

Abstract
Methicillin‐resistant Staphylococcus aureus (MRSA) is a common cause of severe pneumonia and acute respiratory distress syndrome (ARDS). To advance our mechanistic understanding of this important pathogen, we characterized the effects of MRSA‐induced epigenetic modification of histone 3 lysine 9 acetylation (H3K9ac), an activator of gene transcription, on lung endothelial cells (EC), a critical site of ARDS pathophysiology. Chromatin immunoprecipitation and sequencing (ChIP‐seq) analysis revealed that MRSA induces H3K9ac in the promoter regions of multiple genes, with the highest ranked peak annotated to the CYP1A1 gene. Subsequent experiments confirm that MRSA increases CYP1A1 protein and mRNA expression, and its enzymatic activity in EC. Epigenetic inhibitors (C646, RVX‐208) reduce MRSA‐induced CYP1A1 expression and inflammatory responses, including cytokine release and adhesion molecule expression. Inhibition of the Aryl hydrocarbon receptor (Ahr), a known mediator of CYP1A1 expression, blocks MRSA‐induced upregulation of CYP1A1 mRNA and protein expression, enzyme activity, and cytokine release. Reduction of CYP1A1 protein expression by siRNA or inhibition of its activity by rhapontigenin attenuated MRSA‐induced EC permeability and inflammatory responses. In a mouse model of MRSA‐induced acute lung injury (ALI), inhibition of CYP1A1 activity by rhapontigenin improved multiple indices of ALI, including bronchoalveolar lavage (BAL) protein concentration, cytokine levels, and markers of endothelial damage. Analysis of publicly available data suggests upregulation of CYP1A1 expression in ARDS patients compared to ICU controls. In summary, these studies provide new insights into MRSA‐induced lung injury and identify a novel functional role for epigenetic upregulation of CYP1A1 in lung EC during ARDS pathogenesis.
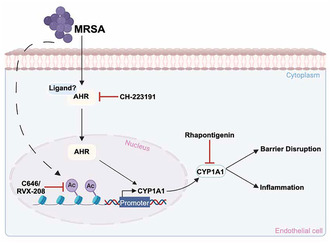
MRSA increases the acetylation of H3K9 within the CYP1A1 promoter region, enhancing its transcription and subsequent expression, and causing lung endothelial barrier disruption and inflammation. Epigenetic inhibitors reduce MRSA‐induced CYP1A1 expression and inflammatory responses. Inhibition of Ahr, a mediator of CYP1A1 expression, also attenuates MRSA‐induced CYP1A1 expression and its enzymatic activity. Inhibiting CYP1A1 activity reduced MRSA‐induced EC permeability, suggesting a novel functional role for epigenetic regulation of CYP1A1 in lung EC during ARDS pathogenesis. This image was generated using BioRender.com.
Abbreviations
- EF
Emotional Function
- HIT‐6
six‐item Headache Impact Test
- ALI
Acute lung injury
- Ahr
Aryl hydrocarbon receptor
- ARDS
Acute Respiratory Distress Syndrome
- AUC
Area under the curve
- BAL
Bronchoalveolar lavage
- BCV
Biological coefficient of variance
- BET
Bromodomain and extraterminal
- BETi
Bromodomain and extraterminal inhibitor
- CBP
CREB‐binding protein
- ChIP‐seq
Chromatin immunoprecipitation and sequencing
- CPM
Counts per million
- CYP1A1
Cytochrome P450 1A1
- EC
Endothelial cells
- ECIS
Electric Cell‐Substrate Impedance Sensing
- EROD
Ethoxyresorufin‐O‐deethylase
- FDR
False discovery rate
- GLM
Generalized linear model
- H3K9ac
Histone H3 lysine 9 acetylation
- HATs
Histone acetyltransferases
- HDACs
Histone deacetylases
- HK‐MRSA
Heat‐killed Methicillin‐resistant Staphylococcus aureus
- HLMVEC
Human lung microvascular endothelial cells
- HPAEC
Human pulmonary artery endothelial cells
- IGV
Integrative Genomics Viewer
- IT
Intratracheal
- LPS
Lipopolysaccharides
- MRSA
Methicillin‐resistant Staphylococcus aureus
- SOFA
Sequential Organ Failure Assessment
- TER
Transendothelial electrical resistance
- TMM
Trimmed mean of M‐values
- WT
Wild‐type
1. INTRODUCTION
Acute respiratory distress syndrome (ARDS) is a life‐threatening cause of respiratory failure for which there is no effective pharmacologic treatment. ARDS occurs when the lungs are severely injured by an acute inflammatory insult, such as infection by bacterial or viral pathogens, and is characterized by diffuse inflammation and vascular leak, resulting in hypoxemic respiratory failure. The most common causes of ARDS are pneumonia and sepsis. 1 Lung endothelial dysfunction represents a pivotal factor in the development of ARDS. The pulmonary endothelium is essential for gas exchange between the blood and tissues while maintaining the integrity of the lung barrier and function of the lungs. 2 During acute lung injury (ALI), the endothelial barrier is compromised, leading to a series of events that ultimately cause respiratory failure. 3 , 4 Understanding the mechanisms underlying lung endothelial barrier dysfunction and its consequences is crucial for the development of new therapeutic strategies against ARDS.
The importance of epigenetic processes in the pathophysiology of inflammatory syndromes such as ARDS and sepsis is being increasingly recognized. 5 , 6 , 7 In particular, recent studies have identified histone modifications induced by ALI‐relevant stimuli, such as lipopolysaccharide (LPS), that alter gene expression. 8 , 9 , 10 Common histone modifications include acetylation and methylation, which alter the histone structure, resulting in gene activation or repression, respectively. 11 , 12 Histone acetylation is controlled by two enzyme families, the histone deacetylases (HDACs), which catalyze histone deacetylation, and histone acetyltransferases (HATs), which catalyze their acetylation. 12 Acetylated Lys residues on histones serve as docking platforms for the bromodomain and extraterminal (BET) proteins, which subsequently “read” these epigenetic signals to promote gene transcription. 13
Several prior studies have linked changes in histone acetylation status to endothelial cell (EC) dysfunction. 14 Here, we characterize the effects of histone modifications on lung EC dysfunction caused by methicillin‐resistant Staphylococcus aureus (MRSA), an antibiotic‐resistant pathogen that is a common cause of sepsis and ARDS. 15 , 16 , 17 , 18 Our recent studies have demonstrated that MRSA is a potent inflammatory stimulus that can cause vascular lung endothelial leakage and inflammation 19 , 20 through mechanisms that are not completely understood. In general, data are sparse concerning the epigenetic effects of MRSA infection, 21 , 22 , 23 with almost no investigation into the epigenetic effects of MRSA on lung EC during ARDS or sepsis. 24
In the current study, ChIP‐seq methodology was employed to characterize the genome‐wide acetylation profile of acetylation at lysine 9 of histone protein 3 (H3K9ac), a modification associated with gene activation, in lung EC challenged with MRSA. ChIP‐seq identified CYP1A1 as a promising epigenetic target in the context of MRSA‐induced injury. CYP1A1 belongs to the cytochrome P450 superfamily of enzymes and metabolizes a wide range of endogenous and exogenous compounds, as well as mediates oxidative stress and other pro‐inflammatory signaling. 25 , 26 The role of CYP1A1 in MRSA‐induced lung endothelial dysfunction and ALI remains poorly understood. Data presented here demonstrate that MRSA upregulates CYP1A1 expression and activity in lung EC through epigenetic modifications, while targeting CYP1A1 promotes endothelial barrier protection against MRSA in vitro and reduces ALI in vivo.
2. MATERIALS AND METHODS
2.1. Reagents
CH‐223191 (Cat# C8124) and C646 (Cat# SML‐0002) were obtained from Sigma‐Aldrich (St. Louis, MO). RVX‐208 (Cat# 16424) and 7‐ethoxyresorufin (Cat# 16122) were obtained from Cayman Chemical (Ann Arbor, MI). Rhapontigenin (Cat# HY‐N2229) was obtained from MedChem Express (New Jersey, USA). Live and heat‐killed (HK) MRSA [USA300 CA‐MRSA wild‐type (LAC) strain] were prepared as previously described. 20
2.2. Cell culture
Human pulmonary artery endothelial cells (HPAEC) and human lung microvascular endothelial cells (HLMVEC) (Lonza, Walkersville, MD) were cultured in EGM‐2 complete medium containing 10% FBS at 37°C in a 5% CO2 incubator. EC passages 6–8 were used for all experiments. Cells were incubated in 2% FBS media for 2 h prior to experimental treatments. For in vitro experiments, lung EC were pretreated with C646 (10 μM), RVX‐208 (5 μM), CH‐223191 (10 μM), or rhapontigenin (5 μM) 1 h prior to treatment with heat‐killed MRSA (2.5 x108 CFU/mL unless otherwise indicated) for the indicated times.
2.3. qPCR
Total RNA was isolated from cells using TRIzol reagent (Zymo Research, Irvine, CA), followed by purification using Direct‐zol RNA miniprep (Zymo Research, Irvine, CA) according to the manufacturer's protocol. RNA was quantified with a NanoDrop spectrophotometer, and 500 ng‐1 μg of total RNA was used to generate cDNA (High‐capacity cDNA reverse transcription kit, Applied Biosystems, Waltham, MA). Quantitative PCR was performed using the CFX384 Touch Real‐Time system (Bio‐Rad, Hercules, CA) utilizing SYBR Green fluorescence (Roche, Mannheim, Germany). The relative mRNA levels were normalized to GAPDH mRNA levels using the ΔΔCt method. Primer sequences are listed below: CYP1A1–FW GCAGATCAACCATGACCAGAAG; RV GATACACTTCCGCTTGCCCA, GAPDH–FW AGGTCGGAGTCAACGGATTT; RV ATGGAATTTGCCATGGGTGG.
