype, the duration of 5 years was longer than the average disease duration of 27.8 months for this subtype [13], which raises questions about whether this extended course is related to the presence of MM2C (sv) pathology. With further cases, a consensus may emerge that the presence of thalamic DWI hyperintensities does not exclude the diagnosis of the MM2C subtype, and our understanding of disease duration may become clearer.
In the typical MM2C subtype, which is characterized by lv pathology, onset usually presents as dementia or cortical symptoms [13]. In contrast, initial presentation with ataxia, as observed in our case, is more commonly observed in the MM2T subtype. Therefore, we considered the possibility of coexistence with the M2T prion strain. However, neuronal loss was not observed in the inferior olivary nucleus, which is typical of MM2T patients [6, 14]. Additionally, PMCA was used to selectively amplify the M2T prion strain [7], but no amplified products were detected. These results rule out the possibility of coexistence of M2T prion strain in this patient.
The MM2C (sv) subtype, identified as the primary pathological subtype in this case, is a newly recognized subtype. Therefore, knowledge regarding its clinical course and pathological findings remains limited. In this patient, the thalamic abnormalities on DWI from the early stage may have been related to the initial symptom of ataxia; however, definitive conclusions cannot be drawn from a single case. Therefore, additional cases of the MM2C (sv) subtype are needed to determine whether ataxia is a characteristic initial symptom, clarify the lesions responsible for this symptom, and assess the impact of thalamic lesions on phenotypes.
During the initial period when the DWI lesion was confined to the thalamus, we observed no obvious ataxia. Subsequently, ataxia progressed rapidly, while cognitive functions remained preserved. The signal intensity of the thalamus decreased over time, which is thought to reflect neuronal death and an increase in extracellular space [15]. Simultaneously, lesions in the cerebral cortex became predominant, which led to a decline in cognitive function, raising suspicion for a CJD diagnosis. Therefore, in cases where isolated thalamic lesions persist, frequent MRI imaging is important for both diagnosis and prediction of subsequent symptoms. Additionally, the persistence of isolated thalamic signal abnormalities may be a predictor of a prolonged disease course.
The pathological findings were also atypical for the MM2C subtype of sCJD. Characteristics of the reported MM2C pathological subtypes and those of the present case are summarized in Table 1. Generally, small vacuoles are considered histopathological features of the M1 prion strain, whereas large vacuoles are associated with the M2C prion strain. However, recently, MM2C with small vacuoles has been reclassified as MM2C (sv) [7]. Our case is compatible with MM2C (sv) pathology in terms of the presence of small vacuoles and characteristic synaptic-type PrP deposits (Table 1). If small vacuoles are observed in sCJD patients with the 129MM genotype, it is important to clarify whether the PrPSc type is type 1 or type 2 by Western blotting to distinguish M1 and M2C (sv) prion strains. Regarding large vacuole pathology, some reported MM2C (lv) cases showed patchy PrP deposits with a few perivacuolar deposits around large vacuoles [10]. Small vacuoles and large vacuoles can coexist, as described in a case of MM2C (sv + lv), featuring small vacuoles with synaptic-type PrP deposition and large vacuoles with perivacuolar PrP staining [7]. In our case, although large vacuoles were absent, patchy-perivacuolar PrP (typically found with large vacuoles) was observed, indicating MM2C (lv) pathology and leading to a final pathological diagnosis of MM2C (sv + lv). As such, M2C (sv) and M2C (lv) prions exhibit different pathologies, but it was not possible to distinguish between them by transmission properties because they showed similar transmission profiles when examined using various PrP knock-in mouse models [7]. Recent transmission data have emerged showing differing infectivity between M2C (sv) and M2C (lv) prions (Kitamoto, unpublished data). Additionally, despite the presence of spongiform changes with small vacuoles, no PrP staining was observed in the thalamus. The absence of PrP deposition in the thalamus might be influenced by postmortem changes but may also represent a characteristic finding related to long-term survival effects in this case.
Table 1.
