

Abstract
The pathway of ammonia disposal in the mammalian organism has been described in 1932 as a metabolic cycle present in the liver in different compartments. In 1958, the first human disorder affecting this pathway was described as a genetic condition leading to cognitive impairment and constant abnormalities of amino acid metabolism. Since then, defects in all enzymes and transporters of the urea cycle have been described, referring to them as primary urea cycle disorders causing primary hyperammonemia. In addition, there is a still increasing list of conditions that impact on the function of the urea cycle by various mechanisms, hereby leading to secondary hyperammonemia. Despite great advances in understanding the molecular background and the biochemical specificities of both primary and secondary hyperammonemias, there remain many open questions: we do not fully understand the pathophysiology in many of the conditions; we do not always understand the highly variable clinical course of affected patients; we clearly appreciate the need for novel and improved diagnostic and therapeutic approaches. This study does look back to the beginning of the urea cycle (hi)story, briefly describes the journey through past decades, hereby illustrating advancements and knowledge gaps, and gives examples for the extremely broad perspective imminent to some of the defects of ureagenesis and allied conditions.
Keywords: ammonia detoxification, primary hyperammonemia, secondary hyperammonemia, urea cycle, ureagenesis
1. INTRODUCTION
The process of urea production or ureagenesis was first described in 1932, 1 in a paper that is still interesting to read even today because it described many aspects of the urea cycle that are still true and of relevance for our basic understanding of this pathway. The first defect in the urea cycle, a deficiency of argininosuccinate lyase (ASL), was described in 1958. 2 Since then, defects in all the six enzymes as well as both transporters required for the flux through the urea cycle have been identified and their genes and proteins studied in depth. 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 In addition, several proteins with supporting roles, for instance, for providing substrates have been identified. 12 , 13 , 14
Progress in patient management occurred on all levels, from neonatal emergency treatment, for which extracorporeal detoxification became feasible, the development of drugs such as nitrogen scavengers in various formulations, the continued improvements of low‐protein diets and essential amino acid supplementation, finally to organ replacement therapies. 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 Despite all this, urea cycle disorders (UCDs) belong to a group of inborn errors of metabolism with an overall still poor prognosis for many of the patients, which is often the result of delayed diagnosis and low awareness of healthcare professionals. 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 Therefore, there is consensus on the necessity to improve early diagnosis in addition to novel therapeutic approaches, rendering necessary a continuous improvement of our basic understanding of the involved biochemical and genetic processes, the development of reliable biomarkers and trustful model systems including digital prediction tools as well as cellular and animal models. Finally, an engagement of both academia and industry will be essential to translate research findings into the clinical reality.
To bring together basic researchers exploring the many unknown areas of urea cycle physiology and pathology with clinicians who care for affected patients, the “2nd International Conference on Ureagenesis Defects and Allied Conditions. Novel Models and Treatment Options” was held in Valencia, Spain, from 16 October 2022 to 20 October 2022. The Special Issue presented here is the proceedings of this meeting, providing a cross‐sectional view on current developments in the field and hopefully stimulating further research activities to complete a journey that is already underway for 90 years.
2. THE BEGINNING OF OUR UNDERSTANDING OF THE PROCESS OF UREA PRODUCTION
The urea cycle, as we know it today, has a history of almost 90 years. This history started with a publication in 1932 by two German researchers. 1 The first author, Dr. Hans Krebs (1900–1981) was at this time working as a medical physician and researcher in Freiburg, Germany; the second author in this publication was in fact his doctoral fellow and coworker, Kurt Henseleit (1907–1973). Hans Krebs was later (in 1953) awarded the Nobel prize for physiology or medicine for the identification and first description of the tricarboxylic acid cycle (also called the “Krebs cycle”). In the year prior to this major achievement, Hans Krebs and Kurt Henseleit performed investigations into the production of urea in animals, hereby laying the ground for the description of the “Krebs–Henseleit‐cycle,” which we nowadays refer to as the urea cycle. 34 When reading these papers today, it becomes obvious how detailed the understanding of the process of urea production (= ureagenesis) in the mammalian organism was already at this time.
Here follow some examples of the content of the first publications dealing with ureagenesis about 90 years ago. 1 , 34 In their publications, Hans Krebs and Kurt Henseleit reported a urea production in the animal body of up to 1.5 mol/day, an amount exceeding the glucose production of 0.6 mol/day. It was inferred that the process of urea production requires intact cells while cell homogenates were not able to produce urea from ammonia. However, Krebs and Henseleit described that cell homogenates are able to produce urea from arginine. This can be seen as an early understanding of the compartmentalization within the cell that was later confirmed when all mitochondrial and cytosolic players of the urea cycle were elucidated. Another interesting observation already made 90 years ago concerns the metabolite ornithine, which was described to increase the production of urea by twofold to fourfold, while not being consumed in this reaction. As well, the rate of ureagenesis in rats was found to be twofold increased in fed versus the fasting state in those animals. Finally, liver was described as the only organ able to produce urea in mammals, while birds were reported as being not able to produce urea since they lack hepatic arginase. Main findings from the first publications on mammalian urea production are listed in Table 1.
TABLE 1.
Main findings from the first publication on mammalian urea production.
|
|
|
|
|
|
|
|
3. ABOUT THE YEARS THAT HAVE PASSED SINCE THEN AND CURRENT “UNKNOWNS”
Despite the many years that have passed since these important discoveries by Hans Krebs and Kurt Henseleit have been made, there remain many “Unknowns” in the field of ureagenesis disorders.