2.4. Chromatin immunoprecipitation (ChIP) and sequencing
HPAEC grown in D150 cell culture dishes were treated with HK‐MRSA or PBS (control) for 30 min (cells from 4 dishes were pooled for each condition). ChIP assay was performed as previously described in detail. 6 For immunoprecipitation of chromatin DNA, we used a ChIP‐seq grade antibody against acetylated Histone H3K9 (EMD Millipore #06‐942, Temecula, CA). Final samples were processed for sequencing at the University of Chicago Genomics Facility using the Illumina HiSEQ system. Sequence data have been uploaded into the Gene Expression Omnibus (accession number GSE274958). Raw reads were aligned to reference genome hg38 using BWA MEM. 27 Apparent PCR duplicates were removed using Picard MarkDuplicates. 28 Peaks were called against the input sample using Macs2, and normalized enrichment tracks were generated using ‐B ‐‐SPMR flags. 29 Normalized enrichment and peak calling tracks were visualized in Integrative Genomics Viewer (IGV). 30 Peaks with a score >5 (−log10 q‐value) were retained. For quantitative comparisons, peaks were merged using bedtools merge. 31 Enrichment levels for merged peak were quantified using featureCounts. 32 Counts for input control samples were subtracted from ChIP‐seq counts after accounting for sequencing depth differences, giving an input‐controlled quantification suitable for differential analysis. Differential expression statistics (fold‐change and p‐value) were computed using edgeR 33 , 34 using a fixed biological coefficient of variance (BCV) of 12%, which was determined as the common dispersion after fitting a two‐factor generalized linear model (GLM) over all samples. Normalized peak enrichment levels were computed as counts per million (CPM) with the trimmed mean of M‐values (TMM) normalization factor from edgeR. p‐values were adjusted for multiple testing using the false discovery rate (FDR) correction of Benjamini and Hochberg. 35
2.5. Ethoxyresorufin‐O‐deethylase (EROD) assay
CYP1A1 activity was measured using the EROD assay as described elsewhere. 36 Briefly, cells were grown in 24‐well plates and treated as indicated for 3 h. After the cells were washed in PBS, 2 μM of 7‐ethoxyresorufin in 50 mM NaHPO4 pH 8.0 was added to each well. After incubating at 37°C for 20 min, 100 μL of the assay supernatant (in triplicates) were transferred to a black 96‐well plate, and fluorescence was measured in a SpectraMax M2e microplate reader (Molecular Devices, San Jose, CA) at excitation wavelength 544 and emission of 590.
2.6. Electric cell‐substrate impedance sensing (ECIS)
Endothelial barrier function was assessed using the ECIS system (Applied Biophysics, Troy, NY), as we have described before. 20 Briefly, HPAEC were seeded onto electrode arrays containing gold film electrodes (8W10E+ arrays) and grown till confluence. Arrays were attached to the ECIS system, and transendothelial electrical resistance (TER) was recorded over time. For data analysis, the area under the curve (AUC) was calculated for each condition for the time period from 2 to 20 h. Data are shown as AUC normalized to that obtained from MRSA‐treated cells.
2.7. siRNA transfection
HPAEC were transfected with Xfect RNA Transfection Reagent (Takara Bio, San Jose, CA) using 100 pmol CYP1A1 (Cat# L‐004790‐00‐0005) or control (Cat# D‐001810‐02‐05) siRNA (Dharmacon, Lafayette, CO) according to manufacturer's protocol.
2.8. Mouse model of MRSA‐induced acute lung injury
All experimental conditions and animal care procedures were approved by the University of Illinois Chicago Animal Care and Use Committee. Wild‐type (WT) C57BL/6 male mice (age 10–12 weeks) were administrated 200 μL rhapontigenin 10 mg/kg (IP) or vehicle (N = 3–5 animals per group). One hour later, mice were anesthetized with ketamine/xylazine (100/5 mg/kg), intubated, and administrated live MRSA (USA300 CA‐MRSA wild‐type LAC strain) intratracheally (IT) at 0.75 × 108 CFU/mouse or an equal volume of PBS (vehicle) as we have previously described. 19 , 20 18 hours later, plasma, bronchoalveolar lavage (BAL) fluid, and lung tissues were collected. 19 , 20 The BAL supernatant was collected after centrifugation at 500 g for 20 min and frozen at −80°C for subsequent analyses. The cell pellet was resuspended in PBS after red blood cell lysis (Thermo Fisher Scientific, Waltham, MA), and BAL cell counts were measured using the Bio‐Rad TC10 automated cell counter. For differential cell counting, cytospins were prepared and stained with Kwik Diff Stain (Thermo Fisher Scientific, Waltham, MA). BAL protein was measured with the BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA). Lungs fixed in formalin were processed for hematoxylin and eosin (H&E) staining by the UIC Research Histology and Research Tissue Imaging cores. Lung tissues on slides were scanned using the Leica Aperio AT2 Scanner at 40x magnification. Images were processed using the Aperio ImageScope (Leica).
2.9. Western blotting
Cell lysates were collected using Cell Lysis Buffer (Cell Signaling, Danvers, MA) containing protease and phosphatase inhibitors (EMD Millipore) and centrifuged at 10 000 g for 5 min at 4°C. Lung tissues were pulverized in liquid nitrogen and resuspended in RIPA buffer (Sigma‐Aldrich, St. Louis, MO) containing protease and phosphatase inhibitors. Samples were sonicated for 10 s, 3 times, then centrifuged at 10 000 g for 10 min at 4°C. Protein concentration in cell or lung tissue lysates was measured with the BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA). Samples containing equal amounts of protein were mixed with 6× Laemmli SDS‐Sample buffer (Boston Bioproducts, Ashland, MA), boiled for 5 min, and subjected to western blotting. Precision Plus Protein Dual Color Standards (BioRad, Des Plaines, IL) and protein samples (from cell and lung lysates or BAL) were separated in 4%–20% SDS‐PAGE gels (Genscript, Piscataway, NJ) and transferred onto nitrocellulose membranes at 100 V for 1 h. Membranes were then blocked for 1 h at room temperature with 5% BSA or 3% milk and incubated with primary antibodies overnight at 4°C. Primary antibodies used were: anti‐CYP1A1 (Cat# 13241‐1‐AP, Proteintech), anti‐VCAM‐1 (Cat# sc‐8304, Santa Cruz Biotechnology, Dallas, Texas), anti‐ICAM‐1 (Cat# A5597, ABclonal Technology, Woburn, MA), anti‐angiopoietin 2 (Cat# A0698, ABclonal Technology, Woburn, MA), and anti‐β‐actin (Cat#HRP‐60008, Proteintech, Rosemond, IL). Dilutions are listed in Table S1. Membranes were incubated with HRP‐conjugated secondary antibodies (Cell signaling) for 1 h at room temperature, and protein bands were detected with enhanced chemiluminescence (Thermo Fisher Scientific, Waltham, MA). Densitometric analysis was performed using Image J (NIH, Bethesda, MD).
2.10. Cytokine measurement
Levels of IL‐6 and IL‐8 (CXCL1/KC for mice) were quantified in cell culture supernatant or BAL fluid using ELISA, according to the manufacturer's protocol (Biolegend, San Diego, CA).
2.11. Data analysis and statistics
Results are expressed as mean ± standard deviation (SD). In vitro experiments were performed at least 3 independent times. Data were analyzed by t‐test or one‐way ANOVA followed by post hoc Tukey test. Data were analyzed and graphed using GraphPad Prism (version 10). p < .05 was considered statistically significant. For the re‐analysis of publicly available gene expression data derived from the tracheal aspirates of 52 mechanically ventilated patients, 37 gene counts and sample metadata were downloaded from Gene Expression Omnibus, accession GSE163426. Differential expression statistics between non‐ARDS and ARDS patient samples were computed using edgeR, 33 using generalized linear models (GLMs) to control for sex and age. Data were derived from patients undergoing mechanical ventilation with COVID ARDS (n = 15), non‐COVID ARDS (n = 32), and control ICU patients without ARDS (n = 5). Normalized expression was computed using the TMM correction from edgeR.
3. RESULTS
3.1. MRSA induces epigenetic changes in human lung EC
Histone lysine acetylation is a central epigenetic alteration that has been linked to gene transcription and functional regulation of endothelial responses to inflammatory stimuli. 14 We therefore first investigated the effects of MRSA exposure on lung EC acetylation at the 9th lysine residue of the histone H3 protein (H3K9ac), which is an important chromatin modification associated with active promoters and gene activation. 14 Human lung EC were exposed to heat‐killed MRSA (HK‐MRSA) for 30 min, chromatin DNA was collected, and then ChIP‐seq analyses were performed. MRSA exposure significantly altered 1605 H3K9ac sites compared to control lung EC, with 68% of these sites increasing after MRSA treatment (Figure 1A). Since H3K9ac is associated with active promoters, H3K9ac peaks were annotated to the nearest transcription start site (TSS) to identify potential genes of interest regulated by MRSA. This analysis identified 105 coding genes or pseudogenes in which H3K9ac was differentially regulated in their promoter regions in response to MRSA [log 2‐fold change > or <1, adjusted p value (FDR) ≤0.05] (Figure 1B). The highest ranked peak was annotated to the CYP1A1 gene (log 2 fold change = 2.40 and FDR = 6.37 × 10−11). MRSA treatment resulted in an increased H3K9ac signal in the CYP1A1 promoter region compared to control conditions (Figure S1). Other highly ranked peaks were annotated to the following coding genes: VIPR1, TONSL, ACP7, ADRB2, CUX1, IL17RE, and PARD3 (top 20 genes are listed in Table S2). Subsequent studies focused on the top identified epigenetic target, CYP1A1, to assess its potential functional role in MRSA‐induced responses. Because H3K9 acetylation is generally associated with increased gene transcription, we first determined the effects of MRSA on CYP1A1 expression in lung EC. We selected a time point later than 30 min to assess the protein and mRNA expression, as these events likely require a delay to reach maximal effects post promoter acetylation. As shown in Figure 1C, CYP1A1 mRNA levels were significantly increased ~12‐fold in HPAEC exposed to MRSA (3 h) compared to control. This effect was not specific to macrovascular HPAEC, as CYP1A1 mRNA levels in microvascular HLMVEC also significantly increased after exposure to MRSA (Figure S2A). CYP1A1 mRNA levels correlated with increased protein expression in both HPAEC (Figure 1D) and HLMVEC (Figure S2B). Furthermore, CYP1A1 enzymatic activity was significantly increased in lung EC by MRSA as measured by the EROD assay (Figure 1E).
FIGURE 1.
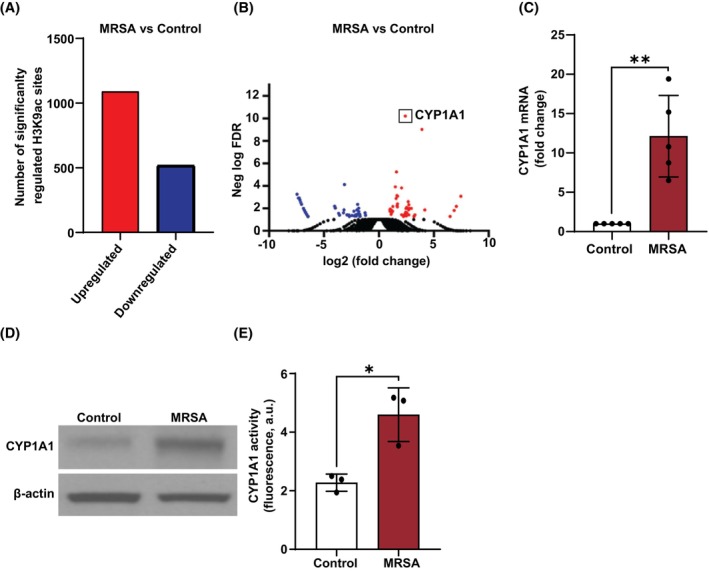
MRSA increases CYP1A1‐associated H3K9 acetylation and expression in lung EC. ChIP‐seq analysis was performed for H3K9 acetylation in HPAEC after exposure to HK‐MRSA (2.5 × 108/mL) for 30 min. (A) Bar graph depiction of the numbers of H3K9ac sites that display significantly increased or decreased acetylation after MRSA (FDR ≤0.05). (B) Volcano plot depicting the –log10 (FDR) of each analyzed H3K9ac site within promoter regions (annotated peaks) versus the corresponding log2 (fold change MRSA vs. Ctr). Red dots represent sites with increased acetylation for MRSA versus Control. Blue dots represent sites with decreased acetylation for MRSA versus Control. The highest ranked peak was annotated to the CYP1A1 promoter. (C–E) HPAEC were treated with HK‐MRSA (2.5 × 108/mL) for 3 h. CYP1A1 (C) mRNA expression, (D) protein expression, and (E) enzyme activity were then assessed. N = 3–5 independent experiments. Data were analyzed using Welch's (data in C) or unpaired t‐test (data in E), *p < .05, **p < .01.
3.2. Histone acetyltransferases mediate CYP1A1 expression and inflammatory signaling in lung EC after MRSA
MRSA is a potent inflammatory stimulus that causes lung EC barrier dysfunction. 19 , 20 Since MRSA increases both H3K9ac levels on the CYP1A1 promoter and CYP1A1 protein expression in lung EC, we next explored the effects of histone acetylation on CYP1A1 expression and inflammatory signaling induced by MRSA. Histone acetylation is mediated by the action of histone acetyltransferases (HATs), enzymes that transfer acetyl groups from acetyl‐CoA to specific lysine residues on histones and other proteins. 38 To characterize the effects of HAT activity on the MRSA response, C646, an inhibitor of the CREB‐binding protein (CBP)/p300 HAT family, was used. In lung EC pretreated with C646, MRSA‐induced CYP1A1 expression was significantly attenuated (Figure 2A), suggesting that the upregulation of H3K9ac levels in the CYP1A1 promoter by MRSA is a critical step for its induction. Importantly, C646 treatment also decreased multiple inflammatory responses to MRSA in lung EC. MRSA significantly increased expression of VCAM‐1 and ICAM‐1 (Figure 2B,C), adhesion molecules that are upregulated upon EC activation, as well as release of pro‐inflammatory cytokines IL‐6 and IL‐8 (Figure 2D,E), while C646 almost completely eliminated these responses. Taken together, these results demonstrate that HAT inhibition by C646 exerts potent anti‐inflammatory effects in lung EC exposed to MRSA and support the hypothesis that epigenetic processes participate in MRSA‐induced injury.
FIGURE 2.
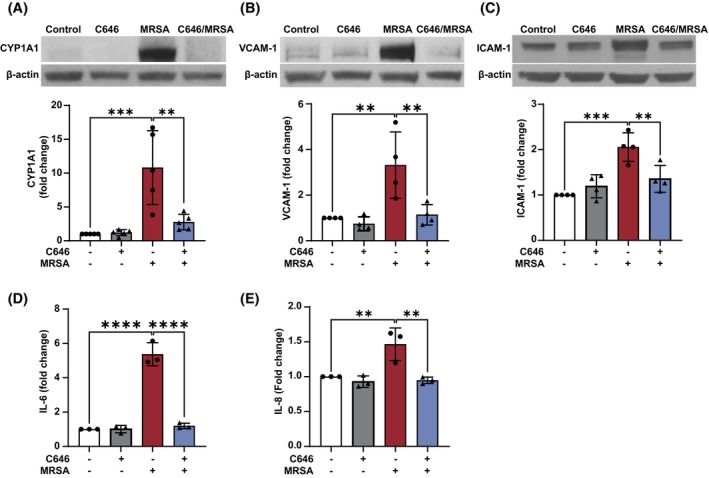
MRSA‐induced CYP1A1 expression and inflammatory responses are regulated by epigenetic HAT activity in lung EC. HPAEC were pretreated for 1 h with the HAT inhibitor C646 (10 μM) prior to HK‐MRSA (2.5 × 108/mL) exposure. Control cells were treated with vehicle (DMSO). Representative western blots and pooled densitometric analyses of EC lysates are shown for (A) CYP1A1 (3 h post MRSA), and (B, C) VCAM‐1 and ICAM‐1 expression (18 h). IL‐6 (D) and IL‐8 (E) levels were measured in EC supernatants using ELISA. N = 3–5 independent experiments. Data were analyzed using one‐way ANOVA, **p < .01, ***p < .001, ****p < .0001.
3.3. Inhibition of BET proteins decreases MRSA‐induced CYP1A1 expression and inflammatory signaling
Bromodomain and extra‐terminal (BET) proteins are epigenetic readers that promote gene expression by binding acetylated lysine residues on histones through their bromodomains. 39 BET inhibitors (BETi) prevent the interaction between the bromodomain and acetyl groups, causing the down‐regulation of certain genes. To further explore the role of histone modifications in MRSA‐induced CYP1A1 expression and inflammation in lung EC, we next employed a BETi that is currently in multiple Phase II/III clinical trials, the pharmaceutical agent RVX‐208 or apabetalone. 40 , 41 Pretreatment with RVX‐208 significantly reduced the effects of MRSA on CYP1A1 protein levels (Figure 3A), VCAM‐1 and ICAM‐1 expression (Figure 3B,C), as well as IL‐6 and IL‐8 release (Figure 3D,E). These data indicate that BET protein activity mediates the epigenetic effects of MRSA on CYP1A1 expression and pro‐inflammatory signaling in lung EC.
FIGURE 3.
