

Summary
There is great interest in discovering new targets for pain therapy since current methods of analgesia are often only partially successful. Although protein kinase C (PKC) enhances nociceptor function, it is not known which PKC isozymes contribute. Here, we show that epinephrine-induced mechanical and thermal hyperalgesia and acetic acid–associated hyperalgesia are markedly attenuated in PKCϵ mutant mice, but baseline nociceptive thresholds are normal. Moreover, epinephrine-, carrageenan-, and nerve growth factor– (NGF-) induced hyperalgesia in normal rats, and epinephrine-induced enhancement of tetrodotoxin-resistant Na+ current (TTX-R INa) in cultured rat dorsal root ganglion (DRG) neurons, are inhibited by a PKCϵ-selective inhibitor peptide. Our findings indicate that PKCϵ regulates nociceptor function and suggest that PKCϵ inhibitors could prove useful in the treatment of pain.
Introduction
Recent evidence suggests that protein kinase C (PKC) regulates nociceptor function. Tumor-promoting phorbol esters, which activate most PKC isozymes (Nishizuka, 1992), sensitize nociceptors (Schepelmann et al., 1993) and enhance, in nociceptors, a current activated by a noxious thermal stimulus (Cesare and McNaughton, 1996). In addition, PKC is an important contributor to diabetic neuropathic hyperalgesia (Ahlgren and Levine, 1994), bradykinin-induced activation and sensitization of nociceptors (McGuirk and Dolphin, 1992), and epinephrine-induced hyperalgesia and nociceptor sensitization (Khasar et al., 1999). However, PKC is a family of at least ten serine-threonine kinases, and while there are functional differences between PKC isozymes (Nishizuka, 1992), it is not known which isozyme contributes to nociceptor responses.
Recent findings suggest that PKCϵ may be important in nociceptor function. PKCϵ is activated by nerve growth factor (NGF) (Ohmichi et al., 1993) and modulates several of NGF’s actions, including activation of mitogen-activated protein (MAP) kinases Erk1 and Erk2 and neurite outgrowth (Hundle et al., 1995, 1997). In addition, NGF is elevated at sites of inflammation (Safieh-Garabedian et al., 1995) and produces hyperalgesia (Lewin et al., 1993). The observations that NGF exerts hyperalgesic effects and that PKCϵ regulates responses to NGF led us to postulate that PKCϵ plays a role in pain signaling.
In the present study, we tested this hypothesis by examining epinephrine- and acetic acid–associated hyperalgesia in PKCϵ mutant mice and epinephrine-, carrageenan-, and NGF-induced hyperalgesia in normal rats treated with a selective peptide inhibitor of PKCϵ. Our findings provide strong evidence that PKCϵ regulates nociceptor function.
Results
Characterization of PKCϵ Mutant Mice
We used embryonic stem (ES) cells to generate PKCϵ mutant mice by homologous recombination (Thompson et al., 1989). A recombination vector was constructed containing mouse genomic PKCϵ sequences harboring a 1.2 kb deletion. Integration of this vector was predicted to result in loss of the start codon (Figure 1A). This vector was transfected into ES cells, and nine clones containing the homologous integration were identified. Two were injected into blastocysts to produce chimeric animals. Three chimeric males mated to C57BL/6J females transmitted the mutation through the germline. Heterozygous progeny were intercrossed, and offspring were typed by Southern analysis for the PKCϵ mutation (Figure 1B).
Figure 1.
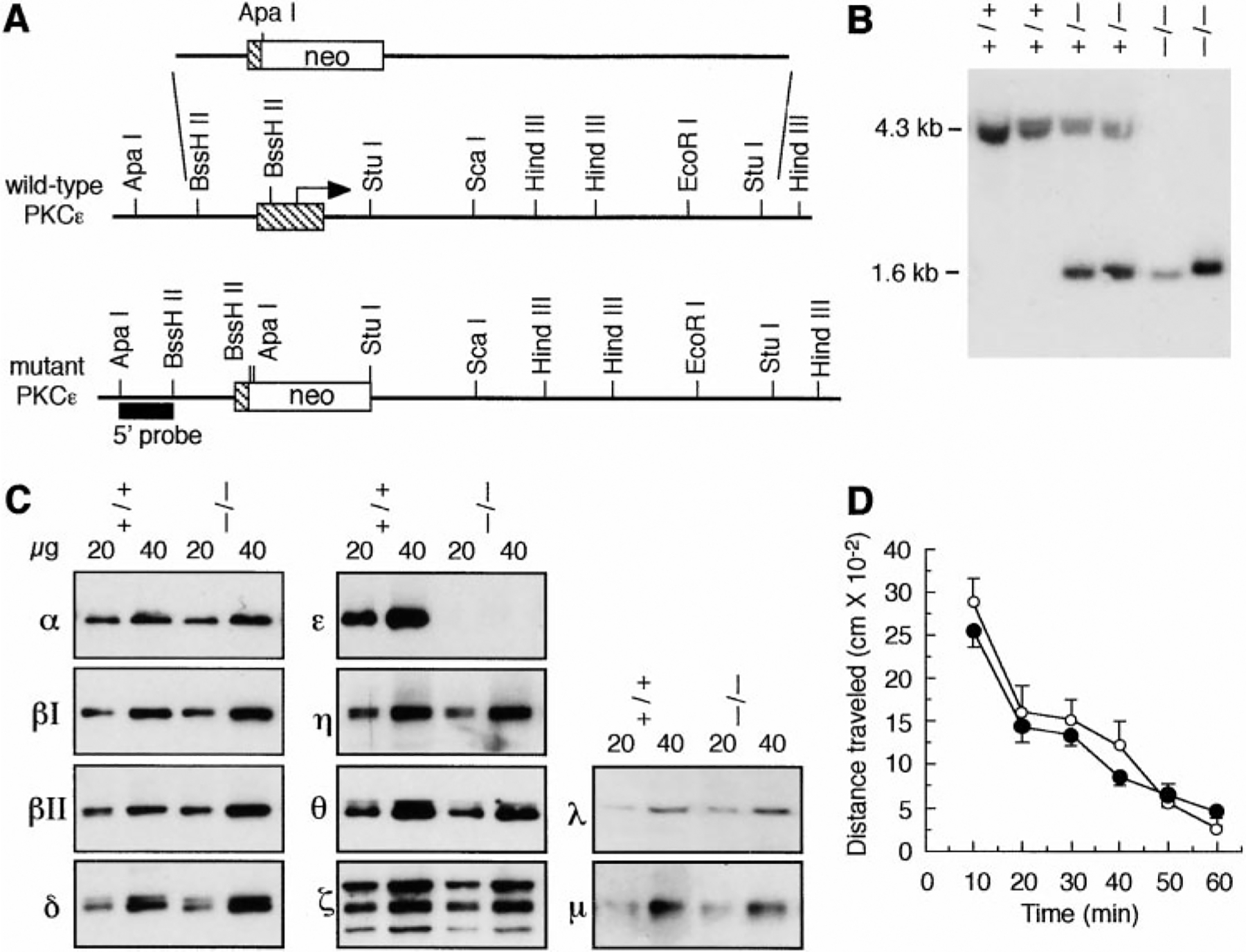
Gene Targeting of Mouse PKCϵ
(A) Schematic structures of wild-type and recombinant loci, the targeting vector, and the 5’ probe used for screening ES cell clones and mice.