We do not fully understand the poor neurological outcome in ASL deficiency, the first described UCD; we do not understand why in this condition even in the absence of hyperammonemia there is often a poor neurological outcome. 35 , 36 , 37 , 38 The recent description of the expression of ASL in some brainstem nuclei contributes to our understanding of the pathophysiology of this condition. 39 , 40 Nevertheless, there remain several open questions: What is the contribution of argininosuccinic acid (ASA) to liver and brain toxicity? Can nitric oxide donors compensate for brain ASL deficiency? Do we need to redefine the role of liver transplantation for this condition? Should gene therapy target liver and brain? How toxic are accumulating guanidino compounds? Is there creatine deficiency and does it matter?
Another example for the “Unknowns” in UCDs is arginase deficiency. This condition often starts in early childhood in a quite different form than other UCDs. 41 Patients are affected by a global developmental delay, progressive spasticity and often seizures, but have a low risk of hyperammonemia. 42 , 43 , 44 Elegant studies exploiting a mouse model of this condition have recently suggested that arginase deficiency is in fact a leukodystrophy of brain and spinal cord. This leukodystrophy could to some extent be rescued by neonatal AAV gene therapy targeting the liver. 45 Still, open questions remain such as concerning the function of arginase deficiency in the central nervous system, specifically in neurons and glia; what levels of arginine are nontoxic; finally, what is the role of other guanidino compounds?
Yet another example for the “Unknowns” concerns the hyperammonemia–hyperornithinemia–homocitrullinuria syndrome (HHH) syndrome caused by defects in SLC25A15, for which there are patients described with a normal outcome and no additional metabolic decompensation after an early neonatal severe metabolic crisis. 46 One possible explanation for this striking situation is the existence of an alternative ornithine transporter that possibly allows a mild phenotype for some patients with HHH syndrome. 47 Supporting this hypothesis is the knowledge about an intronless gene encoding an alternative ornithine transporter with high identity to the SLC25A15 gene. This alternative ornithine transporter has been known for 20 years and is a possible explanation for the overall milder phenotype in this specific transport defect of ureagenesis. 48 However, open questions remain such as concerning the variable clinical phenotype and whether this correlates with the function of the alternative ornithine transporter; can this alternative transporter be induced by pharmacological means; how toxic are ornithine related metabolites such as pyrroline 5‐carboxylate, polyamines, and NO; finally, is there a secondary creatine deficiency due to ornithine inhibition of the enzyme arginine:glycine amidinotransferase?
These were only three examples of the many “Unknowns.” Further examples concern the culprits of UCD neurotoxicity—Is it ammonia or glutamine 49 , 50 , 51 ? Why is the epidemiology of N‐acetylglutamate synthase deficiency so different in North America versus European countries 52 , 53 ? How to explain the various phenotypes in citrin deficiency, a condition in which some patients are sick with a cholestatic liver disease as neonates and infants, followed by a long silent period that may end in a catastrophic deterioration during early adulthood 54 , 55 ? This list could be expanded, but it should illustrate here the high need for additional basic research. This should ideally be performed in teams of people with translational ambition in order to meet the final aim of such activities, which should always be the patient outcome.
In addition to areas where we lack understanding (the “Unknowns”), we have learned on multiple levels about conditions that are related to the primary defects of the urea cycle. While the latter are classified as conditions leading to primary hyperammonemia, there are several conditions that impact on ureagenesis in a secondary way, for instance by inhibition of urea cycle enzymes or by impairing the abundance of substrates required for urea cycle flux. 56 In the following, we exemplify the field of “Ureagenesis Defects and Allied Conditions,” topic of the aforementioned International Conference, by providing an overview and briefly diving into two diverse conditions.
4. UREAGENESIS DEFECTS AND ALLIED CONDITIONS: SIMILARITIES AND DIFFERENCES, SEARCHING FOR EXPLANATIONS
The UCDs comprise defects of any of the six enzymes or of two amino acid transporters, which are primarily involved in the process of ureagenesis from the priming molecules, ammonia, ATP, and bicarbonate. 57 All these genetically determined diseases lead to hyperammonemia. However, other conditions affecting biochemical pathways connected with the urea cycle may impact on the ureagenesis causing hyperammonemia. 56 These so‐called “Urea cycle‐related disorders” include conditions not genetically determined and several inborn errors of metabolism (Table 2). Among the “non‐metabolic” causes, hyperammonemia can be observed in urinary infections caused by urease‐producing bacteria, in abdominal vascular malformations with portosystemic shunts, or can be secondary due to use of drugs—such as chemotherapy agents (e.g., l‐asparaginase), valproic acid, corticosteroids, tyrosine kinase inhibitors and iron chelating agents as deferasinox—all of which can result, albeit with different mechanisms, in a negative effect on the urea cycle function. 58 , 59 , 60
TABLE 2.
Urea cycle‐related disorders.
| Inborn errors of metabolism |
|---|
|
Organic acidurias (PA, MMA, HMGCL, CAVA) |
|
Fatty acid oxidation defects (MCAD, VLCAD, LCHAD MTP, MAD) |
|
Aminoacidopathies (LPI, P5CS HSP, P5CS CL, OAT, HI‐HAs, HHF8, GS) |
|
Mitochondrial disease (DLD, ATPase 6, ATPase 8, TMEM70, PC, SERAC1, ACAD9, MTDPS13, PEPCK) |
| Not genetically determined |
|---|
| Drugs |
| Neoplasia |
| Infections |
| Vascular malformations |
| Acute/chronic illness |
Note: Secondary urea cycle disorders causing hyperammonemia include both inborn errors of metabolism and not genetically determined causes.
Abbreviations: ACAD9, acyl‐CoA dehydrogenase 9; ATPase 6, ATP synthase 6; ATPase 8, ATP synthase 8; CAVA, carbonic anhydrase Va; DLD, dihydrolipoamide dehydrogenase; HMGCL, 3‐hydroxy‐3‐methylglutaryl‐CoA lyase; LCADH MTP, long‐chain3‐hydroxyacyl‐CoA dehydrogenase mitochondrial trifunctional protein; MAD, multiple acyl‐CoA dehydrogenase; MCAD, medium‐chain acyl‐CoA dehydrogenase; MMA, methylmalonic aciduria; MTDPS13, mitochondrial DNA depletion syndrome 13; PA, propionic aciduria; PC, pyruvate carboxylase; PEPCK, phosphoenolpyruvate carboxykinase, cytosolic; SERAC1, 3‐methylglutaconic aciduria with deafness, encephalopathy, and Leigh‐like syndrome; TMEM70, transmembrane protein 70; VLCAD, very long‐chain acyl‐CoA dehydrogenase.
The list of inborn errors, which secondarily affect the urea cycle, include disorders connected to nitrogen metabolism, such as organic acidurias and some aminoacidopathies, or to energy metabolism, as the defects of fatty acid oxidation or those involved in the generation of ATP. 56 In organic acidurias, mainly in propionic and methylmalonic aciduria, the direct toxicity of accumulated compounds on proximal urea cycle enzymes, the reduced free‐CoA availability, and the reduced ATP production, contribute to the urea cycle dysfunction. 61 Some of these pathomechanisms play also a similar negative role in fatty acid oxidation defects and in mitochondrial disorders, which affect the ATP synthesis (e.g., TMEM70, ATPase deficiencies). 62 , 63 In carbonic anhydrase Va (CAVA) deficiency, the intramitochondrial depletion of bicarbonate leads to a proximal impairment of the urea cycle with reduced carbamylphosphate production, 64 , 65 , 66 while in some amino acid‐related disorders, such as pyrroline‐5‐carboxylate synthetase deficiency (P5CSD) 67 , 68 and glutamine synthetase deficiency, 69 , 70 the depletion of essential substrates for the urea cycle may lead to hyperammonemia. 56
Clinically, urea cycle‐related disorders not only share some similarities with primary UCDs, but also present with peculiar manifestations, such as chronic inflammation in lysinuric protein intolerance (LPI), 71 pulmonary hypertension in TMEM70, 72 or spastic paraplegia in P5CSD, as seen in HHH syndrome and in arginase deficiency 73 (Figure 1). Biochemically, besides hyperammonemia, which represents the “fil rouge” linking primary with secondary UCDs, the metabolic profiles are variables, often showing specific abnormalities, which are guiding diagnostics, and which may be helpful in monitoring therapeutic interventions and/or the natural course of the condition, as illustrated in Figure 1.
FIGURE 1.
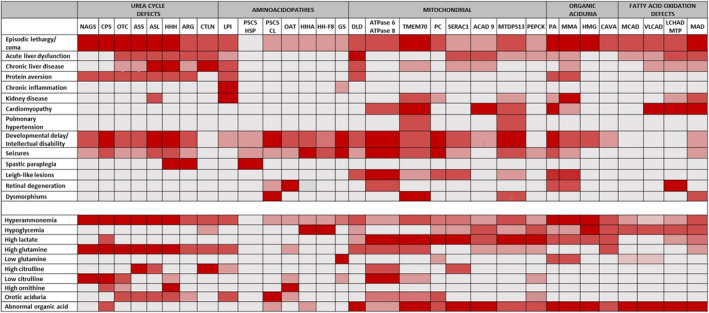
Clinical symptoms (top) and biochemical abnormalities (bottom) of UCDs‐related disorders. ACAD9, acyl‐CoA dehydrogenase 9; ARG, arginase; ASL, argininosuccinate lyase; ASS, argininosuccinate synthetase; ATPase 6, ATP synthase 6 deficiency; ATPase 8, ATP synthase 8 deficiency; CAVA, carbonic anhydrase Va deficiency; CL, Cutis laxa; CPS, carbamoylphosphate synthetase 1; CTLN, citrullinemia type II; DLD, dihydrolipoamide dehydrogenase; GS, glutamine synthetase; HHF8, hyperinsulinemic hypoglycemia, familial 8; HHH, hyperammonemia‐hyperornithinemia‐homocitrullinuria syndrome; HI‐HA, hyperinsulinism–hyperammonemia; HMG, 3‐hydroxy‐3‐methylglutaryl‐CoA lyase aciduria; LCADH MTP, long‐chain 3‐hydroxyacyl‐CoA dehydrogenase mitochondrial trifunctional protein; LPI, lysinuric protein intolerance; MAD, multiple acyl‐CoA dehydrogenase; MCAD, medium‐chain acyl‐CoA dehydrogenase; MMA, methylmalonic aciduria; MTDPS13: mitochondrial DNA depletion syndrome 13; NAGS, N‐acetylglutamate synthase; OAT, ornithine aminotransferase; OTC, ornithine carbamoyltransferase; PA, propionic aciduria; PC, pyruvate carboxylase; PEPCK, phosphoenolpyruvate carboxykinase, cytosolic; P5CS HSP, 1‐pyrroline‐5‐carboxylate synthetase, hereditary spastic paraplegia 9B; P5CS, 1‐pyrroline‐5‐carboxylate synthetase; SERAC1, 3‐methylglutaconic aciduria with deafness, encephalopathy, and Leigh‐like syndrome; TMEM70, transmembrane protein 70; UCDs, urea cycle disorders; VLCAD, very long‐chain acyl‐CoA dehydrogenase.
To shade a light on this complex scenario, we will present two conditions as examples of the connection linking different biochemical and cellular pathways to the urea cycle.
