

Abstract
The relative resistance of human immunodeficiency virus type 1 (HIV-1) primary isolates (PIs) to neutralization by a wide range of antibodies remains a theoretical and practical barrier to the development of an effective HIV vaccine. One model to account for the differential neutralization sensitivity between Pls and laboratory (or T-cell line-adapted [TCLA]) strains of HIV suggests that the envelope protein (Env) complex is made more accessible to antibody binding as a consequence of adaptation to growth in established cell lines. Here, we revisit this question using genetically related PI and TCLA viruses and molecularly cloned env genes. By using complementary techniques of flow cytometry and virion binding assays, we show that monoclonal antibodies targeting the V3 loop, CD4-binding site, CD4-induced determinant of gp120, or the ectodomain of gp41 bind equally well to PI and TCLA Env complexes, despite large differences in neutralization outcome. The data suggest that the differential neutralization sensitivity of PI and TCLA viruses may derive not from differences in the initial antibody binding event but rather from differences in the subsequent functioning of the PI and TCLA Envs during virus entry. An understanding of these as yet undefined differences may enhance our ability to generate broadly neutralizing HIV vaccine immunogens.
Adaptation of a primary isolate (PI) of human immunodeficiency virus type 1 (HIV-1) to persistent growth in established T-cell lines is accompanied by genetic changes in the virus. Because PI viruses are isolated in primary T-lymphocyte culture, and for the most part are unable to productively infect established T-cell lines, powerful selection pressures are exerted upon the PI virus population to obtain variants which grow in T-cell lines. Changes in the viral envelope protein (Env) mediate many of these adaptations (65), although additional changes also affect postentry events. For instance, changes in viral Vpr alter cell cycle control to facilitate persistent growth in continually dividing cell lines (52).
Remarkably, changes in Env that mediate the expanded cell tropism for established T-cell lines also mediate changes in neutralization sensitivity: T-cell line-adapted (TCLA) isolates display increased sensitivity to neutralization by soluble forms of CD4 (sCD4) and by antibodies. This general conclusion comes from numerous cross-sectional comparisons of PI and TCLA viruses, but most convincingly from longitudinal comparisons of the neutralization sensitivity of PI viruses and their derivative TCLA strains (2, 16, 34, 64, 65, 67).
It is this observation of differential neutralization sensitivity that drives the study of T-cell line adaptation. Initial efforts to develop sCD4 for antiviral therapy were thwarted in part by the unexpected resistance to inhibition of PI viruses relative to the TCLA viruses commonly used in earlier studies (10, 11). This differential sensitivity to neutralization again attracted widespread attention in 1993 when it was found that PI viruses were refractory to antibodies elicited by recombinant gp120 vaccine immunogens, antibodies that potently neutralize the infectivity of TCLA viruses (9).
Several theories have been advanced to account for the coincident changes in cell tropism and neutralization sensitivity. Most models suggest that adaptation to growth in T-cell lines involves a facilitation of the initial viral interactions with the cell in order to allow rapid infection in culture and that this facilitation is accomplished through an “opening up” of the trimeric Env complex structure (36). For example, the CD4-binding site of the TCLA Env complex might become relatively more accessible to CD4 binding. This accessibility to cell binding events would carry over to a similar accessibility, and vulnerability, to neutralizing antibodies. According to this model, the resistance of PI viruses to neutralization derives from relative constraints (either steric or dynamic) on antibody binding to the oligomeric Env complex. In fact, several studies have reported differential binding of specific monoclonal antibodies (MAbs) to TCLA versus PI virions and cell surface Envs (4, 56, 57). These studies have compared binding to genetically unrelated PI and TCLA Envs and to Envs that differ significantly at the local MAb binding site. By contrast, we have previously reported equal binding of MAbs to cells infected with genetically related PI and TCLA viruses (65). In other studies, we (42, 43, 68) have demonstrated specific MAb binding to intact and infectious PI virions in the absence of neutralization.
In this report, we revisit the fundamental question: is the differential sensitivity to neutralization of PI and TCLA viruses due to differential antibody binding? We examine the question of antibody binding using two pairs of PI and derivative TCLA viruses. Using three independent and complementary methods, we find equal binding of monoclonal antibodies to neutralization-resistant PI viruses and their neutralization-sensitive TCLA virus derivatives. Therefore, the differential sensitivity of PI and TCLA viruses to neutralization does not arise upon initial antibody binding, but rather reflects differences in downstream events within the functioning PI and TCLA Envs. Our results suggest that a full understanding of virus neutralization and the differential sensitivity of PI and TCLA viruses will require an examination of the dynamic processes whereby the HIV Env mediates virus binding and entry.
MATERIALS AND METHODS
Viruses and molecularly cloned env expression plasmids.
The derivation and characterization of TCLA viruses 168C and 320SI-C3.3 and the respective parental PI viruses 168P and 320SI, have been previously described (see Table 1). Virus stocks were prepared either from acutely infected H9 cell culture (TCLA viruses) or from primary T-lymphocyte culture (PI viruses). It should be noted that these TCLA viruses remain neutralization sensitive and otherwise unaltered when prepared from primary T-lymphocyte culture (65; K. E. Follis and J. H. Nunberg, unpublished). The relative amount of Env per virion (gp 120 to p24) was determined by Western blot analysis of pelleted virions (56, 64) with either an anti-gp 120 MAb or pooled immunoglobulin (Ig) from HIV-infected persons (HIVIG) to detect gp120 or p24, respectively. The signals were quantified using ECL-Plus (Amersham Pharmacia Biotech) and a Fuji FLA-3000G analyzer, and the ratio of gp120 to p24 fluorescence was determined. Virus neutralization assays were performed with 96-well microculture as previously described (16, 28). For this assay, serial dilutions of antibody are incubated with a predetermined amount of virus for 1 h at 37°C, and this mixture is then incubated with U87-CD4-CXCR4 cells (23) for 2 days to allow the expression of HIV proteins. Infected cells (foci) are detected by immunochemical staining with either HIVIG or biotinylated anti-gp120 MAb 50.1 (Repligen Corp.). Similar neutralization results are obtained, for permissive viruses, by using U87-CD4-CCR5 cells (16, 28).
TABLE 1.
Pedigreed PI and TCLA virusesa
| Virus | Isolate type | Coreceptor(s) used | Central V3 loop | Adaptation-associated aa changes | Reference(s) |
|---|---|---|---|---|---|
| 168P | PI | CCR5 and CXCR4 | NIRKRIHIGPGRAFYTTG | 28, 65 | |
| 168C | TCLA | CXCR4 | NIRKRIHIGPGRAFYTTR | V2 I166R, C2 I282N, and V3 G318R | |
| 320SI | PI | CCR5 and CXCR4 | NTRKGIHIGPGRAFYAAR | 16 | |
| 320SI-C3.3 | TCLA | CCR5 and CXCR4 | NTRKGIRIGPGRAFYAAR | V2 I166K and V3 H317R |
Amino acid (aa) changes in the central V3 loop of the TCLA virus are underlined.
High-fidelity XL PCR (rTth and Vent DNA polymerases; PE Applied Biosystems) and oligonucleotide primers envA and envN (18) were used to amplify proviral DNA carrying rev and env genes. All amplifications were from DNA of infected cells, except that 320SI env was isolated from an infectious molecularly cloned provirus, ACH320.2A.1.2, provided by Hanneke Schuitemaker (Central Laboratory of the Netherlands Red Cross) through the NIBSC AIDS Reagent Project (Herts, United Kingdom). PCR products were isolated by unidirectional T/A cloning with the eucaryotic expression vector pCR3.1-Uni (Invitrogen). In all cases, the molecularly cloned TCLA env genes chosen for analysis encode the same adaptation-related amino acid changes as the virus population and recapitulate coreceptor use and neutralization sensitivity phenotypes (16, 28, 65). The specific 168P23 and 168C23 env genes chosen (GenBank accession numbers AF035532 and AF035534, respectively) also differ at random positions unrelated to neutralization sensitivity: gp120 C1 A62V, V1 V150M, V2 K164T, gp41 ectodomain A562T, H587Y, and cytoplasmic domain N810S (28; M. Trahey and J. H. Nunberg, unpublished). The human CD4 cDNA was obtained from R. Axel (through the NIH AIDS Research and Reference Reagent Program) and was subcloned for expression in pcDNA3.1+ (Invitrogen). Green fluorescent protein (GFP) was expressed using the pEGFP-N1 plasmid from Clontech Laboratories, Inc.
Antibodies and related reagents.
MAbs used in these studies are described in Table 2 and were generously provided as indicated. HIVIG was provided by A. Prince (New York Blood Center), and sCD4 was purchased from Progenics Corp. Cellular CD4 was detected in flow cytometry with MAB 379 (R&D Systems) or the non-gp120-competing MAb HCD4-A6 (Devaron, Inc.).
TABLE 2.
