

ABSTRACT
Staphylococcus aureus has become a serious threat to human health. In addition to having increased antibiotic resistance, the bacterium is a master at adapting to its host by evading almost every facet of the immune system, the so-called immune evasion proteins. Many of these immune evasion proteins target neutrophils, the most important immune cells in clearing S. aureus infections. The neutrophil attacks pathogens via a plethora of strategies. Therefore, it is no surprise that S. aureus has evolved numerous immune evasion strategies at almost every level imaginable. In this review we discuss step by step the aspects of neutrophil-mediated killing of S. aureus, such as neutrophil activation, migration to the site of infection, bacterial opsonization, phagocytosis, and subsequent neutrophil-mediated killing. After each section we discuss how S. aureus evasion molecules are able to resist the neutrophil attack of these different steps. To date, around 40 immune evasion molecules of S. aureus are known, but its repertoire is still expanding due to the discovery of new evasion proteins and the addition of new functions to already identified evasion proteins. Interestingly, because the different parts of neutrophil attack are redundant, the evasion molecules display redundant functions as well. Knowing how and with which proteins S. aureus is evading the immune system is important in understanding the pathophysiology of this pathogen. This knowledge is crucial for the development of therapeutic approaches that aim to clear staphylococcal infections.
INTRODUCTION
The Gram-positive bacterium Staphylococcus aureus is considered a commensal bacterium because roughly one-third of the human population is colonized by it without developing disease (1). Colonization occurs in the human nose, whereby the host nasal microbiota plays a major role in promoting or inhibiting S. aureus colonization (2). Despite the fact that S. aureus is considered a commensal, nasal carriage of S. aureus is linked to bacteremia (3). The bacterium may cause a range of infections, from cellulitis and superficial skin disease to abscesses, bacteremia, sepsis, endocarditis, and pneumonia (4). Moreover, S. aureus has been shown to adapt in its interaction with humans by increasing resistance against methicillin and is currently a leading cause of human bacterial disease worldwide. Methicillin-resistant S. aureus (MRSA) was identified in the 1960s as a nosocomial pathogen, when hospitalized patients showed distinct risk factors for acquisition (5). The prevalence of methicillin resistance among nosocomial S. aureus isolates increased from 2.1% in 1975 to 35% in 1991 (6). MRSA epidemiology changed in the 1990s when infections of healthy individuals outside hospitals were reported. These strains, with increased virulence, were the first reports of community-acquired MRSA (7, 8). Now, community-acquired MRSA has been reported as the leading cause of bacterial infections in the bloodstream, skin, soft tissue, and lower respiratory tract in developed countries (9). As a consequence, research interest in the pathophysiology of S. aureus has increased.
This extensive research during the past decades resulted in the identification and characterization of around 40 proteins which are able to evade various processes of the innate and adaptive immune system (10), the so-called immune evasion proteins. A list of these evasion molecules with their abbreviation, what they evade and location on which MGE is listed in Table 1. Proteomics has shown that around 100 to 200 S. aureus proteins are secreted, many with a still unknown function (11). These unknown secreted proteins are potential evasion molecules, demonstrating that the identification and characterization of new evasion proteins is not yet complete and will most likely expand in the future.
TABLE 1.
Abbreviations of staphylococcal immune evasion proteins, what they evade, and on which MGE or paralogous gene cluster they are locateda
| Abbreviation | Full name | Evades processb | MGEc |
|---|---|---|---|
| Aur | Aureolysin | III, V | |
| CHIPS | Chemotaxis inhibitory protein of Staphylococcus | II | IEC-1/ɸSa3 |
| Can | S. aureus collagen adhesion | III | |
| Eap and EapH | Extracellular adherence protein and extracellular adherence protein homologue | I, V | |
| Ecb | Extracellular complement-binding protein | III | IEC-2 |
| Efb | Extracellular fibrinogen-binding protein | III | IEC-2 |
| FLIPr | FPR2 inhibitory protein | II | IEC-2 |
| Hla | Hemolysin-alpha | V | |
| Hlg | Hemolysin-gamma | V | |
| LukS-PV | Panton Valentine leukocidin | V | ɸSa2 |
| Luk-AB | Leukocidin AB | V | |
| Luk-DE | Leukocidin DE | V | |
| Nuc | Nuclease | IV | |
| PSMs | Phenol-soluble modulins | V | |
| Sags | Superantigens | V | IEC1, SaPIn1, vSaβd |
| SAK | Staphylokinase | III, V | IEC-1/ɸSa3 |
| Sbi | Staphylococcal binding of IgG | III | |
| SCIN | Staphylococcal complement inhibitor | III | IEC-1/ɸSa3 |
| ScpA | Staphopain A | II | |
| SdrE | Surface-associated serine-aspartate repeat protein E | III | |
| SelX | Staphylococcal enterotoxin-like X | I | |
| SpA | Staphylococcal protein A | III | |
| SPIN | Staphylococcal peroxidase inhibitor | V | vSaαd |
| SSL | Staphylococcal superantigen-like 1-11 | I, II, III | vSaαd |
| SSL | Staphylococcal superantigen-like 12, 13, 14 | I, II, III | vSaβd |
MGE, mobile genetic element.
The innate immune mechanisms that are inhibited: I, neutrophil extravasation; II, priming, chemotaxis, and activation of neutrophils; III, opsonization and phagocytosis; IV, NET formation; V, bacterial killing by neutrophils.
IEC, immune evasion cluster; SaPI, staphylococcal pathogenicity island.
vSaα and vSaβ are clusters of paralogous genes that have evolved in situ, are nearly universally conserved in S. aureus, and are not MGEs.
Many evasion proteins are targeted against neutrophils. This is not a surprise, because neutrophils are the most prominent leukocytes in blood, covering 60% of the leukocyte population, and play a prominent role in fighting S. aureus infection. They are equipped with various granules with specific content to kill both Gram-negative and Gram-positive bacteria (12, 13). Neutrophils originate and mature in the bone marrow and are then released into the bloodstream. There, these end-stage cells are induced to circulate and migrate toward the site of infection by chemotactic signals produced both by the bacteria themselves and by host cells (13). Upon arrival at the infection site, neutrophils are able to “eat” microorganisms that are opsonized (labeled) by the complement system or immunoglobulins (antibodies). The complement system is especially important in clearing staphylococcal infections. Mice deficient in complement C5 showed decreased clearance of S. aureus after pulmonary and bloodstream infections (14, 15). In addition, antibodies play an important role in fighting staphylococcal infections. Opsonization of bacteria subsequently leads to phagocytosis and, ultimately, killing of microbes, because neutrophils release the content of their antimicrobial granules and produce reactive oxygen species (ROS) (13).
Impaired neutrophil function is linked to staphylococcal infections, as shown in multiple studies. For instance, patients suffering from congenital neutropenia often have severe infections, including staphylococcal infections, which can be fatal (16). Patients with chronic granulomatous disease have defects in their NADPH oxidase, which leads to impaired formation of ROS. Catalase-positive microorganisms in particular, such as S. aureus, cause recurrent infections in these patients because catalase breaks down the bacterial hydrogen peroxide and thereby prevents the generation of ROS (17). Another example where S. aureus infections play a major role is in burn patients. The burn wound is especially susceptible to bacterial colonization and infection, because neutrophils are known to be decreased in burn patients (18, 19). From these observations we can conclude that neutrophil-mediated killing in healthy individuals is crucial in the defense against S. aureus.
After the first response, the acquired immune system comes into the picture. However, this part of the immune system has an insignificant role in acute infection. Nonetheless, antibodies are important in the long term with recurrent staphylococcal infections, but antibodies against staphylococcal proteins do not show effective protection; even though most adult humans have high levels of circulating antibodies against different S. aureus proteins, these are not immunologically protective (20).
In this article we discuss the different facets of the antistaphylococcal innate immune system. First, we describe the mechanisms of neutrophil extravasation through the endothelium. Second, we give an overview of the proteins and receptors involved in neutrophil priming, chemotaxis, and activation. Third, we describe the processes involved in bacterial opsonization and phagocytosis by neutrophils. Fourth, we provide an overview of the processes involved in bacterial killing by neutrophils. After each section we describe the various evasion molecules that interrupt that specific process. Finally, we discuss why S. aureus has evolved so many evasion proteins and the therapeutic implications of these proteins with respect to staphylococcal infections. Thus, we give an overview of what we know so far about S. aureus immune evasion and what lessons we can draw from this knowledge.
NEUTROPHIL EXTRAVASATION THROUGH THE ENDOTHELIUM
Once pathogens cause infections in tissues, neutrophils leave the bloodstream and migrate toward the site of infection. This multistep process is called extravasation and includes neutrophil rolling, crawling and firm adhesion to the endothelial cells, extravasation through the endothelium (diapedesis), and migration to the infection site over a chemokine gradient (21). An overview of this process is shown in Fig. 1.
FIGURE 1.
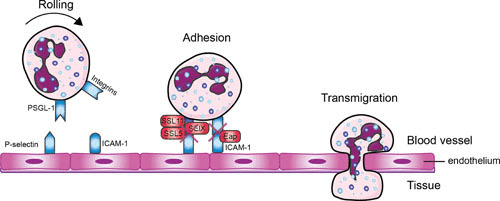
Evading neutrophil extravasation to the infection site. Mechanisms by which S. aureus evades the steps in neutrophil extravasation. Neutrophils start to roll on the activated endothelium, which leads to firm adhesion and subsequently to transmigration through the endothelium. Red boxes indicate staphylococcal proteins, and blue boxes indicate host proteins. Abbreviations: PSGL-1, P-selectin glycoprotein 1; SSL, staphylococcal superantigen-like protein; ICAM-1, intracellular adhesion molecule 1; Eap, extracellular adherence protein; SElX, staphylococcal enterotoxin-like X. The figure was adapted from Servier Medical Art.
First, the neutrophils in the bloodstream slow down near the site of infection. This is initiated by changes on the endothelial surface by inflammatory mediators (e.g., histamine, cytokines, and leukotrienes) (22). The endothelial cells are activated and start to express P-selectin and E-selectin, which interact with P-selectin glycoprotein ligand-1 (PSGL-1) on the surface of neutrophils (23). This leads to the tethering (capturing) of freely moving neutrophils to the endothelial surface as they start to roll along the vessel in the direction of the blood flow. In the second step the neutrophils adhere and “crawl” along the vessel wall by the interaction between intercellular adhesion molecule 1 (ICAM-1) on endothelial cells and β2 integrins (such as LFA-1 and Mac-1) on the surface of phagocytes (24–26). Neutrophils express constitutively high levels of β2 integrins, which undergo conformational change upon either intracellular activation ligand binding (outside-in signaling) or integrin adhesiveness (inside-out signaling). β2 integrins are relocalized to the cell surface upon activation by chemokines (27, 28). The neutrophils enter the arrested state after expressing high levels of ICAM-1. This leads to the initiation of transendothelial cell migration through a paracellular (passing through the endothelium between the cells) or a transcellular (movement through the endothelial cell) pathway. Leukocytes mainly migrate via the paracellular route but follow the transcellular route in the central nervous system and in various inflammatory settings (29). After extravasation, the neutrophils continue their journey toward the infection site via chemotaxis.
EVADING NEUTROPHIL EXTRAVASATION
An overview of the molecules interfering with the extravasation of neutrophils is shown in Fig. 1. S. aureus is able to modulate the first step in neutrophil extravasation, rolling of neutrophils on endothelial cells, by secreting staphylococcal superantigen-like 5 (SSL5). SSL5 directly binds PSGL-1 on leukocytes and human HL-60 leukemia cells. This prevents the interaction of PSGL-1 with its natural ligand, P-selectin, in a sulfation- and sialylation-dependent manner (30, 31). Cocrystallization data revealed SSL5 in complex with tetrasaccharide sialyl Lewis X, a key posttranslational modification of PSGL-1 binding to P-selectin (32). SSL5 is part of a larger group of 14 structurally related proteins (SSL1 to SSL14), which are involved in innate immune evasion. SSL11, of all the SSLs, shares the highest amino acid sequence identity to SSL5 and also shows binding to sialyl Lewis X, as seen in the cocrystal structure of the complex. SSL11 also binds other sialic acid-containing glycoproteins, such as FcαRI, the Fc receptor for IgA (33). S. aureus secretes another immune evasion molecule, named SElX, which also interacts with PSGL-1 in a glycosylation-dependent manner (34). Before the molecular mechanism was identified, SElX was thought to possess superantigenic activity and was associated with a virulence factor in MRSA necrotizing pneumonia (35). Recently, SElX was also found to have the sialylated-glycan-dependent active site, similar to a subfamily in SSLs (SSL2 to SSL4 and SSL6) (36). This leads to binding of neutrophils and monocytes via multiple glycosylated neutrophil surface receptors, thereby disrupting IgG-mediated phagocytosis and contributing to pathogenesis, as revealed in a necrotizing pneumonia rabbit infection model (37, 38).
The second step of extravasation, the adhesion of neutrophils to endothelial cells, is also targeted by S. aureus. The pathogen secretes extracellular adherence protein (Eap) that directly binds ICAM-1 and thereby inhibits neutrophil recruitment to the infection site (39).
PRIMING, CHEMOTAXIS, AND ACTIVATION OF NEUTROPHILS
When neutrophils have migrated through the endothelial barrier, they are primed, recruited toward the site of infection by various chemoattractants, and activated by multiple inflammatory stimulants. Here, we will discuss various sets of proteins and receptors involved in this process. An overview is shown in Fig. 2.
FIGURE 2.
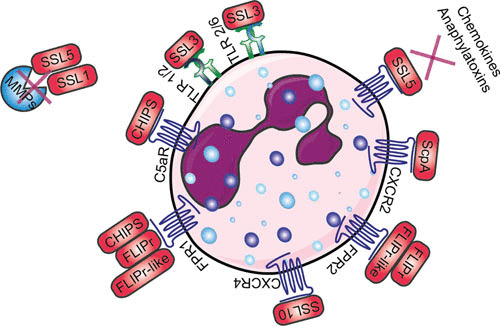
Schematic overview of how S. aureus evades priming, chemotaxis, and activation of neutrophils. Red boxes indicate staphylococcal proteins, and proteins shown in blue indicate host proteins. Abbreviations: TLR, Toll-like receptor; CXCR, chemokine receptor; ScpA, staphopain A; SSL, staphylococcal superantigen-like protein; FPR, formyl peptide receptor; FLIPr, FPR2 inhibitory protein; C5aR, C5a receptor; CHIPS, chemotaxis inhibitory protein of Staphylococcus; MMP, matrix metalloproteinase. The figure was adapted from Servier Medical Art.
