

ABSTRACT
Successful bacterial colonizers and pathogens have evolved with their hosts and have acquired mechanisms to customize essential processes that benefit their lifestyle. In large part, bacterial survival hinges on shaping the transcriptional signature of the host, a process regulated at the chromatin level. Modifications of chromatin, either on histone proteins or on DNA itself, are common targets during bacterium-host cross talk and are the focus of this article.
INTRODUCTION
Chromatin is located within the nuclei of eukaryotic cells and is composed of DNA wrapped around histone proteins. The highly ordered compaction of chromatin is crucial for the different functions encoded by the genetic material. These range from maintaining cell identity and genome integrity to adapting to environmental stimuli and cell replication. At the center of the chromatin language is its structural organization. This depends on the position and reversible covalent modifications to histone proteins and their cross talk with DNA and regulatory proteins. The basic unit of chromatin is the nucleosome, which is composed of an octamer of four histone proteins (H2A, H2B, H3, and H4) around which ∼147 bases of DNA are wrapped, with the linker histone (H1) outside the core structure providing structural integrity to the complex. Nucleosome remodelers are ATP-dependent enzymes that modify the chromatin structure through translocation, eviction, and introduction of histone variants (1, 2), while histone-modifying enzymes introduce reversible covalent posttranslational modifications (PTMs) to histone tails.
Nucleosome remodelers and modifying enzymes regulate chromatin dynamics by repositioning histones, winding and unwinding DNA, and adding and removing PTMs on the N-terminal histone tails that extend from the octamer complex. Each histone (H2A, H2B, H3, and H4), including the linker histone (H1) and histone variants, can be modified at multiple locations along its tail (3–7). The combined activity of remodelers and histone modifiers regulates unraveling and compaction of chromatin, leading to transcriptional regulation. Specific sets of histone marks are associated with opening of the chromatin structure, allowing transcription factors to bind, polymerase II to extend, and gene expression to occur, whereas others are associated with silent genetic regions (8–12).
Additionally, DNA itself can be methylated by DNA methyltransferases, primarily in promoter and enhancer regions preceding transcriptional start sites. DNA methylation is achieved by the covalent transfer of a methyl group to the C-5 position of the cytosine ring of DNA. Removal of this group is thought to be done indirectly through intermediate modifications, as no demethylase enzymes have been identified. DNA methylation results in silencing of the neighboring gene’s expression and is important for cross regulation of histone PTMs (13).
Ultimately, through the intense remodeling of chromatin arises a histone language, which encodes additional regulatory information beyond that present in the DNA sequence (3–7). A large number of histone PTMs have been identified—acetylation, methylation, phosphorylation, ubiquitination, ADP ribosylation, deimination, and proline isomerization—highlighting the complexity of the system (13). The writers and erasers of PTMs are classified by the histone mark they deposit or erase, such as methyltransferases or demethylases, and are usually specific to a given histone residue. The combination of PTMs on a histone tail and the pattern of DNA methylation act as binding platforms onto which “reader” proteins bind and regulate cell processes. For instance, bromodomain proteins have high affinity for acetylated histones and play a role in transcriptional activation, whereas histone methylation at H3K9 will recruit silencing proteins, like heterochromatin protein 1 (HP1), and maintain a repressive genetic environment. DNA methylation is read by methyl-CpG binding proteins, which act as structural proteins to recruit histone deacetylases (HDACs) and ultimately lead to chromatin compaction and gene silencing. Histone PTMs and DNA methylation are therefore crucial in integrating environmental stimuli throughout the cell’s life (13, 14).
Given the key role of chromatin in regulating host transcription, it is not surprising that bacteria have evolved to manipulate it. In this chapter, we focus on the different mechanisms by which bacteria customize host chromatin for their survival, whether it is by indirect of direct targeting of histones, DNA methylation, or even altering DNA integrity.
Histone Modifications in Response to Bacterial Products
Bacterial components are continuously sensed by host cells, and such cross talk is crucial for regulating immune responses. A balanced response must tolerate commensal bacteria in order to maintain homeostasis yet remain reactive to combat invading pathogens. How this delicate balance is achieved is not well understood; however, some evidence points to an integration of bacterial signals at the level of histone modifications to control inflammatory responses.
Proinflammatory Signaling
Sensing of bacterial components, like lipopolysaccharide (LPS), in the cellular milieu occurs in part through pattern recognition receptors, leading to activation of inflammatory responses such as the NF-κB pathway. Activation of Toll-like receptor 4 by LPS triggers NF-κB translocation into the nucleus, where it controls transcription of inflammatory mediators in sequential waves, reflecting the chromatin conformation of the genetic loci regulated (15). Indeed, immediately accessible genes are transcribed first, as they are located in regions characterized by open chromatin and are associated with high levels of H4 acetylation. In fact, all Toll-like receptor 4-responsive genes which are rapidly transcribed are maintained in a basal active state characterized by H3K9 acetylation and H3K4 trimethylation (H3K4me3) (16). Genes in this state then gain H4K5/8/12 acetylation upon activation of the signaling cascade, allowing transcriptional elongation and generation of mature full-length transcripts to occur. In contrast, late-accessibility genes require secondary signaling mediators, such as activation of mitogen-activated protein kinase (MAPK) signaling, and histone modifiers to decompact chromatin in order for NF-κB to bind (17). Therefore, regulation at the chromatin level allows transcriptional fine-tuning of genes in the same pathway. It is in this inflammatory context that pathogens and commensals need to establish their niche. Accordingly, bacteria have developed mechanisms to tamper with host inflammatory responses for their benefit.
Anti-Inflammatory Signaling
In locations such as the gut, skin, oral cavity, and vagina, colonization by the microbiome leads to a high local level of LPS, yet in healthy individuals, strong inflammatory responses are not initiated in this environment. Some reports suggest that cells continuously exposed to LPS become unresponsive to it through mechanisms involving chromatin modifications. For instance, macrophages exposed to LPS once and those exposed multiple times display different histone marks at inflammatory gene loci (18). Upon restimulation with LPS, two classes of gene are revealed: tolerizeable (T) genes, which are transiently silenced, and nontolerizeable (NT) genes, which remain accessible. The promoters of T genes, which include inflammatory cytokines, lose the activatory H3K4me3 mark but maintain H4 acetylation levels. In contrast, the promoters of NT genes, including antimicrobial effectors such as antimicrobial defense proteins, retain H3K4me3 and are reacetylated upon restimulation with LPS. Therefore, multiple exposures to LPS lead to silencing of inflammatory genes while others remain active, and both classes of genes retain a chromatin mark reflecting their LPS encounter.
