

ABSTRACT
Background
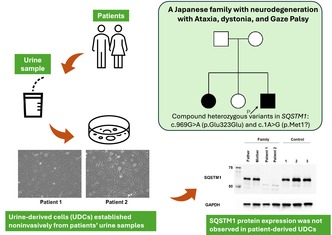
Heterozygous variants of sequestosome‐1 gene (SQSTM1) have been reported in patients with various neurological disorders, whereas biallelic pathogenic variants of SQSTM1 can cause child‐onset and multisystem neurodegeneration, including cerebellar ataxia, dystonia, and vertical gaze palsy (NADGP). Here, we describe two cases of NADGP in a Japanese family.
Methods
We performed clinical and genetic laboratory evaluations of the two patients and their healthy parents.
Results
By whole‐exome sequencing, we identified compound heterozygous variants in SQSTM1(NM_003900.5): c.1A>G p.(Met1?) in the initial codon, and c.969G>A, located at the 3′ end of exon 6, which is novel and seemingly a synonymous but is actually a truncating variant causing aberrant splicing. An SQSTM1 protein expression assay using urine‐derived cells (UDCs) demonstrated that both variants (c.1A>G and c.969G>A) were unable to induce normal splicing of premessenger RNA. Cerebellar ataxia is a characteristic manifestation of this disorder; however, brain magnetic resonance imaging studies have not shown significant cerebellar atrophy. Our patients experienced chorea during adolescence.
Conclusions
Only a few reports have highlighted the presence of chorea; however, our findings suggest that NADGP should be considered as a differential diagnosis of hereditary chorea. This study also demonstrates the utility of UDCs, obtained using noninvasive approaches, in functionally analyzing genetic diseases.
Keywords: GC‐AT intron, NADGP, SQSTM1, UDC, urine‐derived cell
In Japanese siblings with multisystem neurodegeneration, including cerebellar ataxia, dystonia, and vertical gaze palsy, we identified compound heterozygous variants in SQSTM1(NM_003900.5): c.1A>G p.(Met1?) in the initial codon, and c.969G>A. An SQSTM1 protein expression assay using urine‐derived cells (UDCs), which were obtained using noninvasive approaches, demonstrated that both variants (c.1A>G and c.969G>A) were unable to induce normal splicing of premessenger RNA.
1. Introduction
Sequestosome‐1 (SQSTM1, also known as p62) is a stress‐inducible scaffolding protein that plays an essential role in multiple cellular processes, including selective autophagy, apoptosis, and cell survival (Lamark, Svenning, and Johansen 2017). Heterozygous variants of the SQSTM1‐encoded gene (SQSTM1) have been reported in patients with Paget disease of the bone (Laurin et al. 2002), amyotrophic lateral sclerosis, and/or frontotemporal dementia (Hirano et al. 2013), and distal myopathy with rimmed vacuoles (Bucelli et al. 2015). In 2016, homozygous variants of SQSTM1 were initially described as the causative factor for another clinical phenotype, child‐onset multisystem neurodegeneration with ataxia, dystonia, and vertical gaze palsy (NADGP; MIM: 617145) (Haack et al. 2016). Most patients with NADGP have homozygous null variants of SQSTM1, and the transmission pattern is consistent with autosomal recessive inheritance. This suggests that the loss of SQSTM1 function results in the pathogenesis of this disease.
Here, we report on a Japanese family carrying a novel synonymous variant in SQSTM1 (NM_003900.5):c.969G>A,p.Glu323Glu and a previously reported missense variant in the initial codon of SQSTM1 (c.1A>G,p.Met1?), revealed by whole‐exome sequencing (Chacaltana‐Vinas et al. 2024). To evaluate the functional effects of these variants, we investigated the SQSTM1 protein expression levels, which were analyzed using both patient‐ and control‐oriented cell lines established from urine‐derived cells (UDCs).
