

Abstract
Bleeding correlates with disease severity in viral hemorrhagic fevers. We found that the increase in type I interferon (IFN-I) in mice caused by infection with the Armstrong strain of lymphocytic choriomeningitis virus (LCMV; an arenavirus) reduced the megakaryocytic expression of genes encoding enzymes involved in lipid biosynthesis (cyclooxygenase 1 and thromboxane A synthase 1) and a thrombopoietic transcription factor (Nf-e2). The decreased expression of these genes was associated with reduced numbers of circulating platelets and defects in the arachidonic acid synthetic pathway, thereby suppressing serotonin release from δ-granules in platelets. Bleeding resulted when severe thrombocytopenia and altered platelet function reduced the amount of platelet-derived serotonin below a critical threshold. Bleeding was facilitated by the absence of the activity of the kinase Lyn or the administration of aspirin, an inhibitor of arachidonic acid synthesis. Mouse platelets were not directly affected by IFN-I because they lack the receptor for the cytokine (IFNAR1), suggesting that transfusion of normal platelets into LCMV-infected mice could increase the amount of platelet-released serotonin and help to control hemorrhage.
INTRODUCTION
Viral hemorrhagic fevers (VHFs) caused by Arenaviridae, Bunyaviridae, Filoviridae, and Flaviviridae RNA viruses (1, 2) are a symptomatically diverse syndrome in which bleeding may vary from 82% to only 30% of confirmed cases, such as in Lassa (3) or Ebola virus infections (4, 5), respectively. The prognostic value of bleeding is debated (6) because only few patients have been adequately studied (5). In one report, all patients with hemorrhage died, but bleeding was not always the cause of death (4). Thrombocytopenia with endothelial, coagulation, and platelet dysfunction have been reported, but the relationship to bleeding remains undefined (7). The Armstrong (Arm) strain of the lymphocytic choriomeningitis virus (LCMV) is an arenavirus (8) that causes hemorrhagic anemia in platelet-depleted mice mediated by the type I interferon (IFN-I) receptor (which is encoded by Ifnar1) (9). LCMV-infected humans are mostly asymptomatic (10), but transplanted LCMV-infected organs can cause bleeding (11) with clinical manifestations that resemble Argentine hemorrhagic fever caused by the Junín virus (10, 12). Thus, mouse LCMV infection may model the pathogenesis of bleeding in humans infected by arenaviruses and other VHF viruses.
Here, we found that increased IFN-I production in LCMV-infected mice altered gene expression in bone marrow (BM) cells, including megakaryocytes (MKs), and reduced the amount of two key enzymes of the arachidonic acid (AA) and thromboxane A2 (TxA2) signaling pathway. Platelets generated by MKs exposed to IFN-I had impaired activation-induced release from storage granules, particularly dense (δ) granules, and decreased serotonin release. LCMV-infected mice bled when total released serotonin dropped below a critical threshold because of combined thrombocytopenia and reduced platelet activation. IFN-I concentrations equivalent to the highest measured in blood during LCMV infection (13) had no direct effect on platelet function, and hemorrhage could therefore be controlled by transfusion of normal platelets. Understanding this pathogenic mechanism may help improve laboratory evaluation and treatment of patients affected by VHF and potentially other viral infections.
RESULTS
MK stimulation through Ifnar1 causes platelet dysfunction and bleeding in mice
Exposure of human platelets to IFN-I is reported to result in functional impairment (14). However, human proplatelets and blood platelets lack the IFN-I receptor IFNAR1, which is found on MKs and progenitors; accordingly, platelets are not affected by exposure to IFN-α and IFN-β (15). We first confirmed in wild-type (WT) C57BL/6J (B6) mice that IFNAR1 is present on MKs and not on platelets (Fig. 1A) and that mouse IFN-α (300 ng/ml; 15.5 nM)—a high concentration measured in blood after LCMV infection (13)—did not affect platelet aggregation but enhanced expression of interferon-stimulated gene 15 (Isg15) in leukocytes (Fig. 1, B and C).
Fig. 1. IFNAR1 is not expressed on mouse platelets, and IFN-α does not affect platelet function.
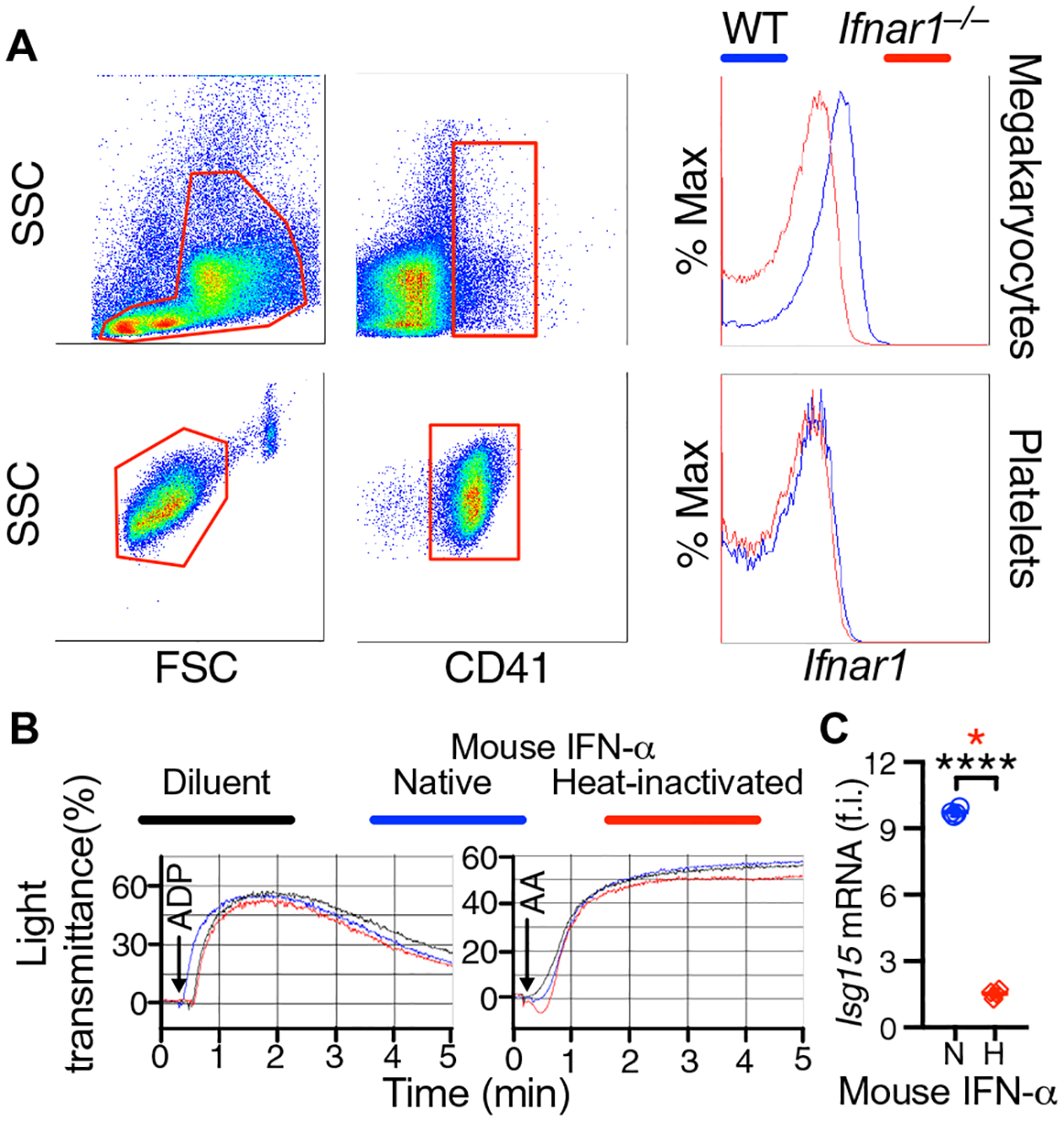
(A) Representative flow cytometric analysis of BM cells and platelets from WT and Ifnar1−/− mice (n = 3 mice per group). Red boxes delimit forward scatter (FSC) and side scatter (SSC) gates analyzed with anti-CD41 (integrin αIIb) and anti-IFNAR1 (IFN α/β receptor subunit 1) antibodies. CD41+ MKs, but not platelets, are Ifnar1+ in WT mice; Ifnar1−/− MKs and platelets are negative controls. (B) Representative WT mouse platelet aggregation (n = 4 mice per group) induced by ADP (left, 5 μM) or AA (right, 75 μM) after 2-hour incubation at room temperature (RT) with native (blue) or heat-inactivated (red) mouse IFN-α (300 ng/ml) or control diluent (black). (C) Effect of native (N) or heat-inactivated (H) mouse IFN-α on IFN-stimulated gene 15 (Isg15) mRNA levels in mononuclear cells from the blood tested in (B). Results (scatterplot with means ± SD; n = 4 mice per group) show fold induction (f.i.) relative to the housekeeping gene Gapdh. Statistical analysis: Two-tailed unpaired t test with Welch’s correction (no assumption of equal variance; black asterisks, ****P < 0.0001) and Mann-Whitney test (red asterisk, *P < 0.05).
This evidence must be reconciled with the role of platelets in the pathogenesis of lethal hemorrhage involving IFNAR1-dependent signaling in LCMV-infected mice (9). To address the problem, we infected Ifnar1−/− and WT mice cross-transplanted with BM cells of the opposite genotype with LCMV in the presence of platelet-depleting antibody (αPLT) (9, 16). In LCMV-infected WT mice, the platelet count decreased to ~200,000/μl by day 3 after infection, which coincided with the peak of viremia, and then started to recover and returned to nearly normal values in ~3 weeks (fig. S1, A and B). Hemorrhage and lethality depend on extreme severity of thrombocytopenia (9). All mice receiving αPLT had blood platelets ≤25,000/μl. However, only LCMV-infected and platelet-depleted WT and WTBM/Ifnar1−/− chimeras developed skin hemorrhage, positive fecal occult blood (FOB), and markedly decreased hematocrit (Fig. 2, A to C), and these mice eventually died between 4 and 7 days after infection (Fig. 2D). All Ifnar1−/− mice, as expected (9), and also all Ifnar1−/− BM/WT chimeras survived LCMV infection and platelet depletion (Fig. 2, A to D). Thus, these results suggest that a BM cell type that is responsive to IFN-I stimulation through IFNAR1 contributes to bleeding and lethality after platelet depletion in LCMV-infected mice.
Fig. 2. Bleeding in LCMV-infected mice is mediated by IFNAR1 expressed on BM cells.