In the gut, metabolic by-products from bacterial growth are potent modulators of host responses and were recently shown to contribute to repression of LPS-inducible inflammatory responses and gut homeostasis. The short-chain fatty acid n-butyrate is produced by commensal gut bacteria and is a potent HDAC inhibitor. In the intestine, butyrate downregulates LPS-mediated inflammatory responses and modulates macrophage function (19). A related study characterized an unusual histone modification regulated by microbiota-derived short-chain fatty acids in the colon. Histone H3 crotonylation, which is an addition of a crotonyl group (C4H5O) to the target lysine, is regulated by class I HDACs and is induced by the microbiota (13, 20; for a review, see reference 21). Therefore, metabolic by-products of the microbiota are potent modifiers of host chromatin and may play an important role in maintaining gut homeostasis.
Similarly to the intestinal tract, the microbiota of the vaginal tract, mainly composed of Lactobacillus spp., is essential to maintaining a homeostatic environment. Lactobacillus gasseri was shown to induce the recruitment of active histone marks (H3 acetylation, H3Kme3, and the H2A.Z histone variant) to the promoter of DEFB1 (encoding human β-defensin-1), an antimicrobial peptide (22). Intriguingly, the related species Lactobacillus reuteri did not. Such studies highlight the idea that maintaining homeostasis is a very delicate process which may even be species specific.
In order for pathogenic bacteria to maintain a long-term presence during chronic infection, they must also use mechanisms to limit the inflammatory response. For this, Pseudomonas aeruginosa generates the quorum-sensing molecule 2-aminoacetophenone, which has anti-inflammatory properties (23). Indeed, treatment with 2-aminoacetophenone prior to infection reduces the expression of proinflammatory cytokines by increased expression and activity of HDAC1 and consequent deacetylation of histone H3 on lysine 18 at promoters of specific targets, such as tumor necrosis factor alpha (TNF-α).
BACTERIAL EFFECTORS TARGETING HISTONE MODIFICATIONS THROUGH SIGNALING EVENTS
In contrast to most colonizing bacteria, pathogens have evolved sophisticated virulence factors which subvert host defenses. Although the mechanisms are diverse, hijacking or interacting with components of host signaling cascades is common to different pathogenic bacteria (24). Targeting of such signaling cascades occurs through direct interaction of bacterial factors with host signaling components, either in the cytoplasm or in the nucleus (Fig. 1).
FIGURE 1.
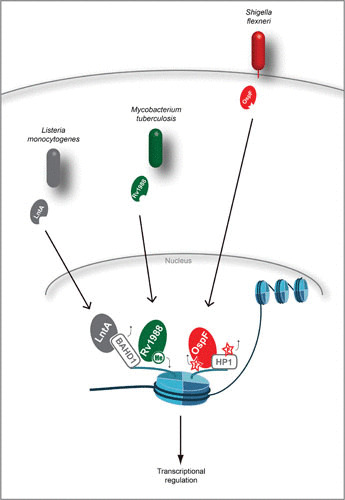
Nuclear effectors targeting histone marks. Secreted effectors from L. monocytogenes, M. tuberculosis, and S. flexneri translocate to the nucleus, where they directly act either upon the nucleosome itself (Rv1988 and OspF), bind chromatin readers to displace them (LntA), or bind chromatin readers to dephosphorylate them (OspF). Small black arrows around modifications indicate whether they are being deposited or removed.
Cytoplasmic Effectors
Mycobacterium tuberculosis
M. tuberculosis is a facultative intracellular pathogen responsible for tuberculosis. During infection, the bacterium dampens the ability of infected macrophages to respond to gamma interferon (IFN-γ) and results in decreased expression of the transcriptional transactivator CIITA, which regulates major histocompatibility complex II (25, 26). In fact, M. tuberculosis blocks IFN-γ-dependent histone acetylation at the CIITA, HLA-DRα, and HLA-DRβ gene promoters. Infection was further shown to induce recruitment of a histone deacetylase complex (Sin3A), leading to histone deacetylation and gene repression. While these findings are not yet attributed to a specific effector, the M. tuberculosis cell wall protein LpqH has been shown to inhibit expression of CIITA, which makes it a putative candidate.
Listeria monocytogenes
L. monocytogenes is a Gram-positive foodborne pathogen that causes listeriosis (27). The internalin B (InlB) gene of L. monocytogenes encodes a factor that binds the host receptor c-Met and activates downstream phosphatidylinositol 3-kinase/AKT signaling (28, 29). Activation of this signaling cascade during infection was shown to hijack the host deacetylase SIRT2, culminating in the translocation of SIRT2 from the cytoplasm to chromatin (30). There, SIRT2 deacetylates H3K18 at the transcriptional start sites of genes repressed during infection. The mechanism by which SIRT2 relocalization occurs was shown to depend on regulation of the phosphorylation status of SIRT2 by two phosphatases, PPM1A and PPM1B. It is this modification of SIRT2 that is crucial for chromatin association and gene regulation (31). Although the genes targeted by SIRT2 need to be further characterized, the activity of SIRT2 at the chromatin level, as well as its dephosphorylation, is essential for a productive Listeria infection both in vitro and in vivo.
Helicobacter pylori
H. pylori is a Gram-negative bacterium causing gastritis and stomach ulcers and is associated with gastric cancer. While the identity of the secreted effector is unknown, H. pylori targets H3S10 and H3T3 for dephosphorylation and H3K23 for deacetylation in a type IV secretion system (T4SS)-dependent manner. In fact, the entire cag pathogenicity island, which contains several virulence factors as well as the Dot/Icm T4SS, is required for chromatin modulations (32, 33). Indeed, mutants with deletions of individual virulence factors, like cytotoxin-associated gene A (cagA) or vacuolating cytotoxin gene A (vacA), fail to dephosphorylate H3S10 (34, 35). On the host side, decreases in H3S10 phosphorylation correlate with cell cycle arrest and inactivation of H3 kinases, mainly VRK1. During late stages of infection, cells reenter the cell cycle and H3S10 phosphorylation reappears (34). Such studies demonstrate that bacteria-mediated histone modifications are associated with other cell processes besides transcription, such as the cell cycle.
Pore formation
The group of toxins known as cholesterol-dependent cytolysin (CDC) are found primarily in Gram-positive bacteria and play crucial roles in virulence. These toxins are generally secreted into the extracellular milieu, where they bind to host plasma membranes in cholesterol-rich areas, oligomerize, and undergo a conformational change to form a large pore (36). The listerial toxin listeriolysin O (LLO) is one member of this family of toxins and was shown to induce H3S10 dephosphorylation and H4 deacetylation. These modifications occur independently of the cell cycle and are associated with the promoter of specific genes such as cxcl2 and dusp4 (37). The signaling cascades known to be induced by LLO (mainly MAPK and NF-κB) are not involved (37); rather, it is potassium efflux through toxin pores which is essential for these chromatin modifications (38).
A recent report showed that the P. aeruginosa T3SS translocon proteins PopB-PopD also induce H3S10 dephosphorylation in a K+ efflux-dependent manner, similarly to LLO (39). These results suggest that the translocon acts as a pore-forming toxin and indicate that such histone modifications could represent a universal host response to a specific type of plasma membrane damage.