2. Materials and Methods
2.1. Case Presentation
Patient 1 (Figure 1A, II‐3), the youngest of three siblings, was a 16‐year‐old boy born to a healthy Japanese father and Filipino mother. His motor and intellectual development fell within a typical range, and his neonatal period passed without any noteworthy events. At 3 years old, he first noticed difficulty in understanding language and clumsiness in his upper extremities. At 9 years old, he underwent extensive medical examinations. An intelligence assessment using the Wechsler Intelligence Scale for Children (first edition) revealed borderline intellectual disability with a full‐scale IQ of 77. Neurological examinations revealed vertical gaze palsy and ataxia. He had choreic involuntary movements involving the trunk and all four limbs. Choreic movements were more common when accompanied by intentional movements, and these movements induced frequent falls while walking. The results of routine blood examinations, cerebrospinal fluid, and nerve conduction studies were all normal. At age 16, his condition was reevaluated in our hospital. His overall clinical course was protracted. He showed total vertical and partial horizontal gaze palsy; however, there were no abnormal findings in the cranial nerves. Both limb and truncal ataxia were evident. The tendon reflexes were normal, and Babinski and Chaddock signs were negative on both sides. Brain magnetic resonance imaging (MRI) results were all normal (Figure 2A–D).
FIGURE 1.
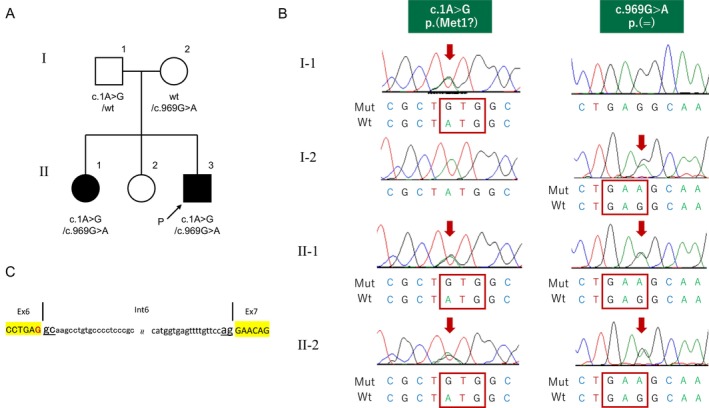
Clinical and genetic features of a family with sequestosome‐1 gene (SQSTM1) variants. (A) Pedigree of the family. (B) Direct nucleotide sequencing of PCR‐amplified DNA of the SQSTM1 gene. The red arrow denotes the substituted bases. (C) Schematic diagram of the DNA sequences of the wild‐type SQSTM1 alleles around c.G969 [indicated by the color red]. ■/●, affected individuals; →, proband; MRI, magnetic resonance imaging.
FIGURE 2.
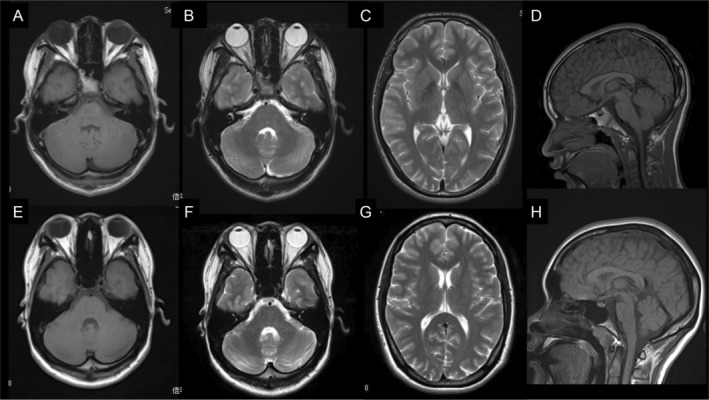
Brain MRI findings of Patient 1 and Patient 2. Brain MRI revealed normal results pertaining to the cerebellar (A–C, E–G) and cerebral hemispheres (D, H). (A–D): Patient 1. (E–H): Patient 2. MRI, magnetic resonance imaging.
Patient 2 (Figure 1A, II‐1) was a 22‐year‐old female. She showed normal development up to the age of 13 years. After which, she began to experience an unsteady gait and chorea. She was referred to our hospital at 18 years of age, mild intellectual disturbance, gaze palsy, gaze directional nystagmus, choric gait disturbance, and cerebellar ataxia were noted. At age 15, brain MRI, nerve conduction study, and ophthalmological examination results were not noteworthy. By the age of 22, there was no worsening of her clinical symptoms or brain MRI results (Figure 2E–H).
