

Abstract
Background
Reactive oxygen species are known to contribute to platelet hyperactivation and thrombosis during aging; however, the mechanistic contribution of the specific oxidative pathway remains elusive.
Objectives
We hypothesized that during aging, endogenous Nox2-NADPH oxidase contributes to platelet reactive oxygen species accumulation and that loss of Nox2 will protect from platelet activation and thrombosis.
Methods
We studied littermates of Nox2 knockout (Nox2-KO) and -wild-type (Nox2-WT) mice at young (3-4 months) and old (18-20 months) age. Within platelets, we examined the expression of subunits of NADPH oxidase and enzyme activity, oxidant levels, activation markers, aggregation, and secretion. We also assessed susceptibility to in vivo thrombosis in 2 experimental models.
Results
While aged Nox2-WT mice displayed increased mRNA levels for Nox2, aged Nox2-KO mice showed an increase in Nox4 mRNA. However, neither the protein levels of several subunits nor the activity of NADPH oxidase were found to be altered by age or genotype. Both aged Nox2-WT and aged Nox2-KO mice exhibited similar enhancement in levels of platelet oxidants, granule release, αIIbβ3 activation, annexin V binding, aggregation and secretion, and a greater susceptibility to platelet-induced pulmonary thrombosis compared with young mice. In a photochemical injury model, adoptive transfer of platelets from aged Nox2-WT or Nox2-KO mice to the aged host mice resulted in a similar time to develop occlusive thrombus in the carotid artery. These findings suggest that loss of endogenous Nox2 does not protect against age-related platelet activation and arterial thrombosis in mice.
Conclusion
We conclude that Nox2 is not an essential mediator of prothrombotic effects associated with aging.
Keywords: aging, NADPH oxidase, platelets, reactive oxygen species, thrombosis
Graphical abstract
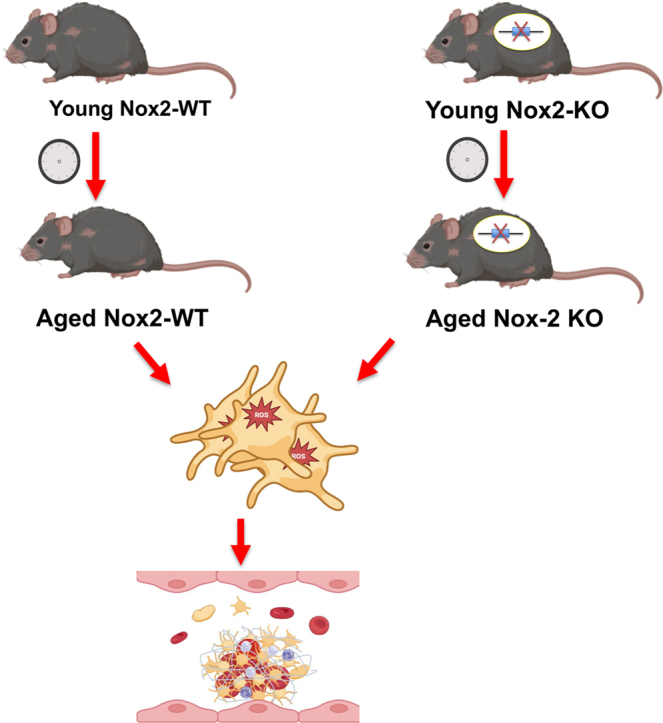
Essentials
-
•
Age-induced thrombosis is known to be mediated via ROS.
-
•
We tested if Nox2 contributes to ROS-mediated prothrombotic phenotype during aging.
-
•
Loss of Nox2 does not inhibit platelet ROS, activation, or thrombosis in aged mice.
-
•
Targeting of mediators other than Nox2 should be considered to limit age-induced thrombosis.
1. Introduction
Aging is an established risk factor for thromboembolic events such as myocardial infarction and stroke [1], but the underlying mechanisms are poorly defined. Recent studies have reported hyperactivation of platelets as an important mechanism of aging-related thrombosis [[2], [3], [4], [5]]. However, the molecular pathways that regulate platelet activation and thrombosis during aging are incompletely understood.
Since reactive oxygen species (ROS) are important mediators of physiological platelet activation [[6], [7], [8], [9], [10], [11]], age-induced elevation in ROS or oxidants within platelets could potentially contribute to platelet hyperactivity. Consistent with this idea, we and others [2,5,12] have previously shown a protective role for antioxidant enzymes such as glutathione peroxidase and superoxide dismutase (SOD) in this phenomenon in aged mice. These reports suggest that during aging, activation of an oxidative pathway could likely contribute to increased platelet ROS, thus causing platelet hyperactivity and thrombosis.
Platelets contain diverse oxidative enzymes, including NADPH oxidase [8,13], cyclooxygenase [14], and xanthine oxidase [15]. However, the mechanistic contributions of these enzymes in platelet activation during aging remain to be determined. Prior studies in aged animals have shown a pathological role of the Nox2 subunit of NADPH oxidase in endothelial dysfunction [16], neovascularization [17], and cerebral vasculature [18]. There is also growing enthusiasm to target Nox2 to control platelet hyperactivation and thrombosis, including aging [13,[19], [20], [21], [22]]. In fact, we previously reported that in aged mice, exogenous treatment of platelets with the NADPH oxidase inhibitor apocynin decreased platelet integrin activation [2], building rationale to consider Nox2-NADPH oxidase as a therapeutic target in age-associated thrombosis. A potential use of NADPH oxidase inhibitors in limiting platelet activation in aged high-risk cardiac patients with aspirin-resistant platelets has also been suggested [22]. However, the inclusion of such a pharmacological approach in clinical practice requires substantial supportive in vivo data in preclinical models. Therefore, the objective of our study was to examine whether genetic loss of Nox2-NADPH oxidase will prevent age-related increased generation of platelet-derived ROS, platelet hyperactivity, and thrombosis. Using mice genetically deficient in Nox2 (Cybb-/y), we report that the loss of Nox2-NADPH oxidase does not protect against aging-induced increases in platelet oxidants, platelet activation, aggregation, secretion, or thrombosis in vivo.
