

Abstract
DESTINY-CRC01 (NCT03384940) was a multicentre, open-label, phase 2 study that investigated the safety and efficacy of trastuzumab deruxtecan (T-DXd) in patients with human epidermal growth factor receptor 2 (HER2)-expressing metastatic colorectal cancer (CRC). The present exploratory biomarker analysis aims to investigate relationships between biomarkers and clinical outcomes in patients with HER2-positive (immunohistochemistry [IHC] 3+ or IHC 2+ and in situ hybridization [ISH] positive) Cohort A (N = 53) of DESTINY-CRC01. Higher levels of HER2 biomarkers in baseline tissue and liquid biopsies, including HER2 status (IHC/ISH), HER2/CEP17 ratio, HER2 ISH signals, HER2 H-score, plasma HER2 (ERBB2) amplification status, HER2 adjusted plasma copy number, and HER2 extracellular domain correlate with antitumor activity (indicated by objective response rate, progression-free survival, and overall survival) of T-DXd. Baseline circulating tumor DNA (ctDNA) analysis suggests antitumor activity of T-DXd in patients who had baseline activating RAS, PIK3CA, or HER2 mutations detected in ctDNA.
Subject terms: Predictive markers, Targeted therapies, Cancer
The phase 2 trial, DESTINYCRC01, previously demonstrated durable activity of trastuzumab deruxtecan (T-DXd) in patients with HER2-positive metastatic colorectal cancer refractory to standard treatment. Here, the authors report the exploratory analysis of the DESTINY-CRC01 trial investigating markers of response or resistance to T-DXd.
Introduction
Colorectal cancer (CRC) is the third most commonly diagnosed cancer worldwide and the second most common contributor to global cancer mortality, representing approximately 10% of all cancer cases and deaths1. Amplification of the HER2 gene (ERBB2) occurs in approximately 5% of patients with metastatic CRC (mCRC) overall2,3, 5% of those with RAS and BRAF wild-type tumors2,4–6, and 17% of those with KRAS mutations2,3.
Recommended first-line and second-line treatment for mCRC is immunotherapy for microsatellite instability high (MSI-H) tumors and chemotherapy in combination with either anti-vascular endothelial growth factor (VEGF) therapy or anti-epidermal growth factor receptor (EGFR) therapy for microsatellite stable (MSS) tumors7. Third-line or later (salvage therapy) options include regorafenib and trifluridine/tipiracil8; however, these treatments have limited clinical benefit after fluoropyrimidine therapy9. Targeted therapies consisting of combined BRAF and EGFR inhibition is the standard second-line option for patients with BRAFV600 mutant tumors7. Human epidermal growth factor receptor 2 (HER2) overexpression and amplification in mCRC has been associated with resistance to anti-EGFR blockade and has been shown to convey worse prognosis compared to HER2-negative tumors10,11.
HER2-targeted therapy has improved outcomes for patients with metastatic breast cancer (mBC) and gastric cancer; however, trastuzumab alone is not effective in HER2-amplified mCRC12. In contrast, dual HER2 inhibition has shown antitumor activity in patients with pretreated HER2 overexpressing or HER2-amplified mCRC who have not received previous HER2-targeted therapy. The phase 2 HERACLES-A trial of trastuzumab plus lapatinib in patients with HER2-positive KRAS exon 2 wild-type mCRC reported an overall objective response rate (ORR) of 28% (95% CI, 14–47%)13, the combination of pertuzumab and trastuzumab in patients with HER2-amplified mCRC in the phase 2a MyPathway study resulted in an ORR of 32% (95% CI, 20–45%)14, and the combination of trastuzumab and tucatinib in the phase 2 MOUNTAINEER study resulted in an ORR of 38.1% (95% CI, 27.7–49.3%)15. However, the HERACLES-B trial, which investigated the HER2 antibody-drug conjugate-based strategy of pertuzumab and trastuzumab emtansine in this patient population, reported an ORR of 9.7% (95% CI, 0–28%)16. Dual HER2 blockade is therefore considered a treatment option for patients with HER2-positive mCRC, especially those with RAS wild-type tumors17.
Trastuzumab deruxtecan (T-DXd) is an antibody-drug conjugate composed of a humanized immunoglobulin G1 monoclonal antibody specifically targeting HER2, a tetrapeptide-based cleavable linker, and a potent topoisomerase I inhibitor payload; the antibody has the same amino acid sequence as trastuzumab18,19. T-DXd is approved in various countries for the treatment of metastatic HER2-positive breast and gastric cancer, HER2-low breast cancer and non–small cell lung cancer with activating HER2 mutations20–22. The open-label, phase 2, DESTINY-CRC01 trial in patients with HER2-expressing mCRC refractory to standard treatment demonstrated promising and durable activity of T-DXd, with an overall confirmed ORR of 45.3% (95% CI, 31.6–59.6%) in patients with HER2-positive tumors and an ORR of 43.8% (95% CI, 19.8–70.1%) among patients who had previously received HER2-targeted therapy23.
Here, we show that known biomarkers in CRC and biomarkers of anti-HER2 therapy and the mechanism of action of T-DXd are correlated with antitumor activity of T-DXd in exploratory biomarker analysis of DESTINY-CRC01. We identify intrinsic and acquired markers of response or resistance to T-DXd and assess clinical outcomes in patients with mCRC who have common oncogenic driver mutations.
Results
Datasets
Overall, 86 patients were enrolled in Cohort A (53 patients; immunohistochemistry [IHC] 3+ or IHC 2+/in situ hybridization [ISH+]), Cohort B (15 patients; IHC 2+/ISH−), or Cohort C (18 patients; IHC 1+) between February 23, 2018, and December 28, 202023. The evaluable circulating tumor DNA (ctDNA) dataset comprised 188 samples, including 132 samples from Cohort A (52 at cycle 1 day 1; 41 at cycle 4 day 1; 39 at the end of treatment), 26 samples from Cohort B (15 at cycle 1 day 1; 11 at the end of treatment), and 30 samples from Cohort C (18 at cycle 1 day 1; 12 at the end of treatment). The evaluable baseline HER2 extracellular domain (HER2ECD) dataset included a total of 74 samples (49 from Cohort A; 7 from Cohort B; 18 from Cohort C), the evaluable HER2 IHC dataset included 86 patients (53 from Cohort A; 15 from Cohort B; 18 from Cohort C) (Fig. 1), and the evaluable HER2 ISH dataset included 85 patients (52 from Cohort A; 15 from Cohort B; 18 from Cohort C). Baseline characteristics of the patients in each biomarker dataset (Supplementary Table S1) were similar to the baseline characteristics of each cohort overall23.