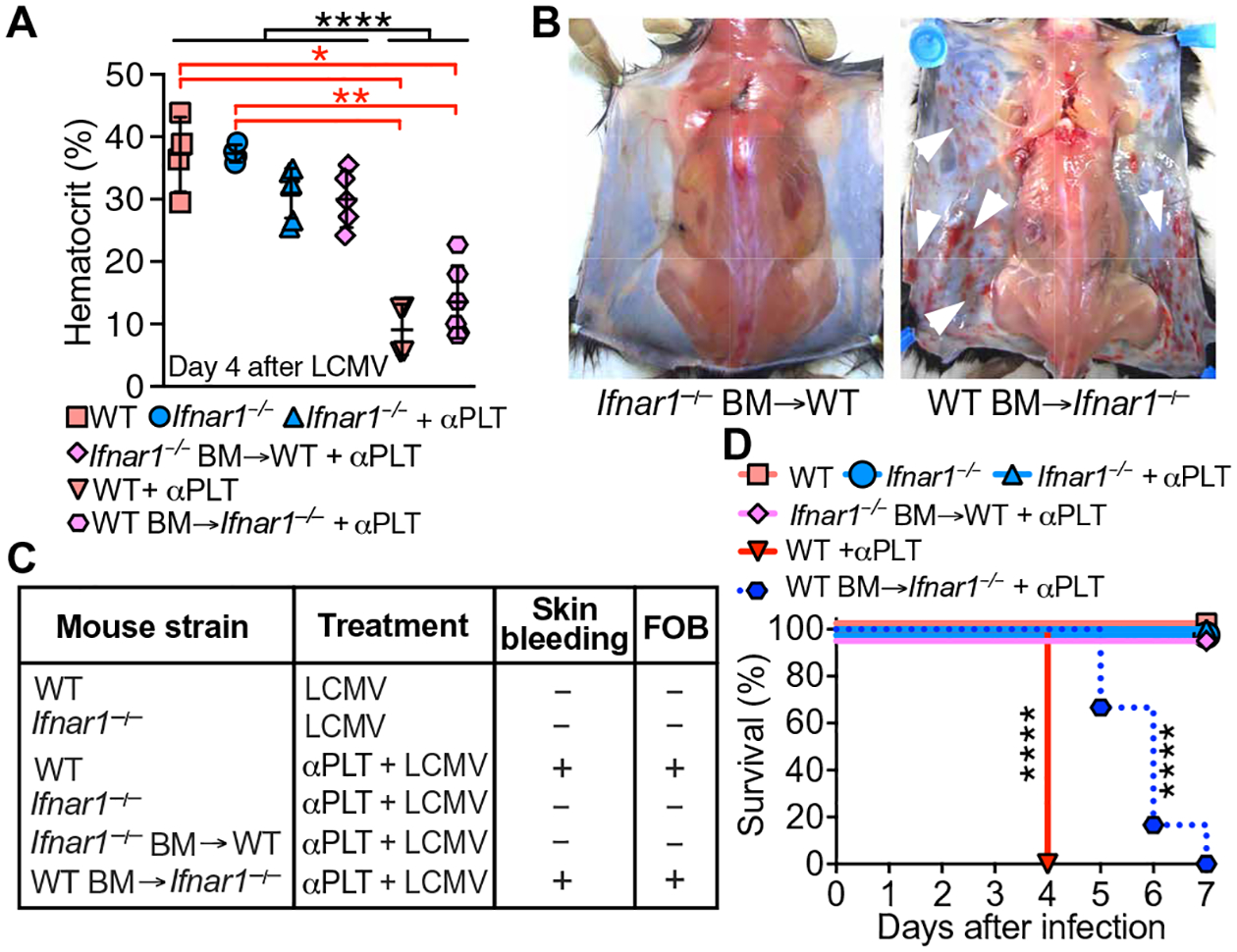
(A) WT mice (n = 4), Ifnar1−/− mice (n = 6), Ifnar1−/− mice with WT BM (n = 6), and WT mice with Ifnar1−/− BM (n = 5) were injected with αPLT followed by 5 × 105–pfu LCMV. Controls (WT and Ifnar1−/−, n = 4 mice per group) received phosphate-buffered saline (PBS) instead of αPLT. Hematocrit in blood from the retro-orbital venous sinus was measured 4 days after infection. Data are shown as scatterplot with means ± SD. (B) Skin bleeding (white arrowheads) or lack thereof in chimeric mice. (C) Summary of the occurrence of skin bleeding and FOB. (D) Survival 7 days after infection without/with platelet depletion. N values for (C) and (D) are the same as in (A). Statistical analysis: (A) one-way analysis of variance (ANOVA) and Tukey’s posttest (black asterisks) or Kruskal-Wallis with Dunn’s posttest (red asterisks); (D) χ2 analysis with Mantel-Cox log-rank test. ****P < 0.0001; **P < 0.01; *P < 0.05.
Defective activation and granular secretion in LCMV-infected mouse platelets
The BM cross-transplantation results suggested that MK targeting by IFN-I stimulation could lead to generation of functionally abnormal platelets. Three days after LCMV infection, the aggregation of B6 mouse platelets stimulated by adenosine 5′-diphosphate (ADP), collagen, and protease-activated receptor 4 activation peptide (Par4-AP) was reduced to ~20 to 40% of preinfection values, improving to ~45 to 60% at day 7 and ~80% (not significantly different from normal) at day 10 (Fig. 3A and fig. S2). In contrast, aggregation induced by AA was still reduced to <5% of preinfection values at day 7, ~40% at day 10, and 60% at day 15, and returned to preinfection level at day 20 (Fig. 3A and fig. S2). Compared with noninfected mice, surface translocation of P-selectin from α-granules was lower 7 days after LCMV infection in WT but not in Ifnar1−/−-stimulated platelets (Fig. 3B). Under the conditions used, AA and Par4-AP induced more P-selectin release than ADP or collagen, but inhibition after LCMV infection was greater after AA stimulation. Like P-selectin, serotonin release from δ-granules was also decreased in infected B6 platelets, but not in Ifnar1−/− platelets. All tested agonists induced comparable release and postinfection inhibition (Fig. 3C). Seven days after LCMV infection, platelet serotonin content was ~50% (Fig. 3D, left), and strong dual-agonist stimulation by ADP/collagen released fourfold less than normal (Fig. 3D, right). Accordingly, LCMV-infected platelets contained fewer and less electron-dense δ-granules, consistent with reduced cargo content (Fig. 3, E and F). Thus, platelets from LCMV-infected mice have impaired α- and δ-granule secretion resulting from defective function mainly of the AA activation pathway. Because platelets are unresponsive to IFN-I stimulation, these abnormalities likely originate in MKs, the sole progenitor of mammalian platelets.
Fig. 3. Platelet function in LCMV-infected mice.
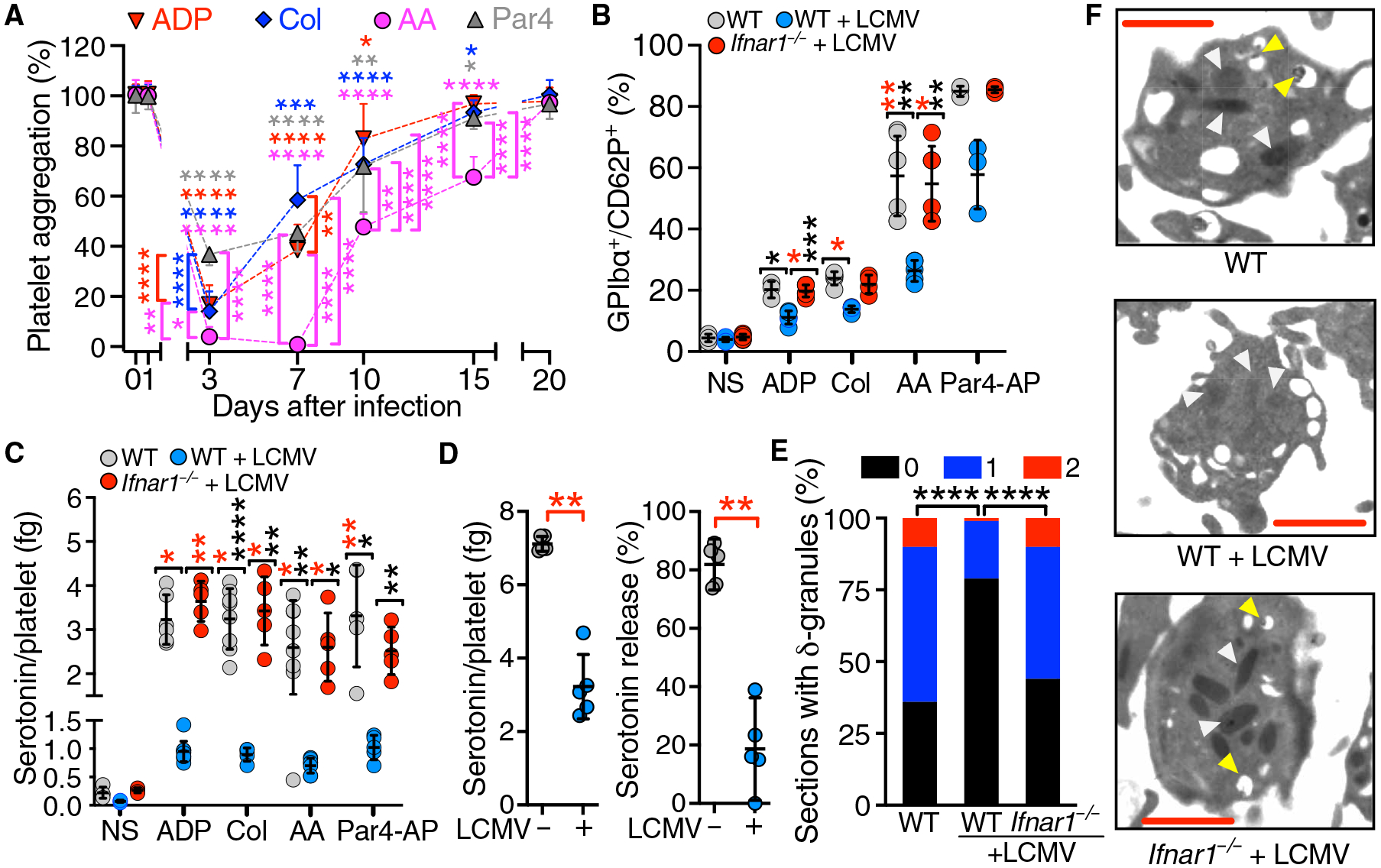
(A) Platelet aggregation (means ± SD; n = 10 mice per group) measured in PRP from LCMV-infected WT mice at the indicated times after infection (day 0). Stimulation by 5 μM ADP, collagen (Col; 4 μg/ml), 75 μM AA, or 40 μM Par4-AP. (B) Blood (n = 3 to 7 mice per group) from infected WT and Ifnar1−/− mice and noninfected WT controls was analyzed by flow cytometry at day 7 after infection for the percentage of GPIbα+/CD62P+ platelets after stimulation as in (A). NS, nonstimulated WT platelets. (C) PRP (n = 4 to 10 mice per group) in the blood described in (B) was analyzed at day 7 after infection for serotonin release. (D) Platelet serotonin content (left) and release after stimulation by ADP and Col (% of total; right) either in mice (n = 5 mice per group) 7 days after infection (+) or in noninfected mice (−). Data in (B) to (D) are shown as scatterplots with means ± SD. (E) Platelets with 0, 1, or 2 δ-granules (% of analyzed) visualized by transmission electron microscopy (TEM) in 41 to 85 platelet sections from mice in (C). (F) TEM visualization of α- and δ-granules indicated by white and yellow arrowheads, respectively. Scale bars, 500 nm. Statistical analysis: (A) Repeated-measures two-way ANOVA with the Geisser-Greenhouse correction for no assumption of sphericity followed by Dunnett’s test for comparisons at different times after infection compared to before infection for each agonist (asterisks at the top of symbols are color coded by agonist) or Tukey’s test for comparisons between agonists at each time after infection (asterisks at the side of symbols are color coded for the agonist with the lower value); (D) two-tailed Mann-Whitney test; (E) contingency table analysis by χ2 test. ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05.