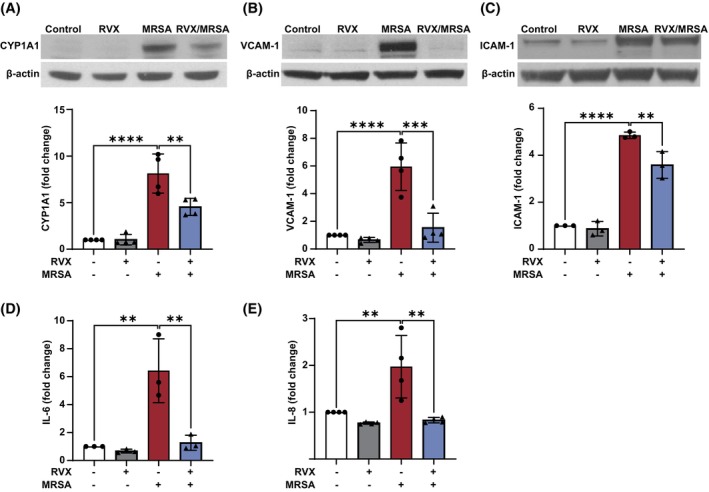
BET inhibition down‐regulates MRSA‐induced CYP1A1 expression and inflammatory responses. HPAEC were pretreated for 1 h with the BET inhibitor RVX‐208 (5 μM) prior to HK‐MRSA (2.5 × 108/mL) exposure. Control cells were treated with vehicle (DMSO). Representative western blots and pooled densitometric analyses of EC lysates are shown for (A) CYP1A1 (3 h post MRSA) and (B, C) VCAM‐1 and ICAM‐1 expression (18 h). IL‐6 (D) and IL‐8 (E) levels were measured in EC supernatants using ELISA. N = 3–4 independent experiments. Data were analyzed using one‐way ANOVA, **p < .01, ***p < .001, ****p < .0001.
3.4. CYP1A1 induction by MRSA is Ahr‐dependent in lung EC
Prior work has demonstrated that CYP1A1 expression is regulated by the ligand‐activated Aryl hydrocarbon receptor (Ahr). 42 To explore the role of Ahr in the effects of MRSA on lung EC, HPAEC were pretreated with the Ahr antagonist CH‐223191 for 1 h prior to MRSA exposure. As depicted in Figure 4A–C, inhibition of Ahr signaling efficiently blocked MRSA‐induced upregulation of CYP1A1 mRNA levels, protein expression, and enzyme activity. In addition, CH‐223191 attenuated MRSA‐induced inflammation as measured by IL‐6 and IL‐8 release (Figure 4D,E), suggesting that Ahr participates in CYP1A1 induction and inflammatory responses after MRSA.
FIGURE 4.
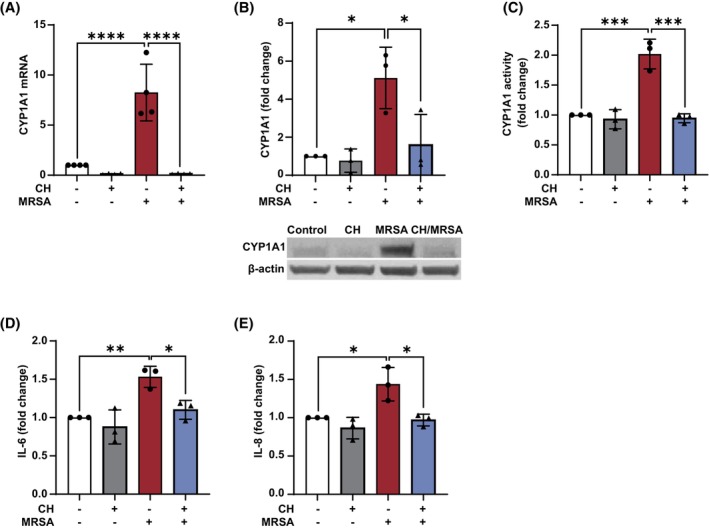
MRSA‐induced CYP1A1 expression and inflammatory responses are mediated by Ahr signaling in lung EC. HPAEC were pretreated for 1 h with the Ahr antagonist CH‐223191 (10 μM) prior to HK‐MRSA (2.5 x 108/mL) exposure. Control cells were treated with vehicle (DMSO). After 3 h, CYP1A1 mRNA levels (A), protein expression (B), and enzyme activity (C) were assessed. (B) Depicted are representative western blots and densitometric analysis. IL‐6 (D) and IL‐8 (E) levels were measured in EC supernatants (6 h after MRSA) using ELISA. N = 3–4 independent experiments. Data were analyzed using one‐way ANOVA *p < .05, **p < .01, ***p < .001, ****p<0.0001.
3.5. MRSA‐induced EC injury is attenuated by CYP1A1 inhibition
To further explore the correlation between CYP1A1 expression in lung EC and MRSA‐induced effects, CYP1A1 levels were reduced by siRNA prior to MRSA challenge. The effects on EC permeability were determined using the ECIS assay, which provides real‐time assessment of barrier function by measuring transendothelial electrical resistance (TER) across the cell monolayer. In control EC, there was no difference in TER values over time between siRNA control and CYP1A1 siRNA transfected cells (Figure 5A). However, MRSA caused a substantial decrease in TER values (i.e., increased EC permeability) that was significantly attenuated by CYP1A1 siRNA, as quantified by determining the area under the curve (for the time period 2–20 h) for these conditions (Figure 5A,B). In addition, CYP1A1 siRNA attenuated aspects of the inflammatory response associated with MRSA, as evidenced by significantly decreased IL‐6 release in response to MRSA compared to siRNA control (Figure 5C).
FIGURE 5.
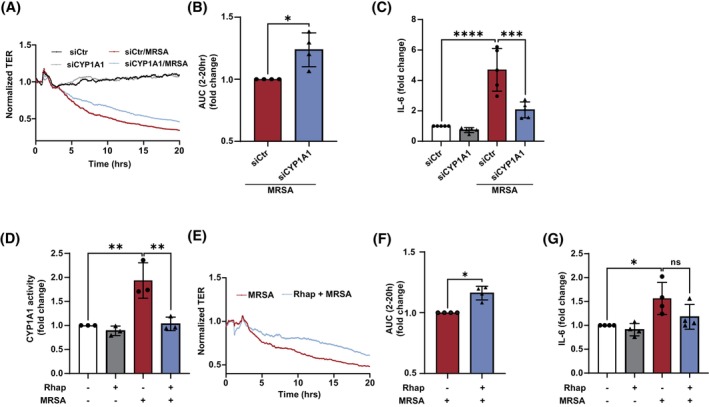
CYP1A1 expression and activity mediate MRSA‐induced EC permeability and inflammatory responses. (A–C) HPAEC were transfected with CYP1A1 siRNA or control for 48 h before HK‐MRSA (2.5 × 108/mL) treatment. EC barrier function was assessed using the ECIS assay. Normalized transendothelial electrical resistance (TER) values were monitored over time. (A) Representative TER tracing, and (B) quantification of the area under the curve during MRSA‐induced EC barrier disruption (higher levels indicate enhanced cumulative barrier function). (C) IL‐6 supernatant levels from MRSA‐treated EC (5 × 108/mL, 6 h) after transfection with CYP1A1 siRNA or control. (D–F) EC were treated with the CYP1A1 inhibitor rhapontigenin (5 μM) or vehicle (DMSO) for 1 h before HK‐MRSA (2.5 × 108/mL). (D) CYP1A1 activity as assessed using the EROD assay. (E, F) EC barrier function was assessed using ECIS. (E) Representative TER tracing, and (F) quantification of the area under the curve during MRSA‐induced EC barrier disruption. (G) IL‐6 levels in EC supernatants after indicated treatments (2.5 × 108/mL, 6 h). N = 3–4 independent experiments. Data were analyzed using Welch's t‐test (B, F) or one‐way ANOVA (C, D, and G), *p < .05, **p < .01, ***p < .001, ****p < .0001.
To assess the role of CYP1A1 enzyme activity in these responses, the small‐molecule CYP1A1 inhibitor rhapontigenin 43 was used. As depicted in Figure 5D, pretreatment with rhapontigenin abolished MRSA‐induced CYP1A1 activity in HPAEC, demonstrating its effectiveness as an inhibitor under these experimental conditions. Similar to reduction of CYP1A1 expression via siRNA, inhibition of CYP1A1 activity by rhapontigenin had no effect on baseline resistance but significantly attenuated the barrier disruptive effects of MRSA (Figure 5E,F). Moreover, rhapontigenin decreased MRSA‐induced IL‐6 release by ~25% compared to vehicle, although this effect did not reach statistical significance (Figure 5G).
3.6. Rhapontigenin attenuates MRSA‐induced lung injury in mice
To determine the functional role of CYP1A1 in vivo, we employed our previously established mouse model of ALI caused by live MRSA. 19 , 20 One hour prior to intratracheal administration of MRSA, animals received either vehicle or the CYP1A1 inhibitor rhapontigenin at 10 mg/kg (IP). Consistent with our in vitro observations, MRSA infection significantly increased CYP1A1 expression in both lung tissue and bronchoalveolar lavage (BAL) fluid (Figure 6A,B). The effects of rhapontigenin on multiple indices of ALI severity were also assessed. Pretreatment with rhapontigenin significantly reduced the BAL protein levels after MRSA by ~29% and trended toward a reduction in BAL neutrophil count (Figure 6C,D) compared to vehicle‐control mice. Histologic assessment of lung tissue with hematoxylin and eosin staining provided further evidence that rhapontigenin treatment reduces MRSA‐induced interstitial thickening, neutrophil infiltration, and edema (Figure 6E).
FIGURE 6.