Fig. 1. Biomarker analysis sets.
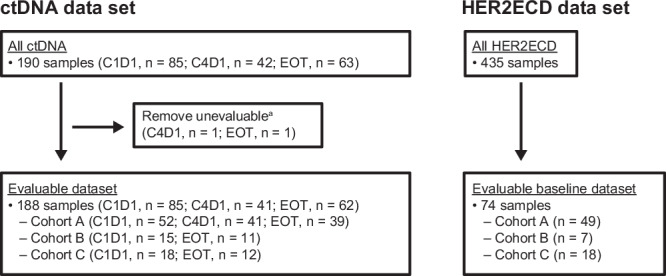
aNo somatic mutation detected. C, cycle; ctDNA, circulating tumor DNA; D, day; EOT, end of treatment; HER2ECD, human epidermal growth factor receptor 2 extracellular domain.
Baseline genomic landscape from ctDNA in HER2-expressing mCRC
Tumor responses measured as the best percentage change in the sum of diameters according to baseline ctDNA genomic landscape in Cohort A (HER2-positive; IHC 3+ or IHC 2+ and ISH+) are shown in Fig. 2. The genomic landscape analysis of ctDNA in all cohorts (Cohort A, Cohort B [IHC 2+ and ISH−], and Cohort C [IHC 1+]) is shown in Supplementary Fig. S1. In Cohort A, no patients had MSI-H or baseline BRAF V600E mutations. Baseline HER2 plasma amplification was detected in 47 of 52 patients who had evaluable ctDNA data. Although patients had to have RAS wild-type tumors to be included in DESTINY-CRC01, according to the eligibility criteria, activating RAS mutations were detected in ctDNA samples from 6 patients in Cohort A. In addition, activating HER2 mutations and activating PIK3CA mutations were detected in ctDNA samples from 8 and 6 patients, respectively. Thirteen patients had a high blood tumor mutational burden (bTMB) status ( ≥ 20 mut/Mb). Most patients had primary tumors located on the left side and had moderately differentiated histology (Fig. 2).
Fig. 2. Antitumor activity of T-DXd as best percentage change from baseline in sum of tumor diameters according to ctDNA genomic landscape at baseline.

Waterfall plot shows the best percentage change in the sum of diameters from baseline, amplifications and mutations of CRC-related genes, bTMB status, histological grade, and primary tumor site. Amplification type was reported from Guardant Health. HER2 gain-of-function mutations were selected based on OncoKB; RAS: mutation at codon 12, 13, 59, 61, 117, or 146. PIK3CA mutation was selected according to previous publication34; PTEN: loss-of-function mutations were selected based on OncoKB. bTMB of ≥ 20 mut/Mb was considered high based on the Guardant Health report. Of 4 patients with evaluable ctDNA data and no data available for best percentage change in the sum of diameters, 2 had focal HER2 plasma amplification, 2 had aneuploidy HER2 plasma amplification, none had HER2 mutation, 1 had RAS mutation, 2 had PIK3CA mutation, 1 had PTEN mutation/loss, 2 had aneuploidy MET amplification, 3 had bTMB ≥ 20 mut/Mb, and 1 had bTMB < 20 mut/Mb. amp, amplification; bTMB, blood tumor mutational burden; ctDNA, circulating tumor DNA; HER2, human epidermal growth factor receptor 2; IHC, immunohistochemistry; ISH, in situ hybridization; mut, mutation.
Relationship between baseline tumor tissue HER2 status and plasma HER2 amplification/adjusted plasma copy number or HER2ECD
Across Cohorts A, B and C, plasma HER2 amplification was detectable for 58 of 86 patients and, of those, 36 were focal and 22 were aneuploidy plasma amplifications. Among 40 patients with HER2 IHC 3+ tumors, 33 (82.5%) had plasma HER2 focal amplification and 5 (12.5%) had plasma HER2 aneuploidy amplification. Among 13 patients with HER2 IHC 2+/ISH+ tumors, 3 (23.1%) had plasma HER2 focal amplification and 6 (46.2%) had plasma HER2 aneuploidy amplification. Absence of plasma HER2 focal amplification was observed in 33 patients who had HER2-low tumors (IHC 2+/ISH− or IHC 1+). Plasma HER2 aneuploidy amplification was detected in 7 of 15 patients with HER2 IHC 2+/ISH− tumors and in 4 of 18 patients with HER2 IHC 1+ tumors. Adjusted plasma copy number (ApCN) according to HER2 IHC/ISH status is shown in Supplementary Fig. S2. A correlation was found between HER2 plasma copy number and both HER2/CEP17 ratio (Spearman r = 0.60) and ApCN (Spearman r = 0.81) (Supplementary Fig. S3). A higher level of plasma HER2ECD was observed in patients with HER2 IHC 3+ tumors (Supplementary Fig. S4).
Association between baseline HER2-related biomarkers and clinical outcomes
The HER2-related biomarkers investigated included HER2 status (IHC/ISH), H-score, ctDNA (plasma HER2 amplification), and HER2ECD. Cutoffs for HER2 biomarker status were defined according to the maximum value of the Youden index (Supplementary Fig. S5). Clinical outcomes (ORR, progression-free survival [PFS], overall survival [OS]) in patients with HER2 levels above or below the cutoffs in baseline tissue samples (IHC/ISH, HER2/CEP17 ratio, average number of HER2 ISH signals, and H-score) and liquid biopsy samples (plasma HER2 amplification status, HER2 ApCN, and HER2ECD) are shown in Figs. 3 and 4. ORR was higher and median PFS (mPFS) and median OS (mOS) were longer in patients with HER2 IHC 3+ versus IHC 2+/ISH+ status and with higher HER2 H-score with respect to an exploratory cutoff (all P < 0.05). For HER2 ISH assessment, ORR was higher and mPFS and mOS were longer in patients with higher HER2 ISH signal or higher HER2/CEP17 ratio with respect to an exploratory cutoff (all P < 0.05). For plasma ctDNA assessment, ORR was higher and mPFS and mOS were longer in patients with focal amplification versus those with aneuploidy/no amplification detected and in patients with higher HER2 ApCN with respect to an exploratory cutoff (all P < 0.01). ORR was higher in patients with higher HER2ECD with respect to an exploratory cutoff (P = 0.023). Results were consistent when the median value was used as the cutoff (Supplementary Fig. S6).