IFN-I alters expression of genes in the AA pathway of platelet activation
In agreement with IFNAR1-mediated MK stimulation causing platelet dysfunction, MKs from LCMV-infected mice had fewer storage granules mostly in the perinuclear zone, with an underdeveloped demarcation membrane system (fig. S3). This was consistent with the platelet phenotype at day 7 after infection (Fig. 3F) when most circulating platelets, which have a life span of 3 to 5 days in mice (17), were derived from IFN-I–stimulated MKs. Therefore, defective AA-induced platelet activation (18) and granule secretion (19) could reflect altered expression of MK genes relevant to TxA2 synthesis. BM expression of genes encoding enzymes essential for AA metabolism (20–22)—thromboxane A synthase 1 (Tbxas1) and prostaglandin/endoperoxide synthase 1 (Ptgs1; also known as cyclooxygenase 1 or Cox-1)—was significantly decreased at day 1 after LCMV infection and returned to preinfection level by days 3 to 10 (Fig. 4, A and B). However, Nf-e2 (23)—which encodes a thrombopoietic transcription factor—was modestly decreased until day 7 and then rapidly increased approximately twofold above baseline by day 10 (Fig. 4C), which was associated with increasing platelet count (fig. S1A). Pro-platelet basic protein (Ppbp) and Cox-2, genes that are not directly involved with platelet production or function, were unchanged or undetectable, respectively (Fig. 4D).
Fig. 4. Expression of platelet-specific genes in LCMV-infected mice.
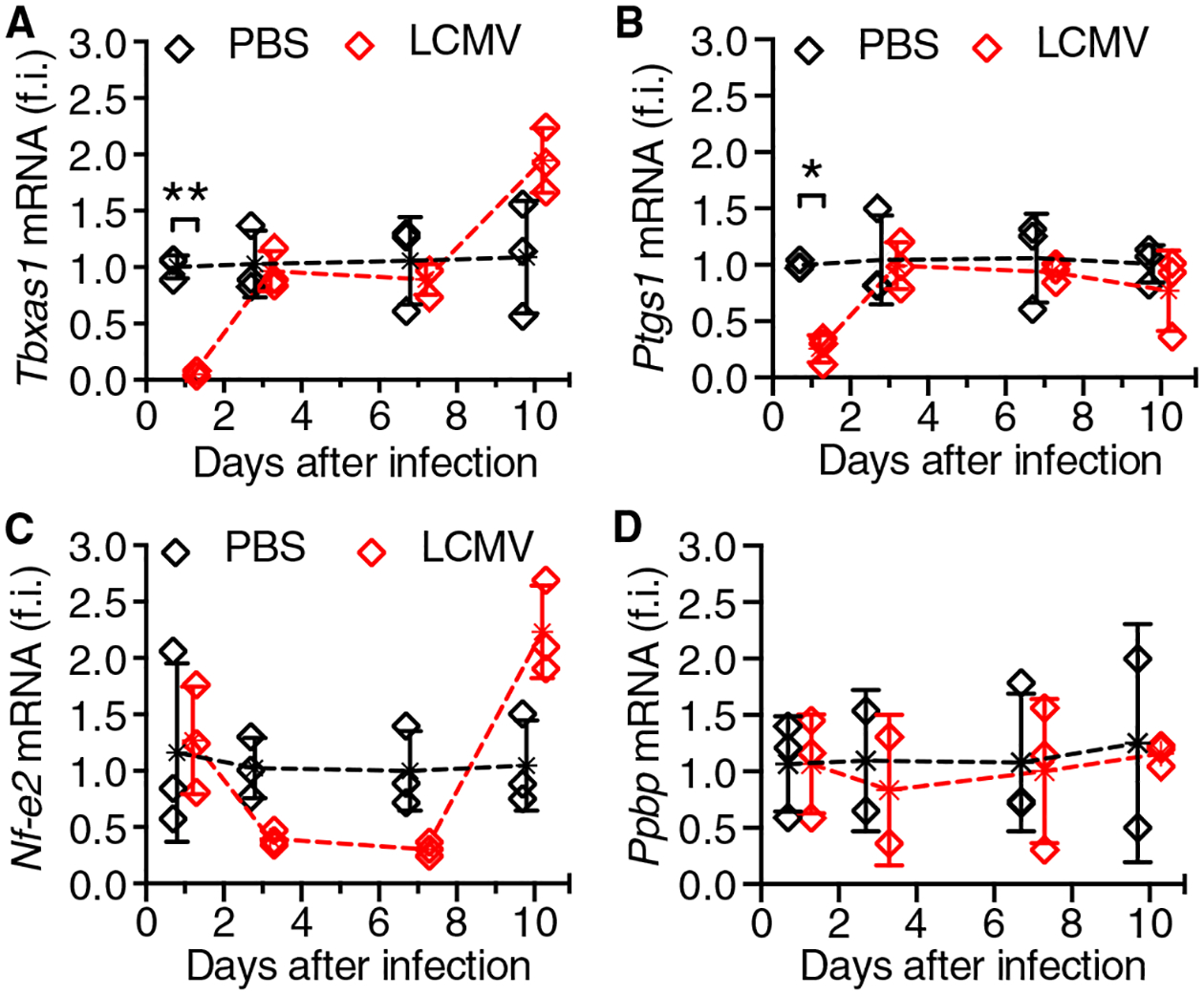
BM cell mRNA from WT mice inoculated with LCMV or PBS was analyzed at different times after infection for expression of Tbxas1 (A), Ptgs1 (also known as Cox-1) (B), Nf-e2 (C), or Ppbp (D). Results (means ± SD; n = 3 mice per group) show fold induction relative to Gapdh. Statistical analysis by repeated-measures two-way ANOVA with the Geisser-Greenhouse correction for no assumption of sphericity followed by Šidák’s posttest for multiple paired comparisons at different time points; only significant differences are shown. *P < 0.05; **P < 0.01.
Aspirin, but not clopidogrel, enhances bleeding in LCMV-infected mice
To further clarify how the AA/TxA2 pathway of platelet activation contributes to LCMV-induced bleeding, we treated mice with acetylsalicylic acid (also known as aspirin) to acetylate directly and irreversibly Cox-1 in platelets and other cell types. This treatment, which reduces TXB2 serum levels because of decreased TxA2 production (24), completely inhibited AA-induced aggregation (fig. S4A). Platelets from aspirin-treated mice had significantly less serotonin release than controls in response to different agonists and significantly less AA-induced release than with all other agonists (Fig. 5A). In addition to serotonin, δ-granules release ADP, which enhances platelet activation through P2Y12 receptors (25, 26). To determine whether this pathway influenced LCMV-induced bleeding, we treated mice with the P2Y12 antagonist clopidogrel from day −3 to day 7 after infection to inhibit ADP-induced platelet aggregation (24). Clopidogrel and aspirin did not affect LCMV replication or LCMV-induced changes in blood leukocyte and platelet counts (fig. S4, B and C) and therefore did not influence the course of infection. Only aspirin-treated mice developed bleeding from day 5 after infection and showed a significantly reduced hematocrit (Fig. 5, B and C), indicating that the P2Y12 feedback pathway activated by released ADP is not required to maintain hemostasis during LCMV infection (27).
Fig. 5. Aspirin, not clopidogrel, exacerbates bleeding in LCMV-infected mice.
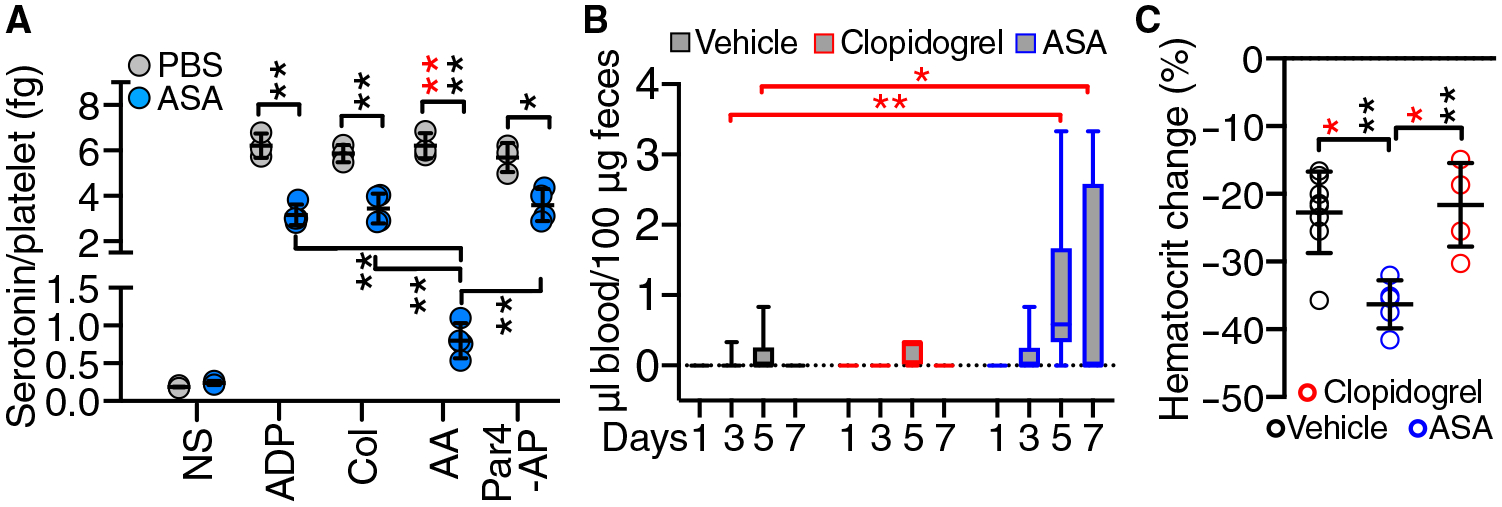
(A) Platelet serotonin release in WT mice treated with aspirin (ASA; 10 mg/kg, intraperitoneally; n = 4) or PBS control (n = 3) for 3 days. The derived PRPs were not stimulated (NS) or stimulated with 75 μM AA, 5 μM ADP, Col (4 μg/ml), or 40 μM Par4-AP (n = 3 to 4 mice per group). (B) FOB at the indicated days after infection in WT mice treated daily with ASA as in (A), clopidogrel (60 μg/day in drinking water), or vehicle control starting 2 days before infection (n = 9 to 16 mice per group). (C) Hematocrit reduction (% of preinfection value) at 7 days after infection in mice (n = 5 to 8 mice per group) treated as in (B). Results are shown as scatterplots with means ± SD (A and C) or as 25th to 75th percentile boxes with minimum-maximum whiskers and line at the median (B). Statistical analysis: (A and C) Brown-Forsythe and Welch ANOVA test with Dunnett’s T3 multiple comparison test (data with normal distribution by Shapiro-Wilk test; black asterisks) or Kruskal-Wallis with Dunn’s posttest (red asterisks). (B) Kruskal-Wallis with Dunn’s posttest. *P < 0.05; **P < 0.01.
