

Abstract
RNA has triggered a significant shift in modern medicine, providing a promising way to revolutionize disease treatment methods. Different therapeutic RNA modalities have shown promise to replace, supplement, correct, suppress, or eliminate the expression of a targeted gene. Currently, there are 22 RNA‐based drugs approved for clinical use, including the COVID‐19 mRNA vaccines, whose unprecedented worldwide success has meant a definitive boost in the RNA research field. Urea cycle disorders (UCD), liver diseases with high mortality and morbidity, may benefit from the progress achieved, as different genetic payloads have been successfully targeted to liver using viral vectors, N‐acetylgalactosamine (GalNAc) conjugations or lipid nanoparticles (LNP). This review explores the potential of RNA‐based medicines for UCD and the ongoing development of applications targeting specific gene defects, enzymes, or transporters taking part in the urea cycle. Notably, LNP‐formulated mRNA therapy has been assayed preclinically for citrullinemia type I (CTLN1), adolescent and adult citrin deficiency, argininosuccinic aciduria, arginase deficiency and ornithine transcarbamylase deficiency, in the latter case has progressed to the clinical trials phase.
Keywords: antisense oligonucleotides, mRNA therapy, ornithine transcarbamylase deficiency, pseudoexon, splicing, urea cycle
RNA therapies for UCD.
1. INTRODUCTION
The urea cycle is a liver‐based pathway enabling the elimination of neurotoxic ammonia produced by deamination of amino acids. Defects in the enzymes or transporters involved in this pathway are collectively known as urea cycle disorders (UCD), rare diseases with an often fatal course despite protein‐restricted dietary management and pharmacological treatments. 1 Liver transplantation remains the only curative treatment to date; therefore, there exists a significant unmet clinical need of novel therapies. In this sense, with liver as target organ, genetic therapies, either DNA or RNA‐based, may provide the opportunity to modify the natural history of these diseases, by correcting genetic defects or supplementing gene expression.
Gene therapy using adenoviral or adeno‐associated vectors has been explored for many years for several UCD enzyme deficiencies, with different clinical trials presently ongoing. 2 In recent years, the development of mRNA biotherapeutics and antisense oligonucleotide (ASO) therapies have modified the perspective of how diseases can be treated, based on the massive clinical success of COVID‐19 mRNA vaccines and the approval of different antisense drugs for rare diseases. 3 , 4 In addition, the emergence of CRISPR/Cas gene editing technologies, with increasing pre‐clinical data and numerous ongoing clinical trials, has opened new personalized treatment opportunities for rare diseases. 5
Even though therapeutic RNA has today become a reality, the RNA molecule was for a long time considered unsuitable for clinical use because of its labile nature and its immunogenic properties. Years of rigorous research and steady progress in chemical modifications 6 , 7 and development of different delivery and cell/tissue targeting modalities 8 , 9 have mainly overcome these limitations. The seminal contributions of Katalin Karikó and Drew Weissman to the research in modifications of the mRNA molecule and its use in vaccines was recently recognized by the award of the 2023 Nobel Prize in Medicine or Physiology. 10
Targeted delivery has been, and in many cases continues to be, one of the main challenges in the clinical translation of RNA drugs. In some cases, RNA medicines can be administered locally, for example intratumoural, intrathecal (for brain delivery, bypassing the blood–brain barrier), or intravitreal. Systemic administration by subcutaneous and intravenous routes largely results in their distribution to liver and to renal clearance. 9 A wide variety of delivery approaches, using viral vectors, ligand conjugates, or nanocarriers, have improved the transport and bioavailability of RNA drugs, achieving safe and effective delivery to target organs. 3 , 8 , 9 For specific delivery to hepatocytes, relevant for UCD, lipid nanoparticles (LNP) and tri‐antennary N‐acetylgalactosamine (GalNAc) conjugation have been shown to be the most effective strategies after systemic administration. 11 GalNAc is a ligand of the asialoglycoprotein receptor, specifically and abundantly expressed on the membrane surface of parenchymal hepatocytes, with rapid internalization and turnover kinetics. LNPs can be modified with targeting moieties for preferential liver distribution and hepatocyte uptake. ApoE‐coated LNPs bind to the LDL receptor expressed on the surface of hepatocytes which mediate cellular uptake. 12 However, given the functional specialization of liver parenchyma known as metabolic zonation, with urea cycle enzymes predominantly expressed in periportal hepatocytes, 13 cell‐type specific targeting is necessary in the context of UCD. This issue has been addressed in different reports using LNP‐mRNA formulations, and results showed a preferential localization of the enzymes encoded by the mRNA molecule (ornithine transcarbamylase‐OTC and arginase 1‐ARG1, in the referred studies) in periportal hepatocytes, with lower immunoreactivity in hepatocytes around central veins. 14 , 15 In another study, using a novel two‐nanoparticle mRNA delivery system comprising a (GalNAc)‐targeted polymer micelle and an LNP‐OTC mRNA formulation, the authors showed rapid and potent liver mRNA expression dependent on both nanoparticles, correlating with a therapeutic benefit in a murine model of OTC deficiency (OTCD). 16 This indirectly indicates that the GalNAc conjugate effectively targets periportal hepatocytes.