Fig. 3. Antitumor activity of T-DXd according to baseline HER2 biomarker status.

Exploratory cutoff values for each HER2 biomarker were defined as the maximum value of the Youden index for ORR. Vertical red dashed line shows the ORR of 45.3% in the overall population for Cohort A. P values are based on two-sided Fisher’s exact test for ORR and those based on two-sided log-rank test for PFS and OS are shown, without adjustment for multiple comparisons. Error bars represent the 95% CI. The exact P values for HER2 H-score for OS, HER2 ISH signal for PFS, HER2 ApCN for PFS, and HER2 ApCN for OS were 0.000175, 0.000394, 0.0000168, and 0.0000991, respectively. Amp, amplification; ApCN, adjusted plasma copy number; HER2, human epidermal growth factor receptor 2; HER2ECD, human epidermal growth factor receptor 2 extracellular domain; IHC, immunohistochemistry; ISH, in situ hybridization; NA, not applicable; ND, not determined; ORR, objective response rate; OS, overall survival; PFS, progression-free survival.
Fig. 4. Probability of PFS according to baseline HER2 biomarker status.

The probability of PFS is shown according to HER2 IHC status (panel A) HER2 biomarkers based on exploratory cutoff values (panels B, C, D, F, G), and type of HER2 gene amplification (panel E). aExploratory cutoff values (panels B, C, D, F, G) were determined using receiver operating characteristics analysis: H-score cutoff = 240; HER2/CEP17 ratio cutoff = 6.30; HER2 ISH signal cutoff = 11.25; HER2 ApCN cutoff = 30.99; serum HER2ECD cutoff = 23.5 ng/mL. bAmplification type (panel E) was reported from Guardant Health. Amp, amplification; ApCN, adjusted plasma copy number; HER2, human epidermal growth factor receptor 2; HER2ECD, human epidermal growth factor receptor 2 extracellular domain; IHC, immunohistochemistry; ISH, in situ hybridization; mPFS, median progression-free survival; NA, not applicable; ND, not determined; PFS, progression-free survival.
Association of clinical outcomes with RAS, PIK3CA, and HER2 mutation status and bTMB in patients with HER2-positive mCRC
Antitumor activity of T-DXd was observed in patients with or without baseline activating RAS, PIK3CA, or HER2 mutations in ctDNA and regardless of bTMB levels (Figs. 5 and 6; Supplementary Table S2). The ORR, mPFS, and mOS in the 6 patients with activating RAS mutations were 33.3%, 4.1 months, and 11.6 months, respectively, whereas in patients without activating RAS mutations the values were 47.8%, 7.6, and 17.3, respectively (Figs. 5 and 6). The ORR, mPFS, and mOS in the 8 patients with activating HER2 mutations were 62.5%, 5.5 months, and 15.9 months, respectively, whereas in patients without activating HER2 mutations, the values were 43.2%, 8.3, and 15.5, respectively (Figs. 5 and 6). The ORR, mPFS, and mOS in the 6 patients with activating PIK3CA mutations were 33.3%, 4.1 months, and 11.6 months, respectively, whereas in patients without activating PIK3CA mutations, the values were 47.8%, 7.3, and 17.3, respectively (Figs. 5 and 6). For the 13 patients with high bTMB status ( ≥ 20 mut/Mb), ORR, mPFS, and mOS were 23.1%, 2.1 months, and 7.1 months, respectively, whereas in patients with low bTMB, the values were 53.8%, 7.6, and 19.9, respectively (Fig. 5).
Fig. 5. Antitumor activity of T-DXd according to mutation in PIK3CA, RAS, and HER2 in ctDNA at baseline and bTMB.

Vertical red dashed line shows the ORR of 45.3% in the overall population for Cohort A. P values are based on two-sided Fisher’s exact test for ORR and those based on two-sided log-rank test for PFS and OS are shown, without adjustment for multiple comparisons. Error bars represent the 95% CI. PIK3CA variants were determined according to published data34. NRAS and KRAS variants were determined as mutation at codon 12, 13, 59, 61, 117, or 146. bTMB ≥ 20 mut/Mb was considered high according to the Guardant Health report. bTMB, blood tumor mutational burden; Mut, mutant; NA, not applicable; ORR, objective response rate; OS, overall survival; PFS, progression-free survival; WT, wild type.
Fig. 6. Probability of PFS according to mutation in PIK3CA, RAS, and HER2 in ctDNA at baseline.

The probability of PFS is shown according to mutation in PIK3CA (panel A), RAS (panel B), or HER2 (panel C) genes. PIK3CA variants were determined according to published data34. NRAS and KRAS variants were determined as mutation at codon 12, 13, 59, 61, 117, or 146. mPFS, median progression-free survival; Mut, mutant; NA, not applicable; PFS, progression-free survival; WT, wild type.
Variant allele fraction (VAF) changes (representing ctDNA clearance) in patients with RAS or PIK3CA activating mutations during T-DXd treatment are shown in Supplementary Fig. S7. RAS and PIK3CA mutation VAFs tended to decrease at cycle 4, day 1, in patients who also had a decreasing sum of target lesion diameter during T-DXd treatment.
Plasma HER2 amplification status at disease progression
Analysis of paired ctDNA samples was performed in 29 patients in Cohort A who discontinued T-DXd treatment because of disease progression and had detectable ctDNA at cycle 1, day 1, and at the end of treatment. Plasma HER2 amplification (focal or aneuploidy) was detected in 27 patients and 2 patients had no detectable plasma HER2 amplification at baseline. Nineteen of the 27 patients (70.4%) with baseline plasma HER2 amplification had plasma HER2 amplification at disease progression and 8 patients (29.6%) did not have detectable plasma HER2 amplification at disease progression (Fig. 7).
Fig. 7. Plasma HER2 amplification status at baseline and at disease progression.

Analysis included 29 patients in Cohort A who discontinued T-DXd because of disease progression and had detectable ctDNA at C1D1 and at EOT. Responder/non-responder status was determined by blinded independent central review. Amp, amplification; C1D1, cycle 1, day 1; ctDNA, circulating tumor DNA; EOT, end of treatment.
Acquired gene mutations at disease progression
Acquired mutations in several genes were observed in plasma ctDNA at disease progression in 12 of the 29 patients in Cohort A who discontinued T-DXd treatment. However, none of the mutations were common across the patient group (Fig. 8; Table 1).