(B) Southern blot of genomic DNA following digestion with ApaI and ScaI, showing fragments corresponding to wild-type (4.3 kb) and targeted (1.6 kb) alleles.
(C) Western blots of DRG tissue from wild-type (+/+) and PKCϵ mutant (−/−) mice, using antibodies to PKC isozymes. Three immunoreactive bands of 65, 74, and 84 kDa were detected with the anti-PKCζ antibody. The 74 kDa band corresponds to what was reported for rat brain PKCζ (Hunter et al., 1995).
(D) Spontaneous locomotor activity and habituation of wild-type (white circles) and PKCϵ mutant (black circles) mice over 1 hr. Data are mean ± SE values from six mice of each genotype.
Using Western blot analysis, we found that PKCϵ immunoreactivity could not be detected in dorsal root ganglia (DRG) (Figure 1C) or in the central nervous system (data not shown) of mutant mice. All other PKC isozymes except PKCγ could be detected in DRG from wild-type mice, and their abundance was similar in wild-type and PKCϵ mutant mice (Figure 1C). From 75 litters of heterozygous breeding pairs, 826 mice were genotyped at weaning: 193 (23%) were wild-type, 441 (54%) were heterozygotes, and 192 (23%) were homozygotes, showing an expected Mendelian ratio (χ2 = 1.83, p = 0.40). Food and water intake were normal in mutant mice (data not shown), and body weight measured daily over 2 weeks in 8-week-old animals was similar in PKCϵ-mutant (24.73 ± 0.9 g, n = 8) and wild-type (23.5 ± 0.9 g, n = 8) mice. Locomotor activity (Figure 1D) and performance on a 3-arm maze (data not shown) were also similar in mutant and wild-type mice, and mutants could not be distinguished from wild-type littermates in the normal cage environment. Mutant mice have survived for over 1 year, and the spontaneous mortality rate among 500 mice of each genotype maintained at least 6 weeks was similar for PKCϵ mutant (2.6%) and wild-type (1.2%) animals (χ2 = 1.28, p = 0.26).
PKCϵ Mutant Mice Show Decreased Epinephrine-Induced and Acetic Acid–Associated Hyperalgesia
To study the role of PKCϵ in mechanical and thermal nociceptor function, we first examined mice treated with epinephrine. Epinephrine-induced hyperalgesia and nociceptor sensitization depend on both PKC and protein kinase A (PKA) (Khasar et al., 1999). We chose to use epinephrine rather than bradykinin, which also activates PKC, because epinephrine acts directly on the primary afferent nociceptor (Khasar et al., 1999), whereas bradykinin can act indirectly (Andreev et al., 1995). We then compared epinephrine-induced responses with responses induced by prostaglandin E2 (PGE2) since PGE2-stimulated sensitization is mediated only by PKA (Taiwo and Levine, 1991).
Mechanical hyperalgesia was evaluated by von Frey hair (VFH) stimulation. The percent withdrawal response to VFH stimulation was similar in wild-type and mutant mice (Figure 2A). However, the increase in withdrawal response following epinephrine injection was significantly reduced in mutant animals. Nevertheless, mutant mice and their wild-type littermates showed a similar increase in response produced by PGE2. Likewise, when thermal stimulation was employed, wild-type and mutant animals demonstrated similar basal thresholds and similar responses to PGE2, but mutant animals displayed a reduced thermal hyperalgesia compared with wild-type littermates after epinephrine administration (Figure 2B). Finally, when evaluated by the acetic acid writhing test (Bjorkman, 1995), mutant mice demonstrated a significantly lower nociceptive score than wild-type animals (Figure 2C).
Figure 2.
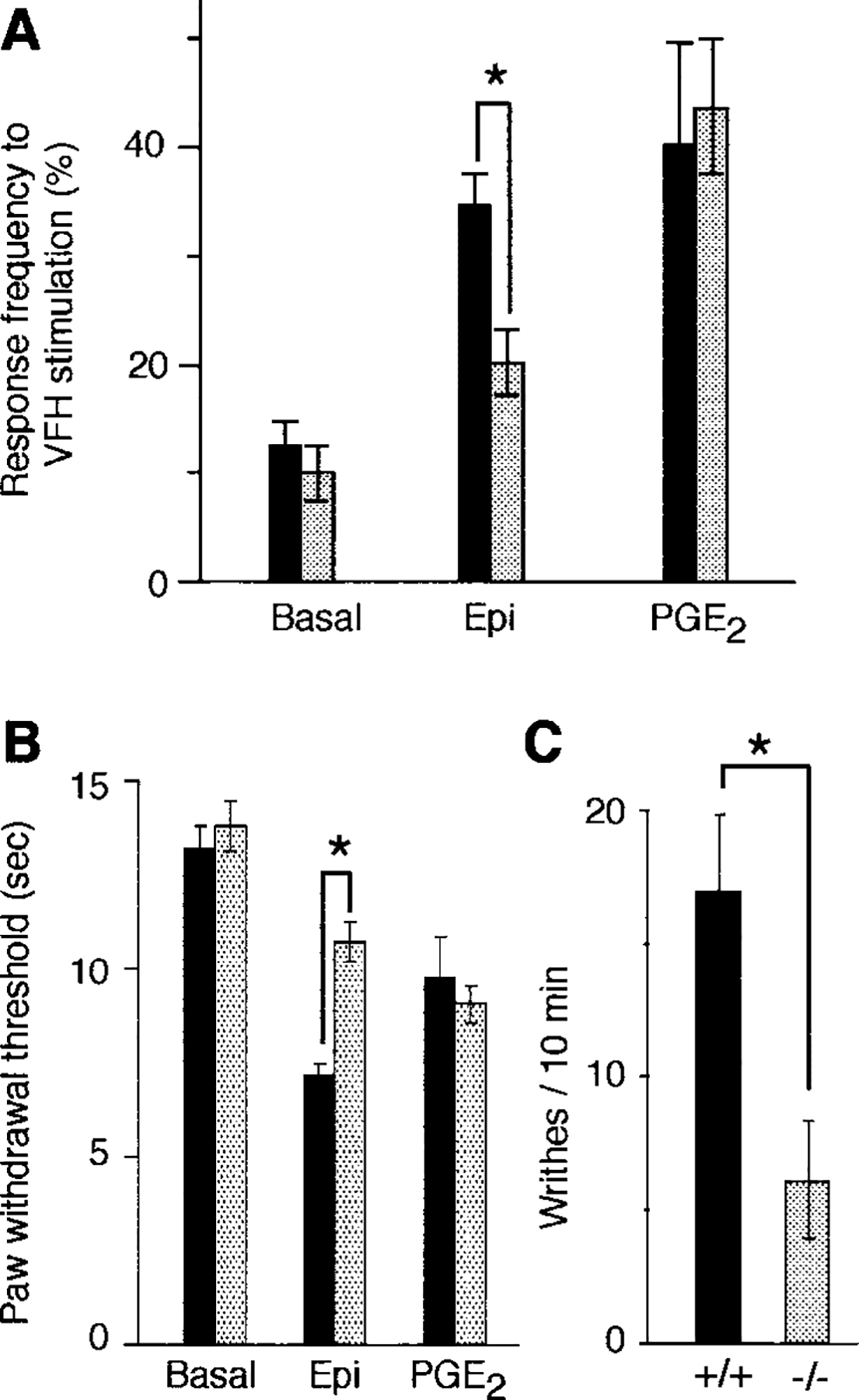
Mechanical and Thermal Nociception and Response to Acetic Acid in PKCϵ Mutant Mice
(A) Mechanical thresholds were measured in eight wild-type (black bars) and eight mutant (gray bars) mice in response to VFH (4 g) stimulation. Responses were measured before (basal) and after intraplantar injection of 100 ng epinephrine (Epi) or PGE2. Asterisk, p < 0.001 (ANOVA and Scheffe F test).