2. Methods
2.1. Mice
All animal studies were approved by the institutional animal care and use committees at the University of Iowa. Male Cybb-/y mice on the C57BL6/J background were obtained from the Jackson Laboratory and bred as described previously by us [23] in the animal facility of the University of Iowa. We generated male littermates of hemizygous wild-type (WT; Cybb+/y or Nox2-WT) and knockout (KO; Cybb-/y or Nox2-KO), and the homozygous littermates of female Cybb-/- (Nox2-KO) and Cybb+/+ (Nox2-WT) for study. Genotyping for the WT and targeted Cybb alleles was performed by polymerase chain reaction. We also studied transgenic mice expressing a chimera of human interleukin 4 receptor (hIL4Rα) and mutated murine GP1bα in a platelet-specific manner (hIL4Rα/GP1bα Tg mouse, which is referred to as hIL4Rα transgenic [hIL4Rα Tg] mouse), and these chimeric mice were generated on a C57BL6/J background [24]. All experiments were conducted in mice between the ages of 3 and 4 months (young cohort) or between 18 and 20 months (aged cohort). Both male and female mice were included in the study design. Mice were studied at 3 to 4 months (young cohort) or 18 to 20 months of age (aged cohort). In vivo studies were performed according to the current Animal Research: Reporting of In Vivo Experiment guidelines (https://arriveguidelines.org/).
2.2. Complete blood counts and platelet preparation
For complete blood count (CBC), mice were anesthetized with isoflurane and bled via the retro-orbital plexus using EDTA-coated tubes, and samples were analyzed using ADVIA 120 (Siemens).
To isolate platelets for flow cytometry and aggregation studies, mice were anesthetized with isoflurane and bled via the retro-orbital plexus [25,26] using a heparin-coated capillary tube, and blood was collected into 3.2% sodium citrate (9:1). For mRNA expression, blood was drawn via carotid artery cannulation into sodium citrate. Platelets were prepared as described previously [5,23]. Briefly, blood was diluted with modified Tyrode’s buffer, pH 7.35, centrifuged at 100 × g for 10 minutes at RT, and platelet-rich plasma was isolated and processed to prepare washed platelets where PGE1 (1 μM) was added to platelet-rich plasma, and samples were centrifuged at 800 × g for 7 minutes to obtain platelet pellets. Pellets were then washed with modified Tyrode’s buffer in the presence of PGE1 (1 μM) and finally resuspended in modified Tyrode’s buffer. For mRNA, platelets were further purified with CD45 and Ter-119 labeled microbeads (Miltenyi Biotec) as described by us [5,23].
2.3. Flow cytometry and aggregometry
Levels of platelet-derived prooxidants were measured using fluorescent dye methods [2,5,23]. Washed platelets (2 × 108/mL) were incubated with 10 μM of the oxidation-sensitive dye, 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA, Molecular Probes), for 10 minutes at 37 °C in the dark. Platelets were then activated with thrombin for 10 minutes, diluted with 1X DPBS, and the fluorescent signal of the oxidized adducts (DCF) was measured by flow cytometry. In some experiments, platelets were incubated with 500 U/mL pegylated (PEG)-catalase prior to incubation with CM-H2DCFDA to measure the peroxide-dependent increase in DCF fluorescence. To measure cellular superoxide levels, platelets were incubated with 5 μM of dihydroethidium (DHE, Molecular probe) for 10 minutes at 37 °C in the dark, then activated with thrombin + convulxin for 10 minutes; samples were diluted, and the DCF was measured by flow cytometry. To probe for mitochondria-derived oxidants, platelets were incubated with 5 μM of MitoSox (Invitrogen) for 10 minutes, activated with thrombin + convulxin for an additional 10 minutes, diluted, and the fluorescent signal was measured by flow cytometry.
Activation of integrin αIIbβ3 and α-granule secretion was evaluated by quantifying JON/A binding and surface expression of P-selectin, respectively. Washed platelets (2 × 108/mL) incubated with 10 μL of PE-conjugated JON/A (1:2, BD Biosciences) or FITC-conjugated rat anti-mouse CD62P (1:5, BD Biosciences) antibody were activated with human thrombin and fixed in 1% paraformaldehyde. Surface phosphatidylserine (PS) exposure was measured by incubating platelets for 15 minutes with annexin V-APC (1:1, Biolegend), followed by activation with thrombin + convulxin for 10 minutes, and a binding buffer (100 μL) was added at the end. All the samples were diluted 10-fold in PBS and analyzed using an LSR Violet flow cytometer (Becton Dickinson).
Platelet aggregation and release of adenosine triphosphate (ATP) from dense granules were measured simultaneously using Chrono-lume Luciferin-Luciferase Reagent (Chrono-log, stock concentration, 0.2 μM luciferase/luciferin) in an Optical/Lumi-Aggregometer (model 700-2, Chrono-Log) as described previously [5]. Briefly, washed platelets (2 × 108/mL) containing 1 mM calcium chloride were incubated with 20 μL of Chrono-lume reagents for 2 minutes at 37 °C. After incubation, thrombin (0.02 U/mL) was added under stirring conditions (1200 rpm) to induce platelet aggregation. Dense granule secretion (ATP release) was monitored in parallel to aggregation. Aggregation and secretion kinetics were calculated as percent (%) increase in light transmittance and luminescence, respectively.
2.4. mRNA, Western blotting, and NADPH oxidase activity
Bead-purified platelets were prepared for these experiments. The mRNA levels for subunits of NADPH oxidase were measured via quantitative polymerase chain reaction in bead-purified platelets, as described previously [23]. The subunit-specific primers were purchased from Thermo Fisher Scientific for Nox1 (Mm00549170_m1), Nox2 (custom designed, AP9HUHD), Nox4 (Mm00627696_m1), p47Phox (Mm00447921_m1), p67Phox (Mm00726636_s1), p22Phox (Mm00514478_m1), and β actin RNA (Mm02619580_g1).