5. LYSINURIC PROTEIN INTOLERANCE AND ITS RELATION TO ARGININE METABOLISM
LPI is a recessive aminoaciduria caused by defective cationic amino acid transport in enterocytes, renal tubular cells, and monocytes/macrophages, due to mutations in SLC7A7, encoding for the y+LAT1 subunit of the y+L amino acid transporter that transfers the cationic amino acids lysine, arginine, and ornithine from the cell to the extracellular space. 74 , 75 Patients display a complex phenotype whose target organs are lung, kidney, bone, liver, spleen, and the immune system. 76 , 77 The transport defect may explain only a minor part of the phenotype, and the mechanisms linking the genetic defect to the clinical manifestations are still obscure. Some of the most severe clinical manifestations (i.e., lupus‐like manifestations, hemophagocytic lymphohistiocytosis, interstitial lung disease, and pulmonary alveolar proteinosis) highlight a major involvement of mononuclear/phagocyte system. 71 The relapse of alveolar proteinosis after lung transplantation in an LPI patient, the success of allogenic hematopoietic stem cell transplantation on LPI‐related immune dysfunction, combined with ex vivo studies on LPI alveolar macrophages supported the pivotal role of macrophages on the pathogenesis of this multifaceted disease. 78 , 79 , 80 , 81 Macrophages may present with different phenotypic subtypes (i.e., non‐activated [M0], activated pro‐inflammatory [M1], and activated anti‐inflammatory [M2]), and an imbalance versus the pro‐inflammatory subtype may explain the deleterious impact on LPI. 82 , 83 Consistent with a dysregulation of macrophages toward the activated pro‐inflammatory M1 phenotype, marked elevation of plasma calprotectin has been found in patients with LPI, as seen in various inflammatory and autoimmune conditions. 84 , 85 In macrophages, arginine metabolism is at the center of two opposite biochemical pathways, which regulate the expression of M1 and M2 phenotypes. 86 , 87 , 88 M1 macrophages metabolize arginine via nitric oxide synthase to NO and citrulline while M2 macrophages via arginase producing ornithine and urea. 88 Interestingly, studies have shown that LPI macrophages displayed a marked increase of arginine content, 89 suggesting that increased intracellular arginine availability may represent the key mechanism promoting the abnormal activation of the (innate) immune system in LPI. Consistent with this hypothesis is the elevation of plasma NO‐3 levels in an LPI patient with active immune‐dysfunction, 90 as an expression of increased NO synthesis from the arginine‐NO pathway.
6. FIBROLAMELLAR HEPATOCELLULAR CARCINOMA
Fibrolamellar hepatocellular carcinoma (FLHCC) is a rare tumor that accounts for less than 1% of all primary liver cancers, mainly affecting young adults below the age of 40 years. 91 Hyperammonemic encephalopathy is a frequent problem in patients with FLHCC even when the liver tissue is still functionally active and not massively destroyed by the neoplasm. Hyperammonemia may appear at disease onset, after chemotherapy treatment or during disease course. 92 , 93 , 94 The common metabolic abnormalities associated to hyperammonemia in FLHCC highlight a profile resembling the most common UCD, ornithine transcarbamylase (OTC) deficiency, displaying elevated plasma glutamine, reduced citrulline, arginine, ornithine, and orotic aciduria. 18 Interestingly, the pathomechanism causing the functional deficiency of the urea cycle is independent of OTC variants and has been associated with a characteristic somatic heterozygous deletion of chromosome 19, causing the chimeric fusion of DNAJB1 and PRKACA genes, observed in most FLHCC. The resulting DNAJB1–PRKACA fusion protein increases in tumor cells the expression of Aurora Kinase A (AURKA), leading to the overexpression of c‐Myc gene, hereby, besides its effects on cell cycle progression, apoptosis, and transformation, also upregulating ornithine decarboxylase (ODC). The increased ODC activity consumes ornithine for polyamines synthesis, which in turn causes a reduced ornithine bioavailability for the urea cycle, thereby resulting in a functional deficiency of the OTC enzyme due to lack of one of its substrates, and finally leading to hyperammonemia. 95 , 96 , 97
These two aforementioned examples, LPI and FLHCC, are not only entirely different in their pathomechanistic characteristics, but are only the tip of the iceberg of the widely heterogeneous panorama of UCD‐related disorders. Therefore, further basic research studies combining bioinformatics with systems biology techniques will help to identify new pathogenic mechanisms and delineate the involvement of specific metabolic pathways with possibly novel therapeutic strategies in UCD‐related disorders.
FUNDING INFORMATION
Work on urea cycle disorders in the University Children's Hospital Zurich is supported by the Swiss National Science Foundation (grants 320030_207965 and CRSII‐222794 to J.H.), the University Research Priority Program ITINERARE, and by Citrin Foundation (grant reference number: RG22005). This work was supported by funding from the Italian Ministry of Health with “Current Research” funds.
ACKNOWLEDGEMENTS
The authors are grateful for all participants of the “2nd International Conference on Ureagenesis Defects and Allied Conditions. Novel Models and Treatment Options,” held in Valencia, Spain in October 2022, as they hugely engaged into considering the future perspectives in the field of ureagenesis defects. We specifically want to express our gratitude to Dr. Vicente Rubio, Valencia, Spain, for organizing this most inspiring conference. The Division of Metabolism is affiliated member of the European Reference Network for hereditary Metabolic Disorders (MetabERN) and partner of the Unified European Registry for Inherited Metabolic Disorders (UIMD) and of the European Registry and network for Intoxication type Metabolic Diseases (E‐IMD). Open access funding provided by Universitat Zurich.
Häberle J, Siri B, Dionisi‐Vici C. Quo vadis ureagenesis disorders? A journey from 90 years ago into the future. J Inherit Metab Dis. 2024;47(6):1120‐1128. doi: 10.1002/jimd.12763
Communicating Editor: Ertan Mayatepek
Contributor Information
Johannes Häberle, Email: johannes.haeberle@kispi.uzh.ch.
Carlo Dionisi‐Vici, Email: carlo.dionisivici@opbg.net.