2.2. Genetic Analysis Using Whole‐Exome Sequencing
Whole‐exome sequencing was performed on the samples collected from Patient 2. Sanger sequencing was used to confirm the SQSTM1 variants in Patients 1 and 2 and their parents.
2.3. SQSTM1 Protein Expression Assay Using Urine‐Derived Cells
Primary cultures of UDCs were obtained from the families of the patients and three healthy controls as previously described (Takizawa, Sato, and Aoki 2020). Briefly, whole urine was centrifuged at room temperature and the cell pellets were resuspended in primary medium. Cell suspensions were seeded onto gelatin‐coated plates and cultured at 37°C in a humid atmosphere (5% CO2). On Day 4, the medium was replaced with growth medium and changed every other day. UDC colonies appeared within 1 week. Once the UDC cultures had become 80%–90% confluent, all cells were split using 0.25% trypsin–EDTA and seeded onto new gelatin‐coated dishes (Passage 1).
Total protein was extracted from the third passage of UDC using radioimmunoprecipitation assay (RIPA) buffer (Thermo Fisher Scientific, Waltham, MA, USA) containing protease inhibitors. Cell lysates were sonicated on ice and centrifuged at 14,000 × g for 15 min at 4°C. The supernatant was collected, and the protein concentration was determined using a bicinchoninic acid (BCA) protein assay kit. Protein samples were mixed with 2× Laemmli sample buffer (Bio‐Rad) and boiled at 70°C for 10 min. Samples containing 3‐μg total protein were separated using the 7% Mini‐Protean TGX gel (Bio‐Rad) and transferred to a polyvinylidene fluoride (PVDF) membrane. Membranes were sequentially incubated with primary and secondary antibodies using the iBind Flex Western Device (Thermo Fisher Scientific). Rabbit anti‐SQSTM1 (1:500; PM045; BML) and mouse anti‐glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH; 1:2000; MAB374; EMD Millipore) were used as primary antibodies. Goat anti‐rabbit IgG (H + L) horseradish peroxidase (HRP) conjugate (1:2000; 1706515; Bio‐Rad) and goat anti‐mouse IgG (H + L) HRP conjugate (1:2000; 1706516; Bio‐Rad) were used as secondary antibodies. Protein expression was detected using the ECL Prime Western Blotting Detection Reagent (Cytiva) and ChemiDoc MP Imaging System (Bio‐Rad).
2.4. Protocol Approval
This study was approved by the Ethics Committee of the Shinshu University School of Medicine (2023‐159). Written informed consent was obtained from all the patients who underwent genetic evaluation.
3. Results
3.1. Genetic Analysis Using Whole‐Exome Sequencing
We identified two heterozygous variants of SQSTM1 in Patient 2, a previously reported missense variant (Chacaltana‐Vinas et al. 2024) c.1A>G (rs1302810798; Figure 1B), and a novel synonymous variant (c.969G>A; Figure 1B,C). The former variant likely affects the start codon of SQSTM1 [p.(Met1?)], leading to the failure of SQSTM1 expression (Chacaltana‐Vinas et al. 2024; Haack et al. 2016). The latter variant appeared to be synonymous but was suspected to cause aberrant splicing since it was located at the 3′ end of exon 6 (Figure 1C). Both nucleotide changes were confirmed by Sanger sequencing in Patient 1. Of the two nucleotide changes, one was inherited from the father and the other from the mother, confirming that Patient 1 had these two variants as a compound heterozygote (Figure 1B). No other relevant variants were detected in the coding regions of other genes, including chorea‐ or ataxia‐related genes. The c.969G>A variant has not been described in databases pertaining to healthy individuals, including the Genome Aggregation Database (https://gnomad.broadinstitute.org/).
