

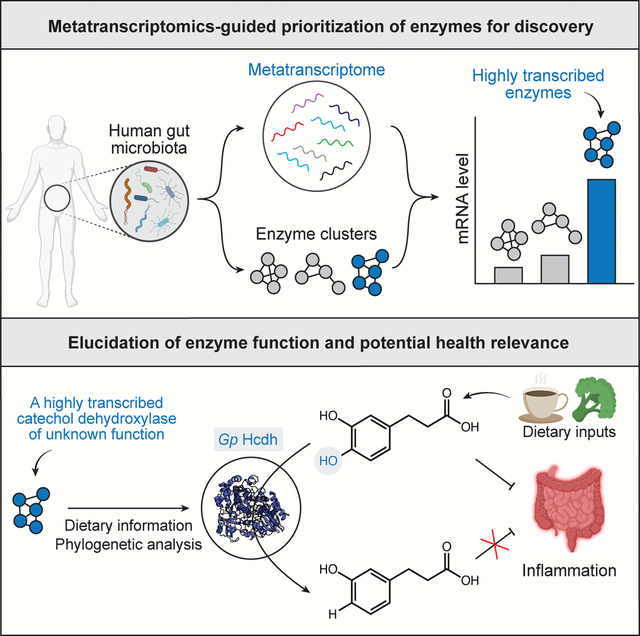
Summary
Gut microbial catechol dehydroxylases are a largely uncharacterized family of metalloenzymes that potentially impact human health by metabolizing dietary polyphenols. Here, we use metatranscriptomics (MTX) to identify highly transcribed catechol dehydroxylase-encoding genes in human gut microbiomes. We discover a prevalent, previously uncharacterized catechol dehydroxylase (Gp Hcdh) from Gordonibacter pamelaeae that dehydroxylates hydrocaffeic acid (HCA), an anti-inflammatory gut microbial metabolite derived from plant-based foods. Further analyses suggest the activity of Gp Hcdh may reduce anti-inflammatory benefits of polyphenol-rich foods. Together, these results show the utility of combining MTX analysis and biochemical characterization for gut microbial enzyme discovery and reveal a potential link between host inflammation and a specific polyphenol-metabolizing gut microbial enzyme.
eTOC
Bae et al. uncover that a highly transcribed member (Gp Hcdh) of the gut bacterial catechol dehydroxylase family metabolizes hydrocaffeic acid, an abundant anti-inflammatory polyphenol. Analyses of human data suggest that the activity of Gp Hcdh may reduce anti-inflammatory activity of foods that are major sources of hydrocaffeic acid.
Graphical Abstract
Introduction
Polyphenols are a diverse group of metabolites (>6,000 reported compounds) characterized by phenol functional groups (Figure 1A). They are abundant in plant-based foods such as vegetables, coffee, and tea1, with an average daily intake >1 gram2,3. Dietary polyphenols have been associated with improved health outcomes, including reduced inflammation, lower risk of cardiovascular diseases, and healthy aging4,5. Importantly, the biological impacts of polyphenols may depend on gut microbial metabolism due to their poor oral bioavailability and extensive modification by the gut microbiota6,7. Indeed, the gut microbiota has been associated with inter-individual variation in the health effects of polyphenols8,9. However, the gut microbial enzymes involved in polyphenol metabolism are largely unknown10.
Figure 1. Most gut bacterial catechol dehydroxylases are uncharacterized.
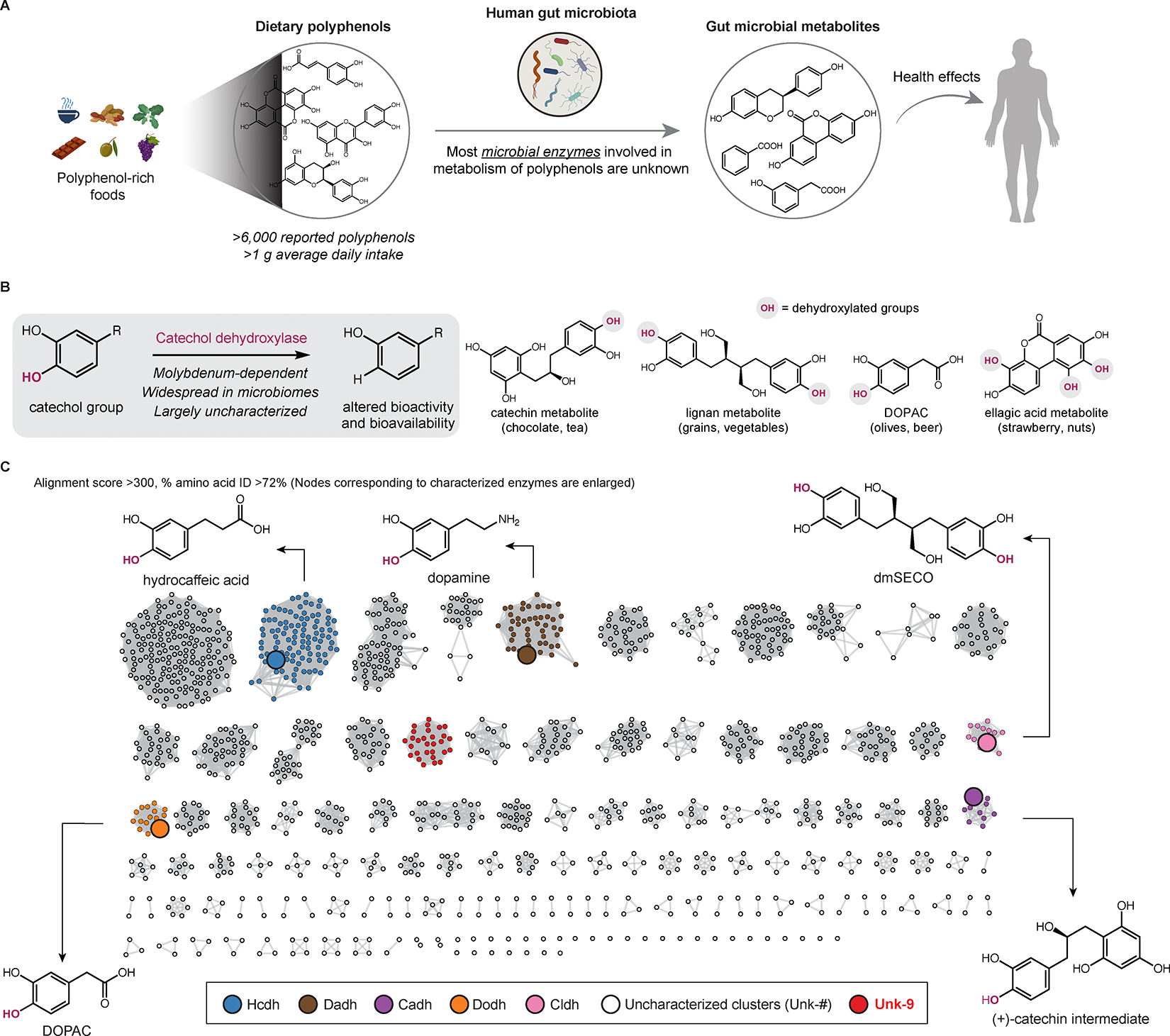
(A) The human gut microbiota metabolizes polyphenols. (B) The general transformation catalyzed by catechol dehydroxylases and example polyphenol substrates. (C) SSN of catechol dehydroxylases found in the human gut. See also Figure S1 and Table S1.
A central reaction in gut microbial polyphenol metabolism is catechol dehydroxylation, in which the para-hydroxyl group of a catechol (1,2-dihydroxylated aromatic ring) is replaced by a hydrogen atom (Figure 1B). Many catechol-containing polyphenols undergo dehydroxylation in the human body11, a process first linked to the gut microbiota in antibiotic-treated rats12. Catechol dehydroxylation often drastically alters the bioactivity and bioavailability of polyphenol metabolites11. Furthermore, this activity is highly variable between humans13–15, which may contribute to interindividual variation in responses to polyphenol consumption. Recent efforts linked catechol dehydroxylation to the molybdenum-dependent catechol dehydroxylase enzyme family and showed individual enzymes are upregulated in response to their specific catechol substrates13,14,16. Despite this progress, only a few catechol dehydroxylases have been biochemically characterized16.
Here, we employ sequence similarity network (SSN) analysis and MTX analysis of human gut microbiome datasets to prioritize highly transcribed, widely distributed catechol dehydroxylases for characterization. We identify a highly transcribed and prevalent catechol dehydroxylase (Gp Hcdh) and determine its substrate as the common dietary polyphenol metabolite HCA. By analyzing a human gut microbiome dataset, we uncover a potential link between this enzyme and a reduction in the anti-inflammatory benefits of consuming polyphenol-rich vegetables. This integrated computational and experimental approach emphasizes the potential of integrating MTX with biochemical studies for discovery of important gut microbial enzymes. This work further demonstrates how knowledge of gut microbial enzyme function can inform mechanistic hypotheses underlying gut microbiota-host interactions.
Results
Most gut bacterial catechol dehydroxylases are uncharacterized
We initially sought to understand the diversity of catechol dehydroxylases in the human gut microbiome. Catechol dehydroxylases are members of the dimethyl sulfoxide (DMSO) reductase superfamily and are predicted to harbor a bis-molybdopterin guanine dinucleotide (bis-MGD) cofactor13,16,17. Following the initial discovery of dopamine dehydroxylase (Dadh) in the gut Coriobacteriia Eggerthella lenta in 201916, four additional catechol dehydroxylases have been linked to the dehydroxylation of specific dietary polyphenols in E. lenta (HCA dehydroxylase (Hcdh) and (+)-catechin dehydroxylase (Cadh)) and another Coriobacteriia Gordonibacter pamelaeae (DOPAC dehydroxylase (Dodh) and catechol lignan dehydroxylase (Cldh))13,14. However, most putative catechol dehydroxylases in Gordonibacter and Eggerthella genomes remain uncharacterized13.
To compile a comprehensive list of gut microbial catechol dehydroxylase homologs, we searched a dataset of >150,000 human-associated metagenome-assembled genomes (MAGs) using protein sequences of the five characterized catechol dehydroxylases as queries18. This search yielded 2,903 unique sequences from Coriobacteriia MAGs (Figure S1A). Multiple homologs were from G. pamelaeae and E. lenta as well as other Coriobacteriia known to dehydroxylate catechols in culture, such as Adlercreutzia equolifaciens and Ellagibacter isourolithinifaciens19–21. Notably, homologs were also found in species not yet reported to perform this metabolism (Figure S1A).
Next, we sought to classify catechol dehydroxylase homologs using sequence similarity network (SSN) analysis22. The minimum alignment score and amino acid (aa) sequence identity were optimized to build an SSN in which catechol dehydroxylases in the same cluster are predicted to share the same biochemical function (Figure S1B and Figure S1C). With a minimum alignment score and aa sequence identity of 300 and 72%, respectively, we separated the five characterized catechol dehydroxylases, which have different substrates, into different clusters (Figure 1C). The resulting SSN contained 126 distinct enzyme clusters.
To investigate whether SSN clusters contain isofunctional enzymes, we examined the dehydroxylating activities of bacterial isolates with sequences from clusters containing characterized enzymes. For example, in addition to sequences from known metabolizers, the Hcdh, Dadh, and Dodh clusters include sequences from cultured bacteria that have not been reported to dehydroxylate their corresponding substrates (Figure S1D). These bacterial cultures dehydroxylated the predicted substrates, suggesting these enzyme clusters are likely isofunctional (Figure S1E). Although we cannot verify all clusters, our SSN broadly reflects the functional diversity of gut microbial catechol dehydroxylases. Notably, our SSN analysis revealed that most catechol dehydroxylase clusters (n=121/126) are of unknown function (Figure 1C and Figure S1F), underscoring the need to prioritize enzymes for discovery.
Metatranscriptomics analysis reveals an uncharacterized catechol dehydroxylase that is highly expressed and prevalent in the human gut
Recognizing that the expression of individual catechol dehydroxylase genes is highly induced by their substrates in bacterial culture (Figure 2A)13,14,16,23, we applied an MTX-based prioritization strategy24,25. We hypothesized that prevalent, highly transcribed catechol dehydroxylases in human gut MTX datasets likely metabolize common, abundant dietary polyphenols as prior studies have shown that in vivo MTX reflects substrate availability for some gut microbial enzymes (Figure 2B)24–27. However, this has not yet been demonstrated for catechol dehydroxylases.
Figure 2. MTX analysis reveals an uncharacterized catechol dehydroxylase that is highly expressed and prevalent in the human gut.