REFERENCES
- 1. Krebs H, Henseleit K. Untersuchungen über die Harnstoffbildung im Tierkörper. Hoppe Seylers Z Physiol Chem. 1932;210:325‐332. [Google Scholar]
- 2. Allan JD, Cusworth DC, Dent CE, Wilson VK. A disease, probably hereditary characterised by severe mental deficiency and a constant gross abnormality of aminoacid metabolism. Lancet. 1958;1(7013):182‐187. [DOI] [PubMed] [Google Scholar]
- 3. Westall RG. Argininosuccinic aciduria: identification and reactions of the abnormal metabolite in a newly described form of mental disease, with some preliminary metabolic studies. Biochem J. 1960;77:135‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tomlinson S, Westall RG. Argininosuccinic aciduria. Argininosuccinase and arginase in human blood cells. Clin Sci. 1964;26:261‐269. [PubMed] [Google Scholar]
- 5. Colombo JP, Richterich R. Urea cycle enzymes in the developing human fetus. Enzymol Biol Clin. 1968;9(1):68‐73. [DOI] [PubMed] [Google Scholar]
- 6. Solitare GB, Shih VE, Nelligan DJ, Dolan TF Jr. Argininosuccinic aciduria: clinical, biochemical, anatomical and neuropathological observations. J Ment Defic Res. 1969;13(3):153‐170. [DOI] [PubMed] [Google Scholar]
- 7. Terheggen HG, Schwenk A, Lowenthal A, Van Sande M, Colombo JP. Argininaemia with arginase deficiency. Lancet. 1969;2(7623):748.4186193 [Google Scholar]
- 8. Freeman JM, Nicholson JF, Schimke RT, Rowland LP, Carter S. Congenital hyperammonemia. Association with hyperglycinemia and decreased levels of carbamyl phosphate synthetase. Arch Neurol. 1970;23(5):430‐437. [DOI] [PubMed] [Google Scholar]
- 9. Wick H, Bachmann C, Baumgartner R, et al. Variants of citrullinaemia. Arch Dis Child. 1973;48(8):636‐641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shih VE. Congenital hyperammonemic syndromes. Clin Perinatol. 1976;3(1):3‐14. [PubMed] [Google Scholar]
- 11. Russell A, Levin B, Oberholzer VG, Sinclair L. Hyperammonaemia. A new instance of an inborn enzymatic defect of the biosynthesis of urea. Lancet. 1962;2(7258):699‐700. [DOI] [PubMed] [Google Scholar]
- 12. Siess EA, Brocks DG, Wieland OH. Distribution of metabolites between the cytosolic and mitochondrial compartments of hepatocytes isolated from fed rats. Hoppe Seylers Z Physiol Chem. 1978;359(7):785‐798. [DOI] [PubMed] [Google Scholar]
- 13. Häussinger D, Kaiser S, Stehle T, Gerok W. Liver carbonic anhydrase and urea synthesis. The effect of diuretics. Biochem Pharmacol. 1986;35(19):3317‐3322. [DOI] [PubMed] [Google Scholar]
- 14. Haussinger D, Lamers WH, Moorman AF. Hepatocyte heterogeneity in the metabolism of amino acids and ammonia. Enzyme. 1992;46(1–3):72‐93. [DOI] [PubMed] [Google Scholar]
- 15. Singh RH, Rhead WJ, Smith W, Lee B, King LS, Summar M. Nutritional management of urea cycle disorders. Crit Care Clin. 2005;21(4 Suppl):S27‐S35. [DOI] [PubMed] [Google Scholar]
- 16. Häberle J, Boddaert N, Burlina A, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012;7:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alfadhel M, Mutairi FA, Makhseed N, et al. Guidelines for acute management of hyperammonemia in the Middle East region. Ther Clin Risk Manag. 2016;12:479‐487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Häberle J, Burlina A, Chakrapani A, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: first revision. J Inherit Metab Dis. 2019;42(6):1192‐1230. [DOI] [PubMed] [Google Scholar]
- 19. Brusilow SW, Valle DL, Batshaw M. New pathways of nitrogen excretion in inborn errors of urea synthesis. Lancet. 1979;2(8140):452‐454. [DOI] [PubMed] [Google Scholar]
- 20. Batshaw ML, Brusilow SW. Treatment of hyperammonemic coma caused by inborn errors of urea synthesis. J Pediatr. 1980;97(6):893‐900. [DOI] [PubMed] [Google Scholar]
- 21. Batshaw ML. Sodium benzoate and arginine: alternative pathway therapy in inborn errors of urea synthesis. Prog Clin Biol Res. 1983;127:69‐83. [PubMed] [Google Scholar]
- 22. Kido J, Matsumoto S, Häberle J, et al. Role of liver transplantation in urea cycle disorders: report from a nationwide study in Japan. J Inherit Metab Dis. 2021;44:1311‐1322. [DOI] [PubMed] [Google Scholar]
- 23. Msall M, Batshaw ML, Suss R, Brusilow SW, Mellits ED. Neurologic outcome in children with inborn errors of urea synthesis. Outcome of urea‐cycle enzymopathies. N Engl J Med. 1984;310(23):1500‐1505. [DOI] [PubMed] [Google Scholar]
- 24. Uchino T, Endo F, Matsuda I. Neurodevelopmental outcome of long‐term therapy of urea cycle disorders in Japan. J Inherit Metab Dis. 1998;21(Suppl 1):151‐159. [DOI] [PubMed] [Google Scholar]
- 25. Nicolaides P, Liebsch D, Dale N, Leonard J, Surtees R. Neurological outcome of patients with ornithine carbamoyltransferase deficiency. Arch Dis Child. 2002;86(1):54‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bachmann C. Outcome and survival of 88 patients with urea cycle disorders: a retrospective evaluation. Eur J Pediatr. 2003;162(6):410‐416. [DOI] [PubMed] [Google Scholar]
- 27. Gropman AL, Batshaw ML. Cognitive outcome in urea cycle disorders. Mol Genet Metab. 2004;81(Suppl 1):S58‐S62. [DOI] [PubMed] [Google Scholar]