Nuclear Effectors
Mycobacterium tuberculosis
In addition to the modulation of cellular pathways in the cytoplasm by M. tuberculosis, the secreted protein Rv1988, which is found exclusively in pathogenic Mycobacterium species, directly targets host chromatin. This effector translocates to the nucleus, where it functions as a methyltransferase, specifically targeting H3R42me2 (40). Rv1988 is required for M. tuberculosis virulence, and it selectively binds to promoter regions of critical immune response genes such as NOX1, NOX4, and NOS2 (required for host reactive-oxygen production). There, it promotes H3R42me3 and represses transcription. Interestingly, expression of Rv1988 is sufficient to confer virulence/pathogenesis in vivo and in vitro to the nonpathogenic species Mycobacterium smegmatis, highlighting the importance of this effector (40).
Shigella flexneri
S. flexneri is a Gram-negative pathogen and is the etiologic agent of dysentery in humans (41). Most S. flexneri virulence factors are secreted through the T3SS, which injects effector proteins directly into the cytoplasm of intestinal epithelial cells (41). One of these, OspF, translocates to the host nucleus upon injection, interrupts MAPK signaling, and binds to the promoter of specific genes involved in inflammatory responses. At the molecular level, OspF is a phosphothreonine lyase that blocks MAPK activation and downstream phosphorylation of histone H3S10 and the chromatin reader HP1γ (42–44). As a result, unphosphorylated HP1 accumulates at promoter sites, thereby blocking interleukin 8 (IL-8) gene transcription. Strikingly, OspF-mediated chromatin modifications and gene repression are specific and target only a subset of genes involved in inflammatory responses. In vivo experiments further show that OspF contributes to blocking neutrophil recruitment to the site of bacterial lesions (42).
Listeria monocytogenes
Independently of InlB and the CDC toxin LLO, Listeria secretes an effector, LntA, which targets the host nucleus. There, it displaces the repressive chromatin reader BAHD1 to activate gene transcription. Upon interaction with lntA, BAHD1 is displaced from chromatin, where H3K9 acetylation occurs and interferon-stimulated gene transcription is activated, leading to IFN-λ expression. In order to fine-tune host inflammatory responses, this process must be tightly regulated by the pathogen, as reflected by the observation that either constitutive expression or absence of LntA is detrimental to infection (45).
BACTERIAL FACTORS MIMICKING HOST CHROMATIN-MODIFYING ENZYMES
SET (suppressor of variegation enhancer of zeste trithorax) domain proteins are ubiquitous in eukaryotes, and this domain can be found in lysine methyltransferases, which can methylate histones in addition to other proteins. Methylated histones at specific residues are associated with different transcriptional states. Silenced genes in heterochromatin regions are marked with H3K9 methylation, whereas active transcription in euchromatin is marked with methylated H3K4 (for a review, see reference 46). To date, secreted SET domain-containing effectors have been found in obligate pathogens, such as Chlamydia trachomatis, Bacillus anthracis, and Legionella pneumophila. Interestingly, the SET domain of secreted bacterial effectors confers methyltransferase activity to bacteria. Due to the lack of histone substrates within bacteria, it is thought that these organisms have hijacked the SET domain to target their hosts (Fig. 2) (47–49).
FIGURE 2.
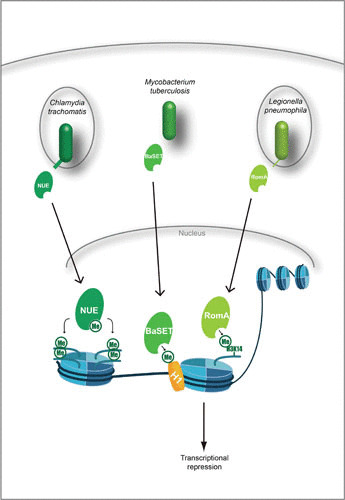
SET domain effectors mediate histone methylation. Effectors of C. trachomatis, M. tuberculosis, and L. pneumophila contain the eukaryotic SET domain. Once translocated to the nucleus, these effectors target histones for direct methylation either globally or at specific residues. For M. tuberculosis and L. pneumophila, this leads to repression of the host immune response and is thought to aid pathogen survival.
Chlamydia trachomatis
While this phenomenon is not fully understood, C. trachomatis is able to increase global methylation of H2B, H3, and H4 through a secreted effector. This protein, NUE, translocates to the nucleus, where it automethylates and increases histone methylation (48). Since C. trachomatis is an obligately intracellular pathogen with a limited repertoire of protein-coding reading frames, global methylation might be essential for reprogramming the host cellular processes to support the intracellular niche of C. trachomatis (50).
Bacillus anthracis
The causative agent of anthrax is B. anthracis, a Gram-positive spore-forming bacterium (51). While anthrax toxins are among the most noted virulence factors of the organism, it also encodes several effector proteins, one of which is BaSET (47, 51). BaSET alters host gene transcription by methylating histone H1 in the promoter regions of NF-κB-controlled genes (the IL-6 gene, c-fos, c-jun, and the TNF-α gene) and counters transcriptional activation by the CREB-binding protein coactivator. Furthermore, BaSET deletion mutants fail to colonize in vivo, in contrast to wild-type bacteria. Therefore, it appears that downregulation of NF-κB host responses by H1 methylation plays a role in survival of the B. anthracis during infection (47).
Legionella pneumophila
L. pneumophila, a facultative intracellular bacterium, uses the T4SS to inject the effector RomA. Once inside the host cell, RomA localizes to the nucleus. There, it induces histone methylation at a site not previously described, H3K14. Interestingly, methylation occurs with a simultaneous decrease in H3K14 acetylation, and thereby, an activating histone mark (acetylation) is replaced with a repressive mark (methylation). Upon infection, 4,870 gene promoter regions are targeted with the H3K14 repressive mark. Specifically, H3K14 methylation damped immunomodulatory components, such as genes coding for TNF-α, IL-6, CXCL1, CXCL2, and Nalp3 (49).
BACTERIAL TARGETING OF DNA
Aside from modifying nucleosome PTMs, bacteria can also target DNA either through methylation or by inducing genotoxicity (Fig. 3). Intriguingly, such effects on DNA are more stable than histone modifications and could have a long-lasting impact on the host.
FIGURE 3.
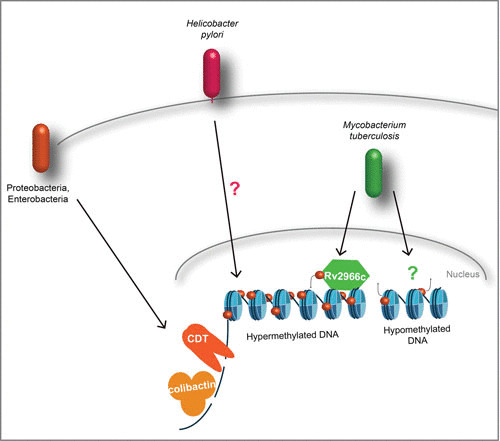
Targeting host DNA. Genotoxins such as CDT and colibactin induce host DNA breaks through either DNase activity (CDT) or DNA cross-linking (colibactin). M. tuberculosis targets host DNA directly for methylation with Rv2966c at non-CpG elements or induces hypomethylation through an unknown effector at CpG islands.
