

Abstract
Background
The clinical management of breast cancer (BC) is mainly based on the assessment of receptor expression by tumour cells. However, there is still an unmet need for novel biomarkers important for prognosis and therapy. The tumour immune microenvironment (TIME) is thought to play a key role in prognosis and therapy selection, therefore this study aimed to describe the TIME in Ethiopian BC patients.
Methods
RNA was isolated from formalin-fixed paraffin-embedded (FFPE) tissue from 82 women with BC. Expression of PAM50 and 54 immune genes was analysed using the Nanostring platform and differentially expressed genes (DEGs) were determined using ROSALIND®. The abundance of different cell populations was estimated using Nanostring’s cell type profiling module, while tumour infiltrating lymphocytes (TILs) were analysed using haematoxylin and eosin (H&E) staining. In addition, the PIK3CA gene was genotyped for three hotspot mutations using qPCR. Kaplan-Meier survival analysis and log-rank test were performed to compare the prognostic relevance of immune subgroups.
Results
Four discrete immune phenotypes (IP1-4) were identified through hierarchical clustering of immune gene expression data. These IPs were characterized by DEGs associated with both immune activation and inhibition as well as variations in the extent of immune infiltration. However, there were no significant differences regarding PIK3CA mutations between the IPs. A downregulation of immune suppressive and activating genes and the lowest number of infiltrating immune cells were found in IP2, which was associated with luminal tumours. In contrast, IP4 displayed an active TME chracterized by an upregulation of cytotoxic genes and the highest density of immune cell infiltrations, independent of the specific intrinsic subtype. IP1 and IP3 exhibited intermediate characteristics. The IPs had a prognostic relevance and patients with an active TME had improved overall survival compared to IPs with a significant downregulation of the majority of immune genes.
Conclusion
Immune gene expression profiling identified four distinct immune contextures of the TME with unique gene expression patterns and immune infiltration. The classification into distinct immune subgroups may provide important information regarding prognosis and the selection of patients undergoing conventional treatments or immunotherapies.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13058-024-01916-4.
Keywords: Ethiopia, Breast cancer, Tumour immune microenvironment, Immune phenotypes, PAM50
Introduction
Breast cancer (BC) is one of the most common female cancer and leading cause of cancer death among women in the vast majority of countries. In 2020, approximately 2.3 million new female BC cases were diagnosed worldwide accounting for almost 25% of cancer cases in women [1]. In several sub-Saharan African countries, including Ethiopia, BC has now become the most commonly diagnosed cancer and the second cause of cancer death among women [2].
BC is a highly heterogeneous disease attributed to distinct genetic alterations in mammary epithelial cells leading to different disease manifestations. It is classified into four intrinsic subtypes (luminal A, luminal B, HER2-enriched and basal-like) based on the expression pattern of hormone receptors (estrogen and/or progesterone receptors; ER/PR), epidermal growth factor receptor 2 (HER2) and additional genomic/transcriptomic profiling [3]. This molecular subtyping has enabled an improved estimation of patients’ risk and treatment stratification for adjuvant hormonal, radiation and/or chemotherapy [4, 5]. Although the molecular subtyping has segregated BC into different prognostic groups, it does not fully represent the heterogeneity concerning genomic features and clinical outcomes. Patients of similar molecular subtypes under identical treatments have a distinct clinical outcome suggesting the need to identify additional parameters to effectively classify patients into different treatment and prognostic groups [6–8].
The tumour microenvironment (TME), consisting of cancer cells, stromal tissue, immune cells, extracellular matrix and soluble mediators has recently emerged as an important factor influencing patients’ prognosis and therapy response [9]. Immune cells as important TME components are found either in the stroma surrounding tumour nests (stromal) or in direct contact with tumour cells (intratumoural) [10]. Expression profiling of the tumour immune microenvironment (TIME) revealed the presence of distinct immune subtypes with a marked difference in prognosis within the molecular subtypes in many populations. A large study from The Cancer Genome Atlas (TCGA) demonstrated a prognostic subgroup of luminal tumours with a differential expression of immune-related genes (IRGs) [11]. Zhu and colleagues have also identified immune subtypes in luminal BC displaying distinct patterns of immune gene expression in the Asian population [12]. A similar pattern of immune-based segregation has also been described in triple-negative BC (TNBC) with significant differences in prognosis between subtypes [13].
The crucial role of the immune cell composition of the TME in prognosis, therapy selection and immunotherapy success demonstrates the high relevance of an in-depth characterization of BC lesions [14]. Based on this assumption, the present study aimed to unravel the architecture of the TME in BC from Ethiopian patients with a focus on its immune landscape and its correlation to clinical and pathological parameters and prognosis.
Materials and methods
Patients and samples
Eighty-two patients, diagnosed with BC between 2013 and 2019 at Tikur Anbessa Specialized Hospital, Addis Ababa, Ethiopia, were enrolled in this study. Formalin-fixed paraffin-embedded (FFPE) samples from each patient were examined by an experienced pathologist using hematoxylin and eosin (H&E) staining to determine the presence of primary cancer cells and to quantify tumour infiltrating lymphocytes (TILs). Immunohistochemical analyses (IHC) of the expression of ER, PR, HER2 and Ki67 are available from the samples. Clinico-pathologic data were retrieved from patients’ medical charts and biopsy reports. Pathologic staging was performed using the American Joint Committee on Cancer (AJCC) TNM system. Patients who had taken neodjuvant chemotherapy were excluded from the study. Due to a significant number of patients lacking follow-up information regarding recurrence, metastasis and overall survival from the medical records, survival status was determined through follow-up phone calls and served as the endpoint measure for this study. This study was approved by the institutional review board (IRB) of the College of Health Sciences of Addis Ababa University (protocol DMIP092/17/17) and the National Research Ethics Review Committee of Ethiopia (protocol MOSHE//RD).
Publicly available validation cohort
A validation data set obtained from The Cancer Genome Atlas (TCGA) was utilized as a validation cohort [15]. The TCGA BRCA data set encompasses RNAseq data of 177 African American BC patients with available clinical and pathological data, such as stage, immunohistochemical receptor expression and patients’ outcome.
RNA extraction
Purification of RNA from FFPE tissues was carried out by Qiagen miRNeasy FFPE Kit (Catalog no. 217504). Up to four sections of FFPE material each with a thickness of 10 μm were used. Deparaffinization was carried out by treating samples with xylene, 100% ethanol and 70% ethanol followed by incubation of the samples with lysis buffer containing proteinase K. All samples were subsequently treated with DNase in order to eliminate contaminating DNA. Buffer RBC and ethanol were added to the lysate and the resulting mixture was finally loaded to RNeasy MinElute spin column in which purified RNA remains attached until subsequent elution of RNA by RNase-free water.
mRNA expression analysis
After the assessment of the quality and quantity, RNA samples meeting the required quality criteria for hybridization reactions on the nCounter platform were subsequently analysed. 25 µg RNA of normal breast tissue (Agilent technologies, MVP™ Total RNA, Human Breast, Catalog Number 540045; pooled from 2 donors) served as a control. Gene expression was determined by nCounter MAX/FLEX platform (Nanostring Technologies, Seattle, WA, US), which is based on direct molecular barcoding of target RNA molecules using colour-coded probe pairs. Hybridization of the probe pairs with RNA was done by incubating 3 µL of the reporter Code Set, 5 µL of hybridization buffer, 5 µL of 400 ng RNA and 2 µL of the Capture ProbeSet overnight. After a washing step to remove excess unbound probes, the RNA/probe complexes were eluted and immobilized in the cartridge for data collection. Counts were read with 555 fields of view (FOV) by the nCounter® Digital Analyzer.