The main types of RNA‐based drugs include (i) single‐stranded ASO, with different modes of action: gene silencing by RNAse H1‐dependent transcript degradation, or steric blocking to modulate splicing, to activate or to inhibit translation; (ii) short interfering RNAs (siRNA), with a double‐stranded structure that after processing are incorporated into the RNA‐induced silencing complex eliciting gene knockdown; (iii) aptamers, single‐stranded nucleic acid molecules, highly 3D structured and with high affinity and specificity for target proteins, modulating their action; (iv) microRNAs (miRNA) inhibitors (which are essentially ASO targeting the corresponding miRNA) and miRNA mimics, precursor double‐stranded RNA molecules that when added exogenously, follow the same mechanism of gene silencing as miRNAs; (v) mRNA, coding for therapeutic or antigenic proteins/peptides; (vi) small activating RNA, a type of double‐stranded RNA that induces target gene upregulation, and (vii) suppressor tRNAs that induce nonsense codon readthrough 17 (Table 1). It should be noted that in this latter modality, a single suppressor tRNA drug may be used to treat multiple diseases, indicated for patients with the same type of nonsense mutations. In UCD, nonsense mutations have a frequency ranging from 7% to 10% (CPS1, OTC, ASL, ASS1, ARG1, and NAGS genes) up to 14% and 31% (SLC25A15 and SLC25A13, respectively) (Human Mutation Database, https://www.hgmd.cf.ac.uk/). In some cases, CRISPR‐based gene editing technologies are included as RNA‐based therapies, as nucleases/base editors are delivered as mRNA, together with chemically synthesized guide RNA 5 , 22 , 23 , 24 (Table 1).
TABLE 1.
RNA therapeutic modalities approved or in development.
| Therapeutic molecule | Mode of action | Stage | Disease (examples) | References |
|---|---|---|---|---|
| ASO | Gene silencing | Approved | ALS, hATTR, FH | [3] |
| SSO | Splice switching | Approved | DMD, SMA | [3] |
| siRNA | Gene silencing | Approved | hATTR, FH, PH1, AHP | [3] |
| Aptamer | Protein binding | Approved | AMD, GA | [3] |
| miRNA inhibitors | Gene upregulation | Clinical trials | Cancer, DM1 | [18] |
| miRNA mimics | Gene silencing | Clinical trials | Cancer | [18] |
| mRNA | Protein expression | Clinical trials | OTCD, PA | [2, 19] |
| saRNA | Gene upregulation | Clinical trials | Cancer | [20] |
| Suppressor tRNA | Stop codon readthrough | Preclinical | CF | [21] |
| CRISPR‐based a | Gene editing |
Preclinical Clinical trials |
PKU, ASA, hATTR, HAE | [22, 23, 24, 25] |
Abbreviations: AHP, acute hepatic porphyria; ALS, amyotrophic lateral sclerosis; AMD, age‐related macular degeneration; ASLD, argininosuccinate lyase deficiency; ASO, antisense oligonucleotide; CF, cystic fibrosis; DMD, Duchenne muscular dystrophy; DM1, myotonic dystrophy 1; FH, familial hypercholesterolemia; GA, geographic atrophy; HAE, hereditary angioedema: hATTR, hereditary amyloidosis transthyretin related; miRNA, microRNA; OTCD, ornithine transcarbamylase deficiency; PA, propionic acidemia; PH1, primary hyperoxaluria type 1; PKU, phenylketonuria; saRNA, small activating RNA; siRNA, small interfering RNA; SMA, spinal muscular atrophy; SSO, splice switching oligonucleotide.
With nuclease/base editors delivered as mRNA.
Currently, 22 RNA‐based drugs are in clinical use, most of them indicated for rare genetic diseases. Overall, 2 aptamers, 5 splice switching ASO, 5 knockdown ASO, 6 siRNAs, and 3 mRNA vaccines have been approved worldwide. They feature varying degrees of nucleic acid modifications, with or without lipid formulation or GalNAc conjugation. 3
In this review, we explore the potential and the ongoing progress of RNA‐based drugs for UCD (Figure 1), focusing on ASO and mRNA therapies, as well as mentioning the advances in gene editing which is in active development using mRNA‐encoded nucleases/editors. To complete the genetic therapeutics scenario for UCD we refer the readers to recent excellent reviews on gene therapy. 2 , 26
FIGURE 1.
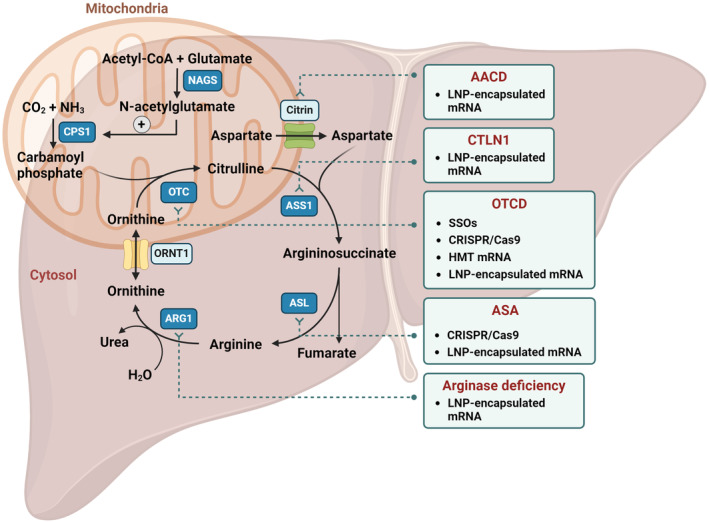
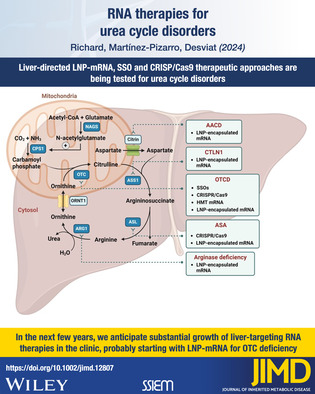
RNA therapies in development for urea cycle defects. AACD, adolescent and adult citrin deficiency; ARG1, arginase; ASA, argininosuccinic aciduria; ASL, argininosuccinate lyase; ASS1, argininosuccinate synthetase; CPS1, carbamoylphosphate synthetase I; CTLN1, citrullinemia type 1; HMT, hybrid mRNA Technology; LNP, lipid nanoparticles; NAGS, N‐acetyl glutamate synthase; ORNT1, ornithine translocase; OTC, ornithine transcarbamylase; OTCD, ornithine transcarbamylase deficiency; SSO, splice switching antisense oligonucleotides. All therapies are in preclinical or clinical stage for UCD and/or other disorders, as specified in Table 1.
2. ASO THERAPY
There are two types of therapeutic ASOs in the market, gapmer ASOs, with a central gap of DNA nucleotides and a phosphorothioate backbone flanked with modified nucleotides on both sides, that result in gene knockdown via RNAse‐H1 recruitment, and fully modified ASO that use a non RNAse H1‐dependent mechanism for sterically blocking access of RNA binding proteins to pre‐mRNA. 17 , 27 This modality is usually focused in blocking splice factors or components of the spliceosome, resulting in an alteration in the splicing outcome. 28 Approved RNAse‐H1 recruiting ASO targeting hepatocytes include eplontersen and inotersen for treatment of transthyretin amyloidosis, volanosersen for familiar chylomicronemia syndrome, and mipomersen for familiar hypercholesterolemia. 3 Splice‐switching antisense oligonucleotides (SSO) have been approved to promote exon inclusion in the central nervous system (nusinersen, spinal muscular atrophy treatment) and for exon skipping, targeting muscle (eteplirsen, golodrisen, viltolarsen, and casimersen for duchenne muscular dystrophy). 3
There are several more therapeutic uses of SSO, that is, preventing aberrant cryptic splice site usage, modifying alternative splicing or preventing pseudoexon inclusion. 29 Some of these approaches have been tested for UCD, as described below.
SSO‐mediated blocking of a cryptic splice site has been tested for the OTC gene variant c.386G >A (p.R129H), identified in patients with OTCD, and also present in the spf/ash mouse model. This mutation, located in the last nucleotide of exon 4, affects the 5′ splice site and was shown to impair splicing in the mouse liver, resulting in the partial use of a cryptic splice site 48 bp downstream exon 4. 30 SSO targeting this cryptic splice site was tested using minigenes, resulting in intron 4 retention, instead of redirecting splicing to the natural 5′ splice site. 31 However, in OTC patients, as well as in minigenes with the human sequences, the variant resulted both in exon 4 skipping and in the use of a different cryptic splice site, at intronic position c.386 + 4. This sequence is included in the 5′ splice site making the SSO approach unfeasible. 31 This example highlights the relevance of the genomic context in the splicing outcome and the challenges in using mouse models for the development of RNA therapies targeting intronic sequences. Nonetheless, SSO may remain a therapeutic modality to be tested for other cryptic splice site activating variants, as has been successfully tested in vitro in different inherited metabolic diseases (IMD). 32 , 33 , 34
Aberrant splicing may also be caused by deep intronic mutations that activate what are known as pseudoexons, given their resemblance to true exons, with potential 3′ and 5′ splice sites. 35 Pseudoexons are not normally spliced into mRNA, probably due to the presence of splicing silencers, or to formation of inhibiting RNA structures. 36 Pseudoexon activation results from a point mutation that activates or creates 3′ or 5′ splicing sites or affects splice regulatory elements. The aberrant pseudoexon inclusion generally results in a transcript with premature termination codons which is degraded by the nonsense‐mediated decay mechanism. 37 Pseudoexon‐activating mutations have been identified in many genetic diseases, including IMD, 32 , 34 , 38 especially as transcriptome analysis is being increasingly included in the genetic diagnosis workflow. 39 , 40 , 41
Several deep intronic variants resulting in aberrant pseudoexon inclusion were also identified in patients with OTCD. 42 , 43 , 44 , 45 Pseudoexons are excellent candidates for SSO therapy, promoting their exclusion from mature mRNA, as natural splice sites of the surrounding exons are intact. Blocking with SSO the splice sites or enhancer elements in the pseudoexon restores normal splicing, as has been demonstrated in many preclinical studies for different gene defects. 32 , 33 , 34 , 46 As an alternative, a recent study reported the successful ex vivo gene editing of hepatocytes from an OTCD patient carrying a pseudoexon activating variant, using dual gRNAs with CRISPR/Cas9 to delete the intronic sequence with the activated splice site. In this case, the CRISPR components (pairs of gRNAs and the nuclease) were nucleofected as a ribonucleoprotein complex and the strategy effectively corrected the urea cycle defect. 45
3. mRNA THERAPY
The success of the mRNA vaccines against SARS‐CoV‐2 has spurred a rapid growth in the field, with over 400 mRNA‐related products currently in development and a sharp increase in investment. 47 Among these, there are more than 30 mRNA‐based products approved or in late‐phase clinical trials, covering a wide range of applications, including: (i) replacement therapy, where mRNA is introduced to provide a functional protein in cases of deficiency or to deliver therapeutic proteins; (ii) vaccination, where mRNA encoding specific antigens is used to generate an immune response for protection; and (iii) cell therapy, where cells are transfected with mRNA ex vivo to modify their phenotype or function before being administered to the patient. 48 , 49
Progress in mRNA modifications, synthesis, purification, and cellular delivery has paved the way for the development of these applications. In recent years, mRNA therapies encapsulated in LNPs have emerged as promising treatments for various UCD. The non‐viral vector‐mediated delivery of mRNA prevents the risk of systemic toxicity elicited by the viral vector and of genotoxicity, as mRNA therapy is transient in nature and has no genome‐integrating properties. Moreover, the synthetic nature of mRNA production simplifies and lowers the cost of GMP‐complied manufacturing compared to other genetic medicines.
The LNP‐formulated mRNA approach has been successfully explored for citrullinemia type 1 (CTLN1) caused by argininosuccinate synthetase 1 (ASS1) deficiency, adolescent and adult citrin deficiency (AACD, previously named citrullinemia type 2 or CTLN2), argininosuccinic aciduria (ASA) caused by argininosuccinate lyase (ASL) deficiency, arginase deficiency caused by ARG1 deficiency and OTCD, as described below (Figure 1).
Recent studies employed LNP‐encapsulated mRNA to treat AACD and ASA. 50 , 51 , 52 In the AACD study, intravenous administration of codon‐optimized hCitrin mRNA successfully induced expression of the citrin protein in the liver of a mouse model of citrin deficiency, resulting in a significant reduction in hepatic citrulline and blood ammonia levels after an oral sucrose challenge and sucrose aversion (key indicators of hCitrin deficiency). 50 Similarly for ASA, the authors used targeted metabolomics and imaging techniques (in vivo positron emission tomography) to demonstrate that ASL mRNA therapy restored ureagenesis and corrected glutathione metabolism in ASL‐deficient mice, suggesting that glutathione biosynthesis in the liver is regulated by nitric oxide availability. 52 In line with these findings, the study by Daly et al. explored the use of nucleoside‐modified mRNA delivered via LNP to treat ASA. The optimized mRNA demonstrated robust in vitro and in vivo expression of ASL protein with minimal immunogenic response and improved the survival in a mouse model of ASA, further suggesting that LNP‐formulated mRNA could be a promising alternative to current therapies. 51
Similar studies employed LNP‐mediated mRNA delivery to address arginase deficiency. 14 , 53 In the first study, the authors demonstrated that codon‐optimized ARG1 mRNA encapsulated in LNPs led to 100% survival of arginase‐deficient mice, restoring urea cycle activity and maintaining normal liver function without signs of hepatotoxicity. 14 In the second study, the authors found that intermittent administration of LNPs carrying ARG1 mRNA significantly improved myelination in the central nervous system and managed ammonia and arginine levels in arginase‐deficient mice. 53
The precision‐cut liver sections (PCLS) model, used by Perocheau et al., 54 provides an innovative alternative to traditional in vitro and in vivo models. This technique maintains the liver's three‐dimensional architecture and cellular environment, offering a more accurate simulation of human metabolic conditions. Compared to organoids, 3D models used extensively for different diseases, PCLS model offers several advantages. First, PCLS is more cost‐effective and less labor‐intensive, making it suitable for high‐throughput therapeutic screening. Additionally, PCLS preserves the 3D tissue architecture and allows for multiple samples from a single organ, significantly reducing the number of animals needed for experiments. In this work, they demonstrated that mRNA therapy, delivered via LNPs, could correct metabolic defects in liver sections affected by CTLN1 and ASA, highlighting the potential of PCLS for preclinical assessment of new therapies. 54
Another noteworthy approach is the Hybrid mRNA Technology (HMT) described by Prieve et al., 16 which combines LNPs and polymer micelles to deliver OTC mRNA for treating OTCD. This dual‐nanoparticle system effectively protected the mRNA in the bloodstream and targeted liver cells, normalizing blood ammonia and orotic acid levels in OTCD mouse models and demonstrating prolonged survival benefits with minimal off‐target effects. 16
Two additional studies have further advanced mRNA therapies for OTCD by using LNP‐formulated human OTC mRNA. 15 , 55 Yamazaki et al. 15 demonstrated that intravenously delivered hOTC mRNA successfully produced functional proteins in the liver, restored the urea cycle, and notably improved survival in OTCD model mice. Their results confirmed dose‐dependent efficacy and safety, suggesting that this approach could be a promising alternative to existing OTCD therapies. 15 Meanwhile, Yu et al. focused on refining diagnostic techniques, showing that plasma OTC activity could effectively serve as a surrogate marker for liver activity, thus potentially reducing the need for invasive liver biopsies. Their study provided a strong foundation for further clinical testing, highlighting the safety and efficacy of their proprietary LUNAR‐OTC in restoring OTC function. 55
Based on preclinical studies, mRNA‐based therapy for OTCD, known as ARCT‐810, has undergone Phase Ia and Ib trials (NCT04416126 and NCT04442347, respectively) demonstrating a promising safety profile. Sponsored by Arcturus Therapeutics, this therapy is advancing to a Phase II clinical trial for adolescent and adult patients with OTCD (NCT05526066), which is currently active, not recruiting (last updated 07/2024, clinicaltrials.gov).