- 28. Nassogne MC, Héron B, Touati G, Rabier D, Saudubray JM. Urea cycle defects: management and outcome. J Inherit Metab Dis. 2005;28(3):407‐414. [DOI] [PubMed] [Google Scholar]
- 29. Kido J, Nakamura K, Mitsubuchi H, et al. Long‐term outcome and intervention of urea cycle disorders in Japan. J Inherit Metab Dis. 2012;35(5):777‐785. [DOI] [PubMed] [Google Scholar]
- 30. Burgard P, Kölker S, Haege G, Lindner M, Hoffmann GF. Neonatal mortality and outcome at the end of the first year of life in early onset urea cycle disorders‐review and meta‐analysis of observational studies published over more than 35 years. J Inherit Metab Dis. 2016;39(2):219‐229. [DOI] [PubMed] [Google Scholar]
- 31. Unsinn C, das A, Valayannopoulos V, et al. Clinical course of 63 patients with neonatal onset urea cycle disorders in the years 2001–2013. Orphanet J Rare Dis. 2016;11(1):116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hediger N, Landolt MA, Diez‐Fernandez C, Huemer M, Häberle J. The impact of ammonia levels and dialysis on outcome in 202 patients with neonatal onset urea cycle disorders. J Inherit Metab Dis. 2018;41(4):689‐698. [DOI] [PubMed] [Google Scholar]
- 33. Kido J, Matsumoto S, Häberle J, et al. Long‐term outcome of urea cycle disorders: report from a nationwide study in Japan. J Inherit Metab Dis. 2021;44(4):826‐837. [DOI] [PubMed] [Google Scholar]
- 34. Krebs H. Urea formation in the animal body. Ergeb Enzymforsch. 1934;3:247‐264. [Google Scholar]
- 35. Baruteau J, Jameson E, Morris AA, et al. Expanding the phenotype in argininosuccinic aciduria: need for new therapies. J Inherit Metab Dis. 2017;40(3):357‐368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Baruteau J, Diez‐Fernandez C, Lerner S, et al. Argininosuccinic aciduria: recent pathophysiological insights and therapeutic prospects. J Inherit Metab Dis. 2019;42(6):1147‐1161. [DOI] [PubMed] [Google Scholar]
- 37. Nagamani SC, Erez A, Lee B. Argininosuccinate lyase deficiency. Genet Med. 2012;14(5):501‐507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Erez A. Argininosuccinic aciduria: from a monogenic to a complex disorder. Genet Med. 2013;15(4):251‐257. [DOI] [PubMed] [Google Scholar]
- 39. Lerner S, Anderzhanova E, Verbitsky S, et al. ASL metabolically regulates tyrosine hydroxylase in the nucleus locus coeruleus. Cell Rep. 2019;29(8):2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lerner S, Eilam R, Adler L, et al. ASL expression in ALDH1A1 neurons in the substantia nigra metabolically contributes to neurodegenerative phenotype. Hum Genet. 2021;140(10):1471‐1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sin YY, Baron G, Schulze A, Funk CD. Arginase‐1 deficiency. J Mol Med. 2015;93(12):1287‐1296. [DOI] [PubMed] [Google Scholar]
- 42. Scholl‐Bürgi S, Baumgartner Sigl S, Häberle J, et al. Amino acids in CSF and plasma in hyperammonaemic coma due to arginase1 deficiency. J Inherit Metab Dis. 2008;31(suppl 2):S323‐S328. [DOI] [PubMed] [Google Scholar]
- 43. Jain‐Ghai S, Nagamani SCS, Blaser S, Siriwardena K, Feigenbaum A. Arginase I deficiency: severe infantile presentation with hyperammonemia: more common than reported? Mol Genet Metab. 2011;104(1–2):107‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huemer M, Carvalho DR, Brum JM, et al. Clinical phenotype, biochemical profile, and treatment in 19 patients with arginase 1 deficiency. J Inherit Metab Dis. 2016;39(3):331‐340. [DOI] [PubMed] [Google Scholar]
- 45. Truong B, Allegri G, Liu XB, et al. Lipid nanoparticle‐targeted mRNA therapy as a treatment for the inherited metabolic liver disorder arginase deficiency. Proc Natl Acad Sci USA. 2019;116(42):21150‐21159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Martinelli D, Diodato D, Ponzi E, et al. The hyperornithinemia–hyperammonemia–homocitrullinuria syndrome. Orphanet J Rare Dis. 2015;10:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tessa A, Fiermonte G, Dionisi‐Vici C, et al. Identification of novel mutations in the SLC25A15 gene in hyperornithinemia‐hyperammonemia‐homocitrullinuria (HHH) syndrome: a clinical, molecular, and functional study. Hum Mutat. 2009;30(5):741‐748. [DOI] [PubMed] [Google Scholar]
- 48. Camacho JA, Obie C, Biery B, et al. Hyperornithinaemia–hyperammonaemia–homocitrullinuria syndrome is caused by mutations in a gene encoding a mitochondrial ornithine transporter. Nat Genet. 1999;22(2):151‐158. [DOI] [PubMed] [Google Scholar]
- 49. Albrecht J, Norenberg MD. Glutamine: a Trojan horse in ammonia neurotoxicity. Hepatology. 2006;44(4):788‐794. [DOI] [PubMed] [Google Scholar]
- 50. Lee B, Diaz GA, Rhead W, et al. Blood ammonia and glutamine as predictors of hyperammonemic crises in patients with urea cycle disorder. Genet Med. 2015;17(7):561‐568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Maestri NE, McGowan KD, Brusilow SW. Plasma glutamine concentration: a guide in the management of urea cycle disorders. J Pediatr. 1992;121(2):259‐261. [DOI] [PubMed] [Google Scholar]