Env-directed MAbsa
| Antibody | Target | Nominal epitopeb | Source | Reference |
|---|---|---|---|---|
| 50.1 | V3 loop | ....RIHIG......... | Repligen Corp. | 63 |
| 58.2 | V3 loop | .......IGPGRAF.... | Repligen Corp. | 63 |
| 59.1 | V3 loop | ........GPGRAF.... | Repligen Corp. | 63 |
| 257-D | V3 loop | ...KRIHI.......... | S. Zolla-Pazner | 19 |
| 268-D | V3 loop | ......HIGPGR...... | S. Zolla-Pazner | 19 |
| 447-52D | V3 loop | ........GPGR...... | S. Zolla-Pazner | 19 |
| IgG1b12 | CD4-bs | D. Burton, from ARRRP | 5 | |
| 559-64D | CD4-bs | S. Zolla-Pazner | 25 | |
| F105 | CD4-bs | M. Posner | 50 | |
| 17b | CD4-i | See reference 27 | J. Robinson | 60 |
| 2F5 | gp41 | gp41...ELDKWA | H. Katinger | 40 |
| F240 | gp41 | gp41...aa 592–606 | M. Posner | 7 |
CD4-bs, CD4-binding site; CD4-i, CD4 induced; aa, amino acids; ARRRP, NIH AIDS Research and Reference Reagent Program.
Nominal MAb epitopes are reported in the associated references. Central V3 loop epitopes are arrayed as in Table 1.
Transfection protocol and analysis of expressed proteins.
FuGENE-6 reagent (Roche Molecular Biochemicals) was used to transfect simian COS-7 cells (American Type Culture Collection) for transient expression. In a typical experiment, 0.8 μg of GFP-expressing plasmid was mixed with 15.2 μg of Env- or CD4-expressing plasmid and applied (with 48 μl of FuGENE-6 reagent in 800 μl of Dulbecco's Modified Eagle Medium [DMEM; Gibco BRL]) to 106 COS-7 cells in a 10-cm-diameter culture dish in 10 ml of DMEM with 10% fetal bovine serum (FBS). Transfected cultures were washed 16 to 18 h later and refed with 10 ml of DMEM with 10% FBS. Transfection efficiencies were determined for Envs by immunochemical staining of cold methanol:acetone (1:1)-fixed monolayers using HIVIG with alkaline phosphate-conjugated goat anti-human IgG antibody (Kirkegaard & Perry Lab.) and PhThalo Red chromogen (Kirkegaard & Perry Lab.) or, for CD4 and GFP, by flow cytometry. Env expression was also assessed by Western blot analysis of cell surface proteins. Cells were biotinylated using NHS-LC-biotin (Pierce Chemical) (31) and lysed on ice in 50 mM Tris (pH 7.5), 150 mM NaCl, and 1% Triton X-100 containing 1 μg each of aprotinin, leupeptin, and pepstatin per ml. Env was immunoprecipitated using 5 μg of HIVIG and protein A-Sepharose (Sigma). Western blot analysis utilized avidin-horseradish peroxidase conjugate (Biomeda) and ECL-Plus detection.
Flow cytometry.
COS-7 cells were used in flow cytometry 40 h after FuGENE-6-mediated cotransfection with GFP and env plasmids. Parallel cultures were cotransfected with GFP and CD4 plasmids to assess nonspecific antibody binding and autofluorescence. For analysis, transfected cell cultures were resuspended using 0.1 mM EDTA in phosphate-buffered saline (PBS). A total of 2 × 105 cells were stained with HIVIG or Env-specific MAbs at 4°C for 20 min in 100 μl of PBS containing 2% FBS and 0.05% sodium azide. Specific MAbs are described in Table 2, and the concentration of each MAb used to assess binding to neutralization-sensitive TCLA Envs and neutralization-resistant PI Envs is listed in the legend to Fig. 3. MAb binding was detected by using either biotinylated anti-mouse IgG or biotinylated anti-human IgG antibody (Jackson ImmunoResearch Laboratories), followed by the use of streptavidin-CyChrome conjugate (PharMingen). Cells were analyzed with a Becton Dickinson FACSCalibur and CELLQuest software (Becton Dickinson Immunocytometry Systems). A double gate was defined by forward versus side scatter and by forward scatter versus the amount of GFP (FL-1). A total of 5,000 events within this gate were collected for analysis. CyChrome-labeled cells were detected in FL-3, and mean fluorescence intensity was determined as mean fluorescence channel (MFC) (CELLQuest). Nonspecific fluorescence, as measured in identically stained CD4- and GFP-cotransfected cells, occupied only the lowest fluorescence channels and was removed using histogram subtraction software (CELLQuest).
FIG. 3.

Summary of MAb binding to 168P and 168C Envs. MAb
binding was determined by flow cytometry as described in Materials and
Methods and quantitated using MFC computation (CELLQuest software). The
respective MFC values for binding to 168P and 168C Envs were normalized
relative to HIVIG binding to account for modest differences in Env
expression. The normalized values for MAb binding to 168P and 168C Envs
are plotted. Among all experiments, MFC values for HIVIG binding to
168P and 168C Envs were 70 ± 35 and 67 ± 28, respectively.
The ratio of HIVIG MFC values within any experiment (1.07 ± 0.39)
was also consistent with the ratio of transfection efficiencies
(1.01 ± 0.13). Representative MAb binding values compared within
one experiment are shown. Purified MAbs were used at a concentration of
10 μg/ml (except for MAbs 58.2 and 59.1, which were used as a 1:100
dilution of ascites fluids), and sCD4 was used at a concentration of 50
μg/ml. At these concentrations, the relative amounts of
neutralization of 168P and 168C, respectively, are shown in parentheses
(as percentages); few if any of the antibody reagents significantly
neutralized 168P. Symbols:
 , 50.1
(0, 95); ⊕, 58.2 (55, 99);
, 50.1
(0, 95); ⊕, 58.2 (55, 99); 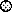 ,
59.1 (20, 70); ○, 257-D (50, 97);
,
59.1 (20, 70); ○, 257-D (50, 97);
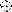 , 268-D (50, 95); ●, 447-52D
(75, 99);
, 268-D (50, 95); ●, 447-52D
(75, 99);
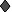 ,
559-64D (45, 95);
,
559-64D (45, 95);
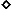 , IgGb12
(25, 55); ⧫, F105 (30, 65); ▴, 17b (30, 80); ▵, sCD4 plus 17b
(not applicable); ■, 2F5 (60, 75); □, F240 (0, 0);
, IgGb12
(25, 55); ⧫, F105 (30, 65); ▴, 17b (30, 80); ▵, sCD4 plus 17b
(not applicable); ■, 2F5 (60, 75); □, F240 (0, 0);
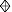 , sCD4
(80, 99 at 10 μg/ml).
, sCD4
(80, 99 at 10 μg/ml).
Quantum PE-Cy5 beads (Flow Cytometry Standards Corp., San Juan, P.R.) were used to convert CyChrome fluorescence intensity to numbers of fluorophores/cell (molecules of equivalent soluble fluorophore). At the instrument settings used in our measurements, we determined a linear relationship: log10(MFC) = [0.811 × log10 (molecules of equivalent soluble fluorophore)] − 2.072.
Virion capture assays.
MAb binding to intact virions was measured using a virion capture enzyme-linked immunosorbent assay (ELISA) as previously described (42, 43). Virus stocks were comprised of cleared cell culture supernatants, and captured HIV p24 core protein was quantitated by a separate p24 antigen capture ELISA (Coulter). All comparisons of PI versus TCLA virus binding were normalized using HIVIG as a nondiscriminating virion capture reagent.
The infectivity of MAb-captured virions was determined in a different format using MAb-coated magnetic beads (Dynal A.S., Oslo, Norway). Streptavidin-coated M-280 DynaBeads were saturated with biotinylated MAb 50.1 in PBS containing 2% FBS or incubated with an equal amount of non-biotinylated MAb 50.1 as a control. For virion capture studies, 0.5 mg of beads (containing approximately 2 μg of biotinylated MAb) was incubated with 168P or 168C virus stocks (containing approximately 200 ng of gp 120). Incubations were carried out with intermittent mixing for 2 h at either 4 or 37°C. The magnetic beads were subsequently washed three times in PBS–2% FBS at 4°C and were then serially diluted onto U87-CD4-CXCR4 cells to assess retained infectivity. Microcultures were rinsed after 2 days of culture, and infected cells were immunochemically stained using HIVIG.
RESULTS
Cross-sectional comparisons of neutralization sensitivity in unrelated PI and TCLA viruses can be confounded by unknown immunochemical differences between genetically disparate viruses. Therefore, we and others (2, 16, 65, 67) have focused on the analysis of pedigreed PI and TCLA viruses, i.e., TCLA viruses derived by stringent adaptation of PI viruses to persistent growth in initially restrictive T-cell lines. We have previously described two pairs of pedigreed PI and TCLA viruses (Table 1); our present studies of MAb binding initially examined one of these pairs, 168P and 168C.
