

Abstract
Introduction
The prognostic role of plasma neurofilament light chain (NfL), phospho-tau, beta-amyloid, and GFAP is still debated in Parkinson’s disease (PD).
Methods
Plasma p-tau181, p-tau231, Aβ1-40, Aβ1-42, GFAP, and NfL were measured by SIMOA in 136 PD with 2.9 + 1.7 years of follow-up and 76 controls. Differences in plasma levels between controls and PD and their correlation with clinical severity and progression rates were evaluated using linear regression analyses.
Results
Patients exhibited similar distribution of plasma biomarkers but higher P-tau181, P-tau231 and lower Aβ1-42 compared with controls. NfL and GFAP correlated with baseline motor and non-motor severity measures. At follow-up, NfL emerged as the best predictor of progression with marginal effect of GFAP and p-tau181 adjusting for age, sex, disease duration, and baseline motor severity.
Conclusion
The present findings confirmed plasma NfL as best predictor of progression in PD, with a marginal role of p-tau181 and GFAP.
Keywords: Parkinson’s disease, Neurofilament light chain, GFAP, Phosphorylated tau, Progression, Plasma biomarkers
Introduction
Parkinson’s disease (PD) is a heterogeneous neurodegenerative disorder characterized by different rates of motor progression and responses to treatment. Several demographic and clinical features have been associated with worse progression in PD in prospective cohorts, including older age, male sex, the akinetic-rigid phenotype, and the presence of autonomic dysfunction [1–4].
It has recently been demonstrated that cerebrospinal fluid (CSF) and plasma neurofilament light chain (NfL) represent the most effective blood marker for predicting disease progression in several neurodegenerative disorders, including Parkinson’s disease (PD) [5–8]. Moreover, glial fibrillary acidic protein (GFAP), a marker of glial activation, has been linked to disease severity and poorer progression in Alzheimer’s disease (AD), frontotemporal dementia (FTD), and the alpha-synucleinopathy dementia with Lewy bodies (DLB) [9–11]. Several large studies suggested that plasma phosphorylated tau (p-tau) and amyloid species may serve as highly reliable markers for identifying Alzheimer’s disease (AD) even in its early stages [12–16]. P-tau species, in particular p-tau181, have been identified as a promising avenue for detecting AD co-pathology and have been shown to possess prognostic value in alpha-synucleinopathies, as recently evidenced in DLB [17, 18]. Nevertheless, the prognostic value of plasma biomarkers in PD remain a topic of debate, and only a limited number of longitudinal findings are currently available in this field.
In this study, we hypothesize that a panel of plasma markers, including p-tau, amyloid species, and GFAP, might increase the known diagnostic ability of NfL to stratify PD patients and predict disease progression over time. To this end, we first compared these markers in PD and age-matched controls and evaluate the possible association with different severity measures at baseline. Furthermore, we evaluated the ability of single and multiple markers to predict motor and non-motor disease progression in PD, adjusting for the effect of baseline severity variables.
Methods
Patient’s selection
Consecutive patients with a clinical diagnosis of PD [19] were evaluated at the outpatient Movement disorder Clinic, Neurology Unit at the University of Brescia, North of Italy. It serves as a tertiary referral center for neurodegenerative disorders and it follows-up approximately 800 subjects with PD on a regular basis. Healthy controls (HC) were selected among patients’ caregivers. This study was approved by the local ethics committee (NP 1471, DMA, last version on December the 7th 2020) and was in conformity with the Helsinki Declaration. Informed consent was obtained from all participants at blood sampling. Levodopa equivalent daily dose (LEDD) was calculated according to standard conversion [20] and the diagnosis was supported by levodopa/dopaminergic response and at least 2 years of clinical follow-up. Only clinically established PD patients [21] were included.
All patients underwent routine blood analyses and magnetic resonance imaging to exclude prominent cortical or subcortical infarcts or brain/iron accumulation or atypical parkinsonian disorders. The following exclusion criteria were applied: (1) dementia at baseline; (2) atypical parkinsonism, at baseline or during follow-up; (3) prominent cortical or subcortical infarcts in structural imaging; (4) other neurologic disorders or medical conditions potentially associated with cognitive deficits; (5) bipolar disorder, schizophrenia, history of drug or alcohol abuse, or impulse control disorder; (6) negative nigrostriatal dopaminergic imaging; (7) recent traumatic events or acute fever/inflammation; (8) kidney disease.
Clinical assessment
At baseline, standardized neurological examination was performed, including the Movement Disorder Society-Unified Parkinson Disease Rating Scale (MDS-UPDRS) [22] and Hoehn and Yahr stage (H and Y) assessment [23]. The following disability milestones were evaluated at baseline and after follow-up for each patient: gait dependency (unable of walking unassisted), recurrent falls (more than 1 fall per month), motor fluctuations (OFF state and/or dyskinesia), and dementia (cognitive impairment causing dependency in ADL).
All patients included in the analyses underwent a clinical and follow-up for at least 2 years and up to 5 years (average follow-up: 2.9 years). The patients were stratified according to the mean annual change in MDS-UPDRS-III; fast progressor were defined as subjects with an MDS change higher than one standard deviation in the cohort (UPDRS-III of > 2 vs ≤ 2 points, respectively) independently from levodopa equivalent daily dose adjustment. The annual changes in MDS-UPDRS and development of disability milestones were considered as linear and dichotomic targets for biomarkers analyses.