3.2. SQSTM1 Protein Expression Analysis in Patients, Their Parents, and Normal Controls
We cultured UDCs from patients, their parents, and three healthy male controls and assessed SQSTM1 expression (Figure 3). All cell lines had good proliferative potential and morphologically similar cell populations. Total protein was extracted from the third passage of cells and immunoblotting was performed. Immunoblotting showed that the SQSTM1 was absent in the UDCs of Patients 1 and 2 (Figure 4). These findings demonstrated that the initial codon variant c.1A>G and the possible splice variant c.969G>A were unable to induce normal protein expression. The c.G969 is a base located at the −1 position of the splicing donor site, and intron 6 is a splice variant of the GC‐AT intron (Figure 1C). Similar to the canonical 5′ splicing site with the GT dinucleotide at the first two bases, the GC‐AG splice sites are processed by the standard U2‐type spliceosome (Thanaraj 2001). When lacking the usual GT dinucleotide, the consensus sequence around the GC donor site, including G at position −1, is needed to maximize the interaction between the donor site and U1 snRNA. Although the consequence of this variant at RNA level is unknown, c.969G>A possibly leads to the aberrant splicing, such as inclusion of a whole or at least a part of intron 6, and results in frame‐shift mutation [p. (Glu324fs)]. SpliceAI‐lookup (https://spliceailookup.broadinstitute.org/) supports our hypothesis that c.969G>A may cause canonical donor loss.
FIGURE 3.
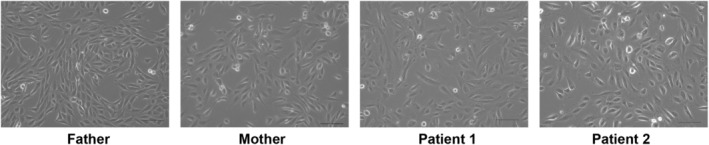
Phase contrast microscopy image of urine‐derived cells (UDCs) during second passage. The images were taken on the second day after passage. Scale bar denotes 200 μm.
FIGURE 4.
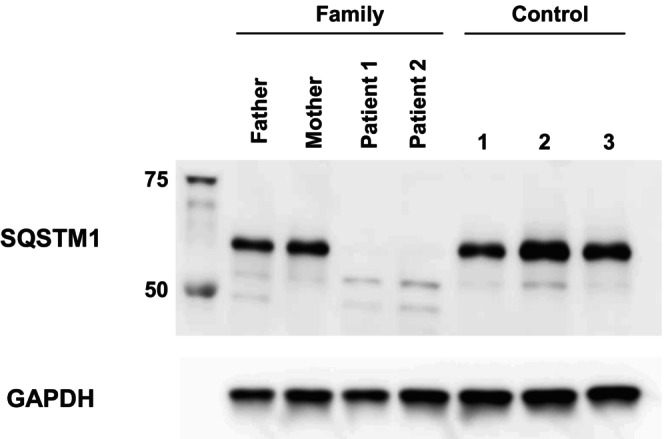
SQSTM1 protein expression was not observed in patient‐derived UDCs. Immunoblotting of SQSTM1 and GAPDH proteins extracted during third passage. Anti‐GAPDH antibody was used as a loading control. SQSTM1, sequestosome‐1; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; UDCs, urine‐derived cells.
4. Discussion
In this study, we report two cases of NADGP in a Japanese family that was confirmed to possess compound heterozygous variants p. [(Met1?)];[(Glu324fs)] in the SQSTM1. To date, 31 patients with genetically confirmed NADGP have shown childhood onset with various degrees of neurological manifestations (Chacaltana‐Vinas et al. 2024; Haack et al. 2016; Kilic et al. 2021; Muto et al. 2018; Vedartham et al. 2019; Zahir et al. 2017; Zúñiga‐Ramírez et al. 2019), including ataxia (93.8%), cognitive decline (96.9%), gaze palsy (90.6%), and dystonia (53.1%) (Table S1). Although our patients exhibited prominent chorea during adolescence, only a small proportion (28.1%) of reported patients with NADGP have involuntary movements. Our patients displayed the majority of NADGP neurological manifestations during their 16 (Patient 1) or 22 (Patient 2) years of follow‐up, but their overall clinical course was protracted. Although multisystem neurodegeneration is suspected to be the pathogenesis of NADGP, neuroradiological studies have shown that cerebral (0/19) and cerebellar (6/19) atrophies are less frequently reported (Table S1). The long‐term clinical course of patients with NADGP has yet to be elucidated.