Characteristic pathological subtypes of MM2-cortical-type sporadic creutzfeldt–jakob disease
| Classification | Prion | Pathological change | PrP deposition pattern |
|---|---|---|---|
| MM2C (lv) | M2C (lv) | Large vacuolation |
Patchy-perivacuolar PrP deposits |
| MM2C (sv) | M2C (sv) | Small vacuolation | Synaptic PrP deposits |
|
MM2C (lv + sv) (reported case (7)) |
M2C (lv) and M2C (sv) |
Large vacuolation and small vacuolation |
Perivascuolar and synaptic PrP deposits |
|
MM2C (lv + sv) (This case) |
M2C (lv) and M2C (sv) | Small vacuolation |
Patchy-perivacuolar and synaptic PrP deposits |
PrP, prion protein; lv, large vacuole; sv, small vacuole
Our case suggests that isolated thalamic diffusion hyperintensities could be an early finding of sCJD with MM2C (sv) pathology, highlighting its significance for diagnosis, prognosis, and infection control, while also reinforcing the necessity for postmortem analysis. The existence of atypical sCJD cases like our case emphasizes the need for increased awareness among physicians regarding the diverse phenotypes of sCJD and the development of more sensitive RT-QuIC assays or new diagnostic biomarkers, which would enable pre-mortem diagnoses of all sCJD cases and ensure thorough monitoring for infection prevention and management.
Acknowledgements
The authors thank the patient and his family for their collaboration. We thank Rachel James, PhD, from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
Abbreviations
- CJD
Creutzfeldt–Jakob Disease
- CSF
Cerebrospinal Fluid
- DWI
Diffusion-Weighted Magnetic Resonance Imaging
- ECD
Ethylcysteinate Dimer
- EEG
Electroencephalogram
- GPDs
Generalized Periodic Discharges
- (lv)
Large Vacuole
- MM2C
MM2-cortical
- MRI
Magnetic Resonance Imaging
- M2T
M2-Thalamic
- PMCA
Protein Misfolding Cyclic Amplification
- PRNP
Prion Protein Gene
- PrP
Prion Protein
- PrPSc
Pathogenic Prion Protein
- PSWCs
Periodic Sharp Wave Complexes
- RT-QuIC
Real-Time Quaking-Induced Conversion
- sCJD
sporadic CJD
- SPECT
Single-Photon Emission Computed Tomography
- (sv)
small vacuole
- 99mTc
Technetium-99 m
Author contributions
MK, HK, MT, MO, KA, HN, KT, SH, SK, MO, HT, and NU provided treatment to the patient and collected the data. HK and MT contributed to the acquisition and analysis of data. MK and KA performed the autopsy and collected data. KS performed CSF analysis. TK performed a pathological evaluation including Western blotting analysis and revised the manuscript. MK wrote the draft. HD and FT analyzed and interpreted the data and revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (M.K.: 21K07419), the Research Committee of Prion Disease and Slow Virus Infection, Health and Labour Sciences Research Grants, and a Grant-in-Aid from the Research Committee of Surveillance and Infection Control of Prion Disease from the Ministry of Health, Labour and Welfare of Japan (T.K. and F.T.).
Data availability
No datasets were generated or analysed during the current study.
Declarations
Ethics approval and consent to participate
Autopsy and gene analysis were performed after written informed consent was obtained from the family of the patient. The experimental protocol was approved by the Independent Review Board of Yokohama City University. All experiments were performed in accordance with institutional guidelines.
Consent for publication
Written informed consent was obtained from the family for publication.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Hiroshi Doi, Email: hdoi@yokohama-cu.ac.jp.
Fumiaki Tanaka, Email: ftanaka@yokohama-cu.ac.jp.