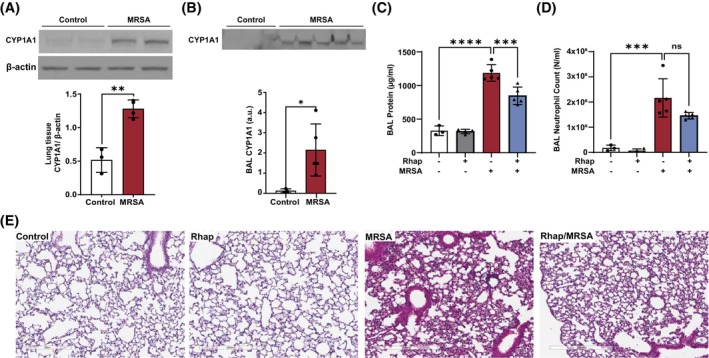
Rhapontigenin attenuates MRSA‐induced ALI in mice. C57BL/6 male mice, 10–12 weeks, were administered vehicle (10% DMSO in saline) or rhapontigenin (IP) at 10 mg/kg for 1 h prior to MRSA (0.75 × 108/mouse) infection (IT). 18 h later, bronchoalveolar lavage (BAL) and lung tissues were collected and analyzed. (A, B) Representative western blots and pooled densitometric analyses for CYP1A1 protein expression in (A) lung tissue and (B) BAL fluid. BAL was assayed for (C) total protein concentration and (D) neutrophil counts. (E) Lung tissue H&E staining. Lung tissues were scanned with a digital slide scanner at 40× and representative images were taken at 20×. Scale bar: 300 μm. Representative lung sections are shown. Each symbol represents an individual mouse. N = 3–5 mice per group. Data were analyzed using an unpaired t‐test or one‐way ANOVA, *p < .05, **p < .01, ***p < .001, ****p < .0001.
Next, levels of inflammatory and endothelial injury markers were assessed in the BAL fluid samples. Rhapontigenin pretreatment significantly attenuated MRSA‐induced upregulation of the inflammatory cytokine IL‐6 in BAL (Figure 7A) but had no substantial effect on BAL KC levels (Figure 7B). Rhapontigenin also significantly attenuated MRSA effects on BAL levels of both sVCAM‐1 and angiopoietin‐2, which are markers of endothelial injury in vivo, 44 compared to vehicle (Figure 7C,D). These data support the hypothesis that CYP1A1 activity participates in ALI pathophysiology caused by MRSA in vivo.
FIGURE 7.
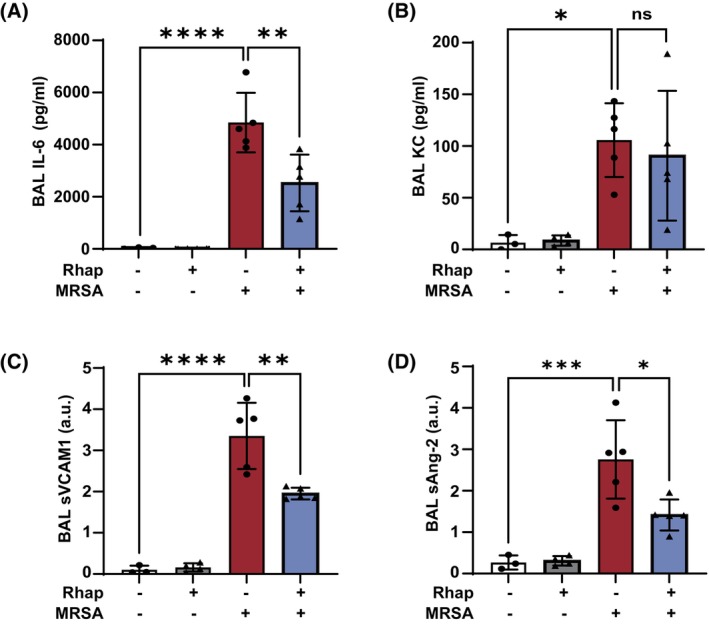
Effects of rhapontigenin on lung inflammatory markers induced by MRSA in mice. C57BL/6 male mice, 10–12 weeks, were administered vehicle (10% DMSO in saline) or rhapontigenin (IP) at 10 mg/kg for 1 h prior to MRSA (0.75 × 108/mouse) infection (IT) for 18 h. (A) IL‐6, (B) KC, (C) sVCAM‐1, and (D) angiopoietin‐2 levels were measured in bronchoalveolar lavage (BAL) fluid. Each symbol represents an individual mouse. N = 3–5 mice per group. Data were analyzed using one‐way ANOVA, *p < .05, **p < .01, ***p < .001, ****p < .0001.
3.7. CYP1A1 expression is increased in the tracheal aspirates of ARDS patients
As an initial screen for potential translational relevance in human disease, we re‐analyzed publicly available data originally published by Sarma et al. 37 in which tracheal aspirates from 52 mechanically ventilated patients were analyzed for gene expression differences between those with ARDS due to COVID‐19 infection, those with ARDS due to other causes, and control ventilated patients (non‐ARDS). Interestingly, both the COVID‐ARDS and non‐COVID‐ARDS patients had significantly elevated CYP1A1 expression in their tracheal aspirates compared to the control patients (p < .01) (Figure 8). When combining the ARDS groups together for further analysis (COVID + non‐COVID), the increase in CYP1A1 expression was even more significant compared to ventilated control patients (p = .003). These data suggest that elevated CYP1A1 levels in the lung may be associated with human ARDS. However, the limited sample size of non‐ARDS patients constrains the generalizability of these findings.
FIGURE 8.
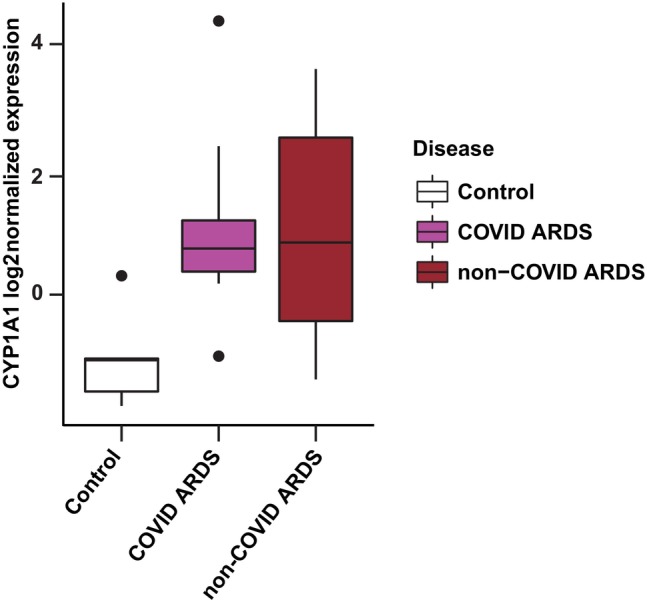
CYP1A1 expression in tracheal aspirates of ARDS patients. Box plot of CYP1A1 log2‐normalized expression in human patient tracheal aspirates derived from publicly available data. 37 The box represents the interquartile range (IQR), with the upper and lower borders indicating the 75th and 25th percentiles, respectively. Outliers are defined as data points exceeding 1.5 times the IQR beyond these percentiles and indicated by dots in the figure. Data were derived from patients undergoing mechanical ventilation with COVID ARDS (n = 15), non‐COVID ARDS (n = 32), and control patients without ARDS (n = 5). Both ARDS groups individually, as well as when combined for analysis, had increased CYP1A1 RNA levels compared to control patients (p < .01). This secondary analysis does not necessarily reflect the opinions and conclusions of the investigators who performed the original study that collected these data. 37
4. DISCUSSION
To summarize the major findings of this study, we have identified a novel functional role for MRSA‐induced epigenetic modifications in mediating lung endothelial dysfunction and ALI (Figure 9). Specifically, the promoter region of CYP1A1, a cytochrome P450 enzyme, is a key target for increased H3K9ac in lung EC following MRSA exposure. Lung EC exposed to MRSA exhibit a significant increase in CYP1A1 mRNA levels, protein expression, and enzymatic activity, which is mediated in part via Ahr signaling. Epigenetic pathway inhibitors attenuate MRSA‐induced CYP1A1 expression and pro‐inflammatory signaling, supporting a critical role for epigenetic modifications in regulating these responses. Importantly, inhibition of CYP1A1 activity or down‐regulation of its expression reduces the effects of MRSA on lung endothelium in vitro, while inhibition of CYP1A1 activity attenuates MRSA‐induced lung injury in mice in vivo. Analysis of publicly available data suggests increased lung expression of CYP1A1 in a small cohort of ARDS patients. Overall, these findings highlight the importance of epigenetic modifications in the development of ALI and suggest CYP1A1 as a novel mediator of bacterial‐induced ALI.
FIGURE 9.