It was suspected that the c.1A>G may cause the loss of start codon of SQSTM1[p.(1Met?)], and c.969G>A may lead to the aberrant splicing and frame‐shift mutation in SQSTM1[p.(Glu324fs)]. In this study, we proved that both variants truly had loss‐of‐function effect by the functional analysis of UDCs collected from the patients and their healthy parents. In recent years, UDC subpopulations possessing stem cell characteristics (high proliferative capacity, pluripotency, and immunomodulatory potential) have attracted attention as novel sources for genetic disease research (Falzarano and Ferlini 2019). In contrast to fibroblasts and myogenic cells, UDC lines can be established noninvasively from urine samples. Thus, UDCs are also a valuable cell source for functional analysis of hereditary diseases. A recent report on global transcriptomics using RNA‐seq of native UDCs showed that 571 of 610 genes known to be involved in neuromuscular diseases are expressed in UDCs (Falzarano et al. 2021), and have already been applied in the functional analysis, including hereditary epidermolysis bullosa (Schosserer et al. 2015), spinal muscular atrophy (Zhang et al. 2017), and Fabry disease (Slaats et al. 2018). Based on the data in this report, SQSTM1 was found to be highly expressed in UDCs; thus, we used UDCs to analyze the pathogenic effect of the two identified variants. To assess whether a seemingly synonymous variant is pathological, it is crucial to examine the transcription and translation of this variant in patient cells. By analyzing the SQSTM1 expressed in UDCs, we demonstrated that both variants in the parental generation induced protein deletion in the offspring and that this loss of SQSTM1 function is likely involved in disease pathogenesis.
Our study identified a novel synonymous SQSTM1 variant likely responsible for the NADGP symptoms experienced by two siblings, and our patients experienced chorea from adolescence; suggesting that NADGP should be considered as a differential diagnosis for hereditary chorea. This study also demonstrates the utility of functional analysis using UDCs obtained by a noninvasive approach. These methods may assist in evaluating the genetic variant effect on genetic diseases, especially those caused by the loss of protein function.
Author Contributions
S.M. and M.S. contributed equally to this article. K.N. and Y.S. conception and design of the study and draft writing. K.H., S.M., Y.I., T.K., and N.M. draft writing and technical support. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Table S1.
Acknowledgments
The authors thank Ms. E. Kise, Ms. T. Kojima, and Ms. A. Sakyu for genetic counseling.
Funding: This study was supported in part by the program for dementia research and development, AMED, under the grant numbers (JP23dk0207066h0001 and 24dk0207066h0002 [Y. Sekijima], and JP24ek0109674, JP24ek0109760, JP24ek0109617, JP24ek0109648, and JP24ek0109677 [N. Matsumoto]); JSPS KAKENHI under the grant numbers JP24K10637 (K. Nakamura), JP23K27520 (S. Miyatake), JP22K15646 (K. Hamanaka), and JP24K02230 (N. Matsumoto); and grants‐in‐aid from the Research Committee on Ataxia and Health Labour Sciences Research Grant from Ministry of Health, Labour and Welfare, under the grant number JPMH23FC1010 (K. Nakamura).
Shinji Masuko and Mitsuto Sato contributed equally to this work.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
References
- Bucelli, R. C. , Arhzaouy K., Pestronk A., et al. 2015. “ SQSTM1 Splice Site Mutation in Distal Myopathy With Rimmed Vacuoles.” Neurology 85, no. 8: 665–674. 10.1212/wnl.0000000000001864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacaltana‐Vinas, C. , Ramirez‐Pajares P., Manrique‐Palomino A., et al. 2024. “A Novel Variant in SQSTM1 Gene Causing Neurodegeneration With Ataxia, Dystonia, and Gaze Palsy in a Peruvian Family.” Movement Disorders Clinical Practice 11, no. 6: 746–748. 10.1002/mdc3.14025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falzarano, M. S. , and Ferlini A.. 2019. “Urinary Stem Cells as Tools to Study Genetic Disease: Overview of the Literature.” Journal of Clinical Medicine 8, no. 5: 627. 