

Significance
Expression of the mitochondrial and nuclear genomes must be tightly coordinated, and imbalance in their expression can lead to metabolic disorders, inflammation, and aging. Here, we report a pathway required for both cytosolic and mitochondrial translation programs that is based on N6AMT1. We show that N6AMT1 is required for mitochondrial RNAse P translation in the cytosol and that its inactivation leads to aberrant mitochondrial RNA processing, mitochondrial defects, and an accumulation of immunogenic double-stranded RNA. Our work brings significant insights in the fields of mitochondrial biogenesis and gene expression, protein synthesis, and innate immunity.
Keywords: mitochondria, translation, RNA processing, OXPHOS, mitochondrial RNA granules
Abstract
Mitochondrial biogenesis relies on both the nuclear and mitochondrial genomes, and imbalance in their expression can lead to inborn errors of metabolism, inflammation, and aging. Here, we investigate N6AMT1, a nucleo-cytosolic methyltransferase that exhibits genetic codependency with mitochondria. We determine transcriptional and translational profiles of N6AMT1 and report that it is required for the cytosolic translation of TRMT10C (MRPP1) and PRORP (MRPP3), two subunits of the mitochondrial RNAse P enzyme. In the absence of N6AMT1, or when its catalytic activity is abolished, RNA processing within mitochondria is impaired, leading to the accumulation of unprocessed and double-stranded RNA, thus preventing mitochondrial protein synthesis and oxidative phosphorylation, and leading to an immune response. Our work sheds light on the function of N6AMT1 in protein synthesis and highlights a cytosolic program required for proper mitochondrial biogenesis.
Most of the mitochondrial proteome is encoded in the nucleus, and only 13 mitochondrial proteins, all of which are required for mitochondrial oxidative phosphorylation (OXPHOS), are derived from the mitochondrial genome (mt-DNA). To ensure the assembly of the respiratory chain complexes and respond to environmental changes, the expression of both genomes must be tightly coordinated. For example, transcriptional mechanisms such as those relying on PGC-1α (peroxisome proliferator-activated receptor gamma coactivator 1-alpha) and NRF1 (nuclear respiratory factor 1), two master regulators of mitochondrial biogenesis, can promote mt-DNA expression by inducing transcription of TFAM (1, 2), a nuclear-encoded rate-limiting factor in mt-DNA replication and transcription. In addition, CLUH (clustered mitochondria protein homologue) and 4E-BPs (eukaryotic translation initiation factor 4E-binding proteins), cytosolic proteins which respond to nutrient deprivation, promote the translation of nuclear-encoded mitochondrial proteins (3, 4).
Within mitochondria, both strands of the mt-DNA molecule are transcribed as complementary polycistronic transcripts containing rRNAs, tRNAs, mRNAs, and noncoding RNAs (ncRNAs). Newly synthesized mt-RNA accumulates in mitochondrial RNA granules (MRGs) (5–7) where the mitochondrial protein-only RNAse P complex (composed of TRMT10C/MRPP1, HSD17B10/MRPP2, and PRORP/MRPP3/KIAA0391) and RNAse Z (ELAC2) process polycistronic mt-RNA by excising tRNAs according to the tRNA punctuation model (8, 9). Blocking mitochondrial RNA processing leads to the retention of newly transcribed mt-RNA in MRGs (5). An integral part of RNA processing is the degradation of ncRNAs by the mitochondrial RNA degradosome (SUPV3L1 and PNPT1), which fine-tunes mitochondrial gene expression and prevents accumulation of the double-stranded RNA (dsRNA) generated through complementarity between the bidirectional mt-RNA transcripts (10, 11). Inborn errors in the machinery of mitochondrial RNA degradation result in mt-dsRNA accumulation, leakage into the cytoplasm, and a chronic interferon response in humans (11).
In two recent genetic screens, we and others have highlighted a possible role for the nucleo-cytoplasmic multisubstrate methyltransferase N6AMT1 in OXPHOS (12, 13). N6AMT1 (also known as HEMK2, KMT9) was originally identified as a putative N6-adenine-specific DNA methyltransferase, but the existence of N6-methyladenosine in human DNA is uncertain (14, 15), and recent in vitro assays as well as structural studies have questioned its DNA methyltransferase activity (16, 17). In bacteria, yeast, and higher eukaryotes, N6AMT1 and its homologues (prmC and Mtq2) methylate translation release factors (RFs) on a highly conserved GGQ motif (18–22). In bacteria, RF1/RF3 methylation increases the speed and accuracy of translation termination (23), while the role of eRF1 methylation in eukaryotes is less clear (21, 24, 25). In yeast, Mtq2 interacts with proteins of the 60S cytosolic ribosomal subunit, and an MTQ2-null strain exhibits defects in ribosome biogenesis, with no defects in translation termination (24). In Drosophila melanogaster, HemK2 depletion leads to ribosome stalling and a decline in mRNA stability (26), while in mice, genetic ablation of N6amt1 is lethal (25), but to date, its impact on mammalian translation has not been investigated. Additional protein methylation substrates have also been identified for N6AMT1, including histone H4, which undergoes lysine 12 monomethylation affecting transcription of cell cycle and cancer-related genes (27, 28), as well as methylation of translation-related factors (29, 30). N6AMT1 may also modulate arsenic-induced toxicity, possibly by converting arsenic derivatives into less toxic forms, and polymorphisms in N6AMT1 are associated with arsenic resistance in Andean women (31, 32). It is currently unclear which of the multiple roles attributed to N6AMT1 accounts for its putative mitochondrial phenotype.
Here, we investigate N6AMT1 and its role in mitochondrial function. We report that N6AMT1 is required for the cytosolic synthesis of two key subunits of the mitochondrial RNA processing machinery. Depletion or catalytic inactivation of N6AMT1 results in the accumulation of unprocessed and double-stranded mt-RNA, leading to decreased mitochondrial protein synthesis, progressive collapse in the mitochondrial respiratory chain, and an interferon response. Our work identifies a mechanism required for the coordinated expression of the nuclear and mitochondrial genomes based on N6AMT1.
Results
N6AMT1 Is a Nucleo-Cytosolic Protein Required for Mitochondrial Gene Expression.
To improve our understanding of N6AMT1 in humans in the context of its previously reported functions, we analyzed the genetic dependency on N6AMT1 across 1,100 cancer cell lines from the Cancer Cell Line Encyclopedia (CCLE) (33). We found that cells are selectively dependent on N6AMT1 (CCLE Chronos score <−0.5 in 35.3% of the cell lines), irrespective of their lineages (Fig. 1A and SI Appendix, Fig. S1A). Comparing the N6AMT1-dependency profile across the 1,100 cancer cell lines with the dependency profiles of 15,677 nonessential genes, we found that N6AMT1 correlated best with nuclear-encoded genes coding for mitochondrial proteins [MitoCarta3.0 genes (34)] (Fig. 1 B and C, SI Appendix, Fig. S1B, and Datasets S1 and S2). This observation supports a possible role for N6AMT1 in mitochondrial function. However, consistent with previous reports (35, 36), we found no colocalization of N6AMT1 with mitochondria using confocal microscopy across multiple cell lines (Fig. 1D and SI Appendix, Fig. S1C).
Fig. 1.
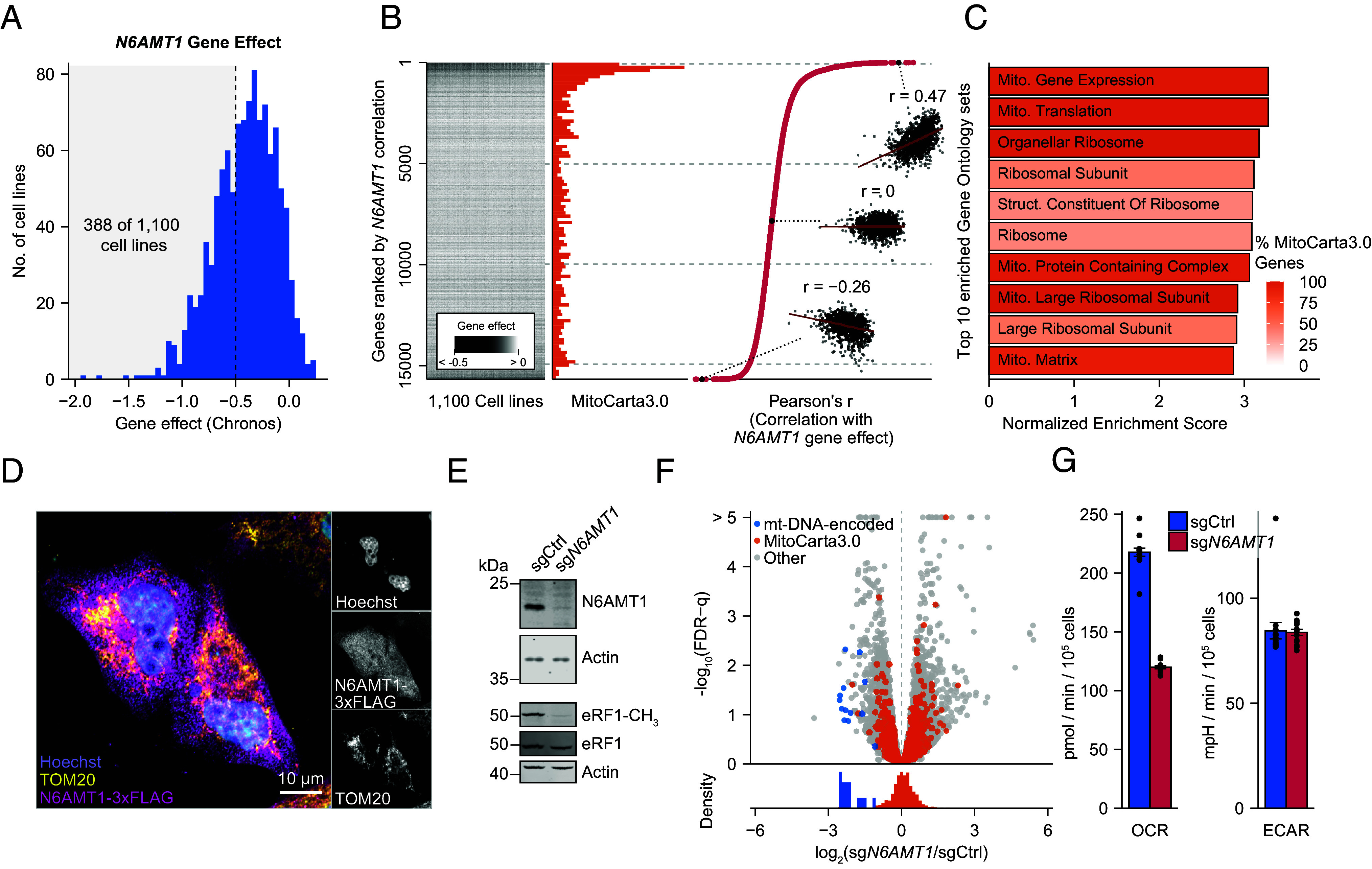
The mitochondrial impact of N6AMT1 is independent of nuclear transcription. (A) Cell line distribution of N6AMT1 Chronos scores across 1,100 cell lines of the CCLE. Density represents the relative number of cell lines with an N6AMT1-gene effect score within the range of each bin. The shaded area represents highly N6AMT1-dependent cell lines (388 of 1,100 cell lines with Chronos score <−0.5). (B) Analysis of N6AMT1 gene effect correlations. Gene-effect scores (Chronos scores; Left) of 15,677 nonessential genes in 1,100 cell lines from the CCLE were correlated with N6AMT1 gene effects and ranked according to Pearson correlation coefficients (Right). The distribution of MitoCarta3.0 genes is indicated in the Middle panel. Inserts show example correlations between N6AMT1 and three arbitrarily selected genes. Pearson correlation coefficients are listed in Dataset S1. (C) Top 10 enriched gene sets from Gene Ontology term-based GSEA of correlation coefficients shown in (B). Color shade indicates the percentage of MitoCarta3.0 genes in each gene set. (D) Immunofluorescence analysis of N6AMT1 localization. HeLa cells were transduced with N6AMT1-3xFLAG cDNA and immunolabeled with anti-FLAG and anti-TOM20 (a mitochondrial marker). Nuclei were visualized by Hoechst staining. (E) Immunoblot analysis of N6AMT1, methylated eRF1 (eRF1-CH3), and total eRF1 protein levels in control (sgCtrl) and N6AMT1-depleted (sgN6AMT1) K562 cells. (F) Transcriptomic analysis of 11,285 transcripts in N6AMT1-depleted K562 cells highlighting MitoCarta3.0 (orange) and mitochondrial DNA (mtDNA)-encoded ORFs (blue). False discovery rate (FDR) was calculated according to the method of Benjamini and Hochberg. n = 3 independent lentiviral transductions per condition. (G) Representative basal oxygen consumption rate (OCR) and ECAR per 10,000 cells in N6AMT1-depleted K562 cells. Bars represent mean values (n = 15 replicate wells, representative of ≥3 independent experiments), and error bars represent SEM.