The dual-coreceptor-utilizing PI virus 168P (ACH168.10) (12, 59) had been adapted to persistent growth in the H9 T-cell line (65). The resulting TCLA 168C env displays a marked preference for use of the CXCR4 (versus CCR5) coreceptor and encodes three adaptation-related amino acid changes (in the V2 loop, C2 domain, and V3 loop) (Table 1), each of which is required for full neutralization sensitivity (Trahey and Nunberg, unpublished). In an earlier study (65), the question of MAb binding to primary T lymphocytes infected with either 168P or 168C was examined, but the interpretation of these studies was confounded by the possibility that binding may have also been to monomeric gp120 that was shed and then captured by cellular CD4, rather than to an authentic cell-associated Env complex. Here, we addressed this question utilizing molecularly cloned 168P and 168C env genes expressed on the surface of CD4-negative COS-7 cells. The molecularly cloned env genes (168P23 and 168C23) encode functional Envs that recapitulate the neutralization and coreceptor phenotypes of the original PI and TCLA virus populations (28). Our initial flow cytometric efforts to quantitate antibody binding to Env on transiently-transfected COS-7 cells were limited, however, by the overlap between the specific fluorescence from transfected cells and the background autofluorescence from nontransfected cells in the population. The resulting “shoulder” of MAb binding makes quantitation problematic.
Flow cytometric analysis of MAb binding.
To separate this background from authentic binding to Env-expressing cells, we developed a strategy in which transfected cells could be analyzed in isolation from nontransfected cells. COS-7 cells were cotransfected with a plasmid expressing GFP and an excess of plasmid expressing Env. We were then able, by flow cytometry, to focus our analysis on GFP-positive cells and thus were able to greatly enrich for cells that also expressed Env. The ratio of Env- to GFP-expressing plasmids (20:1) was optimized such that <95% of GFP-positive cells were also Env positive. Residual background and autofluorescence were determined from binding to cells cotransfected with CD4 and GFP plasmids (20:1); background was uniformly faint and subtraction had little effect on measurements of specific MAb binding.
Clear and symmetric binding histograms were obtained using the GFP cotransfection method (e.g., Fig. 1 and 2). Specific antibody binding was quantified using MFC calculations. These determinations of binding were robust and largely independent of transfection parameters, specific GFP-gating choices, or background subtraction. In replicate measures of antibody binding, MFC values were consistently within 20%. Fluorescent bead standards (Flow Cytometry Standards Corp.) were used to convert MFC values to relative numbers of bound antibody molecules (see Materials and Methods). At the instrument settings used in our antibody binding studies and within the range of typical MFC measurements, and experimental variation of 20% in MFC translates roughly to a 20% variation in the determined number of bound antibody molecules. Specifically, if there were to be a 10-fold reduction in the binding of antibody to PI (versus TCLA) Env, then this would be reflected in a 7-fold reduction in MFC (see Materials and Methods). The sensitivity of this flow cytometric method is more than sufficient to detect binding differences that approach the differences observed in neutralization sensitivity.
FIG. 1.

HIVIG as a nondiscriminating measure of Env expression. COS-7 cells were transfected to express GFP and either 168P Env or 168C Env as described in Materials and Methods. (A) Flow cytometric analysis of HIVIG staining of GFP-positive cells expressing either 168P Env (dark line) or 168C Env (light line). (B) Transfected cultures were biotinylated on the cell surface with NHS-LC-biotin reagent (Pierce), and Env was immunoprecipitated with HIVIG and detected by Western blot analysis using avidin-horseradish peroxidase conjugate. Transfection efficiency was determined by immunochemical staining of fixed monolayers using HIVIG; the percentage of cells stained by HIVIG is indicated.
FIG. 2.




Flow cytometric analysis of MAb binding to 168P and 168C Envs. (A, C, E, and G) Neutralization sensitivity of 168P (dark line) and 168C (light line) viruses to the indicated MAbs was determined using U87-CD4-CXCR4 cells as described in Materials and Methods. Infected cells (foci) were determined by microscopic analysis of immunochemically stained monolayers, and the number of foci was compared to that obtained in the absence of MAb. (B, D, F, and H) COS-7 cells were transfected to express GFP and either 168P Env or 168C Env as described in Materials and Methods. Flow cytometric analysis of MAb binding to GFP-positive cells expressing either 168P Env (dark line) or 168C Env (light line) is illustrated. MAbs were used at the discriminating concentrations determined by neutralization as indicated in the legend to Fig. 3.
In most experiments, transfection efficiencies and Env expression levels were similar for PI and TCLA env plasmids. Cell surface Env expression levels were normalized by flow cytometry using HIVIG as a generic polyclonal reagent that we presumed did not discriminate markedly in binding between PI and TCLA Envs (Fig. 1A). This presumption was confirmed by Western blot analysis of immunoprecipitable Env from detergent lysates of transfected cell cultures (Fig. 1B). This method provides a known nondiscriminatory measure of Env (35) and the concordance among the immunochemical measurement of solubilized monomeric Env, the transfection efficiency, and the flow cytometric analysis of cell-surface Env complex confirmed that HIVIG can be used as a nondiscriminating measure of cell surface PI and TCLA Env expression. Thus, we were able to normalize MFC values across MAb binding studies of independently transfected PI and TCLA Env-expressing cell cultures. Given that PI and TCLA Env expression levels were generally comparable in our studies (see the legend to Fig. 3), normalization had little effect on relative binding values to PI and TCLA Envs.
Note also that proteolytic processing of gp160 was incomplete in these transfected cells (Fig. 1B) and that only approximately 25% of cell surface Env was present as mature gp120. The relative extent of processing was, however, comparable between the PI and TCLA Envs. The extent of gp160 cleavage was not appreciably enhanced by coexpression of furin, one of several Golgi-associated proteases that have been implicated in Env processing (22) and one that has been shown to effectively process secreted gp140 (3).
MAb binding to 168P and 168C Envs.
Using this flow cytometric method, we examined the binding of MAbs directed to key Env targets: the V3 loop, the CD4-binding site, the CD4-induced determinant of gp120, and the ectodomain of gp41 (Table 2). These targets were chosen because they are well-mapped and well-defined neutralizing determinants of TCLA viruses and because they have been suggested to be relatively inaccessible on the PI virus Env complex.
To ensure that MAb binding was assessed at a concentration that is functionally relevant and informative, the neutralization sensitivities of 168P and 168C viruses were determined for each MAb reagent used (Fig. 2A, C, E, and G). In all cases, this PI virus was largely refractory to neutralization. A MAb concentration that clearly distinguished between the neutralization-resistant PI virus and the neutralization-sensitive TCLA virus was chosen (where possible) for flow cytometric binding analysis. In some cases, neutralization of the PI and TCLA viruses differed by over 20-fold at the concentrations used (e.g., MAbs 50.1, 58.2, 257-D, 268-D, 447-52D, and 559-64D). Thus, we were able to obtain accurate flow cytometric quantitation of binding to PI and TCLA Envs at a concentration of MAb that yields dramatically different outcomes in neutralization (Fig. 2B, D, F, and H). A summary comparing MAb binding to 168P PI and 168C TCLA virus Envs is presented in Fig. 3. In all cases, Env-specific MAbs, regardless of specificity, bound equally to neutralization-resistant PI Env and neutralization-sensitive TCLA Env.
One potential variable in studies of gp120-binding MAbs relates to the relative ease with which gp120 can be shed from the cell surface Env complex (38). This same concern may apply, but in reverse, to the binding of gp41-directed MAbs. Although significant amounts of gp120 were found to accumulate in 24-h culture supernatants of cells expressing either 168P or 168C Env, we did not detect loss of gp120 during the short 4°C incubations used in these studies. Comparable amounts of gp120 and gp41 were detected by flow cytometry with cells regardless of prior incubation, including incubation in the presence of sCD4 (data not shown). Thus, gp120 shedding and/or exposure of gp41 was not a confounding variable in the present studies.
The single amino acid change in the V3 loop of 168C did not appear to affect binding to distal epitopes of the V3 MAbs. Although the specific targets for some of the non-V3-directed MAbs are less well defined, it should be remembered that 168P and 168C Envs are very nearly identical in sequence. The slight reduction in sCD4 binding to 168C (Fig. 2H) may be attributable to the amino acid change in the C2 domain near the CD4-binding pocket (I282N) (27), but this difference in sCD4 binding is not seen in the comparison of other PI and TCLA viruses (Fig. 4).
FIG. 4.

Summary of MAb binding to 320SI and 320SI-C3.3 Envs.
Methods and MAb concentrations are as described in the legend to Fig.
3. At these concentrations, the relative amounts of neutralization of
320SI and 320SI-C3.3, respectively, are shown in parentheses (as
percentages); none of the antibody reagents significantly neutralized
320SI. Symbols:
 , 50.1
(0, 45); ⊕, 58.2 (10, 70); ,
59.1 (15, 70);
, 257-D (0, 60);
, 268-D (0, 0); ●, 447-52D
(2, 95);
,
559-64D (20, 30); ◊, lgGb12 (25, 99); ⧫, F105 (10, 70); ▴, 17b
(10, 60); ▵, sCD4 plus 17b (not applicable); ■, 2F5 (40, 99); □,
F240 (0, 0);
, sCD4
(0, 99 at 10 μg/ml). The mean ratio of HIVIG MFC values for 320SI and
320SI-C3.3 Envs was 0.87 ± 0.21, and these values varied among
experiments similarly to those of 168P and 168C.