Biochemical analyses
At the time of assessment, approximately 10 mL venous blood was collected in tubes containing sodium ethylenediaminetetraacetic acid (EDTA) from each subject. Participants were required to fast for at least 2 h prior to collection. The blood samples were centrifuged at 2000 × g at 4 °C for 8 min within 2 h of collection. Plasma supernatant was collected, divided into aliquots, and frozen at − 80 °C until further use. NfL, p-tau181 and p-tau231, Aβ1-40, Aβ1-42, and GFAP concentrations were measured at the Clinical Neurochemistry Laboratory, Sahlgrenska University Hospital, Mölndal, Sweden. Biomarkers concentrations were measured using the Simoa platform (Quanterix, Billerica, MA). For p-tau231, an in-house assay was used as previously described [14]. The other markers were measured using commercially available assays (Quanterix, Billerica, MA). Samples were randomized, blinded, and measured in duplicate using one batch of reagents from the same lot in one round of experiments. Intra-assay coefficients of variation were below 10%. Individuals exhibiting plasma values exceeding five standard deviations of the mean for the entire sample were excluded from the study analysis. Additionally, cases with missing values or outliers were excluded.
Statistical analyses
Differences in demographic features between PD and HC were assessed with t-test for independent samples or Chi-square test for continuous and categorical variables, respectively. Differences within biomarkers levels between PD and HC were evaluated using Student T test for independent variables. Partial correlation analyses adjusted for the effect of age, sex, and disease duration was applied to test significant correlations between biomarkers values and clinical variables (MDS-UPDRS-III, LEDD, and H and Y). Differences in biomarkers level between PD with normal and fast progression were assessed in univariate analyses adjusted for the effect of age, sex, and disease duration. The discriminative ability of the single biomarkers in predicting fast and slow progressors was evaluated through an overall accuracy analysis utilizing the area under the curve (AUC) of a receiver-operating characteristic curve (ROC) and the positive and negative predictive values. The between MDS-UPDRS-III and plasma biomarkers levels was additionally evaluated using a linear regression model adjusted for the effect of age, sex, disease duration, and a principal components regression where all the plasma biomarkers have been decomposed with a principal component analysis, later used as matrix of covariate in a linear model with MDS-UPDRS-III as response.
Differences in time-dependent disability milestones between patients with normal and abnormal biomarkers levels were assessed with Cox regression corrected for age at sampling, disease duration, and sex. Significance was set at p < 0.05 for all the analysis.
Results
Recruitment, and clinical and cognitive baseline features
Out of 190 patients with a clinically confirmed diagnosis of Parkinson’s disease under dopaminergic treatments screened, 136 entered the study (mean age 69.3 ± 9.8 years, mean disease duration 6.5 ± 5.0 years); 78 age-matched controls were included in the analysis (supplementary Fig. 1 for the inclusion flowchart). Table 1 shows the demographics and clinical baseline characteristics and biomarkers level of controls and PD stratified according to the longitudinal follow-up in fast and slow progressors. Compared with HC, PD patients showed higher levels of p-tau231 and p-tau181 and lower levels of Aβ1-42 levels. PD patients with fast motor progression exhibited higher NfL levels compared to both HC and PD with normal motor progression. In the whole cohort and specifically in PD, all the markers correlated with age at sampling and disease duration (except for GFAP) (Fig. 1).
Table 1.
Demographical and biomarkers difference between HC and PD
| HC (n = 78) | PD normal progression (n = 105) | PD fast progression (n = 31) | P | |
|---|---|---|---|---|
| Sex (F) | 54 (69.2%) | 53 (39.0%) | 53 (39.0%) | < 0.001x |
| Age at blood sample | 69.4 ± 7.9 | 68.5 ± 10.0 | 72.0 ± 8.9 | 0.177 |
| Disease duration, y | N.A | 6.2 ± 4.8 | 7.6 ± 5.7 | 0.218 |
| H and Y stage (n) | N.A |
1 (29) 2 (59) 3 (14) 4 (2) 5 (1) |
1 (6) 2 (16) 3 (6) 4 (2) 5 (1) |
0.056 |
| MDS-UPDRS-III | N.A | 20.0 ± 12.4 | 19.1 ± 10.7 | 0.415 |
| Annual MDS-UPDRS-III total score change | N.A | 0.6 ± 2.0 | 6.5 ± 5.4 | < 0.001 |
| LEDD | N.A | 482.0 ± 335.7 | 419.0 ± 215.2 | 0.326 |
| Annual LEDD change | N.A | 12.8 ± 103.3 | 66.8 ± 152.2 | 0.024 |
| Plasma biomarkers | ||||
| p-tau 231 (pg/mL) | 10.2 ± 5.1 | 12.9 ± 7.2 | 14.2 ± 7.4 | 0.004xy |
| p-tau 181 (pg/mL) | 14.3 ± 6.9 | 17.7 ± 8.9 | 19.7 ± 8.9 | 0.002xy |
| Aβ1-40 (pg/mL) | 121.5 ± 45.8 | 124.1. ± 39.4 | 135.9 ± 49.9 | 0.288 |
| Aβ1-42 (pg/mL) | 10.9 ± 3.4 | 8.4 ± 2.7 | 9.2 ± 3.4 | < 0.001xy |
| GFAP (pg/mL) | 117.7 ± 64.9 | 120.4 ± 75.8 | 123.0 ± 63.9 | 0.933 |
| NfL (pg/mL) | 20.1 ± 19.5 | 20.8 ± 10.9.2 | 23.4 ± 15.2 | 0.002yz |
H and Y Hoehn and Yahr stages, LEDD levodopa equivalent daily dose expressed in mg; MDS-UPDRS-III Movement Disorder Society-Unified Parkinson’s disease Rating scale part III, p-tau181 phosphorylated tau 181, p-tau231 phosphorylated tau 231, Aβ1-40 beta-amyloid 1–40, Aβ1-42 beta-amyloid 1–42, GFAP glial fibrillary acidic protein, N.A not applicable, NfL neurofilament light chain, y years, x = HC vs PD normal progression, y = HC vs PD fast progression, z = PD normal progression vs PD fast progression
Fig. 1.

Correlation matrix including age at onset, disease duration, and the biomarkers analyzed in the cohort. p-tau181, phosphorylated Tau 181, p-tau231, phosphorylated Tau 231, Aβ1-40, beta-amyloid 1–40; Aβ1-42, beta-amyloid 1–42; GFAP Glial Fibrillary Acidic Protein, NfL neurofilament light chain; HC healthy controls; SP PD with slow motor progression; FP PD with fast motor progression
In PD, plasma NfL and GFAP exhibited a positive correlation with total MDS-UPDRS-III scores at baseline in analyses adjusted for age, sex, and disease duration (r = 0.207; p = 0.007 and r = 0.163; p = 0.03, respectively). Both biomarkers additionally showed a positive correlation with Hoehn and Yahr stage (r = 0.433; p < 0.001 for NfL; r = 0.173; p = 0.036 for GFAP). The correlation between plasma biomarkers and cognition was evaluated in 96 PD patients with MoCA baseline assessment. In unadjusted analyses, higher levels of p-tau181 and NfL exhibited a negative correlation with MoCA (r = − 0.218, p = 0.048 and r = − 0.329, p = 0.001, respectively), whereas no correlation survived in corrected analyses. Moreover, upon analyzing the correlations between variables without adjusting for age and sex, significant positive associations were observed, particularly between NfL and GFAP (see Fig. 2 and supplementary Table 1).
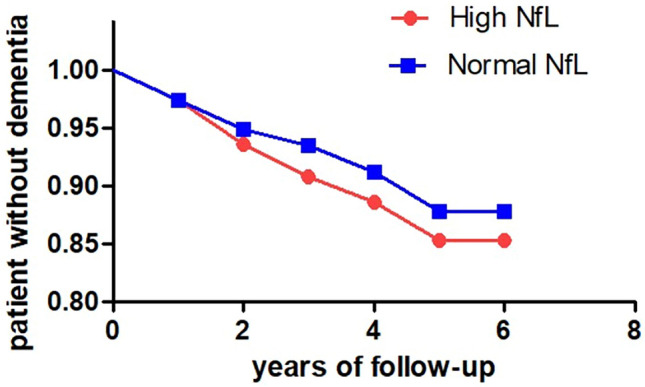
Fig. 2.

Cox regression analysis comparing development of dementia in patients with high NFL levels vs patients with normal NFL levels. NfL neurofilament light chain
Correlation between plasma biomarkers and clinical variables at follow-up
The presence of milestones of disability and the annual change of MDS-UPDRS-III was evaluated in a clinical follow-up (average: 2.9 ± 1.7 years). In the PD group, 32 patients presented falls (14 at baseline, 18 during follow-up with a medium onset 1.3 years after baseline); 16 exhibited progressive inability to walk (2 at baseline, 14 during follow-up with a medium onset of 1.3 years after baseline) and 26 motor fluctuations (6 at baseline, 20 during follow-up) (see supplementary Table 2 for details). Patients with walking impairment (inability to walk unassisted) showed higher baseline mean levels of NfL compared with independent patients (37.5 ± 24.2 vs 21.5 ± 12.5, p < 0.001) with similar levels of other markers. The annual changes in MoCA were evaluated in a subset of 81 patients with at least three consecutive annual assessments. In partial correlation and linear correlation analyses adjusted for age, sex, education, and disease duration, no marker was found to be correlated with worsening of cognition status measured by annual rate change of MoCA.
The levels of plasma biomarkers did not differ between patients with/without falls and motor fluctuations (separately analyzed at baseline and during follow-up). Patients who developed dementia during follow-up (n = 30) exhibited higher levels of NfL compared with non-demented patients, when adjusted for age, sex, and disease duration (20.2 ± 12.1 vs 36.4 ± 22.1, p < 0.001). In Cox regression analysis corrected for sex, age, and disease duration, patients with abnormal plasma NfL levels had a higher risk of developing dementia during follow-up (p = 0.030) (Fig. 3). No other significant result was found in Cox regressions analyzing different biomarkers and disability milestones.
Fig. 3.
