

ABSTRACT
Background
Congenital ocular anomalies (COA) are among the most common causes of visual impairment in children in high‐income countries. The aim of the study is to describe the prevalence of the various COA recorded in European population‐based registries of CA (EUROCAT) participating in the EUROmediCAT consortium.
Methods
Data from 19 EUROmediCAT registries and one healthcare database (EFEMERIS) were included in this descriptive epidemiological study. Cases of COA included live births, FD from 20 weeks gestational age (GA), and termination of pregnancy for fetal anomaly.
Results
The prevalence of total COA was 3.47/10,000 births (95% CI [3.61–3.82]), ranging from 1.41 to 13.46/10,000 depending on the registry. Among COA cases, congenital lens anomalies were the most frequent anomalies (31%), of which over half were single ocular anomalies (presenting with only one ocular anomaly). An/microphthalmia was the second most frequent COA (24%) of which three‐quarters were multiply malformed (associated to extraocular major anomalies). Among single COA cases, 58 were prenatally diagnosed (4%), of which, 58% were diagnosed in the second trimester. Known genetic causes of COA explained 2.5%–25% of COA depending on their class.
Conclusions
This is the first European study describing COA. The detailed prevalence data offered in this study could improve screening and early diagnosis of different classes of COA. As COA are rare, epidemiological surveillance of large populations and accurate clinical descriptions are essential.
Keywords: congenital ocular anomalies, descriptive epidemiological study, epidemiology, Europe, ocular defect
1. Introduction
Data on vision impairment in children are scarce. Worldwide, the leading causes of blindness and moderate to severe vision impairment in children include uncorrected refractive error, cataract, retinopathy of prematurity, congenital ocular anomalies (COA), corneal scarring, and cerebral visual impairment (Burton et al. 2021). COA are among the most common causes of visual impairment in children in developed countries and it has been shown that, for children in higher income countries, cerebral visual impairment and optic nerve anomalies remain the most common causes of severe visual impairment and blindness (Solebo, Teoh, and Rahi 2017). COA detected prenatally or in infancy have rarely been studied: prevalence is estimated between 2.4 and 7.5 per 10,000 births (Stoll et al. 1992; Campbell et al. 2002; Stallings et al. 2018). Some COA have a genetic origin and are most often part of well‐defined genetic syndromes such as CHARGE syndrome (Hsu et al. 2014). However, while a minority of COA has a clear genetic origin, for most patients, the etiology is unknown (Plaisancie et al. 2019, 2018; George, Cogliati, and Brooks 2020; Scanga and Nischal 2014). Moreover, in the past, COA were detected and recorded later, at birth or during the first years of life and registries did not record COA associated with stillbirths or terminations of pregnancy that went undetected (Ondeck et al. 2018). COA can now be detected prenatally by ultrasound and significant advances in ultrasound performance allow the detection of more precise and specific anomalies now reported in congenital anomaly registries and healthcare databases (Ondeck et al. 2018). Furthermore, recent papers demonstrated an interest in ocular monitoring in the antenatal period to optimize diagnostic accuracy, families' expectations, and early treatment (Ondeck et al. 2018; Searle et al. 2018).
The aim of this study is to provide an overview of the epidemiology of the various COA in Europe based on routinely collected data from congenital anomaly registries.
2. Methods
The MEDIKEYE study is a multicentric European study of COA and their causes. This first paper describes the epidemiology of COA, its prevalence, and time of diagnosis. Nineteen population‐based registries that collect information on medication exposures during the first trimester of pregnancy in the EUROmediCAT consortium participated (https://www.euromedicat.eu/). One healthcare database (EFEMERIS, France; http://www.efemeris.fr/) was also included. Each case has at least one major congenital anomaly defined according to the EUROCAT Guide 1.4, using the International Classification of Diseases version 9 (ICD9) or 10 (ICD10) with British Pediatric Association (BPA) extension codes. COA can be classified into: congenital anomalies (CA) of the anterior segment including congenital lens anomalies and congenital glaucoma; eyeball growth and formation defect (anophthalmia/microphthalmia, macrophthalmia, and colobomas); CA of the posterior segment; CA of the eyelids, lacrimal apparatus, and orbit; and dysplasia of the septum and optic tract. We thus categorized COA into different classes according to their ICD Q codes, common embryological origin, and the anatomical structure involved (Table S1).
The MEDIKEYE study has two parts: MEDIKEYE1 reports prevalence and distribution of COA cases across Europe, while MEDIKEYE2 reports on etiologic factors of COA such as perinatal and maternal factors and in utero medication exposure. We present here the results of MEDIKEYE1.
2.1. Study Population
13,607,649 births between 1995 and 2019 were included in the study (Table 1). The study included all cases of major CA among live births (LB), fetal deaths of ≥ 20 completed weeks of gestation (FD), and terminations of pregnancy for fetal anomalies (TOPFA) at any GA. Most registries include cases diagnosed up to 1 year after birth.
TABLE 1.
Prevalence (per 10,000 births) of congenital ocular anomaly (COA) in the studied population by COA class and distribution of congenital ocular anomaly (COA) classes and respective known genetic percentage for each COA class (period: 1995–2019).
| Prevalence Rate MEDIKEYE | Range | Total (%) | Total (%) | % no genetic | % known genetic cause | |
|---|---|---|---|---|---|---|
| Total COAs | 3.71 [95% CI 3.61–3.82] | [1.41–13.46] | N = 6011 | N = 5052 (total number of cases) | Cause known | |
| (total number of anomalies) | ||||||
| Class 1: anophthalmia/microphthalmia | 0.89 [95% CI 0.84–0.94] | [0.38–2.32] | 1231 (20.5%) | 1206 (23.9%) | 75.3 | 24.7 |
| Class 2: congenital cataract and lens anomalies | 1.16 [95% CI: 1.1–1.22] | [0.21–4.15] | 1595 (26.5%) | 1578 (31.2%) | 88.0 | 12.0 |
| Class 3: macrophthalmia | 0.01 [95% CI: 0.01–0.02] | [0.00–0.05] | 18 (0.3%) | 18 (0.4%) | 77.8 | 22.2 |
| Class 4: congenital anomalies of the anterior segment | 0.76 [95% CI: 0.72–0.81] | [0.24–2.80] | 1109 (18.4%) | 1040 (20.6%) | 81.7 | 18.3 |
| Class 5: congenital anomalies of the posterior segment | 0.47 [95% CI: 0.43–0.5] | [0.06–1.91] | 694 (11.5%) | 635 (12.5%) | 82.0 | 18.0 |
| Class 6: congenital glaucoma | 0.28 [95% CI: 0.26–0.31] | [0.06–1.63] | 396 (6.6%) | 387 (7.7%) | 91.2 | 8.8 |
| Class 7: congenital anomalies of the eyelids, lacrimal apparatus, and orbit | 0.54 [95% CI: 0.51–0.59] | [0.10–3.65] | 754 (12.5%) | 741 (14.7%) | 82.5 | 17.5 |
| Class 8: unspecified ocular anomalies | 0.07 [95% CI: 0.05–0.08] | [0.00–0.57] | 92 (1.5%) | 92 (1.8%) | 84.8 | 15.2 |
| Class 9: dysplasia of the septum and optic tract | 0.09 [95% CI: 0.07–0.11] | [0.00–0.66] | 122 (2.0%) | 122 (2.4%) | 97.5 | 2.5 |
| Class 10: colobomas | 0.48 [95% CI: 0.45–0.52] | [0.11–1.96] | 768 (12.8%) | 659 (13.0%) | 78.6 | 21.4 |
| Total births | 13,606,926 | Total COA | 5052 | 4217 (83.5%) | 835 (16.5%) | |
| Subclass: single or complex without associated extra ocular CA | 2406 | 2400 (99.75%) | 6 (0.25%) | |||
| Subclass “ASSOCIATED” (whether single or complex) | 2646 | 1803 (68.2) | 843 (31.8%) | |||
2.2. Covariates
Information (according to the EUROCAT chapter Guide 1.4) on pregnancy and infant characteristics, demographic factors, and socioeconomic status of mother were analyzed. Clinical variables included: sex of the fetus/baby, GA in completed weeks (WGA) (< 28, [28–31], [32–36], [37–41], ≥ 42 weeks), birth outcome (LB, FD, TOPFA, plurality, gravidity, parity, year of birth/termination, maternal age class, birth weight, age at diagnosis of the anomaly). Not all variables were available for all registries and for all birth years included. Registry‐specific variables with > 20% missing data were excluded from the analysis (detailed in Table S2).