Bleeding associated with LCMV infection is increased in mice lacking tryptophan hydroxylase 1 (Tph-1) or the kinase Lyn and reduced by transfusion of normal platelets
We next used Tph-1−/− mice, which lack peripheral serotonin including the platelet δ-granule pool (28). AA-induced platelet serotonin release was significantly lower in LCMV-infected than in noninfected B6 mice and undetectable in Tph-1−/− mice (Fig. 6A), which bled significantly more than WT mice after LCMV infection (Fig. 6B). Transfusion of functionally viable (fig. S5) B6 platelets (~20% of the total in normal blood) on days 2 and 4 after infection reduced bleeding significantly, but not if platelets were pretreated with aspirin (Fig. 6C), which inhibits platelet serotonin release (Fig. 5A). These results confirm that platelets are not affected by IFN-I in vivo and that released serotonin helps control hemorrhage during LCMV infection.
Fig. 6. WT platelets reduce GI bleeding in LCMV-infected Tph-1−/− mice.
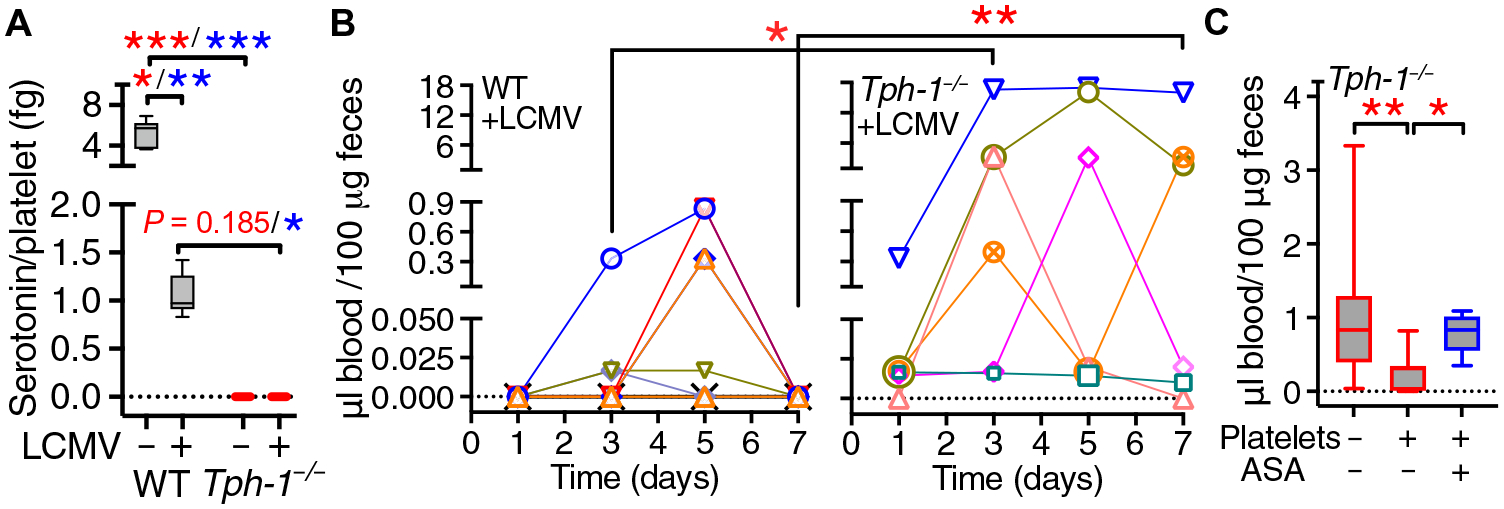
(A) Platelet serotonin release in WT (n = 14) and Tph-1−/− (n = 3) noninfected mice (−LCMV) or WT (n = 7) and Tph-1−/− (n = 3) mice 7 days after LCMV infection (+LCMV). (B) FOB at different postinfection time points in WT (n = 9) and Tph-1−/− mice (n = 6); each symbol represents an individual mouse. (C) On days 2 and 4 after infection, Tph-1−/− mice intravenously received 5 × 108 to 6 × 108 WT platelets suspended in PBS (+), PBS alone (−), or aspirin-treated platelets (+/ASA). FOB was measured on day 7 (n = 13, 9, and 5 mice per group, respectively). Data are shown as 25th to 75th percentile boxes with minimum-maximum whiskers and median (A and C) or individual time course values (B). Statistical analysis: (A and C) Kruskal-Wallis test with multiple paired comparisons performed with the Dunn’s test (red symbols) or the two-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli (blue symbols). (B) Two-way ANOVA followed by Šidák’s posttest for multiple paired comparisons at different time points. *P < 0.05; **P < 0.01; ***P < 0.001.
Signaling through CLEC-2 (C-type lectin-like receptor 2) and GPVI (glycoprotein VI) in platelets is important for vascular integrity during infection and inflammation (29–31). Furthermore, the Src family nonreceptor protein tyrosine kinase Lyn associates with the GPVIFcRγ complex and influences platelet TxA2 production, granule secretion, and aggregation (32, 33). LCMV-infected Lyn−/−mice, like Tph-1−/−, had increased hemorrhage even without platelet depletion (Fig. 7A) and less serotonin release compared with noninfected Lyn−/− mice, but similar serotonin release as infected WT mice (Fig. 7B). Although serotonin has been reported to influence immune and inflammatory responses (34), viral replication, thrombocytopenia, and LCMV-specific “T cell” responses were comparable in LCMV-infected Tph-1−/−, Lyn−/−, and WT mice (fig. S6, A to C), indicating that bleeding tendency differs in mouse strains with similar immune reaction to LCMV infection. Likewise, agonist-induced platelet aggregation was similar in Tph-1−/− and WT mice (fig. S6D), confirming that the absence of peripheral serotonin was not associated with relevant platelet aggregation defects. Of note, platelet GPVI and CLEC-2 expression was decreased by ~30 to 50%, respectively, in infected WT mice (Fig. 7C), suggesting a possible involvement in the pathogenesis of LCMV-induced bleeding.
Fig. 7. Lyn protects LCMV-infected mice from GI bleeding.
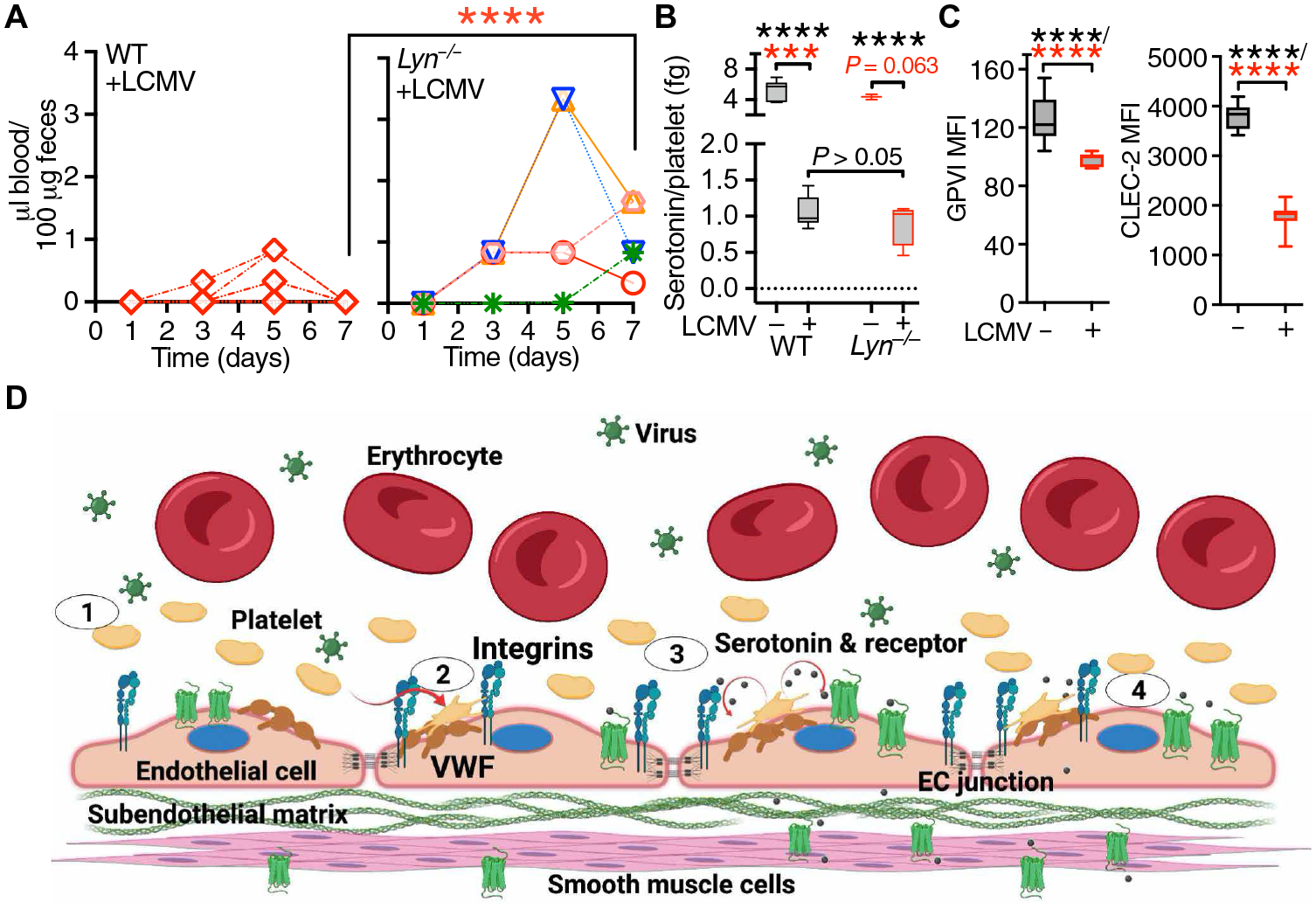
(A) Time course of FOB appearance in WT (n = 9) and Lyn−/− mice (n = 5) after LCMV infection; each symbol represents an individual mouse. Statistical analysis with the Kruskal-Wallis test followed by Dunn’s test for multiple comparisons (red asterisks). (B) Platelet serotonin release in WT (n = 7) and Lyn−/− (n = 3) noninfected mice (−LCMV) or WT (n = 14) and Lyn−/− (n = 4) mice 7 days after LCMV infection (+LCMV). Data, shown as 25th to 75th percentile boxes with minimum-maximum whiskers and median, were analyzed as in (A) (red asterisks) or by one-way ANOVA with Tukey’s posttest (black asterisks). (C) Platelet GPVI and CLEC-2 expression in uninfected WT mice (n = 15) or WT mice 7 days after LCMV infection (n = 9). Data were analyzed by two-tailed unpaired t test with Welch’s correction (black asterisks) and Mann-Whitney test (red asterisks). ***P < 0.001; ****P < 0.0001. (D) Schematic representation (created with BioRender.com) of the prohemostatic role of platelets during LCMV infection. Step 1: Inflammatory stimuli during LCMV infection induce endothelial cell (EC) activation, thereby promoting the release and up-regulation of mediators including von Willebrand factor (VWF) and P-selectin. Step 2: Platelets adhere to VWF on the surface of activated ECs through GPIbα and/or integrin αIIbβ3. These interactions represent the first necessary step for the control of endothelial barrier function by platelets. Step 3: Activated platelets release serotonin from δ-granules; released serotonin activates a subset of 5-hydroxytryptamine (5HT) receptors potentially linked to the activation of the Src kinase Lyn. Step 4: Activated Lyn leads to the tightening of endothelial cell junctions. Platelet-released serotonin can also induce vasoconstriction through effects on smooth muscle cells, further promoting control of vascular permeability.