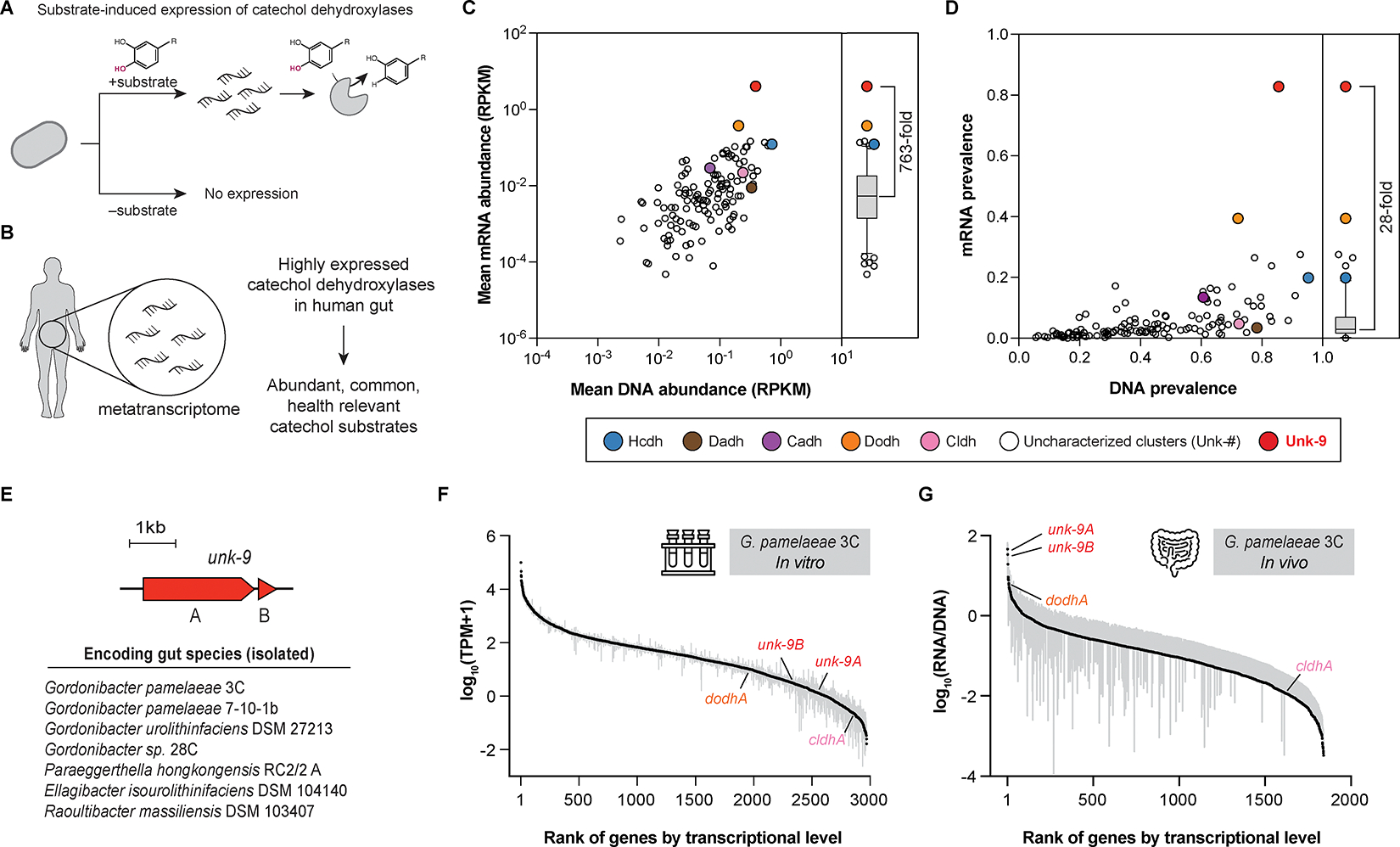
(A) The expression of catechol dehydroxylases is induced by their substrates. (B) MTX-based prioritization of an uncharacterized catechol dehydroxylase. (C–D) Analysis of DNA and mRNA abundance (C) and prevalence (D) of catechol dehydroxylase clusters using the MLVS dataset. Bars represent median. RPKM, reads per kilobase million. (E) List of Unk-9 encoding gut species. (F–G) Ranked median transcriptional levels of the whole protein-coding genes of G. pamelaeae 3C in vitro (F) and in vivo (G), grey bar represents 95% confidence interval. TPM, transcripts per kilobase million. See also Figure S2 and Table S2.
To assess DNA and mRNA levels of catechol dehydroxylase clusters from our SSN in human gut microbiomes, we used metagenomics (MGX) and MTX data from the Men’s Lifestyle Validation Study (MLVS), a cohort of 307 healthy men enrolled in the Health Professionals Follow-Up Study28. Cluster-level quantification showed DNA and mRNA abundances of the clusters were generally correlated (Pearson r = 0.58), consistent with previous findings29,30. Notably, a few outliers, such as the Dodh cluster, showed higher mRNA levels compared to clusters with similar DNA levels (Figure 2C). Dodh transcripts were detected in 40% of the MTX samples, whereas the expression of most clusters (120/126) was detected in <20% of samples (Figure 2D). The substrate of Dodh, 3,4-dihydroxyphenylacetic acid (DOPAC), is a major gut microbial metabolite of quercetin and dopamine and was detected in all urine samples from a previous clinical study31,32. This supports our hypothesis that prominent catechol dehydroxylases can be discovered using MTX-based prioritization.
Our analysis showed certain catechol dehydroxylase clusters containing uncharacterized enzymes were more highly and widely transcribed in the human gut than other clusters. Notably, cluster Unk-9 had the highest mRNA abundance (763-fold higher than the median) and mRNA prevalence (detected in 82.8% of the MTX samples) among all clusters (Figure 2C, Figure 2D, and Figure S2A). Unk-9 also had the highest mRNA/DNA ratio (242-fold higher than the median) (Figure S2B). This trend was confirmed in another dataset from the Human Microbiome Project 2 (HMP2)33, where Unk-9 was the most transcribed catechol dehydroxylase cluster (Figure S2C). Unk-9 did not have the highest DNA abundance and prevalence, emphasizing the importance of MTX in highlighting this enzyme.
Predicted to comprise a catalytic subunit (Unk-9A) that binds the bis-MGD cofactor and an accessory subunit (Unk-9B), Unk-9 is encoded by several cultured gut Coriobacteriia including G. pamelaeae 3C and other Gordonibacter strains, Paraeggerthella hongkongensis RC2/2 A, E. isourolithinifaciens DSM 104140, and Raoultibacter massiliensis DSM 103407 (Figure 2E). Data from our previous MAG search indicated that Unk-9 sequences are globally distributed, found in gut microbiomes from North America, Europe, Asia, as well as from agro-pastoral Mongolian and Malagasy cohorts (Figure S2D).
Knowing that Unk-9 is the most highly expressed catechol dehydroxylase in vivo, we examined its expression in bacterial culture to determine if its substrate is present in culture media. We analyzed the transcriptional levels of all protein-coding genes in the G. pamelaeae 3C strain grown in a modified Brain–Heart Infusion medium (in vitro)14. Interestingly, genes encoding Unk-9A and Unk-9B were among the least expressed ones in vitro (2576th and 2316th out of 2979 genes, respectively) (Figure 2F). In contrast, these genes were the top two most expressed G. pamelaeae 3C genes in the MLVS data (in vivo)28 (Figure 2G), suggesting that Unk-9 expression is induced by an external stimulus present in the gut but not in the culture medium. Altogether, its high in vivo expression and global distribution led us to investigate the biochemical function of Unk-9.
Combining dietary information and phylogenetic analysis of transcriptional regulators reveals that Unk-9 dehydroxylates HCA to support ATP production.
The numerous potential polyphenol substrates for Unk-9 prompted us to deduce candidates using several approaches. First, we correlated Unk-9 levels in gut microbiomes with consumption of specific food groups in the MLVS cohort. We found that Unk-9 DNA abundance correlated with the long-term consumption of cruciferous vegetables and coffee, which contain high levels of HCA precursors such as chlorogenic acid (1.3–3.6 mg/g fresh weight of coffee and up to 0.7 mg/g dry weight of cruciferous vegetables) (Figure 3A and Figure S3A)34,35. When extended to the characterized catechol dehydroxylase clusters, this dietary analysis revealed significant positive correlations with other polyphenol-rich food groups, including fruits (Cadh) and whole grain (Dodh) (Figure S3B). No significant correlation was found between dietary inputs and Unk-9 mRNA abundance, potentially due to factors like differences between long-term diet and rapid transcriptional responses, smaller MTX dataset size, substrate availability in vivo, and post-transcriptional effects. Overall, this analysis suggested that polyphenols from coffee and cruciferous vegetables, such as HCA and its precursors, might be potential substrates for Unk-9.
Figure 3. Combining dietary information and phylogenetic analysis of transcriptional regulators reveals that Unk-9 dehydroxylates HCA to support ATP production.
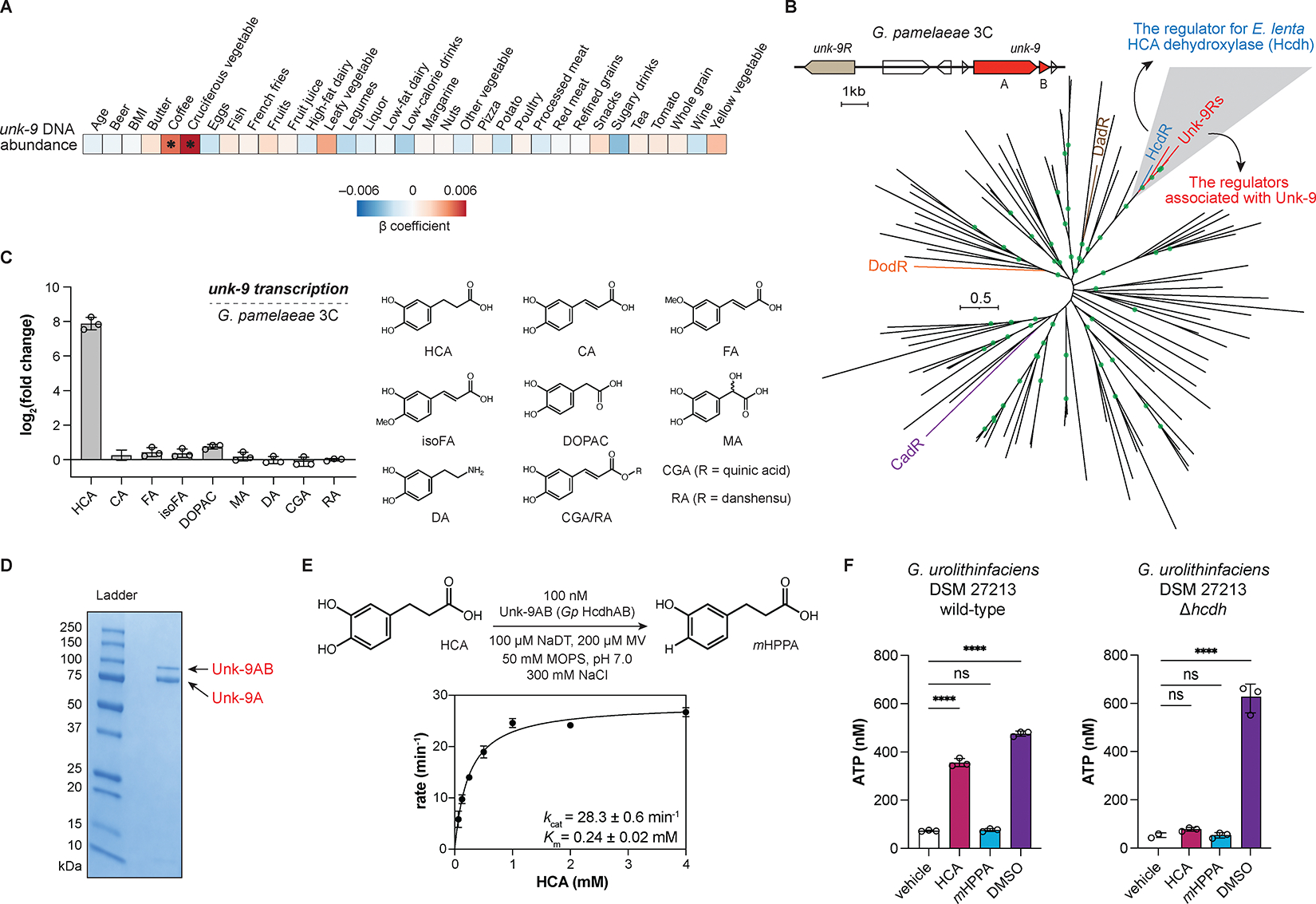
(A) Correlation of unk-9 DNA abundance with dietary inputs from the MLVS cohort. Linear mixed-effects model. *, FDR < 0.10. (B) Maximum-likelihood phylogenetic tree of the membrane-bound LuxR-type transcriptional regulators co-localized with molybdopterin oxidoreductases in the E. lenta DSM 2243 and G. pamelaeae 3C genomes. Branches with bootstrap number > 0.9 are highlighted by green circles. (C) Substrate-specific induction of Unk-9 expression in G. pamelaeae 3C by HCA analogs relative to vehicle measured by RT-qPCR. (D) SDS-PAGE gel image for isolated Gp Hcdh using activity-guided native purification. (E) Michaelis–Menten kinetics of Gp Hcdh using initial rates measured in the first 60 s. (F) ATP production in response to HCA, mHPPA, and DMSO in the cell suspension of G. urolithinfaciens DSM 27213 wild-type and Δhcdh grown anaerobically with HCA. One-way ANOVA followed by Dunnett’s multiple comparisons test with all comparisons made against vehicle control. ns, not significant; ****, adjusted p-value < 0.0001. For all panels, n=3 biologically independent replicates, data presented are mean ± standard deviation. See also Figure S3, Figure S4, Table S1, and Table S4. mHPPA, 3-hydroxyphenyl propionic acid; MV, methyl viologen; NaDT, sodium dithionite.
In parallel, we used knowledge of characterized catechol dehydroxylases to generate hypotheses for the function of Unk-9. First, phylogenetic analysis of the catechol dehydroxylase catalytic subunits revealed that Unk-9 was distant from known enzymes (Figure S3C). We next examined genes that co-localize with unk-9 in bacterial genomes because functionally associated genes are often clustered36. We found that unk-9 is co-localized with a gene encoding a membrane-bound LuxR-type regulator, unk-9R (Figure 3B). This class of transcriptional regulators has recently been shown to regulate expression of their associated genes, including catechol dehydroxylases, in response to their specific substrates23. We therefore reasoned that examining the relationship of these catechol dehydroxylase regulators might provide information about their substrates. Phylogenetic analysis of this transcriptional regulator family revealed that Unk-9R was most closely related to HcdR (44% aa identity, 60% aa similarity), the regulator for E. lenta HCA dehydroxylase (Hcdh) (Figure 3B). This relationship is striking because their associated catechol dehydroxylases, Unk-9 and Hcdh, have low sequence identity (17% aa identity, 29% aa similarity). Altogether, by considering dietary correlations and the similarity between transcriptional regulators, we postulated Unk-9 would dehydroxylate HCA or a related analog.
To identify the precise substrate of Unk-9, we first performed RT–qPCR experiments to measure the transcriptional level of unk-9 in G. pamelaeae 3C in response to HCA and related compounds. We found that HCA induced the transcription of G. pamelaeae 3C unk-9 >250-fold, whereas close analogs did not induce transcription (Figure 3C). HCA dehydroxylation was observed in liquid cultures of G. pamelaeae 3C and several other gut Coriobacteriia strains encoding unk-9, some of which had previously been reported to dehydroxylate HCA (Figure S3D)13,19. We then obtained HCA-dehydroxylating proteins from G. pamelaeae 3C grown with HCA via activity-guided native purification. Proteomic analysis confirmed that the purified enzyme was Unk-9 (Figure 3D, Figure S3E, and Table S4). Assays using the purified Unk-9 enzyme, which we have named G. pamelaeae HCA dehydroxylase (Gp Hcdh), showed robust conversion of HCA to the corresponding dehydroxylated product (Figure S3F), with minimal activity toward related compounds (Figure S3H). Within the DMSO reductase superfamily, Gp Hcdh exhibited a similar enzyme turnover number (kcat) to perchlorate reductase (28.3 vs 27.1 min−1, respectively) (Figure 3E and Figure S3G)37. Additionally, the Km of Gp Hcdh (0.24 mM) was comparable to that of nitrate reductase (0.20 mM)37. Inductively coupled plasma mass spectrometry (ICP-MS) analysis revealed that Gp Hcdh contains equimolar amounts of molybdenum relative to protein, indicating a single bis-MGD cofactor (Figure S3I).