Intrinsic subtyping of BC was carried out using the research-based, 50-gene prediction analysis of microarray (PAM50) subtype predictor [16]. PAM50 was calculated from the nanostring expression data.
PIK3CA genotyping
Genomic DNA (gDNA) was extracted from FFPE tissue samples utilizing the QIAamp® DNA FFPE Tissue Kit. PIK3CA mutations at three hotspot loci—p.E542K (c.1624G > A), p.E545K (c.1633G > A), and p.H1047R (c.3140A > G)—were assessed using the TaqMan® Mutation Detection Assay (Applied Biosystems, Carlsbad, CA 92008 USA). The genotyping procedure involved two experiment types for ΔCT cutoff determination and mutation detection. Each experiment included the detection of both the mutant allele and a corresponding reference gene. PCR mixtures were prepared by combining 20 ng of gDNA with TaqMan® Genotyping Master Mix and the TaqMan® Mutation Detection Assay. The reaction mixture was then added to a PCR plate and processed using the real-time PCR instrument according to the detection protocol. Data analysis was performed in Applied Biosystems®’ real-time PCR instrument software (ABI StepOnePlus v2.1). Tumours were considered wild type (wt) only when all three genotyped sites were congruent with the human DNA reference sequence [17].
Data processing
Data were analysed using ROSALIND® (https://rosalind.bio/) platform (ROSALIND, Inc., San Diego, CA, USA). QC metrics, such as imaging quality, binding density, positive control linearity and limit of detection, were inspected. Normalization of raw counts was executed as specified by Nanostring. Eleven housekeeping genes with lowest coefficient of variation (var. <0.6) were selected following the geNorm algorithm and used as normaliser probes [18]. Data were then normalized against the geometric mean of the selected housekeeping genes and positive control probes as described [19].
Unsupervised hierarchical clustering of the normalized gene expression data was performed using ClustVis, a web tool implementing the pheatmap R package [20]. Expression values were centered and unit variance scaling was applied. Samples were clustered using the correlation distance and Ward linkage.
Identification of differentially expressed genes (DEGs) and cell type profiling
The identification of DEGs between specified groups was performed using ROSALIND®. The Benjamini-Hochberg method was applied for adjustment of p-values in order to correct for multiple comparisons. Genes with |log2 fold-change| > 1.5 and adjusted p-value < 0.05 were defined as DEGs. Abundance of various cell populations was calculated in ROSALIND® using the Nanostring cell type profiling module.
Statistical analysis
Data distributions and frequencies were compared among different groups with oneway ANOVAs, if not stated otherwise, as implemented in GraphPad Prism v9 (GraphPad, San Diego, CA, USA). Fisher-Freeman-Halton exact tests of independence were performed for contingency tables larger than 2 × 2 using IBM SPSS Statistics v26 (IBM Corp., Armonk, NY, USA). Kruskal–Wallis H tests were employed to compare the numbers of TILs among the four IPs using the “stats” library for Scipy/Python [21]. Kaplan–Meier survival analysis was performed to compare the prognostic significance of the immune subgroups and DEG.s Log-rank test was employed as statistical a test.
Results
Clinico-pathologic data of participants
In total eighty-two patients with pathologically confirmed BC were included in this study. Age at diagnosis of participants ranged from 20 to 70 years (median = 40). Early stage tumours (stage I & II) were more frequent in our cohort than late stage tumours (48.8% vs. 33%). The majority (65.8%) of the tumours were highly proliferating and showed a histological grade III. PAM 50-based subtyping of tumours demonstrated a dominance of the luminal subtype (luminal A 34% and luminal B 18%), while the HER2-enriched and basal subtypes were observed with a frequency of 23% and 24.3%, respectively (Table 1). While the majority of molecular subtypes correlated to the IHC groups, some BC samples showed discordant results (Table 1; Fig. 1).
Table 1.
Clinico-pathologic data of participants
| Features | N (%) |
|---|---|
| Age | |
| younger (< 50) | 50 (61%) |
| older (≥ 50) | 19 (23%) |
| Unknown | 13 (16%) |
| Pathologic Stage | |
| Early (stage I & II) | 40 (48.8%) |
| Advanced (stage I & II) | 27 (33%) |
| Unknown | 15 (18.2%) |
| Histological grade | |
| G1 or G2 | 28 (34.1%) |
| G3 | 54 (65.9%) |
| Intrinsic subtype | |
| luminal A | 28 (34.1%) |
| luminal B | 15 (18.3%) |
| HER2-enriched | 19 (23.2%) |
| basal-like | 20 (24.4%) |
| IHC Group | |
|
HR+ HER2− (luA-like) HR+ HER2+ (luB-like) |
41 (50.0) 22 (26.8) |
|
HR− HER2+ (HER2+) HR− HER2− (TNBC) |
7 (8.5) 12 (14.6) |
Fig. 1.

Heatmap of the expression levels of 54 immune-related genes in 82 BC samples. Unsupervised hierarchical clustering of log-transformed expression levels of immune genes identified the four immunophenotype groups IP1 to IP4. Red marks indicate upregulation, blue marks indicate downregulation of immue relatetd genes. Sample annotations are included at the top of the heatmap. HER2: human epidermal growth factor 2. HR: hormone receptor
Identification of BC immune phenotypes
Hierarchical clustering of the gene expression profiles of 54 immune-related genes was performed for 83 samples consisting of 82 BC samples and one sample of normal breast tissue pooled from two healthy donors. Four discrete clusters with distinct gene expression patterns in the TME corresponding to immune phenotypes (IP) 1–4 were identified according to their appearance on the clustering tree (Fig. 1). IP1 consists of 9 samples, IP2 of 31, IP3 of 19 and IP4 of 24 samples, respectively.