For protein expression, a bicinchoninic acid protein assay was performed to quantify total protein in platelet lysates. Samples containing 20 μg of protein were run on a 12% sodium dodecyl sulfate gel and transferred to a PVDF membrane; membranes were blocked using 5% filtered BSA and then stained overnight at 4 °C with primary antibodies for Nox1 (Novus Biosciences, NBP1-31546), Nox4 (Novus Biosciences, NB110–58851), p47phox (Santa Cruz, sc-17844), p67phox (Santa Cruz, sc-374510), or β-actin (Santacruz, Sc-47778) and blots were developed using standard procedure [5].
NADPH oxidase activity assay was performed following an established method [27] with minor modifications. Briefly, NADPH oxidase-induced superoxide generation was measured indirectly via SOD-inhibitable cytochrome C reduction using NADPH as substrate. Bead-purified platelets were lysed in RIPA buffer on ice for 30 minutes, followed by centrifugation for 20 minutes, and protein was quantified via bicinchoninic acid assay. Platelet protein lysate (25 μg) was mixed in a reaction mixture of 250 μL containing 78 μmol/L cytochrome C and 100 μmol/L NADPH, with or without 1000 U/mL SOD. Samples were then incubated in the dark at 37 °C for an hour, and the absorbance at 550 nm was measured. There was no measurable activity in the absence of NADPH or cytochrome C. Change in absorbance in the presence and absence of SOD was calculated and expressed as the amount of superoxide generated.
2.5. Experimental thrombosis
We used 2 experimental models to assess susceptibility to thrombosis in vivo. To assess the susceptibility to pulmonary thrombosis, mice were infused with 0.5 μg/g collagen (Chronolog) retro-orbitally, which resulted in platelet activation-dependent pulmonary thrombosis leading to death [5]. After the infusion of collagen, mice were monitored for up to 10 minutes for symptoms of thrombotic shock, including bradycardia, irregular breathing, and death (defined as the absence of a heartbeat for greater than 60 seconds). If death did not occur within 10 minutes, the experiment was terminated at that time. Carotid artery thrombosis was induced by photochemical injury, as described previously [23,28]. Briefly, the right common carotid artery was transilluminated with a 1.5 mV, 540 nm green laser (Melles Griot). Rose bengal (25 mg/kg) was injected via a femoral vein catheter. Carotid artery blood flow was monitored with a Doppler flow probe for up to 90 minutes after injury or until a stable occlusion developed. Stable occlusion was defined as the time at which blood flow remained absent for ≥10 minutes.
2.6. Adoptive platelet transfer
Platelet transfer was performed as described previously [5]. Blood was collected through carotid cannulation and washed platelets were prepared. The host hIL4Rα Tg mice were injected with 0.5 mg/kg of human CD124 antibody (BD) retro-orbitally to deplete endogenous platelets. A total of 20 μL of blood was obtained pre- and 30 minutes post antibody injection for CBC to quantify circulating platelets. After 2 hours, mice were infused with donor platelets (2.5 × 106/g body weight), and 15 minutes later, a CBC was repeated to ensure repletion of circulating platelets. This was followed by carotid artery thrombosis, as described above.
2.7. Statistical analysis
GraphPad Prism software, version 9.4.1, was used for statistical analysis. Normality testing was performed using the Shapiro–Wilk test. Statistical significance for the data set that showed normal distribution was assessed by using either an unpaired t-test for 2 group comparison or a 2-way analysis of variance (anova) followed by Tukey's multiple comparisons test for multiple group analysis. Data sets not showing normal distribution were analyzed by the Kruskal–Wallis test followed by Dunn’s multiple comparisons. Statistical significance was defined as P < .05. Values are reported as mean ± SE or median with IQR. The survival curve was analyzed using the log-rank (Mantel–Cox) test.
3. Results
3.1. Complete blood count
No alterations in hemoglobin, hematocrit, white blood cell count, or platelet counts were observed between young Nox2-WT and Nox2-KO mice (Supplementary Table). Further, aging did not alter levels of hemoglobin, hematocrit, or white blood cell count. However, aged mice of both genotypes exhibited significantly increased platelet count compared with young mice, which is consistent with our previous reports [2,5].
3.2. mRNA for subunits of NADPH oxidase is altered with age, but protein expression and NADPH oxidase activity do not change
Levels of mRNA for subunits of NADPH oxidase were quantified in bead-purified platelets. First, we did not detect any mRNA for Nox2 in young or aged Nox2-KO mice, confirming the loss of Nox2. The mRNA for Nox2 was found to be significantly increased in aged Nox2-WT mice (P < .05 vs young Nox2-WT; Figure 1A). Aged Nox2-KO but not Nox2-WT mice showed an increase in Nox4 expression (P < .05 vs young Nox2-KO; Figure 1B). Nox1 mRNA showed a modest but nonsignificant decrease with age in mice of both genotypes (Figure 1C). Aged Nox2-WT mice also showed a significant decrease in the regulatory subunits such as p47phox and p67phox (P < .05 vs young Nox2-WT; Figure 1D, E). Finally, we observed a modest but nonsignificant decrease with age in the p22phox, a membranous subunit that associates with NOX subunits for its activity (Figure 1F). However, examination of protein levels for various subunits of NADPH oxidase, including Nox1, Nox4, p47phox, and p67phox, did not show any alterations due to age or genotype (Figure 2A–D). Further, the NADPH oxidase activity within platelets remained similar across the groups (Figure 2E).
Figure 1.

mRNA for Nox2 is increased in platelets from aged mice. Levels of mRNA were measured in bead-purified platelets using quantitative polymerase chain reaction for catalytic subunits (A) Nox2, (B) Nox4, and (C) Nox1, and regulatory subunits (D) p47Phox and (E) p67Phox of NADPH oxidase and for Nox2-associated subunit (F) p22Phox. Relative expression to young Nox2 wild-type (Nox2-WT) is presented. Data are presented as median with IQR and analyzed using the Kruskal–Wallis test with Dunn’s multiple comparisons. N = 6 per group. Nox2-KO, Nox2 knockout.
Figure 2.