, 50.1
(0, 45); ⊕, 58.2 (10, 70); ,
59.1 (15, 70);
, 257-D (0, 60);
, 268-D (0, 0); ●, 447-52D
(2, 95);
,
559-64D (20, 30); ◊, lgGb12 (25, 99); ⧫, F105 (10, 70); ▴, 17b
(10, 60); ▵, sCD4 plus 17b (not applicable); ■, 2F5 (40, 99); □,
F240 (0, 0);
, sCD4
(0, 99 at 10 μg/ml). The mean ratio of HIVIG MFC values for 320SI and
320SI-C3.3 Envs was 0.87 ± 0.21, and these values varied among
experiments similarly to those of 168P and 168C.
In this regard, it is also interesting to note the binding of MAb 17b. This epitope is enhanced upon sCD4 binding (61) and is thought to reflect conformational changes that parallel those within the functioning Env. Based on structural and mutagenic analysis, this site is believed to define an Env interface critical for subsequent binding to a coreceptor (51). In our studies, the level of binding of MAb 17b to 168C was somewhat greater than to 168P (Fig. 3), although this difference between PI and TCLA Envs was not seen in other PI and TCLA viruses (Fig. 4 and reference 58). If PI and TCLA Envs differ markedly in their ability to bind MAb 17b or to undergo the CD4-induced conformational changes that enhance MAb 17b binding, then it was not apparent in these studies.
MAb binding to a second pair of PI and TCLA viruses.
We sought to extend these findings using an additional pair of pedigreed PI and TCLA viruses. We had previously described the functional and genetic changes that accompanied T-cell line adaptation of the molecularly cloned PI virus 320SI (ACH320.2A.1.2) (20, 21). The env gene of the resulting TCLA virus 320SI-C3.3 encodes two adaptation-related amino acid changes (in the V2 and V3 loops; Table 1), and this TCLA Env continues to utilize both CCR5 and CXCR4 coreceptors (16). For the present study, the env genes of 320SI and 320Sl-C3.3 were molecularly cloned using PCR and envA and envN oligonucleotide primers as previously described (28). DNA sequence analysis confirmed the adaptation-associated amino acid changes originally noted in the virus population.
Flow cytometric analysis of binding to GFP-cotransfected cells expressing either 320Sl or 320Sl-C3.3 Env is summarized in Fig. 4. Again, specific MAb binding to the neutralization-resistant PI and neutralization-sensitive TCLA Envs was equal. The sole exception is of interest: MAb 268-D recognizes the nominal epitope HIGPGR (19), which is present on the PI Env but is altered to RIGPGR on the TCLA Env. The observed binding of MAb 268-D to the PI Env and not to the TCLA Env highlights the specificity of the flow cytometric analysis. Other nominal MAb epitopes, such as those of MAbs 50.1 and 257-D, were not entirely predictive of binding to the native Env protein, as has been demonstrated previously (42). Binding of these MAbs to sequences that differ from the nominal epitope (Tables 1 and 2) was detected under saturating conditions, but neutralization potency against the 320SI-C3.3 TCLA virus was clearly reduced relative to, e.g., 168C (see the legend to Fig. 3 and 4). In contrast to what has been observed as sCD4-induced enhancement of MAb 17b binding to other Envs, and to 168P and 168C Envs, sCD4 did not appreciably increase binding of MAb 17b to either 320SI or 320SI-C3.3 Env. This result cannot be attributed to differential sCD4-induced gp120 shedding as, again, we could not detect significant loss of gp120 during incubation at 4°C (data not shown).
Because temperature can play a major role in Env dynamics and interactions (13, 17, 37, 55), we examined whether differences in MAb binding to PI and TCLA Envs might be observed if incubations were performed at physiological temperatures. These efforts were confounded, however, by significant albeit inconsistent losses of membrane proteins, despite the addition of up to 0.2% sodium azide. The loss was not limited to gp120 but included comparable losses in integral membrane proteins such as gp41 and CD4 and occurred in the absence of MAb or sCD4 (data not shown). We presume that these losses reflect membrane blebbing and recycling within the culture (32); much of the membrane protein lost from the GFP-positive cell population at 37°C can be accounted for as a generalized increase in staining of the GFP-negative cell population. Any measure of gp120 shedding at 37°C is confounded by the general loss of membrane proteins. Despite these technical difficulties in quantitation, we could detect no systematic differences in MAb binding to PI (168P or 320SI) versus TCLA (168C or 320SI.C3.3) Envs in any experiment in which MAb or sCD4 was bound at either 22 or 37°C (data not shown). Although the extension of our flow cytometric studies to physiological temperature conditions must remain preliminary, our findings consistently suggest that the extent of MAb binding to PI and TCLA Envs is also equal at 37°C.
MAb binding to intact and infectious PI and TCLA virions.
The studies described above utilized molecularly cloned Envs expressed on the surface of transfected cells. These Envs are fully functional in mediating cell-cell fusion and recapitulate the neutralization and coreceptor phenotypes of the parent virus population (16, 28). We presume that these Envs are presented on the cell surface much as on HIV virions. However, the high level of Env production during transient expression also results in the accumulation of unprocessed gp160 on the cell surface (Fig. 1B), presumably due to saturation of the cellular proteolytic machinery. In actual HIV infection, these immature gp160 molecules are also present but are possibly excluded from the budding virion (14). The structure of the virion Env may also be modified by interaction with virion core proteins (15). Therefore, we wished to confirm our findings of equal binding to PI and TCLA Envs with authentic virion particles. For these studies, we utilized a virion capture ELISA (45) that had previously been used to assess MAb binding to specific virion epitopes (42, 43). In this assay, virion particles are captured onto microtiter wells to which the specific test MAb is bound, and these captured virions are subsequently quantitated using a separate p24 core antigen ELISA. Previous studies have confirmed the antigenic selectivity of this assay and its ability to specifically capture infectious virus particles (7, 42, 43). We used this assay to extend our flow cytometric findings to the Env complex of intact PI and TCLA virions. As in these flow cytometric assays, HIVIG was used as a generic capture reagent to normalize binding of each virus stock. Importantly, MAb incubations in the virion capture format were performed at 37°C.
The relative amount of PI and TCLA virus p24 captured in this assay is shown in Fig. 5. In all cases, the PI virion was efficiently captured by MAbs that were unable to neutralize the PI virus. Where the nominal MAb epitope was conserved between the PI and TCLA Envs, the assay resulted in the isolation of up to twofold-more PI virus than TCLA virus. Although the basis for the greater capture of PI virus is unclear, the finding was consistent between the two pairs of pedigreed viruses. In the case of MAb 268-D, where the nominal epitope is altered on the TCLA virus 320SI-C3.3, capture of this virus was markedly reduced. The nominal epitope of MAb 257-D differs from that of 320SI at a single site and from that of 320SI-C3.3 at one additional amino acid. Although the common difference does not appear to affect virus capture, the additional amino acid change in the TCLA virus may be responsible for the reduction in 320SI-C3.3 binding. It is worth noting that this reduction in binding was not apparent under the saturating antibody conditions used in flow cytometry but is perhaps reflected in the reduced neutralization sensitivity of 320SI-C3.3. These known immunochemical differences provide additional confirmation of the specificity of this assay. In the absence of confounding local immunochemical differences, MAb capture of the PI virion was at least as efficient as that of the TCLA virion.
FIG. 5.

Relative capture of PI and TCLA virions. Virus stocks were prepared from supernatants of acutely infected peripheral blood mononuclear cells (PI virus) or H9 cells (TCLA virus). The virion capture ELISA was performed as described by Nyambi and colleagues (42). V3-loop-directed MAbs 58.2, 268-D, 257-D, and 447-52D were bound to microtiter wells in 100 μl at a concentration of 10 μg/ml; HIVIG was bound at 50 μg/ml. Virions were subsequently captured from 100 μl containing 100 ng of p24 per ml. The amount of virion captured by the specific MAbs is expressed as a percentage of that captured by HIVIG, in order to normalize results across virus stocks. For clarity, we show the relative capture of TCLA virion versus PI virion as the ratio of the HIVIG-normalized percentages. In the experiments shown, the underlying amounts of virion p24 captured by HIVIG were as follows: 168P, 902 pg/ml; 168C, 927 pg/ml; 320SI, 370 pg/ml; and 320SI-C3.3, 257 pg/ml. The amounts of 168P and 320SI PI virus captured by the MAbs were as follows, respectively: 58.2, 299 and 384 pg/ml; 268-D, 439 and 116 pg/ml; 257-D, 665 and 196 pg/ml; and 447-52D, 864 and 180 pg/ml. The relative capture of 168C and 168P is shown in dark bars, and 320SI-C3.3 and 320SI in light bars. As calculable, correspondingly less TCLA virion was captured than PI virion. ∗, 320SI-C3.3 virions whose V3 loop sequence contains an alteration of the nominal MAb epitope.
These virion capture ELISA findings clearly demonstrate that MAbs are able to bind equally to the Env complex in both the TCLA and PI virus populations. These populations are, however, heterodisperse and contain a majority of so-called defective particles—virions which are unable to successfully complete infection (30). To ascertain whether MAbs which distinguish between PI and TCLA viruses by differential neutralization bind comparably to infectious PI and TCLA virions, we utilized MAb-coated magnetic beads to isolate virus and to quantitate captured viral infectivity. Biotinylated MAb 50.1 was attached to streptavidin-coated magnetic beads (M-280 Dynabeads; Dynal) and incubated with virus stocks of either 168P or 168C. Magnetic streptavidin-coated beads which had been preincubated with nonbiotinylated MAb 50.1 served as specificity controls. Beads were subsequently washed extensively and diluted onto U87-CD4-CXCR4 target cells. As shown in Fig. 6, biotinylated MAb 50.1 was able to specifically capture infectious virus from both the PI and TCLA virus populations. In both cases and at either 4 or 37°C, approximately 3 to 5% of the initial viral infectivity was retained on the MAb-coated magnetic beads. Little or no infectivity was retained by control beads that had been mock incubated with nonbiotinylated MAb 50.1. Previous studies by Nyambi et al. (43) have also demonstrated MAb capture of infectious PI virus by using the microtiter well format.
