

Abstract
Background
Keratinocyte carcinomas such as basal cell carcinomas and squamous cell carcinomas are a major burden affecting morbidity and mortality in solid organ transplant recipients (SOTRs). Best treatment includes frequent skin checks for early detection and surgery for high incidence of skin cancers.
Sirolimus is an immunosuppressive drug which may reduce the burden of skin cancer but may be poorly tolerated when given orally. Topical sirolimus has been proven effective at reducing the burden of skin cancers in animal models, and its safety has long been established in children with tuberous sclerosis. A recent 12-week phase II trial of topical sirolimus suggested it was safe and effective at reducing the early signs of skin cancer in the absence of major side effects. The aim of the SiroSkin trial is to determine whether topical sirolimus can fill a major gap in current therapies by reducing the onset and number of new skin cancers thus reducing burden of disease and cost-effectiveness.
Methods
Protocol for a multi-centred phase III, participant- and clinician assessor-blinded, placebo-controlled randomised trial in SOTRs. A minimum 146 participants randomised 1:1 will be treated with 1% topical sirolimus versus placebo applied to the face on a regular basis for 24 weeks. Participation is 24 months in total—24 weeks of treatment and 18 months of follow-up. Outcomes include the number of keratinocyte carcinomas at 24 weeks of treatment compared to placebo and then at 12 and 24 months after initiation of treatment. Analysis will be as per protocol and intention to treat.
Discussion
The results of this trial will inform management strategies for skin cancers in SOTRs and provide evidence for cost-effectiveness.
Trial registration
Clinicaltrials.gov NCT05860881. Registered on June 15, 2023, and on anzctr.org.au (registration number NCT05860881).
Keywords: Sirolimus, Topical sirolimus, Skin cancer, Squamous cell carcinoma, Basal cell carcinoma, Actinic keratosis, Topical 5-fluorouracile, Solid organ transplant recipients
Introduction
Background and rationale {6a}
Keratinocyte carcinomas (KCs) are a major burden affecting mortality and morbidity in solid organ transplant recipients (SOTRs). SOTRs have one to two orders of magnitude higher risk and incidence ratio for developing squamous cell carcinomas (SCC) and basal cell carcinomas (BCC) [1, 2]. This translates to SOTR incidence rates for SCC of 379/1000 patient-years for heart transplant recipients and prevalence of 11% for SCC at any given time for kidney transplant recipients [3, 4]. Similar observations are seen with early skin cancers such as intra-epidermal carcinoma (IEC) and actinic keratosis (AK). In addition to higher rates occurring in SOTRs, BCCs and SCCs have a more aggressive course. Aggressive cancers occur in 2% of heart and lung transplant recipients over 2 years, which is a 66- to 83-fold higher standardised mortality ratio regarding KCs compared to the general population [5, 6]. Currently, the most common treatment for KC is surgical excision with adequate margins. However, this treatment option results in significant disease burden for SOTRs, especially as it is well established that there is an increased risk of additional skin cancers in a field of pre-cancerisation [7, 8]. Multidisciplinary clinics have been established in recent years to enable diagnosis and immediate treatment of skin cancers in SOTRs. One study reviewed 101 patients attending such a clinic in Brisbane, Australia, and recorded over 300 excisions of suspected lesions over 3 months of follow-up [9]. Overall, although surgery for individual KCs resulted in complete remission of individual lesions, it did not prevent the onset of additional cancers in the same field of exposure.
Aside from UV-related primary prevention such as sunscreen and barrier clothing, additional preventive or adjuvant measures have been suboptimal in reducing the onset of new KCs. Field therapy—usually photodynamic therapy or topical 5-fluorouracil (5-FU/Efudix)—of photodamaged skin or actinic keratosis is often recommended for SOTRs. Photodynamic therapy has shown only a clear preventative role in SCC development if performed in unrealistic 4-weekly cycles [10–12]. Topical 5-FU has shown short term benefit with a 75% reduction in SCCs in immunocompetent patients at 1 year [13]. However, this effectiveness is not found at 2–5 years subsequently. In addition, topical 5-FU appears not to prevent the formation of BCCs [13, 14]. There is some deliberation as to whether imiquimod or other immunotherapies should be used in transplant patients. Regardless, their effectiveness is mainly for AKs and SCC rather than BCC.
Oral chemopreventive regimens have been trialled specifically in SOTRs. However, significant side effects have prevented widespread use. Retinoid (acitretin) therapy is an additional adjuvant treatment which prevents occurrence of new SCCs. Doses of 25 to 30 mg per day (0.3 mg/kg) reduce SCCs by 13% in treated groups compared to a 28% increase in placebo groups [15]. Real-world application of acitretin is limited in SOTRs due to high rates (52%) of side effects and withdrawal from trials [16]. Similarly, oral fluorouracil (capecitabine) reduces SCC and BCC incidence (more than 50% reduction) compared to the pre-treatment period [17, 18]. However, 70% of patients experienced significant side effects and dose adjustment with interrupted cycles of therapy. More recently, nicotinamide has been proposed as a chemopreventive measure for KCs, but not in SOTRs [19, 20]. Modulation of the immunosuppression was another strategy investigated to prevent skin cancer formation. Compared to other immunosuppressants, mammalian target of rapamycin inhibitors (mTORi), such as sirolimus (rapamycin) or everolimus, have suggested protective effects on SCC or BCC incidence [21]. In patients with SCCs, changing calcineurin inhibitors to sirolimus resulted in a nearly 50% reduction in SCC risk [22–24]. Unfortunately, these trials of oral sirolimus had poor tolerance with approximately 50% of patients withdrawing from the trial and 94% in the sirolimus group reporting at least one adverse event. The side effects associated with oral sirolimus are commonly difficult to manage and usually result in withdrawal of the drug. These include acne, albuminuria, mouth ulceration, oedema, rash, and pneumonitis. Despite poor tolerance, the benefit oral sirolimus achieved in reducing skin cancer occurrence suggested sirolimus may have specific anticancer properties. To circumvent the side effects of systemic sirolimus therapy, topical sirolimus has recently been explored [24]. Its use has proven effective in reducing the skin cancer burden in animal models and is safe on the face of patients with tuberous sclerosis [25, 26]. In 2019, we conducted a pilot randomised, double-blind, placebo-controlled, phase I–II, feasibility, and safety study of the use of topical sirolimus in chemoprevention of skin cancer (ACTRN12618001961235) [25]. This early 12-week clinical trial suggested topical sirolimus to be safe and effective, as it reduced the early signs of skin cancer without any major side effects, encouraging the pursuit of a larger and longer-term study.
In this proposed phase III randomised, participant- and clinician-blinded, placebo-controlled study, we propose using 1% topical sirolimus applied daily to the face to determine whether this reduces onset of new skin cancers and therefore reduce the burden of disease in terms of number of biopsies, surgeries, hospitalisations, and death.
Objectives {7}
The primary objective of this trial is to determine if 1% sirolimus applied topically to the face on a daily basis for 24 weeks is effective at reducing the number of KCs in the treated area at 6, 12, and 24-months compared with placebo.
The secondary objectives of this trial are whether the intervention, compared with placebo:
Reduces the occurrence of KCs in the treated area
Reduces the number and the occurrence of biopsy-proven SCCs
Reduces the number and occurrence of IECs, BCCs, and subtypes of SCCs or BCCs
Reduces the number of facial AKs
To determine the cost-effectiveness of using topical sirolimus therapy in comparison to the current standard of care, being surgical intervention, in the management of KCs
- To determine the number and occurrence of intervention-related side effects, by type, by reviewing any urgent safety measures (USMs), suspected unexpected serious adverse reaction (SUSARs), serious adverse events (SAEs), and AEs occurring up to 30 days post-EOT, assessed by CTCAE v5.0
- Type
- Grade (1–5)
- Relatedness to treatment (unrelated, possibly related, probably related, definitely related)
The feasibly of the 1% sirolimus treatment in SOTRs by reviewing completion of the 24-week application course, total number of doses applied during the course, cost of treatment (whether topical sirolimus therapy or current standard of care + placebo), quality-adjusted life years (QALY’s), Basal and Squamous Cell Carcinoma Quality of Life (BaSQoL) questionnaire score and EQ-5D-5L questionnaire score
To evaluate the participants’ experience by monitoring side effects, issuing participant surveys (BaSQoL and EQ-5D-5L questionnaires), determining the number of participants who completed the 24-week application course and the number of doses applied during this time.