Protein levels of NADPH oxidase subunits or enzymatic activity in platelets are not altered by Nox2 genotype or age. Protein levels were measured in bead-purified platelets by Western blotting for catalytic subunits (A) Nox1 and (B) Nox4 and regulatory subunits (C) p47Phox and (D) p67Phox of NADPH oxidase. The activity of NADPH oxidase was measured in platelet lysate in the presence of cytochrome C and NADPH, and changes in absorbance in the presence or absence of superoxide dismutase were calculated and expressed as the amount of superoxide generated. Data are presented as median with IQR for A and C and as mean ± SE for B, D, and E. Data were analyzed using the Kruskal–Wallis test with Dunn’s multiple comparisons. N = 4 to 8 per group. AKO; aged Nox2 knockout; AW; aged Nox2 wild-type; Nox2-KO, Nox2 knockout; Nox2-WT, Nox2 wild-type; YKO; young Nox2 knockout; YW, young Nox2 wild-type.
3.3. Nox2 is not a mediator of aging-induced increase in platelet oxidants
Next, we tested how the changes in the expression of NADPH oxidase subunits alter levels of platelet oxidants. First, using the redox-sensitive dye CM-H2DCFDA as a fluorescent probe for total cellular oxidants, we measured DCF fluorescence in the absence and presence of thrombin. At baseline (or resting state) without thrombin activation, we did not detect any differences in DCF fluorescence between young Nox2-WT and young Nox2-KO mice (Figure 3A and Supplementary Figure S1A); however, both aged groups showed significant but similar increases in DCF fluorescence (P < .0001 vs young mice of same genotype). Upon stimulation with 0.02 U/mL and 0.05 U/mL thrombin, there was a dose-dependent increase in fluorescence that was more pronounced in aged groups (Figure 3B, C and Supplementary Figure S1B, C). While we did not detect any differences in DCF fluorescence between young Nox2-WT and Nox2-KO mice at a given dose of thrombin, both aged groups of mice showed significant increases in DCF fluorescence from young mice (P < .0001 for both groups at each dose of thrombin). This age-dependent increase in DCF fluorescence was similar between Nox2-WT and Nox2-KO mice. Further, preincubation of platelets with PEG-catalase lowered the signal across the group (Figure 3D and Supplementary Figure S1D), but the PEG-catalase quenched signal remained high in both the aged groups compared with young cohorts (P < .0001 for both aged groups), suggesting that much of the increase in DCF signal with age is due to peroxide.
Figure 3.

Age-induced increase in cellular oxidants in mice is not prevented by Nox2 deficiency. Washed platelets were prepared from young or aged mice deficient in Nox2 (Nox2-KO) or wild-type littermates (Nox2-WT). Levels of intracellular reactive oxygen species detected by oxidation of CM-H2DCF (DCF fluorescence) were measured in platelets at (A) baseline (resting platelets) and upon stimulation with (B) 0.02 U/mL, (C) 0.05 U/mL thrombin, or (D) 0.05 U/mL thrombin ± pegylated (PEG)-catalase. All the samples were analyzed by flow cytometry, and mean fluorescence intensity (MFI) was measured. Data for A–C are presented as MFI (N = 8 to 9 per group). Data for D are presented as fold change from average young Nox2-WT, where MFI obtained with PEG-catalase treatment was subtracted to show a peroxide-dependent increase in DCF fluorescence (N = 5 per group). Data are presented as mean ± SE and analyzed using 2-way anova with Tukey’s analysis for multiple comparisons.
We next used a relatively specific dye, DHE, to measure total cellular superoxide levels and MitoSox to measure mitochondria-derived superoxide. Our data demonstrate an increase in DHE fluorescence in resting platelets in aged mice relative to young mice (P = .004 for Nox2-WT and P = .002 for Nox2-KO), as well as upon activation with thrombin (P = .02 for Nox2-WT and P = .02 for Nox2-KO; Figure 4A, B and Supplementary Figure S2A, B). MitoSox fluorescence did not show an age-dependent increase in resting platelets, but upon dual stimulation with thrombin + convulxin, both aged Nox2-WT (P = .002) and aged Nox2-KO (P < .0001) displayed increased MitoSox signal relative to the young cohort (Figure 4C, D and Supplementary Figure S2C, D). Together, these data suggest that aging induces the accumulation of platelet oxidants independent of Nox2.
Figure 4.

Age-induced increase in cellular and mitochondrial superoxide in mice is not prevented by Nox2 deficiency. Washed platelets were prepared from young or aged mice deficient in Nox2 (Nox2-KO) or wild-type littermates (Nox2-WT). Levels of intracellular superoxide were detected by oxidation of dihydroethidium (DHE) in platelets at (A) baseline (resting platelets) or upon stimulation with (B) 0.05 U/mL thrombin + 50 ng/mL convulxin. The levels of superoxide in platelet mitochondria were detected by MitoSox staining at (C) baseline or upon stimulation with (D) 0.05 U/mL thrombin + 50 ng/mL convulxin. All the samples were analyzed by flow cytometry and mean fluorescence intensity (MFI) was measured. N = 8 to 9 per group. Data are presented as mean ± SE and analyzed using 2-way anova with Tukey’s analysis for multiple comparisons.
3.4. Aged mice exhibit increased platelet granule release, integrin activation, and PS exposure independent of Nox2
To determine if genetic deletion of Nox2-NADPH oxidase protects from activation of platelets during aging, we examined the expression of granule release, integrin activation, and procoagulant platelet formation in the absence or presence of agonists. At baseline without agonist stimulation, we did not observe any changes due to age or genotype in these platelet activation markers (data only shown as a histogram; Supplementary Figures S3A, S4A, and S5A). By using 2 doses of thrombin (0.02 and 0.05 U/mL), we observed a dose-dependent increase in mean fluorescence for P-selectin (a marker of granule release) within each group. Further, aged Nox2-WT mice showed increased mean fluorescence at both doses compared with young Nox2-WT mice (P < .05 and P < .0001 for low and high doses, respectively; Figure 5A, B and Supplementary Figure S3B, C). Aged Nox2-KO mice also showed an increase in mean fluorescence at both doses of thrombin compared with young Nox2-KO mice (P < .05 and P < .0001, respectively), and these increases were similar to that seen in aged Nox2-WT mice. Activation of αIIbβ3 integrin was also increased to similar extents in both aged groups compared with respective young cohorts when activated with either a low or high dose of thrombin (P < .05 vs young Nox2-WT and P < .01 vs young Nox2-KO; Figure 5C, D and Supplementary Figure S4B, C). Finally, to assess procoagulant platelet formation, we measured PS exposure on the platelet surface via annexin V binding upon activation with a combination of low or high doses of thrombin and convulxin. Aged mice of both genotypes showed significant enhancement in annexin V binding (P < .05 for both doses in aged vs young WT-Nox2 and P < .05 with high dose only in aged vs young Nox2-KO; Figure 5E, F and Supplementary Figure S5B, C).