DISCUSSION
We found that IFN-I targeting of BM cells in LCMV-infected mice decreased the expression of Ptgs1 and Tbxas1 required for AA metabolism and generation of TxA2, a key amplifying signal for platelet activation (20–22). Consequently, circulating platelets, which do not directly respond to IFN-I stimulation, as confirmed here, displayed defective AA-dependent activation and δ-granule serotonin release, indicating that MKs are IFN-I targets in the BM and transfer gene expression reprogramming to generated platelets (35). IFN-I can also increase the expression of indoleamine 2,3-dioxygenase, which would interfere with serotonin production (36), which could be a cause of low platelet serotonin levels in LCMV-infected mice and contribute to reducing total released serotonin. Infected WT mice bled only when platelets were depleted to a count ~10 times lower (≤2 × 104/μl) than that induced by LCMV (9, 37). In contrast, infected Tph-1−/− mice lacking peripheral serotonin bleed without additional depletion, and transfusion of ~6 × 108 viable platelets (one of three of the total number of platelets in the body) restored hemostasis. These findings agree with the notion that serotonin supports endothelial junction stability and prevents erythrocyte extravasation in thrombocytopenic animals (38, 39). Of note, 15 of 17 proteins involved in platelet degranulation are decreased in SARS-CoV-2 infection, and blood serotonin levels are 3.31-fold lower in severe COVID-19 cases than in controls (40).
Normal platelets protected LCMV-infected Tph-1−/− mice from bleeding, which was absent in Tph-1−/− mice without infection. Thus, hemorrhage developed when platelet-released serotonin was below a protective threshold and with the concurrence of other LCMV-dependent alterations. Inflammation induced by IFN-I (41) enhances vascular leakage during infection (42), and endothelial activation influences disease progression in patients with VHF (43, 44). The absence of Ifnar1 in all tissues protects LCMV-infected animals from hemorrhage (9), but BM cross-transplantation did not show protection when Ifnar1 was absent in endothelial cells but present in BM-derived cells. Conversely, mice were protected from hemorrhage if BM cells did not respond to IFN-I. These results indicate that, in addition to MKs, BM-derived cells involved in the initiation and propagation of inflammatory responses dependent on IFN-I stimulation (41, 45) are key determinants of endothelial barrier dysfunction and bleeding during LCMV infection. In this context, platelet inhibitors and vasoactive mediators, such as NO and PGI2 (46–48), released from endothelial cells stimulated by IFN-I may contribute to bleeding.
In the presence of inflammatory stimuli and extreme thrombocytopenia (platelets ≤5% of normal), platelet-mediated protection of vascular integrity is independent of aggregation and thrombus formation, as is the control of intratumor bleeding (49, 50). That platelet aggregation is not involved in IFNAR1-dependent hemorrhage is shown by the lack of effect of clopidogrel, a potent aggregation inhibitor in mice and humans (24), on bleeding in LCMV-infected animals. Two platelet receptors containing the immunoreceptor tyrosine-based activation motif (ITAM)—GPVI/Fcγ complex and CLEC-2—regulate vascular integrity in inflamed tissues (29). Both were substantially, albeit partially, decreased on the platelet membrane of LCMV-infected WT mice. Platelet ability to control endothelial permeability during inflammation is reportedly abolished only when these receptors are completely absent, but our present data cannot exclude that decreased GPVI and CLEC-2 expression contributes to the impairment of endothelial barrier function in LCMV-infected mice.
Like Tph-1−/− mice, LCMV-infected Lyn−/− and aspirin-treated WT mice also bleed without platelet depletion, but this happened independently of serotonin release (Figs. 5A and 7B with Fig. 3C). Thus, Lyn deficiency and aspirin may have effects on vascular function beyond those on platelet activation and δ-granule release. Lyn deficiency may directly destabilize endothelial barrier control (51), whereas ASA may cause acetylation of lysine residues in nitric oxide synthase independently of Cox-1 inhibition (52), resulting in enhanced NO production and reduced platelet adhesion to endothelial cells (53). Decreased platelet adhesion to the vessel wall, particularly in areas of inflammation, could directly decrease barrier function and lower locally released serotonin below the threshold facilitating bleeding. In addition to these effects—and opposite to anti-inflammatory properties that may increase resistance to vascular leakage (54)—aspirin can also alter the gastrointestinal (GI) mucosa causing bleeding (55), although this effect is unlikely at the dose used in our study.
In addition to the effects on platelet functions, IFN-I affects thrombocytopoiesis. LCMV replicates in BM stromal cells but infects <20% of mature MKs (56). Similar to human MKs infected by dengue virus (DENV), IFN-I–dependent up-regulation of IFITM3 could contribute to limit virus spreading (57) and explain the limited infection. Thus, platelet count reduction is unlikely to originate solely from lower MK numbers. Accordingly, we found reduced Tbxas1 and Ptgs1 expression in the BM of infected mice, which normalized within 3 to 7 days after infection in parallel with the platelet count. In animal models, peripheral mast cell–derived serotonin contributes to DENV-induced thrombocytopenia (58). The proposed mechanism involves DENV-dependent serotonin discharge from mast cells and subsequent serotonin-induced platelet activation and removal from the circulation. A similar pathogenic mechanism could be triggered by circulating IgG (immunoglobulin G) immune complexes that activate platelets through the FcγRIIA receptor, leading first to organ-specific platelet sequestration followed by the return of degranulated platelets to the blood (59). These additional mechanisms, which potentially affect both platelet count and function in blood, may be of limited relevance in LCMV infection, because Tph-1−/− mice, which are devoid of peripheral serotonin, have the same course of thrombocytopenia and recovery as B6 controls.
In the mouse LCMV model of arenavirus infection (60, 61), which may reflect mechanisms in human VHF (62, 63), platelet transfusion can reduce bleeding. This finding agrees with serum serotonin levels (64) and platelet activation markers (65) predicting bleeding risk and beneficial effects of platelet transfusion in patients with dengue with platelet counts <20,000/μl (66), although the latter conclusion is still controversial (67, 68). Serious adverse effects of platelet transfusions have also been reported in patients with dengue (69), possibly involving immune complex–mediated activation of the FcγRIIA receptor leading to enhanced platelet activation, serotonin release, and vascular leakage (70). The quality of transfused platelets—particularly levels of activation, microparticle shedding, phosphatidylserine exposure, and ABO blood group matching among other factors—may affect the outcome of treatment (71–76) and explain contradictory results and adverse effects. On the basis of experimental evidence and despite inherent difficulties and limitations, a more controlled and critical evaluation of platelet transfusion to treat bleeding and endothelial barrier dysfunction during viral infection should be reconsidered. Moreover, indications for platelet transfusions in the prevention of bleeding and vascular leakage may vary depending on the virus involved, because direct or indirect viral effects on platelet activation—as in the case of DENV (77) and SARS-CoV-2 (78)—may contribute to pathogenic mechanisms involving platelets.
In conclusion, we demonstrated that the AA/TxA2 pathway of platelet activation leading to serotonin release from δ-granules protected from bleeding induced by IFN-I stimulation. These findings may lead to a better understanding of the pathogenic mechanisms that alter endothelial barrier function during viral infection and other inflammatory conditions (Fig. 7D). By highlighting a quantitative approach to evaluate the role of platelets and platelet transfusion in the prevention and control of bleeding complications, the present results can also help improve the diagnosis and treatment of patients with VHF.
MATERIALS AND METHODS
Study design
Studies were conducted in 8- to 10-week-old male and female WT mice treated with aspirin and clopidogrel, Tph-1−/− mice, and Lyn−/− mice. Sample size to detect significant differences was calculated with previous data, published literature, and power calculation. Statistically significant outliers were excluded from evaluation.
Mice
C57BL/6J mice were purchased from Charles River Laboratories (Calco, LC, Italy) or the Jackson Laboratory (Bar Harbor, ME) or were bred at the Scripps Research Institute. Ifnar1−/− mice were backcrossed against C57BL/6J mice (79, 80). Tph-1−/−(28) and Lyn−/− (81) mice were provided by M. Bader (Max Delbrück Center for Molecular Medicine, Berlin, Germany) and X. Du (University of Illinois at Chicago, IL, USA), respectively. BM chimeras were generated in C57BL/6J or Ifnar1−/− mice lethally irradiated with 1300 rads. On the same day, femurs and tibiae were isolated from euthanized donor mice under sterile conditions, and BM cell suspensions were prepared by centrifugation. Cells (107) were injected in the tail veins of recipient mice, and reconstitution was allowed to proceed for at least 8 weeks before use. In all experiments, mice were age and gender matched. Husbandry and handling of mice conformed to guidelines set by the Institutional Animal Care Committees following the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Mice for BM transplantation were kept in pathogen-free rooms.
Viruses and virus titration
We used the LCMV strain Arm 53b, which was propagated and purified as described (82). Mice were infected by tail vein inoculation of 5 × 105 plaque-forming units (pfu). LCMV viral titers were measured by extracting viral RNA from serum and tissues using QIAamp Viral RNA Mini Kit (Qiagen). Complementary DNA (cDNA) was synthetized using SuperScript cDNA Synthesis Kit (Invitrogen) and used for SYBR Green quantitative real-time polymerase chain reaction (PCR) using the LCMV-specific primers 5′-CTCCTTTCCCAAGAGAAGACTAAG-3′ and 5′-TCCATTTGGTCAGGCAATAAC-3′ (83). A plasmid containing LCMV sequences was used to build a standard curve and viral titer expressed as genome equivalents.
In vivo platelet depletion and peripheral blood values
A mixture of anti-mouse GPIbα rat monoclonal antibodies (αPLT), which depletes platelets in vivo (16, 84), was purchased from Emfret Analytics (Würzburg, Germany). Mice were injected intravenously with αPLT (80 μg per mouse) 3 hours before virus infection. Whole blood was collected from the retro-orbital sinus, and blood cells were counted with an automated cell counter (HeCoVet, Seac-Radim, Firenze, Italy, or ProCyte Dx Hematology Analyzer, Idexx Laboratories, Westbrook, ME, USA).
Fecal occult blood
For rapid detection of fecal blood, we used the Hemoccult II SENSA test (Beckman Coulter Inc., Brea, CA, USA). For quantification, a mixture of fecal pellets and distilled water (30 μg into 100 μl) was vortexed for 5 min and centrifuged at 1100g for 5 min at room temperature (RT); the supernatant was collected for analysis. Before the assay, a 4% (w/v) gum guaiac stock solution in 95% ethanol was diluted 1:10 in glacial acetic acid, and 50 μl was added to the same volume of fecal sample supernatants dispensed in 96-well plates. The reaction was started adding 100 μl of H2O2. After 5-min incubation at RT, plates were then analyzed with a photometer at 405-nm wavelength. A calibration curve was obtained by the analysis of negative fecal samples mixed with known quantities of fresh blood.