Fig. 8. Acquired plasma ctDNA mutations at disease progression in patients with HER2-positive mCRC treated with T-DXd.

Acquired refers to patients with acquired mutations detected only at disease progression. Lost refers to patients with mutations detected only at cycle 1, day 1. Maintained refers to patients with mutations at cycle 1, day 1, and at disease progression. bTMB, blood tumor mutational burden; CBOR, confirmed best overall response; ctDNA, circulating tumor DNA; HER2, human epidermal growth factor receptor 2; mCRC, metastatic colorectal cancer; PD, progressive disease; PFS, progression-free survival; PR, partial response; SD, stable disease.
Table 1.
Acquired ctDNA gene mutations in patients with disease progression (n = 29)
| Patients, n | Gene | Mutation |
|---|---|---|
| 3 | TP53 | Y234C, S241C, R273C |
| 1 | HER2 | D769H |
| 1 | BRAF | V600E, V600M |
| 1 | CASP8 | F338fs |
| 1 | CIC | T2002fs |
| 1 | GRIN2A | Q201* |
| 1 | JAK1 | L1114fs |
| 1 | KEAP1 | F64fs |
| 1 | KMT2D | L671fs |
| 1 | NOTCH2 | N396fs |
| 1 | PIK3CA | R88Q |
ctDNA, circulating tumor DNA.
ctDNA reduction during T-DXd treatment
A relationship was observed between ORR and the level of change in mean VAF (mVAF), a measure of molecular response. Among 2 patients who had complete molecular response (100% reduction in mVAF; ie, nondetectable ctDNA), the ORR was 100% (95% CI, 15.8–100.0). Among 15 patients with major molecular response ( ≥ 90% to < 100% reduction in mVAF), the ORR was 86.7% (95% CI, 59.5–98.3). Among 13 patients with partial molecular response ( ≥ 20% to < 90% reduction in mVAF), the ORR was 46.2% (95% CI, 19.2–74.9). Among 10 patients with absence of molecular response ( < 20% reduction in mVAF), the ORR was 10.0% (95% CI, 0.3–44.5). Similarly, the probability of PFS reduction over time for all molecular response levels was lowest among patients with absence of molecular response (Supplementary Fig. S8).
Retrospective ctDNA analysis according to clinical outcomes (mPFS and ORR) among patients who had at least 16.4% reduction in mVAF at cycle 4, day 1, is shown in Supplementary Fig. S9. The cutoff of 16.4% was defined according to the minimum P value of the log-rank test for PFS.
Clinical outcomes in selected subgroups
Clinical response to T-DXd was observed in patients with HER2-positive mCRC (Cohort A; n = 40 evaluable patients) regardless of CRC histological grade subgroup (poorly, moderately, or well-differentiated tumors according to locally scored histological grade) (Supplementary Fig. S10). Three of four patients who had poorly differentiated tumors responded to T-DXd treatment.
Because topoisomerase I inhibitors are commonly used to treat mCRC and the payload of T-DXd is also a topoisomerase I inhibitor, an exploratory analysis was carried out to investigate whether the interval from prior irinotecan therapy was associated with clinical outcomes. All 53 patients in Cohort A (HER2-positive mCRC) had prior irinotecan treatment; however, four patients for whom the date of the last irinotecan treatment was unknown were excluded from this analysis. A clinical response to T-DXd was observed regardless of the interval from the last irinotecan treatment. There was no clear relationship between the interval from last irinotecan treatment and mPFS (Supplementary Fig. S11).
Discussion
This exploratory biomarker analysis of patients with mCRC treated with T-DXd in the DESTINY-CRC01 trial demonstrated an association between HER2 expression or amplification and antitumor activity of T-DXd. Higher ORR and longer PFS were apparent in patients with higher expression levels of HER2 in tissue biopsy samples at baseline and in patients with plasma HER2 focal amplification and higher levels of HER2 ApCN and HER2ECD in liquid biopsy samples at baseline; however, results should be interpreted with caution due to the small number of patients. Similar findings were previously reported in the HERACLES-A trial of trastuzumab and lapatinib dual-targeted therapy, in which patients with a higher HER2 copy number achieved longer PFS and OS24, and patients with a higher HER2 ApCN achieved higher ORR and longer PFS25. In preclinical models, T-DXd also inhibited cell proliferation in a HER2-dependent manner26. In the current trial, patients with HER2-low expressing mCRC did not respond to T-DXd treatment, whereas in patients with HER2-low mBC in the DESTINY-Breast04 trial (NCT03734029), T-DXd showed superior activity over standard chemotherapy options27. Based on the current results, HER2-positive status defined by IHC/ISH appears to remain the most important biomarker to predict response to T-DXd. However, the exploratory data reported in this study, particularly the blood-based HER2-related biomarkers, also appear to show a correlation with response to T-DXd. Additional studies in larger populations would be required to determine the clinical relevance of these blood-based HER2 biomarkers in guiding T-DXd treatment. The predictive value of potential biomarkers needs to be statistically evaluated in a randomized setting; however, the current results support further investigation of the clinical relevance of HER2-related biomarkers such as plasma HER2 amplification and HER2ECD for patients who do not have adequate tumor tissue for measurement of HER2 IHC status.