With its biochemical function established, we next investigated the biological role of Gp Hcdh for the producing bacterium. Many DMSO reductase superfamily enzymes, including some catechol dehydroxylases in Eggerthella strains, facilitate anaerobic respiration by reducing alternative electron acceptors13,38,39. The similar kinetic parameters between Gp Hcdh and respiratory perchlorate and nitrate reductases further suggest its potential involvement in anaerobic respiration. Thus, we tested whether Gp Hcdh supports growth and ATP production under anaerobic conditions. We observed that HCA, but not its dehydroxylated product, promoted growth of G. pamelaeae 3C similarly to DMSO, another alternative electron acceptor for anaerobic respiration (Figure S4A). This growth coincided with HCA dehydroxylation (Figure S4B). Further, a G. pamelaeae 3C cell suspension at resting state produced ATP when incubated with HCA or DMSO (Figure S4C). To link ATP production to Gp Hcdh activity, we generated a Hcdh deletion mutant (Δhcdh) of G. urolithinfaciens DSM 27213 using a recently developed CRISPR-based genetic tool for Coriobacteriia (Figure S4D)23. The Δhcdh strain lost HCA dehydroxylase activity in culture (Figure S4E), and HCA-induced ATP production was not observed in this strain (Figure 3F). Together, these experiments support the involvement of Gp Hcdh in anaerobic respiration.
Levels of Gp Hcdh correlate with the anti-inflammatory response to cruciferous vegetable consumption
We next investigated potential connections between HCA dehydroxylation and host phenotypes by analyzing gut microbiome datasets. To begin, we considered the well-established anti-inflammatory activity of HCA40. Oral administration of HCA reduced the production of pro-inflammatory cytokines in lipopolysaccharide (LPS)-injected mice41, mice with liver injury42, and rats with dextran sulfate sodium-induced colitis43. HCA significantly reduced inflammatory activities in IL-1β-stimulated human colon fibroblasts43 and LPS-stimulated mouse and human peripheral blood mononuclear cells44,45. In contrast, the anti-inflammatory activity of the HCA dehydroxylation product, 3-hydroxyphenyl propionic acid (mHPPA), was abolished43,44. Therefore, we hypothesized that the dehydroxylating activity of Gp Hcdh might alter the anti-inflammatory benefit of consuming foods that are sources of HCA (Figure 4A).
Figure 4. Levels of Gp Hcdh correlate with the anti-inflammatory response to cruciferous vegetable consumption.
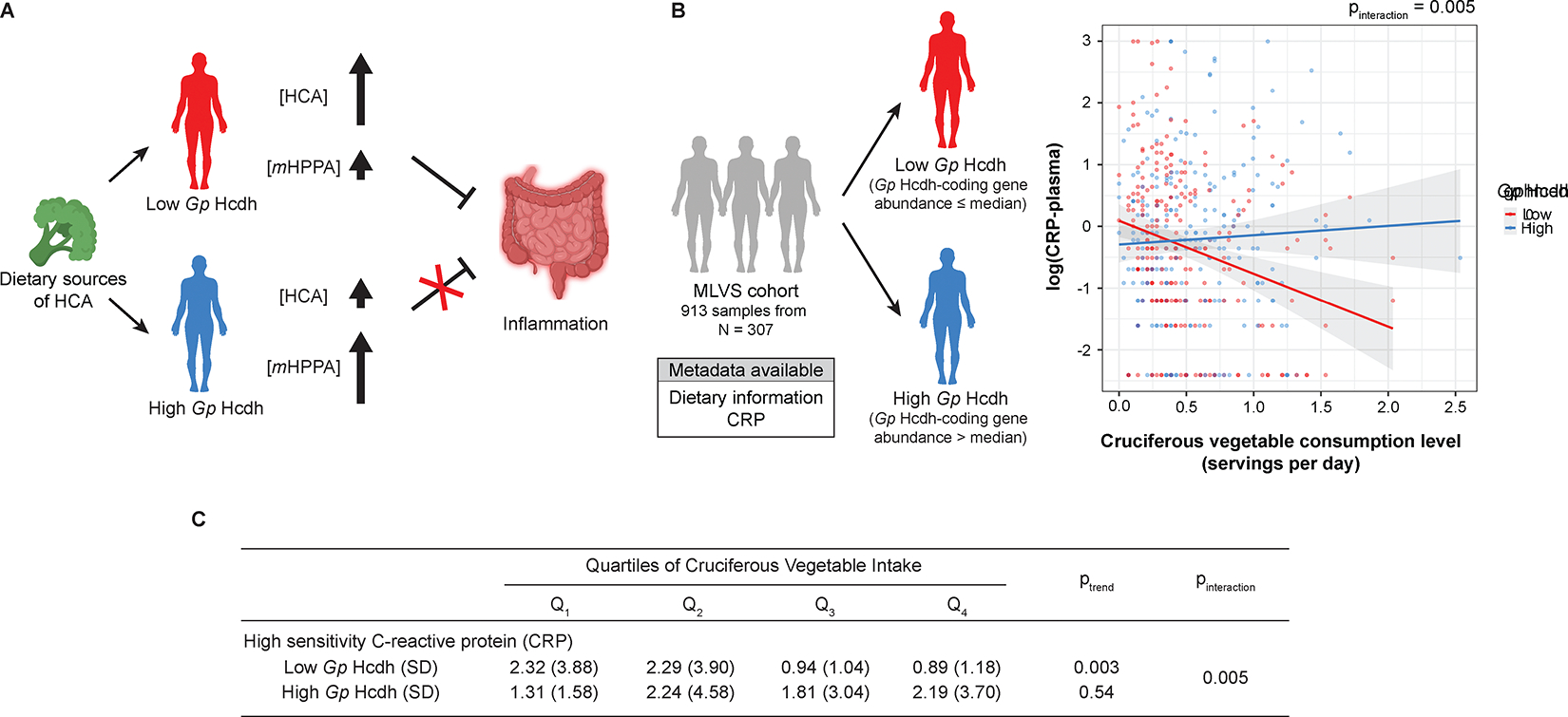
(A) Proposed mechanistic link between Gp Hcdh activity and the anti-inflammatory benefit of cruciferous vegetable consumption. (B) (left) Grouping of the MLVS cohort by Gp Hcdh DNA level. (right) Multivariate linear mixed model analysis for the correlation between cruciferous vegetable consumption and the level of CRP in the low (red trend line) and high (blue trend line) Gp Hcdh groups. Grey areas represent standard error. Panels were created using BioRender.
To investigate this hypothesis, we again analyzed the MLVS data, correlating HCA dehydroxylase abundance with host inflammation markers in the context of consumption of cruciferous vegetables and coffee, which represent the main sources of HCA quantifiable by the available dietary instruments34,35. Given the higher mRNA abundance (33.4-fold) and prevalence (82.8% vs 19.8%) of Gp Hcdh compared to E. lenta Hcdh, we focused on Gp Hcdh levels and their relationship with biomarkers of host inflammation and health. The MLVS samples were divided into two groups based on their Gp Hcdh DNA abundance relative to the median. Within the low Gp Hcdh group, higher cruciferous vegetable intake was associated with reduced levels of plasma C-reactive protein (CRP), a marker of inflammation (Figure 4B and Figure 4C). Conversely, such a correlation was absent among the high Gp Hcdh group (pinteraction = 0.005). We propose this correlation could potentially be explained by the differential activity of Gp Hcdh within the gut microbiotas of subjects, similar to the previously reported microbiota-driven links between dietary fiber and CRP46. Individuals with high levels of Gp Hcdh may dehydroxylate HCA to a higher degree, reducing its anti-inflammatory benefits, whereas those with low levels of the enzyme might not metabolize HCA as extensively. Although mechanistic studies are needed to test causality, our results suggest a potential route by which gut microbial metabolism of a specific polyphenol might influence the impact of diet on human health.
Discussion
It is well-established that highly expressed enzymes in microbial communities perform ecologically and metabolically important functions29,47–50. Though MTX can identify highly transcribed enzymes in microbial communities24,25,28–30,51–55, MTX-based strategies have been underutilized for enzyme discovery because of challenges in acquisition of high-quality MTX and limited examples of successful downstream biochemical characterization of the prioritized genes24,25,53. By integrating MTX and biochemical characterization workflows, we successfully identified a gut bacterial catechol dehydroxylase that metabolizes a common and abundant substrate. This enzyme discovery framework, coupled with the success of related previous efforts24,25,56, should be readily applied to other important gut bacterial enzyme families such as carbohydrate-active enzymes, sulfatases, β-glucuronidases, and glycyl radical enzymes.
A particularly challenging component of enzyme discovery workflows is linking uncharacterized sequences to their biochemical functions24. Catechol dehydroxylases are especially difficult in this regard because of their poorly understood substrate specificity and technical challenges associated with expressing complex metalloenzymes. We overcame these obstacles by investigating the transcriptional regulators controlling expression of catechol dehydroxylases. Individual catechol dehydroxylase genes are colocalized with genes encoding substrate-specific transcriptional regulators, enabling their association23. We found that the regulator of Gp Hcdh was most closely related to the regulator of E. lenta Hcdh, highlighting HCA as a potential substrate. Unlike E. lenta Hcdh, which has three subunits including a catalytic subunit, a subunit with four [4Fe-4S] clusters, and a membrane anchor13, Gp Hcdh has only two subunits. Their catalytic subunits are phylogenetically distant (Figure S3C), differing in length (770 aa for Gp Hcdh and 1193 aa for E. lenta Hcdh) and the presence of a twin-arginine translocation signal peptide. These differences made it challenging to deduce the biochemical function of Gp Hcdh from the sequence of E. lenta Hcdh. Our results indicate that analyses of associated regulators could be effective, underutilized strategy for assigning functions to microbial enzymes.
Finally, we uncovered a striking correlation between reduced levels of Gp Hcdh encoding genes in gut microbiomes and reduced levels of inflammation in subjects with high cruciferous vegetable intake. Our results may be related to previously found microbiota-driven interactions between dietary fibers or the Mediterranean diet and host health46,57. By uncovering a link between host inflammation and a specific gut microbial polyphenol-metabolizing enzyme, our findings set the stage for future mechanistic work to study the role of gut bacterial catechol dehydroxylation in host biology. Discovering the activities of additional polyphenol-metabolizing gut microbial enzymes and their impacts on the host and gut microbiome will be critical in deciphering the health benefits of these important dietary compounds.
Limitations of the study
The causality of the link between Gp Hcdh and host CRP levels requires mechanistic validation in model organisms. Additionally, no significant interactions were observed between Gp Hcdh and other biomarkers of host inflammation. The MLVS cohort sampled healthy participants, so the levels of biomarkers are within a healthy range with low variation, potentially constraining the statistical analysis. Our interaction analysis only considered Gp Hcdh due to its high in vivo transcriptional levels and prevalence compared to E. lenta Hcdh. However, E. lenta Hcdh or dehydroxylases from other organisms may contribute to HCA metabolism in vivo.
STAR Methods
Resource availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Emily P. Balskus (balskus@chemistry.harvard.edu)
Materials Availability
The whole genome sequence of G. urolithinfaciens DSM 27213 Δhcdh strain generated in this study was deposited in the National Center for Biotechnology Information under the accession number of PRJNA1150218. The strain will be available directly from the authors upon reasonable request.