Platelet aggregation and flow cytometry
Blood was collected from the retro-orbital sinus into 1/10th volume of citrate phosphate dextrose (CPD; Sigma-Aldrich, St. Louis, MO) and platelet-rich plasma (PRP) prepared as described (24). The PRP platelet count was adjusted to the lowest value of the day using homologous platelet-poor plasma (PPP). Aggregation in stirred PRP at 37°C was induced by adding the indicated concentrations of ADP, Col, AA (Mascia Brunelli, Milan, Italy) or the Par4-AP AYPGKF (Primm Biotech, Milan, Italy) and monitored by recording changes in light transmittance through the PRP suspension using a Chronolog model 490 aggregometer (Chrono-log Corp., Havertown, PA, USA). For fluorescence-activated cell sorting (FACS) analysis, 1 × 106 platelets were incubated for 15 min at RT in 50 μl of modified Tyrode’s buffer (pH 7.4) containing phycoerythrin-conjugated anti-GPIbα (clone Xia.G5, Emfret Analytics) and fluorescein isothiocyanate–conjugated anti-CD62P (clone RB40.34; BD Pharmingen, San Jose, CA), after which platelet activation was induced by agonists added as for aggregation with 10-min incubation at RT. After stopping the reaction with 300 μl of modified Tyrode’s buffer (pH 6.5), samples were analyzed with a FACSCanto flow cytometer, and data were processed with FlowJo software (version 10.0.7; FlowJo LLC, Ashland, OR).
Ex vivo platelet stimulation with IFN-α
After collecting blood in CPD and counting cells, 500 μl of samples was incubated for 2 hours at RT with PBS containing native or heat-inactivated (90°C for 1 hour) 0.1% bovine serum albumin and recombinant mouse IFN-α (PBL Assay Science, Piscataway, NJ, USA). Total RNA from peripheral blood mononuclear cells (PBMCs) was extracted using RNeasy Mini Kit from Qiagen (Hilden, Germany).
Serotonin release
Blood was collected in CPD to prepare PRP with platelet count adjusted to the lowest value of the day using homologous PPP. Activation was induced by ADP, Col, AA, and Par4-AP for 5 min at RT. Suspensions were centrifuged at 4500g for 5 min at RT, and supernatants were collected and stored at −80°C. Serotonin was measured by high-performance liquid chromatography (EUREKA Lab Division Srl, Chiaravalle, AN, Italy).
Leukocyte preparation from blood and intracellular IFN-γ staining
Single-cell suspensions were prepared from peripheral blood as described (85). Anticoagulated blood and spleen single-cell suspensions were incubated with ACK lysis buffer [0.15 M NH4Cl, 1.0 mM KHCO3, and 0.1 mM Na2EDTA (pH 7.2)] and washed twice with Hanks’ balanced salt solution. Cells were stimulated or not by the immunodominant H-2Db–restricted LCMV glycoprotein peptide GP 33 to 41 (86) and stained for intracellular IFN-γ with Pacific Blue–conjugated anti-CD8 and allophycocyanin-conjugated anti–IFN-γ (clones 53 to 6.7 and XMG1.2, respectively; BD Pharmingen). LCMV-specific CD8+ T cells were quantified by staining PBMC with Pacific Blue–conjugated anti-CD8 and phycoerythrin-conjugated recombinant soluble dimeric H-2Db/Ig fusion protein (BD Pharmingen, CA, USA) complexed with the immunodominant H-2Db–restricted LCMV GP 33 to 41 peptides (87). Samples were analyzed with a FACSCanto flow cytometer, and data were processed with FlowJo.
Tissue RNA analysis
Total RNA isolated from liver, spleen, and BM of WT mice inoculated with LCMV or PBS was analyzed at different times after infection by using the Promega kit Z6211 and DNAse TURBO Thermo AM1907 following the manufacturer’s recommendation. The extracted RNA was subsequently retrotranscribed to cDNA as previously described (88). Quantitative real-time PCR analysis was performed using the ViiA7 Fast Real-Time PCR System (Applied Biosystems). Glyceraldehyde-3-phosphate dehydrogenase (Gapdh), Isg15, and Ppbp mRNAs were quantified by quantitative reverse transcription PCR (89) using the following primers: Gapdh, 5′-TTCACCACCATGGAGAAGGC-3′ and 5′-GGCATGGACTGTGGTCATGA-3′; Isg15, 5′-GGGGGAGAGTCGATCCAG-3′ and 5′-CCCCAGCATCTTCACCTTTA-3′; Ppbp, 5′-CGTTGTTCCCTCCTGGCTCT-3′ and 5′-GGACGATGTAGGTCTGAGTC-3′; and Nf-e2, 5′-GCCCTGTTCAATTCATCCAG-3′ and 5′-GAAAGGGTAAGTGGCTTTGACA-3′. Analysis of Tbxas1, Ptgs1, Ptgs2, and Gapdh mRNAs was performed using TaqMan (gene expression assay nos. Mm00495553_m, Mm00477214_m1, Mm00478374_m1, and Mm99999915_g1, respectively; Applied Biosystems, Foster City, CA). Gene expression was determined as the difference between the threshold cycle (Ct) of the gene of interest and the Ct of Gapdh of the same sample (ΔCt). The fold change expression (fold change) of each gene of interest was calculated over its basal expression in PBS-injected controls of each specific time point by the formula 2-ΔΔCt (90).
Transmission electron microscopy
Blood was collected in CPD and PRP prepared as described (24). One milliliter of PRP was fixed for 15 min at RT by adding one volume of a solution containing 4% paraformaldehyde, 8% glutaraldehyde, and 0.24 M Na cacodylate. After fixation, PRP was centrifuged at 4500g for 10 min at RT, and pellets were stored at 4°C in a solution containing 4% paraformaldehyde, 2% glutaraldehyde, and 0.12 M Na cacodylate until processed. BM cells were isolated from femurs of 7- to 10-week-old NaCl- or LCMV-infected mice by centrifugation and stored at 4°C in 4% paraformaldehyde, 2% glutaraldehyde, and 0.12 M Na cacodylate until processed. For TEM, samples were processed as described (91) and imaged with a ZEISS Leo 912 AB Omega transmission electron microscope (Carl Zeiss, Obercochen, Germany) fitted with a 2k × 2k bottom-mounted slow-scan ProScan camera (ProScan Imaging, Cincinnati, OH) controlled by the Esi VisionPro 3.2 software (Cognex, Natick, MA).
Statistical analysis
Unless otherwise specified, data are expressed as means ± SD, 25th to 75th percentile boxes with minimum to maximum whiskers and line at the median, or contingency summary plot. The significance of differences between two groups was assessed with two-tailed unpaired t test with Welch’s correction (no assumption of equal variance) or nonparametric Mann-Whitney test. The significance of differences between three or more groups was assessed with one- or two-way analysis of variance (ANOVA) followed by Tukey’s, Šidák’s, Dunnett’s, or Dunnett’s T3 test for paired comparisons or nonparametric Kruskal-Wallis test followed by Dunn’s test or two-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli. All statistical analyses were performed with Prism software version 9.3.0 (GraphPad Software Inc., San Diego, CA).
Supplementary Material
Acknowledgments:
We thank D. Pinschewer, M. Cattaneo, G. Poli, and P. Dellabona for the helpful discussions; P. M. Mainetti, M. Raso, I. Fermo, A. Fiocchi, and D. Covarello for technical assistance; M. Panzeri for TEM; Zordan, A. Monestiroli, and E. Daoud for qPCR experiments; and J. Koziol for statistical revision. We would like to acknowledge the PhD program in Basic and Applied Immunology at San Raffaele University because R.A. conducted this study as partial fulfillment of a PhD in Molecular Medicine within that program.
Funding:
This work was supported by grants from the NIH, USA (HL42846, HL117722, and HL135294 to Z.M.R., and AI40696 to L.G.G.); the European Research Council (250219 to L.G.G., and 281648 and 725038 to M.I.); the Italian Ministry of Health (GR-2008-1135776, GR-2008-1138756, and RF-2011-02346754 to G.S.; GR-2011-02347925 to M.I.; and RF-2013-02355209 to L.G.G.); the Italian Association for Cancer Research (18468, 22737, and 22820 to G.S.; 9965, 15350, 19891, and 22737 to M.I.; and 22737 to L.G.G.); and the Lombardy Foundation for Biomedical Research (2015-0010 to M.I.). M.I. was also supported by the European Molecular Biology Organization Young Investigator Program and by a Career Development Award from the Giovanni Armenise-Harvard Foundation. R.A. was supported by a fellowship of MERU Foundation, Italy, at the MERU-Roon Research Center for Vascular Biology of the Scripps Research Institute.
Footnotes
Competing interests: Z.M.R. is founder, president, and CEO of MERU VasImmune Inc., a company interested in the development of products related to the study, diagnosis, and treatments of diseases associated with hemostasis and thrombosis. The other authors declare that they have no competing interests.
Data and materials availability:
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Tph-1−/− mice require a material transfer agreement from Max Delbrück Center for Molecular Medicine, Berlin, Germany.