Since N6AMT1 is involved in histone H4 monomethylation and possibly DNA methylation, we reasoned that it could impact mitochondria through transcriptional regulation in the nucleus. We depleted N6AMT1 using CRISPR/Cas9 in human chronic myelogenous leukemia K562 cells and performed RNA sequencing (RNA-Seq) on total RNA (Fig. 1 E and F, SI Appendix, Fig. S1D, and Dataset S3). Gene set enrichment analysis (GSEA) (37, 38) revealed no trend in the overall transcription of MitoCarta3.0 genes (SI Appendix, Fig. S1D and Dataset S4) and we obtained similar results when we reanalyzed a published RNA-Seq dataset of A549 lung carcinoma cells treated with small interfering RNAs targeting N6AMT1 (28) (SI Appendix, Fig. S1E and Dataset S5). Importantly, and in contrast to earlier studies, we included genes encoded by the mitochondrial genome in our RNA-Seq analysis, allowing us to observe a profound decrease in all 11 mt-mRNAs, corresponding to the 13 mitochondrial open-reading frames (ORFs), in N6AMT1-depleted cells (Fig. 1F). Accordingly, we found decreased respiration in N6AMT1-depleted cells (Fig. 1G). We conclude that N6AMT1 is required for mt-DNA expression and mitochondrial function, even though the protein is not present within mitochondria and is not directly involved in transcriptional regulation of the nuclear genes encoding mitochondrial proteins.
N6AMT1 Depletion Reduces Cytosolic Translation of the mt-RNA Processing Machinery.
Given that a direct role for N6AMT1 in nuclear transcription is unlikely to account for the changes seen in mt-DNA gene expression, we investigated the influence of N6AMT1 on cytosolic translation using ribosome profiling (Ribo-Seq). In K562 cells, N6AMT1 depletion led to no or marginal changes in rRNA abundance, polysome profiles, and ribosome profiles (Fig. 2 A and B and SI Appendix, Fig. S2A). In contrast to earlier studies in yeast and bacteria (23, 24), we observed no appearance of halfmers in our polysome profiles (Fig. 2B) and our meta-analysis of all transcripts in N6AMT1-depleted cells showed no difference in ribosome occupancy on termination codons (Fig. 2C), indicating no global effect on polysome formation or on translation termination, respectively. However, 1,401 individual genes were differentially translated (FDR-q < 0.05) in N6AMT1-depleted cells (Fig. 2D and Dataset S6). Gene ontology analysis revealed several affected pathways (SI Appendix, Fig. S2B and Dataset S7), and among them, we found a significant reduction in the translation of genes involved in mt-RNA metabolism, including PRORP, the catalytic subunit of the mitochondrial RNAse P, and TRMT10C, a methyltransferase also part of the mitochondrial RNAse P complex (Fig. 2D). This was in marked contrast to the RNA-Seq data, which showed no difference in the steady-state levels of mt-RNA metabolism-related transcripts, including in PROPR and TRMT10C (SI Appendix, Fig. S2 B and E). GSEA using the MitoPathways gene sets (34) confirmed “mt-RNA metabolism,” “mt-RNA processing,” and “mt-RNA granules” as the three most depleted mitochondrial pathways (Dataset S8), a finding that we corroborated by analyzing translation efficiency (TE), a metric in which ribosome-protected mRNA fragments are normalized to transcript abundance (39) (Datasets S9 and S10).
Fig. 2.
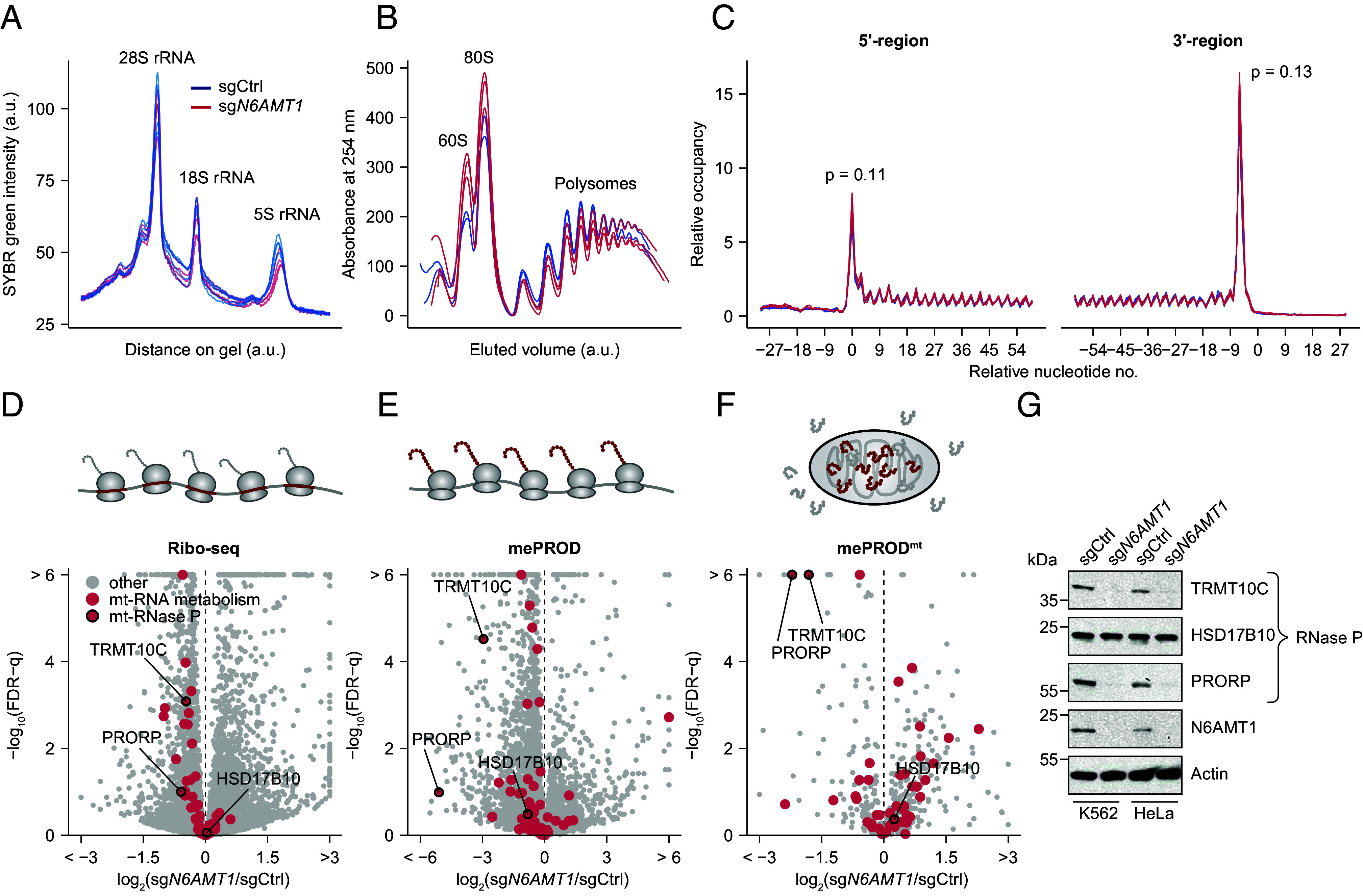
Cytosolic translation of the mt-RNA processing machinery is reduced in N6AMT1-depleted cells. (A) Quantification of RNA agarose gel (shown in SI Appendix, Fig. S2A) comparing control (sgCtrl) to N6AMT1-depleted (sgN6AMT1) K562 cells 10 d post-sgRNA transduction (n = 4 independent lentiviral infections per condition). Each curve represents the SYBR-green intensity across one lane, with the major rRNA peaks indicated. (B) Polysome analysis of control and N6AMT1-depleted K562 cells (n = 2-3 lentiviral infections) 10 d post-sgRNA transduction. Elution volumes are normalized to the 80S peak and the maximal polysome peak to allow direct comparisons between replicates. (C) Metagene analysis of control and N6AMT1-depleted K562 cells 10 d post-sgRNA transduction detected by ribosome profiling (Ribo-Seq). Curves represent ribosome-protected fragments at the indicated nucleotide positions relative to the total number of ribosome-protected fragments (n = 3 independent lentiviral infections per condition, based on 9,600 transcripts). P-values were obtained by Student’s t tests of the 5′ and 3′ peaks, respectively. (D) Transcript ribosome occupancy of 10,793 transcripts detected by ribosome profiling (Ribo-Seq) in N6AMT1-depleted K562 as compared to control cells. n = 3 independent lentiviral transduction per condition. (E and F) Translatome analysis using multiplexed enhanced protein dynamics (mePROD; 4,079 proteins) proteomics (E) and global mitochondrial protein import proteomics (mePRODmt; 495 proteins) (F) in N6AMT1-depleted K562 as compared to control cells. n = 4 independent lentiviral transduction per condition. (G) Protein immunoblot showing mitochondrial RNase P subunit protein levels in control and N6AMT1-depleted K562 and HeLa cells, with antibodies targeting TRMT10C (MRPP1), HSD17B10 (MRPP2), and PRORP (MRPP3).
We next sought to complement our ribosome profiling observations with a protein-based assay and used mePROD (multiplexed enhanced protein dynamic) and its mitochondrial equivalent, mePRODmt. mePROD combines pulsed stable isotope labeling (SILAC) with tandem mass tags, signal amplification, and mass spectrometry to monitor newly synthesized proteins on a proteome-wide scale, while mePRODmt includes a mitochondrial isolation step to quantify more selectively the proteins imported into mitochondria. mePROD revealed N6AMT1-dependent alterations in cytosolic translation that correlated moderately with Ribo-Seq data (Pearson correlation coefficient = 0.42, SI Appendix, Fig. S2C), revealing a subset of mt-RNA-related proteins reduced in N6AMT1-depleted cells, including PRORP and TRMT10C (Fig. 2E, SI Appendix, Fig. S2E, and Dataset S11). We confirmed these observations with mePRODmt data (Fig. 2F, SI Appendix, Fig. S2E, and Dataset S12). Comparing mePROD and mePRODmt revealed no difference in protein import into mitochondria (SI Appendix, Fig. S2D). PRORP and TRMT10C were the two main mt-RNA-related factors depleted in all three datasets, and to confirm our findings, we immunoblotted N6AMT1-depleted cells and observed a significant reduction in the steady-state levels of the two proteins, both in K562 and in HeLa cells, while the third RNase P subunit HSD17B10 was not affected (Fig. 2G). Taken together, our complementary approaches revealed a selective reduction in cytosolic translation of two protein subunits of the mitochondrial RNAse P in N6AMT1-depleted cells.