DNA Methylation
Mycobacterium tuberculosis
Rapid hypomethylation was reported to occur upon in vitro infection of monocyte-derived dendritic cells with M. tuberculosis. Distal enhancer regions upstream of genes known to function as master regulators of the immune response were mainly targeted, with only rare detection at promoter regions (52). Such demethylation was found to correlate with an increase in activatory histone marks and the recruitment of inflammation-activated transcription factors. Although no particular phenotype or specific effector was shown to correlate with hypomethylation, this study shows that demethylation can occur and is dynamically regulated upon bacterial infection.
In contrast to the works cited above, which focused on CpG elements, another study found that a secreted effector of M. tuberculosis, Rv2966c, methylates DNA in regions outside CpG islands (53). This effector is a DNA methyltransferase which requires phosphorylation by either a mycobacterial or a host kinase(s) for activity. Once active, it directly methylates host DNA at CpA and CpT dinucleotides while also binding to histones 3 and 4. Through non-CpG methylation, Rv2966c dampens host transcription at targeted loci, such as H2AFY2 (encoding a macrohistone 2A family member) and GRK5 (encoding a member of the G-protein-coupled receptor kinase family) (53).
These studies clearly show that M. tuberculosis induces differential DNA methylation within host cells by targeting both CpG and non-CpG DNA methylation; however, whether the bacterium has more than one effector to do so remains undetermined.
Helicobacter pylori
Recent work with H. pylori suggests that its presence induces DNA methylation, which is strongly associated with gastric cancer (54–61). However, it is still controversial whether the elevated risk of gastric cancer is directly due to H. pylori-induced DNA methylation or whether it is a result of the inflammatory response to infection. Regardless, it is clear that in response to infection with H. pylori, transient and permanent DNA methylation changes are detected in gastric mucosa. Indeed, H. pylori induces specific DNA hypermethylation patterns in genomic regions termed CpG islands, mainly located in promoter regions and at transcription factor-binding motifs of tumor suppressor genes, such as LOX and HAND1, or inflammatory genes, such as COX2 (59, 61). In addition, several of the CpG islands that undergo DNA methylation during infection remain elevated even after eradication of H. pylori (54). While it is accepted that H. pylori infection increases the risk for gastric cancer, further study is needed to directly link DNA methylation patterns with a predisposition to gastric cancer or define the role of bacterial induced inflammation in this process.
Damaging Chromatin through Bacterial Genotoxins
During infection, it is common for bacteria to induce DNA damage in their host (62–66). Such effects are often indirect, occurring through oxidative stress; however, to date, only a few bona fide bacterial genotoxins have been characterized.
Cytolethal distending toxin
Cytolethal distending toxin (CDT) is a family of proteins found in Gram-negative bacteria, especially in certain members of the Proteobacteria, such as Escherichia coli, Aggregatibacter actinomycetemcomitans, Haemophilus ducreyi, Campylobacter sp., and Helicobacter sp. CDT is functionally conserved in a large number of distantly related pathogenic strains, and except in Salmonella enterica serovar Typhi, CDT is encoded by three genes: cdtA, cdtB, and cdtC (67, 68). Regardless of the microbial source of CDT, cdtB has been shown to be the main gene responsible for toxin activity. Although CdtB was originally described as a cyclomodulin, since intoxicated cells are arrested in their cell cycle, structural analysis of CdtB revealed homology with mammalian DNase I and potentially with inositol lipid phosphatases (67, 69–71). CdtB is an AB2-like toxin; CdtB associates with CdtA and CdtC subunits, showing ricin-like lectin folds that allow the tripartite toxin to enter host cells via endocytosis (68, 70, 72). Interestingly, CDT is the first bacterial toxin known to target the nucleus, where it exhibits DNase activity (72–74). Although other activities for CdtB have been reported, such as phosphatidylinositol phosphatase (71), cell cycle arrest is mainly attributed to its genotoxic activity. CDTs have been reported to induce apoptosis and cell senescence during infection, although the benefit to bacteria of inducing DNA breaks in the host remains mostly unclear. Interestingly, in the context of chronic exposure, the potential role of CDTs in promoting cell transformation has been raised (75–77).
Colibactin
Colibactins are synthesized by several species of Enterobacteriaceae and demonstrate genotoxic activity (78, 79). They are natural products of a “warhead substituted spirobicyclic” structure (80), which are biosynthesized by enzymatic machinery located in a pathogenicity island mainly conserved in virulent bacteria. Each of the 19 genes present in the clb genomic island is essential for the full active genotoxic effect of colibactins, and colibactin is not a unique compound but a mixture of multiple molecules (81). Contact with colibactin-expressing bacteria causes double-strand DNA breaks and eventually cell cycle arrest and death. Recently, the mechanism by which colibactin impacts chromatin integrity was shown to involve DNA interstrand cross-linking, causing replication stress and activation of DNA damage response pathways in intoxicated cells (82). Similarly to the effect of CDTs, a correlation between the presence of bacteria harboring the clb island and human cancers suggests that the colibactin toxin may promote inflammation-triggered colorectal cancer (83).
CONCLUSIONS AND PERSPECTIVES
Overall, many reports clearly indicate that bacteria and bacterial components reprogram the cell epigenetically. However, many questions remain unanswered regarding the role these various chromatin marks play in terms of specificity, regulation, and cellular processes.
How do bacterial effectors target specific histone residues or specific genomic regions? The effectors BaSET, NUE, and RomA all target histones for methylation; however, they each target different a histone(s) and/or residues. This suggests that the effectors are intrinsically capable of recognizing individual histones and tail residues or that their specificity occurs through synergistic interactions with unknown proteins or complexes. Similarly, bacteria target subsets of host genes for histone modifications. How this is achieved is unknown, and additional factors might be required to determine specificity. Therefore, additional work is warranted to fully understand how bacterial factors acquire specificity, whether it is to target a histone residue or a specific genomic region.
What is the impact of chromatin rearrangements on bacterial survival within the host? It is clear that bacteria are able to manipulate host chromatin, and in several cases, these modifications have been shown to affect the survival of the organism within the host (L. monocytogenes, M. tuberculosis, and B. anthracis). However, for other histone marks, their contribution to bacterial replication and niche establishment remains to be further defined. Indeed, the observed chromatin modifications could be a natural response of the host cell to a bacterial encounter and therefore could have no impact on bacterial growth. Thus, to gain a complete picture of chromatin-based bacterium-host interactions, the combination of the epigenetic and transcriptional responses needs to be accounted for. It is possible that future work will define modifications associated with basal responses and those associated with active bacterial manipulation. Further extending these comparisons across species, both commensal and pathogenic, will deepen our understanding of species-dependent histone marks that influence chromatin-based bacterial homeostasis or pathogenesis. Global patterns associating active chromatin remodeling, transcriptional responses, and cellular processes could then begin to be mapped systematically.
Are bacterium-induced histone marks maintained, and do they have a lasting impact on host cells? In the light of infection studies, DNA methylation is proving to be responsive to environmental stimuli; however, the lasting potential of variations in DNA methylation levels needs to be explored. Furthermore, as a clear link between DNA methylation and carcinogenesis has been established, it will be interesting to explore whether bacterium-mediated DNA methylation impacts this process. Similarly, genotoxic stress-causing toxins, depending on the time it takes the host cell to recover, could predispose the host to cancer. A lasting impact of histone modifications on transcriptional regulation of the host is another avenue of interesting studies. Recent studies put forth the idea that innate immune cells retain a memory of past encounters, which would be maintained through histone marks (84–88). Such possibilities have come to light due to the known cross-protective effects of the BCG vaccine, which is associated with H3K27 and H3K4 modifications. Whether bacteria are able to induce such memory or disrupt it remains to be explored.
As we unlock the histone code and the role this language plays in host response during bacterial disease, commensal colonization, and innate immune memory, we will discover novel mechanisms that may give rise to next-generation therapeutics, intelligently designed vaccines, and even medical advancements for microbiome dysbiosis.
ACKNOWLEDGMENTS
We apologize to any colleagues whose work was not included in this review due to space limitations. We thank Orhan Rasid and Emmanuel Lemichez for critical reading of the manuscript. Work in the Chromatin and Infection Group is supported by the Pasteur Institute and the Agence National de la Recherche (ANR-EpiBActIn). Michael Connor is supported by the Pasteur Foundation Fellowship. Work by the genomic plasticity and infection team is supported by the Institut National de la Santé et de la Recherche Médicale (INSERM) (U1151) and by the Agence National de la Recherche (ANR- 15-CE14-0003 and ANR-16-CE15-0006-01).
REFERENCES
- 1.Swygert SG, Peterson CL. 2014. Chromatin dynamics: interplay between remodeling enzymes and histone modifications. Biochim Biophys Acta 1839:728–736 10.1016/j.bbagrm.2014.02.013. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clapier CR, Iwasa J, Cairns BR, Peterson CL. 2017. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat Rev Mol Cell Biol 18:407–422 10.1038/nrm.2017.26. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strahl BD, Allis CD. 2000. The language of covalent histone modifications. Nature 403:41–45 10.1038/47412. [PubMed] [DOI] [PubMed] [Google Scholar]
- 4.Barth TK, Imhof A. 2010. Fast signals and slow marks: the dynamics of histone modifications. Trends Biochem Sci 35:618–626 10.1016/j.tibs.2010.05.006. [PubMed] [DOI] [PubMed] [Google Scholar]
- 5.Li B, Carey M, Workman JL. 2007. The role of chromatin during transcription. Cell 128:707–719 10.1016/j.cell.2007.01.015. [PubMed] [DOI] [PubMed] [Google Scholar]
- 6.Hamon MA, Cossart P. 2008. Histone modifications and chromatin remodeling during bacterial infections. Cell Host Microbe 4:100–109 10.1016/j.chom.2008.07.009. [PubMed] [DOI] [PubMed] [Google Scholar]
- 7.Saunders A, Core LJ, Lis JT. 2006. Breaking barriers to transcription elongation. Nat Rev Mol Cell Biol 7:557–567 10.1038/nrm1981. [PubMed] [DOI] [PubMed] [Google Scholar]
- 8.Tremethick DJ. 2007. Higher-order structures of chromatin: the elusive 30 nm fiber. Cell 128:651–654 10.1016/j.cell.2007.02.008. [PubMed] [DOI] [PubMed] [Google Scholar]
- 9.Kornberg RD, Lorch Y. 1999. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 98:285–294 10.1016/S0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- 10.Kornberg RD. 1974. Chromatin structure: a repeating unit of histones and DNA. Science 184:868–871 10.1126/science.184.4139.868. [PubMed] [DOI] [PubMed] [Google Scholar]
- 11.Thomas JO, Kornberg RD. 1975. An octamer of histones in chromatin and free in solution. Proc Natl Acad Sci USA 72:2626–2630 10.1073/pnas.72.7.2626. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Izzo A, Schneider R. 2016. The role of linker histone H1 modifications in the regulation of gene expression and chromatin dynamics. Biochim Biophys Acta 1859:486–495 10.1016/j.bbagrm.2015.09.003. [PubMed] [DOI] [PubMed] [Google Scholar]
- 13.Sadakierska-Chudy A, Filip M. 2015. A comprehensive view of the epigenetic landscape. Part II: histone post-translational modification, nucleosome level, and chromatin regulation by ncRNAs. Neurotox Res 27:172–197 10.1007/s12640-014-9508-6. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kouzarides T. 2007. Chromatin modifications and their function. Cell 128:693–705 10.1016/j.cell.2007.02.005. [PubMed] [DOI] [PubMed] [Google Scholar]
- 15.Saccani S, Pantano S, Natoli G. 2001. Two waves of nuclear factor κB recruitment to target promoters. J Exp Med 193:1351–1360 10.1084/jem.193.12.1351. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hargreaves DC, Horng T, Medzhitov R. 2009. Control of inducible gene expression by signal-dependent transcriptional elongation. Cell 138:129–145 10.1016/j.cell.2009.05.047. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saccani S, Pantano S, Natoli G. 2002. p38-Dependent marking of inflammatory genes for increased NF-kappa B recruitment. Nat Immunol 3:69–75 10.1038/ni748. [PubMed] [DOI] [PubMed] [Google Scholar]
- 18.Foster SL, Hargreaves DC, Medzhitov R. 2007. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 447:972–978 CORRIGENDUM Nature 451:102 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- 19.Chang PV, Hao L, Offermanns S, Medzhitov R. 2014. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci USA 111:2247–2252 10.1073/pnas.1322269111. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fellows R, Denizot J, Stellato C, Cuomo A, Jain P, Stoyanova E, Balázsi S, Hajnády Z, Liebert A, Kazakevych J, Blackburn H, Corrêa RO, Fachi JL, Sato FT, Ribeiro WR, Ferreira CM, Perée H, Spagnuolo M, Mattiuz R, Matolcsi C, Guedes J, Clark J, Veldhoen M, Bonaldi T, Vinolo MAR, Varga-Weisz P. 2018. Microbiota derived short chain fatty acids promote histone crotonylation in the colon through histone deacetylases. Nat Commun 9:105 10.1038/s41467-017-02651-5. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haberland M, Montgomery RL, Olson EN. 2009. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 10:32–42 10.1038/nrg2485. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yarbrough VL, Winkle S, Herbst-Kralovetz MM. 2015. Antimicrobial peptides in the female reproductive tract: a critical component of the mucosal immune barrier with physiological and clinical implications. Hum Reprod Update 21:353–377 10.1093/humupd/dmu065. [PubMed] [DOI] [PubMed] [Google Scholar]
- 23.Bandyopadhaya A, Tsurumi A, Maura D, Jeffrey KL, Rahme LG. 2016. A quorum-sensing signal promotes host tolerance training through HDAC1-mediated epigenetic reprogramming. Nat Microbiol 1:16174 10.1038/nmicrobiol.2016.174. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alto NM, Orth K. 2012. Subversion of cell signaling by pathogens. Cold Spring Harb Perspect Biol 4:a006114 10.1101/cshperspect.a006114. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, Curry HM, Zwilling BS, Lafuse WP. 2005. Mycobacteria inhibition of IFN-gamma induced HLA-DR gene expression by up-regulating histone deacetylation at the promoter region in human THP-1 monocytic cells. J Immunol 174:5687–5694 10.4049/jimmunol.174.9.5687. [PubMed] [DOI] [PubMed] [Google Scholar]
- 26.Pennini ME, Pai RK, Schultz DC, Boom WH, Harding CV. 2006. Mycobacterium tuberculosis 19-kDa lipoprotein inhibits IFN-gamma-induced chromatin remodeling of MHC2TA by TLR2 and MAPK signaling. J Immunol 176:4323–4330 10.4049/jimmunol.176.7.4323. [PubMed] [DOI] [PubMed] [Google Scholar]
- 27.Cossart P. 2011. Illuminating the landscape of host-pathogen interactions with the bacterium Listeria monocytogenes. Proc Natl Acad Sci USA 108:19484–19491 10.1073/pnas.1112371108. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ireton K, Payrastre B, Cossart P. 1999. The Listeria monocytogenes protein InlB is an agonist of mammalian phosphoinositide 3-kinase. J Biol Chem 274:17025–17032 10.1074/jbc.274.24.17025. [PubMed] [DOI] [PubMed] [Google Scholar]
- 29.Bosse T, Ehinger J, Czuchra A, Benesch S, Steffen A, Wu X, Schloen K, Niemann HH, Scita G, Stradal TE, Brakebusch C, Rottner K. 2007. Cdc42 and phosphoinositide 3-kinase drive Rac-mediated actin polymerization downstream of c-Met in distinct and common pathways. Mol Cell Biol 27:6615–6628 10.1128/MCB.00367-07. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eskandarian HA, Impens F, Nahori MA, Soubigou G, Coppée JY, Cossart P, Hamon MA. 2013. A role for SIRT2-dependent histone H3K18 deacetylation in bacterial infection. Science 341:1238858 10.1126/science.1238858. [PubMed] [DOI] [PubMed] [Google Scholar]
- 31.Pereira JM, Chevalier C, Chaze T, Gianetto Q, Impens F, Matondo M, Cossart P, Hamon MA. 2018. Infection reveals a modification of SIRT2 critical for chromatin association. Cell Reports 23:1124–1137 10.1016/j.celrep.2018.03.116. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kusters JG, van Vliet AHM, Kuipers EJ. 2006. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev 19:449–490 10.1128/CMR.00054-05. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang F, Meng W, Wang B, Qiao L. 2014. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett 345:196–202 10.1016/j.canlet.2013.08.016. [PubMed] [DOI] [PubMed] [Google Scholar]
- 34.Fehri LF, Rechner C, Janssen S, Mak TN, Holland C, Bartfeld S, Brüggemann H, Meyer TF. 2009. Helicobacter pylori-induced modification of the histone H3 phosphorylation status in gastric epithelial cells reflects its impact on cell cycle regulation. Epigenetics 4:577–586 10.4161/epi.4.8.10217. [PubMed] [DOI] [PubMed] [Google Scholar]
- 35.Ding SZ, Fischer W, Kaparakis-Liaskos M, Liechti G, Merrell DS, Grant PA, Ferrero RL, Crowe SE, Haas R, Hatakeyama M, Goldberg JB. 2010. Helicobacter pylori-induced histone modification, associated gene expression in gastric epithelial cells, and its implication in pathogenesis. PLoS One 5:e9875 10.1371/journal.pone.0009875. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dal Peraro M, van der Goot FG. 2016. Pore-forming toxins: ancient, but never really out of fashion. Nat Rev Microbiol 14:77–92 10.1038/nrmicro.2015.3. [PubMed] [DOI] [PubMed] [Google Scholar]
- 37.Hamon MA, Batsché E, Régnault B, Tham TN, Seveau S, Muchardt C, Cossart P. 2007. Histone modifications induced by a family of bacterial toxins. Proc Natl Acad Sci USA 104:13467–13472 CORRECTION Proc Natl Acad Sci USA 104:17555 10.1073/pnas.0702729104. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hamon MA, Cossart P. 2011. K+ efflux is required for histone H3 dephosphorylation by Listeria monocytogenes listeriolysin O and other pore-forming toxins. Infect Immun 79:2839–2846 10.1128/IAI.01243-10. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dortet L, Lombardi C, Cretin F, Dessen A, Filloux A. 2018. Pore-forming activity of the Pseudomonas aeruginosa type III secretion system translocon alters the host epigenome. Nat Microbiol 3:378–386 10.1038/s41564-018-0109-7. [PubMed] [DOI] [PubMed] [Google Scholar]
- 40.Yaseen I, Kaur P, Nandicoori VK, Khosla S. 2015. Mycobacteria modulate host epigenetic machinery by Rv1988 methylation of a non-tail arginine of histone H3. Nat Commun 6:8922 10.1038/ncomms9922. [PubMed] [DOI] [PubMed] [Google Scholar]
- 41.The HC, Thanh DP, Holt KE, Thomson NR, Baker S. 2016. The genomic signatures of Shigella evolution, adaptation and geographical spread. Nat Rev Microbiol 14:235–250 10.1038/nrmicro.2016.10. [PubMed] [DOI] [PubMed] [Google Scholar]
- 42.Arbibe L, Kim DW, Batsche E, Pedron T, Mateescu B, Muchardt C, Parsot C, Sansonetti PJ. 2007. An injected bacterial effector targets chromatin access for transcription factor NF-κB to alter transcription of host genes involved in immune responses. Nat Immunol 8:47–56 10.1038/ni1423. [PubMed] [DOI] [PubMed] [Google Scholar]
- 43.Li H, Xu H, Zhou Y, Zhang J, Long C, Li S, Chen S, Zhou JM, Shao F. 2007. The phosphothreonine lyase activity of a bacterial type III effector family. Science 315:1000–1003 10.1126/science.1138960. [PubMed] [DOI] [PubMed] [Google Scholar]