Figure 5.

Increased platelet activation in aged mice is not mediated through Nox2-NADPH oxidase. Washed platelets were prepared from young or aged mice deficient in Nox2 (Nox2-KO) or wild-type (Nox2-WT) littermates, suspended in Tyrode’s buffer with fluorescent antibody/dye, stimulated with agonists, fixed, and respective fluorescence was measured by flow cytometry. Granule release (P-selectin) and activation of αIIbβ3 were measured upon stimulation with (A and C) 0.02 U/mL thrombin or (B and D) 0.05 U/mL thrombin, respectively. Annexin V binding was quantified upon simultaneous activation with (E) 0.05 U/mL thrombin + 50 ng/mL convulxin or (F) 0.1 U/mL thrombin + 100 ng/mL convulxin. Data are presented as mean ± SE and analyzed with 2-way anova with Tukey’s test for multiple comparisons. N = 7 to 9 per group. MFI, mean fluorescence intensity.
3.5. Aging-induced enhancement of platelet aggregation and ATP release is independent of Nox2
Next, we examined platelet aggregation and secretion simultaneously in response to thrombin (Figure 6A). Upon thrombin activation in young mice, platelet aggregation and dense granule secretion were not found to be altered due to Nox2 deficiency (Figure 6B, C). A significant increase in aggregation was observed in aged Nox2-WT and aged Nox2-KO mice up to 4 minutes compared with young cohorts (P < .0001 vs young Nox2-WT, and P < .01 or P < .0001 vs young Nox2-KO, respectively). This was followed by deaggregation; however, the extent of aggregation remained higher in both aged groups by 6 minutes compared with young cohorts (P < .0001 for both genotypes). The release of ATP from thrombin-activated platelets also increased up to 2 minutes and declined thereafter in all groups, but the level of release remained higher in aged Nox2-WT and aged Nox2-KO mice throughout the duration of the assay (P < .01 or P < .0001 vs young Nox2-WT, and P < .05 or P < .001 vs young Nox2-KO, respectively). There were no differences observed in the magnitude of aggregation or secretion between aged Nox2-WT and aged Nox2-KO mice at any time during the assay.
Figure 6.

Platelets from aged Nox2 wild-type (Nox2-WT) and aged Nox2 knockout (Nox2-KO) mice show similar increases in aggregation and secretion. Washed platelets were prepared from young or aged Nox2-KO and Nox2-WT littermates. (A) Representative tracings for simultaneous aggregation and secretion curves from 4 mice representing each group, where the aggregation curve goes down with an increase in light transmission and the secretion curve goes up with an increase in luminescence. Aggregation curves: black for young Nox2-WT, dark green for young Nox2-KO, dark blue for aged Nox2-WT, and dark red for aged Nox2-KO. Secretion curves: light gray for young Nox2-WT, fluorescent green for young Nox2-KO, light blue for aged Nox2-WT, and orange for aged Nox2-KO mice. (B and C) Quantification of percent aggregation and secretion, respectively, at different time points. Data for B and C are presented as mean ± SE and analyzed using 2-way anova with Tukey’s test for multiple comparisons. N = 7 to 11 per group.
3.6. Nox2 does not mediate platelet-dependent arterial thrombosis in aged mice
Lastly, we examined whether Nox2 deficiency in aged mice is sufficient to protect from susceptibility to in vivo thrombosis. In a model of platelet-dependent pulmonary thrombosis induced by collagen infusion, we observed significantly shortened time to death in aged Nox2-WT and aged Nox2-KO mice compared with young Nox2-WT and young Nox2-KO mice, respectively (P < .05 for both; Figure 7A). The time to death in aged Nox2-WT and aged Nox2-KO mice were not different. Next, in a complementary photochemical injury model, we set out to compare the ability of aged Nox2-WT and Nox2-KO mice to induce occlusive thrombus in vivo in the carotid artery. Unfortunately, aged Nox2-KO mice did not survive the ventilation procedure in this assay. So, we used an alternative strategy (as previously described by us) [5] where we compared the ability of isolated platelets from aged Nox2-WT or aged Nox2-KO mice to develop occlusive thrombi in vivo following adoptive transfer into the aged host hIL4Rα Tg mice. We observed that platelets from either aged Nox2-WT or aged Nox2-KO mice took a similar time to develop either first or stable occlusive thrombus in the carotid artery after photochemical injury in host hIL4Rα Tg mice (Figure 7B).
Figure 7.

Deficiency of Nox2 does not protect from age-induced increased susceptibility to thrombosis. Susceptibility to (A) pulmonary thrombosis after collagen infusion is shown as a survival curve, and data for the survival curve were analyzed using the log-rank (Mantel–Cox) test. (B) Time to first and stable occlusion in host human interleukin 4 receptor transgenic mice after adoptive transfer of platelets from aged Nox2 wild-type (Nox2-WT) or aged Nox2 knockout (Nox2-KO) mice. ∗∗P < .01, where the dotted black line indicates a difference between young and aged Nox2-KO, and the solid black line indicates a difference between young and aged Nox2-WT mice. Data for B are presented as mean ± SE and analyzed with an unpaired t-test. N = 6 to 10 per group.