2.3. Definition and Classification of COA
The EUROmediCAT coding instructions ensure that standard variables, definitions, and coding are used by all registries in the network. All birth outcomes with a code in the “eye anomaly” subgroup were included in the study. Since optic nerve anomalies contribute largely to blindness, the code Q04.4 (Septo‐optic dysplasia of brain) was added (Campbell 2003). Subsequently, eye anomalies were classified into nine classes according to their ICD Q codes, common embryological origin, and the anatomical structure involved (Table S1). We divided class Q11 into two classes: an/microphthalmia (Q11.1 and Q11.2, respectively) and macrophthalmia (Q11.3). We added a separate class for colobomas defined by either code Q12.2 or Q13.0 or the search keyword “colobom” in the free‐text field. After review, the eyelid colobomas were excluded due to different embryologic origin leaving only intraocular colobomas (Tawfik, Abdulhafez, and Fouad 2015; Lingam et al. 2021). When two associated colobomas coexisted (i.e., iris and retina), we considered it as one single defect.
For corneal and iris anomalies, we added two subclasses: (a) corneal disorders (codes Q13.3, Q13.4 plus text description (“corne”)); (b) iris anomalies (codes Q13.0, Q13.1, Q13.2 plus text description (“iris,” “irid”)).
A total of 163 cases were coded as “unspecified” (Q15.8 and Q15.9). Of these, we reclassified 140 cases based on the text description. Twenty‐three cases had no text description.
Cases were classified as single eye anomalies, genetic disorders, teratogenic, or multiple CA according to the EUROCAT multiple congenital anomaly flowchart followed by manual review of potential multiple cases (Garne et al. 2011).
COA cases, were further classified as single cases (only one COA), complex cases (more than one COA), and associated cases (at least one COA as well as other congenital anomalies(s)). Thus, COA cases were classified into four respective classes whether genetic or not.
Single ocular anomaly.
Single associated with extraocular malformation(s).
Complex ocular anomaly.
Complex associated with extraocular malformation(s).
Each case with a COA is counted once, despite the number of eye anomalies present. When a case has a complex COA with > 1 eye anomaly (i.e., anophthalmia and cataract), the malformation was counted in each class. We calculated the prevalence of COA either on the total number of cases or on the total number of COA whether genetic or not.
2.4. Genetic Anomalies
In addition to codes for genetic anomalies, the free‐text field was searched for one of the following terms “GENET,” “HERIT,” or “HEREDIT” and checked manually. Hence, all chromosomal anomalies and monogenic syndromes (whether hereditary or not) appeared in this group. In order to see if a genetic origin was increasing over time as knowledge evolves, we analyzed COA cases with a genetic anomaly by years.
2.5. Descriptive Analysis
We analyzed maternal data for all COA cases including genetic anomalies. In case of a twin pregnancy concordant for COA, we counted it as one case in analyses of maternal characteristics.
Age at discovery was analyzed for cases with single eye anomalies and divided into:
Antenatal period.
At birth.
After birth.
The antenatal period was further classified as first trimester (< 14 WGA), second trimester (14–27 WGA), and third trimester (≥ 28 WGA).
We used estimated fetal weight (EFW) from World Health Organization (WHO) curves related to GA to calculate percentiles (Kiserud et al. 2017). EFW were separate into:
≤ 3rd percentile.
Between the 3rd and the 97th percentile.
≥ 97th percentile.
All cases under 14 WGA were excluded as no reference weight curves were available (n = 12 cases), as well as unknown age cases. The curve of 40 WGA was used for cases aged over 40 WGA. Class 1 and 3 were then analyzed for the distribution in each COA class.
Class 1 was further divided into:
< 1st percentile.
≥ 1st percentile and < 3rd percentile.
2.6. Statistical Analysis
Descriptive statistics used to analyze data were mean (± standard deviation and range [min–max]) for continuous variables and number and percentage for categorical variables. The distribution of each COA was analyzed by year to see changes over time.
To estimate the total prevalence of fetuses/infants with at least one COA in each registry, we used the following:
The prevalence of COA was described by class of COA, by registry, and by year with 95% confidence intervals. Prevalence of COA by year was then compared with the mean EUROCAT prevalence with and without genetic anomalies.
3. Results
A total of 19 registries and one health databases in 15 European countries, contributed 13,607,649 births. The total number of CA cases was 298,405 of which 5052 were COA cases. The 5052 COA cases presented a total of 6011 COA malformations. The total prevalence of COA in our study was 3.71 per 10,000 births (Table 1). Prevalence by registry ranged from 1.41 to 13.46/10,000 births (Table 2, Table S3). The highest prevalence of COA was seen in Wales (13.46/10,000) and the Northern Netherlands (9.77/10,000). Poland had the lowest prevalence of COA (1.41/10,000). Wales had the highest prevalence for all COA classes except for ano/microphthalmia for which Funen (Denmark) had the highest prevalence (2.32/10,000), while the prevalence of an/microphthalmia for all registries combined was 0.89/10,000.
TABLE 2.
Participating registries in the study with cases of congenital ocular anomalies (COA) (period: 1995–2019).
| Center | Years covered | Births covered | Number of COA cases | Prevalence (rate per 10,000 births) |
|---|---|---|---|---|
| Participating European registries | ||||
| Belgium, Antwerp | 1995–2017 | 430,623 | 259 | 6.01 [95% CI: 5.31–6.81] |
| Croatia, Zagreb | 1995–2017 | 142,525 | 31 | 2.18 [95% CI: 1.5–3.13] |
| Denmark, Funen | 1995–2019 | 129,072 | 102 | 7.90 [95% CI: 6.48–9.63] |
| France, Auvergne | 2011–2019 | 117,964 | 82 | 6.95 [95% CI: 5.56–8.67] |
| France, Brittany | 2011–2019 | 308,449 | 182 | 5.90 [95% CI: 5.09–6.84] |
| France, Isle of Reunion | 2005–2018 | 201,889 | 142 | 7.03 [95% CI: 5.95–8.32] |
| France, Paris | 2001–2017 | 445,975 | 172 | 3.86 [95% CI: 3.31–4.49] |
| Germany, Saxony‐Anhalt | 2000–2019 | 348,659 | 129 | 3.70 [95% CI: 3.1–4.41] |
| Ireland, Cork and Kerry | 1996–2019 | 213,581 | 133 | 6.23 [95% CI: 5.23–7.4] |
| Italy, Emilia Romagna | 1995–2019 | 839,295 | 326 | 3.88 [95% CI: 3.48–4.34] |
| Italy, Tuscany | 1995–2019 | 686,906 | 238 | 3.46 [95% CI: 3.04–3.94] |
| Malta | 1996–2019 | 102,481 | 44 | 4.29 [95% CI: 3.16–5.82] |
| Netherlands, Northern | 1995–2019 | 448,476 | 438 | 9.77 [95% CI: 8.88–10.74] |
| Poland, excluding Wielkopolska | 1999–2019 | 6,520,203 | 918 | 1.41 [95% CI: 1.32–1.5] |
| Poland, Wielkopolska | 1999–2019 | 782,301 | 186 | 2.38 [95% CI: 2.05–2.75] |
| Spain, Valencian Region | 2007–2019 | 578,025 | 336 | 5.81 [95% CI: 5.22–6.48] |
| Spain, Basque Country | 2005–2016 | 242,049 | 151 | 6.24 [95% CI: 5.3–7.34] |
| Switzerland, Vaud | 1997–2019 | 180,513 | 96 | 5.32 [95% CI: 4.33–6.52] |
| UK, Wales | 1998–2019 | 729,454 | 982 | 13.46 [95% CI: 12.64–14.34] |
| Participating health care database | ||||
| France, EFEMERIS | 2005–2019 | 158,486 | 105 | 6.63 [95% CI: 5.45–8.05] |
| Total | 1995–2019 | 13,606,926 | 5052 | 3.71 [95% CI: 3.61–3.82] |
As registry‐specific variables with more than 20% missing data were excluded from the analysis, as detailed in Table S2, some variables could not be analyzed satisfactorily, notably maternal illness before and during pregnancy and sociodemographic data. Consequently, the interpretation is limited, and potential differences across Europe, between registries, and between different COA classes cannot be discussed.