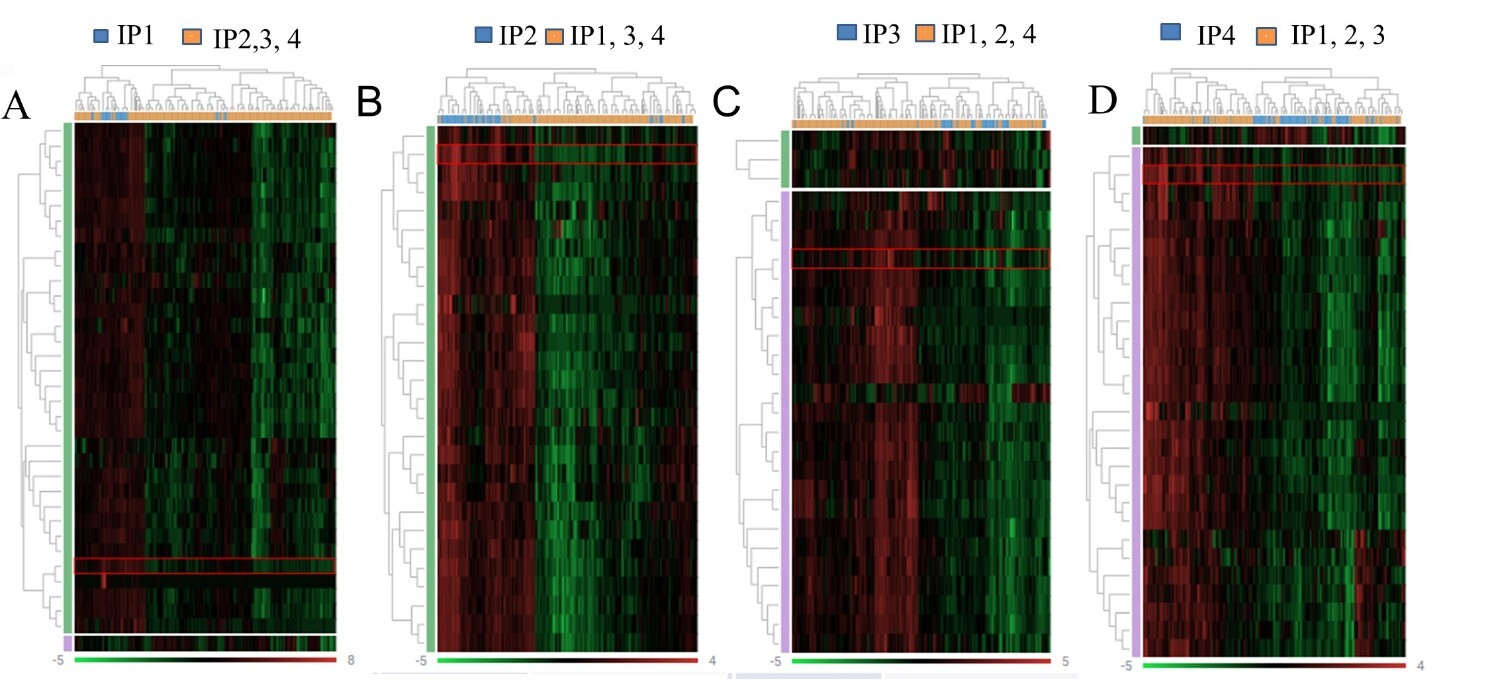
DEG analysis of immune-activating and immune-inhibitory genes demonstrated a different extent of immune activation in the TME between the four IPs (Figs. 1 and 2). IP2 represents an immunologically inert phenotype with a consistent downregulation of immune suppressive as well as immune activating genes. Immune genes encoding effectors cells (CD4, CD8A, CD8B, and CD20), T cell receptor (TCR) signalling pathway (CD3E, CD3G, CD28, ICOS and CD2), immune modulatory molecules (TIGIT, CTLA4, CD274, HLA-E, IDO1, CCR2), antigen processing and presentation (KLRD1, TAP1, TAP2, CD1A), immune activating molecules or receptors (KLRK1, CXCL8, CXCR6), tumour associated macrophage (TAM) markers (CD68, CD163) and the regulatory T cell (Treg) marker (FOXP3) were suppressed in this phenotype (Fig. 2). In contrast, IP4 tumours were marked by highest immune gene expression representing an immunologically active microenvironment, characterized by an up-regulated expression of genes associated with anti-tumoural functions. Genes related to effectors cells (CD4, CD8A, CD8B, CD20), T cell activation (CD69), TCR signalling components (CD3E, CD3G, CD28, ICOS, CD2), antigen processing and presentation (KLRD1, TAP1, TAP2, CD1A) and activating molecules or receptors (KLRK1, CXCR6, FLT3LG) were significantly upregulated in this phenotype compared to the others (IP1-3). Interestingly, next to immune-stimulating molecules, some immune inhibitory molecules, like TIGIT, CTLA4 and CCR2 as well as the Treg marker FOXP3, were also highly expressed in this phenotype.
Fig. 2.

DEG heat map of the distinct immune phenotypes A: IP1 versus IP2, IP3 and IP4; B: IP2 versus IP1, IP3 and IP4; C: IP3 versus IP1, IP2 and IP4; D: IP4 versus IP1, IP2 and IP3. Differentially expressed immune genes between the immune phenotypes were shown. Genes with log2 fold change (FC) ≥ 1.5 and false discovery rate (FDR)-corrected p value < 0.05 were presented. Green: downregulated genes, red: upregulated genes
In addition, two phenotypes with an intermediate level of immune activation were characterized. In IP1, the immune activation genes CD4, NCAM1, CD3G, KLRD1 and the counter-regulatory molecules FOXP3, IDO and HLA-G were differentially suppressed when compared to the other phenotypes. In addition, NTAN1, FURIN and CD276 were upregulated in this phenotype. In the IP3, the majority of the immune genes were normally expressed; genes related with antigen processing and presentation (CD1A, TAP2), inflammation (CXCL8), TCR signalling pathway (CD28) and immune suppression (HLA-G, IDO1) were particularly upregulated in IP3, FGFR4, FLT3LG and CTBS were downregulated.
Immune cell infiltration of the IPs
The extent of immune infiltration was analyzed in the different IPs using cell type-specific profiling and TIL from H&E staining (Fig. 3). In comparison to the other phenotypes, the immune inflamed phenotype (IP4) had the highest log2 abundance score of cytotoxic cells, Treg as well as exhausted phenotypes of immune effector cells (Fig. 3A) with a significantly high TIL counts (H = 8.19, p-value < 0.01) (Fig. 3B). Conversely, the log2 abundance score of infiltrating immune cells was the lowest in IP2 with the lowest TIL count (H = 14.56, p-value < 0.001), whereas IP1 and IP3 exhibit an intermediate level of immune cell infiltration.
Fig. 3.

Distinct distribution of the immune cell subpopulations in the distinct immune phenotypes A: Log2 abundance score of immune cell subpopulations in IP1-4. Nanostring cell type profiling module was used to assess the immune composition of IPs. B: Frequency of TILs. TILs were counted from H&E stained slides. Data are presented as % of TIL in the different IPs
Immune phenotypes and prognosis
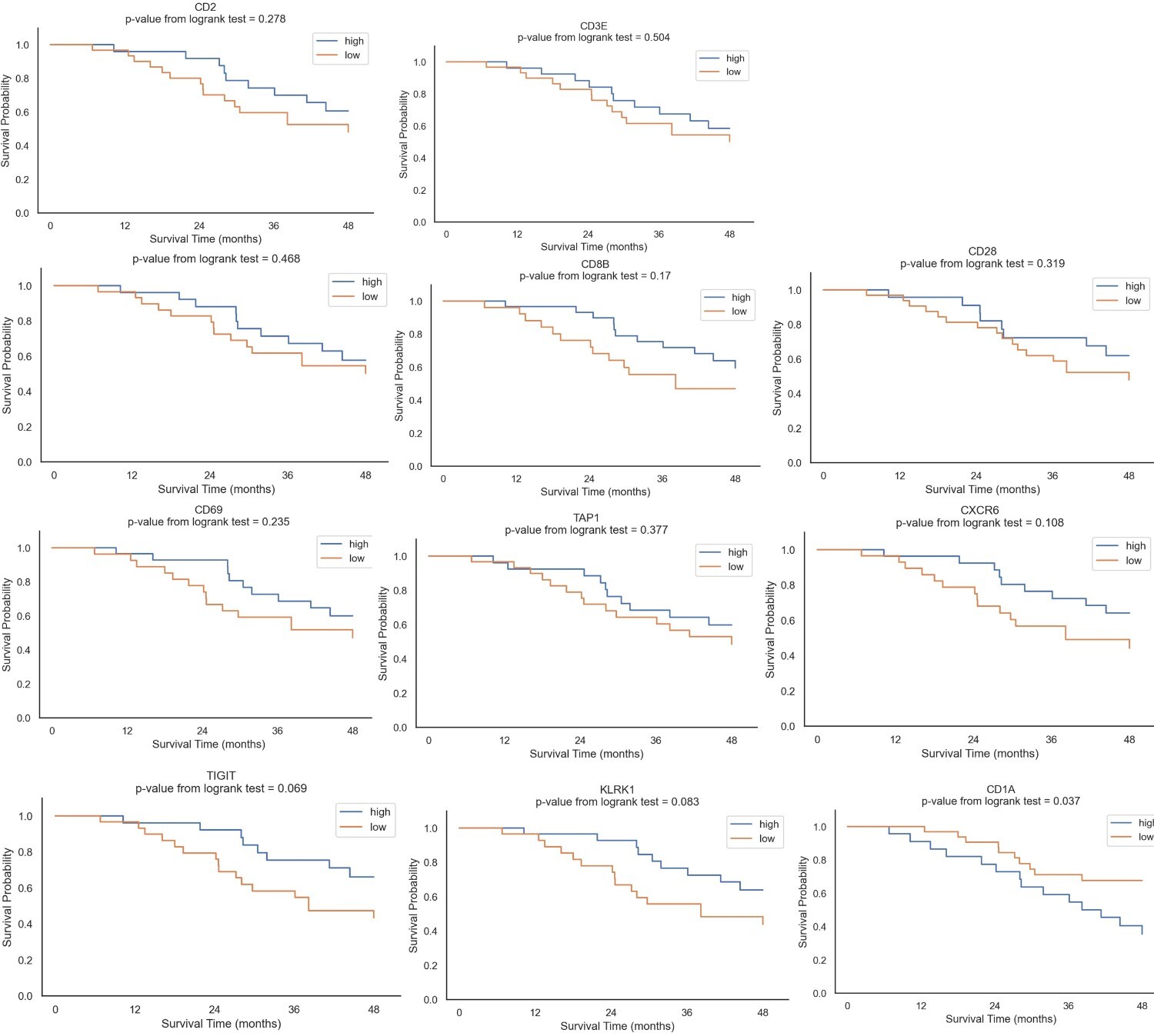
Since the distinct IPs differ in the activation status of the immune cells, we assessed whether the IPs can predict survival probability. Due to the low number of BC patients the IP1 subgroup was excluded from the analysis. Patients with the IP4 type had a longer, but not statistically significant overall survival (OS) than patients with IP2 and IP3 (Fig. 4A). The DEGs from IP4; CD2, CD3E, CD8A, CD8B, CD28, CD69, TAP1, CXCR6, KLRK1 and TIGIT correlated with a higher probability of a longer OS, but due to the limited number of patients involved in each group the difference was not statistically significant. In contrast, among the DEGs a lower expression of CD1A was significantly associated with higher OS (Supplementary Fig. 1).
Fig. 4.

Association of the immune phenotypes with the patients’ survival. A: Kaplan-Meier survival curves between IP4 versus IP2 and IP3. Patients were followed for 60 months and survival probabilities were determined by calculating p values from log rank test. B-G: Kaplan-Meier survival curves derived from TCGA data set of 177 African American BC cases. B: Survival probabilities across the four IPS. C: HR + BC survival within the four IPs. D: HR- BC survival within the four IPs. E: survival between the two IP groups F: HR- BC survival within the two IP groups. G: HR + BC survival within the two IP groups. IP1 & IP2 were categorized as “Immune Activated” while IP3 and IP4 were categorized as “Immune Suppressed”
Validation data set
To further validate our findings, the publicly available BRCA data set from TCGA comprising 177 primary invasive BC cases from African American patients was analysed for the expression of 50 out of 54 genes evaluated in our cohort. Hierarchical clustering of these genes revealed a similar pattern with four distinct IPs, each representing a different immune activation status within the TME (Supplementary Fig. 2).
Differential expression analysis (DEA) revealed that IP1 and IP2 exhibited a significantly immunologically active TME with 28 and 35 immune genes, respectively, being upregulated when compared to the other IPs. In contrast, IP3 and IP4 were characterized by a “cold” immune microenvironment with 26 and 29 immune genes downregulated, respectively (DEG heatmaps shown in supplementary Fig. 3).
We further investigated the prognostic potential of these IPs. The results demonstrated that IPs with an immune-activated microenvironment (IP1 and IP2) were associated with a significantly improved overall survival (OS) compared to the “cold” immune microenvironment IPs (P = 0.0143) (Fig. 4B). Although IP1 and IP2 showed a trend towards a better OS when stratified by hormone receptor (HR) status (ER or PR), this difference did not reach statistical significance (Fig. 4C & D).
To refine our analysis, IP1 and IP2 were combined into a single “immune-activated” phenotype, while IP3 and IP4 were classified as “immune-suppressed.” This reclassification revealed a significantly higher OS in patients with the immune-activated phenotype compared to those with the immune-suppressed phenotype (P = 0.04). Notably, when stratified by HR status, the immune-activated phenotype was associated with significantly better OS in HR-negative cases (P = 0.0274), but not in HR-positive cases (P = 0.126) (Fig. 4D, E & G).
Clinico-pathologic characteristics and PIK3CA mutation within IPs
The different IPs were analysed in relation to the well-known clinico-pathologic features demonstrating the highest frequency of HER2 + tumours in IP4 (50%) and the lowest in IP3 (15.7%) (p < 0.05). IP4 and IP2 with 87.5% and 86.6% have the highest proportion of HR+ tumours, respectively, whereas IP3 has the lowest proportion of HR+ tumours (47.4%). TNBC were dominant in IP3 (47.4%) (Fig. 5A). The analysis of intrinsic BC subtypes revealed that IP1 and IP2 were mainly enriched in luminal subtypes (66.7% and 76.5%, respectively), whereas in IP3 the non-luminal subtypes were dominant with a frequency of 94.7%. Concerning the distribution of intrinsic subtypes, luminal A tumours were the major constituents of IP2 (56.7%), while the basal subtype was predominant in IP3 (84.2%) (Fig. 5B). In IP4, the intrinsic subtypes were equally distributed.
Fig. 5.

Immune phenotypes and clinic-pathologic features of BC. A: Proportion of HR+, HER2 + and TNBC tumours. B: Distribution of the intrinsic subtypes within IPs; C: Frequency of high grade tumours; D: Ki-67 proliferation index; E. Proportion of PIK3CA-mutated tumours: Only 9 tumours were grouped in IP1 and the mutation status could not be determined for the majority of samples (proportion not shown in the figure); F: A box and Whisker plot showing log2 expression value of CD8 in the wt and PIK3CA-mutated tumours
Neither the age nor the pathologic stage was related to different IPs (p = 0.5643 and 0.199, respectively). In contrast, a higher proportion of high grade tumours were found in IP1, IP3 and IP4, whereas IP2 had a similar frequency of high and low grade tumours (P < 0.05) (Fig. 5C). The Ki-67 proliferation index indicated a dormancy of the IP2 subtype, while IP3 had the highest proliferation index (P < 0.05) (Fig. 5D).
Since mutations can generate immunogenic neoantigens leading to immune activation [22], it was analyzed whether mutations in the phosphoinositide 3-kinase CA (PIK3CA) gene are more frequent in the immunologically active phenotype. Analysis of 70/82 BC cases revealed the highest frequencies of PIK3CA-mutated tumors in IP4 (33.3%) and IP2 (31%), respectively without a significant difference between these two extreme IPs. IP3 had a significantly lower proportion of PIK3CA-mutated tumours (6.25%). Interestingly, 75% of the mutations in IP4 occurred within luminal tumours. Mutated tumours have a significantly lower expression of CD8 genes than the wild type BC (Wt mean = 6.3 versus mutated mean = 5.6; p = 0.04), but there was no association with CD4 (Fig. 5E, F).
Differential expression of immune genes across breast cancer subtypes and clinicopathologic features
Immune gene expression of normal breast tissue obtained from 2 pooled samples in different BC subtypes was analysed and compared to that of BCs. Interestingly, no significant differential expression of immune genes (DEIGs) in the 54 immune genes analysed could be identified between control and BC tissues. The control with the breast tissue clustered within IP2 (Fig. 1).
In order to get better insights whether the TME changes with age in BC, the DEIG signatures between the age groups < 50 (younger) versus ≥ 50 (older) were investigated. However, no age-dependent significant DEIGs were detected. Furthermore, analysis of the impact of the expression of immune genes in the TME on prognostic parameters and pathologic features demonstrated that CD20 and GZMB were significantly downregulated in advanced pathologic stages when compared with early BC stages suggesting their use as biomarkers (Fig. 6A). In contrast, no significant DEIGs were found between high grade and low grade tumours.
Fig. 6.

Differentially expressed immune genes in BC subtypes. A: Box and whisker plot showing a differential expression of CD20 and GZMB between early and advanced pathologic stages. B: Volcano plot of DEGs between luminal and non-luminal tumours. Volcano plot is plotted using Graph Pad Prism to show differentially expressed immune genes; FC > 1.5, adjusted P values < 0.05. Each dot represents an immune gene. C: DEG heat map of luminal versus basal like tumours, green shows downregulation and red shows upregulation. D: Box and whisker plot showing a differential expression of CD1A and CTBS between HR + and TNBC. DEG: Differentially expressed genes
Since distinct immune responses have been reported in BC intrinsic subtypes [23, 24], the expression of immune-relevant genes was analysed in the intrinsic subtypes by comparing luminal with non-luminal tumours. Genes associated with antigen processing and presentation (TAP1, TAP2, and CD1A), CTL- and NK cell-mediated cytotoxicity (GZMB, perforin), immune checkpoints (CD274, LAG-3), chemokines, e.g. CXCL8, as well as the immunoregulatory metabolite IDO1 implicated in T cell inhibition [25] were upregulated in non-luminal tumours versus luminal tumours (Fig. 6B). Whereas a comparison of luminal versus basal-like and HER2 + tumors separately revealed downregulation of IDO1, CD1A, CXCL8, GZMB, TAP2, HLA-G, NCR1, LAG3, TAP1, CTLA4, HLA-A, CD274 and CD163 in luminal compared to basal-like tumours, CXCL8 was the only gene that remained downregulated when compared to HER2 + tumours (Fig. 6C). Analysis of the differences in the gene expression patterns between luminal A and luminal B tumours demonstrated that IFITM3 was the only significantly differentially expressed gene (FC = 2.1; p = 0.00095). In non-luminal tumours, a comparatively higher variation in the gene expression profile was found with an upregulation of CD1A, IDO1, HLA-G and TAP2 and a down regulation of NTAN in basal subtypes when compared to HER-2-enriched tumours.
Immune gene expression analysis across the IHC groups showed minimal variation between the subtypes. Only CD1A was downregulated, while CTBS was upregulated in HR + BC compared to TNBC. No significant DEIGs were observed when comparing HR + BC to HER2 + BC (Fig. 6D).
Discussion
In this study four distinct immune subgroups were identified. Similar distinct immune subgroups of BC stratified by the TME immune transcriptome profile was also reported in many studies using different approches [12, 26, 27]. Even though BC was primarily suggested as a general non-immunogenic tumour type, it has been demonstrated that the immune landscape of BC is dynamic and heterogeneous with significant variations observed across BC patients. This study is in line with many other reports demonstrating a spectrum of immune activation status of breast immune microenvironment, in which a subset of BC patients exhibited a T cell-inflamed TME, while the remaining bear an immune intermediate, excluded or desert microenvironment [26–29]. Despite very limited information available about the DEIGs in BC from African women, a recent report on African American women from Carolina Breast Cancer Study also described three immune clusters with gradual immune activation [30].
Interestingly, the immune-activated phenotype (IP4) harboured immune suppressive features with higher proportion of exhausted T cells and increased expression of immune inhibitory molecules compared to other IPs. These results confirm the data of Spranger and co-authors as well as others demonstrating an increased expression of immune inhibitory molecules in a T cell-inflamed melanoma phenotype [31]. The increased expression of these inhibitory pathways in the immune activated phenotype might be a negative feedback mechanism to counterbalance the enhanced immune reaction [31, 32]. In these tumours, anti-tumour immunity was mounted, but the response was not efficient enough to eliminate the tumour. For these tumour types, the therapeutic modulation of the immune response might help to improve the patients’ outcome.