N6AMT1 Is Necessary for mt-RNA Processing, Protein Synthesis, and OXPHOS.
The observed reductions in mt-DNA-encoded mRNAs (Fig. 1F) and in the proteins involved in mitochondrial RNA processing (Fig. 2G) prompted us to characterize further the impact of N6AMT1 on the mitochondrial transcriptome. We used NanoString (40) and a set of mitochondria-specific probes called “MitoString” (41) to quantify mt-mRNAs, unprocessed mt-RNA junctions, and mt-ncRNAs (SI Appendix, Fig. S3 A and B). We observed a dramatic increase in unprocessed mRNA-tRNA and rRNA-tRNA junctions in N6AMT1-depleted cells, affecting both 5′ and 3′ RNA junctions, as well as an accumulation of mt-ncRNAs (Fig. 3A), strongly suggesting a deficiency in mt-preRNA processing, with no effect on mt-DNA copy number (Fig. 3B). To exclude off-target effects, we reintroduced an sgRNA-resistant cDNA of N6AMT1 and observed a full rescue of the phenotype (Fig. 3 A and C). In contrast, we found that a catalytically inactive N6AMT1 mutant (D77A) (24) failed to rescue mt-RNA processing (Fig. 3C and SI Appendix, Fig. S3C), indicating that the catalytic activity of N6AMT1 is required for its function in mt-RNA processing. To test whether PRORP and TRMT10C were limiting in the absence of N6AMT1, we coexpressed both cDNAs in N6AMT1-depleted K562 cells and observed a full or partial rescue of the mt-RNA processing phenotype at all mt-preRNA junctions tested (Fig. 3D and SI Appendix, Fig. S3 D and E).
Fig. 3.
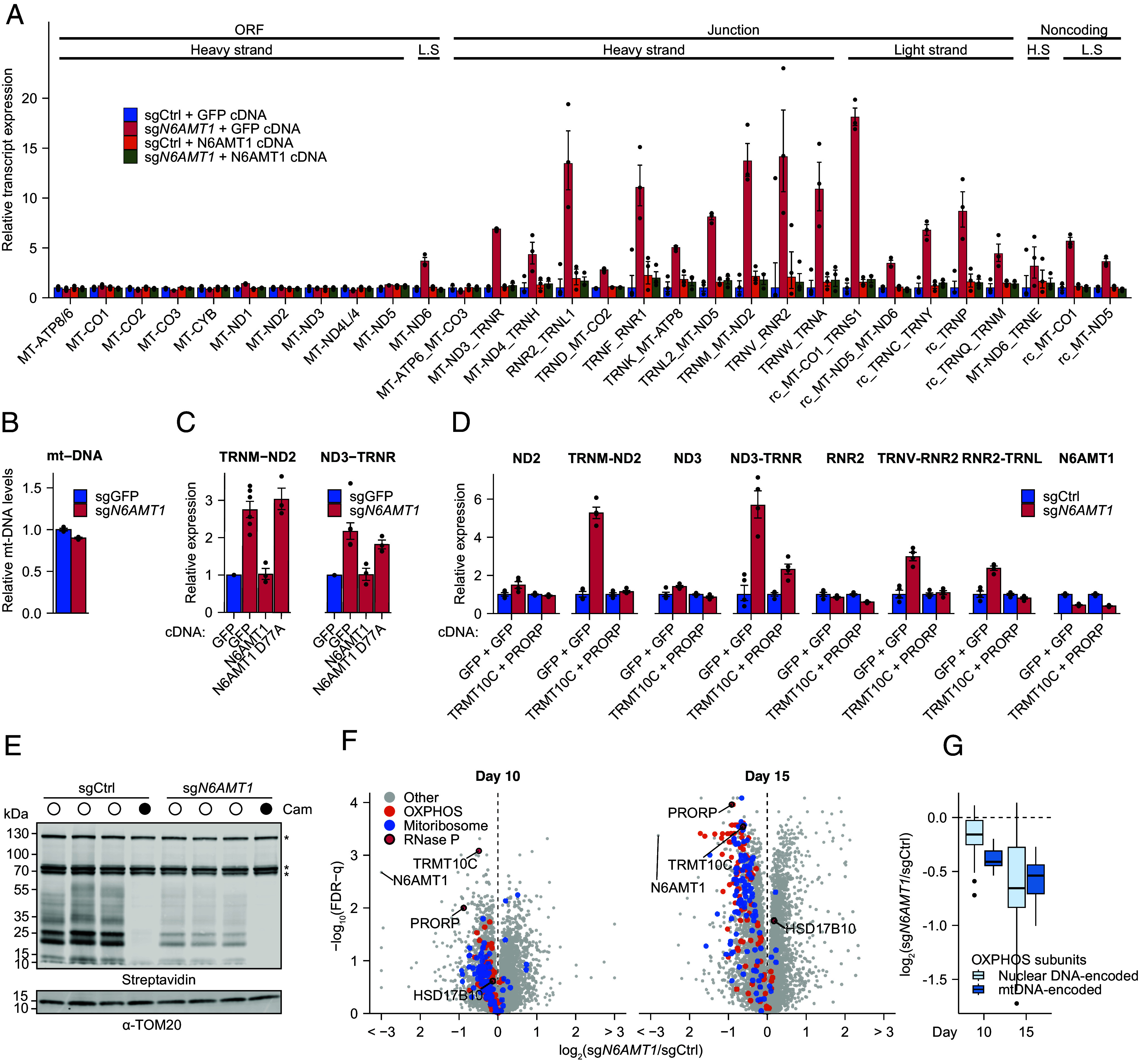
N6AMT1 depletion leads to impaired mt-RNA processing and prevents mitochondrial translation and OXPHOS. (A) MitoString analysis of mitochondrial RNA processing in control (sgCtrl) and N6AMT1-depleted (sgN6AMT1) K562 cells where a control cDNA (coding for GFP) or an sgRNA-resistant cDNA of N6AMT1 was introduced. Bars represent geometric mean values and error bars indicate corresponding SEM (n = 3 independent transduction). L.S.: light strand; H.S.: heavy strand. rc: reverse complement. “ORF,” “Junction,” and “Noncoding” correspond to the regions of the mitochondrial transcriptome targeted by our probes. MitoString probes are described in more detail in SI Appendix, Fig. S3. (B) Relative mt-DNA quantification as determined by qPCR in control or N6AMT1-depleted K562 cells. Relative mt-DNA levels were calculated as the ratio between the expression of the mt-DNA-encoded MT-ND2 gene and the nuclear DNA-encoded 18S rRNA gene. (C) qPCR analysis of unprocessed mt-RNA in control, N6AMT1-depleted cells, and N6AMT1-depleted cells with an sgRNA-resistant N6AMT1 cDNA introduced (wild type or catalytically active mutant D77A) (n = 3 to 6 independent lentiviral infections). (D) qPCR analysis of unprocessed and total mt-mRNA and mt-rRNA in control or N6AMT1-depleted K562 with the introduction of either 2 different GFP cDNAs or TRMT10C + PRORP cDNA (n = 3 independent lentiviral infections). (E) Translation of mitochondrially encoded proteins in control or N6AMT1-depleted K562 cells after labeling with L-Homopropargylglycine in the presence of cycloheximide (a cytosolic translation inhibitor). Chloramphenicol (Cam, a mitochondrial translation inhibitor) is used as control. Asterisks indicate endogenously biotinylated mitochondrial proteins. All mt-DNA-encoded proteins migrate below the 70 kDa marker. (F) Proteome changes of 7,364 quantified proteins in N6AMT1-depleted K562 cells as compared to control, 10 d (Left) or 15 d (Right) following sgRNA transduction (n = 3 independent transduction per condition per day). (G) Proteomics protein abundance of OXPHOS subunits according to the genome (nuclear or mitochondrial (mt-DNA)) that encodes them.
MitoString was originally established and tested by knock-down of 107 mitochondrially localized, predicted RNA-binding proteins (41), allowing us to perform unsupervised clustering to identify which of these genes share similarities with N6AMT1. Among the 107 knockdowns, we found that N6AMT1 depletion clustered best with depletion of subunits from the mitochondrial RNAse P and RNAse Z (SI Appendix, Fig. S3F), thus functionally supporting our translatome results (Fig. 2 D–F).
We then sought to characterize the mitochondrial consequences of low RNase P activity in N6AMT1-depleted cells. Consistent with defects in RNA processing and depletion of mature mt-RNA, we observed a global attenuation of translation by the mitochondrial ribosomes (Fig. 3E). We confirmed this observation using time-resolved quantitative proteomics on N6AMT1-depleted cells, where we observed a strong, gradual depletion of most proteins involved in these processes (Fig. 3F and Dataset S13). Our proteomics analysis also confirmed decreased levels of PRORP and TRMT10C, while HSD17B10 levels were only marginally affected (Fig. 3F). For the respiratory chain, we observed a gradual reduction in most subunits of all OXPHOS complexes, with the exception of the F1 domain of the ATP synthase, which is not mtDNA-encoded and can assemble even when mitochondrial gene expression is impossible (Dataset S13) (42). Consistent with the defect caused by abnormal mt-RNA expression, we noted that at early time points the mt-DNA-encoded OXPHOS subunits, which are all core subunits of the respiratory chain, appeared to be more dramatically reduced than the nuclear DNA-encoded subunits (Fig. 3G). In addition, pathways downstream of mitochondrial gene expression were also progressively decreased at the protein level, as expected from their strict dependency on intact mt-RNA metabolism (SI Appendix, Fig. S3G), while mitochondrial pathways such as “signaling,” “protein import, sorting, and homeostasis,” and “metabolism” were not affected, indicating that mitochondrial functions not related to gene expression or OXPHOS were maintained (SI Appendix, Fig. S3G). We conclude that defects in N6AMT1-dependent synthesis of the mitochondrial RNAse P machinery alter the mitochondrial proteome in a way that leads to a progressive decline in pathways downstream of mt-RNA processing, such as mitochondrial translation and OXPHOS.
Impaired mt-RNA Processing in N6AMT1-Depleted Cells Causes MRG Stress and dsRNA Accumulation.