3.1. COA Distribution
The COA distribution by year (with and without known genetic cause) was analyzed for the homogeneity of our population, as shown in Figure 1. The distribution of the different COA is reported in Table 2. The most frequent COA is congenital cataract (CC) and lens anomalies that account for 31.2% of COA cases. The second most frequent COA is an/microphthalmia which concerned 23.8% of COA cases. CA of the anterior segment account for 20.6% of COA cases, while the frequency of the overall colobomas was 13%.
FIGURE 1.
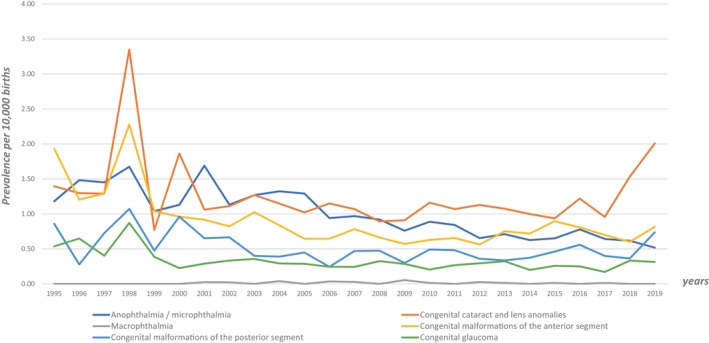
Prevalence (per 10,000 births) of congenital ocular anomaly (COA) with and without genetic anomalies over time (red curve: COA prevalence excluding genetic anomalies; blue curve: COA prevalence including genetic anomalies).
3.2. Genetic Contribution
For all classes of COA, a genetic origin was not reported in the majority of cases (83.5%), ranging from 75.3% to 97.5% depending on the COA class. A genetic origin was recorded most often for an/microphthalmia for which it explains 24.7% of cases. The contribution of a known genetic cause was 2.5% in dysplasia of the septum and optic tract cases. Overall, a genetic origin was specified in around 20% of cases for: macrophtalmia (22.2%), CA of anterior segment (18.3%), CA of posterior segment (18.0%), and colobomas (21.4%). No increase in specific COA classes with genetic origin was observed over time. Among the overall COA cases, only 0.25% of the single or complex COA had a genetic origin reported, whereas 31.8% of COA associated to at least another major extraocular defect had a genetic origin reported. We can thus observe that multiply malformed COA are more likely to have a known genetic cause (either chromosomal or monogenic).
3.3. Single, Associated‐Single, Complex, or Associated‐Complex COA Subclasses
Three‐quarters (74%) of an/microphthalmia are multiple congenital anomaly cases whereas more than half (55.7%) of CC and lens anomalies (Class 2) cases are single (Table 3). In the same way, almost half (47.5%) of congenital glaucoma are single. Around 60% of cases of dysplasia of the septum and optic tract are complex.
TABLE 3.
Distribution of each congenital ocular anomaly (COA) class in the four respective subclasses: (i) single, (ii) associated single, (iii) complex, (iv) associated complex (period: 1995–2019).
| Classes of COA | Results (%) |
|---|---|
| N = 5052 (number of cases) | |
| Anophthalmia/microphthalmia | |
| 166 single (13.8%) | 1206 (23.9%) |
| 659 associated single (54.6%) | |
| 147 complex (12.2%) | |
| 234 associated complex (19.4%) | |
| Congenital cataract and lens anomalies | |
| 878 single (55.7%) | 1578 (31.2%) |
| 403 associated single (25.5%) | |
| 149 complex (9.4%) | |
| 148 associated complex (9.4%) | |
| Congenital malformations of the anterior segment | |
| 302 single (29.0%) | 1040 (20.6%) |
| 361 associated single (34.7%) | |
| 159 complex (15.3%) | |
| 218 associated complex (21.0%) | |
| Congenital malformations of the posterior segment | |
| 123 single (19.4%) | 635 (12.6%) |
| 225 associated single (35.4%) | |
| 115 complex (18.1%) | |
| 172 associated complex (27.1%) | |
| Congenital glaucoma | |
| 184 single (47.5%) | 387 (7.7%) |
| 113 associated single (29.2%) | |
| 56 complex (14.5%) | |
| 34 associated complex (8.8%) | |
| Dysplasia of the septum and optic tract | |
| 36 single (29.5%) | 122 (2.4%) |
| 12 associated single (9.8%) | |
| 72 complex (59.1%) | |
| 2 associated complex (1.6%) | |
| Coloboma | |
| 131 single (19.9%) | 659 (13.0%) |
| 256 associated single (38.8%) | |
| 102 complex (15.5%) | |
| 170 associated complex (25.8%) | |
Of the 2645 associated COA cases, the most frequent associated CA were congenital heart defects (34.03%) and CA of the nervous system (32.89%). In the single COA subgroup, the most frequent associated CA was congenital heart defect (34.51%), while in the complex COA subgroup, the most frequent associated CA concerned the nervous system (34.32%). Other CA, such as orofacial clefts (13.95%), ear, face, and neck CA (11.15%), digestive system CA (10.89%), and urinary CA (12.21%) were comparable in the three groups.
3.4. Fetal Sex
Among different COA classes, we found a trend toward more male cases for: macrophthalmia (66.7%), congenital glaucoma (59.3%), CA of the posterior segment (55.8%), and dysplasia of the septum and optic tract (54.1%), but with no significant difference (Table 4). The sex difference was significant between glaucoma versus the other overall COA (p = 0.016). Such difference was not observed for the other COA classes.
TABLE 4.
Demographic characteristics results and fetal weight analysis by gestational age.