Trial design {8}
The SiroSkin study is a phase III, double-blind, multi-centre, parallel-arm, randomised, placebo-controlled superiority trial. This framework was chosen to determine whether 1% sirolimus is more effective than placebo in preventing or treating the targeted condition.
Participants meeting the eligibility criteria will be randomised in a 1:1 ratio to either the intervention arm (1% sirolimus) or the control arm (placebo). Randomisation is stratified by site and by whether participants have used topical 5-FU on the face as a field therapy within the last 12 month (*Additional Statistical Considerations), allowing us to account for potential differences in baseline skin treatment exposure. Blinding measures are in place to minimise bias, with study personnel, trial staff, and participants blinded to allocation. Only pharmacy personnel and specific designated trial contacts will remain unblinded to facilitate treatment preparation and emergency situations if unblinding is necessary. Figure 1 provides a schematic overview of the trial design, detailing all planned trial visits.
Fig. 1.

SiroSkin trial schema
Methods: participants, interventions, and outcomes
Study setting {9}
This research will be overseen by the Melanoma and Skin Cancer (MASC) research centre, Monash University, and will take place across six locations spanning Victoria (VIC), New South Wales (NSW), and Queensland (QLD) from November 2023 to May 2027. In VIC, it will occur at the Alfred Hospital and Skin Health Institute; in NSW, at Royal Prince Alfred Hospital and Westmead Hospital; and in QLD, at Prince Charles Hospital and Princess Alexandra Hospital.
Eligibility criteria {10}
Inclusion criteria:
Participants may be included in the study if they meet all of the following criteria:
Are aged 18 years or older and able to provide informed consent
Have received an organ transplant equal to or greater than 12 months prior to commencing the trial
Have had at least 1 SCC/BCC in the 5 years prior to commencing the trial
Have at least 5 keratotic lesions on their face at inclusion
Exclusion criteria:
Participants will be excluded from the study for any of the following reasons:
Are currently receiving sirolimus or everolimus orally*
Have a skin cancer on their face requiring excisional surgery**
Have an open wound on their face requiring treatment
Are pregnant or planning to become pregnant in the next 6 months***
Anticipate elective medical events which may prevent daily cream application
Are unable to provide informed consent, complete questionnaires, and attend trial site for visits
Are participating in another clinical trial with an investigational drug/device aiming to reduce skin cancers or affect level of immunosuppression
Planning to move overseas within 2 years
Participants who receive nicotinamide or acitretin can be included in the study.
*Participants are eligible to join the study after ceasing treatment and after a washout period of 16 days for sirolimus and 8 days for everolimus.
**Once treatment of the lesion is completed these participants can be re-screened.
Who will take informed consent? {26a}
Based on research governance in Australia, all members of the research team will undergo training in Good Clinical Practice (GCP). Participants meeting the eligibility criteria will be provided with information about the SiroSkin trial and given the opportunity to discuss their voluntary participation in the trial with their families and doctors. Each participant will sign a participant information consent form prior to any baseline assessment being performed. Once enrolled in the SiroSkin trial, participants will be coded with unique study identifiers.
Patients will attend an appointment with a healthcare professional at the study site, where their eligibility to participate will be assessed. The healthcare professional will explain the study in detail and answer any questions the patients may have. Full written informed consent to participate will then be obtained.
The person obtaining informed consent will be a suitably trained and competent healthcare professional who, in the opinion of the principal investigator (PI), can provide a full, unbiased explanation of the study, including its benefits and risks, to the potential participant.
Additional consent provisions for collection and use of participant data and biological specimens {26b}
We will request consent for review of participants’ medical records as well as for the collection of tumour samples for future genetic research on keratinocyte carcinomas.
Interventions
Explanation for the choice of comparators {6b}
Site staff undertake eligibility screening and randomisation on the MASC Trials web-based randomisation portal available at www.masc.org.au during the ‘screening and baseline’ visit. Participants will be randomised in a 1:1 ratio to either the intervention or control group and will be stratified by site. The randomisation will be stratified, as seen in Fig. 2, by site and use of topical 5-fluorouracil (5-FU/Efudix) in the past 12 months on the face as a field therapy. Site, topical 5-fluorouracil (5-FU/Efudix) use in the past 12 months on the face as a field therapy, baseline risk level, and acitretin use at baseline will be controlled for as covariates in modelling. Covariates to be controlled for at the analysis stage will include baseline risk level—higher (≥ 10 SCCs in the past) vs. lower (< 10 SCCs in the past)—and baseline use of acitretin. The intervention topical cream consists of sirolimus 1% in a vehicle, while the placebo topical cream consists of the vehicle only. The placebo cream consists of a commonly used proprietary base supplied by Medisca pharmaceutical company. The base is designed to maintain stability with the addition of active components (such as sirolimus) and permeation, as well as non-comedogenic and hypoallergenic properties, rendering it appropriate for pharmaceutical purposes. This base is already used in the clinic for the preparation of many compounded creams and is essentially acting as a moisturiser.
Fig. 2.
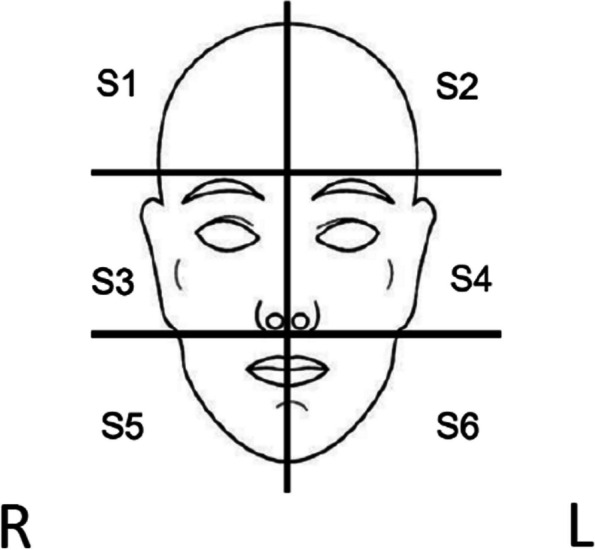
Face sextants numbered 1 to 6, including ears. R, right; L, left
Intervention description {11a}
Both topical creams (intervention and placebo) will be prepared by the compounding pharmacy (Seed Pharmaceuticals a level 2, non-sterile laboratory Pharmacy, located in Brisbane, Australia) according to the formula provided by the study team. All compounding ingredients including the sirolimus powder can be stored at ambient temperature (room temperature) and as such do not require any temperature monitoring (before or after compounding).
The compounding pharmacy is responsible for the compounding, dispensing and distribution of the creams as they will be unblinded to the trial intervention group allocation. Via a trial database automation system, pharmacy staff will receive three individual email notifications to confirm the following: (a) a participant’s treatment allocation, (b) name and address, and (c) confirmation of undetectable blood sirolimus levels at baseline. Receipt of these notifications initiates compounding of the creams (1–2 days) and delivery to the participant (2–3 days). A signature receipt is required on arrival to reduce the possibility of lost packages and delayed delivery. Any unsigned failed-to-deliver packages will be delivered to the nearest postal service for participant collection.
Criteria for discontinuing or modifying allocated interventions {11b}
Dose modification
Permanent dose modifications (i.e. for the duration of the intervention) are not permitted. However, temporary dose adjustments will be permitted for the care of medical and surgical interventions on the face as well as hospital admissions or health interventions preventing the application of the study drug for a short period, as detailed in the section below.
Dose delay
Any adverse event (AE) can be considered as a reason for a dose delay. AEs will be classified as either systemic or cutaneous and related to the face. Dose delay criteria apply for all cutaneous AEs related to the face. For systemic adverse events, a dose delay of up to 2 weeks is acceptable to allow for recovery. In the population of transplant recipients, hospitalisation and medical events are expected. If these events prevent participants from applying the intervention daily, a 2-week delay is acceptable and will not result in the participant being withdrawn from the study.
If a skin cancer develops on the face and requires treatment during the 24-week intervention, the investigator may opt for a dose delay for the entire face for 2 weeks. Alternatively, treatment can continue areas of the face unaffected by the skin cancer treatment. In such cases, the delay on one (or more) sextant(s) of the face will not be considered a delay overall. Interruption of treatment in specific sextants of the face will not exclude the participant from analysis. However, the interrupted sextant/s will not be analysed if they receive less than 8 weeks or if the interruption exceeds 6 weeks in the first 2 months.