FIG. 6.

MAb capture of infectious PI and TCLA virions. Virions were captured from 168P and 168C virus stocks (dark and light symbols, respectively) using streptavidin-coated M-280 Dynabeads to which biotinylated MAb 50.1 had been bound. Infectivity retained by the MAb was assessed by culturing U87-CD4-CXCR4 cells with the extensively washed magnetic beads, and the numbers of infected cell foci were determined as described in Materials and Methods. Incubations of virions with MAb-coated beads were at 4 or 37°C (circle and square symbols), and retained infectivity is compared to that in the initial virus stock used for capture (triangle symbols). Streptavidin-coated beads that were incubated with nonbiotinylated MAb 50.1 (mock) served as specificity controls (data not plotted; all, ≤5 foci/well).
These virion capture findings clearly demonstrate that MAbs are able to bind to the Env complex of both infectious neutralization-resistant PI virions and infectious neutralization-sensitive TCLA virions at 37°C and corroborate results obtained from the flow cytometric analysis of a cell-associated Env complex. Therefore, the differential sensitivity of PI and TCLA viruses to neutralization does not derive from a simple difference in initial MAb binding.
Flow cytometric analysis of MAb binding affinities.
Not only are the extents of initial MAb binding equal between PI and TCLA Envs, but so are the affinities of MAb binding. To derive dissociation constants (Kd) for the binding of anti-Env MAbs, we varied the antibody concentration in the binding reaction and used flow cytometry to determine relative MFC values. These values are equivalent to the relative occupancy value (θ) modeled by Klasse and Moore (26). Thus, MFC = (A/Kd)/(1 + A/Kd), where A is the concentration of free MAb. Under conditions used in the binding assay, where MAb is in vast excess of cell surface Env, A approximates the initial MAb concentration, A0. One can thus approximate Kd by determining the A0 at which the measured MFC is reduced to 50% of the maximum (48, 55). Binding curves obtained using two V3 loop-directed MAbs (50.1 and 447-52D) and cells expressing either 168P or 168C Env are shown in Fig. 7. Approximate Kd values for MAb binding to cell-associated PI and TCLA Envs were equal: 7 nM for MAb 50.1 and 3 nM for MAb 447-52D. At all MAb concentrations tested, the ratio of PI and TCLA MFC values remained constant, allowing further confirmation that the Kd values for MAb binding to the PI and TCLA Envs were approximately equal.
FIG. 7.

Flow cytometric determination of the binding affinity of MAbs 50.1 and 447-52D to 168P and 168C Envs. In this experiment, cotransfected COS-7 cells expressing GFP and either 168P Env (dark lines) or 168C Env (light lines) were stained using the indicated concentrations of MAb 50.1 (A) or 447-52D (B). MFC values for MAb binding to GFP-positive cells were determined as described for Fig. 1 and 2. The percentage of maximal binding was determined for each MAb dilution as the ratio of MFC values.
MAb binding to TCLA, but not PI, Env correlates with neutralization.
It is interesting to note that the binding curves for the two V3-loop-directed MAbs shown in Fig. 7 approximate the curves of TCLA virus neutralization shown in Fig. 2A and D. In the case of the TCLA virus, binding appears to correlate with, and presumably mediate, virus neutralization. These findings are similar to the results of studies of MAb binding to TCLA Envs and Env peptides (6, 48, 62) although, as demonstrated above and by others (7, 43), MAb binding to virions can clearly occur in the absence of even TCLA virus neutralization (e.g., MAb F240). The unique aspect of the present study, however, is that the PI Env complex can be saturated with MAbs that effectively neutralize TCLA viruses, but these saturating concentrations of MAb are not sufficient to affect PI virus neutralization.
DISCUSSION
The remarkable dichotomy in neutralization sensitivity of PI and TCLA viruses raises two related and fundamental questions: what are the mechanisms of antibody-mediated virus neutralization, and how do these differ between PI and TCLA viruses? In other words, by what mechanism do viruses with Env complexes that differ at only 2 or 3 amino acids display such strikingly different patterns of neutralization? Our results clearly demonstrate that these differences in neutralization sensitivity can exist without apparent differences in the initial antibody binding event, and suggest that the differential consequences of MAb binding reflect differences in the subsequent functioning of PI and TCLA Envs in mediating virus fusion and entry.
Previous studies have examined the relationship between MAb binding and the differential neutralization sensitivity of PI and TCLA viruses, but conclusions have been discordant and confounded by methodological limitations. In the present work, we have revisited this question using a panel of well-defined PI and TCLA viruses and three complementary techniques to arrive at a more definitive conclusion. By using molecularly cloned Envs derived from pedigreed PI and TCLA viruses that differ in a very limited number of amino acids, we have reduced to a minimum any local immunochemical differences that may enter into cross-sectional studies of the genetically diverse viruses used by others (4, 56, 57). Adventitious differences arising in one pair of related PI and TCLA viruses were excluded by comparison with a second pair of PI and TCLA viruses. By using CD4-negative cells for Env expression, we have circumvented concerns regarding MAb binding to shed monomeric gp120 that may then be captured by CD4-positive T cells (4, 54, 65, 68). Moreover, by assessing MAb binding to both intact virions and to cell surface-expressed Env, we have attempted to mitigate limitations inherent in each of the methodologies. The concordance of results obtained using cell surface-expressed Env and intact virion particles provides further support to our conclusions and the respective methodologies.
We find that differential neutralization sensitivity of PI and TCLA viruses can exist without demonstrable differences in the extent or affinity of antibody binding. These findings echo those of others who have also demonstrated MAb binding to intact and infectious PI virions and cell-associated PI Env (42, 43, 56, 57, 68). Similarly, there have been reports of MAb binding to TCLA viruses in the absence of a neutralizing outcome (7, 43). What has been lacking in these analyses has been the direct comparison between antigenically related neutralization-resistant PI and neutralization-sensitive TCLA viruses.
Some of these earlier studies also report, as do we, MAb binding to epitopes that have been hypothesized to be buried within the Env complex (7, 42, 43). These observations raise the possibility that the native, membrane-bound Env complex may differ, structurally or dynamically, from the model derived from immunological and structural analyses of soluble Env molecules (gp120 and gp140) (27, 39, 66).
Our findings stand in apparent contradiction to those anticipated by the prevalent model, which suggests that the differences in neutralization sensitivity of PI and TCLA viruses reflect differences in the ability of antibodies to bind to the respective Env complexes (36). This model proposes (i) that adaptation to persistent growth in T-cell lines selects for virions that are able to rapidly bind and enter target cells, (ii) that the structural and/or dynamic changes that facilitate binding to cell receptors concomitantly expose critical neutralizing determinants within the TCLA Env complex, (iii) that binding of antibody to the TCLA Env complex necessarily results in neutralization (48, 49), and (iv) that the relative resistance of PI viruses to neutralization reflects the inaccessibility of the PI Env complex to antibody binding.
Since the initial discovery that PI viruses were resistant to neutralization by gp120 vaccine sera that neutralize TCLA viruses (cited in reference, 9), several alternative explanations have been proposed, but none has been entirely satisfactory. One might imagine that passage of the TCLA virus in the absence of in vivo pressure from antibody may permit the Env to divest itself of unnecessary shielding mechanisms and thereby become more sensitive to neutralization. This explanation is weakened, however, by findings that continued passage of PI viruses in permissive cells (primary T lymphocytes and, for some isolates, MT4 cells) does not select for neutralization sensitivity (16, 46). Alternatively, it has been suggested that PI virions carry more Env spikes than TCLA virions and that this increased spike density may result either in differential shielding of critical PI Env determinants (34, 36, 44), or in an increase in the amount of antibody needed to inactivate a critical number of Env spikes (26). Other studies, however, have not observed consistent differences in spike density (24, 65), and we found that our PI and TCLA virions contain comparable ratios of gp120 Env to p24 Gag protein (Materials and Methods; data not shown). If spike density is comparable between PI and TCLA virions, one might still propose that PI and TCLA virions differ in the minimal level of MAb binding needed to affect neutralization (26). However in our studies, PI viruses remain infectious at MAb concentrations that saturate Env binding. With the discovery of HIV coreceptors, another explanation for differential sensitivity to neutralization was considered, that neutralization sensitivity of PI and TCLA viruses might be related to differential coreceptor utilization (CCR5 or CXCR4, respectively). Multiple studies, however, argue against a role of this dichotomy in coreceptor use in determining neutralization sensitivity (16, 28, 33, 61).