- 52. Batshaw ML, Tuchman M, Summar M, Seminara J. A longitudinal study of urea cycle disorders. Mol Genet Metab. 2014;113(1–2):127‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sancho‐Vaello E, Marco‐Marín C, Gougeard N, et al. Understanding N‐acetyl‐l‐glutamate synthase deficiency: mutational Spectrum, impact of clinical mutations on enzyme functionality, and structural considerations. Hum Mutat. 2016;37(7):679‐694. [DOI] [PubMed] [Google Scholar]
- 54. Hayasaka K. Metabolic basis and treatment of citrin deficiency. J Inherit Metab Dis. 2021;44(1):110‐117. [DOI] [PubMed] [Google Scholar]
- 55. Kido J, Häberle J, Sugawara K, et al. Clinical manifestation and long‐term outcome of citrin deficiency: report from a nationwide study in Japan. J Inherit Metab Dis. 2022;45(3):431‐444. [DOI] [PubMed] [Google Scholar]
- 56. Häberle J. Clinical and biochemical aspects of primary and secondary hyperammonemic disorders. Arch Biochem Biophys. 2013;536(2):101‐108. [DOI] [PubMed] [Google Scholar]
- 57. Häberle J, Rubio V. Disorders of the urea cycle and related enzymes. In: Saudubray JM, Baumgartner MR, García‐Cazorla Á, Walter J, eds. Inborn Metabolic Diseases. Springer; 2022:391‐406. [Google Scholar]
- 58. Shakerdi L, Ryan A. Drug‐induced hyperammonaemia. J Clin Pathol. 2023;76(8):501‐509. [DOI] [PubMed] [Google Scholar]
- 59. Martinelli D, Goffredo BM, Falvella FS, Marano M. Acute hyperammonemia in children under deferasirox treatment: cutting the Gordian knot. Clin Toxicol. 2019;57(5):375‐377. [DOI] [PubMed] [Google Scholar]
- 60. Towerman AS, Guilliams KP, Guerriero R, et al. Hyperammonemia and acute liver failure associated with deferasirox in two adolescents with sickle cell disease. Br J Haematol. 2023;201(4):e30‐e33. [DOI] [PubMed] [Google Scholar]
- 61. Häberle J, Chakrapani A, Ah Mew N, Longo N. Hyperammonaemia in classic organic acidaemias: a review of the literature and two case histories. Orphanet J Rare Dis. 2018;13(1):219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dvorakova V, Magner M, Honzik T. Hyperammonemic crisis in a child with ATP synthase deficiency caused by mtDNA mutation m.8851T>C. Mol Genet Metab Rep. 2015;2:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Žigman T, Šikić K, Petković Ramadža D, et al. ATP synthase deficiency due to m.8528T>C mutation—a novel cause of severe neonatal hyperammonemia requiring hemodialysis. J Pediatr Endocrinol Metab. 2021;34(3):389‐393. [DOI] [PubMed] [Google Scholar]
- 64. Diez‐Fernandez C, Rüfenacht V, Santra S, et al. Defective hepatic bicarbonate production due to carbonic anhydrase VA deficiency leads to early‐onset life‐threatening metabolic crisis. Genet Med. 2016;18(10):991‐1000. [DOI] [PubMed] [Google Scholar]
- 65. van Karnebeek, C. and Häberle J., Carbonic anhydrase VA deficiency, in GeneReviews®, Pagon R.A., Feldman J, Mirzaa GM, et al., Editors. 2015, University of Washington. [PubMed] [Google Scholar]
- 66. van Karnebeek CD, Sly WS, Ross CJ, et al. Mitochondrial carbonic anhydrase VA deficiency resulting from CA5A alterations presents with hyperammonemia in early childhood. Am J Hum Genet. 2014;94(3):453‐461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Marco‐Marin C, Escamilla‐Honrubia JM, Llácer JL, Seri M, Panza E, Rubio V. Δ1‐Pyrroline‐5‐carboxylate synthetase deficiency: an emergent multifaceted urea cycle‐related disorder. J Inherit Metab Dis. 2020;43(4):657‐670. [DOI] [PubMed] [Google Scholar]
- 68. Martinelli D, Häberle J, Rubio V, et al. Understanding pyrroline‐5‐carboxylate synthetase deficiency: clinical, molecular, functional, and expression studies, structure‐based analysis, and novel therapy with arginine. J Inherit Metab Dis. 2012;35(5):761‐776. [DOI] [PubMed] [Google Scholar]
- 69. Häberle J, Görg B, Rutsch F, et al. Congenital glutamine deficiency with glutamine synthetase mutations. N Engl J Med. 2005;353(18):1926‐1933. [DOI] [PubMed] [Google Scholar]
- 70. Häberle J, Shahbeck N, Ibrahim K, Hoffmann GF, Ben‐Omran T. Natural course of glutamine synthetase deficiency in a 3 year old patient. Mol Genet Metab. 2011;103(1):89‐91. [DOI] [PubMed] [Google Scholar]
- 71. Ogier de Baulny H, Schiff M, Dionisi‐Vici C. Lysinuric protein intolerance (LPI): a multi organ disease by far more complex than a classic urea cycle disorder. Mol Genet Metab. 2012;106(1):12‐17. [DOI] [PubMed] [Google Scholar]
- 72. Catteruccia M, Verrigni D, Martinelli D, et al. Persistent pulmonary arterial hypertension in the newborn (PPHN): a frequent manifestation of TMEM70 defective patients. Mol Genet Metab. 2014;111(3):353‐359. [DOI] [PubMed] [Google Scholar]
- 73. Panza E, Martinelli D, Magini P, Dionisi Vici C, Seri M. Hereditary spastic paraplegia is a common phenotypic finding in ARG1 deficiency, P5CS deficiency and HHH syndrome: three inborn errors of metabolism caused by alteration of an interconnected pathway of glutamate and urea cycle metabolism. Front Neurol. 2019;10:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Palacín M, Bertran J, Chillarón J, Estévez R, Zorzano A. Lysinuric protein intolerance: mechanisms of pathophysiology. Mol Genet Metab. 2004;81(suppl 1):S27‐S37. [DOI] [PubMed] [Google Scholar]