A maximum of 3 dose delays per study participant are permissible. Dose delays will not result in an extension of the 6-month treatment period.
If a participant requires a biopsy, skin excision, or spot treatment of a lesion on the face during the trial, interruptions in cream application will be allowed. The face will be divided into 6 sextants (including ears) (see Fig. 2), and cream application will be paused in the corresponding sextant(s) to enable successful treatment and healing. Once healed, sirolimus application to the entire face will resume. A break from application to the whole face will not extend the treatment duration (24 weeks in total, including breaks as permitted by protocol), and sextant(s) not receiving topical application will be documented on the participant’s nightly application recording sheet.
Criteria to resume treatment
Participants may resume treatment when the study treatment-related AE(s) resolve to grade ≤ 1 as per Common Terminology Criteria for Adverse Events v5.0 (NCI CTCAE v5.0) or baseline value, with the following exceptions:
This is the participant’s third dose delay
Participant has a contact hypersensitivity to one of the intervention ingredients as proven by reapplication-triggered contact eczema or by reoccurrence of a lichenoid drug reaction
Participants who have not previously experienced a grade 3 study drug-related skin AE may resume treatment in the presence of grade 2 skin toxicity
Participants with AEs or SAEs unrelated to the drug who have not recovered within 2 weeks of drug delay. This is particularly true regarding systemic AEs
Strategies to improve adherence to interventions {11c}
All study drugs are compounded and distributed by a single compounding pharmacy to minimise batch variation, maintain stock control, and minimise dosage delays due to drug supply.
At the baseline visit, participants are provided with recording sheets and given instructions on how to complete these daily following the application of the study drug. Additionally, participants receive monthly follow-up calls at the end of months 1 and 2 to encourage drug adherence and follow-up on any AEs or SAEs. At the 3- and 6-month clinic visits, daily recording sheets are collected, and cream bottles are weighed to provide an indication of drug adherence.
Relevant concomitant care permitted or prohibited during the trial {11d}
If a participant reports taking any over-the-counter or prescription medications, or if administration of any medication becomes necessary from 14 days prior to sirolimus administration through to the thirty days after the last dose of sirolimus, the name of the medication/therapy, dosage information including dose, route and frequency, date(s) of administration including start and end dates, and reason for use must be recorded. Due to its potential chemo preventive properties, vitamin B3 (nicotinamide) use will be recorded. Use of any other vitamin or herbal supplement will not be considered a concomitant medication. Any concomitant medications administered for an SAE reported after the 30-day follow-up period after the last dose will still be required to be documented.
- The following medications are prohibited during the trial:
- Oral sirolimus
- Oral everolimus
- Agents potentially affecting the development of skin cancers. Examples include immune checkpoint inhibitors such as nivolumab, pembrolizumab, and cemiplimab, chemotherapeutic agents such as capecitabine, 5-FU infusion, or oral 5-FU, and radiation therapy of the face as a field treatment (VMAT)
- The following therapies are permitted in the study:
- Nicotinamide
- Acitretin
- Topical 5-FU
- Adjuvant radiotherapy can be undertaken on the face, following the sextant method described in Fig. 2
- Participating in clinical trials aiming to reduce skin cancers or affect the level of immunosuppression is forbidden
Provisions for post-trial care {30}
Post-trial, all participants will remain under the care of their standard care provider who will have been the referring doctor for participation in this trial.
Outcomes {12}
Primary endpoint:
The primary endpoint of the trial will be the number of KCs on the treated area at the completion of 24 weeks of topical 1% sirolimus then at 12 and 24 months after initiation of treatment.
Secondary endpoints:
The occurrence of keratinocyte carcinomas on the treated area at the completion of 24 weeks of topical 1% sirolimus then at 12 and 24 months after initiation of treatment
The number of biopsy-proven SCC at the completion of 24 weeks of topical 1% sirolimus then upon completion of 12 and 24 months follow-up on the treated area
The occurrence of biopsy-proven SCC at the completion of 24 weeks of topical 1% sirolimus then at 12 and 24 months after initiation of treatment on the treated area
The number of intraepidermal carcinomas (IECs), BCCs, and subtypes of SCCs or BCCs at each of the aforementioned time-points on the treated area
The occurrence of intraepidermal carcinomas (IECs), BCCs, and subtypes of SCCs or BCCs at each of the aforementioned time-points on the treated area
The number of facial actinic keratosis of each participant at recruitment compared to at 3, 6, 12, and 24 months after initiation of treatment on photographic images and counts
The cost-effectiveness of utilising topical sirolimus therapy on SOTRs in comparison to the current standard of care, being surgical intervention, in the management of KCs
- The cost-effectiveness of utilising topical sirolimus therapy on SOTRs in comparison to the current standard of care, being surgical intervention, in the management of KCs
- Type
- Grade (1–5)
- Relatedness to treatment (unrelated, possibly related, probably related, definitely related)
The feasibility of the 1% sirolimus treatment in SOTRs, by reviewing completion of the 24-week application course, total number of doses applied during the course, cost of treatment (whether topical sirolimus therapy or current standard of care + placebo), quality-adjusted life years (QALYs), Basal and Squamous Cell Carcinoma Quality of Life (BaSQoL) questionnaire score, and EQ-5D-5L questionnaire score
To evaluate the participants’ experience by monitoring side effects, issuing participant surveys (BaSQoL and EQ-5D-5L questionnaires), determining the number of participants who completed the 24-week application course and the number of doses applied during this time
Participant timeline {13}
Follow-up visits
Participants will attend the site for study visits at baseline, 3 and 6 months and complete follow-up visits at 12 and 24 months after initiation of treatment. Participation is 2 years in total—24 weeks of treatment and 18 months of follow-up. The length of follow-up for each participant will be 18 months after the end of the 24-week treatment.
Pre-randomisation/registration assessments
Before participants are randomised into the study, the following procedures must be performed, and information obtained to ensure that the patient is eligible for participation:
Participant must provide written informed consent
Review eligibility criteria to ensure all conditions are met
Confirmation of at least 5 keratotic lesions on the face
Medical history (including confirmation of at least one BCC/SCC in past 5 years)
Determination of number of past KC (more or less than 10) and the use of acitretin
For women of child-bearing potential, a blood/urine pregnancy test results up to and including 7 days prior to screening and baseline visit or can be collected from the medical record
Review patients’ participation in any other clinical trials
Assessment at the baseline visit
Once the informed consent procedures have been followed, participant’s medical history and eligibility criteria have been checked, the participant is ready to be randomised onto the study.
Instructions on how to randomise a participant using the randomisation system are explained in the operations manual. Once the participant has been randomised, the arm to which the participant has been allocated to will be shared with the central pharmacy. The participant timeline is shown in Table 1.
Table 1.
Participant timeline

D day, IP investigational product
*Women of childbearing potential (WOCBP) must have a negative serum or urine pregnancy test within 7 days (168 h) prior to their baseline and screening visit (D0) or agree to use adequate methods of contraception for the duration of drug administration
**Blood test for sirolimus levels—collect a 4 ml EDTA tube (purple tube) and record the time the cream was last applied (e.g. 10 pm and date)
aSites are requested to indicate any out-of-range results of biochemistry and haematology tests performed as part of standard-of-care up to and including 3 months prior to the baseline visit and upload de-identified results into the study database
bSites are requested to indicate any out-of-range results of biochemistry and haematology tests performed as part of standard-of-care up to and including 14 days prior to the visit and up to and including 7 days after the visit and upload de-identified results into the study database
The following baseline assessments need to be performed:
Physical examination (including weight)
Skin check
Number of keratotic lesions* on the face, circled and imaged
Full medical history
Review of all existing medical conditions and document use of concomitant medications
Listing of any pre-existing conditions
Quality of life using the BaSQoL
Utility-based QOL using the EQ-5D-5L
Blood test, sirolimus levels appendix V
Collect results for biochemistry and haematology from medical record, if available
Hand out daily recording sheets
Confirm Investigational Product (IP) start date with participant. **
*If standard of care treatment occurs (e.g. cryotherapy of keratotic lesion), the AKs should not be counted and recorded.