Some of the apparent contradictions between our present findings and those anticipated by the prevalent model of Moore and Ho (36) may derive from previous conclusions from studies of antigenically dissimilar PI and TCLA viruses and Envs (4, 36, 56, 57). Others may derive from generalizations to PI viruses of findings obtained using TCLA Envs and their recombinant gp120 and gp140 molecules (35, 48, 54). Whereas we agree with Parren et al. (48) that, among TCLA viruses and their neutralizing MAbs, occupancy of sites on the virion determines neutralization, we question the extrapolation of these findings to PI viruses.
Ultimately, some of the discrepancies between studies may be attributable to universal and insurmountable limits in the ability to definitively correlate measures of MAb binding and virus neutralization. For instance, all studies of MAb binding to cell-associated gp120-gp41 Env complexes are confounded by the presence of uncleaved, nonfunctional gp160 molecules, and this situation is exacerbated in studies using highly expressing transfected cells in which the cell's ability to proteolytically process gp160 is overwhelmed. Although antigenic differences between functional gp120-gp41 and nonfunctional gp160 Env complexes have not been defined, these remain a theoretical concern (47). In the one instance for which structural information is available, the homologous proteolytic cleavage of the influenza virus hemagglutinin (HA) precursor (designated HA0) to mature HA1 and HA2 forms, it has been shown to have only localized effects on HA structure (8). It is unlikely that local structural changes induced by proteolytic cleavage of the preassembled Env oligomer would extend to the broad range of MAb specificities used in this work.
By contrast, little uncleaved gp160 is incorporated into virus particles (reference 14 and data not shown). Therefore, concerns arising from the presence of uncleaved gp160 molecules are minimized in studies of MAb binding to intact virions (42, 43, 56). Such studies also incorporate the structural changes in Env that may derive through interaction with virion core proteins (15).
However, studies of MAb binding to intact virions suffer from the inability to discriminate between MAb binding to infectious virions and to the majority of defective virion particles (30). Thus, one can argue that binding measurements may be unrelated to neutralization of infectious virions. It is largely unknown, however, whether the majority of virions bear inherently defective Env complexes or whether defectiveness reflects a stochastic outcome of attempted infection. Although some virions may lack gp120 as a result of shedding, this mechanism cannot account for the overwhelming bulk of defective virions. Even if one focuses exclusively on MAb binding to infectious particles, as we and others (43) have reported, one cannot exclude the possibility that infectious virions are captured via MAb binding to a defective Env spike. Ultimately, virion stocks contain a range of Env spike densities and functionality. All assays reflect averages within this heterodisperse population, thus making direct comparisons between immunochemical and structural measurements difficult.
This discussion is, of course, not to suggest that immunochemical and functional studies of HIV Env are without merit. Rather, we seek to examine the limits which apply variously to all past and present studies.
If differences in MAb binding affinities do not determine differences in the neutralization sensitivity of PI and TCLA viruses, then what does? Although our equilibrium measurements of binding affinity do not capture the underlying rate constants, it is unlikely that the equal affinities we measure result from compensating differences in on and off rates. Perhaps PI and TCLA Envs assume different conformations or quaternary structures on resting virions, such that initial antibody binding occurs with equal affinity but with different consequences upon subsequent activation of Env. For instance, antibody binding might lock the TCLA Env into a nonfunctioning conformation, whereas the PI Env might be free to bypass this localized perturbation. It is, however, unlikely that these localized differences would be reflected in all MAb specificities examined in the present study. Alternatively, neutralizing MAbs might cross-link adjacent TCLA Env monomers, whereas PI Env complexes might be configured so as to make MAb bridging more difficult. Cross-linking, however, is not required for TCLA virus neutralization (1, 53).
More likely, PI and TCLA Envs may differ in the subsequent conformational changes and/or protein-protein interactions that mediate Env function. We might speculate, for example, that the energetics of receptor binding and subsequent gp120-gp41 conformational changes may differ between PI and TCLA viruses. In the case of PI viruses, these energetics may be sufficient to dislodge bound MAb during Env functioning. Studies are underway to explore the retention of specific MAbs during Env functioning (Trahey and Nunberg, unpublished).
Our findings focus attention on differences that arise subsequent to the initial antibody binding event. If the initial MAb binding event is similar for TCLA and PI virions, then the differential outcome of binding (neutralization or lack thereof) must clearly arise through structural and mechanistic differences between the functioning TCLA and PI Envs. Thus, this report directs future studies towards a more detailed examination of the process of Env-mediated binding, fusion, and entry. Such efforts will shed light onto the mechanisms of virus neutralization and may illuminate the structural basis for the broadly neutralizing antibody response elicited by immunogens comprising the functioning Env (29, 41). From these studies may emerge novel strategies and targets for the development of effective HIV vaccines and antiviral agents.
ACKNOWLEDGMENTS
We are grateful to the following who generously provided antibody reagents: James Robinson, Marshall Posner, Lisa Cavacini, Hermann Katinger, Dennis Burton, Fred Prince, James Hildreth, and Daniel Witt (Repligen Corp.). Other reagents were made available through the NIH AIDS Research and Reference Reagent Program and the NIBSC AIDS Reagent Project of the United Kingdom. Furin cDNA was kindly provided by Gary Thomas (Oregon Health Sciences University). We also thank P. J. Klasse for useful discussions regarding his modeling of virus neutralization.
The present work was supported by Public Health Service grants AI44312 and AI44669 (J.H.N.) and AI32424, AI36085, and HL59725 (S.Z.-P.) and by the Department of Veterans Affairs (S.Z.-P.). Quantitative chemifluorescence analysis of virion proteins was made possible in part by a gift from the J. B. Pendleton Charitable Trust (J.H.N.).
REFERENCES
- 1.Barbas C F, III, Hu D, Dunlop N, Sawyer L, Cababa D, Hendry R M, Nara P L, Burton D R. In vitro evolution of a neutralizing human antibody to human immunodeficiency virus type 1 to enhance affinity and broaden strain cross-reactivity. Proc Natl Acad Sci USA. 1994;91:3809–3813. doi: 10.1073/pnas.91.9.3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beddows S, Louisirirotchanakul S, Cheingsong-Popov R, Easterbrook P J, Simmonds P, Weber J. Neutralization of primary and T-cell line adapted isolates of human immunodeficiency virus type 1: role of V3-specific antibodies. J Gen Virol. 1998;79:77–82. doi: 10.1099/0022-1317-79-1-77. [DOI] [PubMed] [Google Scholar]
- 3.Binley J M, Sanders R W, Clas B, Schuelke N, Master A, Guo Y, Kajumo F, Maddon P J, Olson W C, Moore J P. A recombinant human immunodeficiency virus type 1 envelope glycoprotein complex stabilized by an intermolecular disulfide bond between gp120 and gp41 subunits is an antigenic mimic of the trimeric virion-associated structure. J Virol. 2000;74:627–643. doi: 10.1128/jvi.74.2.627-643.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bou-Habib D, Roderiquez G, Oravecz T, Berman P W, Lusso P, Norcross M A. Cryptic nature of envelope V3 region epitopes protects primary monocytropic human immunodeficiency virus type 1 from antibody neutralization. J Virol. 1994;68:6006–6013. doi: 10.1128/jvi.68.9.6006-6013.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burton D R, Barbas III C F, Persson M A A, Koenig S, Chanock R M, Lerner R A. A large array of human monoclonal antibodies to type 1 human immunodeficiency virus from combinatorial libraries of asymptomatic seropositive individuals. Proc Natl Acad Sci USA. 1991;88:10134–10137. doi: 10.1073/pnas.88.22.10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burton D R, Williamson R A, Parren P W H I. Antibody and virus: binding and neutralization. Virology. 2000;270:1–3. doi: 10.1006/viro.2000.0239. [DOI] [PubMed] [Google Scholar]
- 7.Cavacini L A, Emes C L, Wisnewski A V, Power J, Lewis G, Montefiori D, Posner M R. Functional and molecular characterization of human monoclonal antibody reactive with the immunodominant region of HIV type 1 glycoprotein 41. AIDS Res Hum Retrovir. 1998;14:1271–1280. doi: 10.1089/aid.1998.14.1271. [DOI] [PubMed] [Google Scholar]