Data and Code Availability
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
All original codes have been deposited at Zenodo and is publicly available. DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Gordonibacter pamelaeae 3C | Prof. Peter Turnbaugh | N/A |
| Gordonibacter sp. 28C | Prof. Peter Turnbaugh | N/A |
| Paraeggerthella hongkongensis RC2/2A | Prof. Peter Turnbaugh | N/A |
| Eggerthella lenta A2 | Prof. Peter Turnbaugh | N/A |
| Eggerthella Sinensis DSM 16107 | Prof. Peter Turnbaugh | N/A |
| Adlercreutzia equolifaciens DSM 19450 | DSMZ | N/A |
| Adlercreutzia equolifaciens subsp. celatus DSM 18785 | DSMZ | N/A |
| Slackia heliotrinireducens DSM 20476 | DSMZ | N/A |
| Senegalimassilia anaerobia DSM 25959 | DSMZ | N/A |
| Gordonibacter urolithinfaciens DSM 27213 | DSMZ | N/A |
| Gordonibacter pamelaeae 7-10-1b | DSMZ | N/A |
| Raoultibacter massiliensis DSM 103407 | DSMZ | N/A |
| Gordonibacter urolithinfaciens DSM 27213 hcdh K/O | This paper | N/A |
| Gordonibacter sp. 28C | Prof. Peter Turnbaugh | N/A |
| Paraeggerthella hongkongensis RC2/2A | Prof. Peter Turnbaugh | N/A |
| Eggerthella lenta A2 | Prof. Peter Turnbaugh | N/A |
| Eggerthella Sinensis DSM 16107 | Prof. Peter Turnbaugh | N/A |
| Adlercreutzia equolifaciens DSM 19450 | DSMZ | N/A |
| Adlercreutzia equolifaciens subsp. celatus DSM 18785 | DSMZ | N/A |
| Slackia heliotrinireducens DSM 20476 | DSMZ | N/A |
| Senegalimassilia anaerobia DSM 25959 | DSMZ | N/A |
| Gordonibacter urolithinfaciens DSM 27213 | DSMZ | N/A |
| Gordonibacter pamelaeae 7-10-1b | DSMZ | N/A |
| Raoultibacter massiliensis DSM 103407 | DSMZ | N/A |
| Gordonibacter urolithinfaciens DSM 27213 hcdh K/O | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| 3,4-dihydroxyhydrocinnamic acid (hydrocaffeic acid; HCA) | Sigma-Aldrich | Cat#102601-10G |
| 3-(3-hydroxyphenyl)propionic acid (mHPPA) | Toronto Research Chemicals | Cat#H940090-1g |
| Caffeic acid | Millipore Sigma | Cat#C0625-2G |
| 3,4-dihydroxyphenylacetic acid (DOPAC) | Millipore Sigma | Cat#850217-1G |
| Dopamine | Sigma-Aldrich | Cat#PHR1090-1G |
| Chlorogenic acid | Ambeed | Cat#A163338 |
| trans-ferulic acid | Sigma-Aldrich | Cat#128708-5G |
| trans-isoferulic acid | Sigma-Aldrich | Cat#PHL89717-100MG |
| DL-3,4-dihydroxymandelic acid | Sigma-Aldrich | Cat#151610-500MG |
| Rosmarinic acid | Astatech | Cat#44324 |
| L-arginine monohydrochloride | Sigma-Aldrich | Cat#A5131-500G |
| L-cysteine hydrochloride monohydrate | Sigma-Aldrich | Cat#C7880-100G |
| Sodium formate | Sigma-Aldrich | Cat#247596-100G |
| Methyl viologen | Sigma-Aldrich | Cat#856177-1g |
| Sodium dithionite | Sigma-Aldrich | Cat#157953-5G |
| Ammonium sulfate | Sigma-Aldrich | Cat#157953-5G |
| Sodium molybdate | Sigma-Aldrich | Cat#243655-100G |
| Sodium nitrite | Sigma-Aldrich | Cat#237213-100G |
| SIGMAFAST protease inhibitor tablets | Sigma-Aldrich | Cat#S8830 |
| DNase | Sigma-Aldrich | Cat#DN25-1G |
| Lysozyme | Sigma-Aldrich | Cat#L6876-5G |
| Brain–Heart Infusion (BHI) | Becton Dickinson | Cat#211060 |
| TRIzol | Invitrogen | Cat#15596026 |
| 3-(3-hydroxyphenyl)propionic acid (mHPPA) | Toronto Research Chemicals | Cat#H940090-1g |
| Caffeic acid | Millipore Sigma | Cat#C0625-2G |
| 3,4-dihydroxyphenylacetic acid (DOPAC) | Millipore Sigma | Cat#850217-1G |
| Dopamine | Sigma-Aldrich | Cat#PHR1090-1G |
| Chlorogenic acid | Ambeed | Cat#A163338 |
| trans-ferulic acid | Sigma-Aldrich | Cat#128708-5G |
| trans-isoferulic acid | Sigma-Aldrich | Cat#PHL89717-100MG |
| DL-3,4-dihydroxymandelic acid | Sigma-Aldrich | Cat#151610-500MG |
| Rosmarinic acid | Astatech | Cat#44324 |
| L-arginine monohydrochloride | Sigma-Aldrich | Cat#A5131-500G |
| L-cysteine hydrochloride monohydrate | Sigma-Aldrich | Cat#C7880-100G |
| Sodium formate | Sigma-Aldrich | Cat#247596-100G |
| Methyl viologen | Sigma-Aldrich | Cat#856177-1g |
| Sodium dithionite | Sigma-Aldrich | Cat#157953-5G |
| Ammonium sulfate | Sigma-Aldrich | Cat#157953-5G |
| Sodium molybdate | Sigma-Aldrich | Cat#243655-100G |
| Sodium nitrite | Sigma-Aldrich | Cat#237213-100G |
| SIGMAFAST protease inhibitor tablets | Sigma-Aldrich | Cat#S8830 |
| DNase | Sigma-Aldrich | Cat#DN25-1G |
| Lysozyme | Sigma-Aldrich | Cat#L6876-5G |
| Brain–Heart Infusion (BHI) | Becton Dickinson | Cat#211060 |
| TRIzol | Invitrogen | Cat#15596026 |
| Critical commercial assays | ||
| Direct-Zol RNA MiniPrep Plus Kit | Zymo Research | Cat#R2070 |
| RQ1 RNase-free DNase kit | Promega | Cat#M6101 |
| Luna® Universal One-Step RT-qPCR Kit | New England Biolabs | Cat#E3005S |
| BacTiter-Glo | Promega | Cat#8230 |
| RQ1 RNase-free DNase kit | Promega | Cat#M6101 |
| Luna® Universal One-Step RT-qPCR Kit | New England Biolabs | Cat#E3005S |
| BacTiter-Glo | Promega | Cat#8230 |
| Deposited data | ||
| Gordonibacter urolithinfaciens DSM 27213 hcdh K/O genome | This work | BioProject PRJNA1150218 |
| Metagenome-assembled genomes from human-associated microbiome | Pasolli et al. | N/A |
| Gordonibacter pamelaeae 3C RNA-seq raw data | Bess et al. | BioProject PRJNA450120 |
| Men’s Lifestyle Validation Study (MLVS) | Mehta et al. | BioProject PRJNA354235 |
| Human Microbiome Project 2 (HMP2) | Lloyd-Price et al. | BioProject PRJNA398089 |
| Recombinant DNA | ||
| pXD71Cas10RFP | Addgene | Plasmid #192273 |
| pXD68Kan2 | Addgene | Plasmid #191248 |
| pXD80GuCas3.1 | This work | N/A |
| Software and algorithms | ||
| GraphPad Prism 9 and 10 | GraphPad Software | N/A |
| tblastn | Altschul et al. | N/A |
| PhyloPhlAn 3.0 | Asnicar et al. | https://github.com/biobakery/phylophlan |
| iTOL v6.5.8 | Letunic and Bork | https://github.com/iBiology/iTOL |
| Enzyme Function Initiative’s Enzyme Similarity Tool (EFI-EST) | Zallot et al. | https://efi.igb.illinois.edu/efi-est/ |
| Cytoscape v3.9.1 | Shannon et al. | https://github.com/cytoscape |
| KneadData v0.10.0 | N/A | https://github.com/biobakery/kneaddata |
| ShortBRED | Kaminski et al. | https://github.com/biobakery/shortbred |
| DIAMOND | Buchfink et al. | https://github.com/bbuchfink/diamond |
| MicrobeCensus | Nayfach and Pollard | https://github.com/snayfach/MicrobeCensus |
| Bowtie2 v2.4.2 | Langmead and Salzberg | https://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| HTseq-count v0.13.5 | Anders et al. | https://htseq.readthedocs.io/en/release_0.11.1/index.html |
| Salmon v1.9.0 | Patro et al. | https://combine-lab.github.io/salmon/ |
| MaAsLin2 1.0.0 | Mallick et al. | https://github.com/biobakery/Maaslin2 |
| MAFFT-linsi v7.505 | Katoh and Standley | https://mafft.cbrc.jp/alignment/software/ |
| trimAl v1.4.1 | Capella-Gutiérrez et al. | https://vicfero.github.io/trimal/ |
| iqtree2 v2.1.3 | Minh et al. | https://github.com/iqtree/iqtree2 |
| Diet and MTX analysis pipeline | This paper | https://zenodo.org/records/13390717 |
Experimental model and study participant details
Microbial strains
Gordonibacter pamelaeae 3C, Gordonibacter sp. 28C, Paraeggerthella hongkongensis RC2/2 A, and Eggerthella lenta strains were obtained from Peter Turnbaugh at the University of California, San Francisco. Adlercreutzia equolifaciens DSM 19450, Adlercreutzia equolifaciens subsp. celatus DSM 18785, Slackia heliotrinireducens DSM 20476, Senegalimassilia anaerobia DSM 25959, Gordonibacter urolithinfaciens DSM 27213, Gordonibacter pamelaeae 7–10-1b, and Raoultibacter massiliensis DSM 103407 were purchased from Leibniz Institute DSMZ. Unless otherwise noted, all bacterial culturing was performed in a rigid anaerobic chamber (Coy Laboratory Products) under an atmosphere of 2–4% hydrogen, 2–4% carbon dioxide, and nitrogen as the balance. Hungate tubes were used for anaerobic culture unless otherwise noted (Chemglass, catalog# CLS-4209–01). The human gut Coriobacteriia species were grown at 37 °C on BHI supplemented with 1% L-arginine monohydrochloride (w/v%), 0.05% L-cysteine hydrochloride monohydrate (w/v%), 10 mM sodium formate (BHIrcf medium) unless otherwise noted.
Method details
Genome searches for catechol dehydroxylase homologs
The sequences of the catalytic subunits of the five characterized catechol dehydroxylases (Hcdh, WP_035577003.1; Dadh, WP_086414988.1; Cadh, WP_114556713.1; Dodh, WP_041239388.1; and Cldh, WP_114568826.1) were used as queries for tBLASTn search (search translated nucleotide databases using a protein query) with an e-value cutoff of <0.0001 using a MAG dataset that contains 154,723 MAGs from the human-associated microbiome18,58. Similar to a previous study13, hits with >30% aa identity to any of the queries were considered catechol dehydroxylase homologs. The cutoff was chosen because sequences with lower similarity often more closely resemble other biochemically characterized members of the DMSO reductase superfamily, as assessed by sequence identity. Since the MAG dataset contains truncated protein sequences, protein sequences shorter than 600 amino acids were filtered.
Phylogeny of catechol dehydroxylase-coding species
The MAGs searched in this study were previously assigned to the closest species-level genome bins (SGBs) using PhyloPhlAn 3.059. For each SGB, one representative MAG was selected based on completeness and contamination. Only SGBs whose representative MAGs encode at least one catechol dehydroxylase homolog were considered catechol dehydroxylase-encoding SGBs. The representative MAGs of the catechol dehydroxylase-encoding SGBs were subjected to the construction of a maximum-likelihood phylogenetic tree using PhyloPhlAn 3.0 with the following parameters: medium diversity, fast, default PhyloPhlAn marker database, FastTree and RAxML. The representative MAG of SGB 991 was used as an outgroup to root the tree. The tree was visualized using a free web-based tool, iTOL v6.5.860. SGBs were classified at family level using PhyloPhlAn 3.0 (database: SGB.Jan19). To match SGBs with cultured bacteria, the genomes of cultured Coriobacteriia were assigned to the closest SGBs using PhyloPhlAn 3.0.
SSN analysis of catechol dehydroxylases
The catechol dehydroxylase homologs from the MAG search were submitted to the Enzyme Function Initiative’s Enzyme Similarity Tool (EFI-EST)22. The alignment scores between the characterized catechol dehydroxylases were determined to range from <100 to 291 (Dadh–Cadh pair). As such, initial SSN was generated with minimum alignment scores of 300. The characterized catechol dehydroxylases were initially separated into distinct clusters with a minimum alignment score of 300 and a minimum aa sequence identity of 60%. Manual inspection revealed that the cluster containing E. lenta DSM 2243 Hcdh contained another sequence from E. lenta DSM 2243: ELEN_0497 (60.6% aa identity to Hcdh) (Figure S1B). RT–qPCR experiments showed that HCA upregulated the expression of Hcdh but not that of ELEN_0497 (Figure S1C), suggesting that ELEN_0497 likely does not metabolize HCA. To separate these two sequences, additional SSNs were generated with higher minimum aa sequence identities. ELEN_0497 was separated from the Hcdh cluster at minimum alignment score 300 and minimum aa sequence identity 72% (Figure S1B and Table S1). The final network was then downloaded as 95% representative nodes. Nodes corresponding to the five characterized dehydroxylases are enlarged. The hydroxyl group that is dehydroxylated is colored on the molecular structures. Cytoscape v.3.9.1 was used to visualize and optimize the networks61.
Catechol dehydroxylase activity assay
Turbid 48-hour starter cultures of Coriobacteriia species in BHI medium were diluted 1:100 into 200 μL of BHI medium supplemented with 10 mM sodium formate and 1 mM of a catechol substrate in triplicates. The cultures were anaerobically incubated at 37 °C for three days. For LC–MS/MS analysis of dehydroxylation activity, 40 μL of supernatant was diluted into 160 μL of LC–MS grade methanol, and the precipitates were removed by centrifugation before injection onto LC–MS/MS, as described below. In addition, the consumption of catechol substrates was assessed using a colorimetric assay (Arnow test)62. For the Arnow test, 25 μL of culture supernatant was sequentially added to 25 μL of 0.5 M aqueous HCl, 25 μL of an aqueous solution containing sodium molybdate (0.1 g/mL) and sodium nitrite (0.1 g/mL), and finally 25 μL of 1 M aqueous NaOH. The presence of catechol compounds was indicated by the development of a pink color, and absorbance at 500 nm was measured using a Synergy HTX Multi-Mode Microplate Reader (BioTek).
MGX and MTX quantification
The MGX and MTX datasets of two human studies (MLVS28 and HMP233) were downloaded from the National Center for Biotechnology Information (MLVS: PRJNA354235, HMP2: PRJNA398089). Host-associated reads, low-quality reads, and adapter sequences were removed using KneadData v0.10.0 (default setting). To quantify the MGX and MTX abundance of individual catechol dehydroxylase clusters from the isofunctional SSN, we first used ShortBRED63, a highly specific tool that quantifies peptide markers unique to each cluster. However, most catechol dehydroxylase clusters were not detected using ShortBRED, presumably due to the low abundance of Coriobacteriia. Instead, to detect genes and transcripts encoding these enzymes, we quantified reads mapping on the representative sequence of each cluster. The representative sequence was chosen as the sequence that has the highest sum of alignment scores within each cluster. Singletons were excluded from the analysis because they are poorly aligned with the other sequences. To prevent non-specific read mapping, we also included UniRef50 sequences of the DMSO reductase superfamily (IPR000657) into the list (catechol dehydroxylase homologs were removed from the list manually). Using a BLASTX DIAMOND search (e-value cutoff: 0.0001)64, reads were mapped on the protein sequences. Reads with >72% aa identity to each sequence were counted. If a read was mapped on multiple proteins, the read was counted as a hit for the protein that had the highest identity, which increases the specificity of the quantification. The number of hits was normalized first by the number of the total reads of each sample and the lengths of the proteins to read per kilobase million (RPKM) (Table S2). For further statistical analyses across samples, RPKM was normalized by average genome size (AGS), which was calculated using MicrobeCensus from each MGX samples65. The plot and heatmap were generated using GraphPad PRISM 9.
Transcriptional analysis of G. pamelaeae 3C
For in vivo transcriptional analysis of G. pamelaeae 3C, Bowtie2 v2.4.266 was used to map reads from quality controlled MLVS MGX and MTX data on the whole genome of G. pamelaeae 3C. HTseq-count v0.13.567 was used to count the number of mapped reads. To account for gene copy-number variation, RNA/DNA ratio for each gene and sample was derived using paired MGX and MTX samples (Table S2). For in vitro transcriptional analysis, raw RNA-seq data of G. pamelaeae 3C in mid-exponential growth phase were downloaded from the National Center for Biotechnology Information (PRJNA450120)14. The downloaded RNA-seq data were processed to remove ribosomal RNA, low quality bases, and adapter sequences using KneadData v0.10.0 (--bypass-trf). The processed data were then quantified against the protein coding sequences of the strain using Salmon-quant v1.9.0 (--validateMappings)68. The resulting transcriptional levels of each gene were expressed in transcripts per million (TPM), which was log-transformed using the formula log10(TPM+1) (Table S2). The plots were generated using GraphPad PRISM 9.
Phylogeny of catechol dehydroxylases
The catalytic subunits of representative sequences from the catechol dehydroxylase SSN clusters were first aligned using MAFFT-linsi v7.50569 and then trimmed to 669 aa using trimAl v1.4.1 (-gappyout)70 (Table S1). A maximum-likelihood phylogenetic tree was generated using iqtree2 v2.1.3 (1000 ultrafast bootstraps)71. To enhance the clarity, highly divergent branches were pruned. The tree was visualized using a free web-based tool, iTOL v6.5.860.