Six patients in the current study with HER2 positive CRC were found to harbor activating RAS mutations, even though RAS wild-type status was an inclusion criterion for the DESTINY-CRC01 trial. One of the patients with RAS mutation (NRAS G12D) was enrolled in deviation from the protocol. These are likely acquired mutations because all 6 patients had received anti-EGFR antibody therapy (4 cetuximab, 1 panitumumab, 1 cetuximab and panitumumab) prior to T-DXd treatment and acquired RAS mutations are a known resistance mechanism to EGFR inhibitors28. Antitumor activity of T-DXd was observed in patients with RAS, PIK3CA, or HER2-activating mutations at baseline and regardless of bTMB levels. Although there were signs of potential associations—for example, a reduction in VAFs in patients with RAS or PIK3CA mutations who achieved tumor shrinkage (Supplementary Fig. S7)—the small number of patients with activating mutations of RAS, PIK3CA, and HER2 and high bTMB means that interpretation of these results is limited and further investigation to validate these findings is warranted in a larger study, such as the DESTINY-CRC02 trial (NCT04744831). Nevertheless, the persistence of activity in tumors with these driver mutations in the current study suggests a potentially unique property of T-DXd in comparison to other HER2-targeted therapies. For example, in patient-derived xenografts and matched CRC cell lines, overexpression of mutant alleles of KRAS, BRAF, and PIK3CA conferred resistance to the combination of trastuzumab and lapatinib due to sustained ERK and/or AKT activation29. In the HERACLES-A trial, more than 85% of patients who were refractory to HER2 blockade with trastuzumab plus lapatinib had RAS/BRAF baseline mutations detected in liquid biopsy and, in addition, a role for PIK3CA-AKT pathway activation was suggested as a mechanism of acquired resistance to this dual anti-HER2 blockade30, Furthermore, in the MyPathway trial, 13 of 57 patients (23%) with HER2-amplified mCRC had activating KRAS mutation and 8 patients (14%) had activating PIK3CA mutation. The ORR with pertuzumab plus trastuzumab in the patients with KRAS mutation in the MyPathway trial was 8% (1 of 13 patients) compared with 40% (17 of 43 patients) in patients with wild-type KRAS and the ORR in patients with PIK3CA mutation was 13% (1 of 8 patients) compared with 43% (17 of 40 patients) in patients with wild-type PIK3CA14. The T-DXd cytotoxic payload, DXd, and its bystander effect is unique among HER2 targeted drugs and may partly be responsible for these differences between our study and others. The T-DXd bystander effect that kills adjacent tumor and stromal cells, has been described, with no potential overlapping mechanisms of resistance with the HER2 signaling pathway19,31.
Analysis of on-treatment ctDNA in our study showed an association between clinical outcomes and mVAF reduction. Higher ORR and longer PFS were observed in patients who had a complete molecular response (100% reduction in mVAF) and the lowest responses were observed in patients with absence of molecular response ( < 20% reduction in mVAF). Clinical outcomes (ORR and PFS) seemed to be better if patients had at least a 16.4% decrease in mVAF at cycle 4, day 1, of treatment (Supplementary Fig. S9). Although conclusions cannot be drawn due to the small number of patients, these findings support further investigation of the relationship between changes in mVAF and clinical outcomes in larger trials. Among patients with mCRC undergoing first-line chemotherapy in another study, ctDNA clearance was associated with longer PFS32. In our study of patients with HER2-positive mCRC, 19 of 27 patients who discontinued T-DXd because of disease progression had plasma HER2 amplification at the time of disease progression. This might be an underestimation because of limitations in the technology to detect HER2 plasma amplification in ctDNA samples taken at disease progression with potentially lower ctDNA shedding. Although acquired alterations were observed in several genes at disease progression, including mutations in BRAF, CASP8 (an apoptosis-related gene), and KEAP1 (a reactive oxygen species-related gene) in our study, none were common across the patient population. Patterns of emerging molecular alterations associated with clinical resistance to trastuzumab and lapatinib therapy identified in ctDNA analysis from the HERACLES-A trial suggested that radiographic and genomic evolution patterns could be heterogeneous between target lesions and metastases and response to HER2 blockade may therefore differ30.
Subgroup analysis in the current study indicates that clinical response to T-DXd was observed regardless of the histological grade of mCRC (Supplementary Fig. S10). Three of the 4 patients who had poorly differentiated tumors responded to T-DXd, suggesting anti-tumor activity in patients with aggressive tumors. However, interpretation is limited because the number of patients in each histological grade subgroup was small and histological grades used in the analysis were scored locally using archival samples.
In conclusion, this exploratory analysis identified HER2 biomarkers associated with antitumor activity of T-DXd in patients with mCRC, including those with activating mutations in common mCRC oncogenic driver genes.
Methods
Study design
This research complies with all relevant ethical regulations. Independent ethics committees or institutional review boards at each study site reviewed and approved the protocol23 (Supplementary Table S3). The study was conducted in accordance with the principles of the Declaration of Helsinki, The International Council for Harmonisation guidelines for Good Clinical Practice, and other local regulations where applicable. Written informed consent was provided by all patients before enrollment.
DESTINY-CRC01 (NCT03384940) was an open-label, multicenter, phase 2 trial in patients with HER2-expressing unresectable, recurrent, or metastatic colorectal adenocarcinoma who had received and progressed on at least two previous treatment regimens including fluoropyrimidines, irinotecan, oxaliplatin, and anti-EGFR or anti-VEGF antibodies. Patients had to have RAS and BRAFV600E wild-type tumors according to local assessment. Full enrollment criteria are published23. Patients were grouped into three cohorts according to HER2 expression assessed on archival or recent tumor tissue samples at screening (before treatment) by the central laboratory: Cohort A (IHC 3+ or IHC 2+/ISH+ [HER2-positive]); Cohort B (IHC 2+ and ISH−); Cohort C (IHC 1+).
Patients were treated with T-DXd 6.4 mg/kg by intravenous infusion once every 3 weeks until disease progression (per investigator assessment according to Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST 1.1), investigator decision to discontinue treatment, pregnancy, withdrawal of consent, or death. Tumor assessment was performed by investigators and by Independent Central Review every 6 weeks from cycle 1, day 1, until progressive disease or initiation of new anticancer treatment. Endpoints assessed were cORR, defined as the proportion of patients who had a confirmed best overall response of complete response or partial response at any point from the start of therapy until the patient was withdrawn from the study or started a new anticancer therapy, whichever was earlier, and PFS, defined as the time from the date of the first dose to the earliest disease progression or death23.
The clinical cutoff date used in the current analysis was December 28, 2020.
Biomarker analysis
HER2 testing
Archival or recently obtained biopsied tumor tissue was collected at screening to assess HER2 status. IHC staining for HER2 was performed using PATHWAY anti-HER-2/neu (4B5) Rabbit Monoclonal Primary Antibody (Ventana Medical Systems). Freshly cut 4 µm thick formalin-fixed paraffin-embedded sections were processed on the BenchMark ULTRA staining platform (Ventana Medical Systems). HER2 IHC score (0, 1+, 2+, or 3+) was evaluated by trained pathologists according to the College of American Pathologists/American Society for Clinical Pathology/American Society of Clinical Oncology guidelines for gastric cancer33. H score (0–300) was automatically evaluated by LabVantage laboratory information management system. INFORM HER2 Dual ISH DNA probe cocktail assay (Ventana Medical Systems) was used to provide HER2 amplification status, HER2/CEP17 ratio, and the average number of HER2 signals33.