(B) Thermal nociceptive thresholds in eight wild-type (black bars) and eight mutant (gray bars) mice before and after intraplantar injection of 100 ng epinephrine (Epi) or PGE2. Asterisk, p < 0.001 (ANOVA and Scheffe F test).
(C) Writhing response to acetic acid administration in four wild-type (+/+) and four mutant (−/−) mice. Asterisk, p < 0.05 (Student’s unpaired t test).
A Selective Inhibitor of PKCϵ Decreases Epinephrine-Induced Hyperalgesia in Rats
The studies above suggest that a defect in PKCϵ-mediated signal transduction accounts for reduced epinephrine-induced hyperalgesia. These results were not due to a deficit in motor function since PKCϵ mutant mice demonstrated enhanced withdrawal responses when treated with PGE2. However, these experiments do not exclude the possibility that this phenotype arises from the absence of PKCϵ during development. To address this issue, we examined nociceptive responses in adult rats treated with a selective inhibitor of PKCϵ, so that virtually immediate assessment of hyperalgesia following antagonism of PKCϵ function could be performed. In addition, we treated rats with an inhibitor of multiple PKC isozymes to evaluate possible contributions of other isozymes to nociceptor sensitization.
Upon activation, PKC isozymes translocate to specific intracellular sites, where they appear to bind to anchoring proteins termed RACKs (Mochly-Rosen, 1995). The cotamer protein β’COP has recently been identified as a specific RACK for PKCϵ (Csukai et al., 1997). PKCϵ binds β’COP through its V1 region, and peptides derived from this region selectively inhibit phorbol ester–mediated translocation of PKCϵ in cultured cardiac myocytes (Johnson et al., 1996; Gray et al., 1997), PC12 cells (Hundle et al., 1997), and pancreatic beta cells (Yedovitzky et al., 1997). This inhibition of binding decreases phorbol ester- and norepinephrine-mediated inhibition of contraction in cardiac myocytes (Johnson et al., 1996) and inhibits protection of cardiac myocytes from hypoxia-induced cell death (Gray et al., 1997). It also reduces glucose-stimulated insulin secretion from islet cells (Yedovitzky et al., 1997) and prevents enhancement of NGF-induced neurite outgrowth and MAP kinase activation by phorbol esters in PC12 cells (Hundle et al., 1997). We used such an inhibitory peptide, ϵV1–2 (Johnson et al., 1996; Gray et al., 1997; Yedovitzky et al., 1997) to selectively inhibit PKCϵ in peripheral nerve terminals.
Intradermal injection of ϵV1–2 in normal rats decreased the mechanical hyperalgesia produced by injection of epinephrine into the hindpaw (Figure 3A). There was no effect from a scrambled ϵV1–2 peptide (Johnson et al., 1996). The magnitude of inhibition produced by ϵV1–2 was similar to that produced by the PKC inhibitor bisindoylmaleimide I, which inhibits several PKC isozymes, including PKCϵ (Toullec et al., 1991). Administration of ϵV1–2 had no effect on PGE2-induced hyperalgesia. These results strongly support the conclusion that absence of PKCϵ-mediated signaling in adult neurons is responsible for reduced epinephrine sensitivity in PKCϵ mutant mice. Furthermore, since bisindoylmaleimide I was no more effective in inhibiting epinephrine hyperalgesia than the ϵV1–2 peptide, it is likely that the PKCϵ isozyme alone is responsible for the contribution of PKC to epinephrine-induced hyperalgesia.
Figure 3.
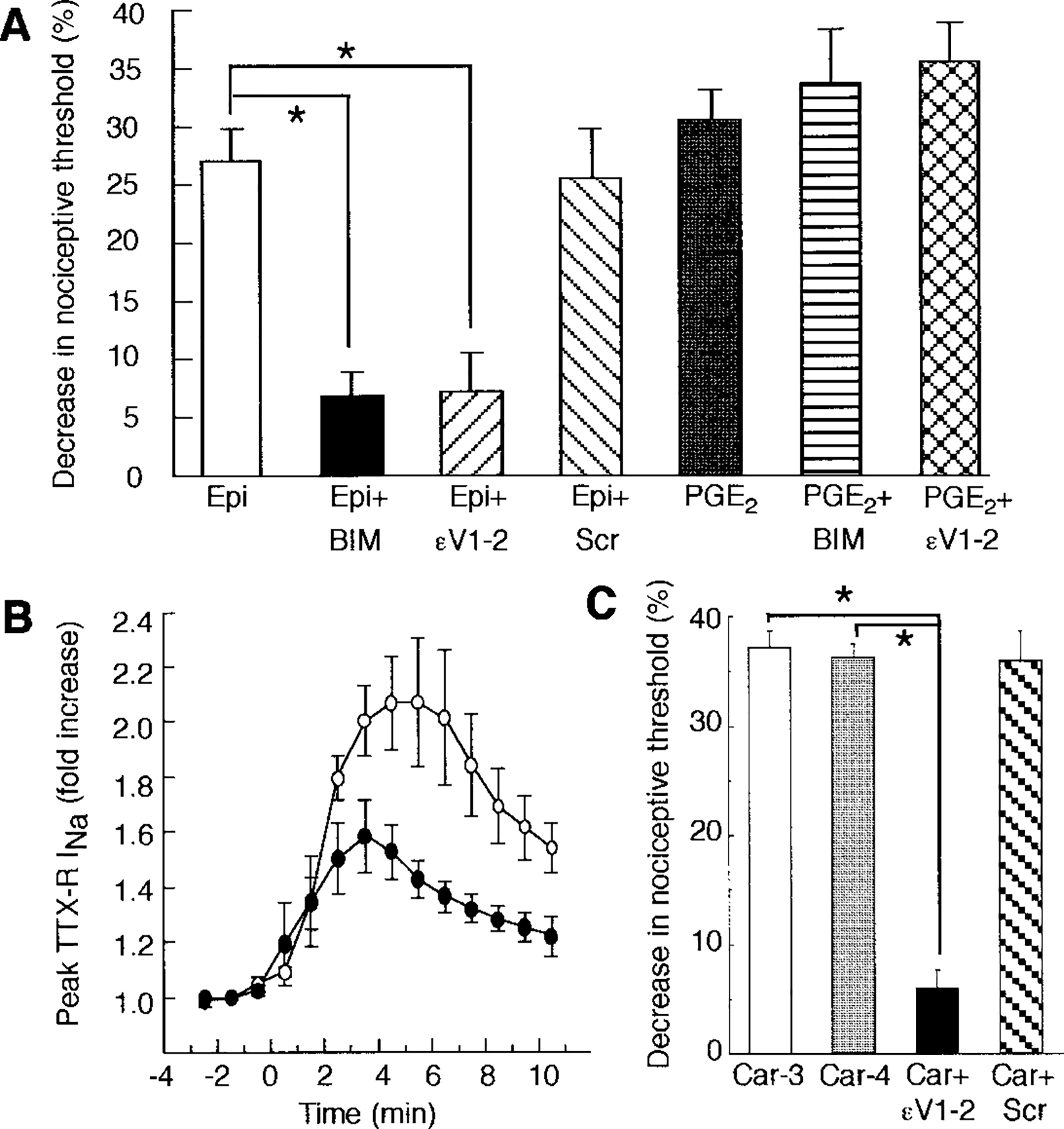
PKC Inhibitors Attenuate Epinephrine- and Carrageenan-Induced Mechanical Hyperalgesia and Epinephrine-Induced Enhancement of TTX-R INa
(A) Rats were treated intradermally with 100 ng epinephrine (Epi) or PGE2 with or without local injection of either 1 μg bisindoylmaleimide I (BIM) or sterile water followed by 1 μg of ϵV1–2 peptide (ϵV1–2; EAVSLKPT) or 1 μg of scrambled ϵV1–2 peptide (Scr; LSETKPAV). The mean ± SE baseline threshold before drug treatment in the group receiving epinephrine was 105.3 ± 2.8 g (n = 8). Data are expressed as the percent change from baseline and are mean + SE values from eight (epinephrine) and ten (PGE2) experiments. Asterisk, p < 0.001 (ANOVA and Scheffe F test).