REFERENCES AND NOTES
- 1.Marty AM, Jahrling PB, Geisbert TW, Viral hemorrhagic fevers. Clin. Lab. Med 26, 345–386 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Bray M, Pathogenesis of viral hemorrhagic fever. Curr. Opin. Immunol 17, 399–403 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Shehu NY, Gomerep SS, Isa SE, Iraoyah KO, Mafuka J, Bitrus N, Dachom MC, Ogwuche JE, Onukak AE, Onyedibe KI, Ogbaini-Emovon E, Egah DZ, Mateer EJ, Paessler S, Lassa Fever 2016 Outbreak in Plateau State, Nigeria—The changing epidemiology and clinical presentation. Front. Public Health 6, 232 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mobula LM, Nathalie M, Clive H, Brantly K, Plyler W, Brown J, Kauffeldt B, Eisenhut D, Cooper LA, Fankhauser J, Clinical manifestations and modes of death among patients with ebola virus disease, Monrovia, Liberia, 2014. Am. J. Trop. Med. Hyg 98, 1186–1193 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ohuabunwo C, Ameh C, Oduyebo O, Ahumibe A, Mutiu B, Olayinka A, Gbadamosi W, Garcia E, Nanclares C, Famiyesin W, Mohammed A, Nguku P, Koko RI, Obasanya J, Adebayo D, Gbadegesin Y, Idigbe O, Oguntimehin O, Nyanti S, Nzuki C, Abdus-Salam I, Adeyemi J, Onyekwere N, Musa E, Brett-Major D, Shuaib F, Nasidi A, Clinical profile and containment of the Ebola virus disease outbreak in two large west African Cities, Nigeria, July–September 2014. Int. J. Infect. Dis 53, 23–29 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McElroy A, Understanding bleeding in Ebola virus disease. Clin. Adv. Hematol. Oncol 13, 29–31 (2015). [PMC free article] [PubMed] [Google Scholar]
- 7.Goeijenbier M, van Wissen M, van de Weg C, Jong E, Gerdes VE, Meijers JC, Brandjes DP, van Gorp EC, Review: Viral infections and mechanisms of thrombosis and bleeding. J. Med. Virol 84, 1680–1696 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oldstone MB, Biology and pathogenesis of lymphocytic choriomeningitis virus infection. Curr. Top. Microbiol. Immunol 263, 83–117 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Iannacone M, Sitia G, Isogawa M, Whitmire JK, Marchese P, Chisari FV, Ruggeri ZM, Guidotti LG, Platelets prevent IFN-α/β-induced lethal hemorrhage promoting CTL-dependent clearance of lymphocytic choriomeningitis virus. Proc. Natl. Acad. Sci. U.S.A 105, 629–634 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peters CJ, Lymphocytic choriomeningitis virus–an old enemy up to new tricks. N. Engl. J. Med 354, 2208–2211 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Fischer SA, Graham MB, Kuehnert MJ, Kotton CN, Srinivasan A, Marty FM, Comer JA, Guarner J, Paddock CD, DeMeo DL, Shieh WJ, Erickson BR, Bandy U, DeMaria A Jr., Davis JP, Delmonico FL, Pavlin B, Likos A, Vincent MJ, Sealy TK, Goldsmith CS, Jernigan DB, Rollin PE, Packard MM, Patel M, Rowland C, Helfand RF, Nichol ST, Fishman JA, Ksiazek T, Zaki SR; LCMV in Transplant Recipients Investigation Team, Transmission of lymphocytic choriomeningitis virus by organ transplantation. N Engl J Med 354, 2235–2249 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Grant A, Seregin A, Huang C, Kolokoltsova O, Brasier A, Peters C, Paessler S, Junín virus pathogenesis and virus replication. Viruses 4, 2317–2339 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merigan TC, Oldstone MB, Welsh RM, Interferon production during lymphocytic choriomeningitis virus infection of nude and normal mice. Nature 268, 67–68 (1977). [DOI] [PubMed] [Google Scholar]
- 14.Bhattacharyya M, Karmohapatra SK, Bhattacharya G, Bhattacharya R, Sinha AK, The role of leucocytes in the acetyl salicylic acid (aspirin) induced nitric oxide synthesis in the production of interferon-alpha, a potent inhibitor of platelet aggregation and a thrombolytic agent. J. Thromb. Thrombolysis 28, 173–184 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Negrotto S, De Giusti CJ, Lapponi MJ, Etulain J, Rivadeneyra L, Pozner RG, Gomez RM, Schattner M, Expression and functionality of type I interferon receptor in the megakaryocytic lineage. J. Thromb. Haemost 9, 2477–2485 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Iannacone M, Sitia G, Isogawa M, Marchese P, Castro MG, Lowenstein PR, Chisari FV, Ruggeri ZM, Guidotti LG, Platelets mediate cytotoxic T lymphocyteinduced liver damage. Nat. Med 11, 1167–1169 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmitt A, Guichard J, Masse JM, Debili N, Cramer EM, Of mice and men: Comparison of the ultrastructure of megakaryocytes and platelets. Exp. Hematol 29, 1295–1302 (2001). [DOI] [PubMed] [Google Scholar]
- 18.FitzGerald GA, Mechanisms of platelet activation: Thromboxane A2 as an amplifying signal for other agonists. Am. J. Cardiol 68, B11–B15 (1991). [DOI] [PubMed] [Google Scholar]
- 19.Rinder CS, Student LA, Bonan JL, Rinder HM, Smith BR, Aspirin does not inhibit adenosine diphosphate-induced platelet α-granule release. Blood 82, 505–512 (1993). [PubMed] [Google Scholar]
- 20.Smith WL, Marnett LJ, DeWitt DL, Prostaglandin and thromboxane biosynthesis. Pharmacol. Ther 49, 153–179 (1991). [DOI] [PubMed] [Google Scholar]
- 21.Wang LH, Kulmacz RJ, Thromboxane synthase: Structure and function of protein and gene. Prostaglandins Other Lipid Mediat 68–69, 409–422 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Smith WL, Meade EA, DeWitt DL, Pharmacology of prostaglandin endoperoxide synthase isozymes-1 and-2. Ann. N. Y. Acad. Sci 714, 136–142 (1994). [DOI] [PubMed] [Google Scholar]
- 23.Shivdasani RA, Rosenblatt MF, Zucker-Franklin D, Jackson CW, Hunt P, Saris CJM, Orkin SH, Transcription factor NF-E2 is required for platelet formation independent of the actions of thrombopoietin/MGDF in megakaryocyte development. Cell 81, 695–704 (1995). [DOI] [PubMed] [Google Scholar]
- 24.Sitia G, Aiolfi R, Di Lucia P, Mainetti M, Fiocchi A, Mingozzi F, Esposito A, Ruggeri ZM, Chisari FV, Iannacone M, Guidotti LG, Antiplatelet therapy prevents hepatocellular carcinoma and improves survival in a mouse model of chronic hepatitis B. Proc. Natl. Acad. Sci. U.S.A 109, E2165–E2172 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dorsam RT, Kunapuli SP, Central role of the P2Y12 receptor in platelet activation. J. Clin. Investig 113, 340–345 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bennett JS, Berger BW, Billings PC, The structure and function of platelet integrins. J. Thromb. Haemost 7 (Suppl. 1), 200–205 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Mendolicchio GL, Zavalloni D, Bacci M, Corrada E, Marconi M, Lodigiani C, Presbitero P, Rota L, Ruggeri ZM, Variable effect of P2Y12 inhibition on platelet thrombus volume in flowing blood. J. Thromb. Haemost 9, 373–382 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walther DJ, Peter JU, Bashammakh S, Hortnagl H, Voits M, Fink H, Bader M, Synthesis of serotonin by a second tryptophan hydroxylase isoform. Science 299, 76 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Watson SP, Herbert JM, Pollitt AY, GPVI and CLEC-2 in hemostasis and vascular integrity. J. Thromb. Haemost 8, 1456–1467 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Navarro-Nunez L, Langan SA, Nash GB, Watson SP, The physiological and pathophysiological roles of platelet CLEC-2. Thromb. Haemost 109, 991–998 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boulaftali Y, Hess PR, Getz TM, Cholka A, Stolla M, Mackman N, Owens III AP, Ware J, Kahn ML, Bergmeier W, Platelet ITAM signaling is critical for vascular integrity in inflammation. J. Clin. Invest 123, 908–916 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho MJ, Pestina TI, Steward SA, Lowell CA, Jackson CW, Gartner TK, Role of the Src family kinase Lyn in TxA2 production, adenosine diphosphate secretion, Akt phosphorylation, and irreversible aggregation in platelets stimulated with γ-thrombin. Blood 99, 2442–2447 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Li Z, Zhang G, Liu J, Stojanovic A, Ruan C, Lowell CA, Du X, An important role of the SRC family kinase Lyn in stimulating platelet granule secretion. J. Biol. Chem 285, 12559–12570 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu H, Denna TH, Storkersen JN, Gerriets VA, Beyond a neurotransmitter: The role of serotonin in inflammation and immunity. Pharmacol. Res, 100–114 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Thon JN, Italiano JE, Platelet formation. Semin. Hematol 47, 220–226 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lood C, Tyden H, Gullstrand B, Klint C, Wenglen C, Nielsen CT, Heegaard NH, Jonsen A, Kahn R, Bengtsson AA, Type I interferon-mediated skewing of the serotonin synthesis is associated with severe disease in systemic lupus erythematosus. PLOS ONE 10, e0125109 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loria GD, Romagnoli PA, Moseley NB, Rucavado A, Altman JD, Platelets support a protective immune response to LCMV by preventing splenic necrosis. Blood 121, 940–950 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sweetman HE, Shepro D, Hechtman HB, Inhibition of thrombocytopenic petechiae by exogenous serotonin administration. Haemostasis 10, 65–78 (1981). [DOI] [PubMed] [Google Scholar]
- 39.Shepro D, Welles SL, Hechtman HB, Vasoactive agonists prevent erythrocyte extravasation in thrombocytopenic hamsters. Thromb. Res 35, 421–430 (1984). [DOI] [PubMed] [Google Scholar]
- 40.Shen B, Yi X, Sun Y, Bi X, Du J, Zhang C, Quan S, Zhang F, Sun R, Qian L, Ge W, Liu W, Liang S, Chen H, Zhang Y, Li J, Xu J, He Z, Chen B, Wang J, Yan H, Zheng Y, Wang D, Zhu J, Kong Z, Kang Z, Liang X, Ding X, Ruan G, Xiang N, Cai X, Gao H, Li L, Li S, Xiao Q, Lu T, Zhu Y, Liu H, Chen H, Guo T, Proteomic and metabolomic characterization of COVID-19 patient sera. Cell 182, 59–72.e15 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kopitar-Jerala N, The role of interferons in inflammation and inflammasome activation. Front. Immunol 8, 873 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baccala R, Welch MJ, Gonzalez-Quintial R, Walsh KB, Teijaro JR, Nguyen A, Ng CT, Sullivan BM, Zarpellon A, Ruggeri ZM, de la Torre JC, Theofilopoulos AN, Oldstone MB, Type I interferon is a therapeutic target for virus-induced lethal vascular damage. Proc. Natl. Acad. Sci. U.S.A 111, 8925–8930 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Basu A, Chaturvedi UC, Vascular endothelium: The battlefield of dengue viruses. FEMS Immunol. Med. Microbiol 53, 287–299 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kunz S, The role of the vascular endothelium in arenavirus haemorrhagic fevers. Thromb. Haemost 102, 1024–1029 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Swiecki M, Colonna M, The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol 15, 471–485 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Faruqi TR, Erzurum SC, Kaneko FT, DiCorleto PE, Role of nitric oxide in poly(I-C)-induced endothelial cell expression of leukocyte adhesion molecules. Am. J. Physiol 273, H2490–H2497 (1997). [DOI] [PubMed] [Google Scholar]
- 47.Eldor A, Fridman R, Vlodavsky I, Hy-Am E, Fuks Z, Panet A, Interferon enhances prostacyclin production by cultured vascular endothelial cells. J. Clin. Invest 73, 251–257 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gomez RM, Pozner RG, Lazzari MA, D’Atri LP, Negrotto S, Chudzinski-Tavassi AM, Berría MI, Schattner M, Endothelial cell function alteration after Junin virus infection. Thromb. Haemost 90, 326–334 (2003). [DOI] [PubMed] [Google Scholar]
- 49.Ho-Tin-Noe B, Boulaftali Y, Camerer E, Platelets and vascular integrity: How platelets prevent bleeding in inflammation. Blood 131, 277–288 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Ho-Tin-Noe B, Goerge T, Cifuni SM, Duerschmied D, Wagner DD, Platelet granule secretion continuously prevents intratumor hemorrhage. Cancer Res 68, 6851–6858 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Han J, Zhang G, Welch EJ, Liang Y, Fu J, Vogel SM, Lowell CA, Du X, Cheresh DA, Malik AB, Li Z, A critical role for Lyn kinase in strengthening endothelial integrity and barrier function. Blood 122, 4140–4149 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jung SB, Kim CS, Naqvi A, Yamamori T, Mattagajasingh I, Hoffman TA, Cole MP, Kumar A, Dericco JS, Jeon BH, Irani K, Histone deacetylase 3 antagonizes aspirin-stimulated endothelial nitric oxide production by reversing aspirin-induced lysine acetylation of endothelial nitric oxide synthase. Circ. Res 107, 877–887 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moncada S, Higgs EA, Nitric oxide and the vascular endothelium. Handb Exp Pharmacol 2006, 213–254 (2006). [DOI] [PubMed] [Google Scholar]
- 54.Dzeshka MS, Shantsila A, Lip GY, Effects of aspirin on endothelial function and hypertension. Curr. Hypertens. Rep 18, 83 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smecuol E, Pinto Sanchez MI, Suarez A, Argonz JE, Sugai E, Vazquez H, Litwin N, Piazuelo E, Meddings JB, Bai JC, Lanas A, Low-dose aspirin affects the small bowel mucosa: Results of a pilot study with a multidimensional assessment. Clin. Gastroenterol. Hepatol 7, 524–529 (2009). [DOI] [PubMed] [Google Scholar]
- 56.Binder D, Fehr J, Hengartner H, Zinkernagel RM, Virus-induced transient bone marrow aplasia: Major role of interferon-alpha/beta during acute infection with the noncytopathic lymphocytic choriomeningitis virus. J. Exp. Med 185, 517–530 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Campbell RA, Schwertz H, Hottz ED, Rowley JW, Manne BK, Washington AV, Hunter-Mellado R, Tolley ND, Christensen M, Eustes AS, Montenont E, Bhatlekar S, Ventrone CH, Kirkpatrick BD, Pierce KK, Whitehead SS, Diehl SA, Bray PF, Zimmerman GA, Kosaka Y, Bozza PT, Bozza FA, Weyrich AS, Rondina MT, Human megakaryocytes possess intrinsic antiviral immunity through regulated induction of IFITM3. Blood 133, 2013–2026 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Masri MFB, Mantri CK, Rathore APS, John ALS, Peripheral serotonin causes dengue virus-induced thrombocytopenia through 5HT2 receptors. Blood 133, 2325–2337 (2019). [DOI] [PubMed] [Google Scholar]
- 59.Cloutier N, Allaeys I, Marcoux G, Machlus KR, Mailhot B, Zufferey A, Levesque T, Becker Y, Tessandier N, Melki I, Zhi H, Poirier G, Rondina MT, Italiano JE, Flamand L, McKenzie SE, Cote F, Nieswandt B, Khan WI, Flick MJ, Newman PJ, Lacroix S, Fortin PR, Boilard E, Platelets release pathogenic serotonin and return to circulation after immune complex-mediated sequestration. Proc. Natl. Acad. Sci. U.S.A 115, E1550–E1559 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zapata JC, Pauza CD, Djavani MM, Rodas JD, Moshkoff D, Bryant J, Ateh E, Garcia C, Lukashevich IS, Salvato MS, Lymphocytic choriomeningitis virus (LCMV) infection of macaques: A model for Lassa fever. Antiviral Res 92, 125–138 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.de la Torre JC, Molecular and cell biology of the prototypic arenavirus LCMV: Implications for understanding and combating hemorrhagic fever arenaviruses. Ann. N. Y. Acad. Sci 1171 (suppl. 1), E57–E64 (2009). [DOI] [PubMed] [Google Scholar]
- 62.Haaskjold YL, Bolkan HA, Krogh KO, Jongopi J, Lundeby KM, Mellesmo S, Garces PS, Josendal O, Opstad A, Svensen E, Fuentes LM, Kamara AS, Riera M, Arranz J, Roberts DP, Stamper PD, Austin P, Moosa AJ, Marke D, Hassan S, Eide GE, Berg A, Blomberg B, Clinical features of and risk factors for fatal Ebola Virus disease, Moyamba District, Sierra Leone, December 2014-February 2015. Emerg. Infect. Dis 22, 1537–1544 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kortepeter MG, Bausch DG, Bray M, Basic clinical and laboratory features of filoviral hemorrhagic fever. J. Infect. Dis 204 (Suppl. 3), S810–S816 (2011). [DOI] [PubMed] [Google Scholar]
- 64.Cui L, Lee YH, Thein TL, Fang J, Pang J, Ooi EE, Leo YS, Ong CN, Tannenbaum SR, Serum metabolomics reveals serotonin as a predictor of severe dengue in the early phase of dengue fever. PLoS Negl. Trop. Dis 10, e0004607 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ojha A, Nandi D, Batra H, Singhal R, Annarapu GK, Bhattacharyya S, Seth T, Dar L, Medigeshi GR, Vrati S, Vikram NK, Guchhait P, Platelet activation determines the severity of thrombocytopenia in dengue infection. Sci. Rep 7, 41697 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Makroo RN, Raina V, Kumar P, Kanth RK, Role of platelet transfusion in the management of dengue patients in a tertiary care hospital. Asian J. Transfus Sci 1, 4–7 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lye DC, Archuleta S, Syed-Omar SF, Low JG, Oh HM, Wei Y, Fisher D, Ponnampalavanar SSL, Wijaya L, Lee LK, Ooi EE, Kamarulzaman A, Lum LC, Tambyah PA, Leo YS, Prophylactic platelet transfusion plus supportive care versus supportive care alone in adults with dengue and thrombocytopenia: A multicentre, open-label, randomised, superiority trial. Lancet 389, 1611–1618 (2017). [DOI] [PubMed] [Google Scholar]
- 68.Rehman M, Ali MA, Siddiqui MA, Prophylactic platelet transfusion in dengue: A dilemma. J. Pak. Med. Assoc 67, 1961 (2017). [PubMed] [Google Scholar]
- 69.Khan Assir MZ, Kamran U, Ahmad HI, Bashir S, Mansoor H, Anees SB, Akram J, Effectiveness of platelet transfusion in dengue fever: A randomized controlled trial. Transfus. Med. Hemother 40, 362–368 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Arman M, Krauel K, Human platelet IgG Fc receptor FcγRIIA in immunity and thrombosis. J. Thromb. Haemost 13, 893–908 (2015). [DOI] [PubMed] [Google Scholar]
- 71.Kansay S, Singh H, Effect of introduction of single-donor apheresis platelets in dengue management: A comparative analysis of two consecutive dengue epidemics. J. Lab. Physicians 10, 173–178 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kostelijk EH, Fijnheer R, Nieuwenhuis HK, Gouwerok CW, de Korte D, Soluble P-selectin as parameter for platelet activation during storage. Thromb. Haemost 76, 1086–1089 (1996). [PubMed] [Google Scholar]
- 73.Kaufman J, Spinelli SL, Schultz E, Blumberg N, Phipps RP, Release of biologically active CD154 during collection and storage of platelet concentrates prepared for transfusion. J. Thromb. Haemost 5, 788–796 (2007). [DOI] [PubMed] [Google Scholar]
- 74.Snyder EL, Hezzey A, Katz AJ, Bock J, Occurrence of the release reaction during preparation and storage of platelet concentrates. Vox Sang 41, 172–177 (1981). [DOI] [PubMed] [Google Scholar]
- 75.Rank A, Nieuwland R, Liebhardt S, Iberer M, Grutzner S, Toth B, Pihusch R, Apheresis platelet concentrates contain platelet-derived and endothelial cell-derived microparticles. Vox Sang 100, 179–186 (2011). [DOI] [PubMed] [Google Scholar]
- 76.Sloand EM, Yu M, Klein HG, Comparison of random-donor platelet concentrates prepared from whole blood units and platelets prepared from single-donor apheresis collections. Transfusion 36, 955–959 (1996). [DOI] [PubMed] [Google Scholar]
- 77.Chao CH, Wu WC, Lai YC, Tsai PJ, Perng GC, Lin YS, Yeh TM, Dengue virus nonstructural protein 1 activates platelets via Toll-like receptor 4, leading to thrombocytopenia and hemorrhage. PLOS Pathog 15, e1007625 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hottz ED, Azevedo-Quintanilha IG, Palhinha L, Teixeira L, Barreto EA, Pão CRR, Righy C, Franco S, Souza TML, Kurtz P, Bozza FA, Bozza PT, Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood 136, 1330–1341 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McClary H, Koch R, Chisari FV, Guidotti LG, Relative sensitivity of hepatitis B virus and other hepatotropic viruses to the antiviral effects of cytokines. J. Virol 74, 2255–2264 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bachmann MF, Oxenius A, Mak TW, Zinkernagel RM, T cell development in CD8−/− mice. Thymic positive selection is biased toward the helper phenotype. J. Immunol 155, 3727–3733 (1995). [PubMed] [Google Scholar]
- 81.Chan VW, Meng F, Soriano P, DeFranco AL, Lowell CA, Characterization of the B lymphocyte populations in Lyn-deficient mice and the role of Lyn in signal initiation and down-regulation. Immunity 7, 69–81 (1997). [DOI] [PubMed] [Google Scholar]
- 82.Guidotti LG, Borrow P, Brown A, McClary H, Koch R, Chisari FV, Noncytopathic clearance of lymphocytic choriomeningitis virus from the hepatocyte. J. Exp. Med 189, 1555–1564 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McCausland MM, Crotty S, Quantitative PCR technique for detecting lymphocytic choriomeningitis virus in vivo. J. Virol. Methods 147, 167–176 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bergmeier W, Rackebrandt K, Schroder W, Zirngibl H, Nieswandt B, Structural and functional characterization of the mouse von Willebrand factor receptor GPIb-IX with novel monoclonal antibodies. Blood 95, 886–893 (2000). [PubMed] [Google Scholar]
- 85.Kakimi K, Lane TE, Wieland S, Asensio VC, Campbell IL, Chisari FV, Guidotti LG, Blocking chemokine responsive to gamma-2/interferon (IFN)-gamma inducible protein and monokine induced by IFN-gamma activity in vivo reduces the pathogenetic but not the antiviral potential of hepatitis B virus-specific cytotoxic T lymphocytes. J. Exp. Med 194, 1755–1766 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kakimi K, Isogawa M, Chung J, Sette A, Chisari FV, Immunogenicity and tolerogenicity of hepatitis B virus structural and nonstructural proteins: Implications for immunotherapy of persistent viral infections. J. Virol 76, 8609–8620 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Isogawa M, Furuichi Y, Chisari FV, Oscillating CD8(+) T cell effector functions after antigen recognition in the liver. Immunity 23, 53–63 (2005). [DOI] [PubMed] [Google Scholar]
- 88.Sitia G, Iannacone M, Aiolfi R, Isogawa M, van Rooijen N, Scozzesi C, Bianchi ME, von Andrian UH, Chisari FV, Guidotti LG, Kupffer cells hasten resolution of liver immunopathology in mouse models of viral hepatitis. PLOS Pathog 7, e1002061 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kapadia SB, Brideau-Andersen A, Chisari FV, Interference of hepatitis C virus RNA replication by short interfering RNAs. Proc. Natl. Acad. Sci. U.S.A 100, 2014–2018 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Livak KJ, Schmittgen TD, Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔ CT Method. Methods 25, 402–408 (2001). [DOI] [PubMed] [Google Scholar]
- 91.Junt T, Schulze H, Chen Z, Massberg S, Goerge T, Krueger A, Wagner DD, Graf T, Italiano JE Jr., R. A. Shivdasani, U. H. von Andrian, Dynamic visualization of thrombopoiesis within bone marrow. Science 317, 1767–1770 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Tph-1−/− mice require a material transfer agreement from Max Delbrück Center for Molecular Medicine, Berlin, Germany.