In the current study, a subset of BCs was marked by the absence of immune responses (IP2) in the TME, where both immune effector and immune regulatory genes were repressed, and thus designated as an immune inert phenotype. This subgroup was predominantly composed of luminal tumours, mainly luminal A breast cancer, and had the lowest Ki-67 proliferation index. In accordance with the present result, lower immune gene expression levels were identified with a higher frequency in BC with ER + and luminal A subtypes by conventional or the Sorlie-Perou intrinsic molecular subtyping methods, respectively [23, 24, 33]. Furthermore, these tumours were characterized by low expression of proliferating genes and low histological grade [34].
Mechanistic studies have shown that T cell-excluded tumours could arise upon disruption of one feature of the cancer-immunity cycle, which extends from antigen recognition by antigen presenting cells, presentation of captured antigen to T cells to homing of activated T cells to the tumour site [35]. Despite the determinants of immune responses are complex, the lower proliferation of such tumours may account in part to their reduced immunogenicity and less immune reaction as described in our study. Higher tumour proliferation might be followed by an enhanced immune response by increasing immunogenic antigens from apoptotic tumour cells [36, 37]. Even though the role of apoptosis in the induction of CD8+ T cell activation was controversially discussed, emerging studies are proving its essential role in immune stimulation. Nowak and co-authors stated an induced apoptosis in tumour bearing mice associated with a tumour regression. This effect was attributed to an enhanced immune response due to the induction of antigen-specific CD8+ T cell proliferation [38]. These results were further confirmed in BC patients from TCGA and METABRIC cohorts by showing a correlation of high apoptotic tumours with a significantly high infiltration of CD8+, CD4+ memory T cells, M1 macrophages and DC [39]. In contrast, in slowly proliferating tumours with less apoptotic cells available antigens capable of priming the adaptive immune response are low resulting in an inert microenvironment.
Another intriguing result was the clustering of the normal breast tissue from healthy donors (used as control) with the immune inert phenotype. Certain level of immune activity is expected in healthy breast as studies had shown the presence immune cells of innate and adaptive immunity with a potential role in immune surveillance. Lymphocytes were the predominant cells with CD8+ T cells being the most common of the lymphocytes. These CD8+ T cells primarily represent effector memory T cells bearing CD45RO+/CD27− phenotype [40–42]. Mammary microbiota might be the source of these antigens for activation [43]. However, during cell transformation, malignant cells release pro-inflammatory mediators and chemoattractants enhancing immune cell infiltration [44]. Consequently, the immune cell content of breast tissue progressively increases from normal breast tissue to BC [40]. However, in our case, no difference was observed in IP2 further signifying the non-immunogenicity of tumours clustered in IP2.
There exists increasing evidence that immune-based subtyping in BC has clinical relevance. For example, Hendrickx and co-authors reported an immune phenotype characterised by the highest levels of immune gene expression, which correlated with an increased overall survival compared to the other immune phenotypes [27]. Similarly, Yao and colleagues identified three immune subsets validated in five BC data sets. The immune-active subtype was consistently associated with a better survival in all data sets [28]. Similarly, the TCGA validation cohort in our study demonstrated an improved overall survival for the immune-activated phenotypes in both the entire patient population and in HR-negative cases when stratified by the hormone receptor (HR) status.
This study enhances our understanding of BC biology among Ethiopian patients, aiding in the stratification of individuals into distinct treatment and prognostic groups. A significant limitation is the small sample size, which affects the robustness of our survival analysis. However, this is an ongoing research effort and currently additional BC patients were recruited to prospectively investigate the prognostic and predictive impact of immune genes expressed in the tumor microenvironment.
Conclusion
Expression profiling has resulted in distinct immune subgroups with a unique gene expression pattern, which provide important prognostic and predictive information for conventional treatments as well as immunotherapy in the management of BC in Ethiopia.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
Supplementary Fig. 1: Differentially upregulated genes in IP4 compared to IP2 and IP3 and their association with survival probability. Gene expression data were normalized and log2 transformed, with the median used to categorize genes into high and low expression groups. Survival probabilities were calculated using p-values derived from the log-rank test conducted in Python
{kind=link}
Supplementary Fig. 2: Heatmap of the expression levels of 50 immune-related genes in 177 BC samples from African Americans. Data were obtained from TCGA database. Unsupervised hierarchical clustering of log-transformed expression levels of immune genes resulted in four immune clusters termed immune phenotypes (IP). Red marks indicate upregulation, blue marks indicate the downregulation of immue relatetd genes
{kind=link}
Supplementary Fig. 3: DEG heat map of the distinct immune phenotypes in the TCGA cohort A: IP1 versus IP2, IP3 and IP4; B: IP2 versus IP1, IP3 and IP4; C: IP3 versus IP1, IP2 and IP4; D: IP4 versus IP1, IP2 and IP3. Differentially expressed immune genes between the immune phenotypes were shown. Genes with log2 fold change (FC) ≥ 1.5 and false discovery rate (FDR)-corrected p value < 0.05 were presented. Green: downregulated genes, red: upregulated genes
Acknowledgements
The authors would like to acknowledge Addis Ababa cancer registry staffs for their cooperation during data collection. The authors also extend their appreciation to Tikur Anbesa Specialized Hospital Oncology and Pathology Department staffs. The authors are grateful for all funders for the financial support. Last but not least, our heartfelt thanks go to Addis Ababa University for covering the tuition fee expense of Meron Yohannes during her PhD study.
Abbreviations
- BC
Breast cancer
- CCL
CC chemokine ligand
- DC
Dendritic cell
- DEG
Differentially expressed gene
- DEIG
Differentially expressed immune gene
- ER
Estrogen receptor
- Fc
Fold-change
- FDR
False discovery rate
- FFPE
Formalin-fixed paraffin-embedded
- FOV
Field of view
- GZMB
Granzyme B
- H&E
Hematoxylin and eosin
- HER2
Epidermal growth factor receptor 2
- HR
Hormone receptor
- IP
Immune phenotype
- IRB
Institutional review board
- IRG
Immune related gene
- PAM50
Prediction analysis of microarray
- PI3K
Phosphoinositide 3-kinases
- PR
Progesterone receptors
- QC
Quality control
- STAT
Signal transducer and activator of transcription
- TAM
Tumor-associated macrophage
- TCGA
The Cancer Genome Atlas
- TCR
T cell receptor
- Th1
T helper type 1
- TIL
Tumor infiltrating lymphocyte
- TME
Tumor microenvironment
- TIME
tumor immune microenvironment
- TNBC
Triple negative breast cancer
- Treg
Regulatory T cell
- Wt
Wild type
Author contributions
MY, TA, BS contributed to study design, sample and data acquisition, analysis, interpretation and writing of the original and final draft. AA, EJK, TA, LT and BS contributed to study concept and design. ZD, TW, EA, YB, MA and YW contributed to sample and data acquisition. ZD, KS, MB and MV carried out the experimental work. BS, CM, CW, KS, EJK, MV, LT and TA contributed to study design, data acquisition, data analysis, data interpretation and editing of the manuscript. MY and PS contributed to data analysis, plotting and statistics. All authors reviewed and approved the final version of the manuscript.