| Demographic characteristics results | Median | Mean | Std dev | |
|---|---|---|---|---|
| Maternal age a (n = 4031) | ||||
| Min | 15 | 30 | 30.06 | 5.86 |
| Max | 50 | |||
| Length of gestation b (N = 3836) | ||||
| Min | 12 | 39 | 37.24 | 5.16 |
| Max | 44 | |||
| Total of pregnancy (gravidity) c (N = 3002) | ||||
| Min | 0 | 1 | 1.29 | 1.57 |
| Max | 19 | |||
| Fetal sex according to COA class | Results effectif (%) | |||
|---|---|---|---|---|
| Male | Female | Indeterminate | Not known | |
| Anophtalmia/microphthalmia | 598 (49.5%) | 586 (48.6%) | 7 (0.6%) | 15 (1.3%) |
| Congenital cataract and lens anomalies | 825 (52.3%) | 750 (47.5%) | 1 (0.1%) | 2 (0.1%) |
| Macrophthalmia | 12 (66.7%) | 6 (33.3%) | 0 (0%) | 0 (0%) |
| Congenital anomalies of the anterior segment | 558 (53.7%) | 480 (46.2%) | 0 (0%) | 2 (0.2%) |
| Congenital anomalies of the posterior segment | 354 (55.9%) | 278 (43.9%) | 0 (0%) | 1 (0.2%) |
| Congenital glaucoma | 229 (59.2%) | 158 (40.8%) | 0 (0%) | 0 (0%) |
| Dysplasia of the septum and optic pathways | 66 (54.1%) | 55 (45.1%) | 0 (0%) | 1 (0.8%) |
| Coloboma | 348 (52.8%) | 311 (47.2%) | 0 (0%) | 0 (0%) |
| Fetal weight analysis by gestational age (ALL) | |
|---|---|
| N = 4585 | |
| < 3rd percentile | 817 (17.8%) |
| 3rd–97th percentile | 3601 (78.5%) |
| > 97th percentile | 167 (3.6%) |
| Intrauterine fetal growth restriction (IUFR) subgroup (817) | |
| 603 < 1st percentile (73.8%) | |
| 214 1st–3rd percentile (26.2%) | |
| Distribution of weight < 3rd percentile for each COA class (N = 817) | |
| Class 1: anophtalmia/microphthalmia | 291 (35.6%) |
| Class 2: congenital cataract and lens anomalies | 195 (23.9%) |
| Class 3: macrophthalmia | 6 (0.7%) |
| Class 4: congenital anomalies of the anterior segment | 177 (21.7%) |
| Class 5: congenital anomalies of the posterior segment | 107 (13.1%) |
| Class 6: congenital glaucoma | 42 (5.1%) |
| Class 9: dysplasia of the septum and optic tract | 8 (1.0%) |
| Class 10: coloboma | 104 (12.7%) |
| Proportion of IUFR ion each COA class | |
| Class 1: anophtalmia/microphthalmia (N = 1106) | 291 (26.3%) |
| Class 2: congenital cataract and lens anomalies (N = 1407) | 195 (13.9%) |
| Class 3: macrophthalmia (N = 17) | 6 (35.3%) |
| Class 4: congenital anomalies of the anterior segment (N = 981) | 177 (18.0%) |
| Class 5: congenital anomalies of the posterior segment (N = 590) | 107 (18.1%) |
| Class 6: congenital glaucoma (N = 350) | 42 (12.0%) |
| Class 9: dysplasia of the septum and optic tract (N = 114) | 8 (7.0%) |
| Class 10: coloboma (N = 634) | 104 (16.4%) |
| Fetal weight analysis by gestational age in LIVE BIRTH only | |
|---|---|
| N = 4379 | |
| < 3rd percentile | 742 (16.9%) |
| 3rd–97th percentile | 3486 (79.6%) |
| > 97th percentile | 151 (3.5%) |
| IUFR subgroup (742) | |
| 542 < 1st percentile (73.1%) | |
| 200 1st–3rd percentile (26.9%) | |
| Distribution of weight < 3rd percentile for each COA class (N = 742) | |
| Class 1: anophtalmia/microphthalmia | 250 (33.7%) |
| Class 2: congenital cataract and lens anomalies | 185 (24.9%) |
| Class 3: macrophthalmia | 3 (0.4%) |
| Class 4: congenital anomalies of the anterior segment | 170 (22.9%) |
| Class 5: congenital anomalies of the posterior segment | 98 (13.2%) |
| Class 6: congenital glaucoma | 42 (5.7%) |
| Class 9: dysplasia of the septum and optic tract | 8 (1.1%) |
| Class 10: coloboma | 94 (12.7%) |
| Proportion of IUFR ion each COA class | |
| Class 1: anophtalmia/microphthalmia (N = 981) | 250 (25.5%) |
| Class 2: congenital cataract and lens anomalies (N = 1383) | 185 (13.4%) |
| Class 3: macrophthalmia (N = 14) | 3 (21.4%) |
| Class 4: congenital anomalies of the anterior segment (N = 964) | 170 (17.6%) |
| Class 5: congenital anomalies of the posterior segment (N = 566) | 98 (17.3%) |
| Class 6: congenital glaucoma (N = 349) | 42 (12.0%) |
| Class 9: dysplasia of the septum and optic tract (N = 105) | 8 (7.6%) |
| Class 10: coloboma (N = 611) | 94 (15.4%) |
| Fetal weight analysis by gestational age (other than livebirth) | |
|---|---|
| N = 206 | |
| < 3rd percentile | 75 (36.4%) |
| 3rd–97th percentile | 115 (55.8%) |
| > 97th percentile | 16 (7.8%) |
| IUFR subgroup (75) | |
| 61 < 1st percentile (81.3%) | |
| 14 1st–3rd percentile (18.7%) | |
| Distribution of weight < 3rd percentile for each COA class (N = 75) | |
| Class 1: anophtalmia/microphthalmia | 41 (54.7%) |
| Class 2: congenital cataract and lens anomalies | 10 (13.3%) |
| Class 3: macrophthalmia | 3 (4.0%) |
| Class 4: congenital anomalies of the anterior segment | 7 (9.3%) |
| Class 5: congenital anomalies of the posterior segment | 9 (12.0%) |
| Class 6: congenital glaucoma | 0 (0%) |
| Class 9: dysplasia of the septum and optic tract | 0 (0%) |
| Class 10: coloboma | 10 (13.3%) |
| Proportion of IUFR ion each COA class | |
| Class 1: anophtalmia/microphthalmia (N = 125) | 41 (32.8%) |
| Class 2: congenital cataract and lens anomalies (N = 24) | 10 (41.6%) |
| Class 3: macrophthalmia (N = 3) | 3 (100%) |
| Class 4: congenital anomalies of the anterior segment (N = 17) | 7 (41.2%) |
| Class 5: congenital anomalies of the posterior segment (N = 24) | 9 (37.5%) |
| Class 6: congenital glaucoma (N = 1) | 0 (0%) |
| Class 9: dysplasia of the septum and optic tract (N = 9) | 0 (0%) |
| Class 10: coloboma (N = 23) | 10 (43.5%) |
One registry excluded.
Two registers excluded.
Four registers excluded.
3.5. Fetal Weight
The fetal birth weight was analyzed for 4585 COA cases (Table 4). EFW was < 3rd percentile for 17.8% of the overall COA cases of which 73.8% were < 1st percentile. EFW was < 3rd percentile for 35.3% of macrophthalmia cases and 26.3% of an/microphthalmia cases, respectively. When we analyzed separately non‐livebirth (TOPFA/stillbirths), an intrauterine fetal growth restriction (IUGR) is found in 100% of macrophthalmia, 43.5% of colobomas, 41.6% of cases with CC and lens anomalies, and 41.2% CA of the anterior segment. Cases with an/microphthalmia account for 54.7% of all EFW < 5th percentile among the overall COA cases in non‐livebirth.
3.6. Time of Diagnosis
Among 1508 single COA cases combined, 58 had a prenatal diagnosis (3.9%), whereas 601 were diagnosed at birth (42.6%) and 849 after birth (53.5%) (Table 5). Among the 58 COA cases discovered antenatally, 36.2% were CC and lens anomalies, 27.6% were an/microphthalmia cases, 15.5% cases were dysplasia of the septum and optic tract, and 8.6% were CA of the posterior segment.
TABLE 5.
Time of the COA diagnosis.
| N = 1508 (cases with single COA) | ||||
|---|---|---|---|---|
| All cases (58 prenatal diagnosis (3.9%); 601 at birth (42.6%); 849 after birth (53.5%)) | Cases discovered at birth | Cases discovered antenatally | Cases discovered after birth | |
| N = 1508 | N = 601 | N = 58 | N = 849 | |
| Class 1: anophtalmia/microphthalmia | 98 (6.5%) | 63 (10.5%) | 16 (27.6%) | 19 (2.2%) |
| Class 2: congenital cataract and lens anomalies | 667 (44.2%) | 284 (47.2%) | 21 (36.2%) | 362 (42.6%) |
| Class 3: macrophthalmia | 1 (0.1%) | 0 (0%) | 0 (0%) | 1 (0.1%) |
| Class 4: congenital malformations of the anterior segment | 217 (14.4%) | 95 (15.8%) | 3 (5.2%) | 119 (14.0%) |
| Class 5: congenital malformations of the posterior segment | 92 (6.1%) | 15 (2.5%) | 5 (8.6%) | 72 (8.5%) |
| Class 6: congenital glaucoma | 149 (9.9%) | 71 (11.8%) | 0 (0%) | 78 (9.2%) |
| Class 7: congenital anomalies of eyelids lacrimal system and orbit | 248 (16.4%) | 67 (11.1%) | 4 (6.9%) | 177 (20.8%) |
| Class 8: unspecified ocular malformation | 9 (0.6%) | 6 (1%) | 0 (0%) | 3 (0.3%) |
| Class 9: dysplasia of the septum and optic pathways | 27 (1.8%) | 0 (0%) | 9 (15.5%) | 18 (2.1%) |
| Class 10: coloboma | 101 (6.7%) | 33 (5.5%) | 2 (3.4%) | 66 (7.8%) |
GA at discovery of the COA was available for 43 single COA cases with a mean age at diagnosis of 23.6 WGA. More than half were diagnosed at the second trimester ultrasound. The distribution of the diagnosis time was 11.6% (n = 5) in first trimester, 58.1% (n = 25) in second trimester, and 30.2% (n = 13) in third trimester. The five cases diagnosed before 14 WGA may have been linked to known family histories of anomalies in the prenatal diagnosis process (with a known genetic anomaly underlying the COA).
A total of 1011 single COA cases were discovered postnatally, of which, the majority (54.20%) were discovered between 1 and 12 months. About 792 cases were diagnosed before age 1 of which almost half of the cases were CCs and lens anomalies (45.08%) followed by CA of eyelids lacrimal system and orbit (16.29%) and CA of the anterior segment (16.16%). Hundred and eighty‐one single COA cases were diagnosed after 1 year of age, mainly CCs and lens anomalies (42.54%) and CA of eyelids lacrimal system and orbit (28.18%).
4. Discussion
In the present European study, the total overall COA prevalence was 3.72 per 10,000 births, which is in accordance with the literature (Stoll et al. 1992; Campbell et al. 2002; Stallings et al. 2018). The highest prevalence was found in Wales at 13.5/10,000 births, whereas the lowest one was 1.4/10,000 births found in Poland registry excluding Wielkopolska. Historically, Wales had to cope with an ecological disaster in the 1990's that could have had a direct impact on the number of CA in this country explaining this gap (Palmer et al. 2005). However, this occurred years before the CARIS registry was started, and no concern raised about a possible increase in ocular anomalies. Moreover, the registry has a scientific collaboration with the local ophthalmology team which enables to routinely report cases from South and West Wales, and their prevalence is likely to be higher due to better data sources for COA mainly of the less obvious COA. This highlights the difficulty in diagnosing and classifying rare CA such as COA and the importance of having an expert center.
We analyzed changes in prevalence over time for each class of COA (shown in Figure 2). The distribution of each malformation by year, showed a tendency for COA prevalence to increase over time for all classes possibly in relation with an increase in COA detection especially since 2017. This is particularly important for CC and lens anomalies. We can also observe a peak in 1998 for almost all classes that could partly be explained by the arrival of Wales in the dataset at that time. COA prevalence distribution over year in our study and in EUROCAT are relatively consistent and superimposable which suggest that results from the MEDIKEYE population can be extrapolated to the whole EUROCAT population. Prevalence rates by year of eye anomalies (using EUROCAT definition of eye anomalies) from all full EUROCAT registries (69 members registries) recorded from 1995 to 2019, are available on EUROCAT website (https://eu‐rd‐platform.jrc.ec.europa.eu/eurocat/eurocat‐data/prevalence_en).
FIGURE 2.
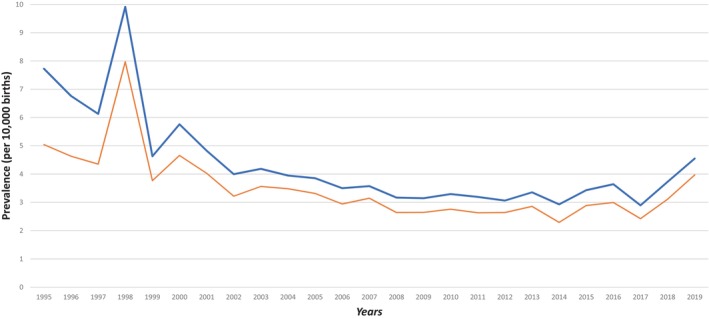
Prevalence per 10,000 births of specific class of congenital ocular anomaly (COA) over time, 1995–2019.
The large covered geographic area and the multiple data sources allowed us to study specific rare COA. Three‐quarters of an/microphthalmia cases were MCA, whereas more than half (55.7%) of CC and lens anomalies cases were single COA. While almost half (47.5%) of congenital glaucoma were single COA, around 60% of cases of dysplasia of the septum and optic tract are complex COA, but almost 90% of them are not associated to extraocular anomalies. These data are of particular interest since they could help clinicians in their clinical practice for the assessment of these specific COA. For example, upon the discovery of an anophthalmia or microphthalmia, a complete extraocular assessment could be carried out. These results will thus have practical implications for genetic and clinical management, both at the individual and community levels.
An IUGR concerned almost 20% of the overall COA cases of which 73.8% were severe < 1st percentile. These cases concerned more than 1/3 of the overall macrophtalmia (35.3%) and more than 1/4 of the overall an/microphthalmia (26.3%). In livebirth, 1/4 of an/microphtlmaia and 21.4% of macrophtalmia were respectively concerned by an IUGR. In livebirth, 17% of the overall COA cases had IUGR while in the non‐livebirth COA cases, 36.4% have IUGR. Among them, IUGR concerned respectively around 40% of CC and lens anomalies, CA of the anterior segment, and posterior segment and colobomas, and 100% of macrophthalmia cases but included only three cases. The fact that a COA is associated with an IUGR is an important factor because, it provides information on the associated/syndromic aspect of a COA in same way extraocular malformations do.
Concerning the diagnosis date (after birth, at birth, or in the antenatal period), the fact that many registries never reported prenatal diagnosis highlights the difficulty to detect COA in the prenatal period even though new antenatal imaging technologies are making considerable progress (Ondeck et al. 2018). However, we showed that many COA can be diagnosed prenatally, such as an/microphthalmia, lens anomalies, DSO, CA of annex and orbit, as well as congenital malformations of the posterior and anterior segments (Table 5). A better evaluation of different classes of COA will improve their detection and their timely management. Indeed, increasing our knowledge of the prenatally detectable COA, will allow their screening and thus their diagnosis performance.
Each COA class will be discussed and compared with what have previously been reported in the literature.
4.1. Anophthalmia/Microphthalmia
According to Schraw et al., an/microphthalmia accounts for approximately 10% of congenital blindness (Schraw et al. 2021). Epidemiologic studies suggest that > 20% of children with an/microphthalmia have a chromosomal abnormality, and around 50% have a pathogenic variant in one of the genes known to be involved in ano/microphthalmia. Chambers et al. reported that 48.2% of an/microphthalmia recorded in the Texas CA Registry from 1999 to 2009 were syndromic and that the most common diagnoses were trisomies 13 and 21 (Chambers et al. 2018). In their study, the overall prevalence of an/microphthalmia was relatively high at of 3.0 per 10,000 LB in contrast to our study were the prevalence was 0.98 per 10,000 births (whether LB or not). In our study, only 24.7% of an/microphthalmia cases with a genetic anomaly having either chromosomal abnormality or a pathogenic variant in one of the known genes involved were recorded. This could be explained by less recent cases for which genetic test was not yet accessible (e.g., 1995) or cases for which genetic diagnosis is made after birth and thus not recorded in the registries (many genetic conditions are diagnosed later in life, whereas the registries in this study mainly followed up children to 1 year of age). Consistent with previous reports, we observed that a large proportion of our cases diagnosed with an/microphthalmia have co‐occurring CA (74% of an/microphthalmia cases were associated with another extraocular major defect). While some of these defects may be part of a known sequence involving an/microphthalmia (e.g., central nervous system defects), other combinations could point to as yet undescribed susceptibility patterns. This is particularly interesting since, as pointed out by Schraw et al., these data show that population‐based congenital anomaly registries are particularly useful for accelerating the discovery of previously uncharacterized malformation syndromes.
4.2. Congenital Cataract and Lens Anomalies
CC is the primary cause of treatable childhood blindness worldwide (Wu et al. 2016). Wu et al. estimated the prevalence of CC at 4.24 per 10,000 people, which was higher than the prevalence found in our study (1.16 per 10,000 births). This is partly explained by the fact that we only have diagnosis at birth or during the first year of life in our study, in contrast to the meta‐analysis which reported CC at any age, and authors report that CC was usually diagnosed after 1 year of age (Wu et al. 2016). In another review, Sheeladevi et al. determined overall prevalence rates of 0.32–22.9 in 10,000 for childhood cataracts and 0.63–9.74 in 10,000 for CC (Sheeladevi et al. 2016). As suggested by authors, numerous factors such as the dynamic genetic infrastructure of societies, socioeconomic and cultural characteristics, access to health services, and the presence of early screening programs cause major differences in the prevalence of CC as well as associated morbidities between populations. Even across Europe, the prevalence of lens anomalies was heterogeneous and ranged from 0.21 to 4.15/10,000 depending on the different registries of our study. Although most cases of CC are described as idiopathic in literature (62%), the prevalence of inherited cataract was reported to be 22% (Taylan Sekeroglu and Utine 2021). In our study, the genetic basis of CC and lens anomalies was known in 12% of cases. As for ano/microphthalmia, this smaller number could be explained by the fact that diagnoses reported in European registries are made at birth or in the first year of life when the genetic diagnosis has not been undertaken or inclusion of cases from years when genetic tests were not yet accessible (e.g., 1995). In CC, anterior segment structures other than the lens are also shown to differ from eyes free of cataract, due to their simultaneous development and mutual interaction during the embryological period (Haargaard et al. 2004). CC may thus be associated with ocular anomalies such as microcornea, microphthalmia, persistent fetal vascularization, glaucoma, and retinal dystrophies. However, in our study, more than half of lens anomalies were Single (55.7%), whereas only 10.8% were complex that is to say associated with other ocular anomalies whether or not they have another extraocular congenital anomaly.
4.3. Macrophthalmia
Apart from the MACOM syndrome (MIM#602499), a rare genetic disease characterized by microcornea, coloboma of the iris and the optic disc, axial enlargement of the globe, staphyloma, and severe myopia with an estimated prevalence < 1/1,000,000, to our knowledge, no studies have yet described the epidemiological characteristics of macrophthalmia. In our study, the prevalence was 1 per 1,000,000 births, with only 18 cases reported. Among them, 66% were associated with another major extraocular defect, and only four cases (22.2%) were single.
4.4. CA Affecting the Anterior Segment
CA of the anterior segment show vast phenotypic and genetic heterogeneity (Reis and Semina 2011). These conditions are characterized by both autosomal dominant and recessive patterns of inheritance often with incomplete penetrance and variable expressivity. Although a number of genetic causes have been identified, many CA of the anterior segment are still of unknown origin as reflected by the only 18.3% of our CA affecting the anterior segment cases reported with a known genetic origin. Among our CA of the anterior segment cases, 55.7% were associated with extraocular defects, whereas 44.3% of them presented COA restricted to the eye (all cases combined whether genetic or not).
4.5. CA Affecting the Posterior Segment
While diseases of the optic nerve are coded elsewhere, the main conditions of the posterior segment included in ICD‐10 Q14 section are congenital malformation of vitreous humor, congenital malformation of retina, congenital malformation of optic disc, and congenital malformation of choroid. These different pathologies are highly heterogeneous and rarely described in the literature. Moreover, the major difficulty in data analysis and interpretation of COA, is that many of them overlap one another, which explains their high frequency in the “complex” classes of our study. Indeed, almost half of the overall CA of the posterior segment (45.2%) were “complex” cases, whether associated with extraocular defects or not. One example which illustrates it well is persistent hyperplastic primary vitreous (PHPV). This specific COA is a complex malformation of the eye characterized by the presence of remnants of the hyaloid and tunica vasculosa lentis systems of blood vessels, with proliferation of fibrovascular tissue behind the lens, and variable degrees of retinal dysplasia. The eye is generally microphthalmic but can also be enlarged. Although PHPV primarily affects the posterior part of the eye, the ciliary body, iris, and lens are involved to a variable extent. They are a common cause of CC. PHPV has also been reported in patients with various genetic diseases (i.e., recessive oculodento‐osseous dysplasia, protein C deficiency, oculopalatocerebral syndrome, or Norrie disease). Nevertheless, in our study the global prevalence was 0.47 per 10,000 births ranging from 0.06 to 1.91 depending on registries. Interestingly, 18% of the overall CA of the posterior segment had a known genetic anomaly, while 62.5% of them were associated with major extraocular defects.
4.6. Congenital Glaucoma
Primary congenital glaucoma (PCG) belongs to the anterior segment anomalies but is analyzed separately in our study as it is coded independently in the ICD‐10 classification. PCG is one of the leading causes of vision loss in children throughout the world. In the United States and Europe, it has been described to affect approximately 0.5–1 in 10,000 LB per year (Forestieri et al. 2019). In our study, its prevalence was 0.48 per 10,000 births. It has been previously reported that approximately 25% of infants with PCG are diagnosed at birth, and over 75% are diagnosed within the first year of life (Allingham, Liu, and Rhee 2009). These defects have been reported to affect males more often than females (Papadopoulos et al. 2007), which is fully in line with what we observed in our study, congenital glaucoma affected males in approximatively 60% of cases. The developmental pathogenesis of childhood glaucoma is variable, and several classification systems have been proposed. The 9th Consensus Report of the World Glaucoma Association classifies childhood glaucoma as either primary or secondary (Weinreb et al. 2013). PCG typically presents in the absence of co‐occurring ocular abnormalities, in contrast to secondary childhood glaucoma, that is pathogenetically heterogeneous and include anterior segment dysgenesis defects. Based on this definition, we could globally distinguish in our study PCG and secondary childhood glaucoma on the “single” or “complex” classes regardless of their extraocular associated defect. More than three‐quarters of the congenital glaucoma were in the single classes (whether associated to extraocular malformations or not) and thus probably PCG, whereas 23.3% of the congenital glaucoma were in the “complex” class, associated with other co‐occurring ocular malformations. It has been reported that PCG is usually no familial (60%–96%) even if PCG can sometimes be inherited (Papadopoulos et al. 2007), which suggests multifactorial causation. This is in accordance with what is observed in our study where only 8.8% of congenital glaucoma have a known genetic origin reported.