- 44.Harouz H, Rachez C, Meijer BM, Marteyn B, Donnadieu F, Cammas F, Muchardt C, Sansonetti P, Arbibe L. 2014. Shigella flexneri targets the HP1γ subcode through the phosphothreonine lyase OspF. EMBO J 33:2606–2622 10.15252/embj.201489244. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lebreton A, Lakisic G, Job V, Fritsch L, Tham TN, Camejo A, Matteï PJ, Regnault B, Nahori MA, Cabanes D, Gautreau A, Ait-Si-Ali S, Dessen A, Cossart P, Bierne H. 2011. A bacterial protein targets the BAHD1 chromatin complex to stimulate type III interferon response. Science 331:1319–1321 10.1126/science.1200120. [PubMed] [DOI] [PubMed] [Google Scholar]
- 46.Dillon SC, Zhang X, Trievel RC, Cheng X. 2005. The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol 6:227 10.1186/gb-2005-6-8-227. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mujtaba S, Winer BY, Jaganathan A, Patel J, Sgobba M, Schuch R, Gupta YK, Haider S, Wang R, Fischetti VA. 2013. Anthrax SET protein: a potential virulence determinant that epigenetically represses NF-κB activation in infected macrophages. J Biol Chem 288:23458–23472 10.1074/jbc.M113.467696. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pennini ME, Perrinet S, Dautry-Varsat A, Subtil A. 2010. Histone methylation by NUE, a novel nuclear effector of the intracellular pathogen Chlamydia trachomatis. PLoS Pathog 6:e1000995 10.1371/journal.ppat.1000995. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rolando M, Sanulli S, Rusniok C, Gomez-Valero L, Bertholet C, Sahr T, Margueron R, Buchrieser C. 2013. Legionella pneumophila effector RomA uniquely modifies host chromatin to repress gene expression and promote intracellular bacterial replication. Cell Host Microbe 13:395–405 10.1016/j.chom.2013.03.004. [PubMed] [DOI] [PubMed] [Google Scholar]
- 50.Elwell C, Mirrashidi K, Engel J. 2016. Chlamydia cell biology and pathogenesis. Nat Rev Microbiol 14:385–400 10.1038/nrmicro.2016.30. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu S, Moayeri M, Leppla SH. 2014. Anthrax lethal and edema toxins in anthrax pathogenesis. Trends Microbiol 22:317–325 10.1016/j.tim.2014.02.012. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pacis A, Tailleux L, Morin AM, Lambourne J, MacIsaac JL, Yotova V, Dumaine A, Danckaert A, Luca F, Grenier JC, Hansen KD, Gicquel B, Yu M, Pai A, He C, Tung J, Pastinen T, Kobor MS, Pique-Regi R, Gilad Y, Barreiro LB. 2015. Bacterial infection remodels the DNA methylation landscape of human dendritic cells. Genome Res 25:1801–1811 10.1101/gr.192005.115. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharma G, Upadhyay S, Srilalitha M, Nandicoori VK, Khosla S. 2015. The interaction of mycobacterial protein Rv2966c with host chromatin is mediated through non-CpG methylation and histone H3/H4 binding. Nucleic Acids Res 43:3922–3937 10.1093/nar/gkv261. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Niwa T, Tsukamoto T, Toyoda T, Mori A, Tanaka H, Maekita T, Ichinose M, Tatematsu M, Ushijima T. 2010. Inflammatory processes triggered by Helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Cancer Res 70:1430–1440 10.1158/0008-5472.CAN-09-2755. [PubMed] [DOI] [PubMed] [Google Scholar]
- 55.Woo HD, Fernandez-Jimenez N, Ghantous A, Degli Esposti D, Cuenin C, Cahais V, Choi IJ, Kim YI, Kim J, Herceg Z. 2018. Genome-wide profiling of normal gastric mucosa identifies Helicobacter pylori- and cancer-associated DNA methylome changes. Int J Cancer 143:597–609 10.1002/ijc.31381. [PubMed] [DOI] [PubMed] [Google Scholar]
- 56.Shin CM, Kim N, Jung Y, Park JH, Kang GH, Kim JS, Jung HC, Song IS. 2010. Role of Helicobacter pylori infection in aberrant DNA methylation along multistep gastric carcinogenesis. Cancer Sci 101:1337–1346 10.1111/j.1349-7006.2010.01535.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shin CM, Kim N, Jung Y, Park JH, Kang GH, Park WY, Kim JS, Jung HC, Song IS. 2011. Genome-wide DNA methylation profiles in noncancerous gastric mucosae with regard to Helicobacter pylori infection and the presence of gastric cancer. Helicobacter 16:179–188 10.1111/j.1523-5378.2011.00838.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 58.Zhang Y, Zhang XR, Park JL, Kim JH, Zhang L, Ma JL, Liu WD, Deng DJ, You WC, Kim YS, Pan KF. 2016. Genome-wide DNA methylation profiles altered by Helicobacter pylori in gastric mucosa and blood leukocyte DNA. Oncotarget 7:37132–37144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pero R, Peluso S, Angrisano T, Tuccillo C, Sacchetti S, Keller S, Tomaiuolo R, Bruni CB, Lembo F, Chiariotti L. 2011. Chromatin and DNA methylation dynamics of Helicobacter pylori-induced COX-2 activation. Int J Med Microbiol 301:140–149 10.1016/j.ijmm.2010.06.009. [PubMed] [DOI] [PubMed] [Google Scholar]
- 60.Maeda M, Moro H, Ushijima T. 2017. Mechanisms for the induction of gastric cancer by Helicobacter pylori infection: aberrant DNA methylation pathway. Gastric Cancer 20(Suppl 1):8–15 10.1007/s10120-016-0650-0. [PubMed] [DOI] [PubMed] [Google Scholar]
- 61.Maekita T, Nakazawa K, Mihara M, Nakajima T, Yanaoka K, Iguchi M, Arii K, Kaneda A, Tsukamoto T, Tatematsu M, Tamura G, Saito D, Sugimura T, Ichinose M, Ushijima T. 2006. High levels of aberrant DNA methylation in Helicobacter pylori-infected gastric mucosae and its possible association with gastric cancer risk. Clin Cancer Res 12:989–995 10.1158/1078-0432.CCR-05-2096. [PubMed] [DOI] [PubMed] [Google Scholar]
- 62.Chumduri C, Gurumurthy RK, Zadora PK, Mi Y, Meyer TF. 2013. Chlamydia infection promotes host DNA damage and proliferation but impairs the DNA damage response. Cell Host Microbe 13:746–758 10.1016/j.chom.2013.05.010. [PubMed] [DOI] [PubMed] [Google Scholar]
- 63.Vielfort K, Söderholm N, Weyler L, Vare D, Löfmark S, Aro H. 2013. Neisseria gonorrhoeae infection causes DNA damage and affects the expression of p21, p27 and p53 in non-tumor epithelial cells. J Cell Sci 126:339–347 10.1242/jcs.117721. [PubMed] [DOI] [PubMed] [Google Scholar]
- 64.Strickertsson JA, Desler C, Martin-Bertelsen T, Machado AM, Wadstrøm T, Winther O, Rasmussen LJ, Friis-Hansen L. 2013. Enterococcus faecalis infection causes inflammation, intracellular oxphos-independent ROS production, and DNA damage in human gastric cancer cells. PLoS One 8:e63147 10.1371/journal.pone.0063147. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Samba-Louaka A, Pereira JM, Nahori MA, Villiers V, Deriano L, Hamon MA, Cossart P. 2014. Listeria monocytogenes dampens the DNA damage response. PLoS Pathog 10:e1004470 10.1371/journal.ppat.1004470. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Toller IM, Neelsen KJ, Steger M, Hartung ML, Hottiger MO, Stucki M, Kalali B, Gerhard M, Sartori AA, Lopes M, Müller A. 2011. Carcinogenic bacterial pathogen Helicobacter pylori triggers DNA double-strand breaks and a DNA damage response in its host cells. Proc Natl Acad Sci USA 108:14944–14949 10.1073/pnas.1100959108. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lara-Tejero M, Galán JE. 2000. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science 290:354–357 10.1126/science.290.5490.354. [PubMed] [DOI] [PubMed] [Google Scholar]