Differences in biomarkers levels in plasma between HC, and slow and fast PD progressors. p-tau181, phosphorylated tau 181, p-tau231, phosphorylated tau 231, Aβ1-40, beta-amyloid 1–40; Aβ 1–42, beta-amyloid 1–42; GFAP glial fibrillary acidic protein, NfL neurofilament light chain
Annual MDS-UPDRS and LEDD change and baseline plasma biomarkers
The relationship between annual change in MDS-UPDRS-III scores and plasma biomarkers was challenged in both partial correlation and linear regression models. NfL was the only plasma marker positively correlated with changes in MDS-UPDRS-III scores in both models (partial correlation r = 0.256; p = 0.003, linear regression model p = 0.02, T 2.2) (Table 2). The correlation between NfL and changes of UPDRS-III were confirmed after exclusion of PD patients with baseline falls (r = 0.223, p = 0.033); this was no longer significant in linear regression analysis (r = 0.139, p = 0.105) (Supplementary Table 3).
Table 2.
Multivariable linear regression model for motor progression defined by annual MDS-UPDRS part III score changes including demographics, clinical baseline variables, and NfL levels
| Model | B | Standard error | Beta | T | Sign |
|---|---|---|---|---|---|
| Constant | 1.107 | 2.898 | 0.382 | 0.703 | |
| Age at sampling | 0.017 | 0.044 | 0.039 | 0.382 | 0.382 |
| Disease duration | 0.096 | 0.097 | 0.112 | 0.987 | 0.325 |
| Gender | − 0.828 | 0.722 | − 0.094 | − 1.147 | 0.254 |
| NfL | 0.067 | 0.029 | 0.233 | 2.265 | 0.025 |
| MDS-UPDRS-III baseline | − 0.091 | 0.033 | − 0.255 | − 2.789 | 0.006 |
| LEDD baseline | − 0.002 | 0.001 | − 0.136 | − 1.337 | 0.184 |
| Falls baseline | 1.914 | 0.836 | 0.189 | 2.289 | 0.024 |
In the first line, “B” refers to unstandardised coefficients, while “Beta” refers to the standardized coefficients
LEDD levodopa equivalent daily dose, MDS-UPDRS-III movement Disorder Society- Unified Parkinson disease Rating Scale, NfL neurofilament light chain
Linear combinations of NfL, p-tau181, p-tau231, Aβ1-40, Aβ1-42, and GFAP into six new variables obtained through principal component decomposition (after standardization) were used as regressors in a linear model with yearly delta MDS-UPDRS-III scores as response and correcting for age, sex, and disease duration. The only significant combination of variables (p = 0.02) was a combination of NfL and GFAP, confirming the association of these variables with delta MDS-UPDRS-III scores (Supplementary Table 4). A similar analysis on the original biomarkers (without standardizing their values) reports that the only significant combination of variables (p = 0.004) consisted of a combination of NfL and p-tau181 (Supplementary Table 5).
Figure 1 shows the different distribution of plasma biomarkers in patients with normal and fast progression, with NfL being the only marker able to differentiate PD patients with different rates of motor change (32.0 ± 22.6 vs 20.1 ± 19.5; p = 0.007). In ROC analyses, NfL demonstrated a positive predictive value of 45% and a negative predictive value of 85% for distinguishing between slow and fast progressors based on UPDRS, with an AUC of 0.65 (95% CI: 0.521–0.770) and a Youden optimal cutoff of 29.6 pg/mL.
Baseline LEDD score and annual changes were not correlated with any of the biomarkers at baseline, and none of them was able to differentiate different trajectories of LEDD changes across the years of follow-up.
Discussion
This prospective study aimed to evaluate the clinical value of a panel of plasma biomarkers for stratifying PD patients and predicting disease progression over time. The findings showed a strong correlation of NfL and GFAP with motor severity at baseline, whereas p-tau181 correlated with cognition only in unadjusted analyses. At follow-up, NfL emerged as the strongest predictor of progression, with only a marginal effect of p-tau181 and GFAP using different statistical approaches.
These findings contribute to expanding the current knowledge regarding the clinical application of plasma biomarkers in PD by evaluating an extended panel of standard plasma markers using Simoa and conducting a clinical follow-up over 3 years. Several recent studies have supported the use of AD-related plasma biomarkers for early identification of AD due to their strong correlation with CSF species [16, 24, 25]. Phosphorylated tau species and GFAP have recently been highlighted as important prognostic markers in AD, atypical parkinsonism, and DLB [26, 27].
Furthermore, CSF AD-related biomarker patterns have been identified as important predictors of progression in PD—especially for cognitive measures [28–32] thus providing a strong rationale for the present study. Compared with age-matched controls, non-demented PD patients exhibited slightly higher levels of GFAP and p-tau species and lower levels of amyloid 1–42. This might indicate a concomitant AD-related pathology in a relevant percentage of PD [25, 29, 31, 33–36]. However, these trends might also indicate unspecific effects of PD pathology on some plasma biomarkers, whereas NfL—the most sensitive neuronal damage marker—appeared to have similar levels compared with controls [6, 7, 37, 38]. The baseline clinical correlation analyses demonstrated a robust correlation between motor severity and NfL, as well as GFAP. This is an intriguing finding, particularly considering the purported role of the latter biomarker in modulating nigrostriatal and cortical alpha-synuclein pathology [39, 40] and its purported utility as a prognostic marker in Asian ethnicity [41]. Conversely, cognitive function at baseline correlated with both NfL and p-tau181 only in unadjusted analyses, in line with the recent works focused on cognition [42].