4. GENE EDITING
Beyond ASO and mRNA therapies, that require lifelong repeated administration, gene editing therapies directed toward the liver have the potential to enable DNA modifications that allow for permanent correction of disease‐causing variants in UCD after a single dose. To date, most of the studies have used adeno‐associated viral (AAV) vectors as delivery agents. In OTCD, which is frequently treated by orthotopic liver transplantation, an ex vivo approach was studied, using primary hepatocytes isolated from an OTC‐deficient patient. 45 These were electroporated with CRISPR components and the edited cells were transplanted in a liver‐humanized model of OTCD, resulting in the restoration of urea cycle, normal liver OTC activity, and a significant phenotypic correction of the disease symptoms, as compared with mice transplanted with unedited hepatocytes. 45
Several in vivo gene editing studies have been undertaken for OTCD using the well‐known Spf ash mouse model. 30 Given the size constraints of AAV vectors, dual‐AAV strategies have been used. Intravenous infusion of one AAV expressing Cas9 and another expressing a guide RNA and a wild‐type donor DNA into newborn Spf ash mice resulted in reversion of the OTC mutation in 10% of hepatocytes and increased survival. 56 However, in adult Spf ash mice gene correction was lower and OTC gene expression was disrupted, leading to lethal hyperammonemia, probably due to different outcomes of the DNA repair mechanisms after Cas9‐mediated double‐stranded breaks. 56
A mutation‐independent approach based on CRISPR‐mediated integration of a functional gene, potentially applicable to all patients irrespective of their mutation, has been tested for different UCDs, both in vitro 57 , 58 and in vivo. 59 There is an ongoing gene editing clinical trial for treatment of OTCD, based on the administration of two AAV vectors, carrying a nuclease targeting the well‐characterized PCSK9 gene locus serving as insertion site and a functional OTC gene, respectively (NCT06255782).
In addition to these advancements using viral vectors, gene editing using LNP‐mediated delivery of mRNA encoding CRISPR tools and a targeting gRNA is gaining importance in the therapeutics landscape for liver diseases. 60 This approach has been tested for ASA, using mRNA encoding an adenine base editor to correct the Finnish founder ASL variant c.1153C >T in human‐induced pluripotent stem cells (hiPSCs). 23 The authors show that in hiPSC‐derived hepatocyte‐like cells, the disrupted urea cycle was effectively rescued. Currently, two clinical trials are ongoing, based on LNP‐Cas9 mRNA delivery to liver, for transthyretin amyloidosis and hereditary angioedema. 22 , 24
5. SUMMARY AND FUTURE PERSPECTIVES
In summary, there is a wealth of preclinical and clinical studies that collectively demonstrate the transformative potential of RNA therapies for treating metabolic disorders of the liver, and specifically, for UCD. The advancements in delivery, therapeutic efficacy, and proposed strategies for developing RNA platforms for each modality and target tissue, offer promising avenues for developing effective treatments reaching the clinic. 3 Effective in vivo viral and non‐viral delivery systems targeting the liver have been developed, although there is still room for improvement. The integration of novel methodologies such as PCLS and HMT further enriches the landscape of RNA therapeutics development. To date, encapsulation in LNPs is a predominant choice, owing to their titratability and ease of manufacturing. However, in terms of delivery, the issue of the urea cycle liver zonation should be specifically addressed, as this will determine the method of choice regarding efficacy, specificity, and safety. Besides, the field of LNP‐mRNA therapies is still relatively young, with limited information regarding long‐term safety. However, LNP optimization for improved efficacy and safety is an area of active research. 61 To date, different companies have multiple LNP‐mRNA drug candidates for non‐immunogenic applications in their pipelines. 61 , 62 Thus, with more LNP‐mRNA non immunogenic drug candidates entering the clinic, it can be envisaged that existing gaps in the knowledge of the clinical applicability of this approach may be closed in the near future.
Another important issue is the identification and selection of relevant biomarkers and/or surrogate markers that accurately reflect meaningful clinical endpoints for each UCD, which should be prioritized in future research. Current preclinical and clinical data on mRNA therapy for UCD suggest a future full of many possibilities, probably starting with OTCD, as it is already in the clinical trials phase and is the most common among UCD. Other UCD with established biomarkers may also be good candidates once the therapeutic LNP‐mRNA platform is optimized.
Other highly active fields of genetic therapy research, such as gene editing, are also showing great potential. This technology has been explored for different liver diseases, including OTCD, 56 , 63 using viral vectors or mRNA‐mediated. In the context of gene therapy, positive preclinical and clinical data in UCD have aroused new excitement and currently, there are several clinical trials for OTCD ongoing. 2 , 26
In the next few years, we anticipate substantial growth of liver‐targeting RNA therapies in the clinic, given the robust productivity of hepatic delivery agents currently available. The balance between benefits and challenges, safety and effectiveness of each therapeutic approach will most probably be clarified, setting the future directions for the development and clinical translation of innovative genetic therapies (DNA or RNA‐based) for UCD.
AUTHOR CONTRIBUTIONS
LRD conceptualized and designed the manuscript. ER and LRD drafted the manuscript. AM‐P drafted the table and figure. All authors revised the manuscript. LRD is the guarantor for the article.
FUNDING INFORMATION
This work was funded by grant PID2022‐137238OB‐100 funded by MICIU/AEI/10.13039/501100011033/ and by ERDF A way of making Europe. Centro de Biología Molecular Severo Ochoa receives an institutional grant from Fundación Ramón Areces.
CONFLICT OF INTEREST STATEMENT
Eva Richard, Ainhoa Martinez‐Pizarro, and Lourdes R. Desviat declare they have no conflict of interest.
INFORMED CONSENT STATEMENT
This article does not contain any studies with human or animal subjects performed by the any of the authors.
ACKNOWLEDGEMENTS
The authors wish to thank the organizers of the “International conference on ureagenesis defects and allied conditions 2022” for their initiative contributing to the collaborative advance in the research in these diseases.