Phylogeny of LuxR-type transcriptional regulators
Sequences of the 10–12 transmembrane LuxR-type regulators encoded by G. pamelaeae 3C and E. lenta DSM 2243 as well as the regulators co-localized with Gp Hcdh homologs in the genomes of G. pamelaeae 7–10-1b, G. sp. 28C, G. urolithinfaciens DSM 27213, P. hongkongensis RC2/2 A, and E. isourolithinifaciens DSM 104140 were first aligned using MAFFT-linsi v7.50569 and then trimmed using trimAl v1.4.1 (-gappyout)70 (Table S1). A maximum-likelihood phylogenetic tree was generated using iqtree2 v2.1.3 (1000 ultrafast bootstraps)71. The tree was visualized using a free web-based tool, iTOL v6.5.860.
RT–qPCR analysis
Method A (G. pamelaeae 3C)
Turbid 54-hour starter cultures of G. pamelaeae 3C in BHI medium were diluted 1:100 into 5 mL of BHIrcf medium in six replicates, and cultures were grown at 37 °C anaerobically. When the optical density at 600 nm (OD600) of the cultures reached 0.2, 50 μL of 100 mM substrate was added at a final concentration of 1 mM to three replicates, and 50 μL of vehicle (dimethylformate) was added to the other three replicates. The cultures were grown at 37 °C until they reached OD600 = 0.5, at which cells were pelleted by centrifugation.
Method B (E. lenta DSM 2243)
Turbid 54-hour grown starter cultures of E. lenta DSM 2243 in BHI medium were diluted 1:100 into 5 mL of BHI medium supplemented with 0.5% L-arginine-HCl (w/v%) and 10 mM sodium formate in six replicates, and cultures were grown at 37 °C anaerobically. When the cultures reached OD600 = 0.2, 50 μL of 100 mM substrate was added at a final concentration of 1 mM to three replicates, and 50 μL of vehicle (dimethylformate) was added to the other three replicates. The cultures were grown at 37 °C until they reached OD600 = 0.5, at which cells were pelleted by centrifugation.
To the bacterial pellets from all methods, 1 mL of TRIzol was added. Total RNA was isolated by bead beating for 5 min (Biospec, mini-beadbeater-12 / Zymo Research BashingBead Lysis Tubes, catalog# S6012–50) and then using the Zymo Research Direct-Zol RNA MiniPrep Plus Kit (catalog# R2070) according to the manufacturer’s protocol. Total RNA was then treated with the Promega RQ1 RNase-free DNase kit (catalog# M6101) according to the manufacturer’s protocol. RT–qPCR assays were performed using the Luna® Universal One-Step RT-qPCR Kit (catalog# E3005S) with 10 ng of purified RNA using a Thermocycler CFX96 Real-Time System (Bio-Rad). The Cq values of target genes were normalized to those of housekeeping genes. Fold changes in transcript levels were calculated by comparing normalized Cq values of substrate-treated samples to those of vehicle-treated samples. The primers used for RT-qPCR experiments are listed in Table S3.
LC–MS/MS methods
Samples were analyzed using a Waters UPLC-QQQ Mass Spectrometer equipped with a CORTECS T3 column (Waters Corp.). The following chromatography conditions were used in all LC–MS/MS assays: Column temperature, 40 °C; Mobile phase A, water (0.1% formic acid); Mobile phase B, acetonitrile (0.1% formic acid); Gradient (percentage denotes the ratio of mobile phase A): 100%, 1 min; 100% to 10%, 1 min; 10%, 0.5 min; 10% to 100%, 0.25 min; 100%, 0.65 min; Flowrate, 0.5 mL/min; Injection volume: 1.0 μL.
For MS/MS, the precursor and daughter ions were detected via Electrospray Ionization (ESI) in negative mode. The transitions and collision energy used to detect specific compounds of interest are listed. (1) HCA: precursor ion m/z, 181.09; daughter ion m/z, 59.03; collision energy, 12 V; cone voltage, 2 V. (2) mHPPA: precursor ion m/z, 165.09; daughter ion m/z, 121.10; collision energy, 12 V; cone voltage, 36 V. (3) Dopamine: precursor ion m/z, 154.17; daughter ion m/z, 106.21; collision energy, 14 V; cone voltage, 58 V. (4) m-Tyramine: precursor ion m/z, 138.18; daughter ion m/z, 77.06; collision energy, 26 V; cone voltage, 26 V. (5) DOPAC: precursor ion m/z, 167.07; daughter ion m/z, 123.10; collision energy, 12 V; cone voltage, 46 V. (6) 3-Hydroxyphenyl acetic acid: precursor ion m/z, 151.08; daughter ion m/z, 107.10; collision energy, 8 V; cone voltage, 30 V.
Activity-guided purification of Gp Hcdh
Growth of cultures for protein purification was performed under strictly anaerobic conditions. The anaerobic atmosphere consisted of 2–4% CO2, 2–4% H2, and N2 as the balance. G. pamelaeae 3C starter cultures were inoculated from frozen glycerol stock into liquid BHI medium and were grown anaerobically without shaking for 48 hours at 37 °C. Expression medium (liquid BHI containing 25 mM L-arginine-HCl and 10 mM sodium formate) was deoxygenated under the anaerobic atmosphere for at least 24 hours, with occasional mixing. The starter cultures were then diluted 1:100 into a total volume of 8 L (4 flasks, 2 L each) of the expression medium. The expression medium was further supplemented with cysteine (6.3 mL of aqueous 1 M L-cysteine-HCl per 2 L of medium, 3 mM final concentration) and HCA (10 mL of aqueous 200 mM HCA per 2 L of medium, 1 mM final concentration). The resulting culture flasks were sealed with anaerobic septa and caps, then incubated at 37 °C for 18 hours, with shaking at 160 rpm. After 18 hours, OD600 usually reached ~0.6. At this point, the growth medium was analyzed for the presence of catechols. The Arnow colorimetric assay yielded a clear solution, indicating an almost complete consumption of HCA.
Activity-guided purification of Gp Hcdh was carried out under air, while keeping all solutions at 4 °C. Cells were pelleted by centrifugation (8k RCF, 15 min) and then suspended in lysis buffer (20 mM Tris, pH 7.5, 500 mM NaCl, 10 mM MgSO4, 1 mM CaCl2, 0.1 mg/mL DNase, 0.5 mg/mL lysozyme, 1 tablet/50 mL SIGMAFAST protease inhibitor) to give a final volume of ~45 mL. Cells were lysed via sonication (Branson Sonifier 450, 25% amplitude, 10 seconds on, 40 seconds off, 4 min total sonication time). The lysate was then clarified via centrifugation (20k RCF, 45 mins). The resulting supernatant (~40 mL) was transferred to a new 50 mL Eppendorf tube, then solid ammonium sulfate (13.2 g) was slowly added to achieve 33% w/v. The tube was inverted gently to dissolve all ammonium sulfate. It was then left to sit at 4 °C for 90 mins, with occasional inverting. The protein precipitate was pelleted by centrifugation (4k RCF, 30 mins). The precipitate was dissolved in ~25 mL of HIC-1M buffer (20 mM Tris, pH 7.5, 1 M ammonium sulfate). The protein solution was then centrifuged to remove insoluble precipitates prior to hydrophobic interaction chromatography (HIC).
The following protein purification steps were performed using an FPLC (Bio-Rad BioLogic DuoFlow System equipped with GE Life Sciences DynaLoop90). To perform HIC, the HIC column (Cytiva HiTrap Phenyl HP, 5 mL) was equilibrated with 15 mL of HIC-1M buffer. The protein solution was loaded at 2 mL/min, followed by an additional 5 mL of HIC-1M buffer. Protein purification was achieved using the following gradient of HIC-1M buffer and HIC-end buffer (20 mM Tris, pH 7.5): 100%, 5 mL; 100% to 0%, 40 mL, 0%, 15 mL (percentage denotes the ratio of HIC-1M); flowrate, 2.5 mL/min; collection volume, 1.25 mL fractions. 25 μL of each fraction was taken into an anaerobic chamber to perform an enzyme activity assay (refer to activity assays during protein purification). Fractions with activity (>10% conversion of HCA to mHPPA) were combined. These fractions typically eluted from the column when the mobile phase was 100% HIC-end buffer. The combined fractions were then subjected to anion exchange chromatography (AEC) without a buffer swap. Analysis of the purified fractions using sodium dodecyl-sulfate polyacrylamide gel electrophoresis showed that the HIC step was most effective in purifying the active protein.
To perform AEC, the AEC column (Cytiva Q HP, 5 mL) was equilibrated with 15 mL of HIC-end buffer. The protein solution was loaded at 2 mL/min, followed by an additional 5 mL of HIC-end buffer. Protein purification was achieved using the following gradient of HIC-end buffer and AEC-end buffer (20 mM Tris, pH 7.5, 1 M NaCl): 100%, 5 mL; 100% to 0%, 60 mL, 0%, 10 mL (percentage denotes the ratio of HIC-end); flowrate, 2.5 mL/min; collection volume, 1.25 mL fractions. 25 μL of each fraction was taken into an anaerobic chamber to perform activity assay. Fractions with high activity (complete conversion of HCA to mHPPA) were combined. The protein solution was concentrated, and the buffer was exchanged to give 20 mM Tris, pH 7.5, 250 mM NaCl (size-exclusion chromatography (SEC) buffer). To perform SEC, the sample was loaded onto the equilibrated SEC column (Cytiva Superdex 200 Increase 10/300 GL). Protein purification was achieved by running 2 column volume of the SEC buffer. Fractions with high activity were combined. Protein yield was 0.03 mg/L of G. pamelaeae 3C culture.
Activity assays during protein purification
Stock solutions of 2 mM HCA, 4 mM methyl viologen dichloride (MV), and 4 mM sodium dithionite (NaDT) were prepared in buffer containing 20 mM Tris, pH 7.5, 250 mM NaCl under anaerobic atmosphere prior to the activity assay. The following assays were set up and conducted in the rigid anaerobic chamber (Coy Laboratory Products). To perform the enzyme activity assay, 25 μL of enzyme solution was added to 2 mM HCA solution (25 μL, 500 μM final substrate concentration) with mixing. This was followed by the addition of 4 mM MV (25 μL, 1 mM final concentrations) and 4 mM NaDT (25 μL, 1 mM final concentrations). The solution typically turns purple after the addition of NaDT. The final solution was sealed and left at room temperature, without shaking, for 20–24 hours. The assay mixture was analyzed using the Arnow colorimetric assay and LC–MS/MS. For the colorimetric assay, 25 μL of the solution was used directly. For LC–MS/MS, 20 μL of the solution was first diluted 1:10 into LC–MS grade methanol, followed by centrifugation to remove precipitates. The supernatant was diluted 1:5 into deionized water prior to analysis.
Proteomics analysis
SDS-PAGE was carried out with a Novex 10–20% Tris-Glycine Mini Protein Gel in Tris-Glycine-SDS buffer (100 V for 20 minutes, followed by 140 V for 50 min, room temperature). Protein samples were prepared with traditional Laemmli buffer without heating. The Precision Plus Protein All Blue Prestained Protein Standards (Bio-Rad) was used as a ladder. The gel band corresponding to the predicted molecular weight of Gp Hcdh was excised and submitted for proteomic analysis. The downstream experiments (sample preparation, mass spectrometry, and MS data analysis) were performed by the Harvard Center for Mass Spectrometry.
Sample preparation
The cut-out gel band was washed twice with 50% aqueous acetonitrile for 5 mins followed by drying in a SpeedVac. The gel was then added to a volume of 100 μL of buffer containing 20 mM tris(2-carboxyethyl)phosphine (TCEP) in 25 mM tetraethylammonium bromide (TEAB) at 37 °C for 45 minutes. After cooling to room temperature, the TCEP solution was removed and replaced with the same volume of 10 mM iodoacetamide Ultra (Sigma) in 25 mM TEAB and kept in the dark at room temperature for 45 mins. Gel pieces were washed with 200 μL of 100 mM TEAB (10 minutes). The gel pieces were then shrunk with acetonitrile. The liquid was then removed followed by swelling with the 100 mM TEAB again and dehydration/shrinking with the same volume of acetonitrile. All liquid was removed, and the gel was completely dried in a SpeedVac for ~20 minutes. 0.06 μg/5 μL of trypsin in 50 mM TEAB was added to the gel pieces and the resulting mixture was placed in a thermomixer at 37 °C for about 15 minutes. 50 μL of 50 mM TEAB was added to the gel slices. The samples were vortexed, centrifuged, and placed back in the thermomixer at 37 °C for digestion. Peptides were extracted with 50 μL of 20 mM TEAB for 20 minutes and 1 change of 50 μL of 5% formic acid in 50% acetonitrile at room temperature for 20 minutes while in a sonicator. All extracts obtained were pooled into an HPLC vial and were dried using a SpeedVac to the desired volume (~50 μL). This sample was used for protein identification by LC–MS/MS, as described below.
Mass spectrometry
Each sample was submitted for a single LC–MS/MS experiment that was performed on an Orbitrap Elite Hybrid Ion Trap-Orbitrap Mass Spectrometer (Thermo Fischer, San Jose, CA) equipped with Waters Aquity with nanopump and nanoLC (Waters Corp. Milford, MA). Peptides were separated using a 100 μm inner diameter microcapillary trapping column packed first with approximately 5 cm of C18 Reprosil resin (5 μm, 100 Å, Dr. Maisch GmbH, Germany) followed by a Waters analytical column. Separation was achieved through applying a gradient from 5–27% acetonitrile with 0.1% formic acid over 90 minutes at 200 nL/min. Electrospray ionization was enabled through applying a voltage of 1.8 kV using a home-made electrode junction at the end of the microcapillary column. The Elite Orbitrap Velos was operated in data-dependent mode for the mass spectrometry methods. The mass spectrometry survey scan was performed in the Orbitrap in the range of 395–1,800 m/z at a resolution of 6 × 104, followed by the selection of the thirty most intense ions (TOP30) for CID-MS2 fragmentation in the ion trap using a precursor isolation width window of 2 m/z, AGC setting of 10,000, and a maximum ion accumulation of 200 ms. Singly charged ion species were not subjected to CID fragmentation.