The motor follow-up assessed with MDS-UPDRS scores, levodopa changes, and disability milestones confirmed NfL as the best predictor of progression for motor progression time, with only marginal impact for GFAP and p-tau181 in PCA-based models. NfL also emerged as the best marker for predicting conversion to dementia, whereas p-tau species, GFAP, and amyloid did not show any effect on linear and dichotomic outcome measures. These findings align with several recent works with different designs focused on p-tau181 and amyloid species [42–44], although the short follow-up and the exclusion of patients without dementia a priori definitively limit the number of patients at risk of conversion of this specific population.
These findings, in line with earlier reports, further highlighted the value of NfL as very sensitive yet unspecific marker of neuronal damage [45]. The clinical relevance of this marker alone—even when adjusting for clinical variables and adopting different models of progression—overshadowed all other more specific glial or AD-related markers in the cohort. These results are relevant for the research community, as they definitively questioned the added value of an extended panel of biomarkers for general stratification of PD instead of NfL alone [6, 33, 46–48]. Nevertheless, the study is exploratory in nature, as the population is lacking in ethnic and genetic diversity, as well as detailed kidney function, and further studies on different populations, comorbidity distributions, and longer follow-up should be conducted. Furthermore, studies with longitudinal assessment of plasma biomarkers changes are important to extend these findings. Another important limitation was the inclusion of consecutive PD patients who were already into the clinical phase of the disease, with a wide distribution of baseline disease duration and a relative stable disease, as highlighted by annual MDS-UPDRS changes lower compared with larger prospective studies [49]. To adjust for baseline severity, this variable was included in all multivariate models using clinically relevant outcome measures listed in PPMI and other prospective longitudinal PD studies [9]. Finally, further studies focused on drug-naïve PD or specific at-risk subpopulation (such as PD-MCI) are warranted to extend these findings and clarify the best combination and relevance of plasma biomarkers as proxies of disease progression in subjects suitable for interventions.
Acknowledgements
The authors would like to thank all the participants for their invaluable contributions.
Author’s contribution
Andrea Pilotto: study concept and design, acquisition of data, analysis and interpretation of data, and drafting/revising the manuscript for content. Nicholas Ashton: acquisition of data, analysis and interpretation of data, and drafting/revising the manuscript for content. Alessandro Lupini: study concept and design, acquisition of data, analysis and interpretation of data, and drafting/revising the manuscript for content. Beatrice Battaglio: acquisition of data, and drafting/revising the manuscript for content. Cinzia Zatti: acquisition of data, and drafting/revising the manuscript for content. Chiara Trasciatti: drafting/revising the manuscript for content. Stefano Gipponi: acquisition of data, and drafting/revising the manuscript for content. Elisabetta Cottini: acquisition of data, and drafting/revising the manuscript for content. Ilaria Grossi: acquisition of data, and drafting/revising the manuscript for content. Alessandro Salvi: acquisition of data, and drafting/revising the manuscript for content. Giuseppina De Petro: acquisition of data, and drafting/revising the manuscript for content. Marina Pizzi: acquisition of data, and drafting/revising the manuscript for content. Antonio Canale: data analysis and interpretation, and drafting/revising the manuscript for content. Kaj Blennow: study concept and design, acquisition of data, analysis and interpretation of data, and drafting/revising the manuscript for content. Henrik Zetterberg: study concept and design, acquisition of data, analysis and interpretation of data, and drafting/revising the manuscript for content. Alessandro Padovani: acquisition of data, analysis and interpretation of data, and drafting/revising the manuscript for content.
Funding
Open access funding provided by Università degli Studi di Brescia within the CRUI-CARE Agreement. The recruitment of patients was partially supported by BIOMANE Project from the Health and Wealth research grants 2016 of the University of Brescia [Grant no. NP 1471, DMA]. Andrea Pilotto has been supported by grants of Airalzh Foundation AGYR2021 Life-Bio Grant, The LIMPE-DISMOV Foundation Segala Grant 2021, the Italian Ministry of University and Research PRIN COCOON (2017MYJ5TH) and PRIN 2021 RePlast, the H2020 IMI IDEA-FAST (ID853981), and Italian Ministry of Health, Grant/Award Number: RF-2018-12366209 and PNRR-Health PNRR-MAD-2022-12376110. Nick Ashton is supported by the Swedish Research Council (#2017-00915), ADDF, USA (#RDAPB-201809-2016615), the Swedish Alzheimer Foundation (#AF-742881), and Hjärnfonden, Sweden (#FO2017-0243). Chiara Trasciatti is supported by the Italian Ministry of University and Research. Kaj Blennov is supported by the Swedish Research Council (#2017-00915), ADDF, USA (#RDAPB-201809-2016615), the Swedish Alzheimer Foundation (#AF-742881), Hjärnfonden, Sweden (#FO2017-0243), the Swedish state under the agreement between the Swedish government and the County Councils, the ALF-agreement (#ALFGBG-715986), the European Union Joint Program for Neurodegenerative Disorders (JPND2019-466-236), and the NIH, USA (grant #1R01AG068398-01). Henrik Zetterberg is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2022-01018 and #2019-02397), the European Union’s Horizon Europe research and innovation program under Grant Agreement no. 101053962, Swedish State Support for Clinical Research (#ALFGBG-71320), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809-2016862), the AD Strategic Fund and the Alzheimer's Association (#ADSF-21-831376-C, #ADSF-21-831381-C, and #ADSF-21-831377-C), the Bluefield Project, the Olav Thon Foundation, the Erling-Persson Family Foundation, Stiftelsen för Gamla Tjänarinnor, Hjärnfonden, Sweden (#FO2022-0270), the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie Grant Agreement no. 860197 (MIRIADE), the European Union Joint Program–Neurodegenerative Disease Research (JPND2021-00694), the National Institute for Health and Care Research University College London Hospitals Biomedical Research Centre, and the UK Dementia Research Institute at UCL (UKDRI-1003).Alessandro Padovani has been supported by grants of the Italian Ministry of University and Research PRIN COCOON (2017MYJ5TH) and PRIN 2021 RePlast, the H2020 IMI IDEA-FAST (ID853981), Italian Ministry of Health, Grant/Award Number: RF-2018-12366209 and PNRR-Health PNRR-MAD-2022-12376110.