4. Discussion
The oxidative enzyme NADPH oxidase is a major mediator of ROS generation in the vasculature, and Nox2 is one of the key catalytic subunits [29]. Since ROS mediates platelet activation, and excessive ROS production during aging could lead to hyperactive platelets and thrombosis [2,5,12], there is growing interest in targeting Nox2 to lower the thrombotic burden in this setting [[19], [20], [21]]. We set out to determine the contribution of Nox2-NADPH oxidase in age-induced platelet ROS/oxidant generation, activation, and thrombosis using mice genetically lacking Nox2-NADPH oxidase. Our novel findings suggest that genetic deficiency of Nox2-NADPH oxidase in mice does not protect from age-induced platelet-oxidant generation, platelet activation, or thrombosis. The strength of our study includes a comprehensive examination of NADPH oxidase subunits and activity, the use of multiple fluorescent dyes to probe for platelet oxidant levels, examination of several markers of platelet activation encompassing granule release, integrin activation, and PS exposure with 2 doses of agonists, the inclusion of 2 models of in vivo thrombosis, and the use of an adoptive platelet transfer approach to compare exclusive effects of Nox2-WT and Nox2-KO platelets in potentiation of in vivo thrombosis in the aged host mice. Using this rigorous approach, our data may argue against the idea that targeting Nox2-NADPH oxidase has the potential to limit thrombotic burden during healthy aging.
In a prior study, we observed that platelets isolated from aged mice, when treated with NADPH oxidase inhibitor apocynin, significantly diminished the age-induced platelet integrin activation [2], suggesting NADPH oxidase as one possible mediator of platelet activation during aging. Another study showed that in the platelets from aged aspirin-resistant patients, expression of Nox2 and p67phox subunits and NADPH-driven production of ROS tended to increase, and aggregation of platelets was significantly decreased by NADPH oxidase inhibitors (apocynin and diphenylene iodonium), whereas these inhibitors had no effect on aspirin-sensitive platelets [22]. These data suggested a therapeutic potential of NADPH oxidase inhibitors in limiting platelet activation and possibly thrombosis in high-risk aged cardiac patients that needs to be validated in the in vivo models. Moreover, pharmacological NOX inhibitors have limited isoform selectivity, so they do not provide a clear understanding of the contribution of individual NOX subunits in each disease state. Therefore, animal models with isoform-specific deletion are critical tools to address the mechanistic role of distinct NADPH oxidase subunits in causing platelet activation or thrombosis in aging.
While the mechanistic role of NADPH oxidase in mediating platelet activation and thrombosis in young mice has raised some controversy in the recent past [13,23,30,31], it may still be an important mediator in age-induced platelet activation and thrombosis. This idea is consistent with previous findings demonstrating that while the antioxidant SOD2 is dispensable for platelet activation in young mice [32], the loss of SOD2 with age exacerbates platelet activation and thrombosis [5]. Therefore, in the present study, we tested the exclusive role of Nox2 in age-induced platelet activation and thrombosis by using mice genetically deficient in Nox2. Washed platelets from the Nox2-WT mice capitulated the aging phenotype and exhibited an increase in platelet oxidants, activation, aggregation, and secretion, as has been shown previously in other aging models by us and other groups [2,3,5,12]. We also observed increased levels of mRNA for Nox2 in the platelets from aged WT mice. However, the aged Cybb-/y mice were not protected from increased platelet-oxidant generation or platelet hyperactivity. We considered that the absence of protection in aged Nox2-KO mice was due to a compensatory increase in Nox4 mRNA that would have maintained prothrombotic effects despite the loss of Nox2. This idea can be addressed in future studies by treating aged mice with NADPH oxidase inhibitor. This notion is also consistent with a study demonstrating that simultaneous inhibition of Nox1, Nox2, and Nox4 is required to inhibit NADPH oxidase-derived ROS generation in platelets [8]. To address this question further, we measured the protein levels of subunits and did not observe any alterations in Nox1, Nox4, p47Phox, or p67phox subunits due to age or genotype. This discrepancy in mRNA and protein levels for subunits may indicate a possible alteration in protein trafficking, turnover, or shedding for these subunits in our model. Importantly, NADPH oxidase activity in the platelets did not show any age- or Nox2-dependent alterations, and this finding aligns with protein expression data. Overall, our data suggest that Nox2 is not an essential mediator for platelet-oxidant generation or activation during murine aging.
Though platelets are considered a major mediator of arterial thrombosis, vessel walls, circulating factors, or shear stress may also contribute significantly, and Nox2 may modulate these factors. Therefore, we next tested the prothrombotic effects due to the loss of Nox2-NADPH oxidase in the in vivo models of experimental thrombosis. In an experimental model of collagen-induced platelet activation-dependent pulmonary thrombosis, compared with young Nox2-WT mice, the aged Nox2-WT mice died more rapidly after collagen infusion, and the deficiency of Nox2 did not protect aged mice from this early mortality. Further, we used the adoptive platelet transfer approach to compare the exclusive potential of platelets from aged Nox2-WT and aged Nox2-KO mice in developing thrombosis in vivo. We observed that platelets from either aged Nox2-WT or aged Nox2-KO mice when transfused to aged host hIL4Rα Tg mice, took a similar time to develop a stable thrombus in the injured carotid artery. So, using 2 in vivo models, our data do not support a critical role for Nox2 in arterial thrombosis.
Finally, though our data in mice do not suggest a role for Nox2 in platelet activation or arterial thrombosis in aging, it does not rule out a potential role for Nox2-derived platelet oxidants in the presence of a cardiovascular risk factor such as obesity [33] hyperlipidemia [34], smoking [35], or COVID-19 [36] and should be investigated rigorously in future studies. In summary, we demonstrate that in the absence of any cardiometabolic disease in aged mice, Nox2 is not the essential mediator for platelet activation or thrombosis. Therefore, oxidative mechanisms of ROS generation other than Nox2 should be considered in otherwise healthy-aged mice. We have previously shown that mitochondria-derived oxidants contribute to the prothrombotic phenotype in aging [5], and in the present model, we also demonstrate an age-induced increase in platelet mitochondria-derived mito-oxidants. Others have also shown aberrant platelet mitochondrial phenotype with age [3,12,37]; therefore, future studies could examine the roles of specific mitochondrial-metabolic pathways in regulating age-associated increase in platelet-oxidant generation, platelet activation, and arterial thrombosis.