**Automated REDCap notifications will be issued to the site coordinator and the participant once the IP has been delivered to the participant. The site coordinator should phone the participant to confirm IP delivery and IP start date.
The above assessments are to be performed prior to randomisation to ensure they reflect true baseline data. Once baseline visit assessments have been performed and eligibility is still current, the participant can be randomised via the MASC randomisation webpage, as described.
Phone visits at months 1 and 2 (D30 and D60 visits)
The following assessments need to be performed:
Verbal check for cream application compliance
Review of any AEs
Review of any SAEs
Changes to concomitant medications
Study visits at months 3 and 6 (D90 and D180 visits)
The following baseline assessments need to be performed:
Skin check
Number of keratotic lesions on the face, circled and imaged
Quality of life using the BaSQoL (month 6 only)
Utility-based QOL using the EQ-5D-5L (month 6 only)
Blood test, sirolimus levels (month 3 only)1
Collect results for biochemistry and haematology from medical record, if available
Collect daily recording sheets
Hand out daily recording sheets (month 3 only)
Check for cream application compliance: recording of pump weight
Review of any AEs
Review of any SAEs
Changes to concomitant medications
Review of participation in other clinical trials
Follow-up visit assessments: duration of 2 years (12 and 24 months)
The following assessments need to be performed at each follow-up visit:
Skin check
Number of keratotic lesions on the face, circled and imaged
Quality of life using the BaSQoL
Utility-based QOL using the EQ-5D-5L
Review of any AEs
Review of any SAEs
Changes to concomitant medications (specifically use of topical 5-FU on face)
Review of participation in other clinical trials
Unscheduled visits
Unscheduled visits and procedures may be conducted at the investigator’s discretion for the safety follow-up of the patient. Such visits and procedures must be fully recorded in source documents regarding the purpose and outcome of the visit/assessment.
Study completion
If a participant has completed the schedule of follow-up visits for 2 years, no further follow-up is necessary. A final study visit will be performed at month 24.
If a participant withdraws consent to participate, please complete the ‘Study Discontinuation Form’ at the final study visit.
Similarly, if a participant dies, please ensure that the ‘Study Discontinuation Form’ is completed and a copy of the death certificate or discharge summary provided. Reason for death will be considered in the final analysis.
Definition of end of trial
The end of the study is defined as the final visit of the last participant.
Sample size {14}
Assuming that all of the IECs detected at 24 weeks in our preliminary study will become invasive at a later stage, we used the average number of IECs per participant to inform the sample size calculation of this trial. Based on that pilot data, in which 12 IECs were diagnosed in 29 placebo participants in 2 years, and 4 IECs were detected in 29 sirolimus participants in the same time period, we assume that the underlying diagnosis rates in the placebo and sirolimus arms are 0.414 and 0.138 SCC/participant respectively over 2 years. Assuming that the IEC rate within each arm is Poisson distributed, to achieve power of 80% with a two-sided type I error rate of 0.05, 47 participants are required to be accrued in each arm with a 1:1 enrolment ratio between arms. Factoring in the expected high level of trial dropouts (36%), we will aim to recruit a minimum of 73 participants per arm, totalling 146 participants.
Recruitment {15}
Patients will be identified and approached by their medical team. They will receive the patient information brochure, and if they show interest in participating, a baseline visit will be arranged to provide further details about the study and obtain their consent.
Assignment of interventions: allocation
Sequence generation {16a}
Randomisation will be carried out by the sponsor at the Monah University, using the MASC Trials web-based randomisation portal available at www.masc.org.au during the ‘screening and baseline’ visit. Participants will be randomised with a 1:1 ratio between arms.
Concealment mechanism {16b}
Allocation to treatment groups is random and is only revealed to the unblinded contacts in the trial through email notifications sent from the web-based randomisation system. Access to sirolimus blood results is only provided to unblinded trials coordinating contacts and only made available to researchers if deemed to be a safety concern for the participant or affecting eligibility status at baseline.
Implementation {16c}
All participants must provide informed written consent prior to randomisation. Delegated site staff will undertake the consent process followed by review of the participant’s medical history ensure all eligibility criteria are met. After confirmation of eligibility, the participant is randomised into the study, and the arm to which the participant has been allocated will be shared with the central pharmacy to facilitate the compounding of the study drug.
Assignment of interventions: blinding
Who will be blinded? {17a}
This study design is double-blind, where neither the participant nor any of the investigators or staff who are involved in the treatment or clinical evaluation of the participants will be aware of the treatment group to which the participant has been assigned. In the event that the treatment assignment for a participant becomes known to the investigator or other study staff involved in the management of trial participants, the trial coordinating centre will be notified immediately. MASC Trials staff and site staff will be blinded to arm allocation.
Pharmacists will know whether participants are receiving the intervention or control, as the pharmacy is not involved in assessing the participants or data collection.
In order to conceal arm allocation, the packaging of the topical cream provided by the pharmacy to the participant will be identical and will not reveal ingredients.
Procedure for unblinding if needed {17b}
If, at any time, knowledge of treatment allocation becomes essential for participant safety, the trial blinding may be broken. The Investigator will notify MASC-RC staff immediately and promptly explain the reason for any unblinding. All unblinding will be documented by recording the date of unblinding and the reason in the relevant case report form. Contact information for breaking the blind in an emergency will be made available to every trial site. Under certain circumstances, unblinding of treatment assignment for a participant may be required following a serious adverse event; for example, if an expedited regulatory report is required.
Data collection and management
Plans for assessment and collection of outcomes {18a}
Personal identifying information (names, dates of birth, contact details) and health information (disease/diagnosis, medical history) will be collected throughout the trial from source documents including the participant’s medical records, hospital charts, questionnaires completed by the participant, histopathology reports, and operation reports as well as the results of diagnostic tests and laboratory results. Source documents pertaining to the trial and signed PICFs must be maintained by investigational sites. Sites must complete all appropriate data entry fields as specified on the forms. The investigator will be asked to confirm the accuracy of completed CRFs by signing forms as indicated. Each site participating in the trial will maintain a site-specific source document plan that will document the source, i.e. original recording, for each discrete data item or category of data items collected for the trial. This source document plan, signed and dated by the site principal investigator, will be prepared prior to the recruitment of the first participant and will be filed in the site’s Investigator Site File.
Data collection methods
Trial data will be recorded in full using electronic CRFs (eCRFs) of the secure online trial-specific web database (REDCap [26]), provided to each site (Table 1) at scheduled visits. Direct entry into eCRFs is the preferred method of data collection, as it is time-efficient and eliminates transcription errors. If a site does not have access to eCRFs, hardcopy forms will be provided. These form source data and will need to be stored at the site.
Quality of life
For QoL assessment, the following publicly available research data collection tools will be used:
Basal and Squamous Cell Carcinoma Quality of Life (BaSQoL) [27]
EuroQol-5 Dimension 5-Level scale (EQ-5D-5L) [28]
The BaSQoL tool is relatively new and assesses health-related quality of life (HRQoL) in participants, covering the experiences of skin cancer treatment, diagnosis-related issues, and long-term behavioural changes. The difference in mean BaSQoL scores between the sirolimus and placebo groups will be used to determine the impact of the intervention on participants’ quality of life. This will be of interest on its own but in addition, we will use the findings to inform the cost-effectiveness analysis as an extra outcome measure. The mean scores for each domain will be calculated; however, the BaSQoL has no summary score or index.
Descriptive statistics (means and standard deviations) will be provided for the BaSQoL domains. Student’s t-tests will identify statistically significant differences between study groups.
Cost-effectiveness
The cost-effectiveness of topical sirolimus will be evaluated by quantifying and comparing the costs and effects in both arms. We will undertake a within-trial cost-effectiveness analysis. Specifically, we will measure and value the costs of the intervention and all healthcare interactions (including cost of sirolimus cream, diagnosis and treatments of skin lesions and managing adverse events) and the benefits of topical sirolimus. Skin procedures and treatments will be collected from clinical chart review and valued using national costing reports. The benefits will be measured by comparing between arms (a) quality-adjusted life years (QALYs), (b) skin-cancer related quality of life using the BaSQoL questionnaire, and (c) the number of skin cancers. The EQ-5D-5L will be applied at each time point and used to calculate QALYs. As costs and EQ-5D-5L scores are typically skewed, we will consider generalised linear models with appropriate family and link functions to deal with non-normal distributions. Cost and QALY data will be combined to calculate an incremental cost-effectiveness ratio (ICER) and net monetary benefit (NMB) statistic from the health system perspective. Seemingly unrelated regression (SUR) will be used, if appropriate, to account for the correlation between the costs and the QALYS. The nonparametric bootstrapping approach will be used to determine the level of sampling uncertainty surrounding the mean ICER by generating 10,000 estimates of incremental costs and benefits. Several sensitivity analyses will be undertaken to explore uncertainties surrounding key parameters in the economic evaluation. The results for complete cost and quality of life data (i.e. those with no missing data) as well as a strict per-protocol analysis of the data will be provided to identify the impact of missing data on the analysis and any sensitivity to protocol violations.