4.7. Dysplasia of the Septum and Optic Tract (DSO)
The incidence of the dysplasia of the septum and optic tract (DSO) has been reported to be 1 in 10,000 LB (Patel et al. 2006). In our study, the prevalence was 0.1 per 10,000 births ranging from 0.00 to 0.66. A previous study reported epidemiology data on DSO from the same network EUROCAT (Garne et al. 2018). Authors showed evidence of under‐reporting in some registries as the prevalence of DSO in Europe was likely to be between 1.9 and 2.5 per 100,000 births. The majority of DSO cases were classified as single cerebral anomalies (77%) and only two cases had a genetic diagnosis. Authors suggested that the anomaly may not be apparent at birth as reflected by the 57% of the postnatal diagnoses that occurred over 1 month after birth. In our study, the time of discovery was available for only 26 DSO cases of which, seven were discovered antenatally, and 19 after birth. No case of DSO was reported to be diagnosed at birth. DSO is described to be equally prevalent in males and females which is mainly what we observed in our study (54% males vs. 45% females). In accordance with what has previously been described in different studies, we observed that DSO were associated with younger maternal age, with 42.3% of our DSO cases having a maternal age < 25 years (Garne et al. 2018; Webb and Dattani 2010). In the literature, a genetic diagnosis can be made in < 1% of the patients (mainly pathogenic variant in HESX1 and SOX2 genes). Interestingly, DSO cases were associated with a known genetic anomaly in 2.5% of our cases which is more than what has been previously reported. However, the small percentage of genetic known cases suggest environmental factors (such as medications, alcohol, young maternal age) may also be involved. Moreover, in our study, 88.5% of DSO cases were either single (29.5%) or complex (59.0%), whereas only 11.4% of DSO were associated with extraocular defects. These data could reflect the known high phenotypic variability of this disorder.
4.8. Colobomas
Ocular coloboma is due to failed closure of the embryonic optic fissure. It has been reported to occur in 0.5–7.5 cases per 10,000 LB (Stoll et al. 1992; Warburg 1993; Gregory‐Evans et al. 2004). In our study, coloboma had a little lower prevalence of 0.49 per 10,000 births maybe due to the difficulties to make an early diagnosis. However, this prevalence was heterogeneous across different registries ranging from 0.11 to 1.96 per 10,000 births. Ocular coloboma is etiologically heterogeneous and can be sporadic or inherited, single or be associated with systemic disorders (Pagon 1981). As a single defect, it is usually inherited as an autosomal dominant disorder, although autosomal recessive inheritance also occurs. In our study, a genetic anomaly was reported in 21.4% of the overall colobomas. Around 20% of colobomas were single and nearly two‐thirds of them were associated with extraocular defects. This information is fundamental for clinicians, as it helps guiding the investigations to be carried out on a patient when a coloboma is discovered.
4.9. Strengths and Limitations
This study is the first one to use a multinational approach in order to make a precise description on COA that enabled us to classify and assess the current situation on COA across Europe. The large sample size provides a reliable basis for estimating the prevalence and characteristics of rare ocular anomalies in the European population. The study sampled a representative population across Europe, which enhances the generalizability of the findings to the broader European context. As the study adhered to standardized definitions and criteria for identifying and classifying ocular anomalies based on ICD‐9/10 classification and using EUROmediCAT infrastructure, it improves the validity and comparability of the results. This study involved collaboration between multiple centers across Europe, which adds strength to the study by capturing regional variations and increasing the overall sample size.
Some limitations can be noted. A few registries did not participate and therefore certain parts of European population are not covered by our study. This limitation has several implications for the generalizability and applicability of the findings. Specific subpopulations or regions that are underrepresented in the study may have different prevalence rates, risk factors, or health outcomes. Europe is characterized by diverse sociodemographic, cultural, and healthcare environments. These factors vary across different European countries and might be influenced by these regional differences, limiting the applicability of the results to parts of Europe not included in the study. Some missing data on certain variables could have not been interpreted and discussed (i.e., many missing data on sociodemographic prevents us from discussing these potential differences across Europe and between registries). Because COA are rare, there may be a risk of underestimating their prevalence, with some COA diagnosed later in childhood and thus missed. Given the rarity of COA, there may be challenges in obtaining complete and accurate data, leading to potential information bias. Rare COA may be challenging to diagnose accurately, leading to potential misclassification. Finally, our study lacks diversity in terms of ethnicity (only restricted to European population) and socioeconomic status, it may not fully reflect the occurrence of COA in different population subgroups of the world.
5. Conclusion
This is the first European study describing all COA recorded in congenital anomaly registries. The prevalence of total COA was 3.47/10,000 births. Congenital lens anomalies and an/microphthalmia were the most frequent COA. As COA are rare diseases, it is necessary to have high epidemiological vigilance over large populations with accurate clinical description of individual cases, such as those provided by congenital anomaly registries since their recording is based on original medical files. A better evaluation of different classes of ocular anomalies could improve their diagnosis and therefore their early management. Our findings underscore the importance of integrating comprehensive prenatal diagnostics to identify COA alongside any associated IUGR or extraocular malformations. This approach not only facilitates early and accurate diagnosis but also informs better genetic counseling by identifying potential syndromic associations. Consequently, families can be better prepared for the potential outcomes and management of these conditions. Moreover, early detection allows for the development of tailored clinical management strategies that address both ocular and systemic manifestations, ultimately improving patient prognosis and quality of life. This integrative methodology highlights the critical role of expert centers in guiding precise diagnosis and personalized treatment plans for rare CA. Furthermore, since most COAs in our study are not associated with a genetic cause, it highlights the need to deepen our understanding of the origins of these anomalies. Other etiologies, such as in utero exposure to prescribed medicines, recreational drugs, environmental pollution, maternal morbidities, and stressors, could be considered.
Author Contributions
C.D., A.C., L.D., M.C.‐S., C.D.‐M., C.H.‐D., and F.F. conceived and designed the analysis. A.J.N., E.B., H.D., M.L., E.G., B.K., N.L., A.R., M.O.M., A.P., M.G., J.B., M.R.K., A.L.B., L.J.E.‐G.‐G., C.C.‐C., M.‐C.A., D.T., S.J., E.D.H., V.N. I.B., F.R., H.R., J.H. and I.P. collected the data and revised the manuscript. A.C., L.D., V.N.G., and C.H.‐D. performed the analysis. C.D. wrote the paper under the supervision of M.C.‐S and C.D.‐M. All the coauthors approved the last version of the manuscript.
Ethics Statement
An ethics statement was not required for this study type as it is based exclusively on data extracted from EUROmediCAT registries and EFEMERIS healthcare database.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Table S1. ICD‐10 code and class of COA.
Table S2. Percentage of missing data per registry for each variable. Missing data > 20% are highlighted.
Table S3. Prevalence of total COA and of the main COA class (1, 2, 4, 5, 7, 10) in the different registries involve in MEDIKEYE.
Acknowledgments
Euromedicat partners for providing their data.
Funding: The authors received no specific funding for this work.
†Deceased
Data Availability Statement
All data generated or analyzed during this study are included in this article. Further enquiries can be directed to the corresponding author.