- 8.Chen J, Lee K H, Steinhauer D A, Stevens D J, Skehel J J, Wiley D C. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell. 1998;95:409–417. doi: 10.1016/s0092-8674(00)81771-7. [DOI] [PubMed] [Google Scholar]
- 9.Cohen J. Jitters jeopardize AIDS vaccine trials. Science. 1993;262:980–981. doi: 10.1126/science.8235635. [DOI] [PubMed] [Google Scholar]
- 10.Daar E S, Ho D D. Relative resistance of primary HIV-1 isolates to neutralization by soluble CD4. Am J Med. 1991;90:22S–26S. doi: 10.1016/0002-9343(91)90407-o. [DOI] [PubMed] [Google Scholar]
- 11.Daar E S, Li X L, Moudgil T, Ho D D. High concentrations of recombinant soluble CD4 are required to neutralize primary human immunodeficiency virus type 1 isolates. Proc Natl Acad Sci USA. 1990;87:6574–6578. doi: 10.1073/pnas.87.17.6574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Jong J-J, Goudsmit J, Keulen W, Klaver B, Krone W, Tersmette M, de Ronde A. Human immunodeficiency virus type 1 clones chimeric for the envelope V3 domain differ in syncytium formation and replication capacity. J Virol. 1992;66:757–765. doi: 10.1128/jvi.66.2.757-765.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doranz B J, Baik S S, Doms R W. Use of a gp120 binding assay to dissect the requirements and kinetics of human immunodeficiency virus fusion events. J Virol. 1999;73:10346–10358. doi: 10.1128/jvi.73.12.10346-10358.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dubay J W, Dubay S R, Shin H-J, Hunter E. Analysis of the cleavage site of the human immunodeficiency virus type 1 glycoprotein: requirement of precursor cleavage for glycoprotein incorporation. J Virol. 1995;69:4675–4682. doi: 10.1128/jvi.69.8.4675-4682.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dubay J W, Roberts S J, Hahn B H, Hunter E. Truncation of the human immunodeficiency virus type 1 transmembrane glycoprotein cytoplasmic domain blocks virus infectivity. J Virol. 1992;66:6616–6625. doi: 10.1128/jvi.66.11.6616-6625.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Follis K E, Trahey M, LaCasse R A, Nunberg J H. Continued utilization of CCR5 coreceptor by a newly-adapted T-cell line-adapted isolate of human immunodeficiency virus type 1. J Virol. 1998;72:7603–7608. doi: 10.1128/jvi.72.9.7603-7608.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu Y K, Hart T K, Jonak Z L, Bugelski P J. Physicochemical dissociation of CD4-mediated syncytium formation and shedding of human immunodeficiency virus type 1 gp120. J Virol. 1993;67:3818–3825. doi: 10.1128/jvi.67.7.3818-3825.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao F, Morrison S G, Robertson D L, Thornton C L, Craig S, Karlsson G, Sodroski J, Morgado M, Galvao-Castro B, von Briesen H, Beddows S, Weber J, Sharp P M, Shaw G M, Hahn B H the WHO and NIAID Networks for HIV Isolation and Characterization. Molecular cloning and analysis of functional envelope genes from human immunodeficiency virus type 1 sequence subtypes A through G. J Virol. 1996;70:1651–1667. doi: 10.1128/jvi.70.3.1651-1667.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gorny M K, Xu J Y, Karwowska S, Buchbinder A, Zolla-Pazner S. Repertoire of neutralizing human monoclonal antibodies specific for the V3 domain of HIV-1 gp120. J Immunol. 1993;150:653–643. [PubMed] [Google Scholar]
- 20.Groenink M, Fouchier R A M, deGoede R E Y, deWolf F, Gruters R A, Cuypers H T M, Huisman H G, Tersmette M. Phenotypic heterogeneity in a panel of infectious molecular human immunodeficiency virus type 1 clones derived from a single individual. J Virol. 1991;65:1968–1975. doi: 10.1128/jvi.65.4.1968-1975.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guillon C, Bedin F, Fouchier R A M, Schuitemaker H, Gruters R A. Completion of nucleotide sequences of non-syncytium-inducing and syncytium-inducing HIV type 1 variants isolated from the same patient. AIDS Res Hum Retrovir. 1995;11:1537–1538. doi: 10.1089/aid.1995.11.1537. [DOI] [PubMed] [Google Scholar]
- 22.Hallenberger S, Moulard M, Sordel M, Klenk H D, Garten W. The role of eukaryotic subtilisin-like endoproteases for the activation of human immunodeficiency virus glycoproteins in natural host cells. J Virol. 1997;71:1036–1045. doi: 10.1128/jvi.71.2.1036-1045.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hill C M, Deng H, Unutmaz D, Kewalramani V N, Bastiani L, Gorny M K, Zolla-Pazner S, Littman D R. Envelope glycoproteins from human immunodeficiency virus types 1 and 2 and simian immunodeficiency virus can use human CCR5 as a coreceptor for viral entry and make direct CD4-dependent interactions with this chemokine receptor. J Virol. 1997;71:6296–6304. doi: 10.1128/jvi.71.9.6296-6304.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karlsson G B, Gao F, Robinson J, Hahn B, Sodroski J. Increased envelope spike density and stability are not required for neutralization resistance of primary human immunodeficiency viruses. J Virol. 1996;70:6136–6142. doi: 10.1128/jvi.70.9.6136-6142.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karwowska S, Gorny M K, Buchbinde A, Gianakakos V, Williams C, Fuerst T, Zolla-Pazner S. Production of human monoclonal antibodies specific for conformational and linear non-V3 epitopes of gp120. AIDS Res Hum Retrovir. 1992;8:1099–1106. doi: 10.1089/aid.1992.8.1099. [DOI] [PubMed] [Google Scholar]
- 26.Klasse P J, Moore J P. Quantitative model of antibody-and soluble CD4-mediated neutralization of primary isolates and T-cell line-adapted strains of human immunodeficiency virus type 1. J Virol. 1996;70:3668–3677. doi: 10.1128/jvi.70.6.3668-3677.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwong P D, Wyatt R, Robinson J, Sweet R W, Sodroski J, Hendrickson W A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.LaCasse R A, Follis K E, Moudgil T, Trahey M, Binley J M, Planelles V, Zolla-Pazner S, Nunberg J H. Coreceptor utilization by human immunodeficiency virus type 1 is not a primary determinant of neutralization sensitivity. J Virol. 1998;72:2491–2495. doi: 10.1128/jvi.72.3.2491-2495.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.LaCasse R A, Follis K E, Trahey M, Scarborough J D, Littman D R, Nunberg J H. Fusion-competent vaccines: broad neutralization of primary isolates of HIV. Science. 1999;283:357–362. doi: 10.1126/science.283.5400.357. [DOI] [PubMed] [Google Scholar]
- 30.Layne S P, Merges M J, Dembo M, Spouge J L, Conley S R, Moore J P, Raina J L, Renz H, Gelderblom H R, Nara P L. Factors underlying spontaneous inactivation and susceptibility to neutralization of human immunodeficiency virus. Virology. 1992;189:695–714. doi: 10.1016/0042-6822(92)90593-e. [DOI] [PubMed] [Google Scholar]
- 31.Lisanti M P, Sargiacomo M, Graeve L, Saltiel A R, Rodriguez-Boulan E. Polarized apical distribution of glycosyl-phosphatidylinositol-anchored proteins in a renal epithelial cell line. Proc Natl Acad Sci USA. 1988;85:9557–9561. doi: 10.1073/pnas.85.24.9557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mack M, Kleinschmidt A, Bruhl H, Klier C, Nelson P J, Cihak J, Plachy J, Stangassinger M, Erfle V, Schlondorff D. Transfer of the chemokine receptor CCR5 between cells by membrane-derived microparticles: a mechanism for cellular human immunodeficiency virus 1 infection. Nat Med. 2000;6:769–775. doi: 10.1038/77498. [DOI] [PubMed] [Google Scholar]
- 33.Montefiori D C, Collman R G, Fouts T R, Zhou J Y, Bilska M, Hoxie J A, Moore J P, Bolognesi D P. Evidence that antibody-mediated neutralization of human immunodeficiency virus type 1 by sera from infected individuals is independent of coreceptor usage. J Virol. 1998;72:1886–1893. doi: 10.1128/jvi.72.3.1886-1893.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moore J P, Burkly L C, Connor R I, Cao Y, Tizard R, Ho D D, Fisher R A. Adaptation of two primary human immunodeficiency virus type 1 isolates to growth in transformed T cell lines correlates with alterations in the response of their envelope glycoproteins to soluble CD4. AIDS Res Hum Retrovir. 1993;9:529–539. doi: 10.1089/aid.1993.9.529. [DOI] [PubMed] [Google Scholar]
- 35.Moore J P, Cao Y, Qing L, Sattentau Q J, Pyati J, Koduri R, Robinson J, Barbas III C F, Burton D R, Ho D D. Primary isolates of human immunodeficiency virus type 1 are relatively resistant to neutralization by monoclonal antibodies to gp120, and their neutralization is not predicted by studies with monomeric gp120. J Virol. 1995;69:101–109. doi: 10.1128/jvi.69.1.101-109.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moore J P, Ho D D. HIV-1 neutralization: the consequences of viral adaptation to growth on transformed T cells. AIDS. 1995;9(Suppl. A):S117–S136. [PubMed] [Google Scholar]
- 37.Moore J P, Klasse P J. Thermodynamic and kinetic analysis of sCD4 binding to HIV-1 virions and of gp120 dissociation. AIDS Res Hum Retrovir. 1992;8:443–450. doi: 10.1089/aid.1992.8.443. [DOI] [PubMed] [Google Scholar]