Use of the data
For all hypothesis tests, a 2-sided p-value of less than 0.05 will be deemed significant. A statistician will be involved throughout the statistical analysis.
The data will be retained long-term following the mandatory archive period (at least 15 years from completion of the trial) for potential use in future related research, also specified in the PICF. For sites located in Queensland, consent for trial participation records relating to consent for the participation of an individual in a research trial will be kept for 25 years. We plan to include the trial in the HeSANDA platform Health Data Australia [29].
Planned subgroup analyses
Subject to availability of sufficient data, the primary and secondary endpoints will be evaluated in the following sub-groups:
Sites
Topical 5-FU use in the past 12 months (Y/N)
Risk level—higher risk vs. lower risk
Acitretin use at baseline (Y/N)
Findings arising from sub-group analyses will be considered to be hypothesis generating only.
Plans to promote participant retention and complete follow-up {18b}
This trial has been designed to promote participant retention to completion and to increase recruitment. Strategies to achieve this include allowing the inclusion of participants with topical 5-FU use within the last 12 months and altering the topical formulation from a gel to a cream to make it more tolerable.
Evaluable participants who do not commence the study treatment (i.e. participant withdraws or is discontinued prior to first application of cream) will be replaced (Fig. 3).
Fig. 3.

Drug discontinuation and participant replacement
Analysis populations
Participants who discontinue the study drug due to treatment-related toxicities will still contribute to the safety analyses, as they will have met the safety endpoint. Where possible, these participants will still undergo ongoing follow-up assessments as per study schedule.
Whole face treatment interruption
Participants will be discontinued from the study and omitted from the ITT analysis if they have:
-
i)
Breaks totalling more than 6 weeks,
-
ii)
A single break of more than 2 weeks; or
-
iii)
More than 3 breaks.
Intention-to-treat (ITT) population
The ITT population includes all participants who were originally allocated to each treatment group, regardless of their adherence to the treatment protocol.
For this study, the ITT population will include:
Participants who have less than 6 weeks of discontinuation during the 24-week intervention period
Participants who discontinue therapy for more than 6 weeks but had compliant intervention for more than 8 weeks
Participants who have whole face treatment interruptions but do not meet the criteria for exclusion listed below
Participants will be excluded from the ITT analysis if they:
Use the intervention for less than 8 weeks over the 24-week period
Have, during the first 2 months of treatment, breaks totalling more than 6 weeks, a single break of more than 2 weeks, or more than 3 breaks
-
2.
Per-protocol (PP) population
The PP population includes only those participants who completed the study according to the protocol without significant deviations. For this study, the PP population will include:
Participants who adhered strictly to the treatment regimen without exceeding the permissible dose delays
Participants who did not exceed the allowed 3 dose delays per study participant
Participants who did not have whole face treatment interruptions totalling more than 6 weeks during the first 2 months or a single break of more than 2 weeks or more than 3 breaks
Participants will be excluded from the PP analysis if they:
Have any significant deviations from the protocol, such as using the intervention for less than 8 weeks over the 24-week period
Have interruptions in treatment exceeding the specified limits (more than 6 weeks in total during the first 2 months, a single break of more than 2 weeks, or more than 3 breaks)
The ITT population is our first endpoint analysis; it includes all participants who were randomised and meet the minimum usage criteria, reflecting a real-world scenario and preserving the benefits of randomisation.
The PP population includes only those participants who strictly adhered to the protocol, providing a measure of efficacy under ideal conditions.
Data management {19}
Overview
The principal investigator is responsible for storing essential trial documents relevant to data management and maintaining a site-specific record of the location(s) of the site’s data management-related essential documents.
The principal investigator is responsible for maintaining adequate and accurate source documents that include all key observations on all participants at their site. Source data will be attributable, legible (including any changes or corrections), contemporaneous, original, accurate, complete, consistent, enduring, and available. Changes to source data (hardcopy and electronic) must be traceable, must not obscure the original entry, and must be explained where this is necessary. A site-specific source document plan will be maintained to indicate the location(s) of source documents.
The principal investigator will also maintain accurate case report forms (CRFs) (i.e. the data collection forms) and be responsible for ensuring that the collected and reported data is accurate, legible, complete, entered in a timely manner, and enduring. To maintain the integrity of the data, any changes to data (hardcopy and electronic) must be traceable, must not obscure the original entry, and must be explained where this is necessary.
Any person delegated to collect data, perform data entry, or sign for data completeness will be recorded on the delegation log and will be trained to perform these trial-related duties and functions.
Storage and access
Hard copy data will be stored by the site in a locked cabinet or locked room in a secure location, accessible to the research team only. Coded CRFs should be stored separately from PICF, as the latter contain identifiable information (i.e. patients’ names).
Electronic data will be securely stored in Monash’s REDCap database system and in files stored in Monash’s network file servers, which are backed up nightly. Files containing private or confidential data will be stored only in locations accessible only by appropriate designated members of the research team.
REDCap is hosted on Monash infrastructure and is subject to the same security and backup regimen as other systems (e.g. the network file servers). Data is backed up nightly to a local backup server, with a monthly backup taken to tape and stored offsite. REDCap maintains an audit trail of data create/update/delete events that is accessible to project users who are granted permission to view it. Access to REDCap will be provided via a Monash user account or (for external collaborators) via a REDCap user account created by the Monash system administrator. The permissions granted to each user within each REDCap project will be controlled by, and will be the responsibility of, the trial team delegated this task by the principal investigator. REDCap has functionality that makes adding and removing users and managing user permissions straightforward. All data transmissions between users and the REDCap server are encrypted. The instructions for data entry to REDCap must be read and the training log signed prior to personnel commencing data entry on REDCap.
Authorised representatives of the sponsoring institution as well as representatives from the HREC, Research Governance Office, and regulatory agencies may inspect all documents and records required to be maintained by the investigator for the participants in this trial. The trial site will permit access to such records.
Confidentiality {27}
The study will be conducted in accordance with applicable Privacy Acts and Regulations. All data generated in this trial will remain confidential. All information will be stored securely at the MASC-Research Centre and will only be available to staff directly involved with the trial.
Personal data identifying trial participants will be held securely at the sites according to local institutional requirements for the purpose of follow-up after the conclusion of the protocol-specified period. Sites may be asked to submit copies of source documents to the MASC-Research Centre, e.g. pathology reports; however, all reports will have participant identifiers redacted. Source documents will be identified through use of a unique participant trial number assigned to the trial participant and initials.
Plans for collection, laboratory evaluation, and storage of biological specimens for genetic or molecular analysis in this trial/future use {33}
Tumour blocks and fresh biopsies
Paraffin-embedded tumour blocks from biopsied/excised lesions (taken as part of standard-of-care) may be collected and stored for future translational work related to the project. Ten Superfrost Plus sections of 5 μm may be provided as an alternative. Samples will be sent to the Translational Research Institute in Brisbane and stored within a secure laboratory with access limited to authorised personnel only. Samples will be stored until no longer required, at which point they will be destroyed by incineration. All samples will be identified by participant ID. This component of the research is optional and subject to sufficient funding.
Statistical methods
Statistical methods for primary and secondary outcomes {20a}
The data will initially be analysed using a Poisson regression model with the number of KCs per participant as the dependent variable and treatment arm (sirolimus vs. placebo) as the independent (predictor) variable. Site, 5-FU use in the past 12 months, baseline risk level (higher risk vs. lower risk), and use of acitretin at baseline will be controlled for as covariates. Overdispersion will be assessed by plotting the standardised residuals against predicted values for the Poisson model and comparing with those for a negative binomial model. If, based on these plots, the negative binomial model is found to improve the fit substantially, then the Poisson regression model will be replaced by a negative binomial model and conclusions will be based on the negative binomial model. The point estimate for the treatment arm effect and its 95% CI will be provided. This method will be repeated for each of the time points of interest (after 24 weeks of treatment and at 12 and 24 months after initiation of treatment).
