

Abstract
Objective—
We investigated the stability, pharmacokinetic, and pharmacodynamic profile of the 2nd generation anti-von Willeband factor aptamer ARC15105.
Methods and Results—
Platelet plug formation was measured by collagen/adenosine diphosphate-induced closure time with the platelet function analyzer-100 and platelet aggregation by multiple electrode aggregometry. Platelet adhesion was measured on denuded porcine aortas and in a flow chamber. Aptamer stability was assessed by incubation in nuclease rich human, monkey, and rat serum for up to 72 hours. Pharmacokinetic and pharmacodynamic profiles were tested in cynomolgus monkeys after IV and SC administration. The median IC100 and IC50 to prolong collagen/adenosine diphosphate-induced closure timewere 27 nmol/L and 12 nmol/L, respectively. ARC15105 (1.3 μmol/L) completely inhibited ristocetin-induced platelet aggregation in whole blood (P<0.001), but also diminished collagen, ADP, arachidonic acid, and thrombin receptor activating peptide-induced platelet aggregation to some extent (P<0.05). ARC15105 (40 nmol/L) decreased platelet adhesion by >90% on denuded porcine aortas (P<0.001), which was comparable to the degree of inhibition obtained with abciximab. ARC15105 (100 nmol/L) also inhibited platelet adhesion to collagen under arterial shear in a flow chamber by 90% (P<0.001). The IV and SC terminal half-lives in cynomolgus monkeys were 67 and 65 hours, respectively, and the SC bioavailability was ≈98%. Allometric scaling estimates the human T1/2 would be ≈217 hours. Pharmacodynamic analysis confirms that ARC15105 inhibits von Willeband factor activity >90% in blood samples taken 300 hours after a 20 mg/kg IV or SC dose in monkeys.
Conclusion—
The potency, pharmacokinetic profile, and SC bioavailability of ARC15105 support its clinical investigation for chronic inhibition of von Willeband factor -mediated platelet activation.
Keywords: adhesion molecules, antiplatelet drugs, arterial thrombosis, coagulation
Von Willebrand Factor (VWF) plays an important role in the initiation of platelet adhesion and aggregation.1,2 VWF is required for normal hemostasis and mediates the adhesion of platelets to sites of vascular damage by binding to specific platelet membrane glycoproteins and constituents of exposed connective tissue.3 VWF is also a carrier protein for blood clotting factor VIII and therefore stabilizes and prevents factor VIII from early inactivation.4 It is released from endothelial cells and platelets following activation by a number of agents including thrombin. VWF plays a role in shear-dependent thrombogenesis, which occurs in stenotic coronary arteries or ruptured atherosclerotic plaque lesions.3 Its levels are also heightened in patients, who experienced adverse cardiac events that are linked to a poorer prognosis.5–7 Conventional therapy of myocardial infarction reduces platelet activation and aggregation, but mostly addresses receptors and targets other than VWF.8 Nevertheless, the central role of VWF in thrombogenesis made it a promising target in research on new antiplatelet therapies that specifically inhibit VWF,9 like the anti-VWF aptamers. ARC1779 is an anti-VWF aptamer, which has been shown to potently and specifically inhibit excessive VWF activity in blood from patients with acute myocardial infarction (AMI),10,11 and clinical proof of mechanism and concept has been demonstrated.12–14 ARC15105 is a new generation chemically modified aptamer with promising effects on shear-induced platelet aggregation in the setting of myocardial infarction. The aim of this study was to find effective dose ranges of ARC15105 that inhibit shear-dependent platelet function and agonist-induced platelet aggregation ex vivo in patients with myocardial infarction and in healthy volunteers for upcoming clinical trials.
Methods
The Ethics Committees of the Medical University of Vienna, of the Harvard Medical School, and of the Montreal Heart Institute approved the study protocols and the studies were conducted in accordance with the Declaration of Helsinki. All animal experiments in this study were approved by the institutional Animal Care and Use Committees of Charles River Laboratories and Montreal Heart Institute. All subjects provided written informed consent before any study-related procedures.
Clinical Study Design
Five healthy study participants were included at the Montreal Heart Institute and 6 participants were included at the Harvard Medical School in the platelet adhesion studies. The clinical study at the Medical University of Vienna used a cross-sectional design, comparing concentration-response curves in patients with AMI with those of healthy volunteers (age-matched to patients with AMI and healthy young volunteers) ex vivo. Forty-two study participants were included in the platelet aggregation study: 21 patients presented with an AMI (13 patients with STEMI and 8 with NSTEMI) being on standard antiplatelet treatment with aspirin and clopidogrel, 2 patients with AMI additionally receiving abciximab and 21 healthy volunteers (12<40 years and 9 age-matched). The inclusion criteria for the study were as follows: written informed consent by the patient; aged 18 years; admission to the Department of Emergency Medicine with AMI in the last 24 hours; willingness to comply with instructions prescribed by the protocol; and healthy aged controls aged ≥18 to 40 with normal findings in the clinical examination and medical history of ECG and vital parameters. Exclusion criteria included history of hemorrhagic diathesis or thromboembolic disease in healthy volunteers and consumption of acetylsalicylic acid or other platelet inhibitors in the past 7 days in healthy volunteers. The analysts were blinded with regard to the patients’ or volunteers’ diagnosis.
Blood Sampling
We collected 40 mL of blood by venipuncture from an antecubital vein into 3.2% citrated tubes (Vacutainer, Becton Dickinson, Vienna, Austria) and hirudin anticoagulated tubes (200U/mL, Dynabyte, Munich, Germany). All samples were processed within 2 hours. AMI patients were recruited from the Department of Emergency Medicine and blood samples were obtained within 24 hours of admittance to the hospital. We spiked blood samples with at least 10 different concentrations of ARC15105 (range: 13 nmol/L–1.3 μmol/L), before running 2 different platelet tests. A predefined algorithm was used to obtain the various aptamer concentrations. Spiked samples were incubated for 10 minutes at 37°C before running the platelet tests.
Oligonucleotide Synthesis and PEG Conjugation
Oligonucleotides were synthesized on an ÄKTA Oligopilot (Amersham Pharmacia Biotech, Piscataway, NJ) using standard phosphoramidite solid-phase chemistry. All phosphoramidites were acquired from Hongene Biotech (Shanghai, China). Aptamers were synthesized with a hexylamine moiety at the 5′-end and conjugated to high molecular weight PEGs from JenKem (Allen, TX) postsynthetically via amine-reactive chemistry. The resulting products were purified by ion exchange and reverse phase HPLC. The sequence of unpegylated aptamer ARC15103 is NH2-mGmGmGmAmCmCmUmAmAmGmAmCmAmCmAmUmGmUmCmCmC-3T, where NH2 is a hexylamine linker, 3T is an inverted deoxythymidine residue, and mN is a 2′-methoxy residue. ARC15104 and ARC15105 have the ARC15103 core sequence and are appended with a 20-kDa or 40-kDa PEG moiety, respectively. The control oligonucleotide15 is a randomized version of an unrelated aptamer with the sequence PEG40K-nh-fC-fU-fC-fC-mA-mG-mA-fC-mA-fC-mA-mG-fC-mG-mG-mA-fU-mG-mA-mA-mA-fU-fC-fC-mG-mG-fC-fC-mA-mG-mA-mG-3T, where PEG40K is a 40-kDa PEG moiety, nh is a hexylamine linker, 3T is an inverted deoxythymidine residue, mN is a 2′-methoxy residue, and fN is a 2′-fluoro residue. PEG-moieties were not considered when assessing the concentrations of active molecules; 1 nmol/L of ARC15105 equals 7.5 ng/mL, whereas 1 nmol/L of ARC1779 corresponds to 13.18 ng/mL.
Impedance Aggregometry
Whole blood aggregation was determined using Multiple Electrode Aggregometry on a new generation impedance aggregometer (Multiplate Analyzer, Verum Diagnostica, GmbH, Munich, Germany). The system detects the electric impedance change due to the adhesion and aggregation of platelets on 2 independent electrode-set surfaces in the test cuvette.16–18 Whole blood was incubated for 10 minutes with and without addition of 1.3 μmol/L ARC15105. A 1:2 dilution of whole blood anticoagulated with hirudin and 0.9% NaCl was stirred at 37°C for 3 minutes in the test cuvettes. Thereafter 5 aggregation-stimulating agonists: ADP (6.4 μmol/L), collagen (3.2 μg/mL), arachidonic acid (0.5 mmol/L), TRAP (30 μmol/L), and ristocetin (0.77 μg/mL) were added to separate samples and the increase in electric impedance was recorded continuously for 6 minutes. The mean values of the 2 independent determinations are expressed as the area under the curve of the aggregation tracing.
Platelet Function Analyzer-100
The platelet function analyzer-100 (Dade Behring, Marburg, Germany) was used for measuring platelet function under high shear rates (5000–6000 s−1). Blood samples collected in 3.2% sodium citrate were placed in a test cartridge and aspirated from the sample reservoir through a capillary (200-μm diameter) under constant negative pressure (indicating high shear stress) toward a biochemically active membrane with a central aperture (150-μm diameter). The membrane is coated with equine type I collagen and adenosine 5′-diphosphate (CADP cartridges). During the test, platelets adhere to collagen-coated membranes, become activated, release their granule contents, and build a platelet thrombus, gradually diminishing and finally arresting the blood flow. The aperture closure time (CT) represents the time in seconds (up to a maximum of 300 seconds) until aperture occlusion by formation of a platelet plug. The reference values for the collagen/adenosine diphosphate-induced closure time (CADP-CT) are 65 to 120 seconds.19,20
Platelet Adhesion to Denuded Porcine Aorta
Platelet isolation and labeling, as well as the perfusion experiments were conducted as described previously.21,22 A 60-mL sample of venous blood from 5 healthy volunteers was anticoagulated with 6 mL of PPACK (D-Phenylalanyl-l-prolyl-l-arginine chloromethyl ketone) in saline (50 nmol/L final concentration, Calbiochem, Quebec, Canada), and a 30-mL sample with anticoagulant citrate dextrose (Baxter, Mississauga, Canada).21,22 The anticoagulant citrate dextrose blood was used to isolate and radiolabel platelets with 111In and resuspended in the remaining 60 mL of the PPACK blood. Fresh porcine aortas (Aggromex, St-Blaise, Canada) were isolated and denuded by lifting and peeling off the intima to expose the subjacent media. The segments were placed into Badimon perfusion chambers with a 1 mm internal diameter×10 mm long. The chambers were placed in parallel in a thermostatically controlled water bath at 37°C, thus permitting simultaneous parallel, pair-wise perfusion over arterial tissues of treated or untreated blood at a high shear (6974/s). Blood (10 mL) was recirculated over the arterial segments for 15 minutes in the flow chambers. A known GPIIb/IIIa antagonist (100 nmol/L of abciximab) and a placebo (physiological saline) were used as controls, and ARC15105 and its predecessor ARC1779 were tested at 40, 80, and 160 nmol/L. The arterial segments were then fixed in formalin 1% and placed in polystyrene tubes for gamma counting to quantify platelet adhesion.23 After compilation of gamma counting, the arterial segments were prepared and observed by scanning electron microscopy.
Platelet Adhesion to Collagen-Associated VWF in Flow Chamber
The potency of ARC15105 in inhibiting platelet adhesion was examined in flow chamber experiments and compared to the previously characterized anti-VWF aptamer ARC1779.22 Binding to a collagen-coated surface was measured in whole blood from 6 healthy volunteers after labeling of platelets and incubating with the aptamers. A 20-mL blood sample, anticoagulated with 90 μmol/L PPACK (Calbiochem, Darmstadt, Germany), was collected from 6 healthy donors who had not taken any medication in the prior 7 days. Platelet-rich plasma was obtained by centrifugation at 100 g for 5 minutes. Platelets were separated by centrifugation at 600 g for 5 minutes in the presence of 2 μg/mL Prostaglandin I2 and washed in modified Tyrode’s Buffer (140 mmol/L NaCl, 0.36 mmol/L Na2HPO4, 3 mmol/L KCl, 12 mmol/L NaHCO3, 5 mmol/L Hepes, and 10 mmol/L glucose, pH 7.3) containing 0.2% BSA. Washed platelets were labeled with 2.5 μg/mL calcein orange. Platelet-poor blood was reconstituted with labeled platelets and remaining plasma at 2×108/mL platelets. Blood was then treated with various concentrations of ARC1779, ARC15105, or the control oligonucleotide for 5 minutes at 37°C (0–250 nmol/L). A shorter incubation time as compared to previously reported experiments with ARC177922 was chosen after preliminary experiments indicated that ARC15105 was more potent. Using a 0.0127-cm silicon rubber gasket, a parallel plate flow chamber (Glycotech, Gaithersburg, MD) was assembled onto 35-mm diameter round glass coverslips, which had been coated with 100 μg/mL collagen type I (Nycomed, Munich, Germany). Perfusion was performed at a shear rate of 1500/s for 3 minutes, which mediates binding of plasma VWF to surfacebound collagen.24 Platelet adhesion to collagen-associated plasma VWF was monitored with an Axiovert 135 inverted microscope (Carl Zeiss, Inc., Thornwood, NY at 32 × and a silicon-intensified tube camera C 2400 (Hamamatsu Photonics, Hamamatsu City, Japan) and analyzed with Image SXM 1.62 (NIH Image, http://rsb.info.nih.gov/nih-image).
Serum Stability Study of ARC15105
The serum metabolic stability of ARC15105 was evaluated in Sprague-Dawley rat, Cynomolgus monkey and human sera at a final concentration of 50 μmol/L. Pooled serum for each species was purchased from Bioreclamation (East Meadow, NY 11554) and used in this study. Test compounds were incubated with serum at 37°C on a shaker (at 150 rpm) for a period up to 72 hours. At each incubation time point (0, 2, 8, 24, 48, and 72 hours), sample aliquots were frozen using liquid nitrogen and stored at −80°C until analysis by HPLC. The stability was expressed as the percentage of intact ARC15105 remaining after the incubation.
Pharmacokinetic Study of ARC15105 in Cynomolgus Monkeys
Three adult male, non-naive, purpose-bred Cynomolgus monkeys (Macaca fascicularis), weight range from 2.5 to 4.0 kg, were selected from a group of stock animals held at a test facility (Maccine Pte Ltd, Singapore). Prior to the study, animals underwent a complete physical examination performed by the attending veterinarian. Prestudy clinical pathology testing (hematology and clinical chemistry) were performed to confirm their suitability for experimental use. The animals were antibody negative for Simian Immunodeficiency Virus (SIV), Simian T-cell Lympotrophic Lentivirus, and Type D Simian Retroviruses. The pharmacokinetics of ARC15105 was studied with an IV to SC crossover design. ARC15105 was administered as a single bolus IV injection of 20 mg/kg (in 0.9% physiological saline) via a peripheral vein. Following dosing, the catheter was flushed with 3 mL saline. After a 3-week wash-out period, 20 mg/kg ARC15105 was administered as a single subcutaneous bolus dose in the leg area.
Analysis of Aptamer Concentrations in Cynomolgus Macaque Plasma and in the Serum Stability Study
Blood samples (≈200 μL) were collected via saphenous venipuncture into tubes containing K2EDTA as an anticoagulant, placed on wet ice, and centrifuged within 30 minutes of collection at approximately 4°C to obtain plasma. The plasma samples were stored frozen at approximately ≤−80°C until analysis for ARC15105 concentration. Prior to analysis, to each aliquot (50 μL) of plasma containing test article was added 25 μL of digestion buffer (60 mmol/L Tris-HCL, pH 8.0, 100 mmol/L EDTA and 0.5% SDS) and 75 μL of proteinase solution (1 mg/mL proteinase K in 10 mmol/L Tris HCl, pH 7.5, 20 mmol/L CaCl2, 10% glycerol v/v). Samples were then incubated at 55°C overnight with shaking. Following the incubation, samples were centrifuged (14000 rpm; 4°C; 15 minutes) and 100 μL of the supernatant withdrawn and transferred to HPLC injection vials. The HPLC system was equipped with a column-temperature-controller, UV detector, and a Dionex DNA PAK PA-100 (4×250 mm) column. The method used a mobile phase elution gradient made from phase A (75% 25 mmol/L sodium phosphate dibasic buffer [pH 7.0] and 25% Acetonitrile) and B (75% 25 mmol/L sodium phosphate dibasic in water [pH 7.0] and 25% Acetonitrile containing 400 mmol/L NaClO4). Flow rate was 0.5 mL/min with column oven temperature set at 80°C. The assay injection volume was approximately 25 μL. The lower limit of quantitation was 0.2 μg/mL with a linear concentration range of 0.2 to 500 μg/mL. The HPLC method was calibrated relative to concentration reference standards of ARC15105 prepared in blank monkey plasma (K2EDTA) and extracted by the same proteinase method used to prepare in vivo samples. All reported concentrations of ARC15105 are based on the mass of aptamer, excluding the mass of PEG.
Analysis of VWF Activity by ELISA
VWF activity, as defined as the amount of active (“free”) VWF with functional A1 domain present, was evaluated with a commercially available quantitative direct ELISA kit that is designed for detection of VWF activity in human citrated plasma (READDS® VWF Activity ELISA test kit, Product No. 10826, Corgenix, Westminster, CO).11–13 In this assay, VWF present in test samples is captured by a mouse monoclonal anti-human VWF antibody that recognizes the functional A1 domain. Captured VWF is subsequently detected with a different, horse radish peroxidase-conjugated mouse polyclonal anti-human VWF. Addition of perborate/3,3′,5,5′-tetramethylbenzidine as substrate for horse radish peroxidase produces a colorimetric end point that is assessed spectrophotometrically for OD450. Absorbance results are compared to a standard curve established with human VWF to determine the plasma VWF activity in the test sample. The lower limit of quantification was determined to be 2%. A 12-point standard curve was run on each plate, along with 12 quality control points (6 concentrations in duplicate). Lower limit of quantitation was determined for each run and was determined, as recommended in Guidance for Industry: Bioanalytical Method Validation, Food and Drug Administration, May 2001, to be the lowest point that was at least 5 times the blank and had a reproducible precision of <20% and accuracy of 80% to 120%. For each run, 4 out of 6 nonzero controls, including lower limit of quantitation, had to have a <20% deviation for the results to be accepted. Percent activity, which was normalized to human plasma, was calculated using the equation: % activity=(ConcentrationAptamer/Concentration0 nmol/L Aptamer Human)*100. Percent inhibition was calculated using the equation: % inhibition=100−([ConcentrationAptamer/Concentration0 nmol/L Aptamer]*100).
There are potential differences in the sites of binding between the VWF A1 free domain and the 2 aptamers, which might make it difficult to compare both aptamers in terms of VWF inhibition. Nevertheless, as the Kd of 1779 and 15105 are both 2 nmol/L, the difference in the affinity for VWF should not impact the assay.
Collagen Binding Assay
Interference of ARC15105 with VWF binding to collagen was tested in a collagen binding assay (CBA) according to the manufacturers’ instruction (Hemochrom Diagnostics, Essen, Germany). Additionally, we tested the effects of polyethylen glycol of various sizes ranging from 200 to 35000 Dalton (at 2 different concentrations 0.7 and 7 μmol/L) on VWF activity in a CBA.
Separation of Free Aptamer From VWF-Bound Aptamer
Plasma pools of healthy volunteers were spiked with ARC15105 concentrations of 0.1 to 10 μmol/L/mL and then centrifuged through membranes with pore sizes of 100 kDa or 300 kDa (Sartorius tubes). The filtrate was then spiked back into normal plasma 1:10 and inhibition of VWF:A1 domains was measured.
Statistical Analyses
We estimated that 10 to 20 age-matched controls and 10 to 20 healthy young volunteers should allow us to establish reliable concentration-effect curves. In patients with myocardial infarction we estimated a minimal sample size of 20. Estimates for IC50 were compared between groups mainly by descriptive statistics. We tested with Kruskal-Wallis ANOVA, Mann Whitney U-test, Spearman’s correlation, and χ2-test and by one-way ANOVA and Dunnett t-test for comparison against control as appropriate. The level of significance was set to a 2-sided probability value (P≤0.05) and was corrected for multiple comparisons according to the Bonferroni adjustment. Commercially available software was used (SPSS Version 18.0; Chicago, IL).
Results
Platelet Adhesion Under High Shear Conditions on Denuded Porcine Aortas and in the Flow Chamber
Platelet adhesion averaged 76×106 platelets/cm2 when porcine aorta was perfused with blood from healthy volunteers under control conditions (Figure 1 A). Preincubation of the blood with abciximab before perfusion reduced platelet adhesion by 94% (4.2×106 platelets/cm2; Figure 1A). Similarly, ARC15105 inhibited platelet adhesion by 93% at the lowest concentration used (40 nmol/L; Figure 1A). The results of the adhesion assay were confirmed by scanning electron microscopy visualization of platelet adhesion. Arterial surfaces exposed to the blood of healthy volunteers accumulated a heavy thrombotic matrix, which was markedly, but similarly, reduced by abciximab and ARC15105 (Figure 1B).
Figure 1.
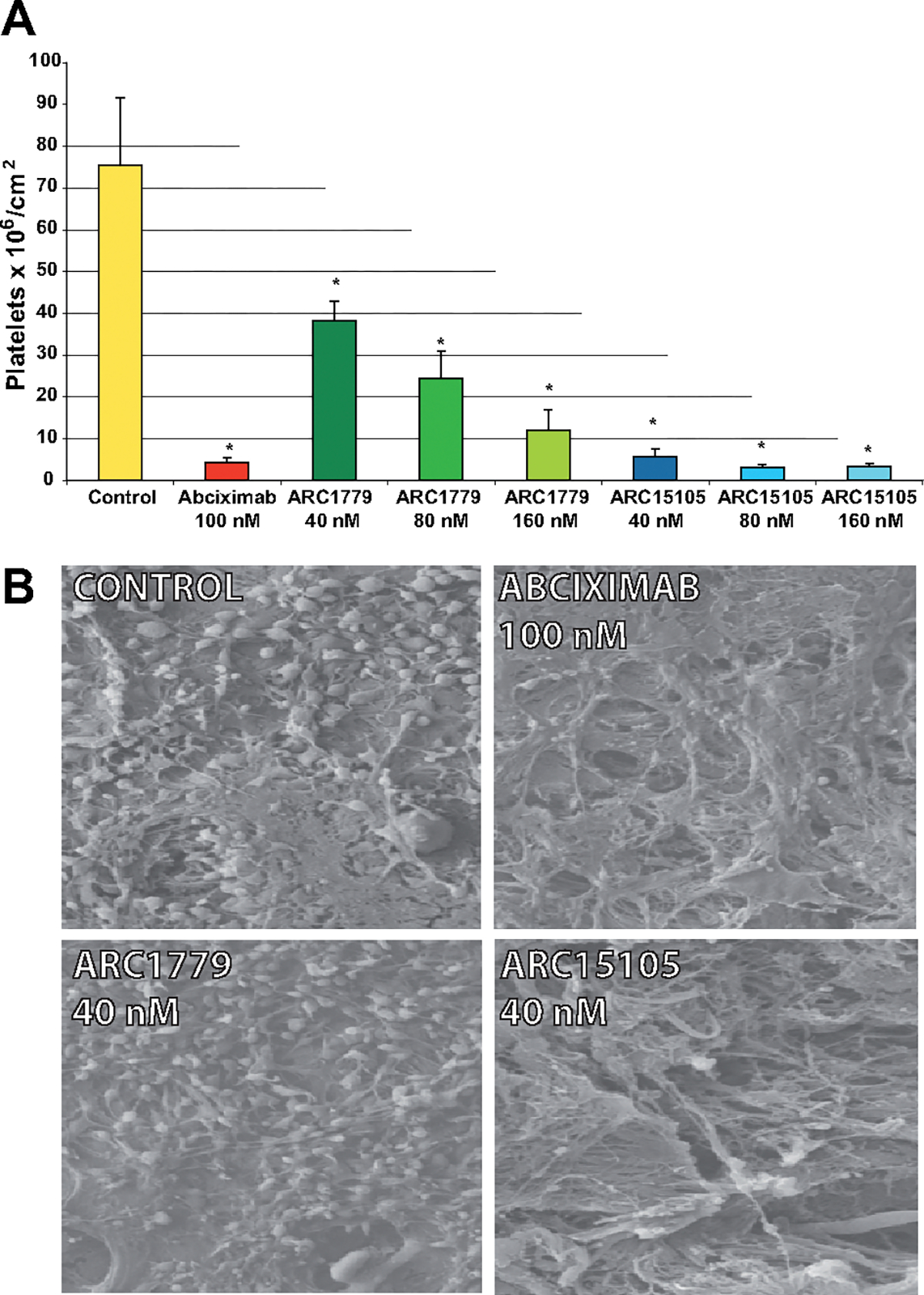
Platelet adhesion on injured porcine arterial segments; A, ARC15105, Arc1779, and abciximab inhibited the adhesion of platelets radiolabeled with 111In on injured porcine arterial segments in perfusion flow chambers. Data are expressed as mean±SEM, n5, *P<0.05 versus control. B, Representative scanning electron microscopy micrographs of the platelet adhesion following exposure of whole blood to injured porcine arterial segments in perfusion flow chambers. Magnification: ×2500.
A control oligonucleotide did not significantly inhibit platelet adhesion to collagen under arterial shear in a flow chamber. The ARC15105 aptamer reduced the area covered by fluorescent platelets to 5±4% (mean±SD; Figure 2; P<0.001).
Figure 2.
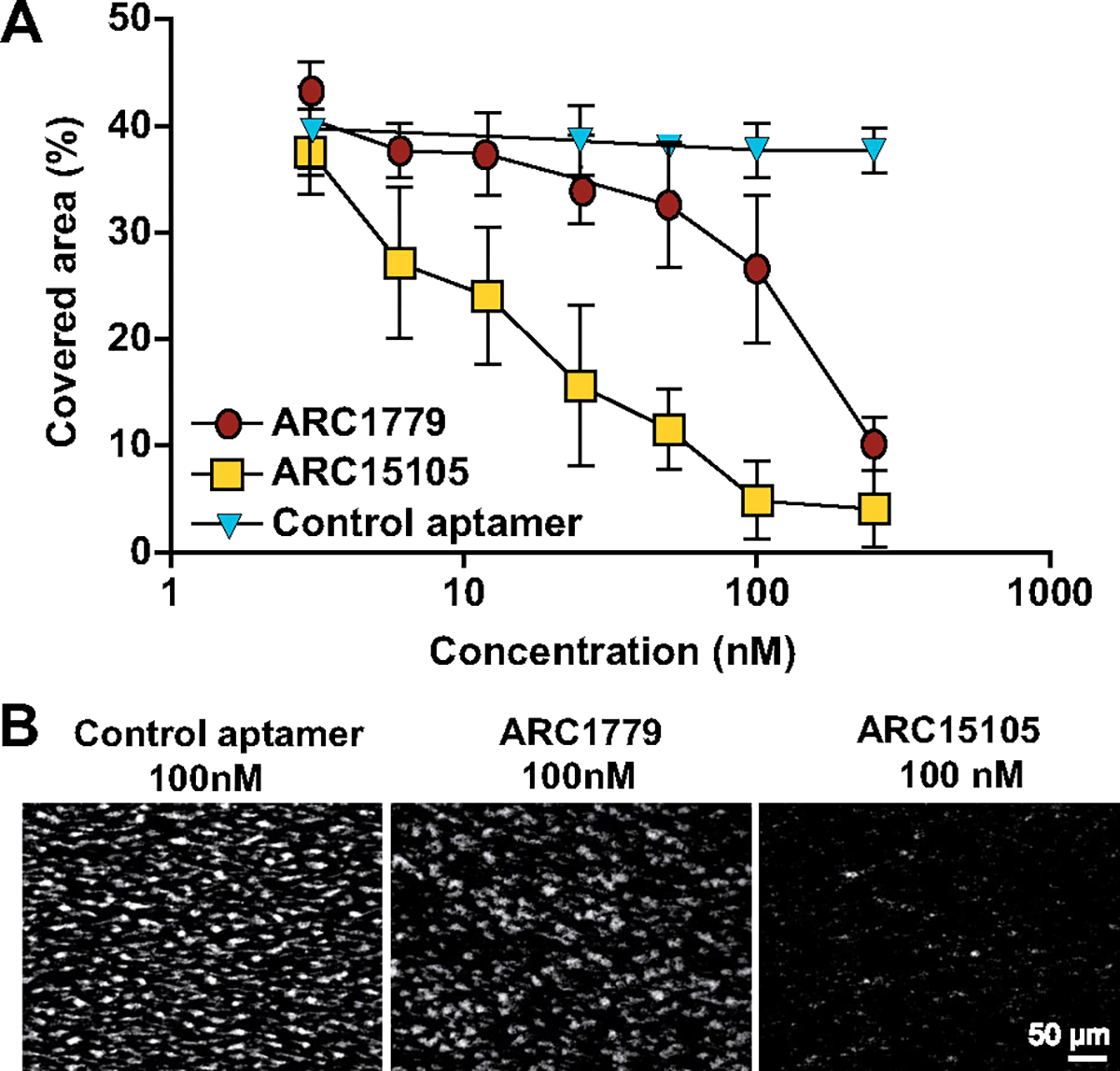
Concentration effect curve of ARC15105 and ARC1779 on platelet adhesion to collagen-bound VWF under arterial shear conditions. A, Data are expressed as mean±SD, n=6. B, Representative images of fluorescent platelets (in white) on the collagen-coated surface. Magnification: ×32; P<0.001 for all comparisons between each group.
Pharmacokinetics of Single Bolus ARC15105 Administered IV and SC in Cynomolgus Monkeys
The high level of in vitro stability translated into a high level of in vivo stability as evidenced by the pharmacokinetic profile ARC15105 displayed in monkeys. The IV and SC terminal half-lives (t1/2) in cynomolgus monkeys were 67 and 65 hours, respectively. The subcutaneous bioavailability was 98% in monkeys. ARC15105 completely inhibited VWF activity after a single 20 mg/kg infusion in monkeys and by >90% after SC injection in blood samples taken at 300 hours (Figure 3).
Figure 3.
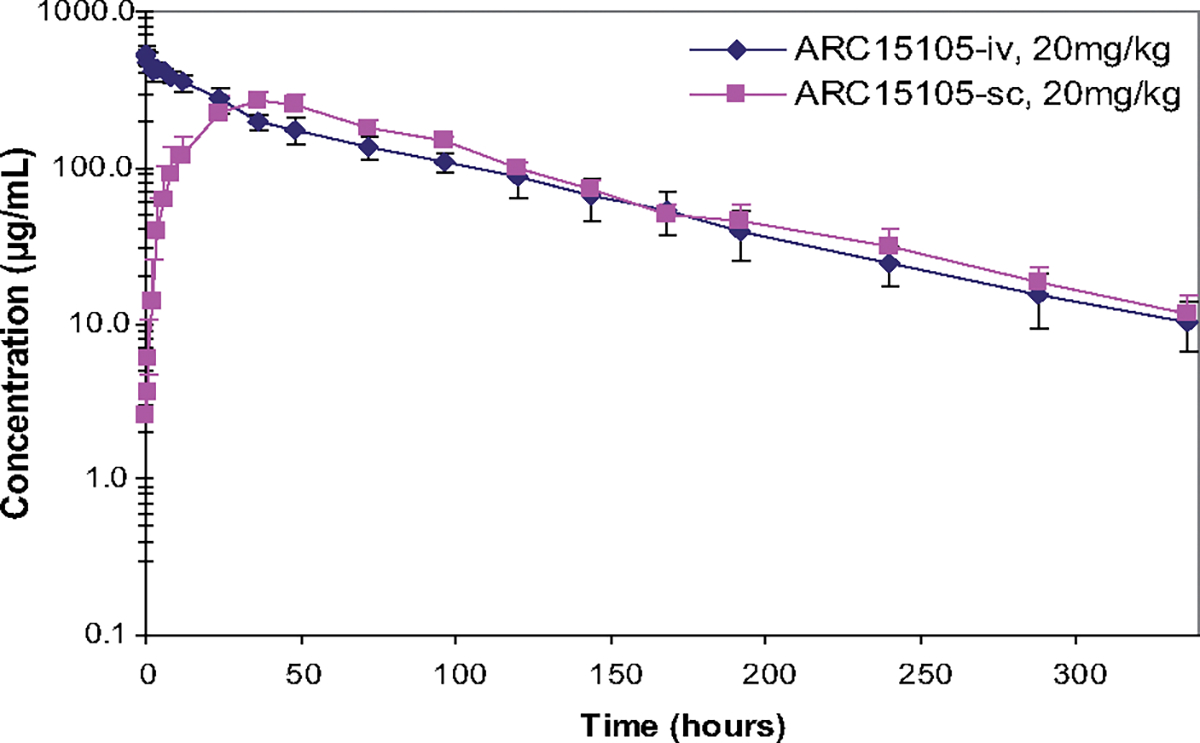
Comparison of the pharmacokinetics of a single bolus of ARC15105 (20 mg/kg) administered intravenously (IV) and ubcutaneously (SC) in 3 cynomolgus monkeys; P<0.05.
ARC15105 (20 mg/kg) inhibited VWF concentrations by 90% in monkeys up to 10 days after SC injection. At that time, plasma concentrations of ARC15105 still ranged from 20 to 40 μg/mL (Figure I in the online-only Data Supplement; Table).
Table.
Pharmacokinetic Parameters of the Aptamer ARC15105 Administered IV and SC
| PK Parameter | Units | 15105 IV Mean | 15105 SC Mean |
|---|---|---|---|
|
| |||
| Cmax (peak concentration) | μg/mL | 537 | 266 |
| AUC last | Hour*μg/mL | 30 200 | 29 422 |
| AUC 0–inf. | Hour*μg/mL | 31 316 | 30 530 |
| T1/2 (half-life) | Hour | 67 | 65 |
| Tmax (time to peak concentration) | Hour | 0.08 | 36 |
| MRT last | Hour | 78 | 96 |
| VSSobs | mL/kg | 59 | Not calculated |
| Vz/F | mL/kg | 69 | 63 |
| CL/F | mL/hour/kg | 0.66 | 0.67 |
| Fsc | % | 100 | 98 |
AUC indicates area under the curve; MRT, mean residence time; VSSobs, observed volume of distribution at steady state; Vz/F, volume of distribution; CL/F, clearance; Fsc, bioavailability.
The safety profile of ARC15105 in monkeys was favorable: there was no evidence for spontaneous bleedings during the whole study period.
Serum Stability of ARC15105 in 3 Species
Over 99% of the parent molecule of ARC15105 remained after 3 days of incubation in nuclease rich sera of rats, nonhuman primates or humans and about 85% remained after incubation in rat serum (Table I in the online-only Data Supplement).
Patient Demographics
All patients with AMI received aspirin, clopidogrel, and heparin. Other frequently administered medications were ACE-inhibitors (80%) and β-blockers (71%). Most AMI patients had well-established cardiovascular risk factors for myocardial infarction like age, overweight, hypertension, hyperlipidemia, diabetes, and smoking (Table II in the online-only Data Supplement). The median age of patients was 65 years, whereas the median age of controls was 45 years. The median BMI was 26 in the AMI population and 23 in controls. The hemoglobin levels and platelet counts were comparable between groups. The leukocyte count was increased in both AMI subgroups.
Platelet Activation Under High Shear Conditions In Vitro: CADP-CT
The median IC100 of ARC15105 (the lowest drug concentration that maximally prolonged CADP-CT) was 27 nmol/L in blood from controls and in blood from patients with AMI, and the median IC50 (the lowest drug concentration that prolonged CADP-CT by 50%) was 12 nmol/L (Figure 4A and 4B). A clear concentration-dependent effect was seen (Figure 4B).
Figure 4.
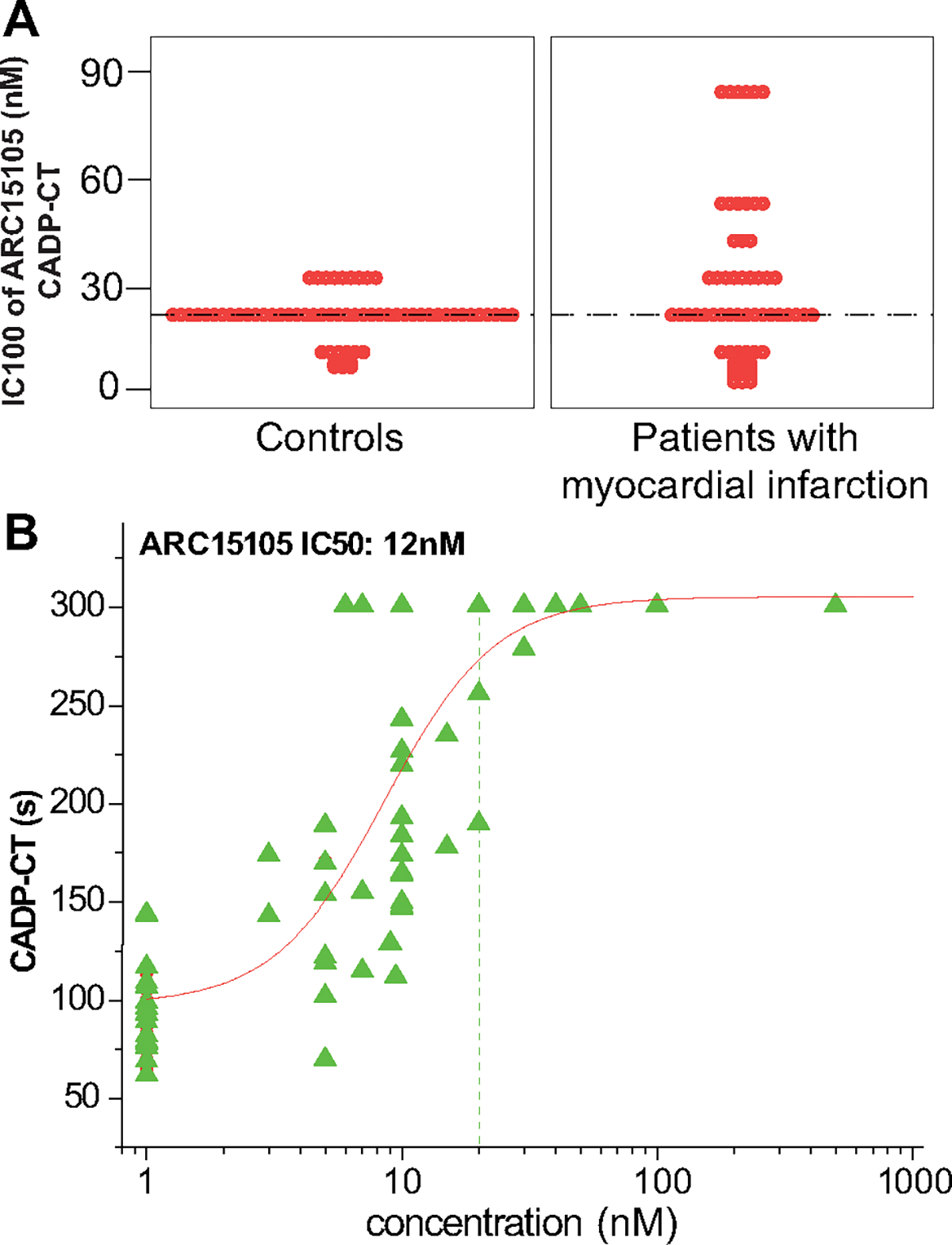
Prolongation in collagen/adenosine diphosphate induced closure time (A) IC100 (the lowest drug concentration that maximally prolonged collagen/adenosine diphosphate induced closure time [CADP-CT]) of ARC15105. B, Concentration-response values and curves of ARC15105-induced prolongation in CADP-CT. IC50 indicates the lowest drug concentration that prolonged CADP-CT by 50%; n42.
Agonist-Induced Platelet Aggregation
ARC15105 significantly reduced agonist-induced platelet aggregation in the following rank order of potency: ristocetin by 100%, arachidonic acid by 44%, collagen and adenosine diphosphate by >30%, and thrombin receptor activating peptide by 14% (Figure 5; P<0.05 for all). The inhibitory effect of ARC15105 on platelet aggregation was significant in controls without any antiplatelet therapy and even in patients with myocardial infarction, who were all pretreated with aspirin, clopidogrel, and heparin (Figure 5). In 2 patients on abciximab, baseline aggregation was already completely inhibited when induced by all agonists except collagen. ARC15105 maximally inhibited collagen-induced platelet aggregation in these 2 patients, who served as positive controls.
Figure 5.
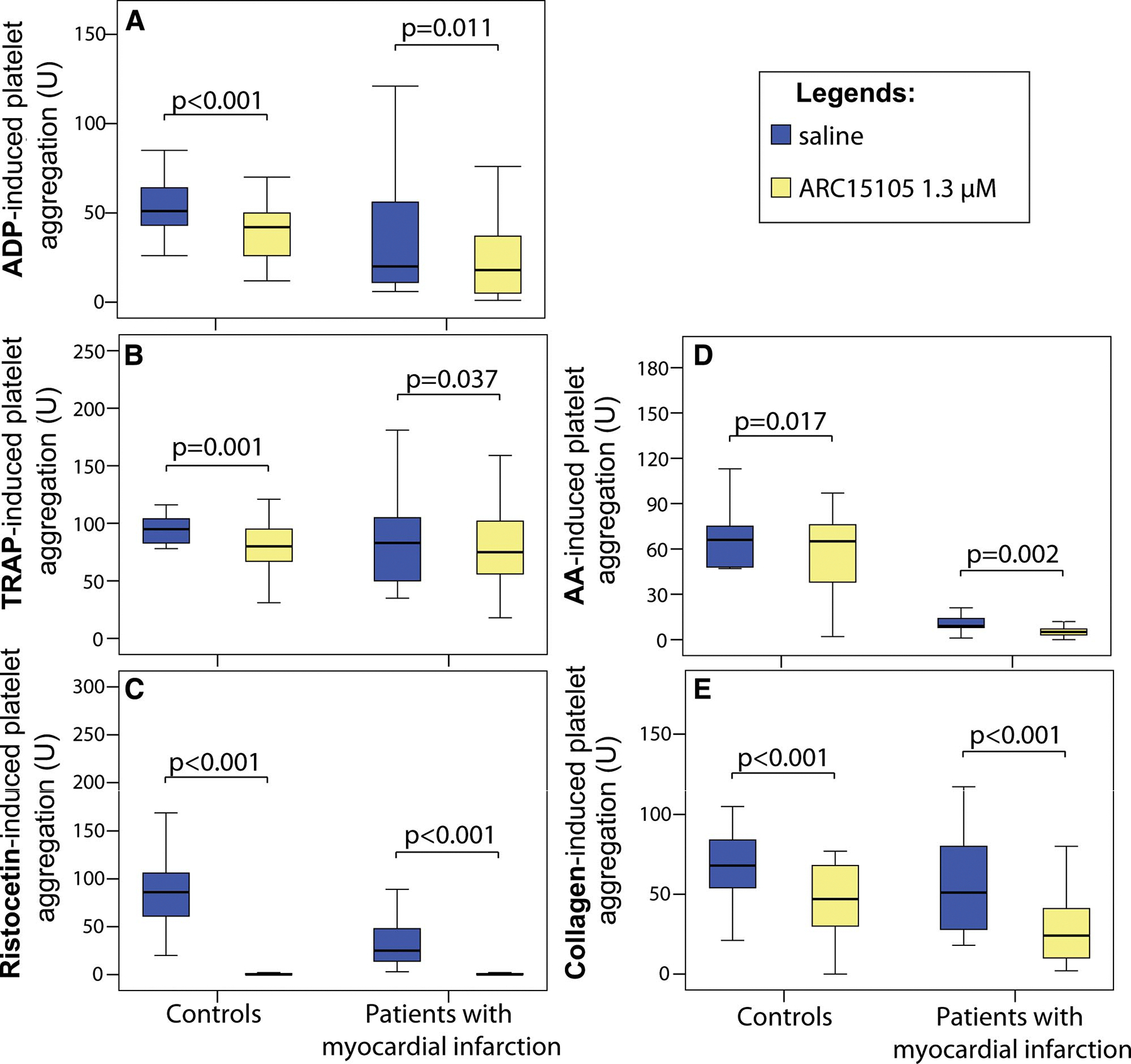
Platelet aggregation to ARC15105 (1.3 μmol/L) induced by various agonists: A, ADP (adenosine diphosphate); B, TRAP (thrombin receptor activating peptide); C, ristocetin; D, AA (arachidonic acid); E, collagen. Data are expressed as median and interquartile range (IQR); n=42.
VWF Activity and the Degree of Inhibition of VWF by ARC15105
Consistent with the literature,2 VWF activity was 2-fold higher in patients with myocardial infarction (238%; IQR: 203% to 251%) as compared to controls (105%; IQR: 86% to 142%). ARC15105 effectively inhibited VWF in the REAADS ELISA with an IC50 of 67 nmol/L (range 27–270 nmol/L) and an IC90 of 270 nmol/L (range 130–667 nmol/L). Similar to a previous study with ARC1779,11 there was no significant difference in the inhibitory concentrations between samples from patients with myocardial infarction as compared to controls (data not shown).
Interference of ARC15105 With VWF Binding to Collagen
ARC15105 inhibited binding of VWF to collagen in a concentration-dependent manner in an enzyme immunoassay: ARC15105 concentrations of 130 nmol/L inhibited VWF binding to collagen (VWF:CBA) by 64%, and the maximal inhibition by 73% was seen at 4 μmol/L (data not shown).
Centrifugation of plasma spiked with ARC15105 at concentrations of 0.1 to 10 μmol/L/mL through membranes (100 kDa or 300 kDa) led to a 3- to 5-fold enrichment in VWF:CBA activity in the remaining nonfiltered plasma, and decreased VWF levels to 0% and 12% to 17%, which passed through 100 kDa and 300 kDa pore sizes, respectively. This is consistent with an estimated size of 250 kDa for the smallest VWF molecules. When the filtrate containing the highest ARC15105 concentrations was spiked into normal plasma, VWF:A1 domains decreased from 77% to 16%, indicating that a significant proportion of ARC15105 is present as free unbound drug in plasma (data not shown).
Comparison of ARC1779 and ARC15105 in Different Test Systems
Platelet aggregation confirmed that the effect of ARC1779 is highly specific for VWF. As expected,11 the inhibitory effect of ARC1779 on ADP, arachidonic acid, ristocetin and thrombin receptor activating peptide-1 was not significant (3% to 11%; data not shown). ARC1779 reduced collagen-induced platelet aggregation by 17%, which was weaker than the effect of ARC15105 (P>0.001; data not shown). This is consistent with the results of the collagen binding assay, which has showed that ARC15105 at concentrations of 130 nmol/L inhibited VWF binding to collagen (VWF:CBA) by 64%, whereas even 2-fold higher ARC1779 concentrations reduced VWF:CBA by maximally 44%.
ARC1779 and ARC15105 concentration dependently reduced platelet adhesion in denuded porcine aortas and in a flow chamber. The effect of ARC15105 was 2-fold greater than the effect of the ARC1779 in denuded porcine aortas (P<0.001; Figure 1). In addition, equimolar concentrations of ARC15105 inhibited platelet adhesion 9.5-fold greater and the area covered by fluorescent platelets 5.4-fold greater compared to ARC1709 (P<0.001; Figure 2) in flow chamber.
Discussion
VWF has become an interesting target for the treatment of arterial thromboembolism.9 We have tested the effects of the anti-VWF aptamer ARC15105 in different complementary assays.
The platelet function analyzer-100 has originally been designed to detect VWF deficiency. Therefore, the platelet function analyzer-100 closure times are prolonged when the VWF activity drops to 50% to 60% of normal. Collagen/ADP induced platelet plug formation was inhibited by 50% (IC50) at 12 nmol/L and complete inhibition (IC100) occurred at 13 to 107 nmol/L of ARC15105 in all samples. These concentrations are in good agreement with (1) a 50% reduction of VWF activity in the ELISA at 67 nmol/L, (2) the almost complete suppression of platelet adhesion to collagen-bound VWF under arterial shear conditions at 100 nmol/L, and (3) the >90% reduction in adhesion of platelets on injured porcine arterial segments in perfusion flow chambers at 40 nmol/L. Hence, these complementary methods are all in good agreement, that effective concentrations of ARC15105 are in the range of <130 nmol/L (1 μg/mL). For comparison, a clinical trial also showed that concentrations of 220 to 380 nmol/L (3–5 μg/mL) ARC1779, which has a 2-fold higher molecular weight, were sufficient to counteract hyper-functional VWF in the setting of von Willebrand disease type 2B.12
Plasma concentrations of the VWF aptamers are in excess of the binding capacity of the VWF, thus the majority of VWF is free unbound and cleared renally. Renal clearance of unbound VWF decreases by increasing PEG sizes. Little is known about the clearance of VWF-bound aptamer. However, at least for ARC15105 it is unlikely a major determinant, because ARC15105 concentration would than be expected to decline with the estimated half life of VWF, which is in the range of 6 to 8 hours.
ARC15105 (1.3 μmol/L) completely suppressed ristocetin induced platelet aggregation in whole blood. Also there was quite a marked decrease in collagen induced aggregation. This can be explained by the fact that collagen/VWF are a ligand/receptor pair. Although, ARC15105 targets the A1 domain, our in vitro experiments showed that ARC15105 reduces VWF collagen binding activity by >66%. This could be due to a potential change in VWF conformation and/or the large size of the drug blocking the VWF/collagen interaction. Interestingly, ARC15105 also slightly inhibited aggregation induced by other agonists. This could potentially reflect a minor role of the VWF/GPIb complex in amplifying aggregation, which may deserve further investigation. As VWF might not contribute to platelet aggregation through this mechanism, the effects observed are surprising and deserve more investigation.
However, overall ARC15105 was highly specific for VWF, particularly when compared to the effects of abciximab, which fully blocked aggregation to all agonists except collagen in the 2 pretreated patients. This high degree of specificity of ARC15105 is in good agreement with the high specificity of the predecessor ARC1779.11,25
The investigated pegylated aptamer ARC15105 has been developed to provide a long-lasting inhibition of VWF after SC injection. Indeed, the formulation provided 98% bioavailability after SC injection, and the half-life is ≈65 hours. This is a substantial increase in bioavailability and half-life as compared to its’ predecessor ARC1779,13 which has a short half-live of approximately 2 to 3 hours. Based on human clearance (CL) and volume of distribution (Vss) estimates extrapolated from allometric scaling, the predicted human t1/2 for ARC15105 would be ≈217 hours. Injection of ARC15105 inhibited VWF activity by >90% in monkey blood samples taken 10 to 12 days after SC injection. Modeling predicts that a single SC bolus of ARC15105 should completely inhibit VWF activity in humans for at least 12 days. As this compound has a long half-life, it may be useful for the treatment of thrombotic microangiopathies. Nevertheless, such a long predicted half-life suggests that reversing activity in case of toxicity could be desirable. Theoretically, complementary antidotes can be developed, VWF concentrates could be given, and plasma exchange has been found to effectively reduce plasma concentrations of aptamers.13,26
In vitro experiments indicate that ARC15105 is not only bound to VWF (plasma levels average about 50 nmol/L), but ARC15105 levels exceed the binding capacity of VWF at higher concentrations; thus ARC15105 is also present as free, unbound molecule. The increased half-life of ARC15105 (>60 hours) relative to ARC1779 (approx. 2–3 hours)27 is a consequence of a 40-kDa PEG as compared to a 20-kDa PEG for ARC1779. Little is known about the in vivo clearance of ARC15105 bound to VWF. However, the long half-life of ARC15105 practically excludes that VWF half-life (estimated <12 hours) is a major determinant of ARC15105 half-life. In contrast, ARC1779 has been shown to slow VWF clearance at least in patients with von Willebrand disease type 2b.12
Dose response curves for ARC1779 indicate that the affinity of ARC1779 to the VWF A1 free domain is comparable between healthy volunteers, patients with TTP, or those with VWF disease.12,13,25 This is an indicator that the capacity of ARC1779 is roughly comparable in subjects with normal, low or high multimer composition.
Efficacy was previously also shown for the predecessor molecule ARC1779 in an electric injury model of carotid arterial thrombosis: the anti-VWF aptamer produced less bleeding in a surgicutt wound at equi-effective concentrations as compared to abciximab.22 This could potentially point toward a larger therapeutic window for anti-VWF aptamers as compared to GPIIb/IIIa inhibitors.
Limitations
The current study has only focused on the functional characterization and pharmacokinetic and pharmacodynamic profile of the new anti-VWF aptamer ARC15105. Further animal or human studies models investigating the efficacy and safety of this compound to treat thrombotic disorders are required.
Conclusion
ARC15105 effectively inhibits VWF when samples from patients with myocardial infarction as well as healthy controls are tested in various assay systems. Its SC bioavailability and long half-life make it a good drug-candidate.
Supplementary Material
Sources of Funding
D.D. and D.D.W. were supported by National Heart, Lung and Blood Institute of the National Institutes of Health grant R01 HL041002.
Footnotes
Disclosures
The study was sponsored by the Archemix Corporation and Robert G. Schaub, Kathleen E. McGinness, P. Shannon Pendergrast, Jou-Ku Chung, and Xianbin Tian are employees of the company. Bernd Jilma was a consultant. Other authors declare no competing financial interests.
The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.111.237529/-/DC1.
Contributor Information
Jolanta M. Siller-Matula, Department of Cardiology, Medical University of Vienna; Austria
Yahye Merhi, Laboratory of Thrombosis and Hemostasis, Montreal Heart Institute and Université de Montréal; Canada.
Jean-François Tanguay, Laboratory of Thrombosis and Hemostasis, Montreal Heart Institute and Université de Montréal; Canada.
Daniel Duerschmied, Immune Disease Institute, Boston, MA; Department of Pediatrics, Harvard Medical School, Boston, MA; Department of Cardiology and Angiology, University Medical Center Freiburg, Freiburg, Germany.
Denisa D. Wagner, Immune Disease Institute, Boston, MA Department of Pediatrics, Harvard Medical School, Boston, MA; Program in Cellular and Molecular Medicine, Children’s Hospital Boston, Boston, MA.
Kathleen E. McGinness, Archemix Corporation, Cambridge, MA
P. Shannon Pendergrast, Archemix Corporation, Cambridge, MA.
Jou-Ku Chung, Archemix Corporation, Cambridge, MA.
Xianbin Tian, Archemix Corporation, Cambridge, MA.
Robert G. Schaub, Archemix Corporation, Cambridge, MA
Bernd Jilma, Department of Clinical Pharmacology, Medical University of Vienna, Vienna, Austria.
References
- 1.Bergmeier W, Chauhan AK, Wagner DD. Glycoprotein Ibalpha and von Willebrand factor in primary platelet adhesion and thrombus formation: lessons from mutant mice. Thromb Haemost. 2008;99:264–270. [DOI] [PubMed] [Google Scholar]
- 2.Spiel AO, Gilbert JC, Jilma B. von Willebrand factor in cardiovascular disease: focus on acute coronary syndromes. Circulation. 2008;117:1449–1459. [DOI] [PubMed] [Google Scholar]
- 3.Sadler JE. Biochemistry and genetics of von Willebrand factor. Annu Rev Biochem. 1998;67:395–424. [DOI] [PubMed] [Google Scholar]
- 4.Wise RJ, Dorner AJ, Krane M, Pittman DD, Kaufman RJ. The role of von Willebrand factor multimers and propeptide cleavage in binding and stabilization of factor VIII. J Biol Chem. 1991;266:21948–21955. [PubMed] [Google Scholar]
- 5.Collet JP, Montalescot G, Vicaut E, Ankri A, Walylo F, Lesty C, Choussat R, Beygui F, Borentain M, Vignolles N, Thomas D. Acute release of plasminogen activator inhibitor-1 in ST-segment elevation myocardial infarction predicts mortality. Circulation. 2003;108:391–394. [DOI] [PubMed] [Google Scholar]
- 6.Fuchs I, Frossard M, Spiel A, Riedmuller E, Laggner AN, Jilma B. Platelet function in patients with acute coronary syndrome (ACS) predicts recurrent ACS. J Thromb Haemost. 2006;4:2547–2552. [DOI] [PubMed] [Google Scholar]
- 7.Thompson SG, Kienast J, Pyke SD, Haverkate F, van de Loo JC. Hemostatic factors and the risk of myocardial infarction or sudden death in patients with angina pectoris. European Concerted Action on Thrombosis and Disabilities Angina Pectoris Study Group. N Engl J Med. 1995;332:635–641. [DOI] [PubMed] [Google Scholar]
- 8.Siller-Matula J, Schror K, Wojta J, Huber K. Thienopyridines in cardiovascular disease: focus on clopidogrel resistance. Thromb Haemost. 2007;97:385–393. [PubMed] [Google Scholar]
- 9.Firbas C, Siller-Matula JM, Jilma B. Targeting von Willebrand factor and platelet glycoprotein Ib receptor. Expert Rev Cardiovasc Ther. 2010;8:1689–1701. [DOI] [PubMed] [Google Scholar]
- 10.Siller-Matula JM, Krumphuber J, Jilma B. Pharmacokinetic, pharmacodynamic and clinical profile of novel antiplatelet drugs targeting vascular disease. British Journal of Pharmacology. 2009;159:502–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spiel AO, Mayr FB, Ladani N, Wagner PG, Schaub RG, Gilbert JC, Jilma B. The aptamer ARC1779 is a potent and specific inhibitor of von Willebrand Factor mediated ex vivo platelet function in acute myocardial infarction. Platelets. 2009;20:334–340. [DOI] [PubMed] [Google Scholar]
- 12.Jilma B, Paulinska P, Jilma-Stohlawetz P, Gilbert JC, Hutabarat R, Knobl P. A randomised pilot trial of the anti-von Willebrand factor aptamer ARC1779 in patients with type 2b von Willebrand disease. Thromb Haemost. 2010;104:563–570. [DOI] [PubMed] [Google Scholar]
- 13.Jilma-Stohlawetz P, Gorczyca ME, Jilma B, Siller-Matula J, Gilbert JC, Knobl P. Inhibition of von Willebrand factor by ARC1779 in patients with acute thrombotic thrombocytopenic purpura. Thromb Haemost. 2011;105:545–552. [DOI] [PubMed] [Google Scholar]
- 14.Jilma-Stohlawetz P, Gilbert JC, Gorczyca ME, Knöbl P, Jilma B. A dose ranging phase I/II trial of the von Willebrand factor inhibiting aptamer ARC1779 in patients with congenital thrombotic thrombocytopenic purpura. Thromb Haemost. 2011;106:539–547. [DOI] [PubMed] [Google Scholar]
- 15.Gutsaeva DR, Parkerson JB, Yerigenahally SD, Kurz JC, Schaub RG, Ikuta T, Head CA. Inhibition of cell adhesion by anti-P–selectin aptamer: a new potential therapeutic agent for sickle cell disease. Blood. 2011;117:727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siller-Matula JM, Christ G, Lang IM, Delle-Karth G, Huber K, Jilma B. Multiple Electrode Aggregometry predicts stent thrombosis better than the VASP assay. J Thromb Haemost. 2010;8:351–359. [DOI] [PubMed] [Google Scholar]
- 17.Siller-Matula JM, Gouya G, Wolzt M, Jilma B. Cross validation of the Multiple Electrode Aggregometry. A prospective trial in healthy volunteers. Thromb Haemost. 2009;102:397–403. [DOI] [PubMed] [Google Scholar]
- 18.Siller-Matula JM, Haberl K, Prillinger K, Panzer S, Lang I, Jilma B. The effect of antiplatelet drugs clopidogrel and aspirin is less immediately after stent implantation. Thromb Res. 2009;123:874–880. [DOI] [PubMed] [Google Scholar]
- 19.Jilma B Platelet function analyzer (PFA-100): a tool to quantify congenital or acquired platelet dysfunction. J Lab Clin Med. 2001;138:152–163. [DOI] [PubMed] [Google Scholar]
- 20.Derhaschnig U, Pachinger C, Jilma B. Variable inhibition of high-shear-induced platelet plug formation by eptifibatide and tirofiban under conditions of platelet activation and high von Willebrand release: a randomized, placebo-controlled, clinical trial. Am Heart J. 2004;147:E17. [DOI] [PubMed] [Google Scholar]
- 21.Arzamendi D, Dandachli F, Theoret JF, Ducrocq G, Chan M, Mourad W, Gilbert JG, Schaub RG, Tanguay JF, Merhi Y. An anti-von Willebrand factor aptamer reduces platelet adhesion among patients receiving aspirin and clopidogrel in an ex vivo shear-induced arterial thrombosis. Clin Appl Thromb Hemost. 2010:doi: 10.1177/1076029610384114. [DOI] [PubMed] [Google Scholar]
- 22.Diener JL, Daniel Lagasse HA, Duerschmied D, Merhi Y, Tanguay JF, Hutabarat R, Gilbert J, Wagner DD, Schaub R. Inhibition of von Willebrand factor-mediated platelet activation and thrombosis by the anti-von Willebrand factor A1-domain aptamer ARC1779. J Thromb Haemost. 2009;7:1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Theoret JF, Bienvenu JG, Kumar A, Merhi Y. P-selectin antagonism with recombinant p-selectin glycoprotein ligand-1 (rPSGL-Ig) inhibits circulating activated platelet binding to neutrophils induced by damaged arterial surfaces. J Pharmacol Exp Ther. 2001;298:658–664. [PubMed] [Google Scholar]
- 24.Ruggeri ZM, Mendolicchio GL. Adhesion mechanisms in platelet function. Circ Res. 2007;100:1673–1685. [DOI] [PubMed] [Google Scholar]
- 25.Mayr FB, Knobl P, Jilma B, Siller-Matula JM, Wagner PG, Schaub RG, Gilbert JC, Jilma-Stohlawetz P. The aptamer ARC1779 blocks von Willebrand factor-dependent platelet function in patients with thrombotic thrombocytopenic purpura ex vivo. Transfusion. 2010;50:1079–1087. [DOI] [PubMed] [Google Scholar]
- 26.Becker RC, Povsic T, Cohen MG, Rusconi CP, Sullenger B. Nucleic acid aptamers as antithrombotic agents: opportunities in extracellular therapeutics. Thromb Haemost. 2010;103:586–595. [DOI] [PubMed] [Google Scholar]
- 27.Gilbert JC, DeFeo-Fraulini T, Hutabarat RM, Horvath CJ, Merlino PG, Marsh HN, Healy JM, Boufakhreddine S, Holohan TV, Schaub RG. First-in-human evaluation of anti von Willebrand factor therapeutic aptamer ARC1779 in healthy volunteers. Circulation. 2007;116:2678–2686. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.