Circulating tumor DNA
Plasma samples for ctDNA analysis were collected at baseline (cycle 1, day 1); cycle 4, day 1; and end of treatment. Plasma ctDNA analysis to explore biomarkers of response or resistance was performed using the Guardant OMNI panel by Guardant Health, which can detect alterations in approximately 500 genes, and bTMB. Possible germline mutations, synonymous mutations, mutations that are not oncogenic (ie, considered likely oncogenic, oncogenic, or predicted oncogenic based on oncoKB) with VAF less than 0.2, and clonal hematopoiesis of indeterminate potential (CHIP) mutations reported by Guardant Health were excluded. The evaluable ctDNA dataset (188 samples) was derived from the total ctDNA dataset (190 samples), excluding those in which somatic mutation was not detected. For cohort A, baseline ctDNA was evaluated in 52 of 53 patients.
For analysis of single nucleotide variants and insertion/deletion mutations, PIK3CA variants were determined according to published data34. NRAS and KRAS variants were considered mutant if there was mutation at codon 12, 13, 59, 61, 117, or 146. HER2 variants were categorized as mutant if variants determined as Gain-of-function and Likely Gain-of-function based on OncoKB. PTEN variants were determined as mutations of Loss-of-function and Likely Loss-of-function based on OncoKB. bTMB ≥ 20 mut/Mb was considered high according to the Guardant Health report. For analysis of plasma HER2 amplification, both focal and aneuploidy amplification detected according to Guardant OMNI pipeline were assessed.
ApCN was used to correct variation in plasma tumor fraction between samples and calculated according to published methods using the following equation25. ApCN = (observed pCN − 2 × [1 − T] / T, where T = 2 × maxVAF[%] / 100. The maximum VAF (maxVAF) for any variant of an individual sample was used to calculate the surrogate tumor fraction (T).
For the analysis of molecular response, mVAF was defined as the mean of VAF of gene mutations (single nucleotide variant/Indel) detected at baseline that were greater than or equal to 0.3. For on-treatment samples, only gene mutations detected at baseline were used for mVAF calculation and were considered as 0 if VAF was less than 0.3.
HER2ECD
Baseline HER2ECD in serum was assessed by enzyme-linked immunosorbent assay (ELISA; Siemens Health Diagnostics).
Statistical analysis
Receiver operating characteristic (ROC) analysis was performed using ORR to determine the exploratory cutoff values for HER2 H-score, HER2 ApCN, HER2/CEP17 ratio, HER2 ISH signal, and HER2ECD. Exploratory cutoff values for each biomarker based on ROC analysis were set at the maximum Youden index (sensitivity + specificity – 1). In cohort A, 5 of the 52 patients did not have HER2 amplification at baseline and the HER2 ApCN was set at a value of 2 for these patients for ROC analysis. Point estimates and two-sided 95% exact binomial CIs were calculated for ORR in each subgroup. The Kaplan-Meier method was used to estimate median event times with two-sided 95% CIs calculated using Brookmeyer and Crowley methods. Exploratory cutoff values (VAF; 16.4%) were selected based on the most significant value for PFS for separating patients into high and low groups. ORR was compared using Fisher’s exact test. Median PFS and OS were estimated by Kaplan-Meier analysis and compared using log-rank test.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Acknowledgements
The authors thank the patients and their families for their participation and the study site staff for their contributions. The study was designed and led by the funder, Daiichi Sankyo, which participated in data collection and analysis. In March 2019, AstraZeneca entered into a collaboration agreement with Daiichi Sankyo for trastuzumab deruxtecan. Both Daiichi Sankyo and AstraZeneca were involved in study oversight and data collection; both funders also assisted in developing the biomarker analysis plan, performing biomarker analysis and data analysis, data interpretation, writing the report, and reviewing the manuscript. Medical writing support was provided by Brian Brennen, PhD, and Sara Duggan, PhD, of ApotheCom and was funded by Daiichi Sankyo, Inc.
Author contributions
S.S.: Principal investigator, intellectual contribution to the development of the DESTINY-CRC01 protocol, ongoing collaborative efforts during conduct of study, data acquisition and quality control, and steering committee member of the DESTINY-CRC01 study. K.R.: Principal investigator, data interpretation, data acquisition and quality control. T.M.: Principal investigator, data acquisition and quality control. K.Y.: Principal investigator, data acquisition and quality control, and steering committee member of DESTINY-CRC01. T.N.: Principal investigator, data acquisition and quality control. E.E.: Principal investigator, data acquisition and quality control. J.R.: Principal investigator, data acquisition and quality control. I.C.: Principal investigator, data acquisition and quality control. M.D.B.: Principal investigator, data acquisition and quality control. H.K.: Principal investigator, data acquisition and quality control. F.S.: Biomarker analysis, protocol (biomarker analysis) design, biomarker data analysis, biomarker data interpretation, and quality control. M.K.: Biomarker analysis, biomarker data analysis, biomarker data interpretation, and quality control. K.I.: Biomarker analysis, biomarker data analysis, biomarker data interpretation, and quality control. Y.K.: Biomarker analysis, biomarker data analysis, biomarker data interpretation, and quality control. I.T.: Management of biomarker assay. D.B.: DESTINY-CRC01 study physician and biomarker data interpretation. K.K.: Development of the DESTINY-CRC01 protocol and biomarker data interpretation. A.G.: Principal investigator, intellectual contribution to the development of the DESTINY-CRC01 protocol, ongoing collaboration and guidance during conduct of study, data interpretation, and steering committee member of the DESTINY-CRC01study. T.Y.: Principal investigator, intellectual contribution to the development of the DESTINY-CRC01 protocol, ongoing collaboration and guidance during conduct of study, data interpretation, data acquisition and quality control, and chief of steering committee of the DESTINY-CRC01 study.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Data availability
Anonymized individual participant data (IPD) on completed studies and applicable supporting clinical trial documents may be available upon request at the Vivli website (https://vivli.org/members/enquiries-about-studies-not-listed-on-the-vivli-platform/). In cases where clinical trial data and supporting documents are provided pursuant to our company policies and procedures, Daiichi Sankyo Companies will continue to protect the privacy of our clinical trial participants. Details on data sharing criteria and the procedure for requesting access can be found at Vivli’s Daiichi Sankyo web page (https://vivli.org/ourmember/daiichi-sankyo). Individual participant data, including data dictionaries, will be available. Documents that will be available include the clinical trial protocol, statistical analysis plan, informed consent form, and clinical study report. Data may be requested after the indication has been approved by major health authorities and the study results are published. The data will be made available to qualified science and medical researchers upon formal request and submission of a research proposal detailing planned analyses. De-identified IPD and relevant clinical trial documents will be shared for the purpose of conducting legitimate research as specified in an approved formal research proposal and may be available upon request via the Vivli Data Sharing Platform at https://vivli.org/. Additional information can be found in Supplementary Table S4. All remaining data can be found in the article or supplementary files.