Funding
The study was supported by the Susan Komen given to Martin-Luther–University Halle-Wittenberg, Medical Faculty (GTDR16378013). It was also supported by Else-Kroener-Foundation through Martin-Luther-University, Halle-Wittenberg, Germany (2018_HA31SP). This study was also supported by a grant from the German Academic Exchange Service to Martin-Luther-University, Halle-Wittenberg, Germany (ID 57216764). Additionally, the study received support from a grant; Hospital partnership through Deutsche Gesellschaft für Internationale Zusammenarbeit funded by the Ministry for Economic Cooperation and Development and the Else-Kroener-Fresenius Foundation (ID 81256434). EJK also received an unrestricted grant from F. Hofmann-La Roche Ltd (27.5.2014). BS received a grant from the German Cancer Aid (Integrate). In addition, data collection budget was partly covered by the Addis Ababa University. The study was conducted independently of any involvement from the funders.
Data availability
All the data sets of this manuscript including the analysed data will be available on reasonable request to the corresponding author.
Declarations
Ethics approval and consent to participate
This study was conducted in accordance with the principles outlined in the Declaration of Helsinki. It was approved by the institutional review board of collage of health sciences of Addis Ababa University (Protocol Number DMIP 092/17/17) and National ethics review committee of Ethiopia (Protocol MOSHE/RD). In addition, sample was collected after written informed consent was obtained from participants.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Barbara Seliger and Tamrat Abebe co-shared last authorship.
References
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer statistics 2020: GLOBOCAN estimates of incidence and Mortality Worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49. [DOI] [PubMed] [Google Scholar]
- 2.Ginsburg O, Bray F, Coleman MP, Vanderpuye V, Eniu A, Kotha R, et al. The global burden of women’s cancers: an unmet grand challenge in global health Europe PMC funders Group. Lancet. 2017;389(10071):847–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perou CM, Sørile T, Eisen MB, Van De Rijn M, Jeffrey SS, Ress CA, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–52. [DOI] [PubMed] [Google Scholar]
- 4.Hennigs A, Riedel F, Gondos A, Sinn P, Schirmacher P, Marmé F et al. Prognosis of breast cancer molecular subtypes in routine clinical care: A large prospective cohort study. BMC Cancer. 2016;16(1):1–9. 10.1186/s12885-016-2766-3 [DOI] [PMC free article] [PubMed]
- 5.Masood S. Breast cancer subtypes: morphologic and biologic characterization. Women’s Heal. 2016;12(1):103–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abe O, Abe R, Enomoto K, Kikuchi K, Koyama H, Masuda H et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: Patient-level meta-analysis of randomised trials. Lancet. 2011;378(9793):771–84. 10.1016/S0140-6736(11)60993-8 [DOI] [PMC free article] [PubMed]
- 7.Curtis C, Shah SP, Chin S, feung, Turashvili G. Europe PMC funders Group the genomic and transcriptomic architecture of 2, 000 breast tumours reveals novel subgroups. 2012;486(7403):346–52. [DOI] [PMC free article] [PubMed]
- 8.Dawson SJ, Rueda OM, Aparicio S, Caldas C. A new genome-driven integrated classification of breast cancer and its implications. EMBO J. 2013;32(5):617–28. 10.1038/emboj.2013.19 [DOI] [PMC free article] [PubMed]
- 9.Li JJ, Tsang JY, Tse GM. Tumor microenvironment in breast cancer—updates on therapeutic implications and pathologic assessment. Cancers (Basel). 2021;13(16). [DOI] [PMC free article] [PubMed]
- 10.Badr NM, Berditchevski F, Shaaban AM. The Immune Microenvironment in breast carcinoma: predictive and prognostic role in the Neoadjuvant setting. Pathobiology. 2020;87(2):61–74. [DOI] [PubMed] [Google Scholar]
- 11.Netanely D, Avraham A, Ben-Baruch A, Evron E, Shamir R. Expression and methylation patterns partition luminal-A breast tumors into distinct prognostic subgroups. Breast Cancer Res. 2016;18(1):1–16. 10.1186/s13058-016-0724-2 [DOI] [PMC free article] [PubMed]
- 12.Zhu B, Tse LA, Wang D, Koka H, Zhang T, Abubakar M, et al. Immune gene expression profiling reveals heterogeneity in luminal breast tumors. Breast Cancer Res. 2019;21(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu S, Qu X, Jiao Y, Hu J, Wang B. Immune classification and Immune Landscape Analysis of Triple-negative breast Cancer. Front Genet. 2021;12:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Denkert C, von Minckwitz G, Darb-Esfahani S, Lederer B, Heppner BI, Weber KE, et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: a pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol. 2018;19(1):40–50. [DOI] [PubMed] [Google Scholar]
- 15.TCGA data. https://www.cbioportal.org/study/summary?id=brca_tcga
- 16.Berchtold E, Vetter M, Gündert M, Csaba G, Fathke C, Ulbrich SE, et al. Comparison of six breast cancer classifiers using qPCR. Bioinformatics. 2019;35(18):3412–20. [DOI] [PubMed] [Google Scholar]
- 17.NCBI RefSeq GRCh38. https://www.ncbi.nlm.nih.gov/refseq/).
- 18.Perkins JR, Dawes JM, Mcmahon SB, Lh Bennett D, Orengo C, Kohl M, SOFTWARE Open Access. BMC Genomics. 2012;13:296. http://www.biomedcentral.com/http://www.bioconductor.org/packages/release/bioc/html/ReadqPCR.html and http://www.bioconductor.org/packages/release/bioc/html/NormqPCR.html [DOI] [PMC free article] [PubMed]
- 19.Nanostring Technologies. Gene Expression Data Analysis Guidelines. 2017;1–23. Available from: www.nanostring.com.