Richard E, Martínez‐Pizarro A, Desviat LR. Exploring RNA therapeutics for urea cycle disorders. J Inherit Metab Dis. 2024;47(6):1269‐1277. doi: 10.1002/jimd.12807
Communicating Editor: Georg Hoffmann
DATA AVAILABILITY STATEMENT
My manuscript has no associated data.
REFERENCES
- 1. Ah Mew N, Simpson KL, Gropman AL, et al. Urea cycle disorders overview. In: Adam MP, Feldman J, Mirzaa GM, et al., eds. GeneReviews®. University of Washington; 1993. [Google Scholar]
- 2. Duff C, Alexander IE, Baruteau J. Gene therapy for urea cycle defects: An update from historical perspectives to future prospects. J Inherit Metab Dis. 2024;47:50‐62. doi: 10.1002/jimd.12609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Androsavich JR. Frameworks for transformational breakthroughs in RNA‐based medicines. Nat Rev Drug Discov. 2024;23:421‐444. doi: 10.1038/s41573-024-00943-2 [DOI] [PubMed] [Google Scholar]
- 4. Oude Blenke E, Schiffelers RM, Mastrobattista E. Oligonucleotides and mRNA therapeutics. In: Crommelin DJA, Sindelar RD, Meibohm B, eds. Pharmaceutical Biotechnology: Fundamentals and Applications. Springer International Publishing; 2024:291‐321. [Google Scholar]
- 5. Villiger L, Joung J, Koblan L, Weissman J, Abudayyeh OO, Gootenberg JS. CRISPR technologies for genome, epigenome and transcriptome editing. Nat Rev Mol Cell Biol. 2024;25:464‐487. doi: 10.1038/s41580-023-00697-6 [DOI] [PubMed] [Google Scholar]
- 6. Khvorova A, Watts JK. The chemical evolution of oligonucleotide therapies of clinical utility. Nat Biotechnol. 2017;35:238‐248. doi: 10.1038/nbt.3765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sahin U, Karikó K, Türeci Ö. mRNA‐based therapeutics—developing a new class of drugs. Nat Rev Drug Discov. 2014;13:759‐780. doi: 10.1038/nrd4278 [DOI] [PubMed] [Google Scholar]
- 8. Belgrad J, Fakih HH, Khvorova A. Nucleic acid therapeutics: successes, milestones, and upcoming innovation. Nucleic Acid Ther. 2024;34:52‐72. doi: 10.1089/nat.2023.0068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hammond SM, Aartsma‐Rus A, Alves S, et al. Delivery of oligonucleotide‐based therapeutics: challenges and opportunities. EMBO mol Med. 2021;13:e13243. doi: 10.15252/emmm.202013243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Callaway E, Naddaf M. Pioneers of mRNA COVID vaccines win medicine Nobel. Nature. 2023;622:228‐229. doi: 10.1038/d41586-023-03046-x [DOI] [PubMed] [Google Scholar]
- 11. Prakash TP, Graham MJ, Yu J, et al. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N‐acetyl galactosamine improves potency 10‐fold in mice. Nucleic Acids Res. 2014;42:8796‐8807. doi: 10.1093/nar/gku531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Akinc A, Querbes W, De S, et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand‐based mechanisms. Mol Ther. 2010;18:1357‐1364. doi: 10.1038/mt.2010.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gebhardt R, Matz‐Soja M. Liver zonation: novel aspects of its regulation and its impact on homeostasis. World J Gastroenterol. 2014;20:8491‐8504. doi: 10.3748/wjg.v20.i26.8491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Truong B, Allegri G, Liu X‐B, et al. Lipid nanoparticle‐targeted mRNA therapy as a treatment for the inherited metabolic liver disorder arginase deficiency. Proc Natl Acad Sci USA. 2019;116:21150‐21159. doi: 10.1073/pnas.1906182116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yamazaki K, Kubara K, Ishii S, et al. Lipid nanoparticle‐targeted mRNA formulation as a treatment for ornithine‐transcarbamylase deficiency model mice. Mol Ther Nucleic Acids. 2023;33:210‐226. doi: 10.1016/j.omtn.2023.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Prieve MG, Harvie P, Monahan SD, et al. Targeted mRNA therapy for ornithine transcarbamylase deficiency. Mol Ther. 2018;26:801‐813. doi: 10.1016/j.ymthe.2017.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martinez‐Pizarro A, Desviat LR. RNA solutions to treat inborn errors of metabolism. Mol Genet Metab. 2022;136:289‐295. doi: 10.1016/j.ymgme.2022.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kp A, Kaliaperumal K, Sekar D. microRNAs and their therapeutic strategy in phase I and phase II clinical trials. Epigenomics. 2024;16:259‐271. doi: 10.2217/epi-2023-0363 [DOI] [PubMed] [Google Scholar]
- 19. Koeberl D, Schulze A, Sondheimer N, et al. Interim analyses of a first‐in‐human phase 1/2 mRNA trial for propionic acidaemia. Nature. 2024;628:872‐877. doi: 10.1038/s41586-024-07266-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yoon S, Rossi JJ. Therapeutic potential of small activating RNAs (saRNAs) in human cancers. Curr Pharm Biotechnol. 2018;19:604‐610. doi: 10.2174/1389201019666180528084059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Albers S, Allen EC, Bharti N, et al. Engineered tRNAs suppress nonsense mutations in cells and in vivo. Nature. 2023;618:842‐848. doi: 10.1038/s41586-023-06133-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gillmore JD, Gane E, Taubel J, et al. CRISPR‐Cas9 in vivo gene editing for transthyretin amyloidosis. N Engl J Med. 2021;385:493‐502. doi: 10.1056/NEJMoa2107454 [DOI] [PubMed] [Google Scholar]