References
- 1.Collie DA, Summers DM, Sellar RJ, Ironside JW, Cooper S, Zeidler M, et al. Diagnosing variant Creutzfeldt–Jakob disease with the pulvinar sign: MR imaging findings in 86 neuropathologically confirmed cases. Am J Neuroradiol. 2003;24:1560–9. [PMC free article] [PubMed] [Google Scholar]
- 2.Kovacs GG, Seguin J, Quadrio I, Höftberger R, Kapás I, Streichenberger N, et al. Genetic Creutzfeldt-Jakob disease associated with the E200K mutation: characterization of a complex proteinopathy. Acta Neuropathol. 2011;121:39–57. [DOI] [PubMed] [Google Scholar]
- 3.Hamaguchi T, Sakai K, Kobayashi A, Kitamoto T, Ae R, Nakamura Y, et al. Characterization of sporadic Creutzfeldt-Jakob disease and history of neurosurgery to identify potential iatrogenic cases. Emerg Infect Dis. 2020;26:1140–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bizzi A, Pascuzzo R, Blevins J, Moscatelli MEM, Grisoli M, Lodi R, et al. Subtype diagnosis of sporadic Creutzfeldt–Jakob disease with diffusion magnetic resonance imaging. Ann Neurol. 2021;89:560–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meissner B, Kallenberg K, Sanchez-Juan P, Collie D, Summers DM, Almonti S, et al. MRI lesion profiles in sporadic Creutzfeldt–Jakob disease. Neurology. 2009;72:1994–2001. [DOI] [PubMed] [Google Scholar]
- 6.Kobayashi A, Iwasaki Y, Takao M, Saito Y, Iwaki T, Qi Z, et al. A novel combination of prion strain co-occurrence in patients with sporadic Creutzfeldt–Jakob disease. Am J Pathol. 2019;189:1276–83. [DOI] [PubMed] [Google Scholar]
- 7.Takeuchi A, Mohri S, Kai H, Tamaoka A, Kobayashi A, Mizusawa H, et al. Two distinct prions in fatal familial insomnia and its sporadic form. Brain Commun. 2019;1:fcz045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akagi A, Iwasaki Y, Mimuro M, Kitamoto T, Yamada M, Yoshida M. Pathological progression of genetic Creutzfeldt–Jakob disease with a PrP V180I mutation. Prion. 2018;12:54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baiardi S, Rossi M, Capellari S, Parchi P. Recent advances in the histo-molecular pathology of human prion disease. Brain Pathol. 2019;29:278–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gelpi E, Klotz S, Vidal-Robau N, Ricken G, Regelsberger G, Ströbel T, et al. Histotype-dependent oligodendroglial PrP pathology in sporadic CJD: a frequent feature of the M2C strain. Viruses. 2021;13:1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Renard D, Castelnovo G, Campello C, Bouly S, Le Floch A, Thouvenot E, et al. Thalamic lesions: a radiological review. Behav Neurol. 2014;2014:e154631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamada M. Research report of CJD Surveillance Committee of the Ministry of Health, Labour and Welfare. research report of CJD Surveillance Committee of the Ministry of Health, Labour and Welfare [Internet]. 2018. http://prion.umin.jp/file/survey_result_H29/H29sougou.pdf. Accessed 15 Jan 1999.
- 13.Hamaguchi T, Sanjo N, Ae R, Nakamura Y, Sakai K, Takao M, et al. MM2-type sporadic creutzfeldt–jakob disease: new diagnostic criteria for MM2-cortical type. J Neurol Neurosurg Psychiatry. 2020;91:1158–65. [DOI] [PubMed] [Google Scholar]
- 14.Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, et al. Classification of sporadic Creutzfeldt–Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224–33. [PubMed] [Google Scholar]
- 15.Eisenmenger L, Porter MC, Carswell C, Thompson A, Mead S, Rudge P, et al. Diffusion-weighted MRI signal abnormality in sporadic CJD increases in extent and intensity with disease duration. JAMA Neurol. 2016;73:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kobayashi A, Mizukoshi K, Iwasaki Y, Miyata H, Yoshida Y, Kitamoto T. Co-occurrence of types 1 and 2 PrPres in sporadic Creutzfeldt-Jakob disease MM1. Am J Pathol. 2011;178:1309–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No datasets were generated or analysed during the current study.