MS data analysis
Raw data were submitted for analysis using Proteome Discoverer 2.4 (Thermo Scientific) software. Assignment of MS/MS spectra was performed using the Sequest HT algorithm by searching the data against a protein sequence database including all entries from the users provided database as well as the Uniprot-Gordonibacter+pamelaeae-filtered-organism_2020.fasta as well as other known contaminants such as human keratins and common lab contaminants. Sequest HT searches were performed using a 20 ppm precursor ion tolerance and requiring each peptides N/C-termini to adhere with Trypsin protease specificity, while allowing up to two missed cleavages. A MS2 spectra assignment false discovery rate (FDR) of 1% on both protein and peptide level was achieved by applying the target-decoy database search. Filtering was performed using a Percolator. Peptide N termini and lysine residues (+229.162932 Da) were set as static modifications while methionine oxidation (+15.99492 Da) was set as variable modification. A MS2 spectra assignment false discovery rate (FDR) of 1% on both protein and peptide level was achieved by applying the target-decoy database search. Filtering was performed using a Percolator (64-bit version).
Determination of enzyme kinetics
All assay concentrations are final concentrations. Anoxic Gp Hcdh and solid substrates were brought into an anaerobic chamber containing N2 and <0.1 ppm O2 (MBRAUN). Substrates were dissolved in pre-reduced dimethyl formate. Assays were setup by adding 100 nM Gp Hcdh to an anoxic buffer (50 mM MOPS, pH 7.0, 300 mM NaCl) containing 200 μM MV, 100 μM NaDT, and 2-fold serial dilutions of HCA (0.0625 to 4 mM). Initial 1 minute rate was measured using a continuous spectrophotometric assay. In this assay, the reductive dehydroxylation of Gp Hcdh was coupled with the oxidation of MV+ (blue, absorption maxima at 605 nm wavelength) to MV2+ (colorless). Each assay was performed in technical triplicates. Fitted parameters are mean ± standard error (n=3) as derived from nonlinear curve fitting to the Michaelis–Menten equation.
Bacterial growth assay
Turbid 48-hour starter cultures of G. pamelaeae 3C in BHI medium were diluted 1:100 into 1 mL of 50% BHI medium containing 10 mM sodium formate and an additive (vehicle, 5 mM HCA, 5 mM mHPPA, or 14 mM DMSO) in triplicates, which were arrayed into a deep-well 96-well plate (Corning, P-2ML-SQ-C-S). The plate was then sealed and anaerobically incubated at 37 °C for three days. 200 μL of aliquots were taken every 24 hours. The growth was assessed by OD600 measurement on a 96-well plate using a Synergy HTX Multi-Mode Microplate Reader (BioTek). To assess dehydroxylation activity, 20 μL of each aliquot was diluted into 180 μL of LC–MS grade methanol, and the precipitates were removed by centrifugation before injection onto LC–MS/MS. HCA and mHPPA were quantified using LC–MS/MS as described above.
ATP quantification assay
Turbid 54-hour starter cultures of G. pamelaeae 3C, G. urolithinfaciens wild-type, or Δhcdh strain in BHI medium were diluted 1:100 into 5 mL of BHI medium supplemented with 10 mM sodium formate in triplicates, and cultures were grown at 37 °C anaerobically for 20 hours. The following steps were then performed under anaerobic conditions. Cells were pelleted by centrifugation, and the supernatant was removed. The cell pellets were washed and resuspended with 0.5 mL of pre-reduced phosphate buffer (40 mM potassium phosphate, 10 mM magnesium chloride, 10 mM sodium formate, pH 7.0). The cell suspensions were incubated in the presence of 1 mM HCA, 1 mM mHPPA, 14 mM DMSO, or vehicle at room temperature for 1 hour. The level of ATP in cell suspensions was quantified using PROMEGA BacTiter-Glo (catalog# 8230) and a Synergy HTX Multi-Mode Microplate Reader (BioTek).
Gp Hcdh knock out using a CRISPR gene editing tool
Plasmid construction was carried out using standard molecular biology techniques, and primers used for plasmid construction are listed in Table S3. To construct an entry plasmid for gene deletion in G. urolithinfaciens DSM 27213, we first introduced tandem terminators flanking expression cassette to a previously reported vector pXD68Kan2 (https://www.addgene.org/191248/). Plasmid pXD68Kan2 backbone was first amplified using primers oXD855 and oXD856 and was ligated to a fragment containing tandem terminators amplified from pXD71Cas10RFP (https://www.addgene.org/192273/) using primers oXD859 and oXD860. The resulting plasmid was then amplified with primers oXD291 and oXD292 to provide plasmid backbone containing terminators flanking expression cassette.
Primers oXD1081 and oXD1181 were used to amplify RFP insert from pXD71Cas10RFP and introduce the flanking G. urolithinfaciens CRISPR repeats. Primers oXD361 and oXD1179 were used to amplify a cumate-inducible genetic part from plasmid pXD70CT3. These two fragments were cloned to pXD68Kan2 backbone to make an entry CRISPR plasmid pXD80GuCas3.0RFP. To make Gp Hcdh-targeting plasmid pXD80GuCas3.1, oXD889 and oXD890 were annealed and phosphorylated and were introduced to pXD80GuCas3.0RFP in replace of the RFP insert using Golden Gate assembly following a previously reported method23.
G. urolithinfaciens DSM 27213 electrocompetent cells were prepared according to a previously reported method23. Briefly, a turbid 48-hour starter culture of each strain in BHIrcf medium was inoculated 1:50 into 40 mL of BHI medium supplemented with 1% L-arginine monohydrochloride (w/v%). When the culture reached OD600 of 0.3, the cells were pelleted by centrifugation, and the supernatant was removed. The cell pellet was washed with 10 mL of ice-cold sterile water three times and then with 5 mL of deoxygenated sterile 10% (v/v) glycerol aqueous solution. Finally, the cell pellet was resuspended in 2.5 mL of deoxygenated sterile 10% (v/v) glycerol aqueous solution and subdivided into 100 μL aliquots, which were flash frozen and stored at −80 °C until use.
For transformation, electrocompetent G. urolithinfaciens cells were electroporated using a MicroPulser Electroporator (BioRad) with 1,000 ng of pXD80GuCas3.1 at 2.5 kV voltage in 1-mm gap width electroporation cuvettes (VWR). 1 mL of pre-reduced BHIrcf medium was immediately added to the electroporated cells and transferred to 1.7 mL Eppendorf tubes. These tubes were brought to anaerobic chamber (Coy Laboratory Products) and incubated at 37 °C for 3 h. The cultures were then plated onto BHI agar plates supplemented with 100 μg/mL kanamycin, 1% L-arginine monohydrochloride (w/v%), and 10 mM sodium formate and grown anaerobically for 3–5 days at 37 °C. Single colonies were picked and inoculated into BHIrcf medium containing 100 μg/mL kanamycin, and grown for 2 days at 37 °C. Each saturated culture was then serially 10-fold diluted onto BHI agar plates supplemented with 100 μg/mL kanamycin, 1% L-arginine-HCl (w/v%), and 10 mM sodium formate with or without 50 μM inducer cumate and grown for 3–4 days. The surviving colonies of pXD80GuCas3.1 on cumate-containing plates were then analyzed individually using colony PCR and Sanger sequencing to confirm the presence of desired mutations. A single clone that contains desired mutation on Gp Hcdh was inoculated into BHIrcf medium containing 100 μg/mL kanamycin to get G. urolithinfaciens Δhcdh strain.
To cure plasmid from the G. urolithinfaciens Δhcdh strain, the strain was passaged in the absence of antibiotics for multiple rounds. The strain was inoculated into BHIrcf medium without antibiotics and let grow for 48 h. The saturated cultures were first inoculated 1:1000 into BHIrcf medium, and then the resulting diluted cultures were further diluted 1:1000 into BHIrcf medium. The 1:106 diluted cultures were grown in the absence of antibiotics anaerobically for 2 days. The saturated cultures were then serially 10-fold diluted onto BHI agar plates supplemented with 1% L-arginine monohydrochloride (w/v%), 10 mM sodium formate, and with or without 100 μg/mL kanamycin and grown for 2–3 days, and CFU counts were performed. Each culture was passaged 2–3 times using the procedure described above to quantify plasmid maintenance. Lastly, single colonies were picked from BHI agar plates supplemented with 1% L-arginine monohydrochloride (w/v%), 10 mM sodium formate without kanamycin to BHIrcf medium with or without 100 μg/mL kanamycin to confirm sensitivity to kanamycin.
Quantification and statistical analysis
Diet analysis
Study population and stool sample collection
The Men’s Lifestyle Validation Study (MLVS) consisted of 914 men aged 45 to 80 years and free from coronary heart disease, stroke, cancer, or major neurological disease at recruitment in 2011. The MLVS study population was recruited from the Health Professionals Follow-up Study (HPFS), an ongoing prospective cohort study of 51,529 US male health professionals initiated in 198672. From 2011 to 2013, 307 participants provided up to two pairs of self-collected stool samples in the MLVS as detailed elsewhere28. In addition, participants provided two fasting blood samples twice, six months apart, during the same period as fecal samples collection as detailed elsewhere57. High-sensitive C-reactive protein concentrations were determined using an immunoturbidimetric high sensitivity assay using reagents and calibrators from Denka Seiken (Niigata, Japan) with assay day-to-day variability between 1 and 2%.
Dietary assessment
In 2012, the MLVS administered two semi-quantitative food frequency questionnaires (SFFQs) developed by Willett and his colleagues73 6-months apart to collect dietary information. Participants reported their usual dietary intake (from never to ≥6 times per day) of a standard portion size (e.g., 0.5 cup of strawberries, 1 banana and 0.5 cup of cooked spinach) over the preceding year / 6 months on each SFFQ. Frequencies and portions of each individual food item were converted to average daily intake for each participant. The reproducibility and validity of these SFFQs in measuring dietary intake have been documented in detail73–76. For this analysis, to minimize measurement error, we calculated cumulative average dietary intake for each participant across the two SFFQs.
Statistical analysis
For per-feature tests between relative abundance of each gene and specific food groups and/or dietary patterns, we leveraged linear mixed effects model in the R package MaAsLin2 1.0.077. All the MaAsLin2 models included identifier of participant as random effects and exposure variable and covariables (age, body mass index [BMI, kg/m2], recent antibiotic use, and Bristol stool score) as fixed effects, with the following formula:
To test the interaction between food intakes and CRP, we applied linear mixed models that include a product term of the food groups and each dichotomized microbial enzyme (e.g., Gp Hcdh > median: Gp Hcdh_binary = 1, Gp Hcdh ≤ median: Gp Hcdh_binary = 0), in addition to their main effects, as below.
The was derived from the product term. These linear mixed models also include identifier of participant as random effects and simultaneously for covariables as fixed effects. Unless otherwise noted, all the p-values were corrected for multiple comparisons using the Benjamini-Hochberg procedure with a threshold of 0.10.
Supplementary Material
Table S2: MGX-MTX quantification results for SSN, related to Figure 2
Table S3: Oligonucleotides used in this study, related to STAR Methods
Table S4: Proteomics IDs from activity-guided protein purification, related to Figure 3
Highlights.
Metatranscriptomics analysis identifies prevalent gut bacterial catechol dehydroxylases
A catechol dehydroxylase from G. pamelaeae (Gp Hcdh) is highly transcribed in vivo
Gp Hcdh dehydroxylates hydrocaffeic acid, an abundant anti-inflammatory polyphenol
Gp Hcdh level correlates with anti-inflammatory activity of cruciferous vegetables
Acknowledgments
This work was funded by the National Science Foundation (Alan T. Waterman Award to E.P.B., CHE-20380529), the National Institutes of Health (U01CA167552, R01HL35464, R35 CA253185, R01DK114034, R01HL122593), and the Biocodex Microbiota Foundation. E.P.B. is a Howard Hughes Medical Institute Investigator. A.T.C. is an American Cancer Society Research Professor. M.B. acknowledges support from the Kwanjeong Educational Foundation. C.L. acknowledges fellowship support from the Jane Coffin Childs Memorial Fund for Medical Research. R.S.M. is a recipient of the Crohn’s and Colitis Foundation Research Fellowship. We thank the Harvard Center for Mass Spectrometry for protein identification and the Harvard Research Computing for computational resources, maintenance, and support. We thank Dr. Vayu Maini Rekdal for insightful discussion and Dr. Beverly Fu and Dr. Antonio Tinoco Valencia for critical reading of the manuscript. This article is subject to HHMI’s Open Access to Publications policy. HHMI lab heads have previously granted a nonexclusive CC BY 4.0 license to the public and a sublicensable license to HHMI in their research articles. Pursuant to those licenses, the author-accepted manuscript of this article can be made freely available under a CC BY 4.0 license immediately upon publication.
Footnotes
Declaration of Interests
Dr. Chan received research support from Zoe Ltd. for a diet-microbiome study unrelated to this manuscript.