- 68.Lara-Tejero M, Galán JE. 2001. CdtA, CdtB, and CdtC form a tripartite complex that is required for cytolethal distending toxin activity. Infect Immun 69:4358–4365 10.1128/IAI.69.7.4358-4365.2001. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Elwell CA, Dreyfus LA. 2000. DNase I homologous residues in CdtB are critical for cytolethal distending toxin-mediated cell cycle arrest. Mol Microbiol 37:952–963 10.1046/j.1365-2958.2000.02070.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 70.Nešić D, Hsu Y, Stebbins CE. 2004. Assembly and function of a bacterial genotoxin. Nature 429:429–433 10.1038/nature02532. [PubMed] [DOI] [PubMed] [Google Scholar]
- 71.Shenker BJ, Boesze-Battaglia K, Scuron MD, Walker LP, Zekavat A, Dlakić M. 2016. The toxicity of the Aggregatibacter actinomycetemcomitans cytolethal distending toxin correlates with its phosphatidylinositol-3,4,5-triphosphate phosphatase activity. Cell Microbiol 18:223–243 10.1111/cmi.12497. [PubMed] [DOI] [PubMed] [Google Scholar]
- 72.Nishikubo S, Ohara M, Ueno Y, Ikura M, Kurihara H, Komatsuzawa H, Oswald E, Sugai M. 2003. An N-terminal segment of the active component of the bacterial genotoxin cytolethal distending toxin B (CDTB) directs CDTB into the nucleus. J Biol Chem 278:50671–50681 10.1074/jbc.M305062200. [PubMed] [DOI] [PubMed] [Google Scholar]
- 73.Frisan T, Cortes-Bratti X, Chaves-Olarte E, Stenerlöw B, Thelestam M. 2003. The Haemophilus ducreyi cytolethal distending toxin induces DNA double-strand breaks and promotes ATM-dependent activation of RhoA. Cell Microbiol 5:695–707 10.1046/j.1462-5822.2003.00311.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 74.Elwell C, Chao K, Patel K, Dreyfus L. 2001. Escherichia coli CdtB mediates cytolethal distending toxin cell cycle arrest. Infect Immun 69:3418–3422 10.1128/IAI.69.5.3418-3422.2001. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guidi R, Guerra L, Levi L, Stenerlöw B, Fox JG, Josenhans C, Masucci MG, Frisan T. 2013. Chronic exposure to the cytolethal distending toxins of Gram-negative bacteria promotes genomic instability and altered DNA damage response. Cell Microbiol 15:98–113 10.1111/cmi.12034. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ge Z, Rogers AB, Feng Y, Lee A, Xu S, Taylor NS, Fox JG. 2007. Bacterial cytolethal distending toxin promotes the development of dysplasia in a model of microbially induced hepatocarcinogenesis. Cell Microbiol 9:2070–2080 10.1111/j.1462-5822.2007.00939.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 77.Graillot V, Dormoy I, Dupuy J, Shay JW, Huc L, Mirey G, Vignard J. 2016. Genotoxicity of cytolethal distending toxin (CDT) on isogenic human colorectal cell lines: potential promoting effects for colorectal carcinogenesis. Front Cell Infect Microbiol 6:34 10.3389/fcimb.2016.00034. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nougayrède J-P, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, Buchrieser C, Hacker J, Dobrindt U, Oswald E. 2006. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 313:848–851 10.1126/science.1127059. [PubMed] [DOI] [PubMed] [Google Scholar]
- 79.Homburg S, Oswald E, Hacker J, Dobrindt U. 2007. Expression analysis of the colibactin gene cluster coding for a novel polyketide in Escherichia coli. FEMS Microbiol Lett 275:255–262 10.1111/j.1574-6968.2007.00889.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 80.Vizcaino MI, Crawford JM. 2015. The colibactin warhead crosslinks DNA. Nat Chem 7:411–417 10.1038/nchem.2221. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vizcaino MI, Engel P, Trautman E, Crawford JM. 2014. Comparative metabolomics and structural characterizations illuminate colibactin pathway-dependent small molecules. J Am Chem Soc 136:9244–9247 10.1021/ja503450q. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bossuet-Greif N, Vignard J, Taieb F, Mirey G, Dubois D, Petit C, Oswald E, Nougayrède JP. 2018. The colibactin genotoxin generates DNA interstrand cross-links in infected cells. mBio 9:e02393-17 10.1128/mBio.02393-17. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Buc E, Dubois D, Sauvanet P, Raisch J, Delmas J, Darfeuille-Michaud A, Pezet D, Bonnet R. 2013. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS One 8:e56964 10.1371/journal.pone.0056964. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, O’Neill LA, Xavier RJ. 2016. Trained immunity: A program of innate immune memory in health and disease. Science 352:aaf1098 10.1126/science.aaf1098. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Netea MG, Quintin J, van der Meer JW. 2011. Trained immunity: a memory for innate host defense. Cell Host Microbe 9:355–361 10.1016/j.chom.2011.04.006. [PubMed] [DOI] [PubMed] [Google Scholar]
- 86.Netea MG, van Crevel R. 2014. BCG-induced protection: effects on innate immune memory. Semin Immunol 26:512–517 10.1016/j.smim.2014.09.006. [PubMed] [DOI] [PubMed] [Google Scholar]
- 87.Grode L, Seiler P, Baumann S, Hess J, Brinkmann V, Nasser Eddine A, Mann P, Goosmann C, Bandermann S, Smith D, Bancroft GJ, Reyrat JM, van Soolingen D, Raupach B, Kaufmann SH. 2005. Increased vaccine efficacy against tuberculosis of recombinant Mycobacterium bovis bacille Calmette-Guérin mutants that secrete listeriolysin. J Clin Invest 115:2472–2479 10.1172/JCI24617. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Arts RJW, Moorlag SJCFM, Novakovic B, Li Y, Wang SY, Oosting M, Kumar V, Xavier RJ, Wijmenga C, Joosten LAB, Reusken CBEM, Benn CS, Aaby P, Koopmans MP, Stunnenberg HG, van Crevel R, Netea MG. 2018. BCG vaccination protects against experimental viral infection in humans through the induction of cytokines associated with trained immunity. Cell Host Microbe 23:89–100.e5 10.1016/j.chom.2017.12.010. [PubMed] [DOI] [PubMed] [Google Scholar]