Primary endpoint
The number of keratinocyte carcinomas (KCs) on the treated area compared with placebo, at the completion of 24 weeks of topical 1% sirolimus then at 12- and 24-month follow-up
The data will be analysed using a Poisson regression model with the number of KCs per participant as the dependent variable and treatment arm (sirolimus vs. placebo) as the independent variable. Site, 5-fluorouracil (5-FU/Efudix) use in the past 12 months, baseline risk level (higher risk (≥ 10 SCCs in the past) vs. lower risk (< 10 SCCs in the past)), and use of acitretin at baseline will be controlled for as covariates. Overdispersion will be assessed by plotting the standardised residuals vs. predicted values for the Poisson model and comparing with those for a negative binomial model. If the negative binomial model improves the fit substantially, it will replace the Poisson regression model, and conclusions will be based on the negative binomial model. The point estimate for the treatment arm effect and its 95% CI will be provided. This method will be repeated for each time point of interest (6, 12, and 24 months after initiation of treatment).
Secondary endpoints
The occurrence of keratinocyte carcinomas (KCs) on the treated area at the completion of 24 weeks of topical 1% sirolimus then at 12- and 24-month follow-up
A logistic regression model will be constructed with the occurrence of one or more KCs (yes/no) as the dependent variable and treatment arm as the independent variable. The stratification variables (baseline risk level, site, 5-fluorouracil use at baseline, and acitretin use at baseline) will be controlled for covariates. The odds ratio of KC occurrence between treatment arms and its 95% CI will be reported at each time point of interest.
The occurrence and number of biopsy-proven SCC at the completion of 24 weeks of topical 1% sirolimus, then at 12- and 24-month follow-up on the treated area
The modelling methods for the primary objective and first secondary objective will be repeated with the number and occurrence of biopsy-proven SCCs as the dependent variable instead of the number and occurrence of KCs.
The occurrence and number of intraepidermal carcinomas (IECs), BCCs, and subtypes of SCCs or BCCs at each of the time points on the treated area
The modelling methods for the primary objective and first secondary objective will be repeated with the number and occurrence of IECs, BCCs, and their subtypes as the dependent variable instead of the number and occurrence of KCs.
The number of facial AK of each participant at recruitment compared to 3, 6, 12, and 24 months on photographic images and counts
The modelling method for the primary objective will be repeated with the number of facial actinic keratoses as the dependent variable instead of the number of KCs. In addition to the covariates controlled for in the primary objective, the number of facial actinic keratoses at baseline will also be controlled for. This modelling exercise will be repeated at each time point of interest (3, 6, 12, and 24 months relative to the start of treatment).
The cost-effectiveness of utilising topical sirolimus therapy on SOTRs
The details of the cost-effectiveness analysis are provided in the dedicated section. A full health economics analysis plan will be developed.
The occurrence and number of intervention-related side effects by type
For both skin dryness and skin irritation, which are the intervention-related AEs of interest, the methods of the primary objective and first secondary objective will be repeated with the number and occurrence of intervention-related AEs as the dependent variable instead of the number and occurrence of KCs.
The number of doses of topical sirolimus (or placebo for the placebo arm) applied during the 24 weeks
A grouped logistic regression model will be constructed at the participant level with the number of doses (of sirolimus or placebo) taken and the number of doses missed per participant as the dependent variable and treatment arm (sirolimus vs. placebo) as the independent variable. Site, 5-fluorouracil (5-FU/Efudix) use in the 12 months immediately prior to randomisation, baseline risk level (higher risk vs. lower risk), and use of acitretin at baseline will be controlled for as covariates.
The number of participants completing the 24-week course
For each treatment arm considered individually, the number of participants in the study sample who complete the 6-month course of treatment will be provided. Based on this, the population estimate of the true underlying proportion of participants expected to complete the course will be estimated as a binary proportion, and its 95% CI will be provided using an exact method.
Basal and Squamous Cell Carcinoma Quality of Life (BaSQoL) questionnaire score
The BaSQoL assessment will be done by the health economics team. The BaSQoL questionnaire will be administered at baseline and at 6, 12, and 24 months post-baseline. A linear mixed-effects model will be constructed with treatment arm and assessment time point as fixed effects and participant as the random effect. The BaSQoL questionnaire score will be the response variable (i.e. the dependent variable). Baseline BaSQoL will be controlled for modelling. A post hoc contrast will be applied to assess the area under the BaSQoL score–time curve (AUC) from the first post-baseline BaSQoL assessment time point until the last BaSQoL assessment time point. This AUC will be compared between arms, and an estimate and 95% CI for the difference between arms will be provided. The p-value for the hypothesis test that the difference in AUC between arms is zero will be provided. The exact same procedure will be followed for the EQ-5D-5L questionnaire score.
Interim analyses {21b}
An interim analysis is scheduled at 12 months after initiation of treatment. This analysis will encompass a comprehensive statistical evaluation specifically focused on AKs. The statisticians will report to the independent data and safety monitoring board (DSMB) and trial management committee (TMC).
Methods for additional analyses (e.g. subgroup analyses) {20b}
Subgroup analyses will be conducted to evaluate the consistency of the treatment effect across various key demographic and clinical subgroups. These subgroups include baseline risk level (higher risk vs. lower risk), treatment site, 5-FU use in the past 12 months (yes vs. no), and acitretin use at baseline (yes vs. no). Interaction terms between the treatment arm and each subgroup variable will be included in the models to assess whether the treatment effect varies significantly across subgroups.
Methods in analysis to handle protocol non-adherence and statistical methods to handle missing data {20c}
Missing data may arise as a result of participants withdrawing from treatment or follow-up, or from missing a scheduled assessment visit. Missing data will not be imputed and participants with missing data points for measurements critical to the assessment of a particular objective at a given time point will be omitted from the assessment of that objective at that time point (case-wise deletion).
Plans to give access to the full protocol, participant-level data, and statistical code {31c}
A data sharing plan will be established in alignment with the Australian New Zealand Clinical Trials Registry (ANZCTR) guidelines. Specifically, this includes provisions for sharing de-identified participant-level data, the full trial protocol, and statistical code, subject to participant consent and any regulatory requirements. Access requests will be reviewed to ensure they align with ethical standards, participant privacy, and the study’s data use agreements.
Oversight and monitoring
Composition of the coordinating centre and trial steering committee {5d}
SiroSkin is coordinated by Melanoma and Skin Cancer Research Centre, Monash University.
This study will closely monitor accrual rates with respect to the feasibility of study completion. Overall and centre-specific accrual rates will be assessed as part of routine reporting (at least annually) to the TMC.
It is acknowledged that recruitment will be slower at the commencement of the study (with fewer sites participating) and will gain momentum as more centres join. Consideration will be given to stopping the trial early if accrual is less than expected or if a clearly more effective therapy has been developed and is available to participants. The trial may also be stopped early if the treatment is shown to work and therefore does not require further testing. The decision to close the study early will be determined by the study chair in consultation with the DSMB and TMC. All investigators and sites will be fully informed of any decision to close the trial early and a full and complete explanation will be provided at that time.
Composition of the data monitoring committee, its role and reporting structure {21a}
The DSMB is an independent group of individuals convened by the trial sponsor (Monash University) to review trial data at regular intervals to monitor the progress and safety of the trial. The principal role of the DSMB is to provide guidance on trial conduct and/or safety by making suggestions to the sponsor or TMC based on ethical or safety reasons to modify, continue, or stop the trial.
Safety oversight will be under the direction of the DSMB. Members of the DSMB will be independent of trial conduct. The DSMB will meet at least annually to assess safety and efficacy data on each arm of the trial. The DSMB will operate under the rules of an approved charter that will be written and reviewed at the organisational meeting of the DSMB. At this time, each data element that the DSMB needs to assess will be clearly defined.