Acknowledgments
We acknowledge the support from the Flow Cytometry Facility, which is a Carver College of Medicine/Holden Comprehensive Cancer Center core research facility at the University of Iowa. The facility is funded through user fees and the generous financial support of the Carver College of Medicine, Holden Comprehensive Cancer Center, and Iowa City Veteran's Administration Medical Center.
Funding
This work was possible through funding from American Heart Association 24POST1195019 https://doi.org/10.58275/AHA.24POST1195019.pc.gr.190825 to A.A. and 23IPA1054531 to J.S., from National Institutes of Health AG049784, AI162778, and HL168630, and Office of Research and Development and Department of Veterans Affairs I01CX001932 to S.D.
Author contributions
A.A. designed and conducted the experiments. G.P., V.K.S., and M.J. also conducted experiments. J.S. provided scientific input on study design and data interpretation. S.D. conceived the idea, directed the project, designed the experiments, interpreted the results, and wrote the manuscript. All authors assisted with the preparation and editing of the manuscript.
Relationship disclosure
There are no competing interests to disclose.
Data availability
The data that support the findings are available from the corresponding author upon reasonable request.
Footnotes
Handling Editor: Carsten Depperman
The online version contains supplementary material available at https://doi.org/10.1016/j.rpth.2024.102597
Supplementary material
References
- 1.Benjamin E.J., Muntner P., Alonso A., Bittencourt M.S., Callaway C.W., Carson A.P., et al. Heart disease and stroke statistics-2019 update: a report from the American Heart Association. Circulation. 2019;139:e56–e528. doi: 10.1161/CIR.0000000000000659. [DOI] [PubMed] [Google Scholar]
- 2.Dayal S., Wilson K.M., Motto D.G., Miller F.J., Jr., Chauhan A.K., Lentz S.R. Hydrogen peroxide promotes aging-related platelet hyperactivation and thrombosis. Circulation. 2013;127:1308–1316. doi: 10.1161/CIRCULATIONAHA.112.000966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davizon-Castillo P., McMahon B., Aguila S., Bark D., Ashworth K., Allawzi A., et al. TNF-alpha driven inflammation and mitochondrial dysfunction define the platelet hyperreactivity of aging. Blood. 2019;134:727–740. doi: 10.1182/blood.2019000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang J., Zhou X., Fan X., Xiao M., Yang D., Liang B., et al. mTORC1 promotes aging-related venous thrombosis in mice via elevation of platelet volume and activation. Blood. 2016;128:615–624. doi: 10.1182/blood-2015-10-672964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sonkar V.K., Eustes A.S., Ahmed A., Jensen M., Solanki M.V., Swamy J., et al. Endogenous SOD2 (superoxide dismutase) regulates platelet-dependent thrombin generation and thrombosis during aging. Arterioscler Thromb Vasc Biol. 2023;43:79–91. doi: 10.1161/ATVBAHA.121.317735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pignatelli P., Pulcinelli F.M., Lenti L., Gazzaniga P.P., Violi F. Hydrogen peroxide is involved in collagen-induced platelet activation. Blood. 1998;91:484–490. [PubMed] [Google Scholar]
- 7.Begonja A.J., Gambaryan S., Geiger J., Aktas B., Pozgajova M., Nieswandt B., et al. Platelet NAD(P)H-oxidase-generated ROS production regulates alphaIIbbeta3-integrin activation independent of the NO/cGMP pathway. Blood. 2005;106:2757–2760. doi: 10.1182/blood-2005-03-1047. [DOI] [PubMed] [Google Scholar]
- 8.Vara D., Mailer R.K., Tarafdar A., Wolska N., Heestermans M., Konrath S., et al. NADPH Oxidases are required for full platelet activation in vitro and thrombosis in vivo but dispensable for plasma coagulation and hemostasis. Arterioscler Thromb Vasc Biol. 2021;41:683–697. doi: 10.1161/ATVBAHA.120.315565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iyer K.S., Dayal S. Modulators of platelet function in aging. Platelets. 2020;31:474–482. doi: 10.1080/09537104.2019.1665641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iyer K.S., Dayal S. Platelet antioxidants: a conundrum in aging. EBioMedicine. 2019;47:29–30. doi: 10.1016/j.ebiom.2019.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gu S.X., Dayal S. Redox mechanisms of platelet activation in aging. Antioxidants (Basel) 2022;11:995. doi: 10.3390/antiox11050995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jain K., Tyagi T., Patell K., Xie Y., Kadado A.J., Lee S.H., et al. Age associated non-linear regulation of redox homeostasis in the anucleate platelet: implications for CVD risk patients. EBioMedicine. 2019;44:28–40. doi: 10.1016/j.ebiom.2019.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delaney M.K., Kim K., Estevez B., Xu Z., Stojanovic-Terpo A., Shen B., et al. Differential roles of the NADPH-oxidase 1 and 2 in platelet activation and thrombosis. Arterioscler Thromb Vasc Biol. 2016;36:846–854. doi: 10.1161/ATVBAHA.116.307308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilson S.J., Cavanagh C.C., Lesher A.M., Frey A.J., Russell S.E., Smyth E.M. Activation-dependent stabilization of the human thromboxane receptor: role of reactive oxygen species. J Lipid Res. 2009;50:1047–1056. doi: 10.1194/jlr.M800447-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wachowicz B., Olas B., Zbikowska H.M., Buczynski A. Generation of reactive oxygen species in blood platelets. Platelets. 2002;13:175–182. doi: 10.1080/09533710022149395. [DOI] [PubMed] [Google Scholar]