10.3390/jcm8050627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falzarano, M. S. , Rossi R., Grilli A., et al. 2021. “Urine‐Derived Stem Cells Express 571 Neuromuscular Disorders Causing Genes, Making Them a Potential In Vitro Model for Rare Genetic Diseases.” Frontiers in Physiology 12: 716471. 10.3389/fphys.2021.716471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haack, T. B. , Ignatius E., Calvo‐Garrido J., et al. 2016. “Absence of the Autophagy Adaptor SQSTM1/p62 Causes Childhood‐Onset Neurodegeneration With Ataxia, Dystonia, and Gaze Palsy.” American Journal of Human Genetics 99, no. 3: 735–743. 10.1016/j.ajhg.2016.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano, M. , Nakamura Y., Saigoh K., et al. 2013. “Mutations in the Gene Encoding p62 in Japanese Patients With Amyotrophic Lateral Sclerosis.” Neurology 80, no. 5: 458–463. 10.1212/wnl.0b013e31827f0fe5. [DOI] [PubMed] [Google Scholar]
- Kilic, M. A. , Kipoglu O., Coskun O., et al. 2021. “Homozygous SQSTM1 Nonsense Variant Identified in a Patient With Brainstem Involvement.” Brain Development 43, no. 10: 1039–1043. 10.1016/j.braindev.2021.06.001. [DOI] [PubMed] [Google Scholar]
- Lamark, T. , Svenning S., and Johansen T.. 2017. “Regulation of Selective Autophagy: The p62/SQSTM1 Paradigm.” Essays in Biochemistry 61, no. 6: 609–624. 10.1042/EBC20170035. [DOI] [PubMed] [Google Scholar]
- Laurin, N. , Brown J. P., Morissette J., and Raymond V.. 2002. “Recurrent Mutation of the Gene Encoding Sequestosome 1 (SQSTM1/p62) in Paget Disease of Bone.” American Journal of Human Genetics 70, no. 6: 1582–1588. 10.1086/340731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto, V. , Flex E., Kupchinsky Z., et al. 2018. “Biallelic SQSTM1 Mutations in Early‐Onset, Variably Progressive Neurodegeneration.” Neurology 91, no. 4: e319–e330. 10.1212/WNL.0000000000005869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schosserer, M. , Reynoso R., Wally V., et al. 2015. “Urine Is a Novel Source of Autologous Mesenchymal Stem Cells for Patients With Epidermolysis Bullosa.” BMC Research Notes 8: 767. 10.1186/s13104-015-1686-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaats, G. G. , Braun F., Hoehne M., et al. 2018. “Urine‐Derived Cells: A Promising Diagnostic Tool in Fabry Disease Patients.” Scientific Reports 8, no. 1: 11042. 10.1038/s41598-018-29240-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa, H. , Sato M., and Aoki Y.. 2020. “Exon Skipping in Directly Reprogrammed Myotubes Obtained From Human Urine‐Derived Cells.” Journal of Visualized Experiments 159: 60840. 10.3791/60840. [DOI] [PubMed] [Google Scholar]
- Thanaraj, T. A. 2001. “Human GC‐AG Alternative Intron Isoforms With Weak Donor Sites Show Enhanced Consensus at Acceptor Exon Positions.” Nucleic Acids Research 29, no. 12: 2581–2593. 10.1093/nar/29.12.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedartham, V. , Sundaram S., Nair S. S., Ganapathy A., Mannan A., and Menon R.. 2019. “Homozygous Sequestosome 1.” Ophthalmic Genetics 40, no. 4: 376–379. 10.1080/13816810.2019.1666414. [DOI] [PubMed] [Google Scholar]
- Zahir, F. R. , Mwenifumbo J. C., Chun H.‐J. E., et al. 2017. “Comprehensive Whole Genome Sequence Analyses Yields Novel Genetic and Structural Insights for Intellectual Disability.” BMC Genomics 18, no. 1: 403. 10.1186/s12864-017-3671-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q. J. , Lin X., Li J. J., et al. 2017. “Application of Urine Cells in Drug Intervention for Spinal Muscular Atrophy.” Experimental and Therapeutic Medicine 14, no. 3: 1993–1998. 10.3892/etm.2017.4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zúñiga‐Ramírez, C. , de Oliveira L. M., Kramis‐Hollands M., et al. 2019. “Beyond Dystonia and Ataxia: Expanding the Phenotype of SQSTM1 Mutations.” Parkinsonism & Related Disorders 62: 192–195. 10.1016/j.parkreldis.2018.12.031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.