Competing interests
The authors declare the following competing interests: S.S. provided medical writing support to Daiichi Sankyo on this manuscript. In the past 36 months, he received payment for participating on data safety monitoring boards or advisory boards for Agenus, AstraZeneca, Bayer, BMS, CheckmAb, Daiichi Sankyo, GSK, Guardant Health, Merck, Novartis, Pierre-Fabre, Roche Genentech, Seagen, and T-One Therapeutics. K.R. provided research support to Daiichi Sankyo for this study. In the past 36 months, he received grants or contracts from Bayer, Merck, Guardant, Hibercell, Daiichi Sankyo, and Seagen; received payments for educational events from Daiichi Sankyo, Bayer, and Seagen; and received payment for participating on data safety monitoring boards or advisory boards from Seagen and Merck. T.M. received grants or contracts in the past 36 months from Amgen, Boehringer Ingelheim, Cimic Shift Zero, Daiichi Sankyo, Merck, Novartis, ONO Pharmaceutical, Pfizer, Syneos Health Clinical, and Eli Lilly and received payments for educational events from Bayer, Bristol Myers Squibb, Chugai, Daiichi Sankyo, Lilly Japan, Merck BioPharma, ONO Pharmaceutical, Sanofi, Taiho, Takeda, and Yakult Honsha. K.Y. reports support from Daiichi Sankyo for the present manuscript, grants from Taiho Pharmaceutical, and payment or honoraria from Daiichi Sankyo Co. Ltd, Chugai Pharmaceutical Co. Ltd, Bristol Myers Squibb, Eli Lilly Japan, Taiho Pharmaceutical Co. Ltd, Ono Pharmaceutical Co. Ltd, Takeda Pharmaceutical Co. Ltd, and Merck Biopharm Co. Ltd. T.N. received honoraria for lectures from Bristol Myers Squibb, Chugai Pharmaceutical, Daiichi Sankyo Pharmaceutical, Eli Lilly Japan, Merck Serono Pharmaceutical, Ono Pharmaceutical, Takeda Pharmaceutical, Taiho Pharmaceutical, and Yakult-Honsha and was a Data Safety Monitoring Board member for Janssen Pharmaceutical. E.E. received consultancy fees from Amgen, Bayer, Hoffmann-La Roche, Merck Serono, MSD, Novartis, Organon, Pierre Fabre, Sanofi, and Servier; received payments for educational events from Amgen, Bayer, Hoffmann-La Roche, Merck Serono, MSD, Novartis, Organon, Pierre Fabre, Sanofi, and Servier; and received payments for participating on advisory boards from Amgen, Bayer, Hoffmann-La Roche, Merck Serono, MSD, Novartis, Organon, Pierre Fabre, Sanofi, and Servier. J.R. reports no conflicts of interest. I.C. has been on advisory boards for Eli Lilly, Bristol Myers Squibb, MSD, Roche, Merck-Serono, AstraZeneca, OncXerna, Pierre Fabre, Boehringer Ingelheim, Incyte, Astellas, GlaxoSmithKline, Sotio, Eisai, Daiichi Sankyo, Taiho, Servier, Seagen, and Turning Point Therapeutics, reports research funding from Eli Lilly and Janssen Cilag, and honorarium from Eli Lilly, Eisai, Servier, Roche, BMS, and Novartis. M.D.B. reports no conflicts of interest. H.K. received grants or contracts in the past 36 months from Bristol Myers Squibb, Eisai Co. Ltd, Kobayashi Pharmaceutical, and Taiho Pharmaceutical; received consultancy fees from Daiichi Sankyo; received payments for educational events from Bayer Yakuhin, Bristol Myers Squibb, Chugai Pharmaceuticals, Daiichi Sankyo, Eli Lilly Japan, Merck Biopharma, MSD, ONO Pharmaceutical, Otsuka, Taiho Pharmaceutical Takeda, Teijin, and Yakult. F.S. reports no conflicts of interest. M.K. reports no conflicts of interest. K.I. is an employee of Daiichi Sankyo RD Novare, which is a subsidiary of Daiichi Sankyo. Y.K is an employee of and has stock options with Daiichi Sankyo. I.T. reports no conflicts of interest. D.B. reports no conflicts of interest. K.K. reports no conflicts of interest. A.G. reports consulting fees from Daiichi Sankyo, Bayer, Merck/MSD, Genentech/Roche, Natera, and BMS, payment/honoraria from Bayer, Genentech/Roche, and Merck/MSD, and participation on a data safety monitoring board or advisory board for Regeneron. T.Y. received grants or contracts from Amgen K.K., Chugai Pharmaceuticals, Daiichi Sankyo, Genomedia, MSD K.K., Nippon Boehringer Ingelheim, ONO Pharmaceutical, Parexel International, Pfizer Japan, Sanofi K.K., Sysmex, and Taiho and received payments for educational events from Bayer Yakuhin, Chugai, Eli Lilly Japan, Merck Biopharma, MSD, ONO Pharmaceutical, and Taiho.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-53223-3.