- 16.Fan L.M., Cahill-Smith S., Geng L., Du J., Brooks G., Li J.M. Aging-associated metabolic disorder induces Nox2 activation and oxidative damage of endothelial function. Free Radic Biol Med. 2017;108:940–951. doi: 10.1016/j.freeradbiomed.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turgeon J., Haddad P., Dussault S., Groleau J., Maingrette F., Perez G., et al. Protection against vascular aging in Nox2-deficient mice: impact on endothelial progenitor cells and reparative neovascularization. Atherosclerosis. 2012;223:122–129. doi: 10.1016/j.atherosclerosis.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 18.Fan L.M., Geng L., Cahill-Smith S., Liu F., Douglas G., McKenzie C.A., et al. Nox2 contributes to age-related oxidative damage to neurons and the cerebral vasculature. J Clin Invest. 2019;129:3374–3386. doi: 10.1172/JCI125173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fuentes E., Gibbins J.M., Holbrook L.M., Palomo I. NADPH oxidase 2 (NOX2): a key target of oxidative stress-mediated platelet activation and thrombosis. Trends Cardiovasc Med. 2018;28:429–434. doi: 10.1016/j.tcm.2018.03.001. [DOI] [PubMed] [Google Scholar]
- 20.Violi F., Pignatelli P. Platelet NOX, a novel target for anti-thrombotic treatment. Thromb Haemost. 2014;111:817–823. doi: 10.1160/TH13-10-0818. [DOI] [PubMed] [Google Scholar]
- 21.Pastori D., Pignatelli P., Carnevale R., Violi F. Nox-2 up-regulation and platelet activation: novel insights. Prostaglandins Other Lipid Mediat. 2015;120:50–55. doi: 10.1016/j.prostaglandins.2015.03.010. [DOI] [PubMed] [Google Scholar]
- 22.Stef G., Csiszar A., Ziangmin Z., Ferdinandy P., Ungvari Z., Veress G. Inhibition of NAD(P)H oxidase attenuates aggregation of platelets from high-risk cardiac patients with aspirin resistance. Pharmacol Rep. 2007;59:428–436. [PubMed] [Google Scholar]
- 23.Sonkar V.K., Kumar R., Jensen M., Wagner B.A., Sharathkumar A.A., Miller F.J., Jr., et al. Nox2 NADPH oxidase is dispensable for platelet activation or arterial thrombosis in mice. Blood Adv. 2019;3:1272–1284. doi: 10.1182/bloodadvances.2018025569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanaji T., Russell S., Ware J. Amelioration of the macrothrombocytopenia associated with the murine Bernard-Soulier syndrome. Blood. 2002;100:2102–2107. doi: 10.1182/blood-2002-03-0997. [DOI] [PubMed] [Google Scholar]
- 25.Pleines I., Eckly A., Elvers M., Hagedorn I., Eliautou S., Bender M., et al. Multiple alterations of platelet functions dominated by increased secretion in mice lacking Cdc42 in platelets. Blood. 2010;115:3364–3373. doi: 10.1182/blood-2009-09-242271. [DOI] [PubMed] [Google Scholar]
- 26.Yang H., Lang S., Zhai Z., Li L., Kahr W.H., Chen P., et al. Fibrinogen is required for maintenance of platelet intracellular and cell-surface P-selectin expression. Blood. 2009;114:425–436. doi: 10.1182/blood-2008-03-145821. [DOI] [PubMed] [Google Scholar]
- 27.Shi Y., Niculescu R., Wang D., Patel S., Davenpeck K.L., Zalewski A. Increased NAD(P)H oxidase and reactive oxygen species in coronary arteries after balloon injury. Arterioscler Thromb Vasc Biol. 2001;21:739–745. doi: 10.1161/01.atv.21.5.739. [DOI] [PubMed] [Google Scholar]
- 28.Wilson K.M., Lynch C.M., Faraci F.M., Lentz S.R. Effect of mechanical ventilation on carotid artery thrombosis induced by photochemical injury in mice. J Thromb Haemost. 2003;1:2669–2674. doi: 10.1111/j.1538-7836.2003.00482.x. [DOI] [PubMed] [Google Scholar]
- 29.Lassegue B., Griendling K.K. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol. 2010;30:653–661. doi: 10.1161/ATVBAHA.108.181610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walsh T.G., Berndt M.C., Carrim N., Cowman J., Kenny D., Metharom P. The role of Nox1 and Nox2 in GPVI-dependent platelet activation and thrombus formation. Redox Biol. 2014;2:178–186. doi: 10.1016/j.redox.2013.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dharmarajah J., Arthur J.F., Sobey C.G., Drummond G.R. The anti-platelet effects of apocynin in mice are not mediated by inhibition of NADPH oxidase activity. Naunyn Schmiedebergs Arch Pharmacol. 2010;382:377–384. doi: 10.1007/s00210-010-0552-3. [DOI] [PubMed] [Google Scholar]
- 32.Fidler T.P., Rowley J.W., Araujo C., Boudreau L.H., Marti A., Souvenir R., et al. Superoxide dismutase 2 is dispensable for platelet function. Thromb Haemost. 2017;117:1859–1867. doi: 10.1160/TH17-03-0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carnevale R., Loffredo L., Sanguigni V., Plebani A., Rossi P., Pignata C., et al. Different degrees of NADPH oxidase 2 regulation and in vivo platelet activation: lesson from chronic granulomatous disease. J Am Heart Assoc. 2014;3 doi: 10.1161/JAHA.114.000920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Magwenzi S., Woodward C., Wraith K.S., Aburima A., Raslan Z., Jones H., et al. Oxidized LDL activates blood platelets through CD36/NOX2-mediated inhibition of the cGMP/protein kinase G signaling cascade. Blood. 2015;125:2693–2703. doi: 10.1182/blood-2014-05-574491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carnevale R., Loffredo L., Pignatelli P., Nocella C., Bartimoccia S., Di Santo S., et al. Dark chocolate inhibits platelet isoprostanes via NOX2 down-regulation in smokers. J Thromb Haemost. 2012;10:125–132. doi: 10.1111/j.1538-7836.2011.04558.x. [DOI] [PubMed] [Google Scholar]
- 36.Carnevale R., Cammisotto V., Bartimoccia S., Nocella C., Castellani V., Bufano M., et al. Toll-like receptor 4-dependent platelet-related thrombosis in SARS-CoV-2 infection. Circ Res. 2023;132:290–305. doi: 10.1161/CIRCRESAHA.122.321541. [DOI] [PubMed] [Google Scholar]
- 37.Braganza A., Corey C.G., Santanasto A.J., Distefano G., Coen P.M., Glynn N.W., et al. Platelet bioenergetics correlate with muscle energetics and are altered in older adults. JCI Insight. 2019;5 doi: 10.1172/jci.insight.128248. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings are available from the corresponding author upon reasonable request.