Data availability
The datasets used are available from the corresponding author on reasonable request.
Declarations
Conflicts of interest
All the authors report no disclosures related to this manuscript. Andrea Pilotto served in the advisory board of Z-cube (technology division of Zambon pharmaceuticals); he received honoraria from Z-cube s.r.l., Biomarin, Zambon, Nutricia and Chiesi Pharmaceuticals. He received research support from Vitaflo Germany and Zambon Italy. Nicholas Ashton has no financial conflicts to disclose. Alessandro Lupini has no financial conflicts to disclose. Beatrice Battaglio has no financial conflicts to disclose. Cinzia Zatti has no financial conflicts to disclose. Chiara Trasciatti has no financial conflicts to disclose. Stefano Gipponi has no financial conflicts to disclose. Elisabetta Cottini has no financial conflicts to disclose. Ilaria Grossi has no financial conflicts to disclose. Alessandro Salvi has no financial conflicts to disclose. Giuseppina De Petro has no financial conflicts to disclose. Marina Pizzi has no financial conflicts to disclose. Antonio Canale has no financial conflicts to disclose. Kaj Blennow has no financial conflicts to disclose. Henrik Zetterberg has served at scientific advisory boards and/or as a consultant for Abbvie, Acumen, Alector, Alzinova, ALZPath, Annexon, Apellis, Artery Therapeutics, AZTherapies, CogRx, Denali, Eisai, Nervgen, Novo Nordisk, Optoceutics, Passage Bio, Pinteon Therapeutics, Prothena, Red Abbey Labs, reMYND, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics, and Wave, has given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure, Biogen, and Roche, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program (outside submitted work). Alessandro Padovani is consultant and served on the scientific advisory board of GE Healthcare, Eli-Lilly and Actelion Ltd Pharmaceuticals, received speaker honoraria from Nutricia, PIAM, Lansgstone Technology, GE Healthcare, Lilly, UCB Pharma, and Chiesi Pharmaceuticals. He is founded by Grant of M1.
Footnotes
Andrea Pilotto, Nicholas J. Ashton and Alessandro Lupini the authors have equally contributed to the work.
References
- 1.Espay AJ, Schwarzschild MA, Tanner CM, Fernandez HH, Simon DK, Leverenz JB et al (2017) Biomarker-driven phenotyping in Parkinson’s disease: a translational missing link in disease-modifying clinical trials. Mov Disord 32:319–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fereshtehnejad S-M, Romenets SR, Anang JBM, Latreille V, Gagnon J-F, Postuma RB (2015) New clinical subtypes of Parkinson disease and their longitudinal progression: a prospective cohort comparison with other phenotypes. JAMA Neurol 72:863–873 [DOI] [PubMed] [Google Scholar]
- 3.Pilotto A, di Schiano Cola F, Premi E, Grasso R, Turrone R, Gipponi S et al (2019) Extrastriatal dopaminergic and serotonergic pathways in Parkinson’s disease and in dementia with Lewy bodies: a 123 I-FP-CIT SPECT study. Eur J Nucl Med Mol Imaging 46:1642–51 [DOI] [PubMed] [Google Scholar]
- 4.Markello RD, Arnatkeviciute A, Poline J-B, Fulcher BD, Fornito A, Misic B (2021) Standardizing workflows in imaging transcriptomics with the abagen toolbox. Elife 10:e72129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boylan LS, Chiò A (2019) Divining progression in Parkinson disease with a blood test: NfL. Neurology 93(11):471–472 [DOI] [PubMed] [Google Scholar]
- 6.Pilotto A, Imarisio A, Conforti F, Scalvini A, Masciocchi S, Nocivelli S et al (2021) Plasma NfL, clinical subtypes and motor progression in Parkinson’s disease. Parkinsonism Relat Disord 87:41–47 [DOI] [PubMed] [Google Scholar]
- 7.Lin C-H, Li C-H, Yang K-C, Lin F-J, Wu C-C, Chieh J-J et al (2019) Blood NfL: a biomarker for disease severity and progression in Parkinson disease. Neurology 93:e1104–e1111 [DOI] [PubMed] [Google Scholar]
- 8.van Rumund A, Aerts MB, Esselink RAJ, Meijer FJA, Verbeek MM, Bloem BR (2018) Parkinson’s Disease Diagnostic Observations (PADDO): study rationale and design of a prospective cohort study for early differentiation of parkinsonism. BMC Neurol 18:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bartl M, Dakna M, Galasko D, Hutten SJ, Foroud T, Quan M et al (2021) Biomarkers of neurodegeneration and glial activation validated in Alzheimer’s disease assessed in longitudinal cerebrospinal fluid samples of Parkinson’s disease. PLoS One 16:e0257372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benedet AL, Milà-Alomà M, Vrillon A, Ashton NJ, Pascoal TA, Lussier F et al (2021) Differences between plasma and cerebrospinal fluid glial fibrillary acidic protein levels across the Alzheimer disease continuum. JAMA Neurol 78:1471–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bolsewig K, van Unnik AAJM, Blujdea ER, Gonzalez MC, Ashton NJ, Aarsland D et al (2024) Association