(B) TTX-R INA, was measured in cells intracellularly perfused through the patch-clamp electrode with 200 μM ϵV1–2 (black circles, n = 4) or 200 μM scrambled ϵV1–2 (white circles, n = 4) for 15 min prior to application of 1 μM epinephrine. There was a significant decrease in the effect of epinephrine in cells treated with ϵV1–2 peptide (p = 0.04 [two-way repeated measures ANOVA]).
(C) Rats were treated intradermally with 1% carrageenan (10 μl per paw) and tested for mechanical hyperalgesia 3 (Car-3, n = 12) or 4 (Car-4, n = 24) hr later. Some animals received a local injection of 1 μg ϵV1–2 (Car + ϵV1–2, n = 6) or scrambled ϵV1–2 (Car + Scr, n = 6) 3.5 hr after carrageenan administration and were tested 30 min later. Asterisk, p < 0.0001 (ANOVA and Scheffe F test).
PKCϵ Inhibition Attenuates Enhancement of Tetrodotoxin-Resistant Sodium Current by Epinephrine
In DRG neurons, epinephrine enhances a tetrodotoxin-resistant sodium current (TTX-R INa) (Khasar et al., 1999) whose activity is increased by agents that act directly on primary afferent nociceptors in vivo to produce hyperalgesia (Gold et al., 1996). Bisindoylmaleimide I attenuates enhancement of TTX-R INa by epinephrine (Khasar et al., 1999), suggesting that this effect of epinephrine is partially PKC dependent. Using whole-cell patch-clamp analysis, we examined the role of PKCϵ in epinephrine sensitization of TTX-R INa in cultured rat DRG neurons. Epinephrine enhanced TTX-R INa by ~2-fold in neurons perfused with the inactive scrambled ϵV1–2 peptide (Figure 3B). Intracellular perfusion of ϵV1–2 peptide reduced this effect of epinephrine by approximately half. These results are consistent with our behavioral data (Figure 3A) and demonstrate a direct, PKCϵ-dependent action of epinephrine on TTX-R INa in primary afferent neurons.
PKCϵ Inhibition Reduces Carrageenan-Mediated Hyperalgesia
In addition to showing less epinephrine-induced hyperalgesia, PKCϵ mutant mice were less responsive than wild-type mice in the acetic acid writhing test. Since the response to acetic acid is reduced by nonsteroidal anti-inflammatory drugs (Jett et al., 1999), these results suggest that PKCϵ may be involved in inflammatory hyperalgesia. To investigate this possibility further, we examined rats treated with intradermal injection of the potent inflammatory agent carrageenan (Ferreira et al., 1988). Carrageenan-evoked mechanical hyperalgesia was maximal 3 hr after injection into the hindpaw (Figure 3C). Intradermal injection of ϵV1–2, 3.5 hr after administration of carrageenan, almost completely reversed this response. There was no effect from the scrambled ϵV1–2 peptide. These results indicate that PKCϵ contributes to inflammatory hyperalgesia.
NGF-Induced Hyperalgesia Is Not Reduced by PKCϵ Inhibition
In PC12 cells, NGF-induced neurite outgrowth and MAP kinase activation are enhanced by overexpression of PKCϵ (Hundle et al., 1995). However, downregulation (Roivainen et al., 1993) or inhibition (Hundle et al., 1997) of PKCϵ does not impair responses to NGF. These results indicate that PKCϵ modulates but is not required for NGF signaling in PC12 cells. To investigate whether PKCϵ is necessary for NGF-induced nociception, we examined rats treated with intradermal injection of NGF.
Rats were injected with 1 μg of either ϵV1–2 or scrambled ϵV1–2 peptide in the dorsal aspect of the hindpaw. After 15 min, all rats received an injection of 1 μg of NGF at the same site in the paw. Repeat doses of each peptide were administered 3 and 5.75 hr after NGF injection. Rats were then tested for mechanical hyperalgesia 6 hr after receiving NGF. NGF decreased the nociceptive threshold by 38.3% ± 3.6% (n = 6) in rats injected with the inactive scrambled ϵV1–2 peptide. In rats treated with the ϵV1–2 peptide, NGF was less effective, decreasing the nociceptive threshold by only 29.9% ± 3.6% (n = 6, p = 0.04, Student’s t test). These results indicate that PKCϵ also contributes to NGF-mediated hyperalgesia.
PKCϵ Expression in DRG Neurons
Since peripheral injection of ϵV1–2 reduced epinephrine-, carrageenan-, and NGF-induced hyperalgesia, we hypothesized that PKCϵ in the peripheral terminals of DRG neurons mediates this effect. That PKCϵ is present in the peripheral processes of sensory neurons was supported by the observation of PKCϵ immunoreactivity in hindpaw skin from normal rats at the dermal–epidermal junction (Figure 4A), where the highest density of afferents in the skin is located (McCarthy et al., 1995; Petruska et al., 1997). Under higher magnification, PKCϵ immunoreactivity in this region was observed in profiles resembling nerve fibers (Figure 4A, insert). In addition, PKCϵ immunoreactivity was found in most small and some medium diameter DRG neurons in wild-type adult mice and rats (Figures 4C, 4D, and 4G). In wild-type mice, 75% ± 4% of all DRG neurons and 93% ± 15% of small neurons (soma area, <570 μm2) (Scroggs et al., 1994) expressed PKCϵ immunoreactivity. Minimal background immunostaining was seen in DRG of PKCϵ mutant mice (Figure 4E). PKCϵ immunoreactivity was also present in the soma and process of cultured DRG neurons that express substance P (Figures 4H and 4I). Therefore, PKCϵ is present in most DRG neurons and in fibers in the skin, as well as in cell bodies and processes of cultured nociceptors.
Figure 4.