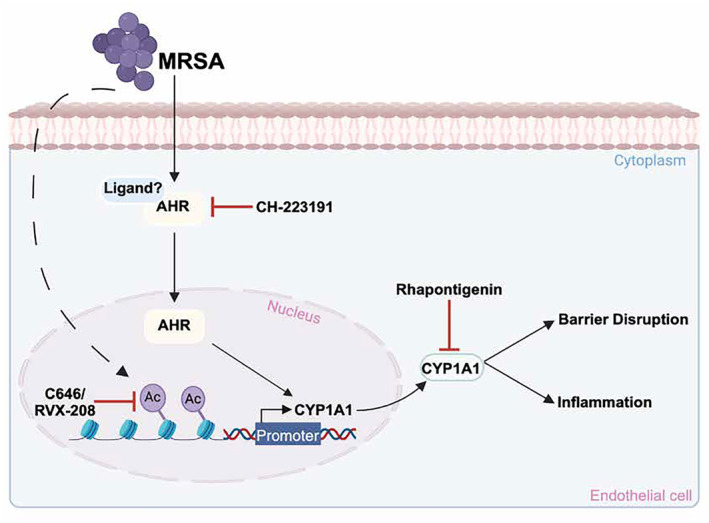
Proposed effects of MRSA on CYP1A1 expression and lung EC function. This schema summarizes observations from the current study in which MRSA induces CYP1A1 expression in lung EC and causes lung endothelial barrier disruption and inflammation. MRSA increases the acetylation of histone H3 at lysine K9 (H3K9) within the CYP1A1 promoter region, enhancing its transcription and subsequent expression. The dashed line indicates that the mechanism by which MRSA induces acetylation is unclear. Inhibition of either histone acetyltransferases (HATs) (C646) or bromodomain and extra‐terminal domain proteins (BETs) (RVX‐208) attenuates CYP1A1 upregulation. Additionally, MRSA activates the aryl hydrocarbon receptor (Ahr) through an unknown ligand, promoting Ahr translocation into the nucleus to further induce CYP1A1 expression. Blocking Ahr (CH‐223191) attenuates CYP1A1 expression and activity. Inhibiting CYP1A1 activity (Rhapontigenin) attenuates MRSA‐induced disruption of the lung endothelial barrier and associated inflammation. This image was generated using BioRender.com.
Emerging research has identified an important role for epigenetic processes in the pathogenesis of ARDS and other lung diseases, particularly in endothelial dysfunction. 14 , 45 , 46 Modifications such as DNA methylation and histone acetylation alter gene expression patterns and can influence the extent of endothelial barrier disruption. 47 , 48 Few studies have characterized the epigenetic alterations caused by MRSA and their role in inflammation. In one study using a mouse model of S. aureus‐induced ALI and sepsis, reduced H3K9/14ac at the Angpt1 gene in the lungs of injured mice was associated with decreased expression of the barrier‐protective and anti‐inflammatory angiopoeitin‐1 protein. 24 In this current study, ChIP‐seq analysis was employed to perform an unbiased genome‐wide profiling of H3K9ac epigenetic patterns in human lung EC exposed to MRSA. H3K9ac modifications are known activators of gene transcription and are generally less studied in lung disease than methylation events. MRSA significantly altered H3K9ac levels at >1500 sites (Figure 1).
Epigenetic modifications are mediated by several enzymes, including HATs, HDACs, and BETs. 38 In lung epithelial cells, down‐regulation of LPS‐induced p300 HAT suppresses NF‐kB inflammatory pathways. 49 In the current study, inhibition of p300/CBP HATs in lung EC attenuated the expression of adhesion molecules (VCAM‐1, ICAM‐1) and pro‐inflammatory cytokine release (IL‐6 and IL‐8) after MRSA (Figure 2). Similar to HATs, several studies report mechanisms by which BETi may alleviate inflammatory pathways in ALI. 50 , 51 Our study demonstrated that inhibition of BET using the pharmaceutical compound RVX‐208 exerted anti‐inflammatory effects on MRSA‐treated lung EC (Figure 3). RVX‐208 has been tested in Phases II and III clinical trials for the treatment of various cardiovascular conditions, 40 , 52 , 53 and also has been suggested as a candidate treatment for COVID‐19‐induced ARDS. 54 Overall, our results demonstrate an important role for histone acetylation in mediating MRSA‐induced endothelial dysfunction and add new evidence about the potential therapeutic utility of epigenetic modifiers in ARDS.
An important observation from the ChIP‐seq analysis is the identification of CYP1A1 as an epigenetic target gene in MRSA‐treated lung EC. CYP1 enzymes have a dual role in both metabolically activating and detoxifying a wide range of carcinogens commonly found in cigarette smoke, air pollution, and other inhaled toxins. 55 In our study, MRSA induced CYP1A1 expression in both lung EC in vitro (Figure 1) and mouse lungs in vivo (Figure 6). CYP1A1 is increased in the lungs by multiple inflammatory stimuli, including hyperoxia, 56 LPS, 57 and cigarette smoke. 58 CYP1A1 levels are also increased in monocytes and plasma of patients with sepsis and positively correlate with the Sequential Organ Failure Assessment (SOFA) scores. 26 The classical pathway of CYP1A1 induction involves the activation of Ahr, 42 which is a ligand‐activated transcription factor. Consistent with this mechanism, the current study demonstrates that inhibition of Ahr resulted in suppression of MRSA‐induced CYP1A1 expression (Figure 4). The mechanisms by which MRSA leads to Ahr activation remain to be determined, though studies suggest bacterial pathogens such as Pseudomonas aeruginosa and Staphylococcus epidermidis activate Ahr through various virulence factors. 59 , 60 A novel finding in our current study is that MRSA‐induced CYP1A1 expression in lung EC is epigenetically mediated. Previously, the CYP1A1 gene was found to be hypomethylated and upregulated in smokers. 61 In another study, hyperoxia‐induced lung injury decreased CYP1A1 gene expression and correlated with H3K27ac loss. 62 Ahr signaling is also associated with histone modifications, including hyperacetylation and trimethylation, that likely play a key role in the epigenetic regulation of CYP1A1. 63 There is substantial evidence that the HATs CBP/p300 are involved in Ahr signaling, 64 and Ahr and p300 directly bind to each other. 65 Considering these prior reports along with our current data, it is likely that histone acetylation changes and CBP/p300 activity are key steps in regulating Ahr‐dependent CYP1A1 expression in MRSA‐exposed lung EC.
Bacterial insults, including by S. aureus, disrupt the endothelial barrier, leading to inflammation and lung dysfunction. 19 , 20 , 66 , 67 The functional significance of CYP1A1 in regulating lung EC responses is not well understood. Previous studies in EC have reported that CYP1A1 downregulation reduces reactive oxygen species (ROS) levels, 48 while its induction is involved in cell cycle arrest through mechanisms that involve Ahr signaling. 24 In the current study, reduction of CYP1A1 expression with siRNA, or inhibition of its enzyme activity in lung EC, attenuated MRSA‐induced endothelial barrier disruption (Figure 5), a key pathogenic event in ARDS. Furthermore, inhibiting CYP1A1 activity reduced MRSA‐induced IL‐6, suggesting that CYP1A1 upregulation is associated with inflammatory EC responses after MRSA. In contrast, overexpression of CYP1A1 in other cell types, including bovine epithelial cells and alveolar macrophages, reportedly reduced inflammation caused by LPS or Mycoplasma hyopneumoniae, respectively. 68 , 69 Interestingly, in another study, MRSA increased CYP1A1 expression in the ileal epithelium of mice, and CYP1A1 global KO mice were protected against gut barrier disruption in this model. 70 Therefore, CYP1A1 appears to have complex roles that may vary depending on cell type and stimulus.
CYP1A1 involvement in ALI is supported by our in vivo results demonstrating that rhapontigenin, an inhibitor of CYP1A1 enzyme activity, significantly attenuated vascular leakage and inflammation in MRSA‐treated mice (Figures 6 and 7). Consistent with these data, rhapontigenin improved survival in septic mice (using intraperitoneal E. coli or CLP models), while overexpression of CYP1A1 increased mortality. 26 In contrast, the same group reported that global CYP1A1 knockout mice had increased lung injury after treatment with intratracheal E. coli or intraperitoneal LPS. 57 CYP1A1 deficiency also potentiated lung damage caused by hyperoxia. 56 In a more recent study, mice lacking endothelial Ahr were more susceptible to lung endothelial barrier disruption following influenza infection, while mice deficient in all 3 CYP1 enzymes (including CYP1A1) were resistant to this type of injury. 71 Since metabolism of ligands mediated by CYP1A1 serves as a negative feedback mechanism that regulates Ahr signaling, 72 it is possible that Ahr expression is maintained at levels that are required to exert its protective effects in mice lacking CYP1A1.
Based on this prior literature and our current findings, we hypothesize that basal levels of CYP1A1 are required for normal lung function and immunity; however, increased CYP1A1 expression is also involved in some inflammatory processes. Therefore, CYP1A1 inhibition may be protective against ALI caused by these stimuli. Given this complexity, a functional role for CYP1A1 in ARDS pathophysiology likely warrants further investigation and is suggested by some preliminary translational observations. These include a genotyping study of 268 pneumonia patients in which ARDS risk was significantly higher in those having the rs2606345 intronic SNP in the CYP1A1 gene, 73 as well as our analysis of publicly available data 37 demonstrating significantly elevated CYP1A1 expression in the tracheal aspirates of ARDS patients compared to mechanically ventilated control subjects (Figure 8).
The current study has some limitations. Although these results demonstrate that CYP1A1 mediates pro‐inflammatory responses to MRSA in lung EC, the specific mechanisms remain to be defined. Given the established role of CYP1A1 in oxidative stress, 25 a reasonable hypothesis is that CYP1A1 contributes to MRSA‐induced EC injury by upregulating ROS levels. Another established function of CYP1A1 is the metabolism of arachidonic acid, resulting in the production of pro‐inflammatory lipid mediators. In macrophages, overexpression of CYP1A1 results in the increased production of 12(S)‐hydroxyeicosatetraenoic acid (12(S)‐HETE), which promotes inflammatory processes. 26 Interestingly, 12‐HETE was recently shown to exert EC barrier disruptive properties. 74 Another limitation of the current study is that the in vivo experiments do not address cell‐specific effects since the CYP1A1 inhibitor rhapontigenin was administrated systemically. It is possible that CYP1A1 inhibition in cell types other than lung EC may contribute to the protective effects observed in MRSA‐induced ALI. However, a recent study elegantly assessed the expression of CYP1A1 in the lungs of mice and reported that its expression was almost exclusively detected in endothelial compartments. 71 These findings, along with our in vitro and in vivo data, strongly indicate that the endothelial CYP1A1 is an important mediator of endothelial dysfunction induced by MRSA. Finally, we analyzed CYP1A1 expression in only a limited number of patient samples (especially the low number of non‐ARDS controls), which constrains the generalizability of these findings. Potential relevance to human disease will be enhanced by further analysis of additional ARDS patient samples for CYP1A1 protein expression and activity.