Dr. Huttenhower is a scientific advisor for Zoe Ltd. The other authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Harborne JB, and Williams CA (2000). Advances in flavonoid research since 1992. Phytochemistry 55, 481–504. 10.1016/S0031-9422(00)00235-1. [DOI] [PubMed] [Google Scholar]
- 2.Huang Q, Braffett BH, Simmens SJ, Young HA, and Ogden CL (2020). Dietary Polyphenol Intake in US Adults and 10-Year Trends: 2007–2016. J. Acad. Nutr. Diet. 120, 1821–1833. 10.1016/j.jand.2020.06.016. [DOI] [PubMed] [Google Scholar]
- 3.Taguchi C, Fukushima Y, Kishimoto Y, Suzuki-Sugihara N, Saita E, Takahashi Y, and Kondo K (2015). Estimated Dietary Polyphenol Intake and Major Food and Beverage Sources among Elderly Japanese. Nutrients 7, 10269–10281. 10.3390/nu7125530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cory H, Passarelli S, Szeto J, Tamez M, and Mattei J (2018). The Role of Polyphenols in Human Health and Food Systems: A Mini-Review. Front. Nutr. 5, 370438. 10.3389/fnut.2018.00087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arts IC, and Hollman PC (2005). Polyphenols and disease risk in epidemiologic studies. Am. J. Clin. Nutr. 81, 317S–325S. 10.1093/ajcn/81.1.317S. [DOI] [PubMed] [Google Scholar]
- 6.Duynhoven J. van, Vaughan EE, Jacobs DM, Kemperman RA, Velzen E.J.J. van, Gross G, Roger LC, Possemiers S, Smilde AK, Doré J, et al. (2011). Metabolic fate of polyphenols in the human superorganism. Proc. Natl. Acad. Sci. USA 108, 4531–4538. 10.1073/pnas.1000098107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Osborn LJ, Claesen J, and Brown JM (2021). Microbial Flavonoid Metabolism: A Cardiometabolic Disease Perspective. Annu. Rev. Nutr. 41, 433–454. 10.1146/annurev-nutr-120420-030424. [DOI] [PubMed] [Google Scholar]
- 8.Li Q, and Van de Wiele T (2021). Gut microbiota as a driver of the interindividual variability of cardiometabolic effects from tea polyphenols. Crit. Rev. Food Sci. Nutr. 63, 1500–1526. 10.1080/10408398.2021.1965536. [DOI] [PubMed] [Google Scholar]
- 9.Manach C, Milenkovic D, Van de Wiele T, Rodriguez-Mateos A, de Roos B, Garcia-Conesa MT, Landberg R, Gibney ER, Heinonen M, Tomás-Barberán F, et al. (2017). Addressing the inter-individual variation in response to consumption of plant food bioactives: Towards a better understanding of their role in healthy aging and cardiometabolic risk reduction. Mol. Nutr. Food Res. 61, 1600557. 10.1002/mnfr.201600557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marino M, Del Bo’ C, Martini D, Porrini M, and Riso P (2020). A Review of Registered Clinical Trials on Dietary (Poly)Phenols: Past Efforts and Possible Future Directions. Foods 9, 1606. 10.3390/foods9111606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Selma MV, Espín JC, and Tomás-Barberán FA (2009). Interaction between Phenolics and Gut Microbiota: Role in Human Health. J. Agric. Food Chem. 57, 6485–6501. 10.1021/jf902107d. [DOI] [PubMed] [Google Scholar]
- 12.Peppercorn MA, and Goldman P (1972). Caffeic Acid Metabolism by Gnotobiotic Rats and Their Intestinal Bacteria. Proc. Natl. Acad. Sci. USA 69, 1413–1415. 10.1073/pnas.69.6.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maini Rekdal V, Nol Bernadino P, Luescher MU, Kiamehr S, Le C, Bisanz JE, Turnbaugh PJ, Bess EN, and Balskus EP (2020). A widely distributed metalloenzyme class enables gut microbial metabolism of host- and diet-derived catechols. eLife 9, e50845. 10.7554/eLife.50845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bess EN, Bisanz JE, Yarza F, Bustion A, Rich BE, Li X, Kitamura S, Waligurski E, Ang QY, Alba DL, et al. (2020). Genetic basis for the cooperative bioactivation of plant lignans by Eggerthella lenta and other human gut bacteria. Nat. Microbiol. 5, 56–66. 10.1038/s41564-019-0596-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Romo-Vaquero M, García-Villalba R, González-Sarrías A, Beltrán D, Tomás-Barberán FA, Espín JC, and Selma MV (2015). Interindividual variability in the human metabolism of ellagic acid: Contribution of Gordonibacter to urolithin production. J. Funct. Foods 17, 785–791. 10.1016/j.jff.2015.06.040. [DOI] [Google Scholar]
- 16.Maini Rekdal V, Bess EN, Bisanz JE, Turnbaugh PJ, and Balskus EP (2019). Discovery and inhibition of an interspecies gut bacterial pathway for Levodopa metabolism. Science 364, eaau6323. 10.1126/science.aau6323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Le C. (Chip), Bae M, Kiamehr S, and Balskus EP (2022). Emerging Chemical Diversity and Potential Applications of Enzymes in the DMSO Reductase Superfamily. Annu. Rev. Biochem. 91, 475–504. 10.1146/annurev-biochem-032620-110804. [DOI] [PubMed] [Google Scholar]
- 18.Pasolli E, Asnicar F, Manara S, Zolfo M, Karcher N, Armanini F, Beghini F, Manghi P, Tett A, Ghensi P, et al. (2019). Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell 176, 649–662.e20. 10.1016/j.cell.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.García-Villalba R, Beltrán D, Frutos D, M., V. Selma M, C. Espín J, and A. Tomás-Barberán F (2020). Metabolism of different dietary phenolic compounds by the urolithin-producing human-gut bacteria Gordonibacter urolithinfaciens and Ellagibacter isourolithinifaciens. Food Funct. 11, 7012–7022. 10.1039/D0FO01649G. [DOI] [PubMed] [Google Scholar]
- 20.Takagaki A, and Nanjo F (2015). Biotransformation of (−)-Epigallocatechin and (−)-Gallocatechin by Intestinal Bacteria Involved in Isoflavone Metabolism. Biol. Pharm. Bull. 38, 325–330. 10.1248/bpb.b14-00646. [DOI] [PubMed] [Google Scholar]
- 21.Takagaki A, and Nanjo F (2015). Bioconversion of (−)-Epicatechin, (+)-Epicatechin, (−)-Catechin, and (+)-Catechin by (−)-Epigallocatechin-Metabolizing Bacteria. Biol. Pharm. Bull. 38, 789–794. 10.1248/bpb.b14-00813. [DOI] [PubMed] [Google Scholar]
- 22.Zallot R, Oberg N, and Gerlt JA (2019). The EFI Web Resource for Genomic Enzymology Tools: Leveraging Protein, Genome, and Metagenome Databases to Discover Novel Enzymes and Metabolic Pathways. Biochemistry 58, 4169–4182. 10.1021/acs.biochem.9b00735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dong X, Guthrie BGH, Alexander M, Noecker C, Ramirez L, Glasser NR, Turnbaugh PJ, and Balskus EP (2022). Genetic manipulation of the human gut bacterium Eggerthella lenta reveals a widespread family of transcriptional regulators. Nat. Commun. 13, 7624. 10.1038/s41467-022-33576-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Bhosle A, Bae S, McIver LJ, Pishchany G, Accorsi EK, Thompson KN, Arze C, Wang Y, Subramanian A, et al. (2022). Discovery of bioactive microbial gene products in inflammatory bowel disease. Nature 606, 754–760. 10.1038/s41586-022-04648-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mehta RS, Mayers JR, Zhang Y, Bhosle A, Glasser NR, Nguyen LH, Ma W, Bae S, Branck T, Song K, et al. (2023). Gut microbial metabolism of 5-ASA diminishes its clinical efficacy in inflammatory bowel disease. Nat. Med. 29, 700–709. 10.1038/s41591-023-02217-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sonnenburg JL, Xu J, Leip DD, Chen C-H, Westover BP, Weatherford J, Buhler JD, and Gordon JI (2005). Glycan Foraging in Vivo by an Intestine-Adapted Bacterial Symbiont. Science 307, 1955–1959. 10.1126/science.1109051. [DOI] [PubMed] [Google Scholar]
- 27.McNulty NP, Yatsunenko T, Hsiao A, Faith JJ, Muegge BD, Goodman AL, Henrissat B, Oozeer R, Cools-Portier S, Gobert G, et al. (2011). The Impact of a Consortium of Fermented Milk Strains on the Gut Microbiome of Gnotobiotic Mice and Monozygotic Twins. Sci. Transl. Med. 3, 106ra106–106ra106. 10.1126/scitranslmed.3002701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mehta RS, Abu-Ali GS, Drew DA, Lloyd-Price J, Subramanian A, Lochhead P, Joshi AD, Ivey KL, Khalili H, Brown GT, et al. (2018). Stability of the human faecal microbiome in a cohort of adult men. Nat. Microbiol. 3, 347–355. 10.1038/s41564-017-0096-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Franzosa EA, Morgan XC, Segata N, Waldron L, Reyes J, Earl AM, Giannoukos G, Boylan MR, Ciulla D, Gevers D, et al. (2014). Relating the metatranscriptome and metagenome of the human gut. Proc. Natl. Acad. Sci. USA 111, E2329–E2338. 10.1073/pnas.1319284111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heintz-Buschart A, May P, Laczny CC, Lebrun LA, Bellora C, Krishna A, Wampach L, Schneider JG, Hogan A, de Beaufort C, et al. (2016). Integrated multi-omics of the human gut microbiome in a case study of familial type 1 diabetes. Nat. Microbiol. 2, 1–13. 10.1038/nmicrobiol.2016.180. [DOI] [PubMed] [Google Scholar]
- 31.Tang Y, Nakashima S, Saiki S, Myoi Y, Abe N, Kuwazuru S, Zhu B, Ashida H, Murata Y, and Nakamura Y (2016). 3,4-Dihydroxyphenylacetic acid is a predominant biologically-active catabolite of quercetin glycosides. Food Res. Int. 89, 716–723. 10.1016/j.foodres.2016.09.034. [DOI] [PubMed] [Google Scholar]
- 32.Zamora-Ros R, Achaintre D, Rothwell JA, Rinaldi S, Assi N, Ferrari P, Leitzmann M, Boutron-Ruault M-C, Fagherazzi G, Auffret A, et al. (2016). Urinary excretions of 34 dietary polyphenols and their associations with lifestyle factors in the EPIC cohort study. Sci. Rep. 6, 26905. 10.1038/srep26905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, Poon TW, Andrews E, Ajami NJ, Bonham KS, Brislawn CJ, et al. (2019). Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655–662. 10.1038/s41586-019-1237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mattila P, and Kumpulainen J (2002). Determination of Free and Total Phenolic Acids in Plant-Derived Foods by HPLC with Diode-Array Detection. J. Agric. Food Chem. 50, 3660–3667. 10.1021/jf020028p. [DOI] [PubMed] [Google Scholar]
- 35.Li Z, Lee HW, Liang X, Liang D, Wang Q, Huang D, and Ong CN (2018). Profiling of Phenolic Compounds and Antioxidant Activity of 12 Cruciferous Vegetables. Molecules 23, 1139. 10.3390/molecules23051139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kountz DJ, and Balskus EP (2021). Leveraging Microbial Genomes and Genomic Context for Chemical Discovery. Acc. Chem. Res. 54, 2788–2797. 10.1021/acs.accounts.1c00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Youngblut MD, Tsai C-L, Clark IC, Carlson HK, Maglaqui AP, Gau-Pan PS, Redford SA, Wong A, Tainer JA, and Coates JD (2016). Perchlorate Reductase Is Distinguished by Active Site Aromatic Gate Residues *. J. Biol. Chem. 291, 9190–9202. 10.1074/jbc.M116.714618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wells M, Kim M, Akob DM, Basu P, and Stolz JF (2023). Impact of the Dimethyl Sulfoxide Reductase Superfamily on the Evolution of Biogeochemical Cycles. Microbiol. Spectr. 11, e04145–22. 10.1128/spectrum.04145-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Little AS, Younker IT, Schechter MS, Bernardino PN, Méheust R, Stemczynski J, Scorza K, Mullowney MW, Sharan D, Waligurski E, et al. (2024). Dietary- and host-derived metabolites are used by diverse gut bacteria for anaerobic respiration. Nat. Microbiol. 9, 55–69. 10.1038/s41564-023-01560-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sova M, and Saso L (2020). Natural Sources, Pharmacokinetics, Biological Activities and Health Benefits of Hydroxycinnamic Acids and Their Metabolites. Nutrients 12, 2190. 10.3390/nu12082190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J, Hodes GE, Zhang H, Zhang S, Zhao W, Golden SA, Bi W, Menard C, Kana V, Leboeuf M, et al. (2018). Epigenetic modulation of inflammation and synaptic plasticity promotes resilience against stress in mice. Nat. Commun. 9, 477. 10.1038/s41467-017-02794-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li R, Xie L, Li L, Chen X, Yao T, Tian Y, Li Q, Wang K, Huang C, Li C, et al. (2022). The gut microbial metabolite, 3,4-dihydroxyphenylpropionic acid, alleviates hepatic ischemia/reperfusion injury via mitigation of macrophage pro-inflammatory activity in mice. Acta Pharm. Sin. B 12, 182–196. 10.1016/j.apsb.2021.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Larrosa M, Luceri C, Vivoli E, Pagliuca C, Lodovici M, Moneti G, and Dolara P (2009). Polyphenol metabolites from colonic microbiota exert anti-inflammatory activity on different inflammation models. Mol. Nutr. Food Res 53, 1044–1054. 10.1002/mnfr.200800446. [DOI] [PubMed] [Google Scholar]
- 44.Monagas M, Khan N, Andrés-Lacueva C, Urpí-Sardá M, Vázquez-Agell M, Lamuela-Raventós RM, and Estruch R (2009). Dihydroxylated phenolic acids derived from microbial metabolism reduce lipopolysaccharide-stimulated cytokine secretion by human peripheral blood mononuclear cells. Br. J. Nutr. 102, 201–206. 10.1017/S0007114508162110. [DOI] [PubMed] [Google Scholar]
- 45.Wang J, Blaze J, Haghighi F, Kim-Schulze S, Raval U, Trageser KJ, and Pasinetti GM (2020). Characterization of 3(3,4-dihydroxy-phenyl) propionic acid as a novel microbiome-derived epigenetic modifier in attenuation of immune inflammatory response in human monocytes. Mol. Immunol. 125, 172–177. 10.1016/j.molimm.2020.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ma W, Nguyen LH, Song M, Wang DD, Franzosa EA, Cao Y, Joshi A, Drew DA, Mehta R, Ivey KL, et al. (2021). Dietary fiber intake, the gut microbiome, and chronic systemic inflammation in a cohort of adult men. Genome Med. 13, 102. 10.1186/s13073-021-00921-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y, Thompson KN, Branck T, Yan Y, Nguyen LH, Franzosa EA, and Huttenhower C (2021). Metatranscriptomics for the Human Microbiome and Microbial Community Functional Profiling. Annu. Rev. of Biomed. Data Sci. 4, 279–311. 10.1146/annurev-biodatasci-031121-103035. [DOI] [PubMed] [Google Scholar]