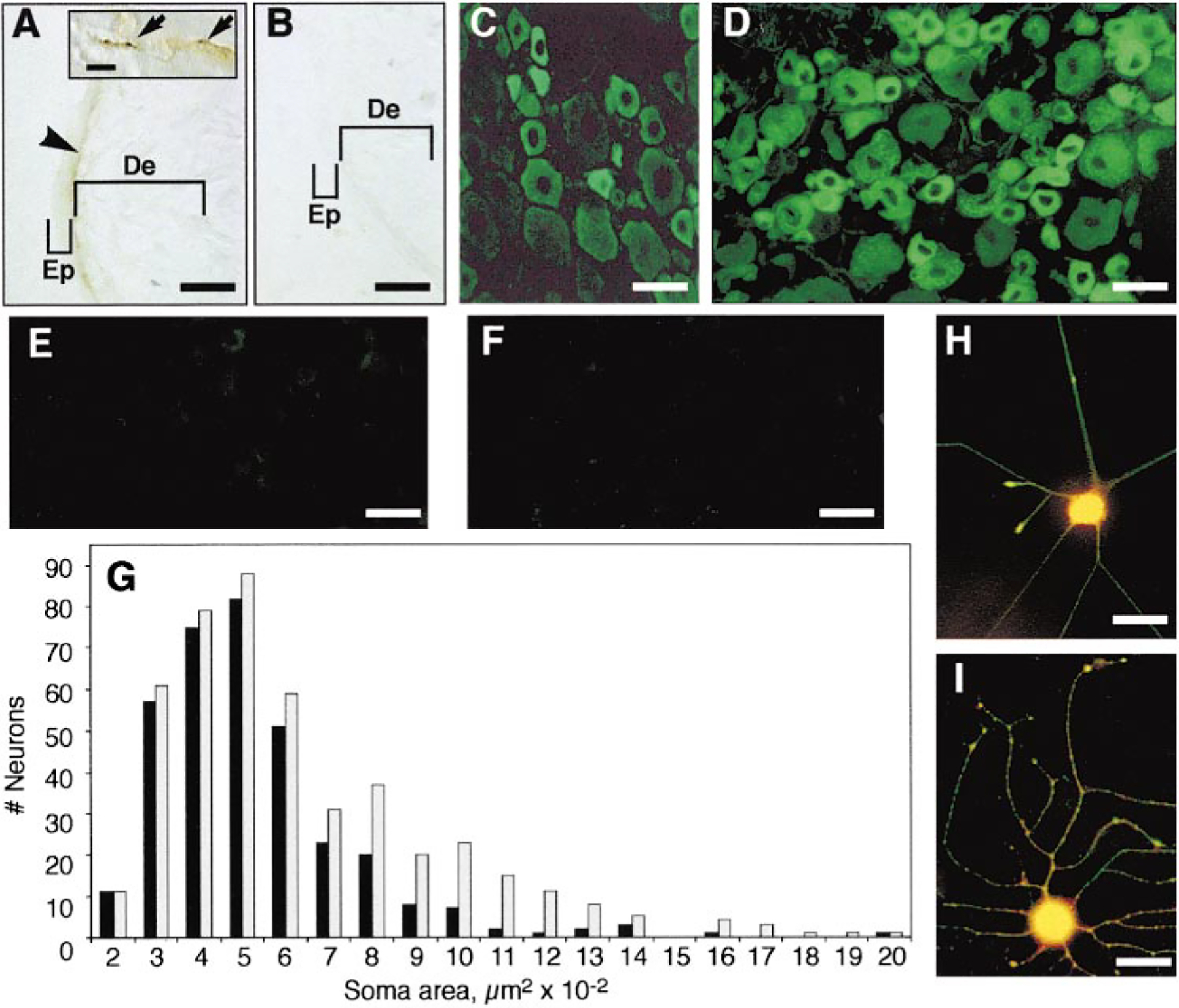
PKCϵ Immunoreactivity in Skin and DRG
Skin from normal rats was incubated with PKCϵ antibody in the absence (A) or presence (B) of PKCϵ antigen peptide. No immunoreactivity was seen when antigen was included in the incubation. PKCϵ-specific immunoreactivity was seen at the dermal–epidermal junction (arrowhead) and in the dermis, with characteristics of nerve fiber staining (inset, arrows). No staining was seen in the epidermis, possibly due to limited sensitivity of the primary antibody or because PKCϵ may be expressed in a subset of nociceptors that does not extend terminals into the epidermis. Representative sections are shown from normal rat (C), wild-type mouse (D), and PKCϵ mutant mouse (E) DRG cells stained with PKCϵ antibody. In (F), wild-type mouse DRG cells stained with secondary antibody alone. (G) shows size–frequency histogram of PKCϵ-expressing (black bars) and all (gray bars) neurons from seven wild-type mouse DRG. DRG neurons from rat (H) and mouse (I) were cultured for 7 hr in 25 ng/ml NGF and stained with antibody to substance P (red) and PKCϵ (green). Yellow color indicates colocalization of both immunoreactivities. No immunostaining was observed in cells incubated with secondary antibodies alone. Scale bar, 50 μm (A–F, H, and I) and 100 μm (inset, [A]). Abbreviations: Ep, epidermis; and De, dermis.
Discussion
For more than a decade, PKC has been suggested to play a role in nociceptive signaling. We now demonstrate the necessary contribution of a specific isozyme of PKC to primary afferent nociceptor function. We found that PKCϵ is required for full expression of epinephrine-induced sensitivity to mechanical and thermal stimuli. Because the magnitude of epinephrine-induced hyperalgesia was similar in rats treated with a nonselective PKC inhibitor and with the PKCϵ inhibitor peptide, PKCϵ may be the only PKC isozyme mediating epinephrine-induced hyperalgesia. The presence of normal PGE2-mediated responses in PKCϵ mutant mice is also consistent with our recent data indicating that PGE2 hyperalgesia and sensitization are not dependent on PKC but require PKA (Khasar et al., 1999). Epinephrine, like PGE2, enhances TTX-R INa, which is important in nociceptor sensitization and in hyperalgesia induced by inflammation (Khasar et al., 1999). The ϵV1–2 peptide reduced the effect of epinephrine on TTX-R INa, indicating that TTX-R INa is a target of PKCϵ as well as PKA. Taken together, our findings suggest that PKCϵ contributes to epinephrine-induced hyperalgesia by enhancing TTX-R INa in primary afferent nociceptors.
Our findings also indicate that PKCϵ is required for full expression of carrageenan-induced hyperalgesia. This suggests a role for PKCϵ in pain due to inflammation and is consistent with evidence linking epinephrine to inflammatory hyperalgesia. For example, carrageenan-induced inflammation increases epinephrine levels (Mikhailov and Rusanova, 1993), and β-adrenergic antagonists reduce carrageenan-induced hyperalgesia (Cunha et al., 1991). Together with our data, these findings suggest that in nociceptors, PKCϵ participates in a β-adrenergic receptor–stimulated pathway that contributes to inflammatory hyperalgesia.
The rationale for the present study was in part our observation that PKCϵ modulates NGF responses in PC12 cells (Hundle et al., 1995, 1997). While NGF is known to induce hyperalgesia (Lewin and Mendell, 1993; Woolf et al., 1994), the mechanisms involved are not known. Therefore, we tested the hypothesis that NGF-induced hyperalgesia is mediated by PKCϵ. We found that inhibition of PKCϵ with the ϵV1–2 peptide decreased NGF-induced mechanical hyperalgesia in rats. Thus, these data provide the first evidence for a signal transduction pathway that underlies NGF-induced hyperalgesia.