- 75. Sebastio G, Sperandeo MP, Andria G. Lysinuric protein intolerance: reviewing concepts on a multisystem disease. Am J Med Genet C Semin Med Genet. 2011;157C(1):54‐62. [DOI] [PubMed] [Google Scholar]
- 76. Noguchi A, Takahashi T. Overview of symptoms and treatment for lysinuric protein intolerance. J Hum Genet. 2019;64(9):849‐858. [DOI] [PubMed] [Google Scholar]
- 77. Mauhin W, Habarou F, Gobin S, et al. Update on lysinuric protein intolerance, a multi‐faceted disease retrospective cohort analysis from birth to adulthood. Orphanet J Rare Dis. 2017;12(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Santamaria F, Brancaccio G, Parenti G, et al. Recurrent fatal pulmonary alveolar proteinosis after heart‐lung transplantation in a child with lysinuric protein intolerance. J Pediatr. 2004;145(2):268‐272. [DOI] [PubMed] [Google Scholar]
- 79. Rotoli BM, Barilli A, Visigalli R, et al. Downregulation of SLC7A7 triggers an inflammatory phenotype in human macrophages and airway epithelial cells. Front Immunol. 2018;9:508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lin Y, Liu Y, Zhu L, et al. Combining newborn metabolic and genetic screening for neonatal intrahepatic cholestasis caused by citrin deficiency. J Inherit Metab Dis. 2020;43(3):467‐477. [DOI] [PubMed] [Google Scholar]
- 81. Kolker S, Garbade SF, Boy N, et al. Decline of acute encephalopathic crises in children with glutaryl‐CoA dehydrogenase deficiency identified by newborn screening in Germany. Pediatr Res. 2007;62(3):357‐363. [DOI] [PubMed] [Google Scholar]
- 82. Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen‐presenting functions. Nat Rev Immunol. 2017;17(6):349‐362. [DOI] [PubMed] [Google Scholar]
- 83. Mills CD. M1 and M2 macrophages: oracles of health and disease. Crit Rev Immunol. 2012;32(6):463‐488. [DOI] [PubMed] [Google Scholar]
- 84. La C, Lê PQ, Ferster A, et al. Serum calprotectin (S100A8/A9): a promising biomarker in diagnosis and follow‐up in different subgroups of juvenile idiopathic arthritis. RMD Open. 2021;7(2):e001646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wang S, Song R, Wang Z, Jing Z, Wang S, Ma J. S100A8/A9 in inflammation. Front Immunol. 2018;9:1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kieler M, Hofmann M, Schabbauer G. More than just protein building blocks: how amino acids and related metabolic pathways fuel macrophage polarization. FEBS J. 2021;288(12):3694‐3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Rodriguez PC, Ochoa AC, Al‐Khami AA. Arginine metabolism in myeloid cells shapes innate and adaptive immunity. Front Immunol. 2017;8:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Rath M, Müller I, Kropf P, Closs EI, Munder M. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front Immunol. 2014;5:532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Barilli A, Rotoli BM, Visigalli R, et al. In lysinuric protein intolerance system y+L activity is defective in monocytes and in GM‐CSF‐differentiated macrophages. Orphanet J Rare Dis. 2010;5:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Mannucci L, Emma F, Markert M, et al. Increased NO production in lysinuric protein intolerance. J Inherit Metab Dis. 2005;28(2):123‐129. [DOI] [PubMed] [Google Scholar]
- 91. Ahmed A, Ata F, Gaber M, et al. Refractory hyperammonemic encephalopathy in fibrolamellar hepatocellular carcinoma, a case report and literature review. Curr Probl Cancer. 2022;46(3):100847. [DOI] [PubMed] [Google Scholar]
- 92. Sethi S, Tageja N, Dave M, et al. Hyperammonemic encephalopathy: a rare presentation of fibrolamellar hepatocellular carcinoma. Am J Med Sci. 2009;338(6):522‐524. [DOI] [PubMed] [Google Scholar]
- 93. Cho J, Chen JCY, Paludo J, et al. Hyperammonemic encephalopathy in a patient with fibrolamellar hepatocellular carcinoma: case report and literature review. J Gastrointest Oncol. 2019;10(3):582‐588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chapuy CI, Sahai I, Sharma R, Zhu AX, Kozyreva ON. Hyperammonemic encephalopathy associated with fibrolamellar hepatocellular carcinoma: case report, literature review, and proposed treatment algorithm. Oncologist. 2016;21(4):514‐520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Honeyman JN, Simon EP, Robine N, et al. Detection of a recurrent DNAJB1‐PRKACA chimeric transcript in fibrolamellar hepatocellular carcinoma. Science. 2014;343(6174):1010‐1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Surjan RC, dos Santos ES, Basseres T, Makdissi FF, Machado MA. A proposed physiopathological pathway to hyperammonemic encephalopathy in a non‐cirrhotic patient with fibrolamellar hepatocellular carcinoma without ornithine transcarbamylase (OTC) mutation. Am J Case Rep. 2017;18:234‐241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Weber G, Queener SF, Morris HP. Imbalance in ornithine metabolism in hepatomas of different growth rates as expressed in behavior of l‐ornithine carbamyl transferase activity. Cancer Res. 1972;32(9):1933‐1940. [PubMed] [Google Scholar]