In summary, this study establishes the functional importance of epigenetic modifications in mediating the pathophysiologic responses of lung EC to MRSA and identifies CYP1A1 as a novel functional effector in this process. These results further our understanding of the molecular mechanisms underlying ARDS pathogenesis and suggest CYP1A1 as a possible therapeutic target. Selective inhibition of histone acetylation represents a promising therapy for mitigating adverse changes in specific promoter regions altered by MRSA; targeting epigenetic mechanisms may be an intriguing future approach for the complex ARDS disease process.
AUTHOR CONTRIBUTIONS
Alison W. Ha performed experiments, analyzed and interpreted data, prepared figures, and drafted the manuscript. Lucille N. Meliton performed experiments, analyzed and interpreted data, prepared figures, and drafted the manuscript. Weiguo Chen and Lichun Wang performed experiments. Mark Maienschein‐Cline analyzed and interpreted data, prepared figures, and edited the manuscript. Jeffrey R. Jacobson interpreted data and edited the manuscript. Eleftheria Letsiou performed experiments, analyzed and interpreted data, prepared figures, drafted and edited the manuscript, and designed the study. Steven M. Dudek conceived and designed the study, edited and revised the manuscript.
DISCLOSURES
The authors have no conflicts of interest in connection with this article.
Supporting information
Figure S1.
Figure S2.
Figure S3.
Table S1.
Table S2.
ACKNOWLEDGMENTS
This work was supported by NIH R01 HL167518 (SD), and P01 HL126609 (SD). A.W.H. was supported by NIH T32 training grant HL144909. E.L is supported by American Heart Association Grant #932176/EL/2022. M.M.C is supported in part by NCATS through Grant UL1TR002003. We thank Dr. Yulia Epshtein for providing excellent technical assistance.
Ha AW, Meliton LN, Chen W, et al. Epigenetic mechanisms mediate cytochrome P450 1A1 expression and lung endothelial injury caused by MRSA in vitro and in vivo. The FASEB Journal. 2024;38:e70205. doi: 10.1096/fj.202401812R
Eleftheria Letsiou and Steven M. Dudek are co‐senior authors.
DATA AVAILABILITY STATEMENT
ChIP‐seq sequence data have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus (GEO) and can be accessed using the accession number GSE274958. Tracheal aspirate RNA sequencing data are also available under the GEO accession number GSE163426. All original western blot images are provided in Figure S3.
REFERENCES
- 1. Bos LDJ, Ware LB. Acute respiratory distress syndrome: causes, pathophysiology, and phenotypes. Lancet. 2022;400:1145‐1156. [DOI] [PubMed] [Google Scholar]
- 2. Huertas A, Guignabert C, Barberà JA, et al. Pulmonary vascular endothelium: the orchestra conductor in respiratory diseases: highlights from basic research to therapy. Eur Respir J. 2018;51:1700745. [DOI] [PubMed] [Google Scholar]
- 3. Vassiliou AG, Kotanidou A, Dimopoulou I, Orfanos SE. Endothelial damage in acute respiratory distress syndrome. Int J Mol Sci. 2020;21:8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Su Y, Lucas R, Fulton DJR, Verin AD. Mechanisms of pulmonary endothelial barrier dysfunction in acute lung injury and acute respiratory distress syndrome. Chin Med J Pulm Crit Care Med. 2024;2:80‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kan M, Shumyatcher M, Himes BE. Using omics approaches to understand pulmonary diseases. Respir Res. 2017;18:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ebenezer DL, Berdyshev EV, Bronova IA, et al. Pseudomonas aeruginosa stimulates nuclear sphingosine‐1‐phosphate generation and epigenetic regulation of lung inflammatory injury. Thorax. 2019;74:579‐591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Graves BT, Munro CL. Epigenetics in critical illness: a new frontier. Nurs Res Pract. 2013;2013:503686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yasutake T, Wada H, Higaki M, et al. Anacardic acid, a histone acetyltransferase inhibitor, modulates LPS‐induced IL‐8 expression in a human alveolar epithelial cell line A549. F1000Res. 2013;2:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lai Y, Li J, Li X, Zou C. Lipopolysaccharide modulates p300 and Sirt1 to promote PRMT1 stability via an SCF(Fbxl17)‐recognized acetyldegron. J Cell Sci. 2017;130:3578‐3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xing S, Nie F, Xu Q, et al. HDAC is essential for epigenetic regulation of Thy‐1 gene expression during LPS/TLR4‐mediated proliferation of lung fibroblasts. Lab Investig. 2015;95:1105‐1116. [DOI] [PubMed] [Google Scholar]
- 11. Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074‐1080. [DOI] [PubMed] [Google Scholar]
- 12. Bates SE. Epigenetic therapies for cancer. N Engl J Med. 2020;383:650‐663. [DOI] [PubMed] [Google Scholar]
- 13. Morgado‐Pascual JL, Rayego‐Mateos S, Tejedor L, Suarez‐Alvarez B, Ruiz‐Ortega M. Bromodomain and Extraterminal proteins as novel epigenetic targets for renal diseases. Front Pharmacol. 2019;10:1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fang Z, Wang X, Sun X, Hu W, Miao QR. The role of histone protein acetylation in regulating endothelial function. Front Cell Dev Biol. 2021;9:672447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Defres S, Marwick C, Nathwani D. MRSA as a cause of lung infection including airway infection, community‐acquired pneumonia and hospital‐acquired pneumonia. Eur Respir J. 2009;34:1470‐1476. [DOI] [PubMed] [Google Scholar]
- 16. Karchmer AW, Bayer AS. Methicillin‐resistant Staphylococcus aureus: an evolving clinical challenge. Clin Infect Dis. 2008;46(Suppl 5):S342‐S343. [DOI] [PubMed] [Google Scholar]
- 17. Napolitano LM, Brunsvold ME, Reddy RC, Hyzy RC. Community‐acquired methicillin‐resistant Staphylococcus aureus pneumonia and ARDS: 1‐year follow‐up. Chest. 2009;136:1407‐1412. [DOI] [PubMed] [Google Scholar]
- 18. Lakhundi S, Zhang K. Methicillin‐resistant Staphylococcus aureus: molecular characterization, evolution, and epidemiology. Clin Microbiol Rev. 2018;31:e00020‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Htwe YM, Wang H, Belvitch P, et al. Group V phospholipase a(2) mediates endothelial dysfunction and acute lung injury caused by methicillin‐resistant Staphylococcus aureus . Cells. 2021;10:1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang L, Letsiou E, Wang H, et al. MRSA‐induced endothelial permeability and acute lung injury are attenuated by FTY720 S‐phosphonate. Am J Physiol Lung Cell Mol Physiol. 2022;322:L149‐L161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maiti A, Jiranek WA. Inhibition of methicillin‐resistant Staphylococcus aureus‐induced cytokines mRNA production in human bone marrow derived mesenchymal stem cells by 1,25‐dihydroxyvitamin D3. BMC Cell Biol. 2014;15:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Modak R, Das Mitra S, Vasudevan M, et al. Epigenetic response in mice mastitis: role of histone H3 acetylation and microRNA(s) in the regulation of host inflammatory gene expression during Staphylococcus aureus infection. Clin Epigenetics. 2014;6:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chang YL, Rossetti M, Gjertson DW, et al. Human DNA methylation signatures differentiate persistent from resolving MRSA bacteremia. Proc Natl Acad Sci USA. 2021;118:e2000663118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bomsztyk K, Mar D, An D, et al. Experimental acute lung injury induces multi‐organ epigenetic modifications in key angiogenic genes implicated in sepsis‐associated endothelial dysfunction. Crit Care. 2015;19:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stading R, Chu C, Couroucli X, Lingappan K, Moorthy B. Molecular role of cytochrome P4501A enzymes inoxidative stress. Curr Opin Toxicol. 2020;20–21:77‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tian LX, Tang X, Zhu JY, et al. Cytochrome P450 1A1 enhances inflammatory responses and impedes phagocytosis of bacteria in macrophages during sepsis. Cell Commun Signal. 2020;18:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. arXiv. 2013; 1303.3997v1 [q‐bio.GN]. [Google Scholar]
- 28. Wysoker ATK, Fennel T. Picard tools version 1.90. 2013.