of plasma amyloid, p-Tau, GFAP, and NfL with CSF, clinical, and cognitive features in patients with dementia with lewy bodies. Neurology 102(12):e209418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mattsson N, Zetterberg H, Janelidze S, Insel PS, Andreasson U, Stomrud E et al (2016) Plasma tau in Alzheimer disease. Neurology 87:1827–1835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Milà-Alomà M, Ashton NJ, Shekari M, Salvadó G, Ortiz-Romero P, Montoliu-Gaya L et al (2022) Plasma p-tau231 and p-tau217 as state markers of amyloid-β pathology in preclinical Alzheimer’s disease. Nat Med 28:1797–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ashton NJ, Pascoal TA, Karikari TK, Benedet AL, Lantero-Rodriguez J, Brinkmalm G et al (2021) Plasma p-tau231: a new biomarker for incipient Alzheimer’s disease pathology. Acta Neuropathol 141:709–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hall S, Janelidze S, Londos E, Leuzy A, Stomrud E, Dage JL et al (2021) Plasma Phospho-tau identifies Alzheimer’s co-pathology in patients with Lewy body disease. Mov Disord 36:767–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pilotto A, Parigi M, Bonzi G, Battaglio B, Ferrari E, Mensi L et al (2022) Differences between plasma and cerebrospinal fluid p-tau181 and p-tau231 in early Alzheimer’s disease. J Alzheimer’s Dis 87:991–997 [DOI] [PubMed] [Google Scholar]
- 17.Gonzalez-Ortiz F, Kac PR, Brum WS, Zetterberg H, Blennow K, and Karikari TK (2023) Plasma phospho-tau in Alzheimer’s disease: toward diagnostic and therapeutic trial applications. In molecular neurodegeneration (Vol. 18, Issue 1). BioMed Central Ltd [DOI] [PMC free article] [PubMed]
- 18.Vrillon A, Bousiges O, Götze K, Demuynck C, Muller C, Ravier A et al (2024) (2024) Plasma biomarkers of amyloid, tau, axonal, and neuroinflammation pathologies in dementia with Lewy bodies. Alzheimers Res Ther 16(1):146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Postuma RB, Berg D, Stern M, Poewe W, Marek K, Litvan I (2015) CME MDS clinical diagnostic criteria for Parkinson’s disease centrality of motor syndrome—Parkinsonism and PD criteria benchmark—the expert examination. Mov Disord 30(12):1591–1601 [DOI] [PubMed] [Google Scholar]
- 20.Tomlinson CL, Stowe R, Patel S, Rick C, Gray R, Clarke CE (2010) Systematic review of levodopa dose equivalency reporting in Parkinson’s disease. Mov Disord 25:2649–2653 [DOI] [PubMed] [Google Scholar]
- 21.Berg D, Adler CH, Bloem BR, Chan P, Gasser T, Goetz CG et al (2018) Movement disorder society criteria for clinically established early Parkinson’s disease. Mov Disord 33:1643–1646 [DOI] [PubMed] [Google Scholar]
- 22.Goetz CG, Tilley BC, Shaftman SR, Stebbins GT, Fahn S, Martinez-Martin P et al (2008) Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov Disord 23:2129–2170 [DOI] [PubMed] [Google Scholar]
- 23.Goetz CG, Poewe W, Rascol O, Sampaio C, Stebbins GT, Counsell C et al (2004) Movement Disorder Society Task Force report on the Hoehn and Yahr staging scale: status and recommendations. Mov Disord 19:1020–1028 [DOI] [PubMed] [Google Scholar]
- 24.Nakamura A, Kaneko N, Villemagne VL, Kato T, Doecke J, Doré V et al (2018) High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature 554:249–254 [DOI] [PubMed] [Google Scholar]
- 25.Chiu P-Y, Yang F-C, Chiu M-J, Lin W-C, Lu C-H, Yang S-Y (2022) Relevance of plasma biomarkers to pathologies in Alzheimer’s disease, Parkinson’s disease and frontotemporal dementia. Sci Rep 12:17919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gonzalez MC, Ashton NJ, Gomes BF, Tovar-Rios DA, Blanc F, Karikari TK et al (2022) Association of plasma p-tau181 and p-tau231 concentrations with cognitive decline in patients with probable dementia with lewy bodies. JAMA Neurol 79:32–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chouliaras L, Thomas A, Malpetti M, Donaghy P, Kane J, Mak E et al (2022) Differential levels of plasma biomarkers of neurodegeneration in Lewy body dementia, Alzheimer’s disease, frontotemporal dementia and progressive supranuclear palsy. J Neurol Neurosurg Psychiatry 93:651–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alves G, Brønnick K, Aarsland D, Blennow K, Zetterberg H, Ballard C et al (2010) CSF amyloid-β and tau proteins, and cognitive performance, in early and untreated Parkinson’s Disease: the Norwegian ParkWest study. J Neurol Neurosurg Psychiatry 81(10):1080–1086 [DOI] [PubMed] [Google Scholar]