The finding that PKCϵ immunoreactivity was present in over 90% of small diameter DRG neurons in wild-type mice indicates that PKCϵ is expressed in nociceptors. Nevertheless, since PKCϵ is widely expressed in the central nervous system (Saito et al., 1993), where it could contribute to the development of hyperalgesia, our findings do not eliminate the possibility that PKCϵ acts by central mechanisms. However, since PGE2-induced hyperalgesia was normal despite attenuation of epinephrine- and inflammation-induced hyperalgesia, it is unlikely that our findings reflect a change in central mechanisms in PKCϵ mutant mice. Furthermore, peripheral intradermal injection of ϵV1–2 peptide was sufficient to reduce epinephrine-induced hyperalgesia in rats.
Recent evidence suggests that another PKC isozyme, PKCγ, is also involved in inflammatory hyperalgesia. In PKCγ null mice, pain responses, tissue swelling, and plasma extravasation are attenuated during the second phase of the formalin test (Malmberg et al., 1997). Since this phase of the response to formalin appears to be driven by inflammation, these findings suggest that loss of PKCγ attenuates pain due to tissue injury. Unlike PKCϵ, PKCγ cannot act by modulating signal transduction in primary afferent nociceptors since it is not expressed in those neurons. However, PKCγ immunoreactivity is present in lamina II of the spinal cord dorsal horn (Malmberg et al., 1997), suggesting that it could modulate pain via spinal mechanisms. Therefore, it appears that at least two members of the PKC family modulate pain responses, but they do so at different levels of the neuraxis.
Local administration of epinephrine exacerbates symptoms in patients with neuropathic pain (Choi and Rowbotham, 1997). Therefore, our findings suggest that epinephrine-induced activation of PKCϵ may be clinically important. Lower scores obtained in the acetic acid writhing test in PKCϵ mutant mice and blockade of carrageenan-induced hyperalgesia by a specific inhibitor of PKCϵ in rats also suggest that PKCϵ contributes to inflammatory pain. Finally, since PKCϵ does not contribute to baseline nociceptive thresholds, and PKCϵ mutant mice appear normal, it may be possible to develop PKCϵ inhibitors that ameliorate pathologic pain without producing serious systemic side effects or interfering with normal nociceptive responses.
Experimental Procedures
Generation of PKCϵ Mutant Mice
PKCϵ was cloned from a lambda FIX II 129 mouse liver genomic library (Stratagene; 946308) with an 813 bp probe (nucleotides 470–1282) from mouse PKCϵ cDNA (Schaap et al., 1989). The exon containing the ATG start codon and encoding amino acids 1–116 of mouse PKCϵ (Schaap et al., 1989) was identified. The homologous recombination vector JD825 was constructed with the vector pNTK, obtained from Dr. R. M. Mortensen (Harvard University). A 1.0 kb BssHII fragment (beginning 60 bp 5’ from the ATG start codon) was cloned into the BamHI site of pNTK between the neo and TK selection markers. A 6.1 kb StuI fragment downstream of the exon was then cloned into the HindIII site of the pNTK construct, adjacent to the neo marker. This construct was designed to delete a 1.2 kb fragment of the pkcϵ gene, including the exon containing the translation initiation codon. The completed construct was linearized with ClaI and transfected by electroporation using a Biorad gene pulsar at 250 V and 500 μF into 129/RF8 ES cells. Recent genotyping of RF8 cells indicates that they are isogenic with 129/SvJae lines (R. Farese, personal communication), not 129/SvTer, as originally reported (Meiner et al., 1996). Clones were selected by culture in the presence of G418 (180 μg/ml) and 1-(2’-deoxy-2’-fluoro-β-D-ara-binofuranosyl)-5-iodouracil (200 nM). Genomic DNA isolated from 400 colonies was digested with ApaI and ScaI restriction enzymes, Southern blotted, and hybridized with a 0.5 kb DNA probe 5’ of the flanking region in JD825. Nine clones were identified that contained the predicted homologous integration. Two were microinjected into embryonic day 3.5 C57BL/6J blastocysts. Male chimeras with 80%−90% agouti coat color were mated with C57BL/6J females, and germline transmission was documented by Southern analysis. Breeding was accomplished using mixed pairs of 10–15 heterozygous hybrid male and female mice. Experiments were performed with wild-type and homozygous mutant littermates.
Western Analysis, Immunohistochemistry, and Immunofluorescence
PKC isozymes were detected in freshly isolated DRG by Western analysis (Hundle et al., 1997) with polyclonal anti-PKC antibodies (0.5 μg/ml) from Santa Cruz Biotechnology (Santa Cruz, CA). Some DRG were dissected and cultured as described (Khasar et al., 1999) and then enriched for neurons and plated onto poly-DL-ornithine and laminin-coated 8-well chamber slides (Nunc, Naperville, IL) in medium containing 25 ng/ml NGF (Lindsay, 1988). After 7 hr in culture, cells were rinsed once in ice-cold phosphate-buffered saline (PBS) (137 mM NaCl, 2.7 mM KCl, 1.47 mM KH2PO4, 8 mM Na2HPO4, 0.5 mM MgCl2, and 0.9 mM CaCl2 [pH 7.2]) and fixed in 2% paraformaldehyde in PBS for 15 min at 4°C. Cells were incubated first in blocking buffer (PBS, 1% BSA, and 0.05% Triton X-100) for 1 hr at 25°C and then in the same buffer containing rabbit anti-PKCϵ anti-body (0.33 μg/ml; Santa Cruz Biotechnology, Santa Cruz, CA) and guinea pig anti-substance P antibody (1:400 dilution; Accurate Chemical, Westbury, NY) overnight at 4°C. Cultures were then incubated at 25°C for 1 hr in buffer containing FITC-conjugated goat anti-rabbit immunoglobulin G (IgG) and Texas Red–conjugated goat anti-guinea pig IgG (Vector Laboratories, Burlingame, CA), both at a 1:500 dilution. Wells were rinsed three times in PBS, coverslips were mounted with Vectashield (Vector Laboratories, Burlingame, CA), and immunofluorescence was observed with a Leitz DMRD microscope (Leica, Wetzlar, Germany).
For tissue sections, anesthetized mice and rats were trans-cardially perfused with PBS, and then 4% paraformaldehyde in PBS. Skin and DRGs were removed and placed in 4% paraformaldehyde for 3 hr, and then in PBS containing 20% sucrose for 24 hr and 40% sucrose for 24 hr. Sections (5–10 μm) were cut with a refrigerated microtome (Leica; SM2000R) and stored at −20°C. DRG sections were treated with methanol:acetone (1:1) for 10 min at −20°C, incubated at 25°C in PBS, 1% normal goat serum, and 0.05% Tween 20 for 1 hr, and then incubated in the same buffer containing anti-PKCϵ (0.33 μg/ml) for 1–2 hr. Sections were visualized with FITC-conjugated goat anti-rabbit IgG. The size of cell bodies was calculated by tracing profiles containing nuclei from digitized photographs of DRG sections with the program NIH Image 1.61. Cells were scored as expressing PKCϵ immunoreactivity if the intensity of immunostaining exceeded background levels observed in DRG specimens from PKCϵ mutant mice. Skin sections were pretreated with 3% H2O2 before incubation with 5% normal goat serum and 0.05% Tween 20 in Superblock (Pierce, Rockford, IL). Specimens were then incubated with anti-PKCϵ antibody (0.2 μg/ml) overnight in PBS, 1% normal goat serum, and 0.05% Tween 20, and immunoreactivity was detected, using the ABC kit (Vector) with diamino-benzamine followed by enhancementwith 0.02% osmium tetroxide.