- 29. Zhang Y, Liu T, Meyer CA, et al. Model‐based analysis of ChIP‐seq (MACS). Genome Biol. 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Robinson JT, Thorvaldsdóttir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841‐842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923‐930. [DOI] [PubMed] [Google Scholar]
- 33. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA‐Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288‐4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57:289‐300. [Google Scholar]
- 36. Manzella C, Singhal M, Alrefai WA, Saksena S, Dudeja PK, Gill RK. Serotonin is an endogenous regulator of intestinal CYP1A1 via AhR. Sci Rep. 2018;8:6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sarma A, Christenson SA, Byrne A, et al. Tracheal aspirate RNA sequencing identifies distinct immunological features of COVID‐19 ARDS. Nat Commun. 2021;12:5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26:5310‐5318. [DOI] [PubMed] [Google Scholar]
- 39. Mujtaba S, Zeng L, Zhou MM. Structure and acetyl‐lysine recognition of the bromodomain. Oncogene. 2007;26:5521‐5527. [DOI] [PubMed] [Google Scholar]
- 40. Tsujikawa LM, Fu L, Das S, et al. Apabetalone (RVX‐208) reduces vascular inflammation in vitro and in CVD patients by a BET‐dependent epigenetic mechanism. Clin Epigenetics. 2019;11:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lu P, Shen Y, Yang H, et al. BET inhibitors RVX‐208 and PFI‐1 reactivate HIV‐1 from latency. Sci Rep. 2017;7:16646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ye W, Chen R, Chen X, et al. AhR regulates the expression of human cytochrome P450 1A1 (CYP1A1) by recruiting Sp1. FEBS J. 2019;286:4215‐4231. [DOI] [PubMed] [Google Scholar]
- 43. Chun YJ, Ryu SY, Jeong TC, Kim MY. Mechanism‐based inhibition of human cytochrome P450 1A1 by rhapontigenin. Drug Metab Dispos. 2001;29:389‐393. [PubMed] [Google Scholar]
- 44. Kulkarni HS, Lee JS, Bastarache JA, et al. Update on the features and measurements of experimental acute lung injury in animals: An official American Thoracic Society workshop report. Am J Respir Cell Mol Biol. 2022;66:e1‐e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ahmad S, Manzoor S, Siddiqui S, et al. Epigenetic underpinnings of inflammation: connecting the dots between pulmonary diseases, lung cancer and COVID‐19. Semin Cancer Biol. 2022;83:384‐398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Joshi AD, Barabutis N, Birmpas C, et al. Histone deacetylase inhibitors prevent pulmonary endothelial hyperpermeability and acute lung injury by regulating heat shock protein 90 function. Am J Physiol Lung Cell Mol Physiol. 2015;309:L1410‐L1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lee HT, Oh S, Ro DH, Yoo H, Kwon YW. The key role of DNA methylation and histone acetylation in epigenetics of atherosclerosis. J Lipid Atheroscler. 2020;9:419‐434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ihezie SA, Mathew IE, McBride DW, Dienel A, Blackburn SL, Thankamani Pandit PK. Epigenetics in blood‐brain barrier disruption. Fluids Barriers CNS. 2021;18:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu Q, Yang H, Xu S, Sun X. Downregulation of p300 alleviates LPS‐induced inflammatory injuries through regulation of RhoA/ROCK/NF‐κB pathways in A549 cells. Biomed Pharmacother. 2018;97:369‐374. [DOI] [PubMed] [Google Scholar]
- 50. Guo X, Olajuyin A, Tucker TA, Idell S, Qian G. BRD4 as a therapeutic target in pulmonary diseases. Int J Mol Sci. 2023;24:13231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Huang M, Zeng S, Zou Y, et al. The suppression of bromodomain and extra‐terminal domain inhibits vascular inflammation by blocking NF‐κB and MAPK activation. Br J Pharmacol. 2017;174:101‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Toth PP, Schwartz GG, Nicholls SJ, et al. Reduction in the risk of major adverse cardiovascular events with the BET protein inhibitor apabetalone in patients with recent acute coronary syndrome, type 2 diabetes, and moderate to high likelihood of non‐alcoholic fatty liver disease. Am J Prev Cardiol. 2022;11:100372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Provencher S, Potus F, Blais‐Lecours P, et al. BET protein inhibition for pulmonary arterial hypertension: a pilot clinical trial. Am J Respir Crit Care Med. 2022;205:1357‐1360. [DOI] [PubMed] [Google Scholar]
- 54. Gilham D, Smith AL, Fu L, et al. Bromodomain and extraterminal protein inhibitor, apabetalone (RVX‐208), reduces ACE2 expression and attenuates SARS‐Cov‐2 infection in vitro. Biomedicine. 2021;9:437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nebert DW, Dalton TP, Okey AB, Gonzalez FJ. Role of aryl hydrocarbon receptor‐mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J Biol Chem. 2004;279:23847‐23850. [DOI] [PubMed] [Google Scholar]
- 56. Lingappan K, Jiang W, Wang L, et al. Mice deficient in the gene for cytochrome P450 (CYP)1A1 are more susceptible than wild‐type to hyperoxic lung injury: evidence for protective role of CYP1A1 against oxidative stress. Toxicol Sci. 2014;141:68‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tian LX, Tang X, Ma W, et al. Knockout of cytochrome P450 1A1 enhances lipopolysaccharide‐induced acute lung injury in mice by targeting NF‐κB activation. FEBS Open Bio. 2020;10:2316‐2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Portal‐Nuñez S, Shankavaram UT, Rao M, et al. Aryl hydrocarbon receptor‐induced adrenomedullin mediates cigarette smoke carcinogenicity in humans and mice. Cancer Res. 2012;72:5790‐5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rademacher F, Simanski M, Hesse B, et al. Staphylococcus epidermidis activates aryl hydrocarbon receptor signaling in human keratinocytes: implications for cutaneous defense. J Innate Immun. 2019;11:125‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Moura‐Alves P, Faé K, Houthuys E, et al. AhR sensing of bacterial pigments regulates antibacterial defence. Nature. 2014;512:387‐392. [DOI] [PubMed] [Google Scholar]
- 61. Tsai PC, Glastonbury CA, Eliot MN, et al. Smoking induces coordinated DNA methylation and gene expression changes in adipose tissue with consequences for metabolic health. Clin Epigenetics. 2018;10:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Coarfa C, Grimm SL, Katz T, et al. Epigenetic response to hyperoxia in the neonatal lung is sexually dimorphic. Redox Biol. 2020;37:101718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schnekenburger M, Peng L, Puga A. HDAC1 bound to the Cyp1a1 promoter blocks histone acetylation associated with Ah receptor‐mediated trans‐activation. Biochim Biophys Acta. 2007;1769:569‐578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fujii‐Kuriyama Y, Mimura J. Molecular mechanisms of AhR functions in the regulation of cytochrome P450 genes. Biochem Biophys Res Commun. 2005;338:311‐317. [DOI] [PubMed] [Google Scholar]
- 65. Tohkin M, Fukuhara M, Elizondo G, Tomita S, Gonzalez FJ. Aryl hydrocarbon receptor is required for p300‐mediated induction of DNA synthesis by adenovirus E1A. Mol Pharmacol. 2000;58:845‐851. [DOI] [PubMed] [Google Scholar]
- 66. Karki P, Ke Y, Tian Y, et al. Staphylococcus aureus‐induced endothelial permeability and inflammation are mediated by microtubule destabilization. J Biol Chem. 2019;294:3369‐3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Meliton AY, Meng F, Tian Y, et al. Oxidized phospholipids protect against lung injury and endothelial barrier dysfunction caused by heat‐inactivated Staphylococcus aureus . Am J Physiol Lung Cell Mol Physiol. 2015;308:L550‐L562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhang WY, Wang H, Qi S, et al. CYP1A1 relieves lipopolysaccharide‐induced inflammatory responses in bovine mammary epithelial cells. Mediat Inflamm. 2018;2018:4093285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fang X, Zhao W, Xu J, et al. CYP1A1 mediates the suppression of major inflammatory cytokines in pulmonary alveolar macrophage (PAM) cell lines caused by Mycoplasma hyponeumoniae . Dev Comp Immunol. 2016;65:132‐138. [DOI] [PubMed] [Google Scholar]
- 70. Ma X, Jin H, Chu X, et al. The host CYP1A1‐microbiota metabolic Axis promotes gut barrier disruption in methicillin‐resistant Staphylococcus aureus‐induced abdominal sepsis. Front Microbiol. 2022;13:802409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Major J, Crotta S, Finsterbusch K, et al. Endothelial AHR activity prevents lung barrier disruption in viral infection. Nature. 2023;621:813‐820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chiaro CR, Patel RD, Marcus CB, Perdew GH. Evidence for an aryl hydrocarbon receptor‐mediated cytochrome p450 autoregulatory pathway. Mol Pharmacol. 2007;72:1369‐1379. [DOI] [PubMed] [Google Scholar]
- 73. Salnikova LE, Smelaya TV, Vesnina IN, Golubev AM, Moroz VV. Genetic susceptibility to nosocomial pneumonia, acute respiratory distress syndrome and poor outcome in patients at risk of critical illness. Inflammation. 2014;37:295‐305. [DOI] [PubMed] [Google Scholar]
- 74. Wang X, Gao L, Xiao L, et al. 12(S)‐hydroxyeicosatetraenoic acid impairs vascular endothelial permeability by altering adherens junction phosphorylation levels and affecting the binding and dissociation of its components in high glucose‐induced vascular injury. J Diabetes Investig. 2019;10:639‐649. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Figure S2.
Figure S3.
Table S1.
Table S2.
Data Availability Statement
ChIP‐seq sequence data have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus (GEO) and can be accessed using the accession number GSE274958. Tracheal aspirate RNA sequencing data are also available under the GEO accession number GSE163426. All original western blot images are provided in Figure S3.