References
- Allingham, R. R. , Liu Y., and Rhee D. J.. 2009. “The Genetics of Primary Open‐Angle Glaucoma: A Review.” Experimental Eye Research 88, no. 4: 837–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton, M. J. , Ramke J., Marques A. P., et al. 2021. “The Lancet Global Health Commission on Global Eye Health: Vision Beyond 2020.” Lancet Global Health 9, no. 4: e489–e551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, C. L. 2003. “Septo‐Optic Dysplasia: A Literature Review.” Optometry 74, no. 7: 417–426. [PubMed] [Google Scholar]
- Campbell, H. , Holmes E., MacDonald S., Morrison D., and Jones I.. 2002. “A Capture‐Recapture Model to Estimate Prevalence of Children Born in Scotland With Developmental Eye Defects.” Journal of Cancer Epidemiology and Prevention 7, no. 1: 21–28. [DOI] [PubMed] [Google Scholar]
- Chambers, T. M. , Agopian A. J., Lewis R. A., et al. 2018. “Epidemiology of Anophthalmia and Microphthalmia: Prevalence and Patterns in Texas, 1999–2009.” American Journal of Medical Genetics. Part A 176, no. 9: 1810–1818. [DOI] [PubMed] [Google Scholar]
- Forestieri, N. E. , Desrosiers T. A., Freedman S. F., et al. 2019. “Risk Factors for Primary Congenital Glaucoma in the National Birth Defects Prevention Study.” American Journal of Medical Genetics. Part A 179, no. 9: 1846–1856. [DOI] [PubMed] [Google Scholar]
- Garne, E. , Dolk H., Loane M., et al. 2011. “Paper 5: Surveillance of Multiple Congenital Anomalies: Implementation of a Computer Algorithm in European Registers for Classification of Cases.” Birth Defects Research. Part A, Clinical and Molecular Teratology 91, no. 1: S44–S50. [DOI] [PubMed] [Google Scholar]
- Garne, E. , Rissmann A., Addor M. C., et al. 2018. “Epidemiology of Septo‐Optic Dysplasia With Focus on Prevalence and Maternal Age—A EUROCAT Study.” European Journal of Medical Genetics 61, no. 9: 483–488. [DOI] [PubMed] [Google Scholar]
- George, A. , Cogliati T., and Brooks B. P.. 2020. “Genetics of Syndromic Ocular Coloboma: CHARGE and COACH Syndromes.” Experimental Eye Research 193: 107940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory‐Evans, C. Y. , Williams M. J., Halford S., and Gregory‐Evans K.. 2004. “Ocular Coloboma: A Reassessment in the Age of Molecular Neuroscience.” Journal of Medical Genetics 41, no. 12: 881–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haargaard, B. , Wohlfahrt J., Fledelius H. C., Rosenberg T., and Melbye M.. 2004. “A Nationwide Danish Study of 1027 Cases of Congenital/Infantile Cataracts: Etiological and Clinical Classifications.” Ophthalmology 111, no. 12: 2292–2298. [DOI] [PubMed] [Google Scholar]
- Hsu, P. , Ma A., Wilson M., et al. 2014. “CHARGE Syndrome: A Review.” Journal of Paediatrics and Child Health 50, no. 7: 504–511. [DOI] [PubMed] [Google Scholar]
- Kiserud, T. , Piaggio G., Carroli G., et al. 2017. “The World Health Organization Fetal Growth Charts: A Multinational Longitudinal Study of Ultrasound Biometric Measurements and Estimated Fetal Weight.” PLoS Medicine 14, no. 1: e1002220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingam, G. , Sen A. C., Lingam V., Bhende M., Padhi T. R., and Xinyi S.. 2021. “Ocular Coloboma—A Comprehensive Review for the Clinician.” Eye 35, no. 8: 2086–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ondeck, C. L. , Pretorius D., McCaulley J., et al. 2018. “Ultrasonographic Prenatal Imaging of Fetal Ocular and Orbital Abnormalities.” Survey of Ophthalmology 63, no. 6: 745–753. [DOI] [PubMed] [Google Scholar]
- Pagon, R. A. 1981. “Ocular Coloboma.” Survey of Ophthalmology 25, no. 4: 223–236. [DOI] [PubMed] [Google Scholar]
- Palmer, S. R. , Dunstan F. D. J., Fielder H., Fone D. L., Higgs G., and Senior M. L.. 2005. “Risk of Congenital Anomalies After the Opening of Landfill Sites.” Environmental Health Perspectives 113, no. 10: 1362–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos, M. , Cable N., Rahi J., and Khaw P. T.. 2007. “The British Infantile and Childhood Glaucoma (BIG) Eye Study.” Investigative Ophthalmology & Visual Science 48, no. 9: 4100–4106. [DOI] [PubMed] [Google Scholar]
- Patel, L. , McNally R. J. Q., Harrison E., Lloyd I. C., and Clayton P. E.. 2006. “Geographical Distribution of Optic Nerve Hypoplasia and Septo‐Optic Dysplasia in Northwest England.” Journal of Pediatrics 148, no. 1: 85–88. [DOI] [PubMed] [Google Scholar]
- Plaisancie, J. , Ceroni F., Holt R., et al. 2019. “Genetics of Anophthalmia and Microphthalmia. Part 1: Non‐syndromic Anophthalmia/Microphthalmia.” Human Genetics 138, no. 8–9: 799–830. [DOI] [PubMed] [Google Scholar]
- Plaisancie, J. , Tarilonte M., Ramos P., et al. 2018. “Implication of Non‐coding PAX6 Mutations in Aniridia.” Human Genetics 137, no. 10: 831–846. [DOI] [PubMed] [Google Scholar]
- Reis, L. M. , and Semina E. V.. 2011. “Genetics of Anterior Segment Dysgenesis Disorders.” Current Opinion in Ophthalmology 22, no. 5: 314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanga, H. L. , and Nischal K. K.. 2014. “Genetics and Ocular Disorders: A Focused Review.” Pediatric Clinics of North America 61, no. 3: 555–565. [DOI] [PubMed] [Google Scholar]
- Schraw, J. M. , Benjamin R. H., Scott D. A., et al. 2021. “A Comprehensive Assessment of Co‐Occurring Birth Defects Among Infants With Non‐Syndromic Anophthalmia or Microphthalmia.” Ophthalmic Epidemiology 28, no. 5: 428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searle, A. , Shetty P., Melov S. J., and Alahakoon T. I.. 2018. “Prenatal Diagnosis and Implications of Microphthalmia and Anophthalmia With a Review of Current Ultrasound Guidelines: Two Case Reports.” Journal of Medical Case Reports 12, no. 1: 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheeladevi, S. , Lawrenson J. G., Fielder A. R., and Suttle C. M.. 2016. “Global Prevalence of Childhood Cataract: A Systematic Review.” Eye 30, no. 1169: 1160–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solebo, A. L. , Teoh L., and Rahi J.. 2017. “Epidemiology of Blindness in Children.” Archives of Disease in Childhood 102, no. 9: 853–857. [DOI] [PubMed] [Google Scholar]
- Stallings, E. B. , Isenburg J. L., Mai C. T., et al. 2018. “Population‐Based Birth Defects Data in the United States, 2011‐2015: A Focus on Eye and Ear Defects.” Birth Defects Research 110, no. 19: 1478–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll, C. , Alembik Y., Dott B., and Roth M. P.. 1992. “Epidemiology of Congenital Eye Malformations in 131,760 Consecutive Births.” Ophthalmic Paediatrics and Genetics 13, no. 3: 179–186. [DOI] [PubMed] [Google Scholar]
- Tawfik, H. A. , Abdulhafez M. H., and Fouad Y. A.. 2015. “Congenital Upper Eyelid Coloboma: Embryologic, Nomenclatorial, Nosologic, Etiologic, Pathogenetic, Epidemiologic, Clinical, and Management Perspectives.” Ophthalmic Plastic & Reconstructive Surgery 31, no. 1: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylan Sekeroglu, H. , and Utine G. E.. 2021. “Congenital Cataract and Its Genetics: The Era of Next‐Generation Sequencing.” Turkish Journal of Ophthalmology 51, no. 2: 107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg, M. 1993. “Classification of Microphthalmos and Coloboma.” Journal of Medical Genetics 30, no. 8: 664–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb, E. A. , and Dattani M. T.. 2010. “Septo‐Optic Dysplasia.” European Journal of Human Genetics 18, no. 4: 393–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb, R. N. , Grajewski A., Papadopoulos M., Grigg J., and Freedman S.. 2013. 9th Consensus Meeting: Childhood Glaucoma. Vancouver, Canada: Kugler Publications. [Google Scholar]
- Wu, X. , Long E., Lin H., and Liu Y.. 2016. “Prevalence and Epidemiological Characteristics of Congenital Cataract: A Systematic Review and Meta‐Analysis.” Scientific Reports 6: 28564. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. ICD‐10 code and class of COA.
Table S2. Percentage of missing data per registry for each variable. Missing data > 20% are highlighted.
Table S3. Prevalence of total COA and of the main COA class (1, 2, 4, 5, 7, 10) in the different registries involve in MEDIKEYE.
Data Availability Statement
All data generated or analyzed during this study are included in this article. Further enquiries can be directed to the corresponding author.