Mitochondrial RNA processing takes place in MRGs (5, 43), where the mitochondrial RNAses P/Z and the degradosome cleave and degrade mt-RNA. In the absence of these events, the accumulation of mt-RNA, often in the form of mt-dsRNA, can trigger an interferon response and inflammation (11). Based on its effects on mt-preRNA and mt-ncRNA processing (Fig. 3A), we hypothesized that N6AMT1 depletion could trigger the accumulation of unprocessed mt-RNA in MRGs, as well as innate immune signaling related to mt-dsRNA. K562 cells lack a large genomic region including the interferon gene locus (44) and, in addition, are poorly compatible with imaging. We therefore used HeLa cells to investigate the MRGs following N6AMT1 depletion, where we also confirmed strong depletion of PRORP and TRMT10C (Fig. 2G). We immunolabeled MRGs with antibodies against FASTKD2, an MRG-specific marker whose abundance was not affected in our proteomics profiles (Dataset S13), as well as against BrU following a short pulse of 5-bromouridine (BrU) to label newly synthesized mt-RNA. We observed a striking accumulation of MRGs in N6AMT1-depleted cells, indicating MRG stress (5) (Fig. 4 A and C). Similarly, and as expected from the accumulation of mt-ncRNA (Fig. 3A), we also observed strong accumulation of dsRNA in mitochondria from N6AMT1-depleted cells, here too forming more abundant, larger and brighter foci following immunolabeling using a dsRNA antibody (Fig. 4 B and D). Finally, we examined the physiological consequences of this mt-RNA accumulation using qPCR and, as expected from previous work on patients’ cells unable to degrade mt-RNA (11), found a transcriptional activation of innate immunity genes characterized by high expression of interferon beta (IFNB1) and interferon-related genes such as USP18, IFIT1, or GBP2 (Fig. 4E). We conclude that the aberrant mt-RNA processing seen in N6AMT1-depleted cells results in the accumulation of unprocessed and double-stranded mt-RNA, with consequences for MRGs, OXPHOS, and immune signaling (Fig. 4F).
Fig. 4.
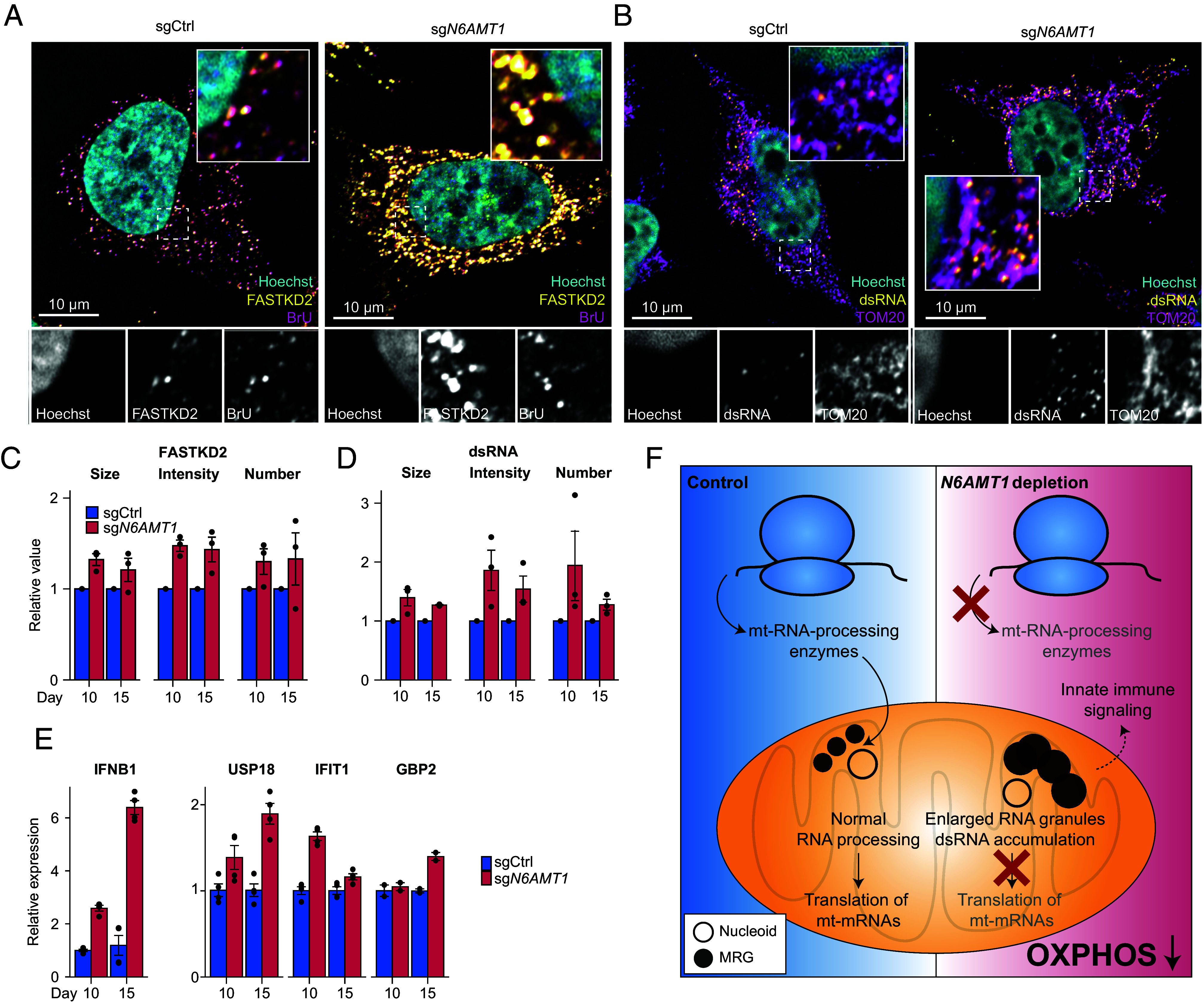
Mitochondrial preRNA and dsRNA accumulate and trigger an interferon response in N6AMT1-depleted cells. (A) Representative images from immunofluorescence analysis of FASTKD2- and bromouridine (BrU)-immunolabeled MRGs in control (sgCtrl) or N6AMT1-depleted (sgN6AMT1) HeLa cells 15 d post transduction. (B) Representative images from immunofluorescence analysis of mitochondria (labeled with antibodies against TOM20) and mitochondrial dsRNA (labeled with J2 dsRNA antibody) in sgCtrl or sgN6AMT1 HeLa cells 15 d post transduction. (C and D) Quantification of FASTKD2 foci (C) and dsRNA foci (D), 10 or 15 d post transduction of HeLa cells with sgCtrl or sgN6AMT1. n = 3 independent experiments, with signal from at least 25 cells per condition. “Number” refers to average number of foci per cell area, “Size” refers to the average foci area, and “Intensity” refers to the average foci intensity. (E) qPCR determination of transcripts of interferon beta (IFNB1) and interferon-inducible USP18, IFIT1, and GBP2 in N6AMT1-depleted HeLa cell lines as compared to controls 10 or 15 d post transduction. n = 4 independent transductions. (F) Proposed role of N6AMT1 in mitochondrial biogenesis. In N6AMT1-depleted cells, translation of mt-RNA-processing enzymes is reduced, leading to accumulation of unprocessed RNA in MRGs. This prevents translation of essential genes involved in mitochondrial gene expression and OXPHOS. In addition, impaired RNA processing leads to the accumulation of double-stranded mt-RNA that activates immune signaling pathways.
Discussion
We report here a cytosolic pathway required for mitochondrial biogenesis that relies on N6AMT1. We show that N6AMT1 is required for the cytosolic translation of two key subunits from the mitochondrial RNA processing machinery, the deficiency of which leads to MRG stress, dsRNA accumulation, immune signaling, and a collapse in OXPHOS (Fig. 4F).
Our results shed light on the cellular function of N6AMT1, a multisubstrate methyltransferase with previously assigned functions ranging from transcription to arsenic metabolism. Using Ribo-Seq and mePROD we established a role for human N6AMT1 in cytosolic protein synthesis. Interestingly, N6AMT1 orthologs in bacteria and yeast are also involved in translation, and while we report that the molecular function of N6AMT1 in humans has diverged, as it does not involve ribosome assembly or translation termination (Fig. 2 A and B), our results nevertheless indicate that N6AMT1 has retained a canonical role in protein synthesis that appears to depend on its catalytic activity (Fig. 3C). Previous work identified translation-related factors, such as RRP1, EIF2BD, and eRF1 (22, 25, 29, 30), as substrates for N6AMT1, while in yeast, Mtq2 interacts with ribosomal proteins (24). Further work will be necessary to clarify which of the potential N6AMT1 methylation targets accounts for its role in translation regulation.
Based on our genome-wide identification of the genes required for mitochondrial function (12), our work reveals a pathway which is necessary for the coordinated expression of the nuclear and mitochondrial genomes. We show that N6AMT1 is required for the translation of PRORP and TRMT10C, two subunits of the mitochondrial RNAse P which are central for mitochondrial gene expression, and which are also implicated in mitochondrial disease (45, 46). Here, we have focused our investigations on these two mitochondrial RNase P subunits and report that their decreased translation impairs mitochondrial RNA processing with two major consequences: 1) mt-mRNAs, mt-rRNAs, and mt-tRNAs remain unprocessed, thus prohibiting mitochondrial ribosome assembly and translation, strongly reducing expression of the 13 mt-DNA-encoded proteins and inhibiting OXPHOS (Fig. 2); 2) mt-RNA accumulates in mitochondria, where it colocalizes with markers of MRGs. Based on genetic depletion, our work thus far points to a role for N6AMT1 in supporting fully functional mitochondria, and it is possible that N6AMT1 may be induced, or activated, during intense mitochondrial biogenesis programs, such as those seen during adipose or muscle differentiation. In the future, it will be interesting to analyze changes in N6AMT1 expression during such processes and to follow the expression of the mitochondrial RNAse P, mt-RNA processing, and the state of MRGs.
The question remains of how, mechanistically, the absence of N6AMT1 leads to a specific reduction in TRMT10C and PRORP. Our immunofluorescence data (Fig. 1D and SI Appendix, Fig. S1C) and earlier work (14, 36, 47) showed no colocalization of N6AMT1 with mitochondria, and previous efforts to characterize mitochondrial proteomes did not localize N6AMT1 in the organelle (48–50). Furthermore, sequence analysis of N6AMT1 predicts no mitochondrial localization (51, 52). Although we cannot rule out the possibility of trace amounts of N6AMT1 in mitochondria, the nucleo-cytosolic localization and the lack of evidence for a mitochondrial localization strongly suggest that N6AMT1 functions through a nonmitochondrial mechanism. Indeed, our Ribo-Seq, mePROD, and mePRODmt observations all point to a significant decrease in the cytosolic translation of TRMT10C and PRORP in N6AMT1-depleted cells, an inhibition we could rescue by exogenously expressing their corresponding cDNAs (SI Appendix, Fig. S3E). Given the role for N6AMT1 in the methylation of the cytosolic translation release factor eRF1 (22) (Fig. 1D), we speculate that the regulation may proceed through a mechanism related to translation termination. In a recent screen, N6AMT1 was identified as necessary for the induction of DDIT3 (CHOP) following induction of the integrated stress response (53). DDIT3 encodes a well-characterized upstream ORF (uORF) in its 5′ untranslated region (54, 55), and since uORFs are also predicted in isoforms of TRMT10C and PRORP mRNAs (56, 57), it is possible that unmethylated eRF1 may affect ribosome release from those uORFs, thereby interfering with downstream translation. Alternatively, a recent study reported a role for Drosophila N6AMT1/HemK2 in preventing the activation of the No-Go Decay pathway following ribosome stalling (26). Future work will be required to clarify the precise mechanism of action of N6AMT1 on cytosolic translation.
Recent work has highlighted the role of both mt-DNA and mt-RNA in triggering inflammation and the interferon response (11, 58, 59). In the case of mt-RNA, depletion or mutations in PNPT1 lead to mt-dsRNA accumulation in mitochondria and its subsequent leakage into the cytosol where it serves as ligand for MDA-5 and RIG-I, leading to both NF-κB and interferon signaling (11). While our K562 cellular model used here for large-scale genetic experiments did not allow us to study interferon signaling, we detected significant upregulation of IFNB1 mRNA as well as interferon-related transcripts in N6AMT1-depleted HeLa cells (Fig. 4E), a result very similar to that observed following PNPT1 depletion or mutation (11).
High levels of N6AMT1 are associated with multiple forms of cancers, and small interfering RNAs to N6AMT1 prevent the proliferation of cancer cells from androgen receptor–dependent and castration-resistant prostate cancer (27), bladder cancer (60, 61), colorectal cancers (62), as well as small cell and non–small cell lung cancer (28). N6AMT1 methyltransferase inhibitors are also being developed (63, 64), and early results show promising activity at blocking tumor cell proliferation. A proposed mechanism for the role of N6AMT1/KMT9 in cancer is the monomethylation of histone H4 that controls transcription of cell cycle regulators and proproliferation factors (27). We have reported here on the role of N6AMT1 in cytosolic translation, mitochondria, and innate immunity. Mitochondria are central to the provision of energy and building blocks that support cell proliferation, including in cancer cells. Based on our observations, we propose that the mechanism by which N6AMT1 promotes cancer cell growth also involves mitochondrial activity. This role is supported by our analysis of N6AMT1 in 1,100 CRISPR/Cas9 screens performed across cancer cell lines from 20 tissues of origin, where we report that the N6AMT1 dependency profile correlates best with dependency on mitochondria (Fig. 1 A–C). Our observations are compatible with earlier studies reporting a function of N6AMT1/KMT9 in epigenetics (27, 62), and, collectively, the evidence points to a dual role for N6AMT1 in both gene expression and protein synthesis. Going forward, it will be important to identify which of these functions underlie cancer cell proliferation, as this could promote the development of novel anti-neoplastic molecules with reduced drug toxicity.
Materials and Methods
Cell Lines.
K562 (CCL-243), HeLa (CCL-2), and 293T (CRL-3216) were obtained from ATCC and reauthenticated by STR profiling at ATCC during the course of this study.
Cell Culture.
Cells were maintained in DMEM containing 1 mM sodium pyruvate (ThermoFisher Scientific) with 25 mM glucose, 10% fetal bovine serum (FBS, ThermoFisher Scientific), and 100 U/mL penicillin/streptomycin (ThermoFisher Scientific) under 5% CO2 at 37 °C. Cells were counted using a ViCell Counter (Beckman), and only viable cells were considered.
Cancer Cell Line Correlation Analysis.
DepMap Public 23Q2 Chronos scores were obtained from the CCLE Dependency Portal (DepMap) (65). Genes annotated as common essentials or with missing values were excluded. Pearson’s correlation coefficients were calculated between N6AMT1 Chronos scores and all other genes using R version 4.3.1 (66). Gene-wise correlation coefficients were exported for GSEA.
Analysis of Published RNA-Seq Datasets.
Count values from dataset GSE131016 were obtained from the NCBI Gene Expression Omnibus data repository and reanalyzed using DESeq2 (67) in R version 4.3.1 (66). For GSEA, log2 fold change shrinkage was performed using the apeglm package version 1.26.1 (68).
GSEA.
CCLE dependency correlation coefficients and log2 (fold-changes) from RNA-Seq, Ribo-Seq, and proteomics analyses were analyzed by GSEA version 4.3.2 (38) using the c5.go.v2023.Hs.symbols (Gene ontology) or MitoPathways (34) gene sets.
Gene-Specific CRISPR/Cas9 Knockouts and cDNA Rescue.
The two best N6AMT1 sgRNAs from the Avana library (69) were ordered as complementary oligonucleotides (Integrated DNA Technologies) and cloned in pLentiCRISPRv2 (70). sgRNAs targeting EGFP (nontargeting) or OR11A1 and OR2M4 (two unexpressed genes in K562 cells) were used as a negative control. Lentiviruses were produced according to Addgene’s protocol, and 24 to 48 h postinfection, cells were selected with 2 µg/mL puromycin (ThermoFisher Scientific) for 2 to 5 d. Cells were then maintained in routine culture media for 10 to 15 d post transduction before analysis. Gene disruption efficiency was verified by immunoblotting. For rescue, a sgRNA-resistant version of N6AMT1-3xFLAG cDNA was synthesized in pWPI (Addgene). The same cDNA was further edited to obtain the D77A variant using site-directed mutagenesis. sgRNA, cDNA, and primer sequences are described in SI Appendix, Table S1.
Mitochondrial and Nuclear DNA Determination.
Mitochondrial and nuclear DNA determination was carried as previously described (34). Approximately 1 × 105 cells from each condition were harvested and lysed in 100 μL mt-DNA lysis buffer (25 mM NaOH and 0.2 mM EDTA) before incubation at 95 °C for 15 min. Then, 100 μL of 40 mM Tris-HCl pH 7.5 was added to neutralize the reaction on ice. Samples were diluted 50×, and the ratio between mitochondrial and nuclear DNA was determined with a CFx96 qPCR machine (Bio-Rad) using probes and primers targeting MT-ND2 and 18S rRNA genes as described in SI Appendix, Table S2. Relative mt-DNA abundance was determined using the ΔΔCt method.
RNA Extraction, Reverse Transcription, and qPCR.
RNA was extracted from total cells with the RNeasy kit (QIAGEN) and digested by DNase I before reverse transcription with murine leukemia virus reverse transcriptase using random primers (Promega). qPCR was performed with and a CFx96 Touch Real-Time PCR system (Bio-Rad) or a LightCycler 480 II (Roche) using probes and primers described in SI Appendix, Table S2. All data were normalized to the expression of TATA-box binding Protein (TBP) using the ΔΔCt method.
Seahorse.
A total of 1.25 × 105 K562 cells were plated on a Seahorse plate in Seahorse XF DMEM media (Agilent) containing 25 mM glucose and 2 mM glutamine (ThermoFisher Scientific). Oxygen consumption and extracellular acidification rates (ECARs) were simultaneously recorded by a Seahorse XFe96 Analyzer (Agilent) using the Mito Stress Test protocol, in which cells were sequentially perturbed by 2 µM oligomycin, 1 µM CCCP, and 0.5 µM antimycin (Sigma). Data were analyzed using Seahorse Wave Desktop Software (Agilent). Data were not corrected for carbonic acid derived from respiratory CO2.
Immunofluorescence and Confocal Microscopy.
For immunofluorescence, cells transduced with N6AMT1-3xFLAG, sgCtrl, or sgN6AMT1 sgRNAs were successively fixed in 4% paraformaldehyde in cell culture media at room temperature for 10 min, permeabilized for 15 min in 0.2% Triton-PBS solution (Sigma-Aldrich, T8787), incubated with primary antibodies (1:200) in 1% PBS-Bovine Serum Albumin (Sigma Aldrich, A9647-100G) for 1 h, washed 3 × 5 min in 1% Bovine Serum Albumin (BSA) in PBS, incubated with fluorophore-coupled secondary antibodies (1:300) and Hoechst (1:1,000) in 1% BSA in PBS for 45 min, washed 3× in PBS, and mounted on a slide using FluorSave (EMD Millipore). Cells were imaged using a Zeiss LSM 880 Airyscan confocal microscope (Zeiss, 518F). For bromouridine (BrU) staining, HeLa cells were pulsed with 5 mM BrU for 30 min before fixation. MRG and dsRNA quantification was performed using an automated pipeline on CellProfiler software. To limit our quantification to the mitochondrial network, nuclei were removed from the analysis, and cell delimitation was performed using a mitochondrial marker. mitodsRED plasmid was from Clontech.
Polyacrylamide Gel Electrophoresis and Immunoblotting.
Cells were harvested, washed in PBS, and lysed for 5 min on ice in RIPA buffer [25 mM Tris pH 7.5, 150 mM NaCl, 0.1% SDS, 0.1% sodium deoxycholate, 1% NP40 analog, 1× protease inhibitor (Cell Signaling), and 1:500 Universal Nuclease (ThermoFisher Scientific)]. Protein concentration was determined from total cell lysates using DC Protein Assay (Bio-Rad). Gel electrophoresis was done on Novex Tris-Glycine gels (ThermoFisher Scientific) before transfer using the Trans-Blot Turbo blotting system and nitrocellulose membranes (Bio-Rad). All immunoblotting was performed in 5% milk powder or 5% BSA in Tris-Buffered Saline (TBS; 20 mM Tris, pH 7.4, and 150 mM NaCl) + 0.1% Tween-20 (Sigma). Washes were done in TBS + 0.1% Tween-20 (Sigma). Specific primary antibodies were diluted 1:1,000-1:5,000 in blocking buffer (TBS + 0.1% Tween-20 and 5% milk powder). Fluorescent- and HRP-coupled secondary antibodies were diluted 1:10,000 in blocking buffer. Membranes were imaged with an Odyssey CLx analyzer (Li-Cor) or chemiluminescence and films.
Antibodies.
Antibodies were N6AMT1 (LSBio LS-C346278), actin (Sigma A1978), eRF1 (Abcam ab31799), FASTKD2 (Proteintech 17464-1-AP), TRMT10C (Sigma, HPA036671), HSD17B10 (Abcam, ab10260), PRORP (Abcam, ab185942), GAPDH (CST, 5174T), bromouridine (Sigma 11170376001), dsRNA (Exalpha, 10010500), and TOM20 (CST, 42406). Antibody against methylated eRF1 (24) was a kind gift from Valérie Heurgué-Hamard (UMR 8261 CNRS/Université Paris Cité, Institut de Biologie Physico-Chimique).
L-Homopropargylglycine Labeling.
Fifteen days post transduction with control sgRNA or N6AMT1-targeted sgRNA, K562 cells were washed with prewarmed PBS and resuspended in methionine-free medium [DMEM, high glucose, no glutamine, no methionine, no cystine (ThermoFisher Scientific) with 200 µM L-cystine, 4 mM glutamine, and 1 mM sodium pyruvate]. Mitochondrial and cytosolic translation was inhibited by incubation with 100 µg/mL chloramphenicol (Sigma-Aldrich) and 50 µg/mL cycloheximide (Sigma-Aldrich), respectively, 10 min before the addition of 500 µM L-homopropargylglycine (Jena Bioscience). After 1.5 h, cells were harvested and washed once in cold PBS and then twice in cold mitochondrial isolation buffer (10 mM HEPES pH 7.4, 210 mM mannitol, and 70 mM sucrose), and mitochondria were isolated as previously described (71). In short, 15 × 106 cells were resuspended in 750 µL MB + 250 U/mL Pierce Universal Nuclease (Thermo Fisher Scientific) and 1× protease inhibitor (Cell Signaling) and lysed with 10 strokes of a 25 G needle. Lysates were centrifuged for 10 min at 500 g, 4 °C, to remove debris and intact cells, and supernatants were further centrifuged 10 min at 10,000 g, 4 °C to pellet mitochondria. Mitochondrial pellets were dissolved in 60 µL RIPA buffer, and protein concentration was determined using a DC Protein Assay (Bio-Rad). Then, 50 µL dissolved mitochondrial pellets were mixed with 50 µL click-reaction mix (10 mM sodium ascorbate (Jena Bioscience); 40 µM picolyl-Azide-PEG4-Biotin (Jena Bioscience); 2.4 mM 2- (4- ( (bis ( (1- (tert-butyl)-1H-1,2,3-triazol-4-yl)methyl)amino)methyl)-1H-1,2,3-triazol-1-yl)acetic acid (Jena Bioscience); 1.2 mM CuSO4 (Jena Bioscience)), incubated 60 min at room temperature, and mixed with 20 µL 5× SDS-PAGE loading buffer [250 mM Tris pH 6.8, 4% sodium dodecyl sulfate, 0.06% (w/v) bromophenol blue, 16% (v/v) beta-mercaptoethanol, and 30% (v/v) glycerol]. Biotinylated proteins were visualized by immunoblotting using IRDye 800CW Streptavidin (LI-COR).
Total Ribosomal RNA (rRNA) Quantification by Gel Electrophoresis.
K562 cells were harvested 10 d post transduction with control sgRNA or N6AMT1-targeted sgRNA, washed in cold PBS, and snap-frozen in liquid nitrogen. RNA was extracted using TRI reagent (Sigma-Aldrich), essentially according to the manufacturer’s instructions, with the inclusion of GlycoBlue Coprecipitant (Invitrogen) for improving the visibility of precipitated RNA. Then, 16 µg purified RNA was mixed with 6× gel loading dye (New England BioLabs) and run on a 1% agarose-TAE (20 mM Tris; 10 mM acetic acid; 0.5 mM EDTA) gel + 0.01% (v/v) SYBR safe DNA Gel Stain (Invitrogen). SYBR green signal intensity was quantified across each lane using ImageJ version 1.53.
Polysome Profiling.
Ten days after viral transduction, control or sgN6AMT1-infected K562 cells were treated with 100 µg/mL cycloheximide 10 min before harvesting. Cells were harvested, washed in PBS, and lysed for 10 min on ice in lysis buffer [50 mM HEPES pH 7.3, 100 mM potassium acetate, 15 mM magnesium acetate, 5% glycerol, 0.5% Triton X-100, 2× protease inhibitor (Cell Signaling), 5 mM DTT, and 100 µg/mL cycloheximide]. Lysates were centrifuged for 9 min at 9,000 g, 4 °C, and RNA concentrations were measured with a Nanodrop ND-2000. Then, 180 µg RNA was loaded on 10 to 50% sucrose gradients in 25 mM HEPES, 100 mM potassium acetate, 5 mM magnesium acetate, and 100 µg/mL cycloheximide and centrifuged 1:45 h at 40,000 g, 4 °C on a Beckman Coulter Optima LE-80K Ultracentrifuge. Gradients were analyzed on an Äkta purifier measuring absorbance at 254 nm. Elution volumes were normalized to volumes at the maximal absorbance (80S peak) and the fourth polysome peak to allow direct comparison between samples.
Next-Generation RNA-Seq and Ribosome Profiling.
Three replicates each of WT and N6AMT1 KO were performed for both RNA-Seq and ribosome profiling. For each replicate, 5 × 106 cells were harvested by centrifugation and flash-frozen in liquid nitrogen. Frozen cell pellets were directly resuspended by pipetting in 200 μL of mammalian footprint lysis buffer [20 mM Tris-Cl pH7.4, 150 mM KCl, 5 mM MgCl2, 1 mM DTT, 1% Triton X-100, 2 units/mL Turbo DNase (Thermo Fisher Scientific), and 0.1 mg/mL cycloheximide (Sigma-Aldrich)]. Lysates were incubated on ice for 10 min and then cleared by centrifugation at 20,000 g for 10 min at 4 °C. Cleared lysates were quantified by a Nanodrop ND-1000 Spectrophotometer (Thermo Fisher Scientific), and aliquots of 1.5 and 0.5 A260 units were flash frozen in liquid nitrogen. For RNA-Seq, 200 µL TRIzol (Thermo Fisher Scientific) was added to an 0.5 A260 aliquot, and RNA was purified with the Zymo Direct-zol RNA miniprep kit according to the manufacturer’s instructions for purifying total RNA. The resulting RNA was quantified by a NanoDrop ND-1000 Spectrophotometer (Thermo Fisher Scientific), and RNA-Seq libraries were prepared using the Illumina TruSeq stranded total RNA-Seq kit with ribozero gold human/mouse/rat rRNA depletion, following the manufacturer's instructions. After final PCR, DNA was quantified by Bioanalyzer High-sensitivity DNA Chip (Agilent) and equimolarly pooled. For ribosome profiling, 1.5 A260 units of lysate was diluted to 200 µL with lysis buffer and digested with 750 units of RNase I (Ambion) in 200 µL lysis buffer at 25 °C for 1 h with 500 rpm agitation in a thermomixer. Reactions were quenched with 200 U SUPERase-in (Thermo Fisher Scientific) and pelleted through a 900 µL sucrose cushion in a TLA100.3 rotor for 1 h at 100,000 rpm. Pellets were resuspended in 300 µL TRIzol (Thermo Fisher Scientific), and RNA was extracted with the Zymo Direct-zol RNA miniprep kit according to the manufacturer’s instructions for purifying total RNA including small RNAs. Fragments were size selected on a 15% TBE-urea PAGE gel (Bio-Rad) with 15 and 36 nt oligos as size markers, cutting a range from 15 to 36 nt. After gel elution, fragments were dephosphorylated with PNK and ligated to preadenylated 3′ linker oBZ407. rRNA depletion was performed with the Ribo-Zero Gold kit (Illumina). Reverse transcription was performed using protoscript II (NEB) and RT primer oBZ408, and RNA was degraded by base hydrolysis. cDNA was gel purified and circularized with Circ Ligase (Lucigen). Libraries were PCR amplified using forward primer oBZ287 (NINI2) and reverse primers bar 11 through 16 (oBZ207-212). PCR was performed with Phusion DNA polymerase, 1 µM of each primer, and 8 cycles. Amplified libraries were purified away from primers on an 8% native TBE acrylamide gel. After extraction, DNA was quantified by Bioanalyzer High-Sensitivity DNA Chip (Agilent) and then equimolarly pooled. All libraries were sequenced on an Illumina HiSeq 2500 at the Johns Hopkins genetic resources core facility with 60 nt single-end reads. RNA-Seq and Ribo-Seq adapters are described in SI Appendix, Tables S3 and S4.
RNA-Seq Mapping.
Fastq files were adaptor stripped using cutadapt with a minimum length of 15 and a quality cutoff of 2 (parameters: -a NNNNNNCACTCGGGCACCAAGGAC –minimum-length = 15 –quality-cutoff = 2). The resulting reads were mapped, using default parameters, with HISAT2 (72) using a GRCh38, release 84 genome and index. Differential expression analysis was performed using DESeq2 (67) with counts generated by overlapping with a GRCh38, release 84 gtf. Volcano plots were generated by plotting the resulting log2 fold change against a -log10 transformation of the FDR-adjusted (Benjamini–Hochberg) P-value.
Ribo-Seq Mapping and Analysis.
Fastq files were adaptor stripped using cutadapt. Only trimmed reads were retained, with a minimum length of 15 and a quality cutoff of 2 (parameters: -a CTGTAGGCACCATCAAT – trimmed-only –minimum-length = 15 –quality-cutoff = 2). Histograms were produced of ribosome footprint lengths and reads were retained if the trimmed size was 26 or 34 nucleotides. The resulting reads were mapped, using default parameters, with HISAT2 (72) using a GRCh38, release 84 genome and index and were removed if they mapped to rRNA or tRNA according to GRCh38 RepeatMasker definitions from UCSC. A full set of transcripts and CDS sequences for Ensembl release 84 was then established. Only canonical transcripts (defined by knownCanonical table, downloaded from UCSC) were retained with their corresponding CDS. Retained reads were then mapped to the canonical transcriptome with bowtie2 using default parameters. For analysis, the P-site position of each read was predicted by riboWaltz (73) and confirmed by inspection. Counts were made by aggregating P-sites overlapping with the CDS and P-sites Per Kilobase Million (PPKMs) were then generated through normalizing by CDS length and total counts for the sample. Differential expression and translational efficiency (ribosome occupancy/mRNA abundance) analysis coupled with RNA-Seq data was performed using DESeq2 (67) with counts generated by overlapping with a GRCh38, release 84 gtf. Volcano plots were generated by plotting the resulting log2 fold change against a −log10 transformation of the adjusted P-value. All metagenes were calculated on a subset of expressed canonical transcripts which had PPKM values greater than 1 across all samples (9,600 transcripts). Within these, P-site depths per nucleotide were normalized to the mean value in their respective CDS. For metagenes around start and stop, the mean of these normalized values was taken for each nucleotide within 30 nt upstream and 60 nt downstream of start or 60 nt upstream and 30 nt downstream of stop.
mePROD and mePRODmt.
K562 cells were infected with sgCtrl or sgN6AMT1 in quadruplicates (two sgRNAs per condition). Ten to eleven days post transduction, cells were resuspended in heavy labeled amino acids (SILAC)-containing medium for 3 h. Cells were collected and washed twice with cold PBS. For mePRODmt, crude mitochondria were obtained as described previously (71). Briefly, pellets were washed once in cold mitochondrial isolation buffer (10 mM HEPES, pH 7.4, 210 mM mannitol, 70 mM sucrose, and 1 mM EDTA) and resuspended in 500 µL cold MIB. Cells were lysed by 30 strokes with a 25 G needle and diluted with 1 mL cold MIB. Lysates were centrifuged at 2,000 g, 5 min, at 4 °C, and the resulting pellets were lysed a second time as described above. Supernatants were pooled and centrifuged at 13,000 g, 10 min at 4 °C, and crude mitochondrial pellets were used in downstream analyses. Lysis, sample preparation, and tandem-mass tag (TMT) labeling were carried out as described previously (74). Equal amounts of TMT-labeled peptides were pooled and fractionated into 8 fractions using a High pH Reversed-Phase Peptide Fractionation Kit (ThermoFisher Scientific) according to the manufacturer’s instructions.
For mass spectrometry analysis, samples were analyzed with settings described earlier (75). Briefly, fractions were resuspended in 2% acetonitrile and 0.1% formic acid and separated on an Easy nLC 1200 (ThermoFisher Scientific) and a 35 cm long, 75 μm ID fused-silica column, which had been packed in house with 1.9 μm C18 particles (ReproSil-Pur, Maisch) and kept at 50 °C using an integrated column oven (Sonation). HPLC solvents were 0.1% formic acid in water (Buffer A) and 0.1% formic acid, 80% acetonitrile in water (Buffer B). Assuming equal amounts in each fraction, 1 µg of peptides was eluted by a nonlinear gradient from 3 to 60% B over 125 min followed by a stepwise increase to 95% acetonitrile in 1 min which was held for another 9 min. After that, peptides were directly sprayed into an Orbitrap Fusion Lumos mass spectrometer equipped with a nanoFlex ion source (ThermoFisher Scientific). Full scan MS spectra (350 to 1,400 m/z) were acquired with a resolution of 120,000 at m/z 100, maximum injection time of 100 ms, and AGC target value of 4 × 105. For targeted mass difference-based runs, the 10 most intense ions with a charge state of 2 to 5 were selected together with their labeled counterparts (Targeted Mass Difference Filter, Arg and lysine delta mass, 1 to 100% partner intensity range with 5 ppm mass difference tolerance), resulting in 20 dependent scans (Top20). Precursors were isolated in the quadrupole with an isolation window of 0.7 Th. MS2 scans were performed in the quadrupole using a maximum injection time of 86 ms, AGC target value of 1 × 105. Ions were then fragmented using HCD with a normalized collision energy of 38% and analyzed in the Orbitrap with a resolution of 50,000 at m/z 200. Repeated sequencing of already acquired precursors was limited by a dynamic exclusion of 60 s and 7 ppm, and advanced peak determination was deactivated.
Raw data were analyzed with Proteome Discoverer 2.4 (ThermoFisher Scientific). SequenceHT node was selected for database searches of MS2-spectra. Human trypsin-digested proteome [Homo sapiens SwissProt database (TaxID:9606, version 2020-03-12)] was used for protein identifications. Contaminants (MaxQuant “contamination.fasta”) were determined for quality control. Fixed modifications were TMT6 (+229.163) at the N terminus, TMT6+K8 (K, +237.177), Arg10 (R, +10.008), and carbamidomethyl (C, +57.021) at cysteine residues. Methionine oxidation (M, +15.995) and acetylation (+42.011) at the protein N terminus were set for dynamic modifications. Precursor mass tolerance was set to 10 ppm and fragment mass tolerance was set to 0.02 Da. Default Percolator settings in Proteome Discoverer were used to filter peptide-spectrum matches (PSMs). Reporter ion quantification was achieved with default settings in consensus workflow. PSMs were exported for further analysis using the DynaTMT package (75). Normalized abundances from DynaTMT were used for statistical analysis after removing common contaminants and peptides with only missing values. Differential expression analysis was performed using peptide-based linear mixed models (75, 76).
Quantitative Steady-State Proteomics.
For sample preparation, samples were digested following a modified version of the iST method (77). Based on tryptophane fluorescence quantification (78), 100 µg of proteins at 2 µg/µL in miST lysis buffer (1% Sodium deoxycholate, 100 mM Tris pH 8.6, 10 mM DTT), were transferred to new tubes. Samples were heated for 5 min at 95 °C, diluted 1:1 (v:v) with water containing 4 mM MgCl2 and benzonase (Merck #70746, 250 U/µL), and incubated for 15 min at RT to digest nucleic acids. Reduced disulfides were alkylated by adding 0.25 sample volumes of 160 mM chloroacetamide (32 mM final) and incubating for 45 min at RT in the dark. Samples were adjusted to 3 mM EDTA and digested with 0.5 µg Trypsin/LysC mix (Promega #V5073) for 1 h at 37 °C, followed by a second 1 h digestion with an additional 0.5 µg of proteases. To remove sodium deoxycholate, two sample volumes of isopropanol containing 1% TFA were added to the digests, and the samples were desalted on a strong cation exchange (SCX) plate (Oasis MCX; Waters Corp., Milford, MA) by centrifugation. After washing with isopropanol/1% TFA, peptides were eluted in 200 µL of 80% MeCN, 19% water, and 1% (v/v) ammonia and dried by centrifugal evaporation. For peptide fractionation, aliquots (1/8) of samples were pooled and separated into 6 fractions by off-line basic reversed-phase (bRP) using the Pierce High pH Reversed-Phase Peptide Fractionation Kit (Thermo Fisher Scientific). The fractions were collected in 7.5, 10, 12.5, 15, 17.5, and 50% acetonitrile in 0.1% triethylamine (~pH 10). Dried bRP fractions were redissolved in 50 µL 2% acetonitrile with 0.5% TFA, and 5 µL was injected for LC-MS/MS analyses. Liquid chromatography–mass spectrometry analysis (LC/MS-MS) was carried out on a TIMS-TOF Pro (Bruker, Bremen, Germany) mass spectrometer interfaced through a nanospray ion source (“captive spray”) to an Ultimate 3000 RSLCnano HPLC system (Dionex). Peptides (200 ng) were separated on a reversed-phase custom packed 45 cm C18 column (75 μm ID, 100 Å, ReproSil-Pur 1.9 µm particles, Maisch, Germany) at a flow rate of 0.250 µL/min with a 2 to 27% acetonitrile gradient in 93 min followed by a ramp to 45% in 15 min and to 90% in 5 min (total method time: 140 min, all solvents contained 0.1% formic acid). Identical LC gradients were used for data-dependent acquisitions (DDA) and data-independent acquisitions (DIA) measurements. For creation of the spectral library, DDA were carried out on the 6 bRP fractions sample pool using a standard TIMS PASEF method (79) with ion accumulation for 100 ms for each survey MS1 scan and the TIMS-coupled MS2 scans. Duty cycle was kept at 100%. Up to 10 precursors were targeted per TIMS scan. Precursor isolation was done with 2 Th or 3 Th windows below or above m/z 800, respectively. The minimum threshold intensity for precursor selection was 2,500. If the inclusion list allowed it, precursors were targeted more than one time to reach a minimum target total intensity of 20,000. Collision energy was ramped linearly based uniquely on the 1/k0 values from 20 (at 1/k0 = 0.6) to 59 eV (at 1/k0 = 1.6). The total duration of a scan cycle including one survey and 10 MS2 TIMS scans was 1.16 s. Precursors could be targeted again in subsequent cycles if their signal increased by a factor 4.0 or more. After selection in one cycle, precursors were excluded from further selection for 60 s. Mass resolution in all MS measurements was approximately 35,000. The DIA method used mostly the same instrument parameters as the DDA method and was as reported previously (79). Per cycle, the mass range 400 to 1,200 m/z was covered by a total of 32 windows, each 25 Th wide and a 1/k0 range of 0.3. Collision energy and resolution settings were the same as in the DDA method. Two windows were acquired per TIMS scan (100 ms) so that the total cycle time was 1.7 s.
Raw Bruker MS data were processed directly with Spectronaut 17.6 (Biognosys, Schlieren, Switzerland). A library was constructed from the DDA bRP fraction data by searching the annotated human proteome (www.uniprot.org) database of 2022 (uniprot_sprot_2022-01-07_HUMAN.fasta). For identification, peptides of 7-52 AA length were considered, cleaved with trypsin/P specificity and a maximum of 2 missed cleavages. Carbamidomethylation of cysteine (fixed), methionine oxidation, and N-terminal protein acetylation (variable) were the modifications applied. Mass calibration was dynamic and based on an initial database search. The Pulsar engine was used for peptide identification. Protein inference was performed with the IDPicker algorithm. Spectra, peptide, and protein identifications were all filtered at 1% FDR against a decoy database. Specific filtering for library construction removed fragments corresponding to less than 3 AA and fragments outside the 300 to 1,800 m/z range. Also, only fragments with a minimum base peak intensity of 5% were kept. Precursors with less than 3 fragments were also eliminated and only the best 6 fragments were kept per precursor. No filtering was done on the basis of charge state and a maximum of two missed cleavages was allowed. Shared (nonproteotypic) peptides were kept. The library created contained 127,456 precursors mapping to 92,961 stripped sequences, of which 88,718 were proteotypic. These corresponded to 7,860 protein groups (7,949 proteins). Of these, 790 were single hits (one peptide precursor). In total 752,440 fragments were used for quantitation. Peptide-centric analysis of DIA data was done with Spectronaut 17.6 using the library described above. Single hits proteins (defined as matched by one stripped sequence only) were kept in the analysis. Peptide quantitation was based on XIC area, for which a minimum of 1 and a maximum of 3 (the 3 best) precursors were considered for each peptide, from which the median value was selected. Quantities for protein groups were derived from interrun peptide ratios based on the MaxLFQ algorithm (80). Global normalization of runs/samples was done based on the median of peptides. The raw output table contained 7,675 quantified protein groups (minimum 1 precursor per group) with an overall data completeness of 94.9%. All subsequent analyses were done with the Perseus software package (version 1.6.15.0) (80). Contaminant proteins were removed, and quantified values were log2-transformed. After assignment to groups, only proteins quantified in at least 4 samples of one group were kept (7,372 protein groups). After missing values imputation (based on normal distribution using Perseus default parameters), t tests were carried out between N6AMT1-depleted and control samples, with Benjamini–Hochberg correction for multiple testing.
NanoString.
A mitochondrial-specific version of NanoString called MitoString was performed as previously described (41). All counts were normalized to TUBB. For the unsupervised clustering, mean expression values from the previously published dataset (41) and the MitoString analysis were combined and hierarchically clustered and visualized using the pheatmap package (81) in R with default clustering settings.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Dataset S08 (XLSX)
Dataset S09 (XLSX)
Dataset S10 (XLSX)
Dataset S11 (XLSX)
Dataset S12 (XLSX)
Dataset S13 (XLSX)
Acknowledgments
We would like to thank past and present members of the Jourdain lab as well as S. Chamois, D. Gatfield, R. Green, V. Heurgué-Hamard, A. Jeltsch, J.C. Martinou, E. Mick, D. Mohr, H.P. Nadimpalli, C.C. Wu, G.L. Xu, the UNIL Protein Analysis Facility, the members of the Center for Functional Proteomics at Goethe University Frankfurt, and the Johns Hopkins Genetic Resources Core Facility. This work was supported by grants from the Swiss NSF (310030_200796 to A.A.J.) and the Hessian Ministry for Arts and Sciences excellence initiative EnABLE (to C.M.) and two EMBO postdoctoral fellowships (ALTF 286-2022 to M.L. and ALTF 944-2022 to E.R.). V.K.M. is an Investigator of HHMI. B.Z. was an HHMI fellow of the Damon Runyon Cancer Research Foundation (DRG-2250-16) during the course of this study.
Author contributions
M.M.F., V.K.M., and A.A.J. designed research; M.M.F., E.R., S.C., M.L., B.Z., D.B., and A.A.J. performed research; C.M. contributed new reagents/analytic tools; M.M.F., E.R., G.E.A., D.B., and A.A.J. analyzed data; and M.M.F. and A.A.J. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
Steady-state proteomics and mePROD proteomics data have been deposited to the ProteomeXchange consortium (www.proteomexchange.org) via the PRIDE partner repository (82) with accession IDs PXD051311 and PXD051180, respectively. RNA sequencing and ribosome profiling data have been deposited to the GEO database with accession IDs GSE267077 and GSE267078, respectively. Uncropped immunoblots are provided in SI Appendix, Figs. S4 and S5.
Supporting Information
References
- 1.Wu Z., et al. , Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98, 115–124 (1999). [DOI] [PubMed] [Google Scholar]
- 2.Virbasius J. V., Scarpulla R. C., Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: A potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc. Natl. Acad. Sci. U.S.A. 91, 1309–1313 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schatton D., et al. , CLUH regulates mitochondrial metabolism by controlling translation and decay of target mRNAs. J. Cell Biol. 216, 675–693 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zid B. M., et al. , 4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila. Cell 139, 149–160 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jourdain A. A., et al. , GRSF1 regulates RNA processing in mitochondrial RNA granules. Cell Metab. 17, 399–410 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antonicka H., Sasarman F., Nishimura T., Paupe V., Shoubridge E. A., The mitochondrial RNA-binding protein GRSF1 localizes to RNA granules and is required for posttranscriptional mitochondrial gene expression. Cell Metab. 17, 386–398 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Jourdain A. A., Boehm E., Maundrell K., Martinou J. C., Mitochondrial RNA granules: Compartmentalizing mitochondrial gene expression. J. Cell Biol. 212, 611–614 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ojala D., Montoya J., Attardi G., tRNA punctuation model of RNA processing in human mitochondria. Nature 290, 470–474 (1981). [DOI] [PubMed] [Google Scholar]
- 9.Holzmann J., et al. , RNase P without RNA: Identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell 135, 462–474 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Szczesny R. J., et al. , Human mitochondrial RNA turnover caught in flagranti: Involvement of hSuv3p helicase in RNA surveillance. Nucleic Acids Res. 38, 279–298 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dhir A., et al. , Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 560, 238–242 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arroyo J. D., et al. , A genome-wide CRISPR death screen identifies genes essential for oxidative phosphorylation. Cell Metab. 24, 875–885 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kramer N. J., et al. , Regulators of mitonuclear balance link mitochondrial metabolism to mtDNA expression. Nat. Cell Biol. 25, 1575–1589 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ratel D., et al. , Undetectable levels of N6-methyl adenine in mouse DNA: Cloning and analysis of PRED28, a gene coding for a putative mammalian DNA adenine methyltransferase. FEBS Lett. 580, 3179–3184 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Schiffers S., et al. , Quantitative LC-MS provides no evidence for m(6) dA or m(4) dC in the genome of mouse embryonic stem cells and tissues. Angew. Chem. Int. Ed. Engl. 56, 11268–11271 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Li W., Shi Y., Zhang T., Ye J., Ding J., Structural insight into human N6amt1-Trm112 complex functioning as a protein methyltransferase. Cell Discov. 5, 51 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woodcock C. B., Yu D., Zhang X., Cheng X., Human HemK2/KMT9/N6AMT1 is an active protein methyltransferase, but does not act on DNA in vitro, in the presence of Trm112. Cell Discov. 5, 50 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakahigashi K., et al. , HemK, a class of protein methyl transferase with similarity to DNA methyl transferases, methylates polypeptide chain release factors, and hemK knockout induces defects in translational termination. Proc. Natl. Acad. Sci. U.S.A. 99, 1473–1478 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heurgue-Hamard V., Champ S., Engstrom A., Ehrenberg M., Buckingham R. H., The hemK gene in Escherichia coli encodes the N(5)-glutamine methyltransferase that modifies peptide release factors. EMBO J. 21, 769–778 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heurgue-Hamard V., et al. , The glutamine residue of the conserved GGQ motif in Saccharomyces cerevisiae release factor eRF1 is methylated by the product of the YDR140w gene. J. Biol. Chem. 280, 2439–2445 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Polevoda B., Span L., Sherman F., The yeast translation release factors Mrf1p and Sup45p (eRF1) are methylated, respectively, by the methyltransferases Mtq1p and Mtq2p. J. Biol. Chem. 281, 2562–2571 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Figaro S., Scrima N., Buckingham R. H., Heurgue-Hamard V., HemK2 protein, encoded on human chromosome 21, methylates translation termination factor eRF1. FEBS Lett. 582, 2352–2356 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Pundir S., Ge X., Sanyal S., GGQ methylation enhances both speed and accuracy of stop codon recognition by bacterial class-I release factors. J. Biol. Chem. 296, 100681 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lacoux C., et al. , The catalytic activity of the translation termination factor methyltransferase Mtq2-Trm112 complex is required for large ribosomal subunit biogenesis. Nucleic Acids Res. 48, 12310–12325 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu P., et al. , Deficiency in a glutamine-specific methyltransferase for release factor causes mouse embryonic lethality. Mol. Cell Biol. 30, 4245–4253 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu F., Suyama R., Inada T., Kawaguchi S., Kai T., HemK2 functions for sufficient protein synthesis and RNA stability through eRF1 methylation during Drosophila oogenesis. Development 151, dev202795 (2024), 10.1242/dev.202795. [DOI] [PubMed] [Google Scholar]
- 27.Metzger E., et al. , KMT9 monomethylates histone H4 lysine 12 and controls proliferation of prostate cancer cells. Nat. Struct. Mol. Biol. 26, 361–371 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Baumert H. M., et al. , Depletion of histone methyltransferase KMT9 inhibits lung cancer cell proliferation by inducing non-apoptotic cell death. Cancer Cell Int. 20, 52 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weirich S., et al. , Distinct specificities of the HEMK2 protein methyltransferase in methylation of glutamine and lysine residues. Protein Sci. 33, e4897 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kusevic D., Kudithipudi S., Jeltsch A., Substrate specificity of the HEMK2 protein glutamine methyltransferase and identification of novel substrates. J. Biol. Chem. 291, 6124–6133 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ren X., et al. , Involvement of N-6 adenine-specific DNA methyltransferase 1 (N6AMT1) in arsenic biomethylation and its role in arsenic-induced toxicity. Environ. Health Perspect. 119, 771–777 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harari F., et al. , N-6-adenine-specific DNA methyltransferase 1 (N6AMT1) polymorphisms and arsenic methylation in Andean women. Environ. Health Perspect. 121, 797–803 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barretina J., et al. , The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rath S., et al. , MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 49, D1541–D1547 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thul P. J., et al. , A subcellular map of the human proteome. Science 356, eaal3321 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Brumele B., et al. , Human TRMT112-methyltransferase network consists of seven partners interacting with a common co-factor. Int. J. Mol. Sci. 22, 13593 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mootha V. K., et al. , PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Subramanian A., et al. , Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ingolia N. T., Ghaemmaghami S., Newman J. R., Weissman J. S., Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geiss G. K., et al. , Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat. Biotechnol. 26, 317–325 (2008). [DOI] [PubMed] [Google Scholar]
- 41.Wolf A. R., Mootha V. K., Functional genomic analysis of human mitochondrial RNA processing. Cell Rep. 7, 918–931 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guerrero-Castillo S., van Strien J., Brandt U., Arnold S., Ablation of mitochondrial DNA results in widespread remodeling of the mitochondrial complexome. EMBO J. 40, e108648 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Borowski L. S., Dziembowski A., Hejnowicz M. S., Stepien P. P., Szczesny R. J., Human mitochondrial RNA decay mediated by PNPase-hSuv3 complex takes place in distinct foci. Nucleic Acids Res. 41, 1223–1240 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Diaz M. O., et al. , Homozygous deletion of the alpha- and beta 1-interferon genes in human leukemia and derived cell lines. Proc. Natl. Acad. Sci. U.S.A. 85, 5259–5263 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Metodiev M. D., et al. , Recessive mutations in TRMT10C cause defects in mitochondrial RNA processing and multiple respiratory chain deficiencies. Am. J. Hum. Genet. 98, 993–1000 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hochberg I., et al. , Bi-allelic variants in the mitochondrial RNase P subunit PRORP cause mitochondrial tRNA processing defects and pleiotropic multisystem presentations. Am. J. Hum. Genet. 108, 2195–2204 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nie D. S., Liu Y. B., Lu G. X., Cloning and primarily function study of two novel putative N5-glutamine methyltransferase (Hemk) splice variants from mouse stem cells. Mol. Biol. Rep. 36, 2221–2228 (2009). [DOI] [PubMed] [Google Scholar]
- 48.Rhee H. W., et al. , Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 339, 1328–1331 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hung V., et al. , Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol. Cell 55, 332–341 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pagliarini D. J., et al. , A mitochondrial protein compendium elucidates complex I disease biology. Cell 134, 112–123 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Claros M. G., Vincens P., Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 241, 779–786 (1996). [DOI] [PubMed] [Google Scholar]
- 52.Almagro Armenteros J. J., et al. , Detecting sequence signals in targeting peptides using deep learning. Life Sci. Alliance 2, e201900429 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fessler E., et al. , A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature 579, 433–437 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jousse C., et al. , Inhibition of CHOP translation by a peptide encoded by an open reading frame localized in the chop 5’UTR. Nucleic Acids Res. 29, 4341–4351 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Palam L. R., Baird T. D., Wek R. C., Phosphorylation of eIF2 facilitates ribosomal bypass of an inhibitory upstream ORF to enhance CHOP translation. J. Biol. Chem. 286, 10939–10949 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McGillivray P., et al. , A comprehensive catalog of predicted functional upstream open reading frames in humans. Nucleic Acids Res. 46, 3326–3338 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Manske F., et al. , The new uORFdb: Integrating literature, sequence, and variation data in a central hub for uORF research. Nucleic Acids Res. 51, D328–D336 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.West A. P., et al. , Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tigano M., Vargas D. C., Tremblay-Belzile S., Fu Y., Sfeir A., Nuclear sensing of breaks in mitochondrial DNA enhances immune surveillance. Nature 591, 477–481 (2021). [DOI] [PubMed] [Google Scholar]
- 60.Koll F. J., et al. , Overexpression of KMT9alpha is associated with aggressive basal-like muscle-invasive bladder cancer. Cells 12, 589 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Totonji S., et al. , Lysine methyltransferase 9 (KMT9) is an actionable target in muscle-invasive bladder cancer. Cancers (Basel) 16, 1532 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Berlin C., et al. , KMT9 controls stemness and growth of colorectal cancer. Cancer Res. 82, 210–220 (2022). [DOI] [PubMed] [Google Scholar]
- 63.Wang S., et al. , Structure-guided design of a selective inhibitor of the methyltransferase KMT9 with cellular activity. Nat. Commun. 15, 43 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen D., et al. , Chemoproteomic study uncovers HemK2/KMT9 as a new target for NTMT1 bisubstrate inhibitors. ACS Chem. Biol. 16, 1234–1242 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tsherniak A., et al. , Defining a cancer dependency map. Cell 170, 564–576.e16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.R Core Team, R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, Vienna, Austria, 2023). https://www.R-project.org/. [Google Scholar]
- 67.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhu A., Ibrahim J. G., Love M. I., Heavy-tailed prior distributions for sequence count data: Removing the noise and preserving large differences. Bioinformatics 35, 2084–2092 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Doench J. G., et al. , Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sanjana N. E., Shalem O., Zhang F., Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Blanco-Fernandez J., Jourdain A. A., Two-step tag-free isolation of mitochondria for improved protein discovery and quantification. J. Vis. Exp. 196, e65252 (2023). [DOI] [PubMed] [Google Scholar]
- 72.Kim D., Langmead B., Salzberg S. L., HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lauria F., et al. , riboWaltz: Optimization of ribosome P-site positioning in ribosome profiling data. PLoS Comput. Biol. 14, e1006169 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Klann K., Tascher G., Munch C., Functional translatome proteomics reveal converging and dose-dependent regulation by mTORC1 and eIF2alpha. Mol. Cell 77, 913–925.e14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schafer J. A., Bozkurt S., Michaelis J. B., Klann K., Munch C., Global mitochondrial protein import proteomics reveal distinct regulation by translation and translocation machinery. Mol. Cell 82, 435–446.e37 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Klann K., Munch C., PBLMM: Peptide-based linear mixed models for differential expression analysis of shotgun proteomics data. J. Cell Biochem. 123, 691–696 (2022). [DOI] [PubMed] [Google Scholar]
- 77.Kulak N. A., Pichler G., Paron I., Nagaraj N., Mann M., Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 11, 319–324 (2014). [DOI] [PubMed] [Google Scholar]
- 78.Wisniewski J. R., Gaugaz F. Z., Fast and sensitive total protein and Peptide assays for proteomic analysis. Anal. Chem. 87, 4110–4116 (2015). [DOI] [PubMed] [Google Scholar]
- 79.Meier F., et al. , Online parallel accumulation-serial fragmentation (PASEF) with a novel trapped ion mobility mass spectrometer. Mol. Cell Proteomics 17, 2534–2545 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tyanova S., et al. , The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016). [DOI] [PubMed] [Google Scholar]
- 81.Kolde R., pheatmap: Pretty Heatmaps (R package version 1.0.12, 2019), https://CRAN.R-project.org/package=pheatmap. Accessed 6 April 2024.
- 82.Perez-Riverol Y., et al. , The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 47, D442–D450 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Dataset S08 (XLSX)
Dataset S09 (XLSX)
Dataset S10 (XLSX)
Dataset S11 (XLSX)
Dataset S12 (XLSX)
Dataset S13 (XLSX)
Data Availability Statement
Steady-state proteomics and mePROD proteomics data have been deposited to the ProteomeXchange consortium (www.proteomexchange.org) via the PRIDE partner repository (82) with accession IDs PXD051311 and PXD051180, respectively. RNA sequencing and ribosome profiling data have been deposited to the GEO database with accession IDs GSE267077 and GSE267078, respectively. Uncropped immunoblots are provided in SI Appendix, Figs. S4 and S5.