Priming occurs when a neutrophil is exposed to a primary priming stimulus which enhances the neutrophil’s functional response (for example, adhesion, phagocytosis, degranulation, and superoxide production [40]), although the priming stimulus does not give the functional response itself (41). Examples of known priming molecules for neutrophils are complement components C3a and C5a (42), interleukin-8 (IL-8) (43), granulocyte colony-stimulating factor (43), tumor-necrosis factor-α (44), and interferon-γ (45).
Chemoattractants are chemokines and bacteria- and complement-derived products that activate phagocytes by interacting with receptors at the surface of neutrophils which belong to the superfamily of G protein-coupled receptors (GPCRs) (46). These GPCRs also belong to the rhodopsin subfamily of GPCRs. Rhodopsin is a seven-transmembrane receptor with seven helical membrane-spanning regions connected by six extramembrane loops, as shown by crystallization (47). The mechanism of stimulation of the GPCR depends on the type of ligand. For example, small peptides (e.g., N-formyl-methionyl-leucyl-phenylalanine [fMLP]) and lipid-derived stimuli (e.g., platelet-activating factor and leukotriene B4) primarily activate their GPCRs through the transmembrane regions. Larger stimulatory molecules (e.g., chemokines and anaphylatoxins) activate GPCRs via a two-step process whereby the stimulus binds the N-terminus of the specific GPCR, whereupon conformational change of the receptor leads to activation by the interaction with the pocket formed by the transmembrane domains (48). CXC chemokine receptors expressed on the surface of various immune cells also belong to the family of GPCRs, and to date, seven receptors have been described (named CXCR1 through CXCR7). They interact with chemokines, a large family of 8- to 12-kDa proteins. The CXC chemokines are one of the most prominent groups and are mostly chemotactic for neutrophils, such as CXCL8 (also known as IL-8) (49).
Newly synthesized bacterial proteins contain formylated methionine, so bacteria secrete a lot of N-formylated proteins and peptides, which were identified as chemoattractants in 1975 (50). These formylated peptides interact with formyl peptide receptors 1 (FPR1) and 2 (FPR2) (FPR2 is also known as FPRL1) on neutrophils (51), both belonging to the GPCR family. The prototype N-formyl-peptide, fMLP, binds with higher affinity to FPR1 than its homologue, FPR2 (52). Neutrophils and monocytes express FPR1 and FPR2, whereas the third homologue, FPR3 (also known as FPRL2), is only expressed in monocytes (52). Staphylococci produce phenol-soluble modulins (PSMs) in addition to formyl peptides. PSMs are not only important secreted staphylococcal toxins, but they are also sensed by FPR2, which leads to activation and attraction of leukocytes (53).
Two complement receptors (CRs) on neutrophils also belong to the family of GPCRs, namely, the C3a receptor (C3aR) and the C5a receptor (C5aR). Their ligands are the small complement components C3a and C5a, formed during complement activation. C5a and C3a are commonly also called anaphylatoxins and are both chemoattractants, acting by attracting phagocytes to the site of infection (54).
Neutrophils also express another class of innate immune receptors that are involved in pathogen recognition, the Toll-like receptors (TLRs). The discovery of these proteins in the mid-1990s revealed that pathogen recognition by the innate immune system is specific, because it recognizes different components of foreign pathogens, which are called pathogen-associated molecular patterns. Pathogen-associated molecular patterns that are important for staphylococcal infections are bacterial lipoproteins (recognized by TLR1, TLR2, and TLR6) and unmethylated CpG sequences in DNA molecules (recognized by TLR9) (55). Ligand binding to the extracellular domains of TLRs causes dimerization of the receptor complexes and triggers recruitment of MyD88 to the intracellular TLR domains, which ultimately leads to the activation of transcription factor NF-κB (56). Most TLRs appear to function as homodimers, although TLR2 forms heterodimers with TLR1 or TLR6. TLR1/2 recognizes triacylated lipoproteins, whereas TLR2/6 specifically responds to diacylated lipoproteins (57). TLRs are not directly involved in chemotaxis, but they contribute to phagocytosis.
Another class of proteins important for controlling inflammation are the matrix metalloproteinases (MMPs). Members of this family of 23 endopeptidases are secreted by numerous cells and are important in the recruitment and migration of neutrophils during bacterial infections. They break down extracellular matrix components so neutrophils can migrate toward the infection, but they also stimulate proinflammatory signals and cleave chemokines. This leads to enhanced inflammation and improved bacterial clearance. The two main neutrophil MMPs are MMP8 (also known as neutrophil collagenase) and MMP9 (also known as neutrophil gelatinase B) (58).
EVADING PRIMING, CHEMOTAXIS, AND ACTIVATION OF NEUTROPHILS
Studies thus far have not shown whether S. aureus can directly modulate neutrophil priming to influence the function of neutrophils. Additional studies are necessary to understand the influence of S. aureus on neutrophil priming (59). However, more knowledge has been gained about the impact of S. aureus on neutrophil chemotaxis and activation. Here, we describe the function of the evasion proteins SSL5, SSL10, SSL3, and SSL4; staphopain A; CHIPS; FPR2 inhibitory protein (FLIPr); and FLIPr-like in their role in evading neutrophil chemotaxis and activation, as shown in Fig. 2.
SSL5 not only binds to PSGL-1 to block neutrophil extravasation, but it also binds glycosylated N-termini GPCRs, thereby inhibiting the ligands that require the N-terminus of the receptor for activation. Therefore, it binds but does not interfere with the activation of FPR1 and FPR2, leukotriene B4 receptor, platelet-activating factor receptor, and nucleotide receptor P2Y2, since ligands of these receptors are small and signal via the transmembrane domains of the GPCRs (60). Pretreating neutrophils with SSL5 blocks the activation induced by C3a and C5a, because chemokines and anaphylatoxins bind the N-termini of their receptors. SSL5 also inhibits activation and neutrophil migration induced by IL-8, which interacts with CXCR1 and CXCR2, but it also targets the response induced by chemokine ligand 1 (CXCL1), which acts only on CXCR2 (60). SSL5 together with SSL1 inhibits neutrophil migration through breakdown of collagen and potentiation of IL-8 by inhibiting cleavage of the neutrophil-specific MMP8 (neutrophil collagenase) and MMP9 (neutrophil gelatinase B) as well as other members of the MMP family (MMP1, 2, 7, 12, 13, and 14) (61, 62). Additionally, SSL5 activates and aggregates platelets by interacting with platelet membrane receptor GPIbα and collagen receptor GPVI, leading to a beneficial environment for staphylococcal colonization (63, 64).
SSL10 targets another chemokine receptor, CXCR4, expressed on neutrophils and cancer cells. It binds to human T-cell acute lymphoblastic leukemia, lymphoma, and cervical carcinoma cell lines and inhibits calcium mobilization and cell migration induced by the ligand of CXCR4, CXCL12 (also known as stromal cell-derived factor-1α) (65).
SSL3 binds the extracellular domain of TLR2 and inhibits its activity on human and murine neutrophils and monocytes (66, 67). The cocrystal structure of SSL3 in complex with TLR2 reveals that SSL3 blocks ligand binding and prevents TLR heterodimerization of TLR1 and TLR6 and downstream signaling by interacting with an already formed TLR2-lipopeptide complex (68). SSL4 shares the highest homology with SSL3 and also blocks TLR2 activation, albeit less potently compared to SSL3. Therefore, it remains unknown whether this is the primary function of SSL4, because SSL4 was also shown to bind myeloid cells in a glycan-dependent manner (66, 69). Recently, antibodies against SSL3 and SSL4 were detected in human serum, showing that both evasion proteins are secreted in vivo. SSL3 is able to bind both murine and human TLR2, so SSL3 was investigated in an intravenous murine infection model, where it was shown that the presence of the gene correlates with high virulence. To circumvent low expression rates of SSL3 in mice, an SSL3-overproducing strain was used (70).
S. aureus also secretes a protease that interferes with chemokine signaling. Staphopain A cleaves the N-terminus of CXCR2 and thereby causes unresponsive neutrophils to undergo activation by CXCR2. The protease also hampers neutrophil migration toward CXCR2 chemokines (71). Mature staphopain A has a broader substrate specificity than CXCR2 alone because it is also able to degrade elastin fibers. However, it is also prone to autolytic degradation, which could explain the loss of activity of staphopain A over time (71, 72).
S. aureus supernatant showed chemotaxis-inhibiting properties by targeting the fMLP and C5a response of neutrophils (73). Later, the protein responsible in the supernatant was identified, and this 14.1-kDa evasion protein was named chemotaxis inhibitory protein of Staphylococcus (CHIPS). CHIPS binds and inhibits the GPCRs FPR1 and C5aR on neutrophils and monocytes. Thus, it blocks the binding of these receptors by their ligands, namely, C5a and fMLP, and thereby impairs the activation and chemotaxis of phagocytes (74, 75). N-terminal peptides of CHIPS inhibited FPR1, while the activity of C5aR was unaffected (76). This showed that the N-terminus of CHIPS is specific for its activity toward FPR1 and that CHIPS probably had another binding site for C5aR. Indeed, CHIPS specifically targets via its C-terminal domain the N-terminus of C5aR through binding to amino acids 10 to 18 and thereby prevents the binding of C5a to its receptor (77, 78). Arginine 44 and lysine 95 of CHIPS in particular appear to be highly important in the antagonism of C5aR (78).
The importance of CHIPS as a potential virulence factor led to the search for homologous excreted proteins in the genome of S. aureus. This resulted in the discovery of the gene encoding FLIPr, which showed 49% similarity to the gene for CHIPS (chp). Similar to CHIPS, FLIPr is able to inhibit FPR1 but can also inhibit FPR2, whereas binding with FPR3 was not observed (79). Later, the evasion protein FLIPr-like was discovered by a BLAST search through sequenced S. aureus genomes with FLIPr as the reference sequence. FLIPr and FLIPr-like are two allelic variants of the same gene, with 73% similarity on the amino acid level. The function of FLIPr-like is also similar to that of FLIPr, because it is an antagonist of both FPR1 and FPR2 (80). Moreover, FLIPr and FLIPr-like also target FcγR, the receptor for IgG, where FLIPr is almost exclusively restricted to class II receptors, with a preference for FcγRIIa, while FLIPr-like binds to most FcγR isoforms (81).
OPSONIZATION AND PHAGOCYTOSIS
Opsonization
The next step in clearing a bacterial infection, after neutrophils have been attracted to the site of infection, is uptake of the pathogens by phagocytes. For this phagocytosis to be effective, bacteria have to be opsonized: coated with components of the complement system, immunoglobulins, or other innate immune components. We will first describe opsonization by the complement system, followed by opsonization by immunoglobulins.
Opsonization by the complement system
Complement is a proteolytic cascade comprising more than 30 proteins in plasma that can (i) opsonize bacteria by depositing complement activation products on the bacterial surface, (ii) attract and activate other immune cells by formation of chemoattractants, and (iii) lyse and kill Gram-negative bacteria directly by the formation of the membrane attack complex (54). The complement system can be activated via three separate pathways that differ in their method of recognition but come together at one central step: formation of C3 convertase, which cleaves C3. One pathway, the lectin pathway, is activated upon recognition of conserved microbial sugars such as ficolins and mannan-binding lectin. The second pathway, the classical pathway, is primarily initiated by the interaction of C1q with antigen-antibody complexes (54). The third pathway, the alternative pathway, is initiated on the surfaces of neutral or positively charged pathogens that do not contain complement inhibitors. Its activation is due to the spontaneous “tick-over” reaction of C3 with water to form hydrolyzed C3, and it exists in an activated state at all times. Importantly, this pathway serves as an amplification loop after C3b is formed on bacterial cells via the lectin and classical pathways (54). C3 convertases are enzyme complexes with proteolytic activity that cleave C3. The classical and lectin C3 convertase is formed by cleavage of C4 by C1s to C4b, after which C4b covalently attaches to the bacterial surface and binds C2. C1s is then able to cleave C2 as well, thereby forming the C3 convertase C4b2a. The alternative pathway C3 convertase is different in that it contains a surface-bound C3b molecule attached to protease Bb, a subunit of factor B. Factor H controls complement activation by stimulating the decay of Bb from the alternative pathway convertase (C3bBb) and is a cofactor for factor I-mediated cleavage and inactivation of C3b (82).
Cleavage of C3 results in the formation of chemoattractant C3a and the deposition of C3b at the bacterial surface. C3b can be cleaved to the proteolytically inactive product iC3b by factor I on the surface of bacteria. iC3b is still able to opsonize bacteria, but it cannot associate with Bb (83). Both C3 convertases can bind an additional C3b molecule to form C5 convertases, and this convertase can cleave C5 into C5a and C5b. C5a is a chemoattractant and has been shown to have a protective role during staphylococcal bloodstream infections in mice (15). C5b together with C6, C7, C8, and multiple copies of C9 form the membrane attack complex (54). The membrane attack complex is able to directly lyse Gram-negative bacteria, whereas Gram-positive bacteria are unaffected due to their thick peptidoglycan layer (84). However, the complement system is important in the opsonization process because it deposits C3b on the surface of Gram-positive bacteria, such as S. aureus. Together, this sequence of events results in rapid and efficient detection and elimination of bacterial invaders.
Opsonization by immunoglobulins
Immunoglobulins (Igs) are a class of proteins that not only enable highly efficient opsonization, but are also involved in agglutination, neutralization of toxins and other virulence factors, and inhibition of adhesion. After binding to antigens on pathogens via their Fab segments, they are recognized by Fc receptors on the surface of phagocytes via their Fc region. The four isotypes of Igs vary in complement activation and are recognized by their own Fc receptor (FcγR, FcαR, FcεR, and FcμR). IgG, IgA, and IgM play roles in controlling infections, whereas IgE is more important in immunity to parasites. IgM is especially effective at opsonization through complement activation due to its polymeric structure (85). IgG consists of four subclasses (IgG1, IgG2, IgG3, and IgG4), and the differences in effector functions between the four IgGs are caused by differences in structure, especially the length and flexibility between the variable Fab segments and the stable Fc segment (86). Therefore the different subclasses of IgG bind differently to C1q, which is at the start of the classical pathway cascade. IgG3 binds the most strongly, whereas IgG1 and IgG2 bind more weakly (IgG1 > IgG2). IgG4 completely lacks the ability to activate the complement system (87).
Phagocytosis
Phagocytosis is a process by which other cells, cell fragments, and microorganisms are engulfed by white blood cells and end up in an internal compartment called the phagosome. As described above, opsonized microorganisms bind to specific receptors on the phagocyte surface. Subsequently, invagination of the cell membrane causes envelopment of the bacterium. This uptake is aided and highly enhanced by factors such as C5a, TLR ligands on the surface of the bacterium, and ligands for C-type lectins (such as DC-SIGN, dectin-1, and the mannose receptor on the surface of neutrophils) (88).
Cross-linking of FcγR by ligand binding on the surface of neutrophils activates several effector functions directed toward killing of pathogens and an inflammatory response (16). FcγRs are members of the immunoglobulin superfamily, and they are capable of binding the Fc region of IgG antibodies. There are several activating receptors (FcγRI/CD64, FcγRIIa/CD32a, FcγRIIc/CD32c, and FcγRIIIa/CD16a), one inhibitory receptor (FcγRIIb/CD32b), and one glycosylphosphatidylinositol-anchored receptor that contains no signal motif (FcγRIIIb/CD16b) (89). Human neutrophils express only two FcγRs, FcγRIIa and FcγRIIIb, and FcγRIIa induces mainly phagocytosis (89).
CR1 (CD35) is found on circulating monocytes, neutrophils, and B-lymphocytes. This receptor binds C4b, C3b, and iC3b and induces phagocytosis (90). CR3 (CD11b/CD18, Mac-1) and CR4 (CD11c/CD18, p150/95) are heterodimeric glycoproteins of the integrin family with a shared β-chain (CD18). They bind the iC3b fragment and to a lesser extent also C3b (91). Stimulation of neutrophils and monocytes via CR3 and CR4 results in induction and enhancement of phagocytosis, degranulation, and generation of ROS (91).
EVADING OPSONIZATION AND PHAGOCYTOSIS
S. aureus has evolved numerous molecules that enable it to evade the different parts of the complement cascade, immunoglobulins, and elements of the phagocytic process. These molecules are illustrated in Fig. 3. Evasion of these components results in a highly effective delay or reduction of the immune response and creates a beneficial situation for the bacterium to survive and multiply within its host.
FIGURE 3.
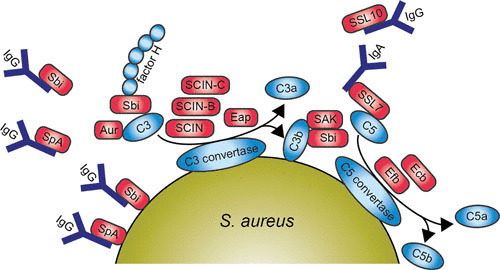
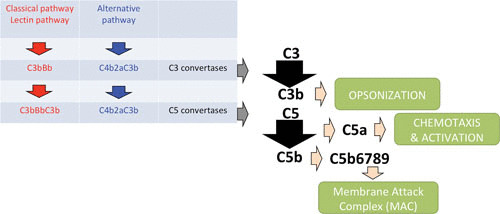
Diagram of the main pathways and components of the human complement system 3a and a schematic representation of S. aureus evading opsonization and phagocytic uptake by neutrophil 3b. Red boxes indicate staphylococcal proteins, and blue boxes indicate host proteins. Abbreviations: IgG, immunoglobulin G; SpA, staphylococcal protein A; Sbi, staphylococcal binding of IgG; SCIN, staphylococcal complement inhibitor; SAK, staphylokinase; Aur, aureolysin; SSL, staphylococcal superantigen-like protein; Efb, extracellular fibrinogen-binding protein; Ecb, extracellular complement-binding protein. The figure was adapted from Servier Medical Art.
Capsule Production
The best way for the bacterium to prevent phagocytosis is to hide the antigenic or immunogenic proteins at the surface of the bacterial cell wall with a polysaccharide capsule. Up to ∼75% of all clinical S. aureus isolates have a capsule or microcapsule whereby most of them contain either capsular polysaccharide 5 (CP5) or capsular polysaccharide 8 (CP8). These bacteria grown under optimal capsule-production conditions showed resistance to opsonophagocytosis and thus killing (92). However, the capsule of S. aureus does not completely block deposition of complement components at the bacterial surface or binding of specific antibodies (93). Other bacteria, such as Streptococcus pneumoniae, produce a very thick capsule (94).
Proteins Targeting Immunoglobulins
The first evasion molecule described to have antiopsonic properties is staphylococcal protein A (SpA). SpA is linked to the staphylococcal cell wall via its LPXTG anchor but can be released upon hydrolysis during growth (95, 96). This protein contains four or five immunoglobulin-binding domains capable of binding the Fc part of IgG, which leads to blockage of FcR-mediated phagocytosis (97). SpA also binds to the Fab domain of variable heavy 3 type B cell receptors of IgM and thus serves as a B cell antigen by stimulating B cell activation (98–100). Consequently, this shows that SpA effectively interferes with the adaptive immune response. Recently, guinea pigs were shown to be a good model to test this therapeutic approach in vivo, which will be important for vaccination strategies in the future (101).
A second IgG-binding protein, S. aureus binder of IgG (Sbi), was identified through a phage display screen against immobilized human IgG (102). Like SpA, Sbi is also expressed at the staphylococcal cell surface and is secreted during bacterial growth (103, 104). Sbi also has two binding targets because it is able to bind to another serum component, β2-glycoprotein I, also known as apolipoprotein H. This was similarly identified by phage display, and the binding site was clearly different from the IgG-binding domain (103). However, in contrast to SpA, Sbi can only interact with the Fc domain of IgG (105). Furthermore, the extracellular region of Sbi consists of four globular domains, where domains I and II are immunoglobulin-binding domains and domains III and IV are able to bind complement component C3 and factor H, forming a stable tripartite complex (Sbi:C3:factor H) (106, 107). Thus, Sbi is a versatile evasion protein, interfering with the innate immune system by binding IgG and apolipoprotein H, as well as the complement components factor H and C3.
SSL10, a multifunctional evasion protein, is yet another protein able to bind IgG1 (108). This leads to the inhibition of complement activation via the classical pathway, and this is the second function of SSL10, since it is also a CXCR4 antagonist. The N-terminus of SSL10 binds to the Fc domain of IgG1, preventing association with C1q and FcγRs and leading to inhibition of FcγR-mediated phagocytosis (109). Furthermore, SSL10 also targets prothrombin and factor Xa to inhibit blood coagulation (110), binds to phosphatidylserine, and recognizes apoptotic cells (111). Finally, S. aureus secretes a serine protease, V8, which is able to cleave immunoglobulins (112).
Proteins Targeting the Complement System
Not all known evasion proteins interfering with opsonization target immunoglobulins. Some of them are specialized in evading the complement system. For example, the secreted metalloprotease aureolysin cleaves the central complement component C3 in a zinc-dependent manner. The cleavage site on C3 of aureolysin differs by two amino acids from the C3 convertase cleavage site generating active C3a′ and C3b′. This suggests that aureolysin mimics C3 convertases, but it also degrades the cleaved C3b′ as well as factors I and H. This cleavage is more effective in vivo than in vitro because it is more effective in serum than without serum. Aureolysin is a secreted protease, and thus it can cleave C3, removing it from the bacterial surface and creating a C3-free microenvironment and thus preventing C3b′ from covalently attaching via its thioester to bacteria (113).
Staphylococcal complement inhibitor (SCIN), initially identified as a prophage-coded protein, inhibits all three complement pathways: the alternative, classical, and lectin pathways. SCIN stabilizes and inhibits surface-bound C3 convertases, and this results in a decline of C3b deposition and release of chemoattractant C5a, which results in the inhibition of phagocytosis (114). Later, SCIN-B, SCIN-C, and open reading frame D were identified in the S. aureus genome. Characterization of these homologues shows that SCIN-B and SCIN-C also inhibit complement, whereas open reading frame D shows no inhibitory activity (115). Cocrystallization studies of SCIN with C3 convertase reveals that SCIN inhibits convertase activity in three ways: (i) SCIN prevents Bb mobility, which prevents Bb from accessing substrate C3 (116); (ii) SCIN may dimerize two C3 convertases and thereby prevent substrate binding (116); (iii) by binding to a single C3b molecule, SCIN slows the rate of factor B loading onto C3b and thus lowers the amount of C3 convertase formation (117). This all leads to inhibition of phagocytosis, C3b deposition, and C5a generation (114). Furthermore, a new SCIN variant was recently identified in equid S. aureus isolates. This equine SCIN has adapted to horses by inhibiting the equine complement system.
Extracellular fibrinogen-binding protein (Efb) and extracellular complement-binding protein (Ecb) are homologous proteins (118). The first characteristic described for Efb was that it binds via its N-terminus to fibrinogen—hence its name (119). Efb can also bind to platelets, as identified by phage display, interfering with platelet aggregation, contributing to virulence in wound infections, and delaying the healing process in rats (120–122). Additionally, the C-terminus of Efb is able to bind C3 and its cleavage products containing the thioester domain (C3d) (123, 124). Efb blocks phagocytosis in vitro of S. aureus by neutrophils in plasma and human whole blood as well as in vivo in a mouse peritonitis model. This evasion protein forms a unique bridge between complement and coagulation systems, since it can bind both C3 and fibrinogen. Efb covers bacteria with a thick layer of fibrinogen, which leads to shielding of surface-bound C3b and antibodies from recognition by phagocytic receptors (125). This mechanism to shield the bacterial surface from phagocytosis together with the formation of a capsule enhances the evasion of S. aureus against phagocytosis (126). It also causes clumping of staphylococcal cells in vivo, thus enhancing agr quorum-sensing (127).
Ecb (also known as Ehp) compared to Efb lacks fibrinogen binding activity, but like Efb, it does inhibit the complement system by binding the C3d domain of C3 (118, 128). This results in blocking of C3 convertases of the alternative pathway and C5 convertases of all three complement pathways (129). The binding of Ecb to C3d is stronger than Efb to C3d, because Ecb contains a second, lower-affinity, C3 binding site. This results in enhanced complement inhibitory effect of Ecb compared to Efb (128). The function of many evasion molecules is restricted to the human host; however, Ecb and Efb efficiently inhibit the complement system in both humans and mice. Therefore, the importance of Ecb and Efb could be examined in vivo in a murine infection model. Mice experienced higher mortality rates in an intravenous model with wild-type bacteria (79%) compared to an isogenic ΔEcbΔEfb mutant (21%). In addition, Ecb and Efb promoted bacterial survival and blocked neutrophil influx in the lungs. These results indicate that Ecb and Efb are essential to S. aureus virulence in vivo and could be attractive targets for vaccine development (130).
S. aureus recruits factor H to its surface with the surface-associated serine-aspartate repeat protein E (SdrE), and this leads to inhibition of the alternative pathway of the complement system (131). Recombinant SdrE recognizes its ligand via a unique “close, dock, lock, and latch” mechanism as determined by crystallographic studies (132). Moreover, factor H bound to SdrE retains its activity for factor I-mediated cleavage of C3b to iC3b (131). Factor I is directly targeted by the full-length cell wall component clumping factor A as well as a secreted moiety of around 50 kDa. Binding of cell wall component clumping factor A to factor I promotes cleavage of C3b to iC3b, which results in disruption of opsonophagocytosis (133, 134).
Extracellular adherence protein (Eap) has a variety of functions in evading the immune system. We already discussed the binding of Eap to ICAM-1 causing impaired function in neutrophil extravasation (39), but Eap is also able to inhibit the complement system. C4b is targeted by Eap, and this leads to inhibition of C2 binding to C4b and thus blocks formation of the active C3 convertase of the lectin and classical pathway (135).
Staphylokinase (SAK) also acts as an antiopsonic immune evasion protein by converting human plasminogen into the active bacterium-bound serine protease plasmin. Plasmin, from its location on the bacterial surface, degrades IgG and C3bf, resulting in decreased phagocytosis by human neutrophils (136).
SSL7 inhibits opsonization in two ways: it selectively binds both IgA and complement C5 (137). The binding of SSL7 to the Fc region of IgA, mediated via its N-terminus, causes interference with antibody recognition (138), whereas its C-terminus binds to C5 and thereby inhibits terminal complement activation and cleavage of C5 by interfering with the binding of C5 to C5 convertases (139). SSL7 was also found to be important in vivo by inhibiting complement-induced neutrophil influx in a murine inflammatory model (139).
S. aureus has also evolved a protein attached to the cell wall that specifically inhibits the classical pathway of the complement system. This protein, S. aureus collagen adhesin (Cna), belongs to the microbial surface component recognizing the adhesive matrix molecule (MSCRAMM) family of adhesins. Cna binds C1q and thus inhibits classical pathway activation. Cna belongs to the structurally related Cna-like family, members of which are found in many other Gram-positive bacterial species. Thus, the function of Cna-like MSCRAMMs as inhibitors of the classical pathway could serve as an immune evasion strategy for numerous Gram-positive pathogens (140).
NEUTROPHIL EXTRACELLULAR TRAP FORMATION
Neutrophils have recently been shown to have an antibacterial defense mechanism whereby they release their DNA, in association with antimicrobial peptides, histones, and proteases, to form a network of extracellular fibers which entrap and kill various microbes. These networks are known as neutrophil extracellular traps (NETs) (141). There are current studies to address the molecular mechanism behind NET formation, but more research is needed to fully understand this phenomenon (142). The formation of NETs in vitro is activated via different proinflammatory stimuli, including hydrogen peroxide, phorbol myristate acetate, lipopolysaccharide, IL-8, and various bacteria, including S. aureus (143). The formation of NETs has also been shown in vivo, where intravascular NETs were observed in the mouse liver during sepsis induced by S. aureus (144).
EVADING NET FORMATION
A secreted nuclease of S. aureus, Nuc, is important in the breakdown of NETs. This has been shown both in vitro and in vivo, whereby an isogenic nuclease knock-out of S. aureus shows impaired degradation of NETs compared to the wild-type strain. This leads to the linkage of nuclease production to delayed bacterial clearance in the lung and increased mortality after intranasal infection in vivo (145). Thus, Nuc helps staphylococci to escape from the extracellular fibers and to avoid getting killed by antimicrobial peptides and proteases. Nuc was also shown to play a role in immune cell death together with secreted adenosine synthase. These two enzymes are able to convert NETs to deoxyadenosine, which triggers the caspase-3-mediated death of immune cells (146).
BACTERIAL KILLING BY NEUTROPHILS
Neutrophils are end-stage cells with a high concentration of antimicrobial proteins safely stored within different granules. They kill pathogens by releasing their granules containing antimicrobial proteins into the phagosome (also called degranulation) and generating ROS (also known as the oxidative burst) upon phagocytosis of bacteria. Phagocytes, especially macrophages, also form reactive nitrogen species via the oxidation of nitric oxide (NO•), which is produced by an inducible nitric oxide synthase (147). The granules of neutrophils can be divided into three groups: the primary or peroxidase-positive granules, the secondary or specific granules, and the tertiary or gelatinase granules. The peroxidase-positive granules are also called azurophilic granules due to their affinity for the basic dye azure A (12). Secretory vesicles contain plasma proteins and are probably formed by endocytosis (12). Azurophilic granules contain a variety of antimicrobial proteins, such as pore-forming peptides (α-defensins), neutrophil serine proteases (NSPs; proteinase 3 and 4, cathepsin G, and elastase), myeloperoxidase (MPO), the bactericidal/permeability-increasing protein, and lysozyme. Secondary granules contain hCAP-18, lactoferrin, neutrophil gelatinase-associated lipocalin, and lysozyme. Tertiary granules contain a number of metalloproteases, such as MMP8 (collagenase) and MMP9 (gelatinase). Secretory vesicles contain plasma proteins and membrane-associated receptors essential at the earliest phase of neutrophil-mediated inflammatory response, such as the extremely rapid upregulation of β2-integrin CD11b/CD18, CR1, and FPR1 and FPR2 (148). However, it should be mentioned that heterogeneity exists between the different granules and that their granule contents overlap, because they are sequentially formed during myeloid cell differentiation (149).
Oxygen-Independent Killing by Neutrophils
Oxygen-independent killing mechanisms by neutrophils greatly contribute to microbial killing (150). Agents that contribute to oxygen-independent microbicidal activity include antimicrobial peptides and proteins such as lactoferrin, lysozyme, α-defensins, cathelicidins such as hCAP-18, azurocidin, proteinase 3, cathepsin G, and neutrophil elastase (12). The C-terminal part of hCAP-18, named LL-37, forms an amphipathic α-helix and has antimicrobial properties against Gram-negative bacteria and Gram-positive bacteria, such as S. aureus (151). LL-37 is effective against both intra- and extracellular S. aureus (152). Other antimicrobial peptides include α-defensins (also known as human neutrophil peptide-1). These peptides are antimicrobial, acting by the formation of multimeric pores and applying this on various microorganisms (153). The structurally related serine proteases (cathepsin G, elastase, and proteinase 3 and 4) have various functions, including the cleavage of bacterial virulence factors and regulation of immune responses by cleaving receptors and chemokines, and they are able to kill bacteria in the extracellular milieu after being secreted (154). The serine proteases also proved to be important in vivo, since mice lacking cathepsin G showed impaired clearance of S. aureus during infection (155). Lactoferrin and calprotectin are chelating proteins; lactoferrin sequesters iron needed for microbial growth, and calprotectin chelates zinc and manganese and thereby inhibits staphylococcal growth (156). Calprotectin is located in the cytoplasm of neutrophils and makes up more than 60% of the proteins in the cytosol (157). Lysozyme is a cationic antimicrobial peptide and is present in all granule subsets, with a peak concentration in secondary granules. Lysozyme cleaves bacterial cell wall peptidoglycan polymers by breaking the β-1,4 glycosidic bonds between N-acetylmuramic acid and N-acetylglucosamine (158). This results in lysis of various bacteria, such as Bacillus subtilis, in human plasma, but has only limited action against staphylococci because of their modified peptidoglycan (159).
Oxygen-Dependent Killing by Neutrophils
Upon phagocytosis of bacteria, the azurophilic granules are the first to fuse with the phagosome, and this leads to assembly of the five essential parts of the NADPH oxidase complex and activation of the complex (160). Active NADPH oxidase transfers electrons across the phagosomal membrane from cytosolic NADPH to intraphagosomal molecular oxygen to produce superoxide, which is compensated for by an influx of cations or protons. Although superoxide has limited direct microbicidal capacity, the molecule is involved in generating secondary derived ROS such as hypochlorous acid, chloramines, hydroxyl radicals, and singlet oxygen, which directly contribute to polymorphonuclear microbicidal activity. The superoxide anion is converted to hydrogen peroxide spontaneously or with the help of superoxide dismutase. MPO catalyzes the reaction of hydrogen peroxide with chloride to form the highly bactericidal agent hypochlorous acid (161, 162). Chlorine, chloramines (e.g., monochloramines, dichloramines, and taurine chloramine), singlet oxygen, hydroxyl radicals, and ozone are subsequently formed in secondary reactions and are also potent antimicrobial compounds (163).
The role of MPO in intracellular bacterial killing is controversial. In the early 1970s it was thought that MPO was the main factor in killing of S. aureus by hypochlorous acid production, because MPO-deficient patients showed a 10-fold decrease in killing bacteria after phagocytosis (164). However, this process is incompletely understood, and reproducible results are lacking. Later, MPO was shown to contribute poorly to the pulmonary defense when comparing wild-type mice with MPO-deficient mice infected with S. aureus. In contrast, MPO-deficient mice showed a significant decrease in clearing other microorganisms (e.g., Candida albicans, Candida tropicalis, Pseudomonas aeruginosa) compared to wild-type mice (165). Back then it was believed that the pH inside the phagosome dropped to 4.0 (166). Later, the pH inside the phagosome was believed to be 7.8 and to drop to 6.9 after 15 minutes of phagocytosis (167) or even to 5.7 after 60 minutes (168). Recently, data revealed that the pH becomes more alkaline due to K+ influx, which is optimal for granule proteases (155). This brings into question the role of MPO in bacterial killing because an alkaline pH results in a virtual absence of the peroxidase and chlorinating activities of MPO (169). It is possible that MPO has two functions in vivo: (i) peroxidase activity at pH ∼6 when the neutrophil is unable to fully engulf an organism and (ii) superoxide dismutase (161) or catalase (170) activity in an alkaline milieu in a fully enclosed vacuole with a pH of ∼9 to favor conditions for the granule proteases (e.g., proteinase 3, cathepsin G, and elastase) (171). MPO could also protect the microbicidal enzymes against oxidative damage, so both oxygen-dependent and oxygen-independent machineries inside the phagosome synergize for most effective killing of microbes. In our own experiments we have demonstrated that the MPO-dependent part of killing of S. aureus in neutrophils is 26% in the first 60 minutes (172).
EVADING KILLING
In this section we give an overview of the evasion proteins and pigments that are involved in staphylococcal survival of oxygen-dependent and -independent killing. These proteins are summarized in Fig. 4. Then the various staphylococcal toxins will be discussed. An overview of these toxins is shown in Fig. 5.
FIGURE 4.
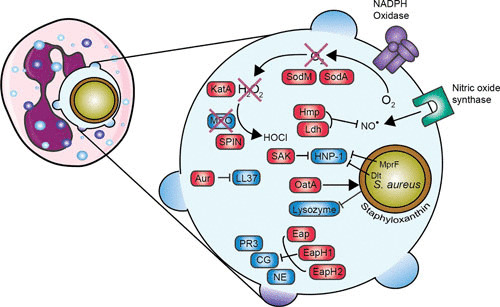
Overview of evasion proteins that are involved in evading neutrophil killing. Enlargement of the phagosome is shown on the right. Red boxes indicate staphylococcal proteins, and blue boxes indicate host proteins. Staphyloxanthin provides a protective shield, KatA neutralizes hydrogen peroxide (H2O2) into water (H2O) and oxygen (O2), and SPIN inhibits MPO activity. MprF and the Dlt operon lead to an increase in positive charge of the bacterial surface. Abbreviations: SOD, superoxide dismutase; SAK, staphylokinase; KatA, catalase; MPO, myeloperoxidase; SPIN, staphylococcal peroxidase inhibitor; Aur, aureolysin; Hmp, flavohemoglobin; Ldh, l-lactate dehydrogenase; Eap, extracellular adherence protein; EapH, extracellular adherence protein homologue; PR3, proteinase 3; CG, cathepsin G; NE, neutrophil elastase. The figure was adapted from Servier Medical Art.
FIGURE 5.
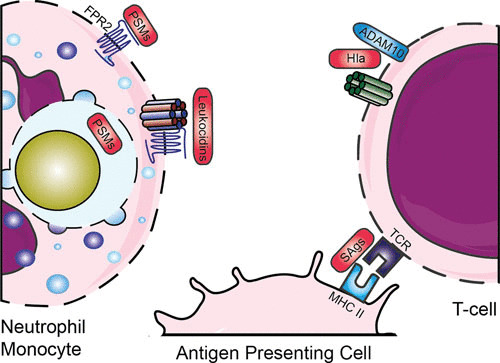
Evasion by staphylococcal toxins. Various leukocidins bind specific GPCRs, after which they form a pore and lyse host cells. PSMs are released inside the phagosome and can bind via FPR2. SAgs cross-link major histocompatibility complex class II and T-cell receptors. Abbreviations: GPCR, G-protein-coupled receptor; FPR, formyl protein receptor; PSMs, phenol-soluble modulins; Hla, hemolysin-alpha; SAgs, superantigens; MHC II, major histocompatibility complex II; TCR, T-cell receptor. The figure was adapted from Servier Medical Art.
Evading Oxygen-Dependent Killing by Neutrophils
S. aureus’s species name is derived from the fact that this bacterium has “a jacket” of the golden pigment staphyloxanthin. This staphyloxanthin serves as an antioxidant and is protective in killing by hydrogen peroxide and singlet oxygen. Bacteria lacking staphyloxanthin showed impaired survival in vitro and in vivo (173). The genes crtM (dehydrosqualene synthase) and crtN (dehydrosqualene desaturase) are essential in the biosynthetic pathway to produce staphyloxanthin. Therefore, synthetic inhibitors of CrtM are interesting new virulence factor-based therapies that act by targeting the biosynthesis of staphyloxanthin (174, 175).
S. aureus has developed other proteins that contribute to the resistance against ROS. The bacterium produces two superoxide dismutases, sodA and sodM, that convert the harmful superoxide radicals into hydrogen peroxide and oxygen (176). Isogenic sodA and/or sodM knockout bacteria had reduced virulence in a mouse abscess model, showing that both sodA and sodM contribute to the virulence of S. aureus (176). Probably, sodA has the major SOD activity in S. aureus throughout all growth stages, whereas sodM becomes active under oxidative stress during the late exponential and stationary growth phases (177). The hydrogen peroxide generated by the neutrophil is degraded into water and oxygen by KatA, a staphylococcal catalase, which is described to be a major virulence factor (178). However, studies conducted with a catalase mutant strain showed no difference in virulence in a murine abscess infection model (179). Another catalase-like protein produced by S. aureus is alkyl hydroperoxide reductase (AhpC). KatA catalase appears to be important for protection against external oxidative stress, whereas AhpC clears endogenously produced hydrogen peroxide. In that study, AhpC and KatA were not required for virulence of S. aureus, but AhpC and/or KatA are important for nasal colonization (180). Another virulence factor, at first unidentified, regulated via the SaeR/S two-component system was suspected to play a role in the decrease of hydrogen peroxide and hypochlorous acid production. This factor was independent of SOD and catalase, since their expression is not regulated by SaeR/S (181). Later, this factor was identified as staphylococcal peroxidase inhibitor (SPIN), a secreted protein of 8.4 kDa which is able to bind and inhibit MPO. From crystallographic studies, we know that SPIN acts as a molecular plug that prevents access of substrate hydrogen peroxide to the MPO active site. Despite the controversy concerning the importance of MPO in bacterial clearance, SPIN protects S. aureus from MPO-mediated killing (172).
S. aureus has also evolved two proteins to resist the stress of nitric oxide produced in activated phagocytes. Flavohemoglobin scavenges host-derived NO•, and the NO•–inducible l-lactate dehydrogenase catalyzes the production of l-lactate, which maintains redox homeostasis during nitrosative stress (182, 183).
Evading Oxygen-Independent Killing by Neutrophils
S. aureus contains two independent loci (dltABCD and mprF) that affect susceptibility to defensins and other cationic pore-forming peptides by modifying the net charge of the cell wall envelope. The dlt operon is responsible for the incorporation of d-alanine into teichoic acids, and this leads to a decrease in negative charge on the bacterial surface and tolerance of high concentrations of positively charged antimicrobial peptides (184). A dlt knockout mutant was more susceptible to killing by defensin peptides and human neutrophils, even in the absence of a functional respiratory burst. As a control, there was no difference in killing between wild-type and dlt knockout bacteria by monocytes, which do not produce defensins (185). The mprF (multiple peptide resistance factor) gene encodes a lysylphosphatidylglycerol synthetase which is involved in modification of membrane phosphatidylglycerol with l-lysine, which also results in a reduction of the negatively charged membrane surface (186, 187). In contrast to most defensins, cathelicidin-derived bactericidal peptides, such as LL-37, have substantial activity against staphylococci. However, S. aureus has also evolved a protein to evade the antimicrobial effects of LL-37 by degrading and thereby inactivating LL-37 by the staphylococcal metalloproteinase aureolysin (which is also involved in the opsonization process by cleaving C3) (188). As described above, lysozyme degrades the cell wall peptidoglycan layer. Nonetheless, S. aureus resists lysozyme by expressing the enzyme O-acetyltransferase A (OatA). OatA causes O-acetylation of the peptidoglycan, and this leads to resistance to the muramidase activity of lysozyme. OatA is regarded as a virulence factor since it is expressed only in pathogenic, lysozyme-resistant staphylococci (for example S. aureus, S. epidermidis, and S. lugdunensis) (189, 190).
Eap inhibits the activity of NSPs (neutrophil elastase, cathepsin G, and proteinase 3). Its homologues, EapH1 and EapH2, each contain one EAP domain, whereas the earlier described Eap usually contains four or five EAP repeats. A single EAP domain impairs the function of NSPs; therefore, EapH1 and EapH2 also inhibit NSP activity (191). Recently, the interaction between EAP and NSPs was investigated, and despite the homology between EapH1 and EapH2, they form different complexes with NSPs (192). S. aureus protects its own immune evasion arsenal from degradation and cleavage by inhibiting these NSPs (193). For example, the N-terminus of SPIN is susceptible to proteolysis by NSPs, which results in a loss of function to inhibit MPO, but it is protected again by Eap. SAK is not only involved in evading opsonization by cleaving plasminogen to active plasmin (136), but it also binds and inhibits α-defensin (human neutrophil peptide 1). Human neutrophil peptide 1 is an important antibacterial peptide in neutrophil granules. This inhibition affects intracellular killing (194).
Escape by Toxins
S. aureus also protects itself by the secretion of toxins (Fig. 5). Toxins can directly lyse and thereby kill (immune) cells by disrupting the cell membrane and protecting the pathogen both before and after engulfment by neutrophils. These lytic toxins can be divided into two groups based on their structure: the β-barrel pore-forming toxins such as α-hemolysin and leukocidins and the small α-helical peptides such as the PSMs. A different group of toxins, the superantigens, activate T-cells by cross-linking major histocompatibility complex class II and T-cell receptors, resulting in massive nonspecific activation of T-cells. This temporary overactivation of a large population of T-cells causes secondary inhibition of the adaptive immune system and thereby contributes to immune escape.
β-Barrel pore-forming toxins target eukaryotic cells expressing specific factors. Hemolysin-alpha (Hla, also known as α-toxin) is secreted as a monomer, which forms a homo-heptameric pore upon binding to its receptor, the zinc-dependent metalloprotease ADAM-10 (195). Hla also upregulates ADAM10 on endothelial and epithelial cells, causing disruption of the endothelial and epithelial barrier (196, 197). Hla does not lyse granulocytes (e.g., neutrophils), but it does lyse other blood cells, such as erythrocytes, macrophages, and subpopulations of lymphocytes (198). Other β-barrel pore-forming toxins are leukocidins. These toxins consist of two independent secreted monomers known as F (fast) and S (slow) and form octamer pores of four F and four S subunits in membranes of host cells (199). Currently, seven S. aureus leukocidins have been described, including five in human-associated strains: LukAB (also known as LukGH), LukSF (also known as PVL), LukED, and two γ-hemolysins (HlgAB and HlgCB). Two animal-associated leukocidins (LukPQ [200] and LukMF′ [201]) were recently added to the arsenal (202). The receptors for these leukocidins have recently been identified, and this explains the cellular tropism and biological consequences of these pore-forming toxins during infection (203). Their receptors, all GPCRs and almost exclusively chemoattractant receptors, are located on various immune cells, but mainly on phagocytes. This indicates that the major role of these toxins is in immune evasion. They are designed to kill those cells that are crucial in eliminating staphylococci. HlgAB and LukED, however, also target the Duffy antigen receptor for chemokines expressed on erythrocytes, which leads to release of hemoglobin and promotes bacterial growth (204). Furthermore, LukAB has been shown to play a role in intracellular lysis of neutrophils, thereby promoting bacterial replication and outgrowth (205). Interestingly, the presence of these leukocidins in the genome of S. aureus is highly diverse. For example, the HlgACB and lukAB genes are located in the core genome and are present in more than 95.5% of human S. aureus isolates, whereas PVL is located on a phage and is found in fewer than 2% of all clinical isolates, but it is found in the majority of community-acquired MRSA strains in the United States. The presence of the PVL genes (lukS-PV and LukF-PV) in staphylococci correlates with the occurrence of necrotizing pneumonitis, a severe and devastating form of bacterial pneumonia (206).
The other class of toxins is PSMs, which are small α-helical peptides. These peptides are associated with enhanced virulence in community-acquired MRSA and are produced at high concentrations. PSMs contain a common amphipathic α-helical region which is responsible for disrupting the cell membrane; nonetheless, they are categorized in two groups: the β-type PSMs (around 44 amino acids long) and the shorter α-type PSMs (20 to 25 amino acids long), which have more enhanced toxic characteristics (207). The function of PSMs is inhibited by serum lipoproteins, which are present in large quantities in human serum (208). Therefore, their function is mainly intracellular, where they are able to lyse phagocytes from the inside out. The critical concentration of PSM is easily reached inside the condensed phagosome (209). Through the interaction between PSMs and FPR2 (see “Opsonization,” above), PSMs can not only lyse neutrophils, but can also attract and activate leukocytes via FPR2 (53).
S. aureus also encodes a variety of toxins (the superantigens) that modulate the adaptive immune system. Around 20 serologically distinct staphylococcal superantigens are known, which are divided into enterotoxins, enterotoxin-like, and toxic shock syndrome toxin-1. Their nomenclature is differentiated on the basis of proven emetic activity when orally ingested (enterotoxins SEA-E, G-J, and S-T) or unconfirmed activity (enterotoxin-like SElK-R, U and U2, V, and X-Y) (210). Superantigens cross-link major histocompatibility complex class II and T-cell receptors. This causes massive nonspecific activation of T-cells and an increase of many cytokines to toxic levels, which leads to damage of tissue and organs. This is called the toxic shock syndrome, and it is often lethal (211). The SSLs were first annotated as SET proteins for “staphylococcal enterotoxin-like” but were later renamed staphylococcal superantigen-like (SSL) since these proteins lack enterotoxic activity (210). Even though their enterotoxic activity is lacking, the SSLs have distinct functions in evading the innate immune system.
DISCUSSION AND CONCLUSION
Staphylococcal Innate Immune Evasion
Bacteria adapt to their environment. Pathogenic bacteria adapt to survival in their host. In the human host this means that they have evolved various virulence strategies to overcome innate and adaptive immunity. These virulence strategies are (i) camouflage of the microbial surface to avoid immune recognition by the production of an inert capsule, often composed of sugars, (ii) hiding from the immune system by adapting to an intracellular lifestyle in immune cells or other cells, and (iii) secretion of small proteins that inhibit specific elements of the immune system (212). These approaches all have advantages and disadvantages. Production of a capsule requires a considerable amount of energy. In addition, at some stages during infection, the bacteria need to remove their capsule, for example, for adherence to host cells, thereby exposing themselves to the immune system. For example, pneumococcal capsules are 60 nm thick, covering all surface structures on the bacterium. Hiding from the immune system through survival inside cells is also a challenge owing to the presence of multiple microbicidal enzymes and proteins inside these cells. Moreover, discovery of intracellular TLRs, NOD-like receptors, and inflammasomes revealed that bacteria can also be recognized inside cells (55). Therefore, the production of small secreted inhibitory proteins is, by itself or in combination with the other strategies, a strong evasion strategy to survive within the host and works extracellularly as well as intracellularly. S. aureus has extensively evolved proteins to evade all facets of the initial attack by the host immune system.
S. aureus remains a leading cause of bacterial infections and has an increasing risk for human health with the rise of antibiotic resistance in community-acquired strains. Also, the financial burden is growing due to increasing pressure on the health care system. Research into immune evasion molecules is very important, since these proteins determine staphylococcal virulence and pathogenesis. Understanding the mode of action of these evasion molecules will help us to find new therapeutics for prophylaxis or will improve treatment against S. aureus, since a functional vaccine is still lacking.
So Many Evasion Molecules: Luxury or Necessity?
To date, the functions of around 40 immune evasion proteins have been identified, and this number is still growing. These evasion molecules are present in clinical isolates and are expressed in the human host, since healthy carriers and noncarriers in adults and even young children have antibodies against these proteins (20, 213). Some of these evasion molecules were detected by high-resolution mass spectrometry from nasal polyp tissue from patients with chronic rhinosinusitis (214). One question arising from this observation is Why is S. aureus expressing so many evasion molecules? It seems a waste of time and energy for the bacterium to produce and secrete all of these proteins. Most of them target different specific parts of the immune system. We think that this demonstrates the strength, the significance, and the redundancy of our innate immune system. A very efficient and redundant immune system can only be overcome by a redundant counterattack. Moreover, other microbes have different methods to avoid recognition and killing by the immune system. As said before, a thick capsule is an efficient method to hide and sterically hinder access to the highly immunogenic surface, for example, masking the binding of C3 fragments to the complement receptor. This leads to decreased opsonization, phagocytosis, and subsequently, killing of the microorganisms by neutrophils (94). Gram-negative bacteria such as Neisseria meningitides and Gram-positive bacteria such as group A Streptococcus and S. pneumoniae are known to produce this thick capsule (94, 215, 216). A typical capsule is encoded by at least 15 large glycosyltranferases that are encoded by 3 to 4% of all base pairs in the genome, because the average size of a glycosyltransferase is between 200 and 300 kDa. Evasion proteins are much smaller, typically between 10 and 30 kDa, and encoding 40 of these therefore amounts to less than 1% of the whole S. aureus genome. Also, the production and maintenance of a thick polysaccharide capsule consumes a large amount of energy. Therefore, the production of 40 small proteins that directly attack the immune system is cheaper for the bacterium than the strategy of making a thick capsule.
Another explanation for the enormous number of different immune evasion proteins is variability in the human’s defense. The arsenal of immune evasion molecules makes it easier to fight the immune system of different individuals with variable antigens and genomic variation in their immune response. In addition, the expression of the various evasion molecules is tightly regulated via multiple regulation systems, and this results in differential expression during different growth stages. Interestingly, some of these relatively small molecules have multiple functions. The total number of secreted proteins in S. aureus is 100 to 200, and many of these proteins still have no known function (11). Thus, it is likely not only that new evasion molecules will be identified in the future, but also that new functions will be associated with already known proteins (11).
Despite the fact that S. aureus produces so many evasion molecules, the majority of colonized individuals will not suffer a staphylococcal infection. This tells us how complex and efficient our own immune system is. Invading microorganisms can be cleared within minutes by our innate immune system, possibly before some immune evasion proteins are produced. The pathogens are recognized by pattern recognition receptors and opsonized by components of the complement system and immunoglobulins. These immunoglobulins from the adaptive immune system are also able to neutralize bacterial components, such as superantigens, toxins, and other immune evasion proteins. The opsonized bacteria are efficiently ingested by neutrophils and killed by oxidative and nonoxidative proteins, which are safely stored inside their granules.
In conclusion, it seems that there is a delicate balance between the immune system and S. aureus in their ongoing battle to fight each other. However, S. aureus is gaining ground due to the rise of antibiotic resistance and community-acquired and highly virulent strains.
Evasion Proteins and Therapeutic Strategies
Over the past 2 decades we have gained in-depth knowledge about the existence, the importance, and the molecular mode of action of many immune evasion proteins. Now, what can we do with this knowledge? As illustrated below, one crucial goal is to find a treatment specifically against staphylococcal infections, but these immune evasion proteins can also be used to dampen the immune response in inflammatory diseases.
Combating S. aureus with (new) antibiotics is a challenge, since the bacterium has the extraordinary ability to develop resistance against antibiotics and the discovery of new antibiotics is declining (7). Vaccination is a strategy that could solve many of the problems that we encounter with antibiotic resistance. A few vaccine candidates have been tested in clinical trials, but none of them successfully passed phase III studies (217). For example, the vaccine candidates against the relatively small capsule of S. aureus, capsular polysaccharides types 5 and 8, have not passed clinical trials yet (218). This is possibly due to the presence of natural nonopsonic antibodies to another S. aureus cell surface polysaccharide, poly-N-acetylglucosamine, in human serum, and this interferes with the vaccine (219). Other vaccines targeting a single cell surface-associated antigen have failed clinical trials, for example, the iron surface determinant B (IsdB). Antibody against IsdB showed good tolerance in healthy volunteers, but in phase III studies patients administered with this vaccine were more susceptible to developing S. aureus infections and were five times more likely to die than unvaccinated patients with S. aureus infections (220). A recent overview of vaccine candidates and the status of their clinical trials was described, and this shows that although there is still ongoing research in this field, no functional vaccine will be available in the near future (217).
Another way to neutralize evasion molecules is by the generation of small inhibitory peptides or neutralizing antibodies via passive immunization, which is short-term immunity provided by antibodies obtained outside the body. For example, a CCR5 antagonist often used in HIV treatment, maraviroc, also protects T-cells and myeloid cells from LukED-mediated toxicity (221). Thus, these types of drugs could also be used as therapeutic alternatives against staphylococcal infections. Also, it might be a good approach to target one of the toxins produced by S. aureus. Currently, there are three antistaphylococcal monoclonal antibodies in clinical trials. Two of them target the secreted virulence factor alpha-toxin, which has recently been shown to protect against S. aureus-induced pneumonia (222). Unfortunately, so far, antibody-based therapy has not passed clinical trial phase II. Earlier research focused on single antigens displayed at the bacterial surface, but recently, this has moved toward agents targeting multiple S. aureus proteins, for example, immune evasion molecules and intracellular reservoirs of the bacterium (223). For example, the monoclonal antibody 6D4 interferes with the activity of SCIN, but it is likely that this will not be the only target in antistaphylococcal therapy since SCIN-deficient S. aureus strains still cause infections (224). Nevertheless, despite the lack of a functional monoclonal antibody/antibodies at this moment, antibody-based therapy is a promising therapeutic agent to use against staphylococcal infections in the future (225).
The fact that, so far, the development of an antistaphylococcal vaccine has failed can be attributed to several factors. Vaccines targeting single antigens without adjuvants were insufficient to give protection. This is likely due to the secretion of the manifold evasion molecules. Therefore, research focus has switched to developing a vaccine against a combination of proteins. Also, the lack of a highly reliable animal model is another factor and is partially caused by immune evasion molecules since many of the evasion proteins have high-affinity binding capacity to only human targets. These high-affinity protein-protein interactions between virulence factor and target cause a lack of affinity in targets in other, less related hosts. Because multiple immune evasion proteins are restricted to the human host, it leads to difficulties in studying S. aureus infections in vivo, such as in mice. Therefore, in vivo studies of SCIN, CHIPS, SAK, and PVL are difficult. Some isolated virulence factors are, however, not human specific and do allow studies in other species, for example, Ecb and Efb in a murine infection model (130). The genes located in the core variable genome are immobile and generally have a broad species specificity, whereas mobile genetic element-encoded immune evasion proteins (e.g., pathogenicity islands and prophages) are highly species specific (226). The third reason for the failure of previous vaccination attempts is the existence of immune evasion by itself, which leads to lower local immunity. Even if the vaccine raises sufficient opsonizing antibodies during this lowered local immunity, the induced antibodies cannot confer protection by the lack of an effector system (such as complement and neutrophils). A way to overcome this is to incorporate evasion molecules into the vaccine and raise neutralizing antibodies against these evasion molecules. Inhibiting the inhibitor and restoring local immunity could be a solution to solve vaccination issues in the future. Nevertheless, redundancy of immune evasion molecules and a good animal model remain obstacles to be overcome.
The evasion molecules can also be used as anti-inflammatory therapy in diseases where there is a deflecting immune activation. This is a growing area of research due to the increased prevalence of staphylococcal infections in Western countries. There are numerous inflammatory diseases, for example, allergy, asthma, autoimmune diseases, vasculitis, and inflammatory bowel disease. Many evasion molecules inhibit the central players in human inflammatory diseases, such as some GPCRs, complement components, and TLRSs, and are therefore interesting therapeutic targets (227–229). This approach has already been used in several studies; for example, SCIN-derived peptides were screened for complement inhibition through phage display to develop new complement-directed therapies (230). However, the evasion molecules are immunogenic by themselves. Thus, we need something else that is based on these evasion proteins, such as small peptides or derivatives which are no longer immunogenic. The (co)crystal structure is known from many evasion molecules, and this shows us where the exact binding interface at the amino acid level is between the staphylococcal protein and its binding partner of the immune system (226). This is very helpful in designing (structured) peptides.
Another way to use peptides derived from evasion molecules is in anticancer strategies. Some of these immune receptors promote cancer growth, such as FPR1 in brain tumors. Thus, the staphylococcal inhibitor of FPR1, CHIPS, is a potential anticancer drug. Recent research with mice expressing human FPR1 on astrocytoma cells treated with CHIPS showed increased survival of the mice and reduced tumor growth (231).
Immune Evasion Cluster 1/ɸSa3: a Mobile Element on a Quintuple Converting Bacteriophage
CHIPS, SCIN, and SAK (136) are small, secreted proteins that play a crucial role in the staphylococcal defense against the human innate immune system (complement and neutrophils) (46). The genes for SAK (sak), CHIPS (chp), and SCIN (scn), and also for the superantigen SEA (sea), are clustered on an innate immune evasion cluster (ɸSa3) in S. aureus, an 8-kb region at the 3′ end of β-hemolysin-converting bacteriophages. When this phage inserts into the β-hemolysin gene, it inactivates the gene itself, and the bacterium can no longer produce β-hemolysin. In addition, the bacterium now can produce four virulence/evasion factors, which have roles in different aspects of the pathogenetic strategy of the organism, as described above. Therefore, these phages are known as quintuple converting phages (226). When studying the occurrence of this mobile element in different S. aureus strains, it becomes immediately clear that it is strictly limited to human strains and is absent in, for example, bovine strains (226). Further, when studying the functional interaction between the four evasion molecules present on this element, it becomes clear that all four proteins act only on human targets, never on animal receptors or molecules. This is true for CHIPS in its interaction with C5aR and FPR, is true for SCIN in its interaction with the complement components C3b and Bb, and is true for SAK and even for the superantigen SEA. CHIPS does not work on animal neutrophils, and SEA does not work on animal T-cells. SCIN does not work in animal sera (116). From the molecular data from crystallography and nuclear magnetic resonance studies between evasion molecules and targets, we can now deduce the action on the precise amino acids in the host (78, 115). Taken together, the conclusions from epidemiological studies of various animals and humans, the functional studies conducted with sera and isolated cells versus recombinant bacterial proteins, and the structural data generated over the years are very clear: this mobile element and all of the components located on it are strictly human specific. It can transform a staphylococcus into a human pathogen. This also illustrates the importance of these virulence factors in the pathophysiology of infections in humans.
Concluding Remarks on Biological Warfare between Staphylococci and their Hosts
Our immune system can rapidly clear bacteria from our body after infection. However, infecting bacteria do not idly stand by when confronted by the host immune response. The following bacterial immune evasion strategies are often employed by pathogenic bacteria to resist our immune system.
Form a capsule.
Invade host cells and survive within them (intracellular lifestyle).
Change surface/change recognition molecules.
Secrete modulators/inhibitors/immune evasion molecules.
Hiding within an inert capsule is the hallmark of pneumococci. Mycobacteria are renowned for their capacity to hide within host cells. Staphylococci are the masters of surface modulation and immune evasion by secreted proteins. On the one hand, in the past 15 years, these phenomena have been studied extensively in S. aureus; on the other hand, in the next decades, it is likely to become clear how far this understanding can be extended to other bacteria. For any pathogenic bacterium, we know that there are still hundreds of nonannotated secreted proteins.
Staphylococcal immune evasion is very important for the bacterium, especially to fight the immune system during acute infection. Therefore, most evasion proteins are directed against this initial attack by neutrophils. This review gave an overview of the various evasion proteins and their function(s) during infection. Interestingly, the repertoire is still expanding with the recent discovery of novel proteins and new functions of existing evasion molecules. It is an important field of research, since functional therapeutics against staphylococcal infections are still lacking, while the economic burden increases. However, with the current speed of experiments, our knowledge is growing every year, and most likely it will only be a matter of time before we find the appropriate therapeutics for prophylaxis or treatment of this extremely clever bacterium.
ACKNOWLEDGMENTS
We thank Kobus Bosman and Seline Zwarthoff for useful discussions and critical review of the manuscript.
This work was supported by ZonMw grant 205200004 from the Netherlands Organization for Health Research and Development (to J.A.G.v.S.).
REFERENCES
- 1.Gorwitz RJ, Kruszon-Moran D, McAllister SK, McQuillan G, McDougal LK, Fosheim GE, Jensen BJ, Killgore G, Tenover FC, Kuehnert MJ. 2008. Changes in the prevalence of nasal colonization with Staphylococcus aureus in the United States, 2001-2004. J Infect Dis 197:1226–1234 10.1086/533494. [PubMed] [DOI] [PubMed] [Google Scholar]
- 2.Krismer B, Weidenmaier C, Zipperer A, Peschel A. 2017. The commensal lifestyle of Staphylococcus aureus and its interactions with the nasal microbiota. Nat Rev Microbiol 15:675–687 10.1038/nrmicro.2017.104. [PubMed] [DOI] [PubMed] [Google Scholar]
- 3.von Eiff C, Becker K, Machka K, Stammer H, Peters G. 2001. Nasal carriage as a source of Staphylococcus aureus bacteremia. N Engl J Med 344:11–16 10.1056/NEJM200101043440102. [PubMed] [DOI] [PubMed] [Google Scholar]
- 4.Lowy FD. 1998. Staphylococcus aureus infections. N Engl J Med 339:520–532 10.1056/NEJM199808203390806. [PubMed] [DOI] [PubMed] [Google Scholar]
- 5.Barrett FF, McGehee RF Jr, Finland M. 1968. Methicillin-resistant Staphylococcus aureus at Boston City Hospital. Bacteriologic and epidemiologic observations. N Engl J Med 279:441–448 10.1056/NEJM196808292790901. [PubMed] [DOI] [PubMed] [Google Scholar]
- 6.Panlilio AL, Culver DH, Gaynes RP, Banerjee S, Henderson TS, Tolson JS, Martone WJ, System NNIS. 1992. Methicillin-resistant Staphylococcus aureus in U.S. hospitals, 1975-1991. Infect Control Hosp Epidemiol 13:582–586 10.2307/30148460. [PubMed] [DOI] [PubMed] [Google Scholar]
- 7.DeLeo FR, Otto M, Kreiswirth BN, Chambers HF. 2010. Community-associated meticillin-resistant Staphylococcus aureus. Lancet 375:1557–1568 10.1016/S0140-6736(09)61999-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeLeo FR, Diep BA, Otto M. 2009. Host defense and pathogenesis in Staphylococcus aureus infections. Infect Dis Clin North Am 23:17–34 10.1016/j.idc.2008.10.003. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diekema DJ, Pfaller MA, Schmitz FJ, Smayevsky J, Bell J, Jones RN, Beach M, SENTRY Partcipants Group. 2001. Survey of infections due to Staphylococcus species: frequency of occurrence and antimicrobial susceptibility of isolates collected in the United States, Canada, Latin America, Europe, and the Western Pacific region for the SENTRY Antimicrobial Surveillance Program, 1997-1999. Clin Infect Dis 32(Suppl 2):S114–S132 10.1086/320184. [PubMed] [DOI] [PubMed] [Google Scholar]
- 10.Goldmann O, Medina E. 2017. Staphylococcus aureus strategies to evade the host acquired immune response. Int J Med Microbiol 308:625–630. [PubMed] [DOI] [PubMed] [Google Scholar]
- 11.Kusch H, Engelmann S. 2014. Secrets of the secretome in Staphylococcus aureus. Int J Med Microbiol 304:133–141 10.1016/j.ijmm.2013.11.005. [PubMed] [DOI] [PubMed] [Google Scholar]
- 12.Faurschou M, Borregaard N. 2003. Neutrophil granules and secretory vesicles in inflammation. Microbes Infect 5:1317–1327 10.1016/j.micinf.2003.09.008. [PubMed] [DOI] [PubMed] [Google Scholar]
- 13.Rigby KM, DeLeo FR. 2012. Neutrophils in innate host defense against Staphylococcus aureus infections. Semin Immunopathol 34:237–259 10.1007/s00281-011-0295-3. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cerquetti MC, Sordelli DO, Ortegon RA, Bellanti JA. 1983. Impaired lung defenses against Staphylococcus aureus in mice with hereditary deficiency of the fifth component of complement. Infect Immun 41:1071–1076. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.von Köckritz-Blickwede M, Konrad S, Foster S, Gessner JE, Medina E. 2010. Protective role of complement C5a in an experimental model of Staphylococcus aureus bacteremia. J Innate Immun 2:87–92 10.1159/000247157. [PubMed] [DOI] [PubMed] [Google Scholar]
- 16.Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. 2012. Neutrophil function: from mechanisms to disease. Annu Rev Immunol 30:459–489 10.1146/annurev-immunol-020711-074942. [PubMed] [DOI] [PubMed] [Google Scholar]
- 17.Winkelstein JA, Marino MC, Johnston RB Jr, Boyle J, Curnutte J, Gallin JI, Malech HL, Holland SM, Ochs H, Quie P, Buckley RH, Foster CB, Chanock SJ, Dickler H. 2000. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore) 79:155–169 10.1097/00005792-200005000-00003. [DOI] [PubMed] [Google Scholar]
- 18.Cook N. 1998. Methicillin-resistant Staphylococcus aureus versus the burn patient. Burns 24:91–98 10.1016/S0305-4179(97)00114-9. [DOI] [PubMed] [Google Scholar]
- 19.Church D, Elsayed S, Reid O, Winston B, Lindsay R. 2006. Burn wound infections. Clin Microbiol Rev 19:403–434 10.1128/CMR.19.2.403-434.2006. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verkaik NJ, de Vogel CP, Boelens HA, Grumann D, Hoogenboezem T, Vink C, Hooijkaas H, Foster TJ, Verbrugh HA, van Belkum A, van Wamel WJ. 2009. Anti-staphylococcal humoral immune response in persistent nasal carriers and noncarriers of Staphylococcus aureus. J Infect Dis 199:625–632 10.1086/596743. [PubMed] [DOI] [PubMed] [Google Scholar]
- 21.Vestweber D. 2007. Adhesion and signaling molecules controlling the transmigration of leukocytes through endothelium. Immunol Rev 218:178–196 10.1111/j.1600-065X.2007.00533.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 22.Kolaczkowska E, Kubes P. 2013. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 13:159–175 10.1038/nri3399. [PubMed] [DOI] [PubMed] [Google Scholar]
- 23.Moore KL, Patel KD, Bruehl RE, Li F, Johnson DA, Lichenstein HS, Cummings RD, Bainton DF, McEver RP. 1995. P-selectin glycoprotein ligand-1 mediates rolling of human neutrophils on P-selectin. J Cell Biol 128:661–671 10.1083/jcb.128.4.661. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P. 2006. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med 203:2569–2575 10.1084/jem.20060925. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith CW, Marlin SD, Rothlein R, Toman C, Anderson DC. 1989. Cooperative interactions of LFA-1 and Mac-1 with intercellular adhesion molecule-1 in facilitating adherence and transendothelial migration of human neutrophils in vitro. J Clin Invest 83:2008–2017 10.1172/JCI114111. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diamond MS, Staunton DE, de Fougerolles AR, Stacker SA, Garcia-Aguilar J, Hibbs ML, Springer TA. 1990. ICAM-1 (CD54): a counter-receptor for Mac-1 (CD11b/CD18). J Cell Biol 111:3129–3139 10.1083/jcb.111.6.3129. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim M, Carman CV, Springer TA. 2003. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science 301:1720–1725 10.1126/science.1084174. [PubMed] [DOI] [PubMed] [Google Scholar]
- 28.Jones DH, Anderson DC, Burr BL, Rudloff HE, Smith CW, Krater SS, Schmalstieg FC. 1988. Quantitation of intracellular Mac-1 (CD11b/CD18) pools in human neutrophils. J Leukoc Biol 44:535–544 10.1002/jlb.44.6.535. [PubMed] [DOI] [PubMed] [Google Scholar]
- 29.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. 2007. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 7:678–689 10.1038/nri2156. [PubMed] [DOI] [PubMed] [Google Scholar]
- 30.Bestebroer J, Poppelier MJJG, Ulfman LH, Lenting PJ, Denis CV, van Kessel KPM, van Strijp JA, de Haas CJ. 2007. Staphylococcal superantigen-like 5 binds PSGL-1 and inhibits P-selectin-mediated neutrophil rolling. Blood 109:2936–2943. [PubMed] [DOI] [PubMed] [Google Scholar]
- 31.Walenkamp AME, Bestebroer J, Boer IGJ, Kruizinga R, Verheul HM, van Strijp JA, de Haas CJ. 2010. Staphylococcal SSL5 binding to human leukemia cells inhibits cell adhesion to endothelial cells and platelets. Cell Oncol 32:1–10. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Somers WS, Shaw GD, Camphausen RT. 2001. Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P- and E-selectin bound to SLeX and PSGL-1 (Cell 103:3 (467-479)). Cell 105:971 10.1016/S0092-8674(01)00399-3. [DOI] [PubMed] [Google Scholar]
- 33.Chung MC, Wines BD, Baker H, Langley RJ, Baker EN, Fraser JD. 2007. The crystal structure of staphylococcal superantigen-like protein 11 in complex with sialyl Lewis X reveals the mechanism for cell binding and immune inhibition. Mol Microbiol 66:1342–1355 10.1111/j.1365-2958.2007.05989.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 34.Fevre C, Bestebroer J, Mebius MM, de Haas CJC, van Strijp JA, Fitzgerald JR, Haas PJ. 2014. Staphylococcus aureus proteins SSL6 and SElX interact with neutrophil receptors as identified using secretome phage display. Cell Microbiol 16:1646–1665 10.1111/cmi.12313. [PubMed] [DOI] [PubMed] [Google Scholar]
- 35.Wilson GJ, Seo KS, Cartwright RA, Connelley T, Chuang-Smith ON, Merriman JA, Guinane CM, Park JY, Bohach GA, Schlievert PM, Morrison WI, Fitzgerald JR. 2011. A novel core genome-encoded superantigen contributes to lethality of community-associated MRSA necrotizing pneumonia. PLoS Pathog 7:e1002271 10.1371/journal.ppat.1002271. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baker HM, Basu I, Chung MC, Caradoc-Davies T, Fraser JD, Baker EN. 2007. Crystal structures of the staphylococcal toxin SSL5 in complex with sialyl Lewis X reveal a conserved binding site that shares common features with viral and bacterial sialic acid binding proteins. J Mol Biol 374:1298–1308 10.1016/j.jmb.2007.09.091. [PubMed] [DOI] [PubMed] [Google Scholar]
- 37.Tuffs SW, James DBA, Bestebroer J, Richards AC, Goncheva MI, O’Shea M, Wee BA, Seo KS, Schlievert PM, Lengeling A, van Strijp JA, Torres VJ, Fitzgerald JR. 2017. The Staphylococcus aureus superantigen SElX is a bifunctional toxin that inhibits neutrophil function. PLoS Pathog 13:e1006461 10.1371/journal.ppat.1006461. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Langley RJ, Ting YT, Clow F, Young PG, Radcliff FJ, Choi JM, Sequeira RP, Holtfreter S, Baker H, Fraser JD. 2017. Staphylococcal enterotoxin-like X (SElX) is a unique superantigen with functional features of two major families of staphylococcal virulence factors. PLoS Pathog 13:e1006549 10.1371/journal.ppat.1006549. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chavakis T, Hussain M, Kanse SM, Peters G, Bretzel RG, Flock J-I, Herrmann M, Preissner KT. 2002. Staphylococcus aureus extracellular adherence protein serves as anti-inflammatory factor by inhibiting the recruitment of host leukocytes. Nat Med 8:687–693 10.1038/nm728. [PubMed] [DOI] [PubMed] [Google Scholar]
- 40.Ellis TN, Beaman BL. 2004. Interferon-γ activation of polymorphonuclear neutrophil function. Immunology 112:2–12 10.1111/j.1365-2567.2004.01849.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swain SD, Rohn TT, Quinn MT. 2002. Neutrophil priming in host defense: role of oxidants as priming agents. Antioxid Redox Signal 4:69–83 10.1089/152308602753625870. [PubMed] [DOI] [PubMed] [Google Scholar]
- 42.Skjeflo EW, Christiansen D, Espevik T, Nielsen EW, Mollnes TE. 2014. Combined inhibition of complement and CD14 efficiently attenuated the inflammatory response induced by Staphylococcus aureus in a human whole blood model. J Immunol 192:2857–2864 10.4049/jimmunol.1300755. [PubMed] [DOI] [PubMed] [Google Scholar]
- 43.Mitchell GB, Albright BN, Caswell JL. 2003. Effect of interleukin-8 and granulocyte colony-stimulating factor on priming and activation of bovine neutrophils. Infect Immun 71:1643–1649 10.1128/IAI.71.4.1643-1649.2003. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rainard P, Riollet C, Poutrel B, Paape MJ. 2000. Phagocytosis and killing of Staphylococcus aureus by bovine neutrophils after priming by tumor necrosis factor-alpha and the des-arginine derivative of C5a. Am J Vet Res 61:951–959 10.2460/ajvr.2000.61.951. [PubMed] [DOI] [PubMed] [Google Scholar]
- 45.Edwards SW, Say JE, Hughes V. 1988. Gamma interferon enhances the killing of Staphylococcus aureus by human neutrophils. J Gen Microbiol 134:37–42. [DOI] [PubMed] [Google Scholar]
- 46.Bestebroer J, De Haas CJC, Van Strijp JA. 2010. How microorganisms avoid phagocyte attraction. FEMS Microbiol Rev 34:395–414 10.1111/j.1574-6976.2009.00202.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 47.Chalovich JM, Eisenberg E. 2005. G protein-coupled receptor rhodopsin. Biophys Chem 257:2432–2437. [Google Scholar]
- 48.Allen SJ, Crown SE, Handel TM. 2007. Chemokine: receptor structure, interactions, and antagonism. Annu Rev Immunol 25:787–820 10.1146/annurev.immunol.24.021605.090529. [PubMed] [DOI] [PubMed] [Google Scholar]
- 49.Tecchio C, Cassatella MA. 2016. Neutrophil-derived chemokines on the road to immunity. Semin Immunol 28:119–128 10.1016/j.smim.2016.04.003. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schiffmann E, Corcoran BA, Wahl SM. 1975. N-formylmethionyl peptides as chemoattractants for leucocytes. Proc Natl Acad Sci U S A 72:1059–1062 10.1073/pnas.72.3.1059. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dahlgren C, Gabl M, Holdfeldt A, Winther M, Forsman H. 2016. Basic characteristics of the neutrophil receptors that recognize formylated peptides, a danger-associated molecular pattern generated by bacteria and mitochondria. Biochem Pharmacol 114:22–39 10.1016/j.bcp.2016.04.014. [PubMed] [DOI] [PubMed] [Google Scholar]
- 52.Le Y, Oppenheim JJ, Wang JM. 2001. Pleiotropic roles of formyl peptide receptors. Cytokine Growth Factor Rev 12:91–105 10.1016/S1359-6101(01)00003-X. [DOI] [PubMed] [Google Scholar]
- 53.Kretschmer D, Gleske A-K, Rautenberg M, Wang R, Köberle M, Bohn E, Schöneberg T, Rabiet M-J, Boulay F, Klebanoff SJ, van Kessel KA, van Strijp JA, Otto M, Peschel A. 2010. Human formyl peptide receptor 2 senses highly pathogenic Staphylococcus aureus. Cell Host Microbe 7:463–473 10.1016/j.chom.2010.05.012. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gasque P. 2004. Complement: a unique innate immune sensor for danger signals. Mol Immunol 41:1089–1098 10.1016/j.molimm.2004.06.011. [PubMed] [DOI] [PubMed] [Google Scholar]
- 55.Kawai T, Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11:373–384 10.1038/ni.1863. [PubMed] [DOI] [PubMed] [Google Scholar]
- 56.O’Neill LA, Bowie AG. 2007. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol 7:353–364 10.1038/nri2079. [PubMed] [DOI] [PubMed] [Google Scholar]
- 57.Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik SG, Lee H, Lee JO. 2007. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 130:1071–1082 10.1016/j.cell.2007.09.008. [PubMed] [DOI] [PubMed] [Google Scholar]
- 58.Rodríguez D, Morrison CJ, Overall CM. 2010. Matrix metalloproteinases: what do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim Biophys Acta 1803:39–54 10.1016/j.bbamcr.2009.09.015. [PubMed] [DOI] [PubMed] [Google Scholar]
- 59.Guerra FE, Borgogna TR, Patel DM, Sward EW, Voyich JM. 2017. Epic immune battles of history: neutrophils vs. Staphylococcus aureus. Front Cell Infect Microbiol 7:286 10.3389/fcimb.2017.00286. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bestebroer J, van Kessel KPM, Azouagh H, Walenkamp AM, Boer IGJ, Romijn RA, van Strijp JA, de Haas CJC. 2009. Staphylococcal SSL5 inhibits leukocyte activation by chemokines and anaphylatoxins. Blood 113:328–337 10.1182/blood-2008-04-153882. [PubMed] [DOI] [PubMed] [Google Scholar]
- 61.Itoh S, Hamada E, Kamoshida G, Takeshita K, Oku T, Tsuji T. 2010. Staphylococcal superantigen-like protein 5 inhibits matrix metalloproteinase 9 from human neutrophils. Infect Immun 78:3298–3305 10.1128/IAI.00178-10. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Koymans KJ, Bisschop A, Vughs MM, van Kessel KPM, de Haas CJC, van Strijp JA. 2016. Staphylococcal superantigen-like protein 1 and 5 (SSL1 & SSL5) limit neutrophil chemotaxis and migration through MMP-inhibition. Int J Mol Sci 17:1–16 10.3390/ijms17071072. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.de Haas CJC, Weeterings C, Vughs MM, de Groot PG, Van Strijp JA, Lisman T. 2009. Staphylococcal superantigen-like 5 activates platelets and supports platelet adhesion under flow conditions, which involves glycoprotein Ibalpha and α IIb β 3. J Thromb Haemost 7:1867–1874 10.1111/j.1538-7836.2009.03564.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 64.Hu H, Armstrong PCJ, Khalil E, Chen YC, Straub A, Li M, Soosairajah J, Hagemeyer CE, Bassler N, Huang D, Ahrens I, Krippner G, Gardiner E, Peter K. 2011. GPVI and GPIBα mediate staphylococcal superantigen-like protein 5 (SSL5) induced platelet activation and direct toward glycans as potential inhibitors. PLoS One 6:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Walenkamp AME, Boer IGJ, Bestebroer J, Rozeveld D, Timmer-Bosscha H, Hemrika W, van Strijp JA, de Haas CJC. 2009. Staphylococcal superantigen-like 10 inhibits CXCL12-induced human tumor cell migration. Neoplasia 11:333–344 10.1593/neo.81508. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bardoel BW, Vos R, Bouman T, Aerts PC, Bestebroer J, Huizinga EG, Brondijk THC, van Strijp JA, de Haas CJ. 2012. Evasion of Toll-like receptor 2 activation by staphylococcal superantigen-like protein 3. J Mol Med (Berl) 90:1109–1120 10.1007/s00109-012-0926-8. [PubMed] [DOI] [PubMed] [Google Scholar]
- 67.Yokoyama R, Itoh S, Kamoshida G, Takii T, Fujii S, Tsuji T, Onozaki K. 2012. Staphylococcal superantigen-like protein 3 binds to the Toll-like receptor 2 extracellular domain and inhibits cytokine production induced by Staphylococcus aureus, cell wall component, or lipopeptides in murine macrophages. Infect Immun 80:2816–2825 10.1128/IAI.00399-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Koymans KJ, Feitsma LJ, Brondijk THC, Aerts PC, Lukkien E, Lössl P, van Kessel KPM, de Haas CJC, van Strijp JA, Huizinga EG. 2015. Structural basis for inhibition of TLR2 by staphylococcal superantigen-like protein 3 (SSL3). Proc Natl Acad Sci U S A 112:11018–11023 10.1073/pnas.1502026112. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hermans SJ, Baker HM, Sequeira RP, Langley RJ, Baker EN, Fraser JD. 2012. Structural and functional properties of staphylococcal superantigen-like protein 4. Infect Immun 80:4004–4013 10.1128/IAI.00764-12. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Koymans KJ, Goldmann O, Karlsson CAQ, Sital W, Thänert R, Bisschop A, Vrieling M, Malmström J, van Kessel KPM, de Haas CJC, van Strijp JAG, Medina E. 2017. The TLR2 antagonist staphylococcal superantigen-like protein 3 acts as a virulence factor to promote bacterial pathogenicity in vivo. J Innate Immun 9:561–573 10.1159/000479100. [PubMed] [DOI] [PubMed] [Google Scholar]
- 71.Laarman AJ, Mijnheer G, Mootz JM, van Rooijen WJM, Ruyken M, Malone CL, Heezius EC, Ward R, Milligan G, van Strijp JA, de Haas CJC, Horswill AR, van Kessel KPM, Rooijakkers SHM. 2012. Staphylococcus aureus staphopain A inhibits CXCR2-dependent neutrophil activation and chemotaxis. EMBO J 31:3607–3619 10.1038/emboj.2012.212. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nickerson N, Ip J, Passos DT, McGavin MJ. 2010. Comparison of staphopain A (ScpA) and B (SspB) precursor activation mechanisms reveals unique secretion kinetics of proSspB (staphopain B), and a different interaction with its cognate Staphostatin, SspC. Mol Microbiol 75:161–177 10.1111/j.1365-2958.2009.06974.x. [DOI] [PubMed] [Google Scholar]
- 73.Veldkamp KE, Heezius HCJM, Verhoef J, van Strijp JAG, van Kessel KPM. 2000. Modulation of neutrophil chemokine receptors by Staphylococcus aureus supernate. Infect Immun 68:5908–5913 10.1128/IAI.68.10.5908-5913.2000. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.de Haas CJC, Veldkamp KE, Peschel A, Weerkamp F, Van Wamel WJ, Heezius EC, Poppelier MJ, Van Kessel KP, van Strijp JA. 2004. Chemotaxis inhibitory protein of Staphylococcus aureus, a bacterial antiinflammatory agent. J Exp Med 199:687–695 10.1084/jem.20031636. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Postma B, Poppelier MJ, van Galen JC, Prossnitz ER, van Strijp JA, de Haas CJC, van Kessel KPM. 2004. Chemotaxis inhibitory protein of Staphylococcus aureus binds specifically to the C5a and formylated peptide receptor. J Immunol 172:6994–7001 10.4049/jimmunol.172.11.6994. [PubMed] [DOI] [PubMed] [Google Scholar]
- 76.Haas P-J, de Haas CJC, Kleibeuker W, Poppelier MJJG, van Kessel KPM, Kruijtzer JA, Liskamp RM, van Strijp JA. 2004. N-terminal residues of the chemotaxis inhibitory protein of Staphylococcus aureus are essential for blocking formylated peptide receptor but not C5a receptor. J Immunol 173:5704–5711 10.4049/jimmunol.173.9.5704. [PubMed] [DOI] [PubMed] [Google Scholar]
- 77.Postma B, Kleibeuker W, Poppelier MJJG, Boonstra M, Van Kessel KPM, Van Strijp JA, de Haas CJC. 2005. Residues 10-18 within the C5a receptor N terminus compose a binding domain for chemotaxis inhibitory protein of Staphylococcus aureus. J Biol Chem 280:2020–2027 10.1074/jbc.M412230200. [PubMed] [DOI] [PubMed] [Google Scholar]
- 78.Haas PJ, de Haas CJC, Poppelier MJJC, van Kessel KPM, van Strijp JA, Dijkstra K, Scheek RM, Fan H, Kruijtzer JA, Liskamp RMJ, Kemmink J. 2005. The structure of the C5a receptor-blocking domain of chemotaxis inhibitory protein of Staphylococcus aureus is related to a group of immune evasive molecules. J Mol Biol 353:859–872 10.1016/j.jmb.2005.09.014. [PubMed] [DOI] [PubMed] [Google Scholar]
- 79.Prat C, Bestebroer J, de Haas CJC, van Strijp JA, van Kessel KPM. 2006. A new staphylococcal anti-inflammatory protein that antagonizes the formyl peptide receptor-like 1. J Immunol 177:8017–8026 10.4049/jimmunol.177.11.8017. [PubMed] [DOI] [PubMed] [Google Scholar]
- 80.Prat C, Haas P-J, Bestebroer J, de Haas CJC, van Strijp JA, van Kessel KP. 2009. A homolog of formyl peptide receptor-like 1 (FPRL1) inhibitor from Staphylococcus aureus (FPRL1 inhibitory protein) that inhibits FPRL1 and FPR. J Immunol 183:6569–6578 10.4049/jimmunol.0801523. [PubMed] [DOI] [PubMed] [Google Scholar]
- 81.Stemerding AM, Köhl J, Pandey MK, Kuipers A, Leusen JH, Boross P, Nederend M, Vidarsson G, Weersink AYL, van de Winkel JGJ, van Kessel KPM, van Strijp JA. 2013. Staphylococcus aureus formyl peptide receptor-like 1 inhibitor (FLIPr) and its homologue FLIPr-like are potent FcγR antagonists that inhibit IgG-mediated effector functions. J Immunol 191:353–362 10.4049/jimmunol.1203243. [PubMed] [DOI] [PubMed] [Google Scholar]
- 82.Ferreira VP, Pangburn MK, Cortés C. 2010. Complement control protein factor H: the good, the bad, and the inadequate. Mol Immunol 47:2187–2197 10.1016/j.molimm.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cunnion KM, Hair PS, Buescher ES. 2004. Cleavage of complement C3b to iC3b on the surface of Staphylococcus aureus is mediated by serum complement factor I. Infect Immun 72:2858–2863 10.1128/IAI.72.5.2858-2863.2004. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Müller-Eberhard HJ. 1986. The membrane attack complex of complement. Annu Rev Immunol 4:503–528 10.1146/annurev.iy.04.040186.002443. [PubMed] [DOI] [PubMed] [Google Scholar]
- 85.Wellek B, Hahn H, Opferkuch W. 1976. Opsonizing activities of IgG, IgM antibodies and the C3b inactivator-cleaved third component of complement in macrophage phagocytosis. Agents Actions 6:260–262 10.1007/BF01972219. [PubMed] [DOI] [PubMed] [Google Scholar]
- 86.Mantovani B. 1975. Different roles of IgG and complement receptors in phagocytosis by polymorphonuclear leukocytes. J Immunol 115:15–17. [PubMed] [PubMed] [Google Scholar]
- 87.Radaev S, Sun P. 2013. Structural recognition of immunoglobulins by Fcγ receptors, p 131–144. In Ackerman ME, Nimmerjahn F (ed), Antibody Fc: Linking Adaptive and Innate Immunity. Academic Press, San Diego, CA. [Google Scholar]
- 88.Stuart LM, Ezekowitz RAB. 2005. Phagocytosis: elegant complexity. Immunity 22:539–550 10.1016/j.immuni.2005.05.002. [PubMed] [DOI] [PubMed] [Google Scholar]
- 89.Rosales C. 2017. Fcγ receptor heterogeneity in leukocyte functional responses. Front Immunol 8:280 10.3389/fimmu.2017.00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.McGreal E, Gasque P. 2002. Structure-function studies of the receptors for complement C1q. Biochem Soc Trans 30:1010–1014 10.1042/bst0301010. [PubMed] [DOI] [PubMed] [Google Scholar]
- 91.Todd RF III. 1996. The continuing saga of complement receptor type 3 (CR3). J Clin Invest 98:1–2 10.1172/JCI118752. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.O’Riordan K, Lee JC, Riordan KO, Lee JC. 2004. Staphylococcus aureus capsular polysaccharides. Clin Microbiol Rev 17:218–234 10.1128/CMR.17.1.218-234.2004. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cunnion KM, Lee JC, Frank MM. 2001. Capsule production and growth phase influence binding of complement to Staphylococcus aureus. Infect Immun 69:6796–6803 10.1128/IAI.69.11.6796-6803.2001. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rajagopal M, Walker S. 2015. Envelope structures of Gram-positive bacteria. Curr Top Microbiol Immunol 404:1–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schneewind O, Model P, Fischetti VA. 1992. Sorting of protein A to the staphylococcal cell wall. Cell 70:267–281 10.1016/0092-8674(92)90101-H. [DOI] [PubMed] [Google Scholar]
- 96.Becker S, Frankel MB, Schneewind O, Missiakas D. 2014. Release of protein A from the cell wall of Staphylococcus aureus. Proc Natl Acad Sci U S A 111:1574–1579 10.1073/pnas.1317181111. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Forsgren A, Sjöquist J. 1966. “Protein A” from S. aureus. I. Pseudo-immune reaction with human gamma-globulin. J Immunol 97:822–827. [PubMed] [PubMed] [Google Scholar]
- 98.Falugi F, Kim HK, Missiakas DM, Schneewind O. 2013. Role of protein A in the evasion of host adaptive immune responses by Staphylococcus aureus. MBio 4:e00575-13 10.1128/mBio.00575-13. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Goodyear CS, Silverman GJ. 2003. Death by a B cell superantigen: in vivo VH-targeted apoptotic supraclonal B cell deletion by a staphylococcal toxin. J Exp Med 197:1125–1139 10.1084/jem.20020552. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pauli NT, Kim HK, Falugi F, Huang M, Dulac J, Henry Dunand C, Zheng N-Y, Kaur K, Andrews SF, Huang Y, DeDent A, Frank KM, Charnot-Katsikas A, Schneewind O, Wilson PC. 2014. Staphylococcus aureus infection induces protein A-mediated immune evasion in humans. J Exp Med 211:2331–2339 10.1084/jem.20141404. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kim HK, Falugi F, Thomer L, Missiakas DM, Schneewind O. 2015. Protein A suppresses immune responses during Staphylococcus aureus bloodstream infection in guinea pigs. MBio 6:e02369-14 10.1128/mBio.02369-14. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhang L, Jacobsson K, Vasi J, Lindberg M, Frykberg L. 1998. A second IgG-binding protein in Staphylococcus aureus. Microbiology 144:985–991 10.1099/00221287-144-4-985. [PubMed] [DOI] [PubMed] [Google Scholar]
- 103.Zhang L, Jacobsson K, Ström K, Lindberg M, Frykberg L. 1999. Staphylococcus aureus expresses a cell surface protein that binds both IgG and glycoprotein I. Microbiology 145:177–183 10.1099/13500872-145-1-177. [PubMed] [DOI] [PubMed] [Google Scholar]
- 104.Ebner P, Prax M, Nega M, Koch I, Dube L, Yu W, Rinker J, Popella P, Flötenmeyer M, Götz F. 2015. Excretion of cytoplasmic proteins (ECP) in Staphylococcus aureus. Mol Microbiol 97:775–789 10.1111/mmi.13065. [PubMed] [DOI] [PubMed] [Google Scholar]
- 105.Atkins KL, Burman JD, Chamberlain ES, Cooper JE, Poutrel B, Bagby S, Jenkins AT, Feil EJ, van den Elsen JM. 2008. S. aureus IgG-binding proteins SpA and Sbi: host specificity and mechanisms of immune complex formation. Mol Immunol 45:1600–1611 10.1016/j.molimm.2007.10.021. [PubMed] [DOI] [PubMed] [Google Scholar]
- 106.Burman JD, Leung E, Atkins KL, O’Seaghdha MN, Lango L, Bernadó P, Bagby S, Svergun DI, Foster TJ, Isenman DE, van den Elsen JMH. 2008. Interaction of human complement with Sbi, a staphylococcal immunoglobulin-binding protein: indications of a novel mechanism of complement evasion by Staphylococcus aureus. J Biol Chem 283:17579–17593 10.1074/jbc.M800265200. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Haupt K, Reuter M, van den Elsen J, Burman J, Hälbich S, Richter J, Skerka C, Zipfel PF. 2008. The Staphylococcus aureus protein Sbi acts as a complement inhibitor and forms a tripartite complex with host complement factor H and C3b. PLoS Pathog 4:e1000250 10.1371/journal.ppat.1000250. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Itoh S, Hamada E, Kamoshida G, Yokoyama R, Takii T, Onozaki K, Tsuji T. 2010. Staphylococcal superantigen-like protein 10 (SSL10) binds to human immunoglobulin G (IgG) and inhibits complement activation via the classical pathway. Mol Immunol 47:932–938 10.1016/j.molimm.2009.09.027. [PubMed] [DOI] [PubMed] [Google Scholar]
- 109.Patel D, Wines BD, Langley RJ, Fraser JD. 2010. Specificity of staphylococcal superantigen-like protein 10 toward the human IgG1 Fc domain. J Immunol 184:6283–6292 10.4049/jimmunol.0903311. [PubMed] [DOI] [PubMed] [Google Scholar]
- 110.Itoh S, Yokoyama R, Kamoshida G, Fujiwara T, Okada H, Takii T, Tsuji T, Fujii S, Hashizume H, Onozaki K. 2013. Staphylococcal superantigen-like protein 10 (SSL10) inhibits blood coagulation by binding to prothrombin and factor Xa via their γ-carboxyglutamic acid (Gla) domain. J Biol Chem 288:21569–21580 10.1074/jbc.M113.451419. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Itoh S, Yokoyama R, Murase C, Takii T, Tsuji T, Onozaki K. 2012. Staphylococcal superantigen-like protein 10 binds to phosphatidylserine and apoptotic cells. Microbiol Immunol 56:363–371 10.1111/j.1348-0421.2012.00452.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 112.Prokesová L, Potuzníková B, Potempa J, Zikán J, Radl J, Hachová L, Baran K, Porwit-Bobr Z, John C. 1992. Cleavage of human immunoglobulins by serine proteinase from Staphylococcus aureus. Immunol Lett 31:259–265 10.1016/0165-2478(92)90124-7. [DOI] [PubMed] [Google Scholar]
- 113.Laarman AJ, Ruyken M, Malone CL, van Strijp JA, Horswill AR, Rooijakkers SHM. 2011. Staphylococcus aureus metalloprotease aureolysin cleaves complement C3 to mediate immune evasion. J Immunol 186:6445–6453 10.4049/jimmunol.1002948. [PubMed] [DOI] [PubMed] [Google Scholar]
- 114.Rooijakkers SHM, Ruyken M, Roos A, Daha MR, Presanis JS, Sim RB, van Wamel WJB, van Kessel KPM, van Strijp JAG. 2005. Immune evasion by a staphylococcal complement inhibitor that acts on C3 convertases. Nat Immunol 6:920–927 10.1038/ni1235. [PubMed] [DOI] [PubMed] [Google Scholar]
- 115.Rooijakkers SHM, Milder FJ, Bardoel BW, Ruyken M, van Strijp JA, Gros P. 2007. Staphylococcal complement inhibitor: structure and active sites. J Immunol 179:2989–2998 10.4049/jimmunol.179.5.2989. [PubMed] [DOI] [PubMed] [Google Scholar]
- 116.Rooijakkers SHM, Wu J, Ruyken M, van Domselaar R, Planken KL, Tzekou A, Ricklin D, Lambris JD, Janssen BJC, van Strijp JA, Gros P. 2009. Structural and functional implications of the alternative complement pathway C3 convertase stabilized by a staphylococcal inhibitor. Nat Immunol 10:721–727 10.1038/ni.1756. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Garcia BL, Ramyar KX, Tzekou A, Ricklin D, McWhorter WJ, Lambris JD, Geisbrecht BV. 2010. Molecular basis for complement recognition and inhibition determined by crystallographic studies of the staphylococcal complement inhibitor (SCIN) bound to C3c and C3b. J Mol Biol 402:17–29 10.1016/j.jmb.2010.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jongerius I, Köhl J, Pandey MK, Ruyken M, van Kessel KPM, van Strijp JA, Rooijakkers SHM. 2007. Staphylococcal complement evasion by various convertase-blocking molecules. J Exp Med 204:2461–2471 10.1084/jem.20070818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bodén MK, Flock JI. 1994. Cloning and characterization of a gene for a 19 kDa fibrinogen-binding protein from Staphylococcus aureus. Mol Microbiol 12:599–606 10.1111/j.1365-2958.1994.tb01046.x. [DOI] [PubMed] [Google Scholar]
- 120.Palma M, Shannon O, Quezada HC, Berg A, Flock JI. 2001. Extracellular fibrinogen-binding protein, Efb, from Staphylococcus aureus blocks platelet aggregation due to its binding to the α-chain. J Biol Chem 276:31691–31697 10.1074/jbc.M104554200. [DOI] [PubMed] [Google Scholar]
- 121.Heilmann C, Herrmann M, Kehrel BE, Peters G. 2002. Platelet-binding domains in 2 fibrinogen-binding proteins of Staphylococcus aureus identified by phage display. J Infect Dis 186:32–39 10.1086/341081. [DOI] [PubMed] [Google Scholar]
- 122.Palma M, Nozohoor S, Schennings T, Heimdahl A, Flock JI. 1996. Lack of the extracellular 19-kilodalton fibrinogen-binding protein from Staphylococcus aureus decreases virulence in experimental wound infection. Infect Immun 64:5284–5289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lee LYL, Höök M, Haviland D, Wetsel RA, Yonter EO, Syