

Abstract
A wide variety of systemic pathologies, including infectious and autoimmune diseases, are accompanied by joint pain or inflammation, often mediated by circulating immune complexes (ICs). How such stimuli access joints and trigger inflammation is unclear. Whole-mount synovial imaging revealed PV1+ fenestrated capillaries at the periphery of the synovium in the lining–sublining interface. Circulating ICs extravasated from these PV1+ capillaries, and nociceptor neurons and three distinct macrophage subsets formed a sentinel unit around them. Macrophages showed subset-specific responses to systemic IC challenge; LYVE1+CX3CR1+ macrophages orchestrated neutrophil recruitment and activated calcitonin gene-related peptide+ (CGRP+) nociceptor neurons via interleukin-1β. In contrast, major histocompatibility complex class II+CD11c+ (MHCII+CD11c+) and MHCII+CD11c– interstitial macrophages formed tight clusters around PV1+ capillaries in response to systemic immune stimuli, a feature enhanced by nociceptor-derived CGRP. Altogether, we identify the anatomical location of synovial PV1+ capillaries and subset-specific macrophage–nociceptor cross-talk that forms a blood–joint barrier protecting the synovium from circulating immune challenges.
Subject terms: Innate immunity, Monocytes and macrophages, Neuroimmunology, Systemic lupus erythematosus
Why joints are highly responsive to systemic inflammation is unknown. Hasegawa et al. sought to address this question, developing a whole-mount imaging system of the entire synovium to profile the vascular, neuronal and immune components.
Main
Synovial joints are the target of antigen-specific autoimmune responses in diseases such as rheumatoid arthritis (RA)1, but joint pain (arthralgia), or even overt inflammation, is a relatively common manifestation of viral and bacterial infections in distant unrelated organs (for example, bacterial enteritis or streptococcal pharyngitis). This phenomenon is thought, at least in part, to be mediated by circulating microbial antigen–IgG immune complexes (ICs)2–4. Similarly, in systemic lupus erythematosus, an autoimmune disease characterized by IgG IC deposition, arthritis is a common symptom5. Thus, synovial joints act as a ‘barometer’ of systemic inflammation, manifesting as arthralgia or arthritis.
The synovial membrane lines the joint cavity, is the source of synovial fluid and contains several fibroblast and macrophage subsets6,7. It is highly vascular, supplies oxygen and nutrients to adjacent avascular articular cartilage and contains pain fibers8–12. The synovium is also involved in joint pathology, with proliferative pannus arising at the synovia–bone interface, eroding bone in RA13–15.
There is increasing interest in the interplay between neuronal signals and tissue immune cells16–18, but whether or how the triggering of arthralgia-generating pain neurons in response to circulating stimuli is influenced by synovial immune cells or how immune cells in turn might be affected by pain is currently unknown. Here, we established a whole-mount imaging system for the synovium to gain a comprehensive understanding of the spatial organization of the constituent immune, vascular and neuronal components. We investigated the responses of these components to circulating stimuli and addressed the question of why joints are responsive to many systemic pathologies.
Results
PV1+ fenestrated capillaries at the periphery of the synovium allow access to circulating stimuli
To interrogate the synovial vasculature via which circulating stimuli gain access to the synovium, we analyzed endothelial cells from single-cell RNA-sequencing (scRNA-seq) data of mouse synovium6 (Extended Data Fig. 1a). Genes encoding adhesion molecules, such as Sele, Selp and Icam1, were mainly expressed on Ackr1+ postcapillary venules, where immune cells extravasate19 (Extended Data Fig. 1b). Capillary endothelial cells were also evident and, when considered in isolation, comprised two subsets (Fig. 1a). Plvap was exclusively expressed in cluster 1 and was among the top ten differentially expressed genes (DEGs) when comparing these two capillary cell clusters (Fig. 1b,c and Extended Data Fig. 1c). PV1 (encoded by Plvap) is a molecular component of fenestral diaphragms, conferring vascular permeability20,21, and its expression is indicative of fenestrated capillaries.
Extended Data Fig. 1. PV1+ capillaries with distinct molecular signatures localised at the L-SL interface of the synovium.

(a) Uniform manifold approximation and projection (UMAP) visualizations of Pecam1+ endothelial cells in synovium. ScRNA-seq data from GSE145286. (b) Dot plots showing the scaled gene expression and percentage of cells expressing genes for cell markers and adhesion molecules in endothelial cell populations from scRNA-seq analysis. (c) Top 10 differentially expressed genes between two cell populations in synovial capillary endothelial cells. (d) Anatomic localization of the synovium from the frontal view and dissection protocol from the lateral view. P: patella, PL: patella ligament, FP: fat pad, Sy: synovium, Fe: femur, Ti: tibia. (e) Schematic depicting tissue preparation, imaging and iterative bleaching extends multiplexity (IBEX) protocol. (f) 3D reconstruction of representative confocal images of whole mount synovium. Arrowheads indicate PV1+ capillaries at the lining-sublining (L-SL) interface (Z-stack images). Bars, 200 and 50 μm. Images are representative of at least three independent experiments with similar results. (g) Schematic diagram showing the protocol. (h) Representative confocal images of sections of skin and lung from wild type mice injected i.v. with 70 and 2000 kDa Dextran (300 μg 70 kDa Dextran and 150 μg 2000 kDa Dextran) 1 h prior to analysis. Images are representative of at least three independent experiments with similar results. (i) 3D reconstruction of representative confocal images of whole mount synovium from wild type mice injected i.v. with fluorescently labeled microbeads of different sizes 1 h prior to analysis. Arrows indicate the sites where microbeads extravasated. Bars, 200 and 50 μm. Images are representative of at least three independent experiments with similar results.
Fig. 1. PV1+ fenestrated capillaries in the L–SL interface at the peripheral area of the synovium allow circulating stimuli to access the synovium.

a, Uniform manifold approximation and projection (UMAP) visualization of synovial capillary endothelial cells extracted from CD31+ endothelial cells (Extended Data Fig. 1a,b). scRNA-seq data are from GSE145286. b, Volcano plots showing DEGs between two clusters of synovial capillary endothelial cells. c, UMAP visualization of synovial capillary endothelial cells expressing Plvap. Color bar shows the expression level. d, Representative confocal images of sections of knee joints; BM, bone marrow; M, meniscus; L, patella ligament; Syn, synovium; n = 3 mice for each group. e, Schematic diagram showing the protocol to dissect whole synovium from knee joints. The red dashed outline indicates the area of synovium dissected. Fe, femur; Ti, tibia; P, patella; Fi, fibula; Prox, proximal; Lat, lateral; Dis, distal. f, Three-dimensional reconstruction of representative confocal images of the indicated layer of whole-mount synovium. Quantification of the PV1+ area among CD31+ area at the indicated layers is shown on the bottom right; n = 5 mice for each group. g, Three-dimensional reconstruction of representative confocal images and density map of the L–SL interface of whole-mount synovium. Quantification of the PV1+ area among CD31+ area at the indicated compartments is shown on the bottom right; n = 5 mice. h, Representative confocal images of sections of knee joints from WT mice injected i.v. with 70- and 2,000-kDa dextran (300 μg of 70-kDa dextran (Dex70k) and 150 μg of 2,000-kDa dextran (Dex2000k)) 1 h before analysis. Arrowheads indicate the area where 70- and 2,000-kDa dextran merged; CL, cruciate ligament; Epi. GP, epiphyseal growth plate; scale bars, 500 and 100 μm (inset). i, Quantification of the extravasated area in each tissue; n = 4 to 6 mice for each group. j, Pie graph showing the percentage of distance between 2,000-kDa dextran and the ERTR7+ lining layer of the synovium in the section images; n = 4 mice. k, Three-dimensional reconstruction of representative confocal images of whole-mount synovium from WT mice injected i.v. with 70- and 2,000-kDa dextran 1 h before analysis; scale bar, 100 μm. l, Three-dimensional reconstruction of representative confocal images of whole-mount synovium from WT mice injected i.v. with fluorescently labeled microbeads of different sizes (25 μl of each FluoSphere carboxylate-modified microsphere dissolved in PBS) 1 h before analysis. Arrowheads indicate the sites where microbeads extravasated; scale bars, 500 and 50 μm (inset). Quantification of the area and capillary microbeads extravasated is shown on the bottom right; n = 3 to 5 mice for each group. m, Three-dimensional reconstruction of representative confocal images of whole-mount synovium from WT mice injected i.v. with OVA–AF647;rabbit polyclonal anti-OVA (RaOVA) ICs (40 μg of OVA–AF647 + 150 μg of RaOVA) 2 h before analysis. Arrowheads indicate sites where ICs extravasated; scale bars, 200 and 100 μm (z-stack images). n, Density map of a three-dimensional reconstruction of representative confocal images of whole-mount synovium from WT mice injected i.v. with microbeads or ICs. Data in d, f and l (right) were analyzed by two-tailed t-test. The center compartment was used as a control group in one-way analysis of variance (ANOVA) with a Dunnett’s test for g. Data in i and l (left) were analyzed by one-way ANOVA with Tukey’s post hoc test, and data in b were analyzed by two-tailed Wilcoxon rank-sum test. Data in d, f, g, i and l are shown as mean ± s.e.m. Images in k, m and n are representative of at least three independent experiments with similar results.
The synovium can be divided into a lining layer containing synovial fibroblasts, with overlying macrophages that interface with the synovial cavity, and a sublining layer with additional macrophage populations7,22–24. Sagittal section imaging showed that PV1+ capillaries were specifically located at the interface of the lining and sublining layers, termed hereafter as the lining–sublining (L–SL) interface (Fig. 1d). Although sagittal sections have been used as the gold standard to define the spatial organization of synovial cells, this method provides a limited snapshot of the synovial membrane as a whole, which stretches around the entire joint. Indeed, our knowledge of the global arrangement of synovial cells, particularly in homeostasis, is surprisingly rudimentary. To address these limitations, we developed a protocol to stereotactically dissect the entire synovium of knee joints (Fig. 1e and Extended Data Fig. 1d,e) and combined whole-mount synovial imaging with an iterative bleaching and staining protocol25 to enable multiparameter imaging. This revealed a dense PV1– vascular network in the sublining layer and confirmed that PV1 was predominantly expressed on capillaries at the L–SL interface (Fig. 1f and Extended Data Fig. 1f). When considering the L–SL interface across the entire area of the synovium, we found that PV1+ capillaries were not uniformly distributed but rather were abundant at the periphery of the synovium in proximity to adjacent bones (Fig. 1g).
To test the functional importance of the distribution of PV1 capillaries, we administered 70-kDa and 2,000-kDa dextran intravenously (i.v.) and collected organs 1 h later. Although 70-kDa dextran highly extravasated at the diaphysial side of the growth plate in bone marrow, 2,000-kDa dextran was evident in the synovium (Fig. 1h,i and Extended Data Fig. 1g,h), predominantly in the L–SL interface (Fig. 1j,k). Similarly, i.v. administration of fluorescently labeled microbeads showed that 0.2-μm microbeads extravasated exclusively from PV1+ capillaries in the L–SL interface in the periphery of healthy synovium (Fig. 1l), whereas 2-μm microbeads were excluded (Extended Data Fig. 1i). Finally, we used a clinically relevant challenge; i.v. ICs (ovalbumin (OVA) opsonized with a polyclonal anti-OVA IgG26) also extravasated into the local synovial tissue from PV1+ capillaries (Fig. 1m,n). Together, these data indicate that circulating stimuli readily gain access to the healthy synovium via highly permeable PV1+ capillaries located at the L–SL interface in the periphery of the synovium.
Three subsets of macrophages with distinct distribution patterns line synovial PV1+ capillaries
We next sought to interrogate which immune cells localized to these sites of potential vulnerability around PV1+ capillaries. ERTR7+ lining fibroblasts evident in sagittal sections (Fig. 2a) formed a uniform, tightly knit lining layer in the synovial whole-mount images (Fig. 2b), but CD68+ macrophages showed increased density in the periphery of the synovium (Fig. 2b and Extended Data Fig. 3a). No extravascular synovial T or B cells were detectable (Fig. 2c). We therefore focused on further characterizing the synovial macrophage populations.
Fig. 2. Three subsets of macrophages with distinct distribution patterns line synovial PV1+ capillaries.

a, Representative confocal images of sections of healthy knee joints; BM, bone marrow; Syn, synovium; L, patella ligament; scale bar, 200 μm. b, Three-dimensional reconstruction of representative confocal images of whole-mount synovium; scale bars, 200 (left) and 300 μm (right). c, Three-dimensional reconstruction of representative confocal images of whole-mount synovium; scale bars, 200 and 10 μm. d, Gating strategy and flow cytometric analysis of three subsets of synovial macrophages with indicated cell surface markers. Shaded regions indicate staining with isotype controls. Data are representative of at least two independent experiments with similar results. The asterisks (*) indicate that fluorophores for LYVE1 were changed to apply the indicated antibodies. Representative confocal images of each subset of macrophages are shown on the right. e, Three-dimensional reconstruction of representative confocal images of whole-mount synovium from a vertical angle. The two dashed lines are (1) the border of lining layer and L–SL interface, and (2) L–SL interface and sublining layer. Cap., capillary; Art., arteriole. f, Three-dimensional reconstruction of representative confocal images of whole-mount synovium from a vertical angle; scale bars, 100 μm. Quantification of the percentage of CD11c+MHCII+ and CD11c–MHCII+ macrophages (Mac) attached to PV1+CD31+ or PV1−CD31+ vessels; n = 3 mice for each group. Data represent mean ± s.e.m. g, Three-dimensional reconstruction of representative confocal images at the indicated layers of whole-mount synovium. The pie graphs show the percentages of the three types of macrophages in the indicated layers of the synovium; n = 3 mice. h, Three-dimensional reconstruction of representative confocal images of whole-mount synovium (left) and density maps of three subsets of macrophages (middle); scale bar, 200 μm. The numbers and densities of each macrophage type in the indicated compartment of the synovium are shown on the right; n = 4 mice for each group. i, Representative confocal images of sections of healthy knee joints; M, meniscus; C.Lig, crescent ligament; SC, synovial cavity; n = 3 mice for each group. Data represent mean ± s.e.m. Data in f and i were analyzed by two-tailed t-test. Images in a–e are representative of at least two independent experiments with similar results.
Extended Data Fig. 3. IC uptake and localisation of macrophages expressing FcγRII/lll in the synovium.

(a) 3D reconstruction of representative confocal images of whole mount synovium. Bars, 50 (left) and 10 μm (right). Images are representative of at least three independent experiments with similar results. (b) 3D reconstruction of representative confocal images of whole mount synovium from Ms4a3Cre-RosaTdT mice. Bars, 50 μm. (c) Gating strategy and flow cytometric analysis of OVA-IC uptake by synovial macrophages from wild type mice injected i.v. with OVA-AF647;RaOVA (40 μg OVA-AF647 + 150 μg RaOVA) 2 h prior to analysis. Plots are representative of 3 mice. (d) Schematic of FcγR A:I ratios. (e) Representative plot profile intensity of cells (right) as indicated by the white line shown in the 3D reconstruction of representative confocal images of L-SL interface of whole mount synovium (left), showing five representative cells consisting of MHCII+ cells and Lyve1+ cells. Images are representative of at least two independent experiments with similar results. (f) 3D reconstruction of representative confocal images of indicated layer of whole mount synovium. Bars, 50 μm. Arrowheads indicate FcγRll/lll+ area at L-SL interface. (g) Schematic depicting tissue preparation, clearing, and imaging protocol of human synovium. (h) Representative confocal images of sections of human synovium. CD55+ area represents lining fibroblasts. Bars, 100 and 20 μm. Images are representative of at least two independent experiments with similar results.
scRNA-seq data showed that synovial macrophages (Cd68+Adgre1+) can be divided into a Cx3cr1+ population (previously described as lining macrophages7), which also express Lyve1, Timd4 and Folr2, an H2-Ab1+ population (previously described as interstitial macrophages7) and a subpopulation within the H2-Ab1+ population expressing Itgax (encoding CD11c; Extended Data Fig. 2a–c). Unsupervised assessment of myeloid surface markers using flow cytometry also showed that myeloid cells in the healthy synovium can be divided into major histocompatibility complex class II+CD11c– (MHCII+CD11c–), MHCII+CD11c+ and MHCII–LYVE1+ clusters, which also express CX3CR1 (Extended Data Fig. 2d,e). These data informed the gating strategy subsequently used in our study characterizing three subsets of synovial macrophages, CD11c+IAIE(MHCII)+, CD11c–MHCII+ and MHCII–Ly6C−LYVE1+CX3CR1+cells, which were phenotypically and morphologically distinct (Fig. 2d). MHCII–LYVE1+ macrophages expressed CX3CR1 and TIM4, whereas CD206 and CD64 were expressed in all three populations.
Extended Data Fig. 2. Gating strategy for healthy synovial macrophages.

(a) UMAP visualisations of the healthy synovium and (b, c) myeloid cells (red circle) in the same dataset. ScRNA-seq data from GSE145286. (d, e) tSNE plot of surface marker expressions of CD11b + Gr1− cells in the healthy synovium using flow cytometry. Data from 6 mice were concatenated.
To investigate the spatial distribution of macrophages relative to PV1+ capillaries, we performed whole-mount synovial imaging vertically at the edge of whole-mounted synovium. This showed that α-smooth muscle actin+ (αSMA+) arterioles give rise to PV1+ capillaries at the L–SL interface, which were surrounded by LYVE1+ macrophages (Fig. 2e). CD11c–MHCII+ macrophages lined both PV1– and PV1+ vasculature, whereas CD11c+MHCII+ mononuclear phagocytes (MNPs) specifically localized to PV1+ vessels (Fig. 2f). Therefore, all three macrophage subsets were found in proximity to PV1+ capillaries in the L–SL interface (Fig. 2g). When considering the synovium as a whole, lining macrophages varied in their distribution density, with far fewer cells in the central region (Fig. 2h). In the sublining layer, CD11c+MHCII+ MNPs predominantly localized in the periphery, whereas CD11c–MHCII+ macrophages were uniformly distributed throughout the synovium (Fig. 2h). Sagittal sections confirmed that the density of lining macrophages and the thickness of the lining layer were significantly greater at the peripheral area near the meniscus between the femur and tibia than at the central area (Fig. 2i).
Bulk RNA-seq of the sorted macrophage subsets showed that they were transcriptionally distinct, with LYVE1+CX3CR1+ macrophages expressing markers of lining macrophages (Cx3cr1, Vsig4 and Sparc) and CD11c+MHCII+ and CD11c–MHCII+ macrophages demonstrating transcriptional similarity to interstitial macrophages (H2-Ab1 and Retnla7; Fig. 3a–d). Compared to reference transcriptional signatures obtained from macrophages activated with different stimuli27, CD11c+MHCII+ MNPs were enriched for gene sets associated with M2 stimuli (interleukin-4 (IL-4) and IL-13), whereas LYVE1+CX3CR1+ macrophages were enriched for tumor necrosis factor (TNF) and high-density lipoprotein stimulation signatures (Fig. 3e,f). Pathway analysis showed that LYVE1+CX3CR1+ macrophages expressed genes related to endocytosis and the lysosome, whereas the MHCll+ subsets were enriched for gene sets related to cell adhesion and antigen processing and presentation (Fig. 3g).
Fig. 3. Three subsets of synovial macrophages show distinct transcriptomes and ontogenies.

a, Illustration of the experimental protocol. b, Principal component analysis (PCA) of three subsets of synovial macrophages by RNA-seq; n = 3 mice for each plot and n = 9 mice for each population. c, Volcano plots showing DEGs between LYVE1+CX3CR1+, MHCII+CD11c− and MHCII+CD11c+ macrophages from WT mice; Padj, adjusted P value. d, Heat map of the expression of canonical macrophage genes (normalized values) and dendritic cell markers in LYVE1+CX3CR1+, MHCII+CD11c− and MHCII+CD11c+ macrophages from bulk RNA-seq analysis. e, Heat map of single-sample gene set enrichment analysis (ssGSEA) of three synovial macrophage subsets by RNA-seq. The signature genes from a previously published dataset (Xue et al.27) describing the transcriptional programs activated with 28 different stimuli were used; TPP, TNF+PGE2+P3C; IFN, interferon; PA, palmitic acid; LPS, lipopolysaccharide; TNF, tumor necrosis factor; GC, glucocorticoid; HDL, high-density lipoprotein; P3C, Pam3CysSerLys4; OA, oleic acid; Lia, linoleic acid; LA, lauric acid; sLPS, standard lipopolysaccharide; upLPS, ultrapure lipopolysaccharide. f, Quantification of ssGSEA scores for signaling pathways of the indicated stimuli for each subset; n = 3 mice for each plot and n = 9 mice for each population. Data represent mean ± s.e.m. g, Heat map of ssGSEAs of three synovial macrophage subsets with KEGG enrichment analysis (scaled normalized values). h, Illustration of the experimental protocol; FCM, flow cytometry. i, Flow cytometric analysis of MS4A3–tdTomato positivity of the indicated macrophage subsets from 10-week-old mice; n = 3 mice for each group. Data represent mean ± s.e.m.; NC, negative control; mono, monocytes; PC, positive control. j, Three-dimensional reconstruction of representative confocal images of whole-mount synovium and density map of MS4A3–tdTomato. Images are representative of three animals with similar results. Data in f were analyzed by one-way ANOVA with Tukey’s post hoc test, and data in c were analyzed by Wald test.
We next investigated the kinetics of synovial macrophage replenishment by circulating monocytes using Ms4a3Cre-RosaTdT mice. Ms4a3 is specifically expressed by granulocyte–monocyte progenitors, and this model efficiently fate maps monocytes and granulocytes but not lymphocytes or tissue dendritic cells28. In accordance with published reports showing that MHCII+ macrophages are replaced faster than MHCII– macrophages in other organs28, in the synovium, almost half of MHCII+ macrophages were tdTomato+, whereas most of LYVE1+CX3CR1+ macrophages were tdTomato– (Fig. 3h,i). An equivalent proportion of MHCII+CD11c+ MNPs were tdTomato+ compared to MHCII+CD11c– macrophages, indicating that MHCII+CD11c+ MNPs have an ontogenic profile similar to macrophages rather than dendritic cells. Whole-mount synovial imaging of Ms4a3Cre-RosaTdT mice showed that tdTomato+ cells were predominantly distributed at the periphery of the synovium (Fig. 2j and Extended Data Fig. 3b).
Synovial macrophages sample circulating ICs and present antigens
Given their proximity to PV1+ capillaries, we next investigated the capacity of synovial macrophages to take up circulating ICs in vivo. Two hours following i.v. administration, ICs extravasated from PV1+ capillaries at the L–SL interface (Fig. 4a,b), and uptake was evident mainly in LYVE1+CX3CR1+ macrophages, consistent with a previous report24. CD11c+MHCII+ and CD11c–MHCII+ macrophages were also capable of internalizing ICs, with a greater IC uptake than OVA alone (Fig. 4c,d and Extended Data Fig. 3c). Notably, CD11c+MHCII+ MNPs were also able to internalize and present circulating peptide (Eα; Fig. 4e–g), consistent with their enrichment for antigen processing and presentation gene sets (Fig. 3g).
Fig. 4. Synovial macrophages sample circulating ICs, and MHCII+ macrophages present antigens.

a, Three-dimensional reconstruction of representative confocal images of the indicated layers and vertical views of whole-mount synovium from WT mice injected i.v. with OVA–AF647;RaOVA (40 μg of OVA–AF647 + 150 μg of RaOVA) 2 h before analysis. Quantification of the percentage of OVA-IC+ area within PV1+ and PV1−CD31+ area and the percentage of OVA-IC+ area in the lining layer, L–SL interface and sublining layer is shown on the right; n = 4 mice for each group. b, Schematic diagram showing the protocol. c, Pie graph showing the mean percentage of OVA-IC+ macrophage subsets among all OVA-IC+ cells; n = 3 mice. d, Scatter plots of mean fluorescence intensity (MFI) of OVA–AF647 and OVA–AF647;RaOVA; n = 3 mice for each group. e, Schematic diagram showing the protocol of antigen presentation in vivo using the Eα:YAe system. f,g, Flow cytometric analysis (f) and quantification (g) of YAe MFI of the indicated macrophage subsets from mice injected i.v. with Eα divided by that observed in macrophages from mice injected with PBS. Shaded regions indicate mice injected with PBS control; n = 3 mice for each group. h, Flow cytometric analysis of different types of synovial macrophages with indicated FcγRs. Cyan regions indicate staining with isotype controls. Scatter plots show the MFI ratio of each FcγR and isotype controls on each subset; n = 3 mice for each group. i, Three-dimensional reconstruction of representative confocal images of the indicated layers of whole-mount synovium; scale bars, 50 μm. Quantification of the percentage of FcγRllb+ area in the indicated layers is shown on the right; n = 4 mice for each group. j, Flow cytometric analysis of different types of synovial macrophages with the indicated FcγRs before and 24 h after IC injection. Gray regions indicate staining with isotype controls. The FcγR A:I ratios were calculated according to MFI ratios of activating (FcγRlll and FcγRlV) and inhibitory FcγRllb before and 24 h after IC injection on each subset; n = 3 mice for each group. k, Three-dimensional reconstruction of representative confocal images of whole-mount human synovium; scale bars, 100 (left) and 30 μm (right); L, lining layer; SL, sublining layer; SC, synovial cavity. l, Three-dimensional reconstruction of representative confocal images of whole-mount human synovium. Quantification of PV1+ area among CD31+ area in each layer; n = 3 individuals for each group. m, Three-dimensional reconstruction of representative confocal images of whole-mount human synovium. Quantification of LYVE1+ and HLA-DR+ area in the visual field is shown on the right; n = 3 individuals for each group. n, Three-dimensional reconstruction of representative confocal images of whole-mount human synovium; scale bars (right), 100 μm. Images are representative of at least two independent experiments with similar results. The arrowheads indicate the merged area for LYVE1 and CD32B. Data in a and m were analyzed by two-tailed t-test, and data in d and g–i were analyzed by one-way ANOVA with Tukey’s post hoc test. Data in a, d, g–j, l and m are shown as mean ± s.e.m.
Because Fcγ receptors (FcγRs) bind to the Fc portion of IgG to mediate the cellular effector responses to ICs, we analyzed their expression in synovial macrophages. Of note, the relative expression of activating receptors (FcγRIII/FcγRIV) and the inhibitory receptor (FcγRIIb) determines the activation threshold of a cell when encountering IgG ICs, termed the A:I ratio (Extended Data Fig. 3d)29. Flow cytometric analysis showed that LYVE1+CX3CR1+ macrophages expressed higher levels of FcγRIIb and FcγRIII than the other two subsets, but FcγRIV expression was absent in synovial macrophages (Fig. 4h). Whole-mount synovium imaging showed FcγRII/FcγRIII expression mostly at the L–SL interface on LYVE1+ macrophages (Fig. 4i and Extended Data Fig. 3e,f). Circulating ICs led to downregulation of FcγRIII expression in all three subsets, whereas the expression of the inhibitory receptor (FcγRIIb) was maintained (Fig. 4j), thereby increasing the activation threshold and preventing excessive (and potentially damaging) responses to circulating ICs.
To determine if this microarchitectural arrangement was present in human synovium, we optimized a whole-mount imaging system for human knee joint synovium using clearing-enhanced 3D (Ce3D), a tissue clearing solution (Extended Data Fig. 3g)30. Beneath the CD55+ lining layer fibroblasts6 (Fig. 4k and Extended Data Fig. 3h), an abundant PV1+ capillary network was present (Fig. 4l) and was tightly covered with LYVE1+ and HLA-DR+ macrophages and some LYVE1+ macrophages expressing CD32B (FCGR2B), whereas fat pad capillaries were scantily lined with perivascular macrophages (Fig. 4m,n), analogous to our observations in mouse synovium.
Systemic IC challenge induced distinct responses in synovial macrophage subsets
To further interrogate synovial macrophage responses to ICs, we performed bulk RNA-seq on sorted subsets following i.v. IC challenge in vivo in wild-type (WT) and FCGR2B-deficient (Fcgr2b−/−) mice (Fig. 5a). IC challenge was associated with increased Fcgr2b expression in LYVE1+CX3CR1+ macrophages in WT mice, consistent with a negative feedback loop (Fig. 5b). Less than 3% of DEGs were shared among the three macrophage subsets in both WT and FCGR2B-deficient mice, indicating subset-specific responses to circulating IC challenge (Fig. 5c). CD11c+MHCII+ and CD11c–MHCII+ macrophages shared more DEGs, including Fcgr1, Mmp19, Irf7 and Cxcl13, than LYVE1+CX3CR1+ macrophages (Fig. 5c and Extended Data Fig. 4a). Pathway enrichment analysis using upregulated genes showed that LYVE1+CX3CR1+ macrophages were enriched with gene sets related to neutrophil migration, whereas CD11c+MHCII+ macrophages were enriched with gene sets related to cell adhesion and migration after IC stimulation (Fig. 5d).
Fig. 5. Systemic IC challenge induces distinct responses in synovial macrophage subsets.

a, Schematic diagram showing the protocol for bulk RNA-seq. b, Volcano plot showing DEGs due to OVA-IC stimulation in LYVE1+CX3CR1+ macrophages from WT mice by RNA-seq; Sig., significantly. c, Venn diagram showing the number of common DEGs affected by IC stimulation between LYVE1+CX3CR1+, MHCII+CD11c− and MHCII+CD11c+ macrophages in WT and Fcgr2b−/− mice. d, Gene ontogeny (GO) analysis of DEGs specific to each macrophage type with all the DEGs of three macrophage subsets as the background gene list; commun., communication; stim., stimulation; Pos, positive; O/E, observed/expected. e, Number of DEGs in each synovial macrophage from WT and Fcgr2b−/− mice and common DEGs in both strains. f, Heat map of the expression of chemokines (scaled normalized values) with or without IC injection in LYVE1+CX3CR1+ macrophages from WT and Fcgr2b−/− mice. g, Ratio of mean Cxcl1 expression in LYVE1+CX3CR1+, MHCII+CD11c− and MHCII+CD11c+ macrophages from WT and Fcgr2b−/− mice injected i.v. with or without ICs; stim/unstim, stimulated/unstimulated. h, CXCL1 and CXCL2 enzyme-linked immunosorbent assay (ELISA) of the synovial digestion from Fcgr2b−/− mice with or without IC injection; n = 5 (CXCL1) and 4 (CXCL2) mice for each group. i, Schematic diagram showing the protocol. j, Flow cytometry quantification of synovial neutrophils (Ly6G+ gates) from WT and Fcgr2b−/− mice injected i.v. with PBS, OVA or OVA;RaOVA 6 h before analysis; n = 3 (WT) and n = 6 (Fcgr2b−/−) mice. k, Three-dimensional reconstruction of representative confocal images of whole-mount synovium depicting an MHCII+ macrophage cluster around PV1+ capillaries; scale bars, 200 (left), 50 (top right) and 100 μm (bottom right). Images are representative of at least two independent experiments with similar results. l, Number of MHCII+ macrophage clusters with a diameter of >30 μm in the whole-mount synovium in 4- and 52-week-old mice; n = 5 (4-week-old) and 6 (52-week-old) mice; w.o., weeks old; scale bars, 100 μm. m, Schematic diagram showing the protocol of systemic challenges. n,o, Number and images of MHCII+ macrophage clusters with a diameter of >30 μm in the whole-mount synovium in mice injected i.v. with PBS or OVA-IC over 2 consecutive days and analyzed 24 h after the last injection; n = 5 and 7 mice for each group. p, Number of MHCII+ macrophage clusters in mice infected orally with PBS or 5 × 106 S. enterica serovar Typhimurium and analyzed after 3 weeks; n = 5 mice for each group; OG, oral gavage. The arrowheads in l, o and p mark macrophage clusters. q, Number of MHCII+ macrophage clusters in mice inoculated with two doses of 4 × 107 uropathogenic E. coli into the bladder and analyzed after 3 weeks; n = 5 mice for each group. Data in h, l and o–q were analyzed by two-tailed t-test, data in j were analyzed by one-way ANOVA with Tukey’s post hoc test, and data in b were analyzed by Wald test. Data are shown as mean ± s.e.m. in h, j, l and n–q.
Extended Data Fig. 4. Depletion of FcγRllb alters the response to IC in each synovial macrophage subset.

(a) Volcano plots showing DEGs due to OVA-IC stimulation in MHCII+CD11c− and MHCII+CD11c+ macrophages from wild type mice by RNA-seq. Wald test was used. (b) Volcano plots showing DEGs due to OVA-IC stimulation in Lyve1 + , MHCII+CD11c − , and MHCII+CD11c+ macrophages from Fcgr2b − /− mice by RNA-seq. Wald test was used. (c) Gene ontogeny (GO) analysis of DEGs specific to each macrophage subset with all the DEGs of 3 subsets as the background gene list. (d) Heat map of expression of chemokines with or without IV IC injection in MHCII+CD11c− and MHCII+CD11c+ macrophages from wild type and Fcgr2b − /− mice (scaled normalized values).
FcγRIIb deficiency was associated with a greater increase in the number of DEGs in LYVE1+CX3CR1+ macrophages than in WT cells (Fig. 5e and Extended Data Fig. 4b), consistent with the high FcγRIIb expression detectable in this population. Because leukocyte activation- and neutrophil migration-associated genes were enriched in LYVE1+CX3CR1+ cells after i.v. IC challenge (Extended Data Fig. 4c), we further probed the expression of chemokines. We found a greater IC-induced upregulation of the neutrophil-recruiting chemokine Cxcl1 in LYVE1+CX3CR1+ and CD11c+MHCII+ macrophages in Fcgr2b−/− mice than in WT mice (Fig. 5f,g and Extended Data Fig. 4d), supporting the conclusion that FcγRllb is important for limiting neutrophil entry after IC challenge. We confirmed a significant increase in CXCL1 and CXCL2 protein expression and infiltrating neutrophils within the synovium of Fcgr2b−/− mice following i.v. IC challenge (Fig. 5h–j).
Interestingly, given the enrichment of cell adhesion and migration gene sets in activated CD11c+MHCII+ macrophages, we occasionally noted clusters of MHCII+ macrophages tightly entwined around PV1+ capillaries in unchallenged synovium, with LYVE1+ macrophages absent from these aggregates (Fig. 5k). The number of these aggregates increased with age (Fig. 5l). We therefore hypothesized that they may arise in response to chronic circulating stimuli, including ICs, or those present in the context of inflammation/infection in distant organs. To test this, we challenged mice with i.v. IC twice over a period of 48 h (Fig. 5m). This resulted in the appearance of MHCII+ macrophage aggregates at 72 h (Fig. 5n,o). Furthermore, oral challenge with Salmonella enterica serovar Typhimurium, a colitogenic bacteria31, also induced synovial macrophage aggregates (Fig. 5p). However, chronic urinary tract infection with uropathogenic Escherichia coli did not induce macrophage aggregates in the synovium (Fig. 5q), suggesting that organ infection variably influences joints.
We next assessed the contribution of circulating monocytes to macrophage aggregate formation. A recent study32 established an in vivo protocol to fluorescently label intravascular leukocytes that were shielded from subsequent rounds of intravascular labeling once they entered tissues. We applied this system in our chronic IC stimulation model to assess the monocyte contribution to synovial macrophage aggregates. We administered i.v. ICs together with phycoerythrin (PE)-labeled anti-CD45 48 h before synovial collection, followed by administration of i.v. ICs and AF488-labeled anti-CD45 24 h before analysis (Extended Data Fig. 5a). Although we detected both 24- and 48-h time stamp signals in the spleen, synovial macrophage aggregates were not labeled with either 24- or 48-h time stamps (Extended Data Fig. 5b). Furthermore, using monocyte reporter Ms4a3Cre-RosaTdT mice, synovial macrophage aggregates were not labeled with tdTomato (Extended Data Fig. 5c), together indicating that these synovial macrophage clusters are derived from a local macrophage pool rather than circulating monocytes.
Extended Data Fig. 5. Monocyte contribution in the formation of MHCll+ macrophage clusters.

(a) Schematic depicting the experiment. (b) 3D reconstruction of representative confocal images of whole mount synovium and sectional images of spleen after IC and anti-CD45 antibody injection. Bars, 200 (synovium) and 100 μm (spleen). Data represents mean ± SEM. Two-tailed t test was used. (c) 3D reconstruction of representative confocal images of whole mount synovium from Ms4a3Cre-RosaTdT mice. Bars, 200 μm. Data represents mean ± SEM.
We next asked whether fibroblasts play any role as secondary effectors, responding to cues produced by FcγR-expressing synovial macrophages after IC challenge (Extended Data Fig. 6a). Bulk RNA-seq of flow-sorted fibroblasts showed 417 IC-induced genes in synovial fibroblasts (Extended Data Fig. 6b,c), including Cxcl1, Cxcl13 and Ccl8 (Extended Data Fig. 6d). Enrichment analysis also showed that inflammatory response gene pathways, including interferon and IL-6–JAK–STAT3 signaling pathways, were upregulated following IC challenge (Extended Data Fig. 6e).
Extended Data Fig. 6. Transcriptomic changes of synovial fibroblast post systemic IC challenge.

(a) Schematic depicting the protocol. (b) Principal component analysis of synovial fibroblasts by RNA-Seq. n = 3 mice for each population. (c) Volcano plot showing differentially expressed genes in synovial fibroblasts post IC challenge. Wald test was used. (d) Heatmap of inflammatory cytokines and chemokines of synovial fibroblasts by RNA-seq. (e) Gene set enrichment analysis of the bulk RNA-seq data.
Together, these data show distinct responses of synovial macrophage subsets to circulating IC challenge, with LYVE1+CX3CR1+ macrophages poised to trigger neutrophil recruitment but held in check by FcγRllb expression. By contrast, CD11c–MHCII+ and CD11c+MHCII+ macrophages can present circulating antigens and respond to systemic immune stimuli, including ICs, by forming tight clusters around fenestrated capillaries, thus forming a physical barrier that might limit the spread of potentially harmful blood-borne cargo into the joint. In addition, synovial fibroblasts may function as secondary effectors, responding to macrophage cues.
Synovial macrophages activate nociceptors with IL-1β and nociceptors reciprocally enhance macrophage responses through CGRP
As joint pain is one of the most common features of systemic challenges, we next sought to determine the anatomical relationship between PV1+ capillaries and neurons. Sympathetic tyrosine hydroxylase+ (TH+) neurons primarily colocalized with αSMA+ arterioles in the sublining layer (Extended Data Fig. 7a), whereas calcitonin gene-related peptide+ (CGRP+) nociceptor neuronal fibers branched into the L–SL interface around PV1+ capillaries (Fig. 6a and Extended Data Fig. 7b). Quantification of the spatial location of macrophages, PV1+ vessels, PV1–CD31+ vessels and CGRP+ fibers (Extended Data Fig. 8) indicated that approximately 30–40% of each macrophage subset were in direct contact with PV1+ vessels with LYVE1+CX3CR1+ macrophages, and CD11c+ MNPs preferentially localized to PV1+ vessels compared to PV1– vessels (Fig. 6b). The average distance between CGRP+ fibers and macrophages was within 30 μm, and CD11c+ MNPs had the highest overlapped volume with CGRP+ fibers (Fig. 6c).
Extended Data Fig. 7. Distribution of TH+ and CGRP+ fibres in the synovium using IBEX.

(a) 3D reconstruction of representative confocal images of whole mount synovium stained for tyrosine hydroxylase (TH). Bars, 100 μm. Images are representative of at least three independent experiments with similar results. (b) 3D reconstruction of representative confocal images of iterative bleaching extends multiplexity (IBEX) of whole mount synovium. Bars, 100 μm. Four Z-stack images of IBEX of whole mount synovium (bottom). Quantification of the penetration depth of CGRP+ and TH+ fibres as a percentage among total fibres for each subset (right). n = 3 mice for each group. (c) Representative confocal images of L4 dorsal root ganglia (DRG) from mice intravenously injected with OVA or IC. Bars, 100 μm. n = 5 mice for each group. (d) Representative confocal images of L4 DRG from mice intraperitoneally injected with isotype control or anti-CSF1R antibody. Bars, 100 μm. n = 4 mice for each group. (e) Enlarged images for each macrophage subset in Fig. 6e. (f) Flow cytometry plot showing the number of monocytes from mice intraperitoneally injected with isotype control or anti-CSF1R antibody 72 h prior to assessment. Data represents mean ± SEM and two-tailed t test was used for in b, c, d, f.
Fig. 6. Synovial macrophages activate nociceptors in part through IL-1β.

a, Three-dimensional reconstruction of representative confocal images of whole-mount synovium; scale bars, 100 μm. Arrows indicate CGRP+ neurons in z-stack images; n = 5 mice for each group. b, Three-dimensional reconstruction of representative confocal images of whole-mount synovium using the Surface module in Imaris and quantification of the distance between each macrophage subset and vessels; n = 7 mice for each group. Each plot indicates the mean value of each mouse. c, Three-dimensional reconstruction of representative confocal images of whole-mount synovium using the Surface module in Imaris and quantification of the distance or overlapped volume ratio between each macrophage subset and CGRP+ fibers; n = 12 mice for each group. Each plot indicates the mean value of each mouse. d, Representative confocal images of lumbar (L4) and thoracic (T13) DRG in mice injected i.v. with OVA or OVA-IC and analyzed after 6 h; n = 7 (T13) and 11 (L4) mice for each group; Tubβ3, tubulin-β3. The arrowheads indicate the merged area for NP2 and CGRP. e, Three-dimensional reconstruction of representative confocal images of whole-mount synovium from mice injected intraperitoneally (i.p.) with 400 μg of anti-CSF1R or isotype control antibody and analyzed after 72 h. Quantification of the percentage of area covered by each macrophage subset in the synovium is shown on the right; n = 4 (isotype) and 3 (anti-CSF1R) mice. f, Representative confocal images of L4 DRG in mice injected i.v. with OVA or OVA-IC 72 h after i.p. injection with 400 μg of anti-CSF1R; n = 4 (OVA) and 6 (IC) mice. g, Illustration of experimental protocol; SN, supernatant. h, CGRP ELISA of DRG culture SN stimulated with SN from OVA- or IC-stimulated synovial explants from indicated mice or directly stimulated with OVA or IC; n = 3 to 5 mice for each group. i, Heat map of the expression of potential candidates responsible for immune-driven pain in bulk RNA-seq data from IC-stimulated LYVE1+CX3CR1+ macrophages (scaled normalized values). j, CGRP ELISA of DRG culture supernatants stimulated with supernatants from IC-stimulated synovial explants. Indicated neutralizing antibodies (4 μg ml–1) or A438079 (100 μM) was added to synovial SN before adding to DRG neurons. Diflofenac (200 μM) was added with IC when stimulating synovial explants; n = 6 (IC), 4 (CXCL1), 7 (IL-1β), 7 (A438079) and 5 (Coxi) mice. k, IL-1β concentration of the synovial digestion measured by cytometric bead array from Il1bfl/fl and Cx3cr1CreERIl1bfl/fl mice 12 h after i.p. injection of 2 mg per kg (body weight) lipopolysaccharide (LPS); n = 5 mice for each group. l, Representative confocal images of T13 and L4 DRG in Il1bfl/fl and Cx3cr1CreERIl1bfl/fl mice injected i.v. with OVA-IC. Mice were i.p. injected with tamoxifen twice 48 h apart 2 weeks before IC injection and analyzed 6 h after IC injection; n = 10 (Il1bfl/fl) and 9 (Cx3cr1CreERIl1bfl/fl) mice. Data in a, b, d–f, h, k and l were analyzed by two-tailed t-test. The IC group was used as a control group in a one-way ANOVA with Dunnett’s post hoc test in j. Data in c were analyzed by one-way ANOVA with Tukey’s post hoc test. Data in a–f, h and j–l represent mean ± s.e.m.
Extended Data Fig. 8. Protocol for quantifying spatial distribution of three types of macrophages, vessels, and CGRP+ fibres.

Multi-parameter imaging of whole mount synovium was reconstructed with each fluorescent staining and quantitative analysis was performed.
CGRP+ neuronal cell bodies were evident in the L4 dorsal root ganglia (DRG) that innervates the knee joint33 (Extended Data Fig. 7c). Systemic IC injection induced activation of CGRP+ nociceptors in L4 but not T13 DRGs, as evidenced by neuronal pentraxin 2 (NP2) expression (Fig. 6d), an activity-dependent immediate early gene product34 (Extended Data Fig. 9) that facilitates excitatory responses in peptidergic DRG neurons, including CGRP+ afferents35. The proportion of CGRP+ neuronal bodies in the L4 DRGs did not change with IC injection nor anti-CSF1R treatment (Extended Data Fig. 7c,d).
Extended Data Fig. 9. Validation of anti-NP2 antibody.
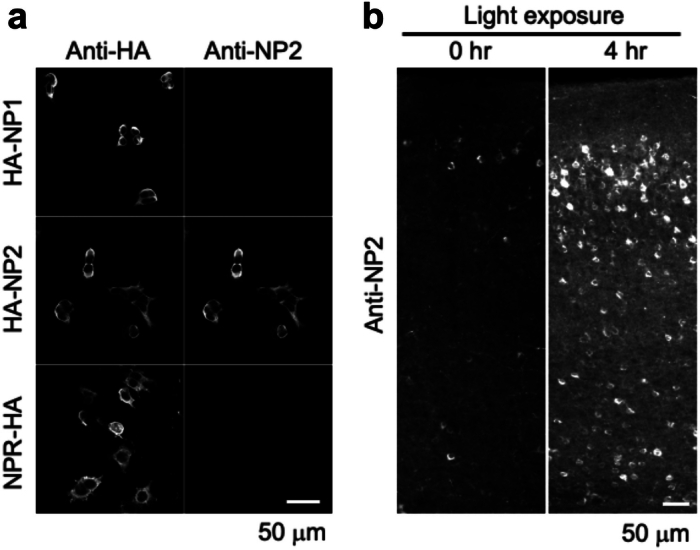
(a) Immunocytochemical analysis of HA-tagged NP1, NP2 and NPR (full-length) expressed in HEK293 cells. Newly raised anti-NP2 antibody showed selective signals for NP2. (b) Specific immunostaining of endogenous NP2 in the visual cortex after the light stimulation. Anti-NP2 antibody detected the increase of NP2 protein expression in all layers of visual cortex 4 hours after light exposure compared with 0 hr exposure control. Bars, 50 μm.
Because neurons do not express FcγRs and are therefore incapable of direct detection of circulating ICs, we hypothesized that adjacent FcγR-expressing macrophages relay their presence and activate CGRP+ nociceptors. To test this, we used anti-CSF1R that preferentially depleted LYVE1+ macrophages (Fig. 6e and Extended Data Fig. 7e,f). This attenuated the activation of CGRP+ nociceptors (Fig. 6f), confirming potential cross-talk between macrophages and CGRP+ nociceptors. Because anti-CSF1R potentially depletes macrophages in other tissues, we added supernatants from synovial whole-mount explants stimulated with OVA or IC to DRG neurons to validate a specific effect of synovial macrophages ex vivo (Fig. 6g). This induced the release of CGRP from DRG neurons, an effect absent in IC-stimulated synovium dissected from anti-CSF1R-treated mice (Fig. 6h). We next used our bulk RNA-seq data from IC-stimulated LYVE1+CX3CR1+ macrophages to prioritize inhibitors for testing in our ex vivo DRG culture model. This analysis identified several candidate molecules upregulated in IC-stimulated macrophages that have previously been described to mediate immune cell interactions with neurons17,36, including Cxcl1, Tnf and Il1β (Fig. 6i), and neutralization of IL-1β partly suppressed the excitation of DRG neurons (Fig. 6j and Extended Data Fig. 10a). Because Cx3cr1CreER mice label lining macrophages in naive animals7,24, we crossed Cx3cr1CreER mice with Il-1βfl/fl mice to ablate IL-1β in LYVE1+CX3CR1+ macrophages. Cre recombinase activation of Cx3cr1CreERIl-1βfl/fl mice resulted in a marked reduction of IL-1β production in the synovium (Fig. 6k), and we observed a reduction in nociceptor activation in L4 DRGs but not in nonjoint innervating T13 DRGs, demonstrating a role for synovial lining macrophage-derived IL-1β in activating nociceptor neurons (Fig. 6l).
Extended Data Fig. 10. CGRP induces distinct transcriptional changes in MHCII+CD11c− and MHCII+CD11c+ macrophages.

(a) CGRP ELISA of DRG culture supernatants stimulated with supernatants from IC stimulated synovial explant. TNF neutralizing antibodies (4 μg/ ml) or isotype control were added to synovial supernatants before adding to DRG neurons. n = 4 mice for each group. Data represents mean ± SEM. Two-tailed t test was used. (b) Illustration of receptor activity-modifying protein (RAMP) association on calcitonin receptor-like receptor (CLR). (c) Expression of marker genes for macrophages and neuropeptide receptors in healthy synovium. scRNA-seq data from GSE145286. (d) Principle component analysis (PCA) of MHCII+CD11c− and MHCII+CD11c+ macrophages from synovial explant of WT mice treated with 100 nM CGRP or control for 4 hours by RNA-seq. n = 3 mice were compiled for each plot and n = 18 mice for each population. (e) Volcano plots showing differentially expressed genes in MHCII+CD11c− and MHCII+CD11c+ macrophages with CGRP stimulation. Wald test was used.
Finally, we asked whether cross-talk between immune cells and nociceptor neurons in the synovium may also involve reciprocal signals from neurons to macrophages. scRNA-seq data of healthy synovium6 showed that Ramp1, Ramp2 and Calcrl were the most highly expressed among neuropeptide receptor transcripts and Ramp1, which forms a complex with calcitonin receptor-like receptor to form the CGRP receptor, was particularly expressed in MHCII+ macrophages (Fig. 7a and Extended Data Fig. 10b,c). Bulk RNA-seq data confirmed Ramp1 expression in MHCII+ macrophages, particularly the CD11c+ population (Fig. 7b). To investigate the functional importance of CGRP receptor expression in MHCII+ synovial macrophages, we stimulated whole synovial explants with CGRP for 4 h and sorted both MHCII+ macrophage subsets for bulk RNA-seq (Fig. 7c). We observed robust transcriptional responses to CGRP stimulation (Extended Data Fig. 10d), with some genes increased in both subsets, but almost half of the DEGs were unique to each macrophage subset (Fig. 7d and Extended Data Fig. 10e). In CD11c+MHCII+ MNPs, CGRP induced upregulation of Fcgr2b, Fcgr3 and Tlr4, and gene sets related to cell adhesion and extracellular matrix organization, including collagen genes (Fig. 7d–f). We next treated mice with BIBN4096, an antagonist of RAMP1 signaling. Pretreatment with BIBN4096 partially suppressed MHCII+ macrophage cluster formation and collagen deposition, the latter visualized by second harmonic generation around the macrophage clusters induced by consecutive i.v. IC challenges (Fig. 7g). Together, these data show that following i.v. IC challenge, LYVE1+CX3CR1+ macrophages in the synovium activate nociceptors with IL-1β, and, in turn, nociceptors enhance CD11c+MHCII+ macrophage responses to IC through CGRP to coordinate a synovial defense system against circulating challenges (Fig. 7h).
Fig. 7. Nociceptors reciprocally enhance synovial macrophage responses through CGRP.

a, Dot plots showing the average expression levels and the percentage of cells from each cluster expressing genes for neuropeptide receptors. scRNA-seq data are from GSE145286; EPI, epinephrine; Ach, acetylcholine; VIP, vasoactive intestinal polypeptide; SP, substance P; EC, endothelial cell; fibro, fibroblast; Av., average. b, Heat map of expression of genes for neuropeptide receptors in LYVE1+CX3CR1+, MHCII+CD11c− and MHCII+CD11c+ macrophages from bulk RNA-seq analysis (normalized values). Ramp1 and Calcrl, which encode proteins that form the CGRP receptor complex, are highlighted in red font. c, Illustration of the experimental protocol for bulk RNA-seq analysis. d, Venn diagram showing the number of distinct and common DEGs affected by CGRP stimulation between MHCII+CD11c− and MHCII+CD11c+ macrophages. e, Gene ontogeny (GO) analysis of DEGs specific to each macrophage type with all the DEGs of two macrophage subsets as the background gene list. Fisher’s exact test was used; BP, biological process. f, Heat map of expression of genes related to extracellular matrix organization and fibroblast proliferation with CGRP stimulation in MHCII+CD11c− and MHCII+CD11c+ macrophages (scaled normalized values). The vertical dashed lines mark the border of control and CGRP groups. g, Number of MHCII+ macrophage clusters with a diameter of >30 μm and second harmonic generation (SHG) on the clusters in the whole-mount synovium in mice injected i.v. with OVA-IC over 2 consecutive days after treatment with BIBN4096 (BIBN; 500 μg per kg (body weight) i.p.) or vehicle; n = 11 mice for each group. Data are shown as mean ± s.e.m. and were analyzed by two-tailed t-test. h, Schematic diagram of the immune-histochemical microarchitecture of the entire synovium and the sentinel unit surrounding PV1+ capillaries; ECM, extracellular matrix.
Discussion
Joint pain or inflammation is a common and early feature of several systemic diseases. These include autoimmune diseases, such as systemic lupus erythematosus, adult onset Still’s disease and inflammatory bowel disease, as well as infection in organs distant to the musculoskeletal system, including enteric or genitourinary infections, streptococcal pharyngitis and viral infections, for example, parvovirus2–4. The pathogenic mediators vary, but circulating ICs or microbial antigens have been implicated37. However, why joints are highly responsive to systemic inflammation is unknown. We sought to address this question, developing a whole-mount imaging system of the entire synovium to profile the vascular, neuronal and immune components. This revealed that highly permeable PV1+ capillaries were specifically located at the L–SL interface in the periphery of the synovium, enabling entry of circulating stimuli into the joint. We found that this area of vulnerability was occupied by three subsets of macrophages that demonstrated distinct responses to systemic IC challenge and interacted with nociceptor neurons, forming a blood–joint barrier (BJB) to defend joint tissue.
The location of fenestrated capillaries and their defensive macrophage–nociceptor sentinel unit in the periphery of the synovium at the synovia–bone interface is particularly interesting because this is the site of onset and formation of destructive granulomatous pannus in RA13,15. RA is characterized by circulating antibodies, including rheumatoid factor (IgM antibodies specific for the Fc portion of IgG) and anti-citrullinated protein IgG1. Our study identifies a fenestrated capillary bed that would enable access of autoantibodies or IC to the synovium at the very site of pannus formation. We found that adjacent macrophages expressed FcγRs, enabling responses to circulating IC and autoantibodies that might access the synovium via the fenestrated vasculature. Indeed, the inhibitory receptor FcγRIIb was a critical regulator of IC-induced activation of LYVE1+CX3CR1+ macrophages, with neutrophil recruitment to the synovium evident in its absence. Our data are consistent with human studies in RA showing that individuals with an FCGR2B polymorphism associated with receptor dysfunction demonstrate increased joint damage38.
The presence of highly permeable PV1+ capillaries in the synovium provides a mechanistic explanation for the joint manifestations of infections affecting organs distal to the musculoskeletal system. Previous studies have identified the presence of bacterial rRNA in a wide spectrum of inflammatory joints39 and viral nucleic acids in viral-associated arthritis40. Together, this suggests that the synovium is accessible to circulating microbes or microbial components. Clinically, reactive arthritis is characterized by a neutrophil-rich synovial effusion41, consistent with our observation that PV1+ capillary-associated synovial macrophages avidly produce neutrophil-recruiting chemokines in response to circulating stimuli. In addition, we found that S. enterica serovar Typhimurium colitis promoted macrophage cluster formation around PV1+ capillaries in the synovium, potentially reflecting the pathology of reactive arthritis.
Using the Ms4a3Cre-RosaTdT model, we assessed the contribution of circulating monocytes to synovial macrophages under steady-state conditions. Compared to other organs that have no monocyte contribution (microglia, Kupffer cells and Langerhans cells), fast replacement (gut and dermis) or slow replacement (kidney and spleen)28, synovial macrophages have heterogeneous kinetics, with the MHCII+ macrophage pool having a greater monocyte contribution than LYVE1+CX3CR1+ lining macrophages. Whole-mount synovial imaging also showed that monocytes preferentially replace synovial macrophages in the synovia–bone interface where proliferative pannus arises in RA, which further highlights the importance of PV1+ fenestrated capillaries as sites where joint pathology is initiated.
The activation of nociceptors alerts humans to the presence of harmful stimuli, leading to pain42. There has been recent interest in potential interactions between nociceptors and immune cells, including via neuropeptides, mainly in the context of infectious diseases where bacteria or fungi can directly activate nociceptors16,43–45. By contrast, our study shows that synovial macrophages sense local IC challenge and stimulate nociceptors, an effect mediated in part through IL-1β. IL-1β has also been previously implicated in augmenting pain sensitivity by promoting prostaglandin production46, upregulating transient receptor potential cation channel subfamily V member 1 (ref. 47) and generating action potentials in a p38 MAP kinase-dependent manner48. Our results suggest that IL-1 receptor antagonists may be a useful therapy for joint pain in a variety of IC-mediated diseases. Other candidates responsible for immune-driven joint pain should be explored in future studies. Furthermore, we found that this immune-driven nociceptor activation led to CGRP production, which in turn affected CD11c+MHCII+ MNPs, increasing the expression of IC and pathogen-associated molecular pattern receptors, such as Fcgr2b, Fcgr3 and Tlr4. This heightened ability to detect and be activated by circulating stimuli reinforces a physical barrier to defend the area of vulnerability around PV1+ capillaries. Among the interstitial macrophages, CD11c–MHCII+ macrophages and CD11c+MHCII+ MNPs showed distinct spatial localization (Fig. 2f), transcriptional profiles in the homeostatic state (Fig. 3b) and responses to IC and CGRP stimulation (Figs. 5c and 7d–f).
Finally, perhaps the most interesting question is why a BJB equipped to sense circulating stimuli and telegraph their presence via pain has evolved. It is tempting to speculate that there might be a selective advantage to such a warning system at both an individual and species level. First, arthralgia might reduce mobility and thereby preserve energy for immune responses. Second, reduced movement may limit the transmission of infectious diseases to other individuals. Similar concepts have been proposed to explain why some infection-associated cytokines lead to increased anxiety and reduced socialization49.
Together, our data show how the unique anatomical arrangement of cells in the peripheral region of the synovium provides access to circulating immune stimuli through fenestrated capillaries and how this is policed by interacting macrophages and nociceptor neurons. Our findings have implications for the development of treatment strategies for immune-driven arthralgia.
Methods
Mice
WT mice (C57BL/6J background) were bred in-house or purchased from The Jackson Laboratory. Transgenic mice expressing Venus enhanced yellow fluorescent protein under the control of the Itgax promoter were a gift from M. Nussenzweig (Rockefeller University). Fcgr2b−/− mice were kindly provided by J. Ravetch (Rockefeller University) and S. Bolland (US National Institutes of Health and US National Institute of Allergy and Infectious Diseases)50. Il1bfl/fl floxed mice were crossed with tamoxifen-inducible B6.129P2(C)-Cx3cr1tm2.1(cre/ERT2)Jung/J mice (The Jackson Laboratory, 020940) as described before51. Cre recombinase activity was induced by two i.p. injections of tamoxifen (2 mg 100 μl–1, Sigma-Aldrich, dissolved in corn oil) 48 h apart in 5- to 7-week-old male mice 2 weeks before the experiment.
In all experiments, both male and female mice were used. For all in vivo experiments, 8- to 20-week-old mice were used unless otherwise mentioned. Mice were maintained under specific pathogen-free conditions at a Home Office-approved facility with controlled humidity and temperature with a 12-h light/12-h dark cycle in the United Kingdom. All procedures were ethically approved by the University of Cambridge Animal Welfare and Ethical Review Body and were performed in accordance with the United Kingdom Animals (Scientific Procedures) Act of 1986 under the authority of a UK Home Office Licence.
Human samples
Human synovial specimens were obtained from individuals with osteoarthritis undergoing replacement surgery or synovectomy with prior ethical approval (18/NW/0545) and informed consent at Addenbrooke’s Hospital, Cambridge. Samples were obtained from two male and three female donors aged 57–83. Participant compensation was not applicable.
Generation and administration of small circulating ICs
All compounds were administered i.v. via tail vein injection using a 30-G insulin syringe (Becton Dickinson). ICs were prepared in vitro by incubating AF647–OVA (2 mg ml–1 in PBS; Invitrogen, O34784) with rabbit polyclonal anti-OVA antiserum (3.7 mg ml–1; Sigma-Aldrich, C-6534) at a 1:1 molar ratio (40 μg of OVA + 150 μg of RaOVA) for 60 min in a 37 °C water bath26.
Infectious models
Mice were infected orally with approximately 5 × 106 colony-forming units of S. enterica serovar Typhimurium (stain BRD509). Twenty-four hours before infection, mice were gavaged with 20 mg of streptomycin (Bio Basic, SB0494) in 100 μl of PBS to aid colonization. Colony-forming unit counts were verified by streaking the inoculums on to agar plates postgavage. Mice were culled 21 days later by rising CO2 concentration.
Urinary tract infection was induced in experimental mice as previously described52. Briefly, under isoflurane anesthesia (Baxter), the perineum was cleaned with ethanol, and the urethra was catheterized using 0.28 × 0.60 mm polyethylene tubing (Instech Laboratories) lubricated in sterile Instillagel (CliniMed). In total, 4 × 107 colony-forming units in 100 μl of prepared uropathogenic E. coli (strain UTI89) were inoculated into the bladder during one session. Repeated inoculation was performed with 2-h intervals to increase the likelihood of ascending pyelonephritis. Mice were culled 21 days later by rising CO2 concentration.
Isolation of synovium leukocytes from tissues
After death by anesthesia, the right auricles of the mice were cut, and 10 ml of prewarmed 1× PBS was injected into the left ventricle for perfusion. Perfusion was omitted in experiments designed to assess blood samples.
After removal of the skin, the quadriceps femoris muscles were carefully removed. Attachment of the synovium to the bare area of femur was observed by pinching and lifting up the patella with tweezers under a stereoscopic microscope (Stemi 2000-CS, Zeiss). The bone–synovium and meniscus–synovium interface was carefully dissected throughout the knee joint without damaging the bone, and the patella was removed at the end. For flow cytometry analysis, whole-mount synovial tissues were digested with 2 mg ml–1 type I collagenase in RPMI and incubated at 37 °C for 45 min. Disaggregated tissue elements were passed through a 70-µm cell strainer.
Flow cytometry
Measurements were performed on an CytoFLEX LX (Beckman Coulter) and analyzed with FlowJo software (Tree Star). Sorting was performed on an FACSAria Fusion (Becton Dickinson). Single-cell suspensions were incubated with Zombie Aqua (Biolegend) or Viakrome 808 fixable viability dye (Beckman Coulter) diluted 1:250 in PBS for 15 min at 4 °C. Samples were centrifuged and resuspended in FACS buffer supplemented with anti-CD16/CD32 (Biolegend) diluted 1:50, followed by staining with antibodies for 15 min at 4 °C. Antibodies used in this study are listed in Supplementary Table 1.
Confocal and multiphoton microscopy of mouse samples
Dissected knee joints were fixed in fixative solution made with 9% Glyoxal (Sigma-Aldrich, 128465) and 40% Antigenfix (Diapath, P0016) overnight at 4 °C. For sagittal section imaging, samples were decalcified in 14% EDTA solution for 12 days, followed by 4 h in 30% sucrose in PBS. EDTA was replaced every 72 h. Then, 30-μm sections were blocked and permeabilized in 0.1 M Tris containing 0.3% Triton X-100 (MilliporeSigma), 1% normal mouse serum, 1% normal donkey serum and 1% bovine serum albumin (BSA; R&D Systems). Samples were stained for 2 h at room temperature (RT) in a wet chamber with the appropriate antibodies, washed three times in PBS and mounted in Fluoromount-G (Southern Biotech). For whole-mount imaging, the knee synovium was dissected, as described above, and blocked and permeabilized in 0.1 M Tris containing 0.1% Triton X-100, 1% normal mouse serum, 1% normal donkey serum and 1% BSA in 0.5-ml Eppendorf safe-lock tubes. Samples were stained for 12 h at 4 °C with the appropriate antibodies in 0.5-ml Eppendorf safe-lock tubes, washed three times extensively in PBS and mounted in Fluoromount-G. Images were acquired using a TCS SP8 inverted confocal microscope with a scan format of 512 × 512 (Leica Microsystems) or TCS SP8 3X gated STED confocal inverted microscope (Leica Microsystems) with a ×40/1.3-NA oil or ×40/1.1-NA water objective. Second harmonic generation images were acquired using a Zeiss 710 NLO upright multiphoton microscope equipped with a ×20 water objective and infrared lasers (880 nm) driven by a Ti sapphire laser. Raw imaging data were processed, and a Gaussian filter was applied using Imaris (Bitplane). The antibodies used in this study are listed in Supplementary Table 1.
Confocal microscopy of human samples
Human synovial specimens were fixed in a fixative solution made with 9% Glyoxal (Sigma-Aldrich, 128465) and 40% Antigenfix (Diapath, P0016) at 4 °C. For sagittal section imaging, samples were placed in 30% sucrose in PBS for 4 h and embedded in OCT embedding matrix (CellPath). Sections were incubated with primary antibodies for 2 h at RT and washed three times in PBS. Sections were then incubated with the appropriate fluorochrome-labeled secondary antibodies for 1 h at RT, washed in PBS and mounted in Fluoromount-G. For whole-mount imaging, synovium samples were dissected into 1 × 1 cm pieces and blocked and permeabilized in 0.1 M Tris containing 0.3% Triton X-100, 1% normal donkey serum, 1% BSA and 0.1% saponin (Sigma). Samples were incubated with primary antibodies for 12 h at 4 °C and washed three times extensively in PBS. Samples were then incubated with the appropriate fluorochrome-labeled secondary antibodies for 12 h at 4 °C and washed three times in PBS. Samples were then embedded in Ce3D solution30 for 12 h to clear the tissue and mounted in Ce3D solution. Images were acquired using a TCS SP8 inverted confocal microscope or TCS SP8 3X gated STED confocal inverted microscope on a ×40/1.3-NA oil or ×40/1.1-NA water objective. Raw imaging data were processed, and a Gaussian filter was applied using Imaris (Bitplane). Antibodies used in this study are listed in Supplementary Table 1.
Iterative staining
Iterative staining of sections was performed as previously described25,53. Following acquisition of initial images, the coverslip was removed, and slides were washed three times in PBS to remove mounting medium. Bleaching of the fluorochromes was achieved using a 1 mg ml–1 solution of lithium borohydride in water (Acros Organics) for 15 min at RT. The slides or whole-mount synovium were washed three times in PBS before staining with a different set of antibodies. The process was repeated up to three times. Raw imaging data were processed using Imaris (Bitplane), and Hoechst or CD31 was used as fiducial for the alignment of subsequent images.
Dissection and culture of mouse DRG neurons
DRG neurons were bilaterally excised under a dissection microscope as previously described54. For section images, lumbar (L4) or thoracic (T13) DRGs were embedded in OCT embedding matrix, and sections were subsequently made. For culture systems, DRGs were digested with the combined 1.25 mg ml–1 collagenase A + 2.5 mg ml–1 Dispase ll solution for 30 min at 37 °C. Sensory neurons were cultured on laminin-coated 96-well plates in Neurobasal medium (Thermo Fisher Scientific) supplemented with 1% GlutaMAX (Thermo Fisher Scientific), 50 ng ml–1 NGF 2.5S (Gibco) and B-27 supplement (Gibco) for 36 h to remove glial and axon debris that float in the medium. For CGRP release assays, ICs were prepared in vitro by incubating OVA with rabbit polyclonal anti-OVA (1 μg of OVA + 18.5 μg of RaOVA) for 30 min in a 37 °C water bath, spinning down the pellet and washing with PBS three times. Synovium explants were stimulated with OVA or ICs in 200 μl of RPMI for 6 h, and DRG neurons were incubated with the supernatants for 2 h at 37 °C and 5% CO2. After incubation, the DRG supernatant was collected and used to quantify the concentration of CGRP using an ELISA kit (Antibodies.com) according to the manufacturer’s instructions.
CGRP stimulation assay from synovium explants
Whole-mount synovium was dissected from WT mice and incubated with CGRP (100 nM) or vehicle for 4 h. After incubation, the synovium was digested as described above, and macrophages were sorted into 350 μl of Buffer RLT Plus (Qiagen) using a FACSAria Fusion (Becton Dickinson).
In vivo BIBN4096 treatment
The impact of CGRP signaling on the outcome of systemic IC challenge was evaluated by treating mice with the RAMP1 antagonist BIBN4096 (Tocris), as described previously16. Mice were treated by i.p. injection of BIBN4096 (0.5 mg per kg (body weight)) or vehicle 2 h before i.v. IC injection for 2 d in a row, and knee synovium was dissected 48 h after the last injection.
Cytometric bead array
Concentrations of IL-1β were determined by BD Cytometric Bead Array using the Mouse IL-1β Enhanced Sensitivity Flex Set (562278) according to the manufacturer’s instructions. Bilateral synovial whole mounts were minced in 500 μl of PBS per animal, and samples were acquired using a BD FACSVerse flow cytometer. The results were analyzed by FCAP Array v3 software (BD).
Bulk RNA-seq library construction and sequencing
The indicated cell types in each experiment were lysed using RLT Plus buffer (Qiagen), vortexed, snap-frozen on dry ice and stored at −80 °C. To extract RNA from cell lysates, an RNeasy Plus Micro kit (Qiagen) was used according to the manufacturer’s instructions. Genomic DNA contamination was removed using Optimal DNA depletion columns (Qiagen). Purified RNA was eluted in nuclease-free water (Ambion) and stored at −80 °C. To assess the quality and concentration of purified RNA, an RNA Pico chip (Applied Biosystems) on a Bioanalyzer 2000 (Applied Biosystems) was used according to the manufacturer’s instructions. For library preparation, a SMARTer Stranded Total RNA-seq Mammalian Pico Input kit v3 (Takara) was used according to the manufacturer’s instructions. Library size was assessed using a High Sensitivity DNA chip (Applied Biosystems) on a Bioanalyzer 2000 (Applied Biosystems) according to the manufacturer’s instructions. Bulk RNA-seq was performed using a Novaseq 6000 (Illumina) on a 2 × 150 bp sequencing run. Pooled libraries were demultiplexed using Casava (Illumina). Fastq files from sequencing libraries were trimmed of the first three nucleotides on the R1 strand. Contaminating adaptor sequences and poor-quality bases were removed using Trim Galore (Babraham Bioinformatics). Libraries were only trimmed for quality. The quality of the resulting files was assessed by FastQC and aligned to the mm10 genome using HISAT2.
Subsequent RNA-seq analysis was performed in the R statistical environment, as described before55, with RStudio 2022.02.2. Resulting data are available on the Gene Expression Omnibus (GEO) under accession numbers GSE247475, GSE247476, GSE247477 and GSE272541. Reads were counted and assigned to genes using the Featurecount function from the Rsubread package, and differential expression analysis was performed using DESeq2 with an appropriate design matrix according to the default workflow. Batch effects were removed using the sva package. Figures were plotted using the ggplot2 and pheatmap packages and Prism software. Gene ontology enrichment testing was performed using topGO. GSEA (https://www.gsea-msigdb.org/gsea) was conducted using GSEA v4.3.0 according to developer’s instructions with the preranked option and classic setting. KEGG gene sets were downloaded from the Molecular Signatures Database.
Analysis of public scRNA-seq datasets
For the analysis of the dataset from GSE145286 (ref. 6), raw data from WT mice were downloaded from the GEO archive, and count data were imported into R. After quality control, normalization and dimensional reduction, gene expression was visualized by UMAP. For Extended Data Fig. 1a–c, clusters of endothelial cells were extracted with Pecam1 and Cdh5 expression and reclustered. Resulting clusters were annotated using canonical marker gene expression (Pecam1, Sema3g, Hey1, Podxl, Ackr1, Vwf and Rgcc), and capillary endothelial cells were further extracted for Fig. 1a–c. Figures were plotted using the EnhancedVolcano and DotPlot functions. For Fig. 7a, cells were clustered using canonical marker genes (Cd68, Lyve1, H2-Ab1, Cd55, Prg4, Thy1 and Pecam1).
Development and validation of anti-NP2
Anti-NP2 was newly raised by guinea pigs immunized with peptides Gly 107–Leu 429 (UniProtKB entry O70340).
For immunocytochemistry, HEK293 tsA201 cells (a gift from R. Horn, Thomas Jefferson University) were cultured in high-glucose DMEM (Sigma-Aldrich) containing 10% fetal bovine serum (HyClone), 50 U ml–1 penicillin, 50 mg ml–1 streptomycin (Invitrogen) and 2 mM l-glutamine at 10% CO2 and 37 °C. Cells were transfected with HA-tagged mouse NP1, NP2 or NPR in pCAGGS using Lipofectamine 2000 (Invitrogen). The following day, transfected cells were fixed with 4% paraformaldehyde (PFA) in PBS for 15 min and washed with PBS three times. After blocking/permeabilization with 3% BSA (Sigma-Aldrich) in PBS containing 0.1% Triton X-100 (Sigma-Aldrich) for 30 min, cells were stained with primary antibodies to the HA tag (mouse, 1:1,000, BAbCo, MMS-101P) and NP2CC2 (guinea pig; final concentration of 1 μg ml–1) for 2 h at RT, followed by washing with PBS and incubation with the respective secondary antibodies (Alexa 488 (Invitrogen) and Cy3 (Jackson ImmunoResearch Laboratories) to the respective primary antibody; 1:1,000) for 30 min. After washing with PBS, coverslips were mounted on a glass slide with Fluoromount-G (Thermo Fisher Scientific). Fluorescence was detected using an SD-OSR microscope (Olympus).
For immunohistochemistry, mice were housed on a standard light cycle (0800–2000 h) before placement into constant darkness for 7 days. Mice were transcardially perfused with 4% PFA in 0.1 M sodium phosphate buffer (pH 7.2) for 10 min under deep pentobarbital anesthesia in the dark (0 h = control condition) or light-exposed for 4 h (4 h = light-stimulated condition) before death, followed by postfixation of dissected brain samples with 4% PFA in PBS for 2 h at 4 °C. Free-floating sagittal sections (50-μm thickness) from brains fixed in 4% PFA were prepared with a microslicer (DTK-1000, Dosaka EM). Following washing of the sections with PBS containing 0.1% Triton X-100 (wash buffer), sections were subsequently treated with 10% donkey serum for 30 min at RT, incubated with a mixture of primary antibodies (1 μg ml–1) diluted in wash buffer overnight, washed three times, incubated with species-specific secondary antibody (Alexa 488, Invitrogen; 1:200) in wash buffer and washed again three times. Finally, sections were attached to glass slides and mounted with VECTASHIELD (Vector Laboratories). Fluorescence images of NP2 were obtained in the visual cortex using a confocal microscope (SD-OSR, Olympus) and a ×10 objective.
Image analysis
Image analysis was performed using Imaris 9.9.1, ImageJ2 version 2.14.0/1.54f or QuPath as detailed below.
For quantification of the density of each macrophage in the indicated compartments in the synovium, whole-mount synovium images for each macrophage subset were opened in QuPath, and the positions (x and y axes) of each macrophage were quantified using the ‘Cell detection’ function. The x and y axes of the center of the synovium were subtracted from the position of each macrophage, and the distance from center was calculated. Macrophage densities were calculated by dividing the number of cells by the indicated area. Density maps were created following the step in the ‘Cell detection’ function using Qupath.
For area quantification, the Imaris analysis tool ‘percentage of material above threshold colocalization’ was used to detect the area of dextran 70 kDa, dextran 2,000 kDa, PV1 over CD31, fluorescently labeled microbeads over PV1 or CD31, CD32b and CGRP. For comparison of different layers of the synovium from whole-mount images, the lining layer was defined by the presence of ERTR7+ lining fibroblasts or lining macrophages.
For quantification of penetration depth of synovial sensory fibers, the outermost layer of the synovium (lining layer) was determined by ERTR7 staining, which identifies lining fibroblasts in the synovium. z-Stack images of synovial whole mounts were created, and the distances between each fiber (CGRP+ and TH+ fibers) and the ERTR7+ outermost layer were measured and averaged per mouse.
For quantification of the number of clusters of MHCll+ macrophages in the whole-mount synovium images, clusters were defined as MHCll+ macrophages attached together to form a granulomatous structure with a diameter of >30 μm.
For analysis of the proximity of each macrophage subset to PV1+ capillaries, PV1– capillaries and CGRP+ nociceptor neurons, we used the Surface module of Imaris (Bitplane)56. Briefly, we performed multiparameter imaging of whole-mount synovium using confocal microscopy and reconstructed fluorescent staining of MHCll, CD11c, LYVE1, PV1, CD31 and CGRP separately as three-dimensional volumes using the Surface module and smooth parameters of 1.99 μm for vessels, 1.00 μm for cells and 0.50 μm for neurons. Under the function ‘Set voxel intensity inside surface to 0’, we used PV1 signals as a mask to determine CD31 staining outside of the PV1 volume (defined as ‘CD31+PV1− volume’) and CD11c signals as a mask to determine MHCll staining outside of the CD11c volume (defined as ‘MHCll+CD11c− volume’). Threshold for absolute intensity and filter for number of voxels were selected to cover signals of the region of interest, and surfaces that do not cover the region of interest were removed. We then quantified the shortest distance between the surface of each macrophage and specific capillaries or neurons, percentage of cells in direct contact with each structure and overlapped volume ratio between each macrophage and neurons. The percentage of cells in the indicated interval of distance and the mean value per animal were calculated.
Statistical analysis
The results are shown as single data points in a scatter dot plot and as mean ± s.e.m. Between-group differences were determined using a two-tailed t-test. A one-way ANOVA with Tukey’s post hoc test was used for comparisons among three or more groups, and a one-way ANOVA with Dunnett’s test was used for comparisons of the mean of each column to the mean of a control column. Statistical analyses were performed using GraphPad Prism 8.4.1 (GraphPad Software). Animal and human sample sizes are indicated on scatter dot plots unless mentioned in the figure legend.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41590-024-02011-8.