- 23. Jalil S, Keskinen T, Juutila J, et al. Genetic and functional correction of argininosuccinate lyase deficiency using CRISPR adenine base editors. Am J Hum Genet. 2024;111:714‐728. doi: 10.1016/j.ajhg.2024.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Longhurst HJ, Lindsay K, Petersen RS, et al. CRISPR‐Cas9 in vivo gene editing of KLKB1 for hereditary angioedema. N Engl J Med. 2024;390:432‐441. doi: 10.1056/NEJMoa2309149 [DOI] [PubMed] [Google Scholar]
- 25. Villiger L, Rothgangl T, Witzigmann D, et al. In vivo cytidine base editing of hepatocytes without detectable off‐target mutations in RNA and DNA. Nat Biomed Eng. 2021;5:179‐189. doi: 10.1038/s41551-020-00671-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baruteau J, Brunetti‐Pierri N, Gissen P. Liver‐directed gene therapy for inherited metabolic diseases. J Inherit Metab Dis. 2024;47:9‐21. doi: 10.1002/jimd.12709 [DOI] [PubMed] [Google Scholar]
- 27. Crooke ST, Baker BF, Crooke RM, Liang X‐H. Antisense technology: an overview and prospectus. Nat Rev Drug Discov. 2021;20:427‐453. doi: 10.1038/s41573-021-00162-z [DOI] [PubMed] [Google Scholar]
- 28. Havens MA, Hastings ML. Splice‐switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016;44:6549‐6563. doi: 10.1093/nar/gkw533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kuijper EC, Bergsma AJ, Pijnappel WWMP, Aartsma‐Rus A. Opportunities and challenges for antisense oligonucleotide therapies. J Inherit Metab Dis. 2021;44:72‐87. doi: 10.1002/jimd.12251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hodges PE, Rosenberg LE. The spfash mouse: a missense mutation in the ornithine transcarbamylase gene also causes aberrant mRNA splicing. Proc Natl Acad Sci USA. 1989;86:4142‐4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rivera‐Barahona A, Sánchez‐Alcudia R, Viecelli HM, et al. Functional characterization of the spf/ash splicing variation in OTC deficiency of mice and man. PLoS One. 2015;10:e0122966. doi: 10.1371/journal.pone.0122966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martínez‐Pizarro A, Leal F, Holm LL, et al. Antisense oligonucleotide Rescue of Deep‐Intronic Variants Activating Pseudoexons in the 6‐Pyruvoyl‐Tetrahydropterin synthase gene. Nucleic Acid Ther. 2022;32:378‐390. doi: 10.1089/nat.2021.0066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Perez B, Vilageliu L, Grinberg D, Desviat LR. Antisense mediated splicing modulation for inherited metabolic diseases: challenges for delivery. Nucleic Acid Ther. 2014;24:48‐56. doi: 10.1089/nat.2013.0453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Spangsberg Petersen US, Dembic M, Martínez‐Pizarro A, et al. Regulating PCCA gene expression by modulation of pseudoexon splicing patterns to rescue enzyme activity in propionic acidemia. Mol Ther—Nucl Acids. 2024;35:102101. doi: 10.1016/j.omtn.2023.102101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vaz‐Drago R, Custodio N, Carmo‐Fonseca M. Deep intronic mutations and human disease. Hum Genet. 2017;136:1093‐1111. doi: 10.1007/s00439-017-1809-4 [DOI] [PubMed] [Google Scholar]
- 36. Dhir A, Buratti E. Alternative splicing: role of pseudoexons in human disease and potential therapeutic strategies. FEBS J. 2010;277:841‐855. [DOI] [PubMed] [Google Scholar]
- 37. Lykke‐Andersen S, Jensen TH. Nonsense‐mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol. 2015;16:665‐677. doi: 10.1038/nrm4063 [DOI] [PubMed] [Google Scholar]
- 38. Perez B, Rodriguez‐Pascau L, Vilageliu L, et al. Present and future of antisense therapy for splicing modulation in inherited metabolic disease. J Inherit Metab Dis. 2010;33:397‐403. doi: 10.1007/s10545-010-9135-1 [DOI] [PubMed] [Google Scholar]
- 39. Cummings BB, Marshall JL, Tukiainen T, et al. Improving genetic diagnosis in mendelian disease with transcriptome sequencing. Sci Transl Med. 2017;9:eaal5209. doi: 10.1126/scitranslmed.aal5209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gonorazky HD, Naumenko S, Ramani AK, et al. Expanding the boundaries of RNA sequencing as a diagnostic tool for rare mendelian disease. Am J Hum Genet. 2019;104:1007. doi: 10.1016/j.ajhg.2019.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Truty R, Ouyang K, Rojahn S, et al. Spectrum of splicing variants in disease genes and the ability of RNA analysis to reduce uncertainty in clinical interpretation. Am J Hum Genet. 2021;108:696‐708. doi: 10.1016/j.ajhg.2021.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Engel K, Nuoffer JM, Muhlhausen C, et al. Analysis of mRNA transcripts improves the success rate of molecular genetic testing in OTC deficiency. Mol Genet Metab. 2008;94:292‐297. doi: 10.1016/j.ymgme.2008.03.009 [DOI] [PubMed] [Google Scholar]