Adverse event reporting and harms {22}
For the purpose of this trial, only skin-related AEs will be collected up to 30 days after the end of treatment. In the event that sirolimus blood levels are > 20 ng/ml,a SAE may need to be reported also in the participant meets SAE criteria (any AE fatal, life-threatening, requiring in-patient hospitalisation, results in persistent or significant disability or incapacity, is a congenital anomaly or birth defect, or is a grade 4 (NCI CTCAE v5.0) toxicity). SOTRs are an unwell target population and are expected to develop a number of unrelated AEs. This was evidenced in the pilot study, where a substantial number of AEs reported were determined to be unrelated to the sirolimus cream intervention. To avoid unnecessary capture of unrelated AEs, investigators have sought the guidance of members of MASC Trials DSMB to set these guidelines. The operations manual will guide sites with regard to the collection of AEs.
AE reporting is not required for the following circumstances:
Conditions that are present at screening and do not deteriorate will not be considered adverse events
Abnormal laboratory values will not be considered adverse events unless deemed clinically significant by the investigator and documented as such
The AE will be described in the source documents (e.g. medical record) and captured on the CRF and will include:
The onset date, duration, date of resolution
Severity (mild, moderate or severe—what is the impact on the participant’s daily life?)
Seriousness (i.e. is it an SAE?)
Any action taken, (e.g. treatment, follow-up tests)
The outcome (recovery, recovered with sequalae, death, continuing, worsening)
The likelihood of the relationship of the AE to the trial treatment (unrelated, possible, probable, definite)
Narrative description of the event
For SAEs, the name of staff member completing the SAE form and participant trial ID
Copies of relevant source documents should be provided (if available). If all details are not available at the time of the initial report, a follow-up report must be sent as soon as possible.
Changes in the severity of an AE will be reported. AEs characterised as intermittent will be documented for each episode. All AEs will be followed to adequate resolution, where possible.
Assessing the relatedness (causality) of a participant’s AE
All adverse events must have their relationship to trial intervention assessed by the investigator who evaluates the adverse event based on temporal relationship and his/her clinical judgment. The degree of certainty about causality will be graded using the categories below. In a clinical trial, the trial product should always be at least ‘possibly related’.
The relationship of the event to the trial intervention will be assessed as per the Table 2.
Table 2.
Glossary of adverse event relatedness

Abbreviation: AE, adverse event
Frequency and plans for auditing trial conduct {23}
The trial will be monitored using a risk-based approach, as per NHMRC Guidance ‘Risk-based management and monitoring of clinical trials involving therapeutic goods’ (dated 2018; https://www.nhmrc.gov.au/guidelines-publications/eh59).
Full details of trial site monitoring are documented in the data management and monitoring plan. This plan describes in detail who will conduct the monitoring, at what frequency monitoring will be done, at what level of detail monitoring will be performed, and the distribution of monitoring reports.
Monitoring for this trial will be performed by MASC Trials’ staff. Site monitoring is scheduled annually for this trial (also subject to funding and recruitment rate and at the discretion of the TMC).
The investigational site will provide direct access to all trial-related source data/documents and reports for the purpose of monitoring and auditing by Monash University (MASC-RC) and inspection by local and regulatory authorities.
Plans for communicating important protocol amendments to relevant parties (e.g. trial participants, ethical committees) {25}
Every care has been taken in the preparation of this protocol; however, corrections or amendments may be necessary. Changes and amendments to the protocol will be circulated to investigators in the trial. Approval of amendments by the HREC is required prior to their implementation. In some instances, an amendment may require a change to the participant information sheet and/or consent form. The investigator must receive approval/advice of the revised consent form prior to implementation of the change. In addition, changes to the eCRFs, if required, will be incorporated in the amendment.
Dissemination plans {31a}
We plan to disseminate the results of this research through a variety of channels, including academic publications, conference presentations, and updates on the trial registry. We plan to disseminate the results of this research through a variety of channels, including academic publications, conference presentations, and updates on the trial registry. De-identified trial data will be made available through the Health Studies Australian National Data Asset (HeSANDA) program, a national infrastructure that enables researchers to access and share data from health studies, including clinical trials, cohort studies, and other research data.
Discussion
Mammalian target of rapamycin inhibitors (mTORi), such as sirolimus (rapamycin) or everolimus, are immunosuppressive but do not increase the risk of SCC or BCC. In participants with SCCs, changing calcineurin inhibitors to sirolimus resulted in a nearly two-fold reduction in SCC risk [6–8].
Although the exact mechanism by which sirolimus can reduce skin cancers remains unclear, it is proposed that inhibition of the mTOR pathway will have anti-proliferative effects, reducing the size of mutant epidermal clones [30] and therefore the epidermal mutation load [31].
However, oral sirolimus is not always well tolerated. Using the topical form, if providing similar levels of protection, would be a major benefit for transplant recipients at high risk of SCC [22].
Topical sirolimus has however been widely and safely used in paediatric settings in the context of tuberous sclerosis, a genetic condition artificially activating the mTOR [23].
Therefore, the risk of SAEs with topical sirolimus appears minimal. These consist in irritation and eventually contact allergy and do not seem to include some of the major side effects of oral sirolimus. The potential benefit of reducing the number of superficial skin cancers by more than threefold is potentially a life-changing event, as it will reduce the number of surgeries needed in the long term. The hope is that this benefit extends to all invasive forms of skin cancer.
Trial status
The trial received HREC approval on April 18, 2023. The current protocol is version 6.0, dated June 03, 2024. The first participant was recruited on February 9, 2024. At the time of submission, this trial has not completed participant recruitment (60 participants out of 146 planned). At the current recruitment rate, we anticipate completing recruitment by April 2025.
Acknowledgements
The authors would like to thank the MASC trials for their precious input, and the staff at every participating SiroSkin site for their valuable contributions to this research.
Abbreviations
- 5-FU
Fluorouracil
- AE
Adverse event
- BaSQoL
Basal and Squamous Cell Carcinoma Quality of Life
- BCC
Basal cell carcinoma
- CI
Chief investigator
- CRF
Case report form
- DSMB
Data safety monitoring board
- eCRF
Electronic case report form
- EQ-5D-5L
EuroQol 5-dimensional
- HREC
Human Research Ethics Committee
- IEC
Intraepidermal carcinoma
- KC
Keratinocyte carcinoma
- mTORi
Mammalian target of rapamycin inhibitors
- NHMRC
National Health and Medical Research Council
- PICF
Patient information and consent form
- QALY
Quality-adjusted life year
- QOL
Quality of life
- SAE
Serious adverse event
- SCC
Squamous cell carcinoma
- SOTR
Solid organ transplant recipient
- TMC
Trial management committee
Authors’ contributions {31b}
KK is the chief investigator; he conceived the study, led the proposal and protocol development, obtained funding, and is supervising the conduct of the study. LD and ER contributed to the study design and to development of the proposal. DD, DC, AT, EK, HS, PS, AC, PFP, and JK have provided intellectual input to the conduct of the trial and are executing the trial on a day-to-day basis. LC provided the health economics analysis, and AH was the lead trial methodologist. All authors have reviewed, edited and approved the final version.
Funding {4}
This research program will be funded by the Medical Research Future Fund-Clinical Trials Activity Initiative (MRFF2015150). LD has received scholarships from the French Society of Dermatology (ArchiMed and CapFinance), La Ligue contre le Cancer, the CEDEF, and the European Association of Dermatology and Venereology. She has received honoraria for educational events from la Roche Posay. These funders were not involved in this study.
Data availability {29}
This document constitutes the full protocol. Datasets and statistical code used in this study will be available from the Melanoma and Skin Cancer Research Centre-Monash University on reasonable request following the completion of the trial.
Declarations
Ethics approval and consent to participate {24}
The study was approved by Metro South Human Research Ethics Committee on 18 April 2023 (HREC/2023/QMS/95325). All participants will have to provide written informed consent before participation.
Consent for publication {32}
This is not applicable. No identifying images or other personal or clinical details of participants are presented here or will be presented in reports of the trial results. The participant information materials and informed consent form are available from the corresponding author on request. All authors listed above meet the authorship criteria according to the latest guidelines of the International Committee of Medical Journal Editors, and all authors agree with the manuscript.
Competing interests {28}
The authors declare that they have no competing interests.