- 38.Moore J P, McKeating J A, Weiss R A, Sattentau Q J. Dissociation of gp120 from HIV-1 virions induced by soluble CD4. Science. 1990;250:1139–1142. doi: 10.1126/science.2251501. [DOI] [PubMed] [Google Scholar]
- 39.Moore J P, Sodroski J. Antibody cross-competition analysis of the human immunodeficiency virus type 1 gp120 exterior envelope glycoprotein. J Virol. 1996;70:1863–1872. doi: 10.1128/jvi.70.3.1863-1872.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muster T, Steindl F, Purtscher M, Trkola A, Klima A, Himmler G, Rüker F, Katinger H. A conserved neutralizing epitope on gp41 of human immunodeficiency virus type 1. J Virol. 1993;67:6642–6647. doi: 10.1128/jvi.67.11.6642-6647.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nunberg J H, Follis K E, Trahey M, LaCasse R A. Turning a corner on HIV neutralization? Microbes Infect. 2000;2:213–221. doi: 10.1016/s1286-4579(00)00267-7. [DOI] [PubMed] [Google Scholar]
- 42.Nyambi P N, Gorny M K, Bastiani L, van der Groen G, Williams C, Zolla-Pazner S. Mapping of epitopes exposed on intact human immunodeficiency virus type 1 (HIV-1) virions: a new strategy for studying the immunologic relatedness of HIV-1. J Virol. 1998;72:9384–9391. doi: 10.1128/jvi.72.11.9384-9391.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nyambi P N, Mbah H A, Burda S, Williams C, Gorny M K, Nadas A, Zolla-Pazner S. Conserved and exposed epitopes on intact, native, primary human immunodeficiency virus type 1 virions of group M. J Virol. 2000;74:7096–7107. doi: 10.1128/jvi.74.15.7096-7107.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O'Brien W A, Mao S H, Cao Y, Moore J P. Macrophage-tropic and T-cell line-adapted chimeric strains of human immunodeficiency virus type 1 differ in their susceptibilities to neutralization by soluble CD4 at different temperatures. J Virol. 1994;68:5264–5269. doi: 10.1128/jvi.68.8.5264-5269.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Orentas R J, Hildreth J E. Association of host cell surface adhesion receptors and other membrane proteins with HIV and SIV. AIDS Res Hum Retrovir. 1993;9:1157–1165. doi: 10.1089/aid.1993.9.1157. [DOI] [PubMed] [Google Scholar]
- 46.Orloff S L, Bandea C I, Kennedy M S, Allaway G P, Maddon P J, McDougal J S. Increase in sensitivity to soluble CD4 by primary HIV type 1 isolates after passage through C8166 cells: association with sequence differences in the first constant (C1) region of glycoprotein 120. AIDS Res Hum Retrovir. 1995;11:335–342. doi: 10.1089/aid.1995.11.335. [DOI] [PubMed] [Google Scholar]
- 47.Parren P W H I, Burton D R. HIV-1 antibody—debris or virion? Nat Med. 1997;3:366–367. doi: 10.1038/nm0497-366d. [DOI] [PubMed] [Google Scholar]
- 48.Parren P W H I, Mondor I, Naniche D, Ditzel H J, Klasse P J, Burton D R, Sattentau Q J. Neutralization of human immunodeficiency virus type 1 by antibody to gp120 is determined primarily by occupancy of sites on the virion irrespective of envelope specificity. J Virol. 1998;72:3512–3519. doi: 10.1128/jvi.72.5.3512-3519.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parren P W H I, Moore J P, Burton D R, Sattentau Q J. The neutralizing antibody response to HIV-1: viral evasion and escape from humoral immunity. AIDS. 1999;13(Suppl A):S137–S162. [PubMed] [Google Scholar]
- 50.Posner M R, Cavacini L A, Emes C L, Power J, Byrn R. Neutralization of HIV-1 by F105, a human monoclonal antibody to the CD4 binding site of gp120. J Acquir Immune Defic Syndr. 1993;6:7–14. [PubMed] [Google Scholar]
- 51.Rizzuto C D, Wyatt R, Hernandez-Ramos N, Sun Y, Kwong P D, Hendrickson W A, Sodroski J. A conserved HIV gp120 glycoprotein structure involved in chemokine receptor binding. Science. 1998;280:1949–1953. doi: 10.1126/science.280.5371.1949. [DOI] [PubMed] [Google Scholar]
- 52.Rogel M E, Wu L I, Emerman M. The human immunodeficiency virus type 1 vprgene prevents cell proliferation during chronic infection. J Virol. 1995;69:882–888. doi: 10.1128/jvi.69.2.882-888.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Samuelsson A, Yari F, Hinkula J, Ersoy O, Norrby E, Persson M A. Human antibodies from phage libraries: neutralizing activity against human immunodeficiency virus type 1 equally improved after expression as Fab and IgG in mammalian cells. Eur J Immunol. 1996;26:3029–3034. doi: 10.1002/eji.1830261231. [DOI] [PubMed] [Google Scholar]
- 54.Sattentau Q J, Moore J P. Human immunodeficiency virus type 1 neutralization is determined by epitope exposure on the gp120 oligomer. J Exp Med. 1995;182:185–196. doi: 10.1084/jem.182.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sattentau Q J, Moore J P, Vignaux F, Traincard F, Poignard P. Conformational changes induced in the envelope glycoproteins of the human and simian immunodeficiency viruses by soluble receptor binding. J Virol. 1993;67:7383–7393. doi: 10.1128/jvi.67.12.7383-7393.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stamatatos L, Cheng-Mayer C. Structural modulations of the envelope gp120 glycoprotein of human immunodeficiency virus type 1 upon oligomerization and differential V3 loop-epitope exposure of isolates displaying distinct tropism upon virion-soluble receptor binding. J Virol. 1995;69:6191–6198. doi: 10.1128/jvi.69.10.6191-6198.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sullivan N, Sun Y, Li J, Hofmann W, Sodroski J. Replicative function and neutralization sensitivity of envelope glycoproteins from primary and T-cell line-passaged human immunodeficiency virus type 1 isolates. J Virol. 1995;69:4413–4422. doi: 10.1128/jvi.69.7.4413-4422.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sullivan N, Sun Y, Sattentau Q, Thali M, Wu D, Denisova G, Gershoni J, Robinson J, Moore J, Sodroski J. CD4-induced conformational changes in the human immunodeficiency type 1 gp120 glycoprotein: consequences for virus entry and neutralization. J Virol. 1998;72:4694–4703. doi: 10.1128/jvi.72.6.4694-4703.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tersmette M, Gruters R A, deWolf F, deGoede R E Y, Lange J M A, Schellekens P T A, Goudsmit J, Huisman H G, Miedema F. Evidence for a role of virulent human immunodeficiency virus (HIV) variants in the pathogenesis of acquired immunodeficiency syndrome: studies on sequential HIV isolates. J Virol. 1989;63:2118–2125. doi: 10.1128/jvi.63.5.2118-2125.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thali M, Moore J P, Furman C, Charles M, Ho D D, Robinson J, Sodroski J. Characterization of conserved human immunodeficiency virus type 1 gp120 neutralization epitopes exposed upon gp120-CD4 binding. J Virol. 1993;67:3978–3988. doi: 10.1128/jvi.67.7.3978-3988.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Trkola A, Ketas T, KewalRamani V N, Endorf F, Binley J M, Katinger H, Robinson J, Littman D R, Moore J P. Neutralization sensitivity of human immunodeficiency virus type 1 primary isolates to antibody and CD4-based reagents is independent of coreceptor usage. J Virol. 1998;72:1876–1885. doi: 10.1128/jvi.72.3.1876-1885.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.VanCott T C, Bethke F R, Polonis V R, Gorny M K, Zolla-Pazner S, Redfield R R, Birx D L. Dissociation rate of antibody-gp120 binding interactions is predictive of V3-mediated neutralization of HIV-1. J Immunol. 1994;153:449–459. [PubMed] [Google Scholar]
- 63.White-Scharf M E, Potts B J, Smith L M, Sokolowski K A, Rusche J R, Silver S. Broadly neutralizing monoclonal antibodies to the V3 region of HIV-1 can be elicited by peptide immunization. Virology. 1993;192:197–206. doi: 10.1006/viro.1993.1022. [DOI] [PubMed] [Google Scholar]
- 64.Willey R L, Martin M A, Peden K W C. Increase in soluble CD4 binding to and CD4-induced dissociation of gp120 from virions correlates with infectivity of human immunodeficiency virus type 1. J Virol. 1994;68:1029–1039. doi: 10.1128/jvi.68.2.1029-1039.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wrin T, Loh T P, Charron-Vennari J, Schuitemaker H, Nunberg J H. Adaptation to persistent growth in the H9 cell line renders a primary isolate of human immunodeficiency virus type 1 sensitive to neutralization by vaccine sera. J Virol. 1995;69:39–48. doi: 10.1128/jvi.69.1.39-48.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wyatt R, Kwong P D, Desjardins E, Sweet R W, Robinson J, Hendrickson W A, Sodroski J. The antigenic structure of the HIV gp120 envelope glycoprotein. Nature. 1998;393:705–711. doi: 10.1038/31514. [DOI] [PubMed] [Google Scholar]
- 67.Zhang Y J, Fredriksson R, McKeating J A, Fenyo E M. Passage of HIV-1 molecular clones into different cell lines confers differential sensitivity to neutralization. Virology. 1997;238:254–264. doi: 10.1006/viro.1997.8812. [DOI] [PubMed] [Google Scholar]
- 68.Zolla-Pazner S, O'Leary J, Burda S, Gorny M K, Kim M, Mascola J, McCutchan F. Serotyping of primary human immunodeficiency virus type 1 isolates from diverse geographic locations by flow cytometry. J Virol. 1995;69:3807–3815. doi: 10.1128/jvi.69.6.3807-3815.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]