- 20.https://cran.r-project.org/package=pheatmap
- 21.Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods. 2020;17(3):261–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Efremova M, Finotello F, Rieder D, Trajanoski Z. Neoantigens generated by individual mutations and their role in cancer immunity and immunotherapy. Front Immunol. 2017;8:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Griguolo G, Dieci MV, Paré L, Miglietta F, Generali DG, Frassoldati A et al. Immune microenvironment and intrinsic subtyping in hormone receptor-positive/HER2-negative breast cancer. npj Breast Cancer. 2021;7(1). [DOI] [PMC free article] [PubMed]
- 24.Li J, Wu J, Han J. Analysis of Tumor Microenvironment heterogeneity among breast Cancer subtypes to identify subtype-specific signatures. Genes (Basel). 2023;14(1). [DOI] [PMC free article] [PubMed]
- 25.Dill EA, Dillon PM, Bullock TN, Mills AM. IDO expression in breast cancer: an assessment of 281 primary and metastatic cases with comparison to PD-L1. Mod Pathol. 2018;31(10):1513–22. 10.1038/s41379-018-0061-3 [DOI] [PubMed]
- 26.Tekpli X, Lien T, Røssevold AH, Nebdal D, Borgen E, Ohnstad HO et al. An independent poor-prognosis subtype of breast cancer defined by a distinct tumor immune microenvironment. Nat Commun. 2019;10(1). [DOI] [PMC free article] [PubMed]
- 27.Hendrickx W, Simeone I, Anjum S, Mokrab Y, Bertucci F, Finetti P et al. Identification of genetic determinants of breast cancer immune phenotypes by integrative genome-scale analysis. Oncoimmunology. 2017;6(2):1–19. 10.1080/2162402X.2016.1253654 [DOI] [PMC free article] [PubMed]
- 28.Yao J, Li S, Wang X. Identification of breast Cancer Immune subtypes by analyzing bulk tumor and single cell transcriptomes. Front Cell Dev Biol. 2022;9:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amara D, Wolf DM, van ’t Veer L, Esserman L, Campbell M, Yau C. Co-expression modules identified from published immune signatures reveal five distinct immune subtypes in breast cancer. Breast Cancer Res Treat. 2017;161(1):41–50. [DOI] [PubMed] [Google Scholar]
- 30.Hamilton AM, Hurson AN, Olsson LT, Walens A, Nsonwu-Farley J, Kirk EL et al. The Landscape of Immune Microenvironments in Racially Diverse Breast Cancer Patients. Cancer Epidemiol Biomarkers Prev. 2022;31(7):1341–50. https://aacrjournals.org/cebp/article/31/7/1341/705157/The-Landscape-of-Immune-Microenvironments-in [DOI] [PMC free article] [PubMed]
- 31.Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up-regulation of PD-L1, IDO, and Tregs in the melanoma tumor microenvironment is driven by CD8 + T cells. Sci Transl Med. 2013;5(200):1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bedognetti D, Hendrickx W, Ceccarelli M, Miller LD, Seliger B. Disentangling the relationship between tumor genetic programs and immune responsiveness. Curr Opin Immunol. 2016;39:150–8. 10.1016/j.coi.2016.02.001 [DOI] [PubMed]
- 33.Goldberg J, Pastorello RG, Vallius T, Davis J, Cui YX, Agudo J, et al. The immunology of hormone receptor positive breast Cancer. Front Immunol. 2021;12:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yersal O, Barutca S. Biological subtypes of breast cancer: prognostic and therapeutic implications. World J Clin Oncol. 2014;5(3):412–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10. [DOI] [PubMed] [Google Scholar]
- 36.Bai M, Agnantis NJ, Kamina S, Demou A, Zagorianakou P, Katsaraki A, et al. In vivo cell kinetics in breast carcinogenesis. Breast Cancer Res. 2001;3(4):276–83. [PMC free article] [PubMed] [Google Scholar]
- 37.Lee JS, Kim HS, Jung JJ, Kim YB, Park CS, Lee MC. Correlation between angiogenesis, apoptosis and cell proliferation in invasive ductal carcinoma of the breast and their relation to tumor behavior. Anal Quant Cytol Histol. 2001;23(2):161–8. [PubMed] [Google Scholar]
- 38.Nowak AK, Lake RA, Marzo AL, Scott B, Heath WR, Collins EJ, et al. Induction of Tumor Cell apoptosis in vivo increases Tumor Antigen Cross-Presentation, Cross-priming rather than cross-tolerizing host tumor-specific CD8 T cells. J Immunol. 2003;170(10):4905–13. [DOI] [PubMed] [Google Scholar]
- 39.Murthy V, Oshi M, Tokumaru Y, Endo I, Takabe K. Increased apoptosis is associated with robust immune cell infiltration and cytolytic activity in breast cancer. Am J Cancer Res. 2021;11(7):3674–87. http://www.ncbi.nlm.nih.gov/pubmed/34354867%0Ahttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC8332871 [PMC free article] [PubMed]
- 40.Goff SL, Danforth DN. The Role of Immune Cells in Breast Tissue and Immunotherapy for the Treatment of Breast Cancer. Clin Breast Cancer. 2021;21(1):e63–73. 10.1016/j.clbc.2020.06.011 [DOI] [PMC free article] [PubMed]
- 41.Degnim AC, Brahmbhatt RD, Radisky DC, Hoskin TL, Stallings-Mann M, Laudenschlager M, et al. Immune cell quantitation in normal breast tissue lobules with and without lobulitis. Breast Cancer Res Treat. 2014;144(3):539–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zumwalde NA, Haag JD, Sharma D, Mirrielees JA, Wilke LG, Gould MN et al. Analysis of Immune Cells from Human Mammary Ductal Epithelial Organoids Reveals Vδ2 + T Cells That Efficiently Target Breast Carcinoma Cells in the Presence of Bisphosphonate. Cancer Prev Res. 2016 1;9(4):305–16. [DOI] [PMC free article] [PubMed]
- 43.Parida S, Sharma D. The power of small changes: Comprehensive analyses of microbial dysbiosis in breast cancer. Biochim Biophys Acta - Rev Cancer. 2019;1871(2):392–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao D, Cazares LH, Fish EN. CCL5-CCR5 interactions modulate metabolic events during tumor onset to promote tumorigenesis. BMC Cancer. 2017;17(1):834. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1: Differentially upregulated genes in IP4 compared to IP2 and IP3 and their association with survival probability. Gene expression data were normalized and log2 transformed, with the median used to categorize genes into high and low expression groups. Survival probabilities were calculated using p-values derived from the log-rank test conducted in Python
Supplementary Fig. 2: Heatmap of the expression levels of 50 immune-related genes in 177 BC samples from African Americans. Data were obtained from TCGA database. Unsupervised hierarchical clustering of log-transformed expression levels of immune genes resulted in four immune clusters termed immune phenotypes (IP). Red marks indicate upregulation, blue marks indicate the downregulation of immue relatetd genes
Supplementary Fig. 3: DEG heat map of the distinct immune phenotypes in the TCGA cohort A: IP1 versus IP2, IP3 and IP4; B: IP2 versus IP1, IP3 and IP4; C: IP3 versus IP1, IP2 and IP4; D: IP4 versus IP1, IP2 and IP3. Differentially expressed immune genes between the immune phenotypes were shown. Genes with log2 fold change (FC) ≥ 1.5 and false discovery rate (FDR)-corrected p value < 0.05 were presented. Green: downregulated genes, red: upregulated genes
Data Availability Statement
All the data sets of this manuscript including the analysed data will be available on reasonable request to the corresponding author.