

Dear Editor,
Mayotte, an island in the Indian Ocean, has the highest fertility rate of all French overseas departments. Some 40% of its population is of foreign origin, and 84% live below the poverty line. Xeroderma pigmentosum (XP) is a rare autosomal recessive disorder characterized by extreme sensitivity to ultraviolet (UV) radiation, predisposing affected individuals to various photoinduced malignancies. Mayotte and the neighbouring Comoros Islands report a high prevalence of XP (1). Given the critical need for UV protection in these patients and the extended waiting times for genetic analysis, early clinical screening is imperative.
We report the case of a 9-month-old girl, born to non-consanguineous parents from Anjouan, Comoros, whose 5-year-old brother was diagnosed with XP. The pregnancy and neonatal periods were uneventful, and no screening for XP was conducted antenatally or at birth. She presented with no previous medical history and was in good general health. There were no signs of photophobia or conjunctivitis. Clinical examination revealed multiple scattered, millimetric hypochromic macules localized exclusively to photo-exposed areas (Fig. 1A). The lesions were neither desquamating nor pruritic, and there were no signs of oozing, insensitivity, or papules. Dermoscopic examination (Fig. 1B, C) revealed multiple millimetric hypopigmented areas on the face but not on the trunk. Given her brother’s XP diagnosis and the clinical presentation, a genetic test was ordered. The family was strongly advised on photoprotection measures. Two months later, genetic analysis confirmed the c.2251-1G > C mutation of the XPC gene, which is prevalent in this region (1). At the follow-up visit, clinical examination revealed an increase in the size and number of hypopigmented macules (Fig. 2).
Fig. 1.
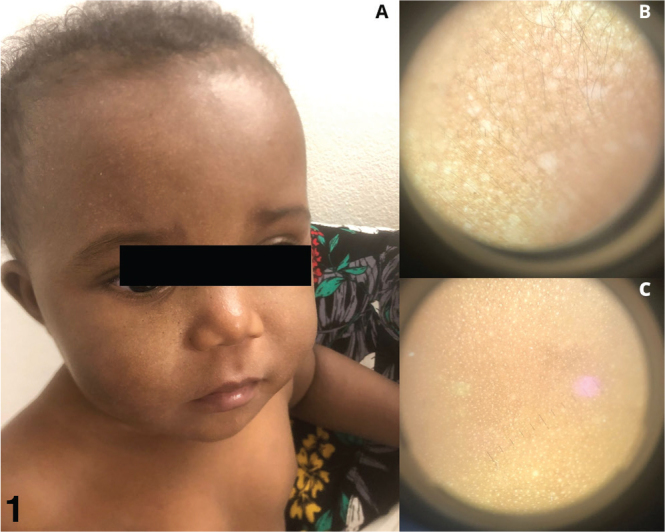
Clinical and dermoscopic description in December 2021. (A) discrete hypochromic macules of the face, (B) dermoscopic appearance of the face, (C) dermoscopic appearance of the back.
Fig. 2.
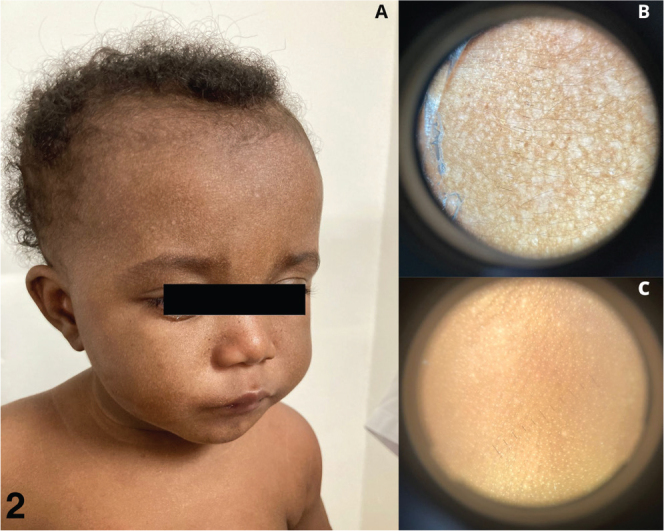
Clinical and dermoscopic description in February 2022. (A) hypochromic macules of the face, (B) dermoscopic aspect of the face, (C) dermoscopic aspect of the back.
XP progresses in 3 stages: (i) subtle, persistent erythema, which can be challenging to diagnose on phototypes V and VI (the predominant skin types in Mayotte), or recurrent actinic erythema; (ii) evident dyschromia appearing from 12 months of age; and (iii) the development of skin cancers (2). The dyschromia consists of small, pigmented and hypopigmented lenticular macules with poorly defined borders. These actinic lesions, manifestations of premature skin ageing, result from localized melanocyte depletion, leading to hypopigmentation (3). Hyperpigmented lesions, described as lentigines or ephelides, alternate with these hypopigmented areas, contributing to poikiloderma.
Photophobia can present as early as 6 months, sometimes mistaken for viral or bacterial conjunctivitis (4). However, this early ocular involvement may be missed, underscoring the importance of thorough skin examinations to detect precursor signs of XP. Dermoscopy, although not widely discussed in the literature for this purpose, may prove valuable as a screening tool for early XP diagnosis.
In a 2017 study from Mayotte, Ventéjou et al. (1) described 18 XP patients with a median age of 12.9 years (7 females, 11 males), most originating from Anjouan. Since then, 19 XP cases have been followed in Mayotte, 3 in Anjouan, and 7 in Reunion Island. The absence of a resident dermatologist, the lack of health education, and the frequent turnover of healthcare professionals in Mayotte present challenges for managing chronic conditions like XP. Educational posters (Figs 1, 2) and dermatoscopes could be distributed across healthcare centres to raise awareness and enable earlier detection of XP.
In conclusion, the presence of multiple millimetric hypopigmented macules in photo-exposed areas in infants should prompt consideration of an XP diagnosis. Dermoscopy can aid in early clinical recognition of XP while awaiting genetic confirmation, facilitating the timely implementation of photoprotection measures.
ACKNOWLEDGEMENT
Ethics statement: The mother of the patient in this manuscript has given written informed consent to publication of her case details.
REFERENCES
- 1.Ventéjou S, Bagny K, Waldmeyer J, Cartault F, Machet L, Osdoit S. Skin cancers in patients of skin phototype V or VI with xeroderma pigmentosum type C (XP-C): a retrospective study. Ann Dermatol Venereol 2019; 146: 192–203. 10.1016/j.annder.2018.11.013 [DOI] [PubMed] [Google Scholar]
- 2.Santiago KM, Castro LP, Neto JPD, de Nóbrega AF, Pinto CAL, Ashton-Prolla P, et al. Comprehensive germline mutation analysis and clinical profile in a large cohort of Brazilian xeroderma pigmentosum patients. J Eur Acad Dermatol Venereol 2020; 34: 2392–2401. 10.1111/jdv.16405 [DOI] [PubMed] [Google Scholar]
- 3.Lehmann J, Schubert S, Emmert S. Xeroderma pigmentosum: diagnostic procedures, interdisciplinary patient care, and novel therapeutic approaches. J Dtsch Dermatol Ges 2014; 12: 867–872. 10.1111/ddg.12419 [DOI] [PubMed] [Google Scholar]
- 4.Moriwaki S, Kanda F, Hayashi M, Yamashita D, Sakai Y, Nishigori C; Xeroderma pigmentosum clinical practice guidelines revision committee . Xeroderma pigmentosum clinical practice guidelines. J Dermatol. 2017; 44: 1087–1096. 10.1111/1346-8138.13907 [DOI] [PubMed] [Google Scholar]