Supplementary information
Supplementary Table 1.
Source data
Statistical source data.
Statistical source data.
Statistical source data.
Statistical source data.
Statistical source data.
Statistical source data.
Statistical source data.
Statistical source data.
Statistical source data.
Statistical source data.
Acknowledgements
We thank E. Johnston for technical assistance with dissecting DRGs, A. Vogt and R. Al Hosni for assistance with preparing human samples and J. Villar for assistance with collecting mouse samples. T.H. is supported by Human Frontier Science Program (LT000028/2021-L) and a Kennedy Trust senior research fellowship (KENN 23 24 15). R.S. is supported by the Wellcome Trust (215378/Z/19/Z). A.J.H. is supported by Versus Arthritis clinical research fellowship (22293). M.R.C. is supported by the National Institute of Health Research, Cambridge Biomedical Research Centre and a Wellcome Investigator Award (220268/Z/20/Z). K.S. was supported by JSPS KAKENHI (JP16H06280).
Extended data
Author contributions
T.H. and M.R.C. designed the study. T.H., C.Y.C.L., N.R., A.J.H., A.F. and R.S. performed experiments. A.W.M. and M.A.B. provided human synovium specimens. K.S., M.Y. and M.W. developed and validated the antibody to NP2. N.L., K.T. and A.D. supported experiments using Cx3cr1CreERIl1bfl/fl mice. Z.L. and F.G. supported experiments using Ms4a3Cre-RosaTdT mice. T.H. and M.R.C. wrote the manuscript.
Peer review
Peer review information
Nature Immunology thanks Clifford Woolf and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: S. Houston, in collaboration with the Nature Immunology team.
Data availability
The datasets analyzed during the current study are available from the corresponding author upon reasonable request. Access to raw RNA-seq data related to this study is available through GEO (accession numbers GSE247475, GSE247476, GSE247477 and GSE272541). For the reanalysis of mouse synovium scRNA-seq, we obtained the dataset from GEO (accession number GSE145286). The KEGG gene set is available from Molecular Signatures Database (https://www.gsea-msigdb.org/gsea/msigdb/index.jsp). Source data are provided with this paper.
Code availability
RNA-seq data analysis was performed in R using field-standard, previously reported packages and indicated parameters, which are described in the Methods and with the relevant source publications cited where appropriate. No custom code beyond adaptation of existing software packages was used in this study. The code is available on reasonable request from the corresponding authors.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Tetsuo Hasegawa, Email: th647@cam.ac.uk.
Menna R. Clatworthy, Email: mrc38@medschl.cam.ac.uk
Extended data
is available for this paper at 10.1038/s41590-024-02011-8.
Supplementary information
The online version contains supplementary material available at 10.1038/s41590-024-02011-8.
References
- 1.Smolen, J. S., Aletaha, D. & McInnes, I. B. Rheumatoid arthritis. Lancet388, 2023–2038 (2016). [DOI] [PubMed] [Google Scholar]
- 2.García-Kutzbach, A., Chacón-Súchite, J., García-Ferrer, H. & Iraheta, I. Reactive arthritis: update 2018. Clin. Rheumatol.37, 869–874 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Mackie, S. L. & Keat, A. Poststreptococcal reactive arthritis: what is it and how do we know? Rheumatology43, 949–954 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Franssila, R. & Hedman, K. Viral causes of arthritis. Best. Pract. Res. Clin. Rheumatol.20, 1139–1157 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Ceccarelli, F. et al. Joint involvement in systemic lupus erythematosus: from pathogenesis to clinical assessment. Semin. Arthritis Rheum.47, 53–64 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Wei, K. et al. Notch signalling drives synovial fibroblast identity and arthritis pathology. Nature582, 259–264 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Culemann, S. et al. Locally renewing resident synovial macrophages provide a protective barrier for the joint. Nature572, 670–675 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simkin, P. A. The human knee: a window on the microvasculature. Tissue Barriers3, e970465 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simkin, P. A. & Bassett, J. E. Pathways of microvascular permeability in the synovium of normal and diseased human knees. J. Rheumatol.38, 2635–2642 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Knight, A. D. & Levick, J. R. Effect of fluid pressure on the hydrauric conductance of interstitium and fenestrated endothelium in rabbit knee. J. Physiol.360, 311–332 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mapp, P. I. Innervation of the synovium. Ann. Rheum. Dis.54, 398–403 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith, M. D. The normal synovium. Open Rheumatol. J.5, 100–106 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schett, G. & Gravallese, E. Bone erosion in rheumatoid arthritis: mechanisms, diagnosis and treatment. Nat. Rev. Rheumatol.8, 656–664 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hasegawa, T., Kikuta, J. & Ishii, M. Imaging of bone and joints in vivo: pathological osteoclastogenesis in arthritis. Int. Immunol.33, 679–686 (2021). [DOI] [PubMed] [Google Scholar]
- 15.Hasegawa, T. et al. Identification of a novel arthritis-associated osteoclast precursor macrophage regulated by FoxM1. Nat. Immunol.20, 1631–1643 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Pinho-Ribeiro, F. A. et al. Bacteria hijack a meningeal neuroimmune axis to facilitate brain invasion. Nature615, 472–481 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baral, P., Udit, S. & Chiu, I. M. Pain and immunity: implications for host defence. Nat. Rev. Immunol.19, 433–447 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Straub, R. H. & Pongratz, G. Role of peripheral nerve fibres in acute and chronic inflammation in arthritis. Nat. Rev. Rheumatol.9, 117–126 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Thiriot, A. et al. Differential DARC/ACKR1 expression distinguishes venular from non-venular endothelial cells in murine tissues. BMC Biol.15, 45 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stan, R. V. et al. The diaphragms of fenestrated endothelia: gatekeepers of vascular permeability and blood composition. Dev. Cell23, 1203–1218 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carloni, S. et al. Closing during intestinal inflammation. Science448, 439–448 (2021). [DOI] [PubMed] [Google Scholar]
- 22.Lee, D. et al. Cadherin-11 in synovial lining formation and pathology in arthritis. Science315, 1006–1010 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Croft, A. P. et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature570, 246–251 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zec, K. et al. Macrophages in the synovial lining niche initiate neutrophil recruitment and articular inflammation. J. Exp. Med.220, e20220595 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Radtke, A. J. et al. IBEX: a versatile multiplex optical imaging approach for deep phenotyping and spatial analysis of cells in complex tissues. Proc. Natl Acad. Sci. USA117, 33455–33465 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clatworthy, M. R. et al. Immune complexes stimulate CCR7-dependent dendritic cell migration to lymph nodes. Nat. Med.20, 1458–1463 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xue, J. et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity40, 274–288 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu, Z. et al. Fate mapping via MS4A3—expression history traces monocyte-derived cells. Cell178, 1509–1525 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Smith, K. G. C. & Clatworthy, M. R. FcγRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nat. Rev. Immunol.10, 328–343 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li, W., Germain, R. N. & Gerner, M. Y. High-dimensional cell-level analysis of tissues with Ce3D multiplex volume imaging. Nat. Protoc.14, 1708–1733 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hapfelmeier, S. & Hardt, W. D. A mouse model for S. typhimurium-induced enterocolitis. Trends Microbiol.13, 497–503 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Kirschenbaum, D. et al. Time-resolved single-cell transcriptomics defines immune trajectories in glioblastoma. Cell187, 149–165 (2024). [DOI] [PubMed] [Google Scholar]
- 33.Aso, K. et al. Nociceptive phenotype of dorsal root ganglia neurons innervating the subchondral bone in rat knee joints. Eur. J. Pain18, 174–181 (2014). [DOI] [PubMed] [Google Scholar]
- 34.O’Brien, R. J. et al. Synaptic clustering of AMPA receptors by the extracellular immediate- early gene product Narp. Neuron23, 309–323 (1999). [DOI] [PubMed] [Google Scholar]
- 35.Kanehisa, K. et al. Neuronal pentraxin 2 is required for facilitating excitatory synaptic inputs onto spinal neurons involved in pruriceptive transmission in a model of chronic itch. Nat. Commun.13, 2367 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hasebe, R. et al. ATP spreads inflammation to other limbs through crosstalk between sensory neurons and interneurons. J. Exp. Med.219, e20212019 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Toivanen, P. & Toivanen, A. Two forms of reactive arthritis? Ann. Rheum. Dis.58, 737–741 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Radstake, T. R. D. J. et al. The functional variant of the inhibitory Fcγ receptor IIb (CD32B) is associated with the rate of radiologic joint damage and dendritic cell function in rheumatoid arthritis. Arthritis Rheum.54, 3828–3837 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Kempsell, K. E. et al. Reverse transcriptase–PCR analysis of bacterial rRNA for detection and characterization of bacterial species in arthritis synovial tissue. Infect. Immun.68, 6012–6026 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moore, T. Parvovirus-associated arthritis. Curr. Opin. Rheumatol.12, 289–294 (2000). [DOI] [PubMed] [Google Scholar]
- 41.Hønge, B. L., Hermansen, M. L. F. & Storgaard, M. Reactive arthritis after COVID-19. BMJ Case Rep.14, 2020–2022 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Basbaum, A. I., Bautista, D. M., Scherrer, G. & Julius, D. Cellular and molecular mechanisms of pain. Cell139, 267–284 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cohen, J. A. et al. Cutaneous TRPV1+ neurons trigger protective innate type 17 anticipatory immunity. Cell178, 919–932 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chiu, I. M. et al. Bacteria activate sensory neurons that modulate pain and inflammation. Nature501, 52–57 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maruyama, K. et al. Nociceptors boost the resolution of fungal osteoinflammation via the TRP channel–CGRP–JDP2 axis. Cell Rep.19, 2730–2742 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Cunha, T. M. et al. A cascade of cytokines mediates mechanical inflammatory hypernociception in mice. Proc. Natl Acad. Sci. USA102, 1755–1760 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ebbinghaus, M. et al. The role of interleukin-1β in arthritic pain: main involvement in thermal, but not mechanical, hyperalgesia in rat antigen-induced arthritis. Arthritis Rheum.64, 3897–3907 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Binshtok, A. M. et al. Nociceptors are interleukin-1β sensors. J. Neurosci.28, 14062–14073 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Filiano, A. J. et al. Unexpected role of interferon-γ 3 in regulating neuronal connectivity and social behaviour. Nature535, 425–429 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bolland, S. & Ravetch, J. V. Spontaneous autoimmune disease in FcγRIIB-deficient mice results from strain-specific epistasis. Immunity13, 277–285 (2000). [DOI] [PubMed] [Google Scholar]
- 51.Helyes, Z. et al. Transfer of complex regional pain syndrome to mice via human autoantibodies is mediated by interleukin-1-induced mechanisms. Proc. Natl Acad. Sci. USA116, 13067–13076 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suchanek, O. et al. Tissue-resident B cells orchestrate macrophage polarisation and function. Nat. Commun.14, 7081 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Richoz, N. et al. Distinct pathogenic roles for resident and monocyte-derived macrophages in lupus nephritis. JCI Insight7, e159751 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perner, C. & Sokol, C. L. Protocol for dissection and culture of murine dorsal root ganglia neurons to study neuropeptide release. STAR Protoc.2, 100333 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Banham, G. et al. Bromodomain inhibitors modulate FcγR-mediated mononuclear phagocyte activation and chemotaxis. Front. Immunol.13, 885101 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bissen, D., Kracht, M. K., Foss, F. & Acker-Palmer, A. Expansion microscopy of mouse brain organotypic slice cultures to study protein distribution. STAR Protoc.3, 101507 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1.
Statistical source data.
Statistical source data.
Statistical source data.
Statistical source data.
Statistical source data.
Statistical source data.
Statistical source data.
Statistical source data.
Statistical source data.
Statistical source data.
Data Availability Statement
The datasets analyzed during the current study are available from the corresponding author upon reasonable request. Access to raw RNA-seq data related to this study is available through GEO (accession numbers GSE247475, GSE247476, GSE247477 and GSE272541). For the reanalysis of mouse synovium scRNA-seq, we obtained the dataset from GEO (accession number GSE145286). The KEGG gene set is available from Molecular Signatures Database (https://www.gsea-msigdb.org/gsea/msigdb/index.jsp). Source data are provided with this paper.
RNA-seq data analysis was performed in R using field-standard, previously reported packages and indicated parameters, which are described in the Methods and with the relevant source publications cited where appropriate. No custom code beyond adaptation of existing software packages was used in this study. The code is available on reasonable request from the corresponding authors.