- 48.Bashiardes S, Zilberman-Schapira G, and Elinav E (2016). Use of Metatranscriptomics in Microbiome Research. Bioinform. Biol. Insights 10, BBI.S34610. 10.4137/BBI.S34610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shi Y, Tyson GW, Eppley JM, and DeLong EF (2011). Integrated metatranscriptomic and metagenomic analyses of stratified microbial assemblages in the open ocean. ISME J. 5, 999–1013. 10.1038/ismej.2010.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ojala T, Häkkinen A-E, Kankuri E, and Kankainen M (2023). Current concepts, advances, and challenges in deciphering the human microbiota with metatranscriptomics. Trends Genet. 39, 686–702. 10.1016/j.tig.2023.05.004. [DOI] [PubMed] [Google Scholar]
- 51.Pascal Andreu V, Augustijn HE, van den Berg K, van der Hooft JJJ, Fischbach MA, and Medema MH (2021). BiG-MAP: an Automated Pipeline To Profile Metabolic Gene Cluster Abundance and Expression in Microbiomes. mSystems 6, e00937–21. 10.1128/mSystems.00937-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim KH, Park D, Jia B, Baek JH, Hahn Y, and Jeon CO (2022). Identification and Characterization of Major Bile Acid 7α-Dehydroxylating Bacteria in the Human Gut. mSystems 7, e00455–22. 10.1128/msystems.00455-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mello BL, Alessi AM, Riaño-Pachón DM, deAzevedo ER, Guimarães FEG, Espirito Santo MC, McQueen-Mason S, Bruce NC, and Polikarpov I (2017). Targeted metatranscriptomics of compost-derived consortia reveals a GH11 exerting an unusual exo-1,4-β-xylanase activity. Biotechnol. Biofuels 10, 254. 10.1186/s13068-017-0944-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takasaki K, Miura T, Kanno M, Tamaki H, Hanada S, Kamagata Y, and Kimura N (2013). Discovery of Glycoside Hydrolase Enzymes in an Avicel-Adapted Forest Soil Fungal Community by a Metatranscriptomic Approach. PLoS One 8, e55485. 10.1371/journal.pone.0055485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.He B, Jin S, Cao J, Mi L, and Wang J (2019). Metatranscriptomics of the Hu sheep rumen microbiome reveals novel cellulases. Biotechnol. Biofuels 12, 153. 10.1186/s13068-019-1498-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Y, Thompson KN, Huttenhower C, and Franzosa EA (2021). Statistical approaches for differential expression analysis in metatranscriptomics. Bioinformatics 37, i34–i41. 10.1093/bioinformatics/btab327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang DD, Nguyen LH, Li Y, Yan Y, Ma W, Rinott E, Ivey KL, Shai I, Willett WC, Hu FB, et al. (2021). The gut microbiome modulates the protective association between a Mediterranean diet and cardiometabolic disease risk. Nat. Med. 27, 333–343. 10.1038/s41591-020-01223-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Altschul SF, Gish W, Miller W, Myers EW, and Lipman DJ (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 59.Asnicar F, Thomas AM, Beghini F, Mengoni C, Manara S, Manghi P, Zhu Q, Bolzan M, Cumbo F, May U, et al. (2020). Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat. Commun. 11, 2500. 10.1038/s41467-020-16366-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Letunic I, and Bork P (2021). Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. 10.1093/nar/gkab301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 13, 2498–2504. 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Arnow LE (1937). COLORIMETRIC DETERMINATION OF THE COMPONENTS OF 3,4-DIHYDROXYPHENYLALANINETYROSINE MIXTURES. J. Biol. Chem. 118, 531–537. 10.1016/S0021-9258(18)74509-2. [DOI] [Google Scholar]
- 63.Kaminski J, Gibson MK, Franzosa EA, Segata N, Dantas G, and Huttenhower C (2015). High-Specificity Targeted Functional Profiling in Microbial Communities with ShortBRED. PLoS Comput. Biol. 11, e1004557. 10.1371/journal.pcbi.1004557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Buchfink B, Xie C, and Huson DH (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 65.Nayfach S, and Pollard KS (2015). Average genome size estimation improves comparative metagenomics and sheds light on the functional ecology of the human microbiome. Genome Biol. 16, 51. 10.1186/s13059-015-0611-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Anders S, Pyl PT, and Huber W (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Katoh K, and Standley DM (2013). MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 30, 772–780. 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Capella-Gutiérrez S, Silla-Martínez JM, and Gabaldón T (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, and Lanfear R (2020). IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 37, 1530–1534. 10.1093/molbev/msaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grobbee DE, Rimm EB, Giovannucci E, Colditz G, Stampfer M, and Willett W (1990). Coffee, Caffeine, and Cardiovascular Disease in Men. N. Engl. J. Med. 323, 1026–1032. 10.1056/NEJM199010113231504. [DOI] [PubMed] [Google Scholar]
- 73.WILLETT WC, SAMPSON L, STAMPFER MJ, ROSNER B, BAIN C, WITSCHI J, HENNEKENS CH, and SPEIZER FE (1985). REPRODUCIBILITY AND VALIDITY OF A SEMIQUANTITATIVE FOOD FREQUENCY QUESTIONNAIRE. Am. J. Epidemiol. 122, 51–65. 10.1093/oxfordjournals.aje.a114086. [DOI] [PubMed] [Google Scholar]
- 74.Feskanich D, Rimm EB, Giovannucci EL, Colditz GA, Stampfer MJ, Litin LB, and Willett WC (1993). Reproducibility and validity of food intake measurements from a semiquantitative food frequency questionnaire. J. Am. Diet. Assoc. 93, 790–796. 10.1016/0002-8223(93)91754-E. [DOI] [PubMed] [Google Scholar]
- 75.Rimm EB, Giovannucci EL, Stampfer MJ, Colditz GA, Litin LB, and Willett WC (1992). Reproducibility and Validity of an Expanded Self-Administered Semiquantitative Food Frequency Questionnaire among Male Health Professionals. Am. J. Epidemiol. 135, 1114–1126. 10.1093/oxfordjournals.aje.a116211. [DOI] [PubMed] [Google Scholar]
- 76.Weng L, Bockstaele DRV, Wauters J, Marck EV, Plum J, Berneman ZN, and Merregaert J (2003). A Novel Alternative Spliced Chondrolectin Isoform Lacking the Transmembrane Domain Is Expressed during T Cell Maturation *. J. Biol. Chem. 278, 19164–19170. 10.1074/jbc.M300653200. [DOI] [PubMed] [Google Scholar]
- 77.Mallick H, Rahnavard A, McIver LJ, Ma S, Zhang Y, Nguyen LH, Tickle TL, Weingart G, Ren B, Schwager EH, et al. (2021). Multivariable association discovery in population-scale metaomics studies. PLoS Comput. Biol. 17, e1009442. 10.1371/journal.pcbi.1009442. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2: MGX-MTX quantification results for SSN, related to Figure 2
Table S3: Oligonucleotides used in this study, related to STAR Methods
Table S4: Proteomics IDs from activity-guided protein purification, related to Figure 3
Data Availability Statement
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
All original codes have been deposited at Zenodo and is publicly available. DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Gordonibacter pamelaeae 3C | Prof. Peter Turnbaugh | N/A |
| Gordonibacter sp. 28C | Prof. Peter Turnbaugh | N/A |
| Paraeggerthella hongkongensis RC2/2A | Prof. Peter Turnbaugh | N/A |
| Eggerthella lenta A2 | Prof. Peter Turnbaugh | N/A |
| Eggerthella Sinensis DSM 16107 | Prof. Peter Turnbaugh | N/A |
| Adlercreutzia equolifaciens DSM 19450 | DSMZ | N/A |
| Adlercreutzia equolifaciens subsp. celatus DSM 18785 | DSMZ | N/A |
| Slackia heliotrinireducens DSM 20476 | DSMZ | N/A |
| Senegalimassilia anaerobia DSM 25959 | DSMZ | N/A |
| Gordonibacter urolithinfaciens DSM 27213 | DSMZ | N/A |
| Gordonibacter pamelaeae 7-10-1b | DSMZ | N/A |
| Raoultibacter massiliensis DSM 103407 | DSMZ | N/A |
| Gordonibacter urolithinfaciens DSM 27213 hcdh K/O | This paper | N/A |
| Gordonibacter sp. 28C | Prof. Peter Turnbaugh | N/A |
| Paraeggerthella hongkongensis RC2/2A | Prof. Peter Turnbaugh | N/A |
| Eggerthella lenta A2 | Prof. Peter Turnbaugh | N/A |
| Eggerthella Sinensis DSM 16107 | Prof. Peter Turnbaugh | N/A |
| Adlercreutzia equolifaciens DSM 19450 | DSMZ | N/A |
| Adlercreutzia equolifaciens subsp. celatus DSM 18785 | DSMZ | N/A |
| Slackia heliotrinireducens DSM 20476 | DSMZ | N/A |
| Senegalimassilia anaerobia DSM 25959 | DSMZ | N/A |
| Gordonibacter urolithinfaciens DSM 27213 | DSMZ | N/A |
| Gordonibacter pamelaeae 7-10-1b | DSMZ | N/A |
| Raoultibacter massiliensis DSM 103407 | DSMZ | N/A |
| Gordonibacter urolithinfaciens DSM 27213 hcdh K/O | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| 3,4-dihydroxyhydrocinnamic acid (hydrocaffeic acid; HCA) | Sigma-Aldrich | Cat#102601-10G |
| 3-(3-hydroxyphenyl)propionic acid (mHPPA) | Toronto Research Chemicals | Cat#H940090-1g |
| Caffeic acid | Millipore Sigma | Cat#C0625-2G |
| 3,4-dihydroxyphenylacetic acid (DOPAC) | Millipore Sigma | Cat#850217-1G |
| Dopamine | Sigma-Aldrich | Cat#PHR1090-1G |
| Chlorogenic acid | Ambeed | Cat#A163338 |
| trans-ferulic acid | Sigma-Aldrich | Cat#128708-5G |
| trans-isoferulic acid | Sigma-Aldrich | Cat#PHL89717-100MG |
| DL-3,4-dihydroxymandelic acid | Sigma-Aldrich | Cat#151610-500MG |
| Rosmarinic acid | Astatech | Cat#44324 |
| L-arginine monohydrochloride | Sigma-Aldrich | Cat#A5131-500G |
| L-cysteine hydrochloride monohydrate | Sigma-Aldrich | Cat#C7880-100G |
| Sodium formate | Sigma-Aldrich | Cat#247596-100G |
| Methyl viologen | Sigma-Aldrich | Cat#856177-1g |
| Sodium dithionite | Sigma-Aldrich | Cat#157953-5G |
| Ammonium sulfate | Sigma-Aldrich | Cat#157953-5G |
| Sodium molybdate | Sigma-Aldrich | Cat#243655-100G |
| Sodium nitrite | Sigma-Aldrich | Cat#237213-100G |
| SIGMAFAST protease inhibitor tablets | Sigma-Aldrich | Cat#S8830 |
| DNase | Sigma-Aldrich | Cat#DN25-1G |
| Lysozyme | Sigma-Aldrich | Cat#L6876-5G |
| Brain–Heart Infusion (BHI) | Becton Dickinson | Cat#211060 |
| TRIzol | Invitrogen | Cat#15596026 |
| 3-(3-hydroxyphenyl)propionic acid (mHPPA) | Toronto Research Chemicals | Cat#H940090-1g |
| Caffeic acid | Millipore Sigma | Cat#C0625-2G |
| 3,4-dihydroxyphenylacetic acid (DOPAC) | Millipore Sigma | Cat#850217-1G |
| Dopamine | Sigma-Aldrich | Cat#PHR1090-1G |
| Chlorogenic acid | Ambeed | Cat#A163338 |
| trans-ferulic acid | Sigma-Aldrich | Cat#128708-5G |
| trans-isoferulic acid | Sigma-Aldrich | Cat#PHL89717-100MG |
| DL-3,4-dihydroxymandelic acid | Sigma-Aldrich | Cat#151610-500MG |
| Rosmarinic acid | Astatech | Cat#44324 |
| L-arginine monohydrochloride | Sigma-Aldrich | Cat#A5131-500G |
| L-cysteine hydrochloride monohydrate | Sigma-Aldrich | Cat#C7880-100G |
| Sodium formate | Sigma-Aldrich | Cat#247596-100G |
| Methyl viologen | Sigma-Aldrich | Cat#856177-1g |
| Sodium dithionite | Sigma-Aldrich | Cat#157953-5G |
| Ammonium sulfate | Sigma-Aldrich | Cat#157953-5G |
| Sodium molybdate | Sigma-Aldrich | Cat#243655-100G |
| Sodium nitrite | Sigma-Aldrich | Cat#237213-100G |
| SIGMAFAST protease inhibitor tablets | Sigma-Aldrich | Cat#S8830 |
| DNase | Sigma-Aldrich | Cat#DN25-1G |
| Lysozyme | Sigma-Aldrich | Cat#L6876-5G |
| Brain–Heart Infusion (BHI) | Becton Dickinson | Cat#211060 |
| TRIzol | Invitrogen | Cat#15596026 |
| Critical commercial assays | ||
| Direct-Zol RNA MiniPrep Plus Kit | Zymo Research | Cat#R2070 |
| RQ1 RNase-free DNase kit | Promega | Cat#M6101 |
| Luna® Universal One-Step RT-qPCR Kit | New England Biolabs | Cat#E3005S |
| BacTiter-Glo | Promega | Cat#8230 |
| RQ1 RNase-free DNase kit | Promega | Cat#M6101 |
| Luna® Universal One-Step RT-qPCR Kit | New England Biolabs | Cat#E3005S |
| BacTiter-Glo | Promega | Cat#8230 |
| Deposited data | ||
| Gordonibacter urolithinfaciens DSM 27213 hcdh K/O genome | This work | BioProject PRJNA1150218 |
| Metagenome-assembled genomes from human-associated microbiome | Pasolli et al. | N/A |
| Gordonibacter pamelaeae 3C RNA-seq raw data | Bess et al. | BioProject PRJNA450120 |
| Men’s Lifestyle Validation Study (MLVS) | Mehta et al. | BioProject PRJNA354235 |
| Human Microbiome Project 2 (HMP2) | Lloyd-Price et al. | BioProject PRJNA398089 |
| Recombinant DNA | ||
| pXD71Cas10RFP | Addgene | Plasmid #192273 |
| pXD68Kan2 | Addgene | Plasmid #191248 |
| pXD80GuCas3.1 | This work | N/A |
| Software and algorithms | ||
| GraphPad Prism 9 and 10 | GraphPad Software | N/A |
| tblastn | Altschul et al. | N/A |
| PhyloPhlAn 3.0 | Asnicar et al. | https://github.com/biobakery/phylophlan |
| iTOL v6.5.8 | Letunic and Bork | https://github.com/iBiology/iTOL |
| Enzyme Function Initiative’s Enzyme Similarity Tool (EFI-EST) | Zallot et al. | https://efi.igb.illinois.edu/efi-est/ |
| Cytoscape v3.9.1 | Shannon et al. | https://github.com/cytoscape |
| KneadData v0.10.0 | N/A | https://github.com/biobakery/kneaddata |
| ShortBRED | Kaminski et al. | https://github.com/biobakery/shortbred |
| DIAMOND | Buchfink et al. | https://github.com/bbuchfink/diamond |
| MicrobeCensus | Nayfach and Pollard | https://github.com/snayfach/MicrobeCensus |
| Bowtie2 v2.4.2 | Langmead and Salzberg | https://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| HTseq-count v0.13.5 | Anders et al. | https://htseq.readthedocs.io/en/release_0.11.1/index.html |
| Salmon v1.9.0 | Patro et al. | https://combine-lab.github.io/salmon/ |
| MaAsLin2 1.0.0 | Mallick et al. | https://github.com/biobakery/Maaslin2 |
| MAFFT-linsi v7.505 | Katoh and Standley | https://mafft.cbrc.jp/alignment/software/ |
| trimAl v1.4.1 | Capella-Gutiérrez et al. | https://vicfero.github.io/trimal/ |
| iqtree2 v2.1.3 | Minh et al. | https://github.com/iqtree/iqtree2 |
| Diet and MTX analysis pipeline | This paper | https://zenodo.org/records/13390717 |