Footnotes
To ensure site staff is blinded to a participant’s treatment allocation, they will not receive sirolimus blood results. However, any participant with therapeutic sirolimus levels will be flagged for clinical review. The specific process will be worked through with the sites, as it may be different for individual sites. Participants found to have sirolimus blood levels ≥ 4 ngAQ/ml (within therapeutic levels) will be flagged for medical review and have AE reported. Blood sirolimus levels > 20 ng/ml are considered an overdose and potentially a serious safety issue.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Moloney FJ, Comber H, O’Lorcain P, et al. A population-based study of skin cancer incidence and prevalence in renal transplant recipients. Br J Dermatol. 2006;154(3):498–504. [DOI] [PubMed] [Google Scholar]
- 2.Bouwes Bavinck JN, Hardie DR, Green A, et al. The risk of skin cancer in renal transplant recipients in Queensland, Australia. A follow-up study. Transplantation. 1996;61(5):715–21. [DOI] [PubMed] [Google Scholar]
- 3.Iannacone MR, Sinnya S, Pandeya N, et al. Prevalence of skin cancer and related skin tumors in high-risk kidney and liver transplant recipients in Queensland, Australia. J Invest Dermatol. 2016;136(7):1382–6. [DOI] [PubMed] [Google Scholar]
- 4.Ong CS, Keogh AM, Kossard S, et al. Skin cancer in Australian heart transplant recipients. J Am Acad Dermatol. 1999;40(1):27–34. [DOI] [PubMed] [Google Scholar]
- 5.Na R, Grulich AE, Meagher NS, et al. De novo cancer-related death in Australian liver and cardiothoracic transplant recipients. Am J Transplant. 2013;13(5):1296–304. [DOI] [PubMed] [Google Scholar]
- 6.Veness MJ, Quinn DI, Ong CS, et al. Aggressive cutaneous malignancies following cardiothoracic transplantation: the Australian experience. Cancer. 1999;85(8):1758–64. [PubMed] [Google Scholar]
- 7.Richmond-Sinclair NM, Pandeya N, Ware SW, et al. Incidence of basal cell carcinoma multiplicity and detailed anatomic distribution: longitudinal study of an Australian population. J Invest Dermatol. 2009;129(2):323–8. [DOI] [PubMed] [Google Scholar]
- 8.Whiteman DC, Thompson BS, Thrift AP, et al. A model to predict the risk of keratinocyte carcinomas. J Invest Dermatol. 2016;136(6):1247–54. [DOI] [PubMed] [Google Scholar]
- 9.Papier K, et al. Management of organ transplant recipients attending a high-throughput skin cancer surgery and surveillance clinic in Queensland. Br J Dermatol. 2019;180(3):631–6. [DOI] [PubMed] [Google Scholar]
- 10.Apalla Z, Sotiriou E, Chovarda E, et al. Skin cancer: preventive photodynamic therapy in patients with face and scalp cancerization. A randomized placebo-controlled study. Br J Dermatol. 2010;162(1):171–5. [DOI] [PubMed] [Google Scholar]
- 11.de Graaf YG, Kennedy C, Wolterbeek R, et al. Photodynamic therapy does not prevent cutaneous squamous-cell carcinoma in organ-transplant recipients: results of a randomized-controlled trial. J Invest Dermatol. 2006;126(3):569–74. [DOI] [PubMed] [Google Scholar]
- 12.Willey A, Mehta S, Lee PK. Reduction in the incidence of squamous cell carcinoma in solid organ transplant recipients treated with cyclic photodynamic therapy. Dermatol Surg. 2010;36(5):652–8. [DOI] [PubMed] [Google Scholar]
- 13.Weinstock MA, Thwin SS, Siegel JA, et al. Chemoprevention of basal and squamous cell carcinoma with a single course of fluorouracil, 5%, cream: a randomized clinical trial. JAMA Dermatol. 2018;154(2):167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perrett CM, McGregor JM, Warwick J, et al. Treatment of post-transplant premalignant skin disease: a randomized intrapatient comparative study of 5-fluorouracil cream and topical photodynamic therapy. Br J Dermatol. 2007;156(2):320–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bavinck JN, Tieben LM, Van der Woude FJ, et al. Prevention of skin cancer and reduction of keratotic skin lesions during acitretin therapy in renal transplant recipients: a double-blind, placebo-controlled study. J Clin Oncol. 1995;13(8):1933–8. [DOI] [PubMed] [Google Scholar]
- 16.George R, Weightman W, Russ GR, et al. Acitretin for chemoprevention of non-melanoma skin cancers in renal transplant recipients. Australas J Dermatol. 2002;43(4):269–73. [DOI] [PubMed] [Google Scholar]
- 17.Endrizzi B, Ahmed RL, Ray T, et al. Capecitabine to reduce nonmelanoma skin carcinoma burden in solid organ transplant recipients. Dermatol Surg. 2013;39(4):634–45. [DOI] [PubMed] [Google Scholar]
- 18.Jirakulaporn T, Endrizzi B, Lindgren B, et al. Capecitabine for skin cancer prevention in solid organ transplant recipients. Clin Transplant. 2011;25(4):541–8. [DOI] [PubMed] [Google Scholar]
- 19.Chen AC, Martin AJ, Choy B, et al. A phase 3 randomized trial of nicotinamide for skin-cancer chemoprevention. N Engl J Med. 2015;373(17):1618–26. [DOI] [PubMed] [Google Scholar]
- 20.Allen NC, Martin AJ, Snaidr VA, et al. Nicotinamide for skin-cancer chemoprevention in transplant recipients. N Engl J Med. 2023;388(9):804–12. [DOI] [PubMed] [Google Scholar]
- 21.Asgari MM, Arron AT, Warton EM, et al. Sirolimus use and risk of cutaneous squamous cell carcinoma (SCC) in solid organ transplant recipients (SOTRs). J Am Acad Dermatol. 2015;73(3):444–50. [DOI] [PubMed] [Google Scholar]
- 22.Euvrard S, Morelon E, Rostaing L, et al. Sirolimus and secondary skin-cancer prevention in kidney transplantation. N Engl J Med. 2012;367(4):329–39. [DOI] [PubMed] [Google Scholar]
- 23.Hoogendijk-van den Akker JM, Harden PN, Hoitsma AJ, et al. Two-year randomized controlled prospective trial converting treatment of stable renal transplant recipients with cutaneous invasive squamous cell carcinomas to sirolimus. J Clin Oncol. 2013;31(10):1317–23. [DOI] [PubMed] [Google Scholar]
- 24.Wataya-Kaneda M, Nakamura A, Tanaka M, et al. Efficacy and safety of topical sirolimus therapy for facial angiofibromas in the tuberous sclerosis complex : a randomized clinical trial. JAMA Dermatol. 2017;153(1):39–48. [DOI] [PubMed] [Google Scholar]
- 25.Chong S, Wong HY, Althabteh A, et al. Chemoprevention of cutaneous squamous cell carcinoma and its precursors in solid organ transplant recipients using topical sirolimus: a randomized, double-blind, placebo-controlled pilot trial. J Am Acad Dermatol. 2022;87(5):1163–6. [DOI] [PubMed] [Google Scholar]
- 26.Harris PA, Taylor R, Thielke R, et al. Research electronic data capture (REDCap)-a metadata-driven methodology andworkfow process for providing translational research informatics support. J Biomed Informat. 2009;42(2):377–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu WY, Waalboer-Spuij R, Bremer R, et al. Validation of the english Basal and Squamous Cell Carcinoma Quality of Life (BaSQoL) questionnaire. Dermatol Surg. 2020;46(3):327–34. [DOI] [PubMed] [Google Scholar]
- 28.Devlin NJ, Brooks R. EQ-5D and the EuroQol group: past, present and future. Appl Health Econ Health Policy. 2017;15(2):127–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seidler AL, Willson ML, Aberoumand M, et al. The changing landscape of clinical trials in Australia. Med J Aust. 2023;219(5):192–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roy E, Wong HY, Villani R, et al. Regional variation in epidermal susceptibility to UV-induced carcinogenesis reflects proliferative activity of epidermal progenitors. Cell Rep. 2020;31(9):107702. [DOI] [PubMed] [Google Scholar]
- 31.Wong HY, Lee RC, Chong S, Kapadia S, et al. Epidermal mutation accumulation in photodamaged skin is associated with skin cancer burden and can be targeted through ablative therapy. Sci Adv. 2023;9(19):2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This document constitutes the full protocol. Datasets and statistical code used in this study will be available from the Melanoma and Skin Cancer Research Centre-Monash University on reasonable request following the completion of the trial.