- 43. Kumar RD, Burrage LC, Bartos J, et al. A deep intronic variant is a common cause of OTC deficiency in individuals with previously negative genetic testing. Mol Genet Metab Rep. 2021;26:100706. doi: 10.1016/j.ymgmr.2020.100706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ogino W, Takeshima Y, Nishiyama A, et al. Mutation analysis of the ornithine transcarbamylase (OTC) gene in five Japanese OTC deficiency patients revealed two known and three novel mutations including a deep intronic mutation. Kobe J Med Sci. 2007;53:229‐240. [PubMed] [Google Scholar]
- 45. Zabulica M, Srinivasan RC, Akcakaya P, et al. Correction of a urea cycle defect after ex vivo gene editing of human hepatocytes. Mol Ther. 2021;29:1903‐1917. doi: 10.1016/j.ymthe.2021.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sangermano R, Garanto A, Khan M, et al. Deep‐intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet Med. 2019;21:1751‐1760. doi: 10.1038/s41436-018-0414-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang F, Zuroske T, Watts JK. RNA therapeutics on the rise. Nat Rev Drug Discov. 2020;19:441‐442. doi: 10.1038/d41573-020-00078-0 [DOI] [PubMed] [Google Scholar]
- 48. Damase TR, Sukhovershin R, Boada C, Taraballi F, Pettigrew RI, Cooke JP. The limitless future of RNA therapeutics. Front Bioeng Biotechnol. 2021;9:628137. doi: 10.3389/fbioe.2021.628137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hou X, Zaks T, Langer R, Dong Y. Lipid nanoparticles for mRNA delivery. Nat Rev Mater. 2021;6:1078‐1094. doi: 10.1038/s41578-021-00358-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cao J, An D, Galduroz M, et al. mRNA therapy improves metabolic and behavioral abnormalities in a murine model of Citrin deficiency. Mol Ther. 2019;27:1242‐1251. doi: 10.1016/j.ymthe.2019.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Daly O, Mahiny AJ, Majeski S, et al. ASL mRNA‐LNP therapeutic for the treatment of Argininosuccinic aciduria enables survival benefit in a mouse model. Biomedicine. 2023;11:1735. doi: 10.3390/biomedicines11061735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gurung S, Timmermand OV, Perocheau D, et al. mRNA therapy corrects defective glutathione metabolism and restores ureagenesis in preclinical argininosuccinic aciduria. Sci Transl Med. 2024;16:eadh1334. doi: 10.1126/scitranslmed.adh1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Khoja S, Liu X‐B, Truong B, et al. Intermittent lipid nanoparticle mRNA administration prevents cortical dysmyelination associated with arginase deficiency. Mol Ther Nucleic Acids. 2022;28:859‐874. doi: 10.1016/j.omtn.2022.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Perocheau D, Gurung S, Touramanidou L, et al. Ex vivo precision‐cut liver slices model disease phenotype and monitor therapeutic response for liver monogenic diseases. F1000Res. 2023;12:1580. doi: 10.12688/f1000research.142014.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yu H, Brewer E, Shields M, et al. Restoring ornithine transcarbamylase (OTC) activity in an OTC‐deficient mouse model using LUNAR‐OTC mRNA. Clin Transl Discov. 2022;2:e33. doi: 10.1002/ctd2.33 [DOI] [Google Scholar]
- 56. Yang Y, Wang L, Bell P, et al. A dual AAV system enables the Cas9‐mediated correction of a metabolic liver disease in newborn mice. Nat Biotechnol. 2016;34:334‐338. doi: 10.1038/nbt.3469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lee PC, Truong B, Vega‐Crespo A, et al. Restoring ureagenesis in hepatocytes by CRISPR/Cas9‐mediated genomic addition to arginase‐deficient induced pluripotent stem cells. Mol Ther Nucleic Acids. 2016;5:e394. doi: 10.1038/mtna.2016.98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nitzahn M, Truong B, Khoja S, et al. CRISPR‐mediated genomic addition to CPS1 deficient iPSCs is insufficient to restore nitrogen homeostasis. Yale J Biol Med. 2021;94:545‐557. [PMC free article] [PubMed] [Google Scholar]
- 59. Wang L, Yang Y, Breton C, et al. A mutation‐independent CRISPR‐Cas9‐mediated gene targeting approach to treat a murine model of ornithine transcarbamylase deficiency. Sci Adv. 2020;6:eaax5701. doi: 10.1126/sciadv.aax5701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ginn SL, Christina S, Alexander IE. Genome editing in the human liver: Progress and translational considerations. Prog Mol Biol Transl Sci. 2021;182:257‐288. doi: 10.1016/bs.pmbts.2021.01.030 [DOI] [PubMed] [Google Scholar]
- 61. Vlatkovic I. Non‐immunotherapy application of LNP‐mRNA: maximizing efficacy and safety. Biomedicine. 2021;9:530. doi: 10.3390/biomedicines9050530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Masarwy R, Stotsky‐Oterin L, Elisha A, Hazan‐Halevy I, Peer D. Delivery of nucleic acid based genome editing platforms via lipid nanoparticles: clinical applications. Adv Drug Deliv Rev. 2024;211:115359. doi: 10.1016/j.addr.2024.115359 [DOI] [PubMed] [Google Scholar]
- 63. Adlat S, Vázquez Salgado AM, Lee M, Yin D, Wangensteen KJ. Emerging and potential use of CRISPR in human liver disease. Hepatology. 2023. doi: 10.1097/HEP.0000000000000578 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
My manuscript has no associated data.