Behavioral Tests
Animal care and handling procedures were in accordance with University of California, San Francisco and National Institutes of Health guidelines. All behavioral testing was done between 10 am and 4 pm. Sprague-Dawley rats (200–250 g) were obtained from Bantin-Kingman, Fremont, CA. Peptides were administered to rats by intradermal injection into the dorsal aspect of the hindpaw following injection of sterile water, which was used to provide hypotonic shock and to allow entry of the peptides into cells (Keeney and Linn, 1990; Lepers et al., 1990; Schulz, 1990).
For mechanical nociceptive testing in rats, the nociceptive flexion reflex was quantified with an Ugo Basile analgesymeter (Stoelting, Chicago, IL) (Aley and Levine, 1997a, 1997b). Baseline mechanical thresholds were determined before drug treatment and then in three trials performed 15, 20, and 25 min after administration of a hyperalgesic agent. The paw withdrawal threshold was calculated as the mean of these three readings.
Mechanical nociceptive thresholds in mice were measured as the withdrawal response frequency to five applications of a 4 g VFH (Stoelting, Chicago), before and after intraplantar injection of the test agent (Kinnman and Levine, 1995). Thermal thresholds in mice were determined by the Hargreave test (Aley et al., 1996), and chemical nociception in mice was determined by the acetic acid writhing test (Ward and Takemori, 1983).
Spontaneous locomotor activity in mice was measured in 17 × 17 inch Plexiglas chambers located in sound-attenuating cubicles and equipped with two sets of 16 pulse-modulated infrared photobeams to record X–Y ambulatory movements at a 100 ms resolution (Med Associates, Lafayette, IN). Mice were handled and weighed daily for 1 week prior to activity testing. On the test day, mice were weighed and placed immediately in the activity chambers. Horizontal distance traveled (cm) was recorded for 1 hr.
Cell Culture and Electrophysiology
Primary cultures ofadult rat lumbar DRGs were prepared, and TTX-R INa was examined by whole-cell voltage-clamp recording as described previously (Khasar et al., 1999). The ϵV1–2 and scrambled ϵV1–2 peptides were intracellularly perfused from the electrode for 15 min prior to bath application of 1 μM epinephrine. Throughout each experiment, the amplitude of the peak inward current was normalized to the average current measured during the 3 min prior to application of epinephrine. The time course of changes in the mean normalized current ± SE was plotted with data binned into 1 min intervals.
Acknowledgments
We thank R. V. Farese, Jr. and E. Sande for valuable help in the early phases of this work. We also thank S. Lim for excellent technical assistance. This work was supported by grants from the United States Public Health Service to J. D. L. (NS21647) and R. O. M. (AA10036) and by funds contributed by the Ernest Gallo Clinic and Research Center to R. O. M.
References
- Ahlgren SC, and Levine JD (1994). Protein kinase C inhibitors decrease hyperalgesia and C-fiber hyperexcitability in the streptozotocin-diabetic rat. J. Neurophysiol 72, 684–692. [DOI] [PubMed] [Google Scholar]
- Aley KO, and Levine JD (1997a). Different mechanisms mediate development and expression of tolerance and dependence for peripheral mu-opioid antinociception in rat. J. Neurosci 17,8018–8023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aley KO, and Levine JD (1997b). Dissociation of tolerance and dependence for opioid peripheral antinociception in rats. J. Neurosci 17, 3907–3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aley KO, Reichling DB, and Levine JD (1996). Vincristine hyperalgesia in the rat: a model of painful vincristine neuropathy in humans. Neuroscience 73, 259–265. [DOI] [PubMed] [Google Scholar]
- Andreev NY, Dimitrieva N, Koltzenburg M, and McMahon SB (1995). Peripheral administration of nerve growth factor in the adult rat produces a thermal hyperalgesia that requires the presence of sympathetic post-ganglionic neurones. Pain 63, 109–115. [DOI] [PubMed] [Google Scholar]
- Bjorkman R (1995). Central antinociceptive effects of non-steroidal anti-inflammatory drugs and paracetamol. Experimental studies in the rat. Acta Anaesthesiol. Scand Suppl. 103, 1–44. [PubMed] [Google Scholar]
- Cesare P, and McNaughton P (1996). A novel heat-activated current in nociceptive neurons and its sensitization by bradykinin. Proc. Natl. Acad. Sci. USA 93, 15435–15439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi B, and Rowbotham MC (1997). Effect ofadrenergic receptor activation on post-herpetic neuralgia pain and sensory disturbances. Pain 69, 55–63. [DOI] [PubMed] [Google Scholar]
- Csukai M, Cehn C-H, De Matteis MA, and Mochly-Rosen D (1997). Cotamer protein β’-COP, a selective binding protein (RACK) for epsilon protein kinase C. J. Biol. Chem 272, 29200–29206. [DOI] [PubMed] [Google Scholar]
- Cunha FQ, Lorenzetti BB, Poole S, and Ferreira SH (1991). Interleukin-8 as a mediator of sympathetic pain. Br. J. Pharmacol 104, 765–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira SH, Lorenzetti BB, Bristow AF, and Poole S (1988). Interleukin-1 beta as a potent hyperalgesic agent antagonized by a tripeptide analogue. Nature 334, 698–700. [DOI] [PubMed] [Google Scholar]
- Gold MS, Reichling DB, Shuster MJ, and Levine JD (1996). Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proc. Natl. Acad. Sci. USA 93, 1108–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray MO, Karliner JS, and Mochly-Rosen D (1997). A selective epsilon–protein kinase C antagonist inhibits protection of cardiac myocytes from hypoxia-induced cell death. J. Biol. Chem 272, 30945–30951. [DOI] [PubMed] [Google Scholar]
- Hundle B, McMahon T, Dadgar J, and Messing RO (1995). Overexpression of ϵ-protein kinase C enhances nerve growth factor–induced phosphorylation of mitogen-activated protein kinases and neurite outgrowth. J. Biol. Chem 270, 30134–30140. [DOI] [PubMed] [Google Scholar]
- Hundle B, McMahon T, Dadgar J, Chen C-H, Mochly-Rosen D, and Messing RO (1997). An inhibitory fragment derived from protein kinase C ϵ prevents enhancement of nerve growth factor responses by ethanol and phorbol esters. J. Biol. Chem 272,15028–15035. [DOI] [PubMed] [Google Scholar]
- Hunter SE, Seibenhener ML, and Wooten MW (1995). Atypical ζ-protein kinase C displays a unique developmental expression pattern in rat brain. Dev. Brain 85, 239–248. [DOI] [PubMed] [Google Scholar]
- Jett MF, Ramesha CS, Brown CD, Chiu S, Emmett C, Voronin T, Sun T, O’Yang C, Hunter JC, Eglen RM, and Johnson RM (1999). Characterization of theanalgesic and anti-inflammatory activities of ketorolac and its enantiomers in the rat. J. Pharmacol. Exp. Ther 288, 1288–1297. [PubMed] [Google Scholar]
- Johnson JA, Gray MO, Chen C-H, and Mochly-Rosen D (1996). A protein kinase C translocation inhibitor as isozyme-specific antagonist of cardiac function. J. Biol. Chem 271, 24962–24966. [DOI] [PubMed] [Google Scholar]
- Keeney S, and Linn S (1990). A critical review of permeabilized cell systems for studying mammalian DNA repair. Mutat. Res 236, 239–252. [DOI] [PubMed] [Google Scholar]
- Khasar SG, McCarter GM, and Levine JD (1999). Epinephrine produces a β-adrenergic receptor-mediated mechanical hyperalgesia and in vitro sensitization of rat nociceptors. J. Neurophysiol 81, 1104–1112. [DOI] [PubMed] [Google Scholar]
- Kinnman E, and Levine JD (1995). Sensory and sympathetic contributions to nerve injury–induced sensory abnormalities in the rat. Neuroscience 64, 751–767. [DOI] [PubMed] [Google Scholar]
- Lepers A, Cacan R, and Verbert A (1990). Permeabilized cells as a way of gaining access to intracellular organelles: an approach to glycosylation reactions. Biochimie 72, 1–5. [DOI] [PubMed] [Google Scholar]
- Lewin GR, and Mendell LM (1993). Nerve growth factor and nociception. Trends Neurosci. 16, 353–359. [DOI] [PubMed] [Google Scholar]
- Lewin GR, Ritter AM, and Mendell LM (1993). Nerve growth factor–induced hyperalgesia in the neonatal and adult rat. J. Neurosci 13, 2136–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay RM (1988). Nerve growth factors (NGF, BDNF) enhance axonal regeneration but are not required for survival of adult sensory neurons. J. Neurosci 8, 2394–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmberg AB, Chen C, Tonegawa S, and Basbaum AI (1997). Preserved acute pain and reduced neuropathic pain in mice lacking PKCγ. Science 278, 279–283. [DOI] [PubMed] [Google Scholar]
- McCarthy BG, Hsieh ST, Stocks A, Hauer P, Macko C, Corn-blath DR, Griffin JW, and McArthur JC (1995). Cutaneous innervation in sensory neuropathies: evaluation by skin biopsy. Neurology 45, 1848–1855. [DOI] [PubMed] [Google Scholar]
- McGuirk SM, and Dolphin AC (1992). G-protein mediation in nociceptive signal transduction: an investigation into the excitatory action of bradykinin in a subpopulation of cultured rat sensory neurons. Neuroscience 49, 117–128. [DOI] [PubMed] [Google Scholar]
- Meiner VL, Cases S, Myers HM, Sande ER, Bellosta S, Schambelan M, Pitas RE, McGuire J, Herz J, and Farese RV Jr. (1996). Disruption of the acyl-CoA: cholesterol acyltransferase gene in mice: evidence suggesting multiple cholesterol esterification enzymes in mammals. Proc. Natl. Acad. Sci. USA 93,14041–14046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhailov VV, and Rusanova AG (1993). The interrelationship of the catecholamine and protein content of the tissue of the subman-dibular salivary glands and the mucosa during the secretory cycle in chronic inflammation of the oral soft tissues. Bull. Eksp. Biol. Med 116, 472–474. [PubMed] [Google Scholar]
- Mochly-Rosen D (1995). Localization of proteinkinases byanchoring proteins: a theme in signal transduction. Science 268, 247–251. [DOI] [PubMed] [Google Scholar]
- Nishizuka Y (1992). Intracellular signaling by hydrolysis of phospho-lipids and activation of protein kinase C. Science 258, 607–614. [DOI] [PubMed] [Google Scholar]
- Ohmichi M, Zhu G, and Saltiel AR (1993). Nerve growth factor activates calcium-insensitive protein kinase C-ϵ in PC-12 rat pheo-chromocytoma cells. Biochem. J 295, 767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petruska JC, Streit WJ, and Johnson RD (1997). Localization of unmyelinated axons in rat skin and mucocutaneous tissue utilizing the isolectin GS-I-B4. Somatosens. Mot. Res 14, 17–26. [DOI] [PubMed] [Google Scholar]
- Roivainen R, McMahon T, and Messing RO (1993). Protein kinase C isozymes that mediate enhancement of neurite outgrowth by ethanol and phorbol esters in PC12 cells. Brain Res. 624, 85–93. [DOI] [PubMed] [Google Scholar]
- Safieh-Garabedian B, Poole S, Allchorne A, Winter J, and Woolf CJ (1995). Contribution of interleukin-1 beta to the inflammation-induced increase in nerve growth factor levels and inflammatory hyperalgesia. Br. J. Pharmacol 115, 1265–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito N, Itouji A, Totani Y, Osawa I, Koide H, Fujisawa N, Ogita K, andTanaka C (1993). Cellularand intracellularlocalization of ϵ-subspecies of protein kinase C in the rat brain; presynaptic localization of the ϵ-subspecies. Brain Res. 607, 241–248. [DOI] [PubMed] [Google Scholar]
- Schaap D, Parker PJ, Bristol A, Kriz R, and Knopf J (1989). Unique substrate specificity and regulatory properties of PKC-ϵ: a rationale for diversity. FEBS Lett. 243, 351–357. [DOI] [PubMed] [Google Scholar]
- Schepelmann K, Messlinger K, and Schmidt RF (1993). The effects of phorbol ester on slowly conducting afferents of the cat’s knee joint. Exp. Brain Res 92, 391–398. [DOI] [PubMed] [Google Scholar]
- Schulz I (1990). Permeabilizing cells: some methods and applications for the study of intracellular processes. Methods Enzymol. 192, 280–300. [DOI] [PubMed] [Google Scholar]
- Scroggs RS, Todorovic EG, Anderson AP, and Fox J (1994). Variation in IH, IIR, and ILEAK between acutely isolated adult rat dorsal root ganglion neurons of different size. J. Neurophysiol 71, 271–279. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, and Levine JD (1991). Further confirmation of the role of adenyl cyclase and of cAMP-dependent protein kinase in primary afferent hyperalgesia. Neuroscience 44, 131–135. [DOI] [PubMed] [Google Scholar]
- Thompson S, Clarke AR, Pow AM, Hooper ML, and Melton DW (1989). Germ line transmission and expression of a corrected HPRT gene produced by gene targeting in embryonic stem cells. Cell 56, 313–321. [DOI] [PubMed] [Google Scholar]
- Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F, et al. (1991). The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J. Biol. Chem 266, 15771–15781. [PubMed] [Google Scholar]
- Ward SJ, and Takemori AE (1983). Relative involvement of mu, kappa and delta receptor mechanisms in opiate-mediated antinociception in mice. J. Pharmacol. Exp. Ther 224, 525–530. [PubMed] [Google Scholar]
- Woolf CJ, Safieh-Garabedian B, Ma Q-P, Crilly P, and Winters J (1994). Nerve growth factor contributes to the generation of inflammatory sensory hypersensitivity. Neuroscience 62, 327–331. [DOI] [PubMed] [Google Scholar]
- Yedovitzky M, Mochly-Rosen D, Johnson JA, Gray MO, Ron D, Abramovitch E, Cerasi E, and Nesher R (1997). Translocation inhibitors define specificity of protein kinase C isoenzymes in pancreatic β-cells. J. Biol. Chem 272, 1417–1420. [DOI] [PubMed] [Google Scholar]