- 29.Liu C, Cholerton B, Shi M, Ginghina C, Cain KC, Auinger P et al (2015) CSF tau and tau/Aβ42 predict cognitive decline in Parkinson’s disease. Parkinsonism Relat Disord 21:271–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abdelnour C, Ferreira D, Oppedal K, Cavallin L, Bousiges O, Wahlund LO et al (2020) The combined effect of amyloid-β and tau biomarkers on brain atrophy in dementia with Lewy bodies. Neuroimage Clin 27:102333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schrag A, Siddiqui UF, Anastasiou Z, Weintraub D, Schott JM (2017) Clinical variables and biomarkers in prediction of cognitive impairment in patients with newly diagnosed Parkinson’s disease: a cohort study. Lancet Neurol 16:66–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Terrelonge M, Marder KS, Weintraub D, Alcalay RN (2016) CSF β-amyloid 1–42 predicts progression to cognitive impairment in newly diagnosed Parkinson disease. J Mol Neurosci 58:88–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Y, Dou K, Xue L, Li X, Xie A (2022) Neurofilament light as a biomarker for motor decline in Parkinson’s disease. Front Neurosci 16:959261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Compta Y, Martí MJ, Ibarretxe-Bilbao N, Junqué C, Valldeoriola F, Muñoz E et al (2009) Cerebrospinal tau, phospho-tau, and beta-amyloid and neuropsychological functions in Parkinson’s disease. Mov Disord 24:2203–2210 [DOI] [PubMed] [Google Scholar]
- 35.Ferman TJ, Aoki N, Boeve BF, Aakre JA, Kantarci K, Graff-Radford J et al (2020) Subtypes of dementia with Lewy bodies are associated with α-synuclein and tau distribution. Neurology 95:e155–e165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Montine TJ, Shi M, Quinn JF, Peskind ER, Craft S, Ginghina C et al (2010) CSF Aβ42 and tau in Parkinson’s disease with cognitive impairment. Mov Disord 25:2682–2685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hansson O, Janelidze S, Hall S, Magdalinou N, Lees AJ, Andreasson U et al (2017) Blood-based NfL: a biomarker for differential diagnosis of parkinsonian disorder. Neurology 88:930–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mollenhauer B, Dakna M, Kruse N, Galasko D, Foroud T, Zetterberg H et al (2020) Validation of serum neurofilament light chain as a biomarker of Parkinson’s disease progression. Mov Disord 35:1999–2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park Y, Paneque A, Cole PD (2024) Tau, Glial fibrillary acidic protein, and neurofilament light chain as brain protein biomarkers in cerebrospinal fluid and blood for diagnosis of neurobiological diseases. Int J Mol Sci 25(12):6295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dai Y, Bi M, Jiao Q, DuYanJiang XCH (2024) Astrocyte-derived apolipoprotein D is required for neuronal survival in Parkinson’s disease. NPJ Parkinsons Dis 10(1):143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin J, Ou R, Li C, Hou Y, Zhang L, Wei Q et al (2023) Plasma glial fibrillary acidic protein as a biomarker of disease progression in Parkinson’s disease: a prospective cohort study. BMC Med 21(1):420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Batzu L, Rota S, Hye A, Heslegrave A, Trivedi D, Gibson LL et al (2022) Plasma p-tau181, neurofilament light chain and association with cognition in Parkinson’s disease. NPJ Parkinsons Dis 8:154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pagonabarraga J, Pérez-González R, Bejr-Kasem H, Marín-Lahoz J, Horta-Barba A, Martinez-Horta S et al (2022) Dissociable contribution of plasma NfL and p-tau181 to cognitive impairment in Parkinson’s disease. Parkinsonism Relat Disord 105:132–138 [DOI] [PubMed] [Google Scholar]
- 44.Mizutani Y, Ohdake R, Tatebe H, Higashi A, Shima S, Ueda A et al (2024) Associations of Alzheimer’s-related plasma biomarkers with cognitive decline in Parkinson’s disease. J Neurol 270(11):5461–5474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arslan B, Zetterberg H (2023) Neurofilament light chain as neuronal injury marker–what is needed to facilitate implementation in clinical laboratory practice? Clin Chem Lab Med 61:1140–1149 [DOI] [PubMed] [Google Scholar]
- 46.Aamodt WW, Waligorska T, Shen J, Tropea TF, Siderowf A, Weintraub D et al (2021) Neurofilament light chain as a biomarker for cognitive decline in Parkinson disease. Mov Disord 36:2945–2950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bäckström D, Linder J, Jakobson Mo S, Riklund K, Zetterberg H, Blennow K et al (2020) NfL as a biomarker for neurodegeneration and survival in Parkinson disease. Neurology 95:e827–e838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mollenhauer B, Caspell-Garcia CJ, Coffey CS, Taylor P, Singleton A, Shaw LM et al (2019) Longitudinal analyses of cerebrospinal fluid α-Synuclein in prodromal and early Parkinson’s disease. Mov Disord 34:1354–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Holden SK, Finseth T, Sillau SH, Berman BD (2018) Progression of MDS-UPDRS scores over five years in de novo Parkinson disease from the Parkinson’s progression markers initiative cohort. Mov Disord Clin Pract 5:47–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used are available from the corresponding author on reasonable request.