References
- 1.Sung, H., et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021). [DOI] [PubMed]
- 2.Ross, J. S. et al. Targeting HER2 in colorectal cancer: the landscape of amplification and short variant mutations in ERBB2 and ERBB3. Cancer124, 1358–1373 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siena, S. et al. Targeting the human epidermal growth factor receptor 2 (HER2) oncogene in colorectal cancer. Ann. Oncol.29, 1108–1119 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sawada, K. et al. Prognostic and predictive value of HER2 amplification in patients with metastatic colorectal cancer. Clin. Colorectal Cancer17, 198–205 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Nam, S. K. et al. BRAF, PIK3CA, and HER2 oncogenic alterations according to KRAS mutation status in advanced colorectal cancers with distant metastasis. PLoS One11, e0151865 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strickler, J. H., Yoshino, T., Graham, R. P., Siena, S. & Bekaii-Saab, T. Diagnosis and treatment of ERBB2-positive metastatic colorectal cancer: A review. JAMA Oncol.8, 760–769 (2022). [DOI] [PubMed] [Google Scholar]
- 7.Morris, V. K. et al. Treatment of metastatic colorectal cancer: ASCO guideline. J. Clin. Oncol.41, 678–700 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riedesser, J. E., Ebert, M. P. & Betge, J. Precision medicine for metastatic colorectal cancer in clinical practice. Ther. Adv. Med Oncol.14, 17588359211072703 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Voutsadakis, I. A. A systematic review and meta-analysis of Trifluridine/Tipiracil plus Bevacizumab for the treatment of metastatic colorectal cancer: Evidence from real-world series. Curr. Oncol.30, 5227–5239 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sartore-Bianchi, A. et al. HER2 positivity predicts unresponsiveness to EGFR-targeted treatment in metastatic colorectal cancer. Oncologist24, 1395–1402 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raghav, K., et al. Validation of HER2 amplification as a predictive biomarker for anti–epidermal growth factor receptor antibody therapy in metastatic colorectal cancer. JCO Precision Oncol.3, 1–13 (2019). [DOI] [PubMed]
- 12.Wong, A. H. N., Ma, B. & Lui, R. N. New developments in targeted therapy for metastatic colorectal cancer. Ther. Adv. Med Oncol.15, 17588359221148540 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tosi, F. et al. Long-term clinical outcome of trastuzumab and lapatinib for HER2-positive metastatic colorectal cancer. Clin. Colorectal Cancer19, 256–262.e252 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Meric-Bernstam, F. et al. Pertuzumab and trastuzumab for HER2-amplified metastatic colorectal cancer (MyPathway): an updated report from a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol.20, 518–530 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strickler, J. H. et al. Tucatinib plus trastuzumab for chemotherapy-refractory, HER2-positive, RAS wild-type unresectable or metastatic colorectal cancer (MOUNTAINEER): a multicentre, open-label, phase 2 study. Lancet Oncol.24, 496–508 (2023). [DOI] [PubMed] [Google Scholar]
- 16.Sartore-Bianchi, A. et al. Pertuzumab and trastuzumab emtansine in patients with HER2-amplified metastatic colorectal cancer: the phase II HERACLES-B trial. ESMO Open5, e000911 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cervantes, A. et al. Metastatic colorectal cancer: ESMO clinical practice guideline for diagnosis, treatment and follow-up. Ann. Oncol.34, 10–32 (2023). [DOI] [PubMed] [Google Scholar]
- 18.Ogitani, Y. et al. DS-8201a, a novel HER2-targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T-DM1. Clin. Cancer Res22, 5097–5108 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Ogitani, Y., Hagihara, K., Oitate, M., Naito, H. & Agatsuma, T. Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 antibody-drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci.107, 1039–1046 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.von Arx, C. et al. The evolving therapeutic landscape of trastuzumab-drug conjugates: future perspectives beyond HER2-positive breast cancer. Cancer Treat. Rev.113, 102500 (2023). [DOI] [PubMed] [Google Scholar]
- 21.Daiichi Sankyo, Inc. ENHERTU® (fam-trastuzumab deruxtecan-nxki) For Injection, For Intravenous Use. in Daiichi Sankyo, Inc. (ed. 11/2022) (14, Basking Ridge, NJ, 2022).
- 22.Daiichi Sankyo Europe GmbH. Enhertu 100 Mg Powder For Concentrate For Solution For Infusion. in Daiichi Sankyo Europe GmbH (ed. 2023) (44, Munich, Germany, 2023).
- 23.Yoshino, T. et al. Final results of DESTINY-CRC01 investigating trastuzumab deruxtecan in patients with HER2-expressing metastatic colorectal cancer. Nat. Commun.14, 3332 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sartore-Bianchi, A. et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol.17, 738–746 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Siravegna, G. et al. Plasma HER2 (ERBB2) copy number predicts response to HER2-targeted therapy in metastatic colorectal cancer. Clin. Cancer Res25, 3046–3053 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Takegawa, N. et al. fam-] trastuzumab deruxtecan, antitumor activity is dependent on HER2 expression level rather than on HER2 amplification. Int J. Cancer145, 3414–3424 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Modi, S. et al. Trastuzumab deruxtecan in previously treated HER2-low advanced breast cancer. N. Engl. J. Med387, 9–20 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Misale, S. et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature486, 532–536 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lazzari, L. et al. Patient-derived xenografts and matched cell lines identify pharmacogenomic vulnerabilities in colorectal cancer. Clin. Cancer Res25, 6243–6259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siravegna, G. et al. Radiologic and genomic evolution of individual metastases during HER2 blockade in colorectal cancer. Cancer Cell34, 148–162.e147 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Singh, A. P. et al. Evolution of the systems pharmacokinetics-pharmacodynamics model for antibody-drug conjugates to characterize tumor heterogeneity and in vivo bystander effect. J. Pharm. Exp. Ther.374, 184–199 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim, S. et al. Dynamic changes in longitudinal circulating tumour DNA profile during metastatic colorectal cancer treatment. Br. J. Cancer127, 898–907 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.College of American Pathologists. CAP-ASCP-ASCO HER2 Testing And Clinical Decision Making In Gastroesophageal Adenocarcinoma Guideline. 2022 (College of American Pathologists, Northfield, IL, 2016).
- 34.Baselga, J. et al. Relationship between tumor biomarkers and efficacy in EMILIA, a Phase III study of trastuzumab emtansine in HER2-positive metastatic breast cancer. Clin. Cancer Res22, 3755–3763 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Anonymized individual participant data (IPD) on completed studies and applicable supporting clinical trial documents may be available upon request at the Vivli website (https://vivli.org/members/enquiries-about-studies-not-listed-on-the-vivli-platform/). In cases where clinical trial data and supporting documents are provided pursuant to our company policies and procedures, Daiichi Sankyo Companies will continue to protect the privacy of our clinical trial participants. Details on data sharing criteria and the procedure for requesting access can be found at Vivli’s Daiichi Sankyo web page (https://vivli.org/ourmember/daiichi-sankyo). Individual participant data, including data dictionaries, will be available. Documents that will be available include the clinical trial protocol, statistical analysis plan, informed consent form, and clinical study report. Data may be requested after the indication has been approved by major health authorities and the study results are published. The data will be made available to qualified science and medical researchers upon formal request and submission of a research proposal detailing planned analyses. De-identified IPD and relevant clinical trial documents will be shared for the purpose of conducting legitimate research as specified in an approved formal research proposal and may be available upon request via the Vivli Data Sharing Platform at https://vivli.org/. Additional information can be found in Supplementary Table S4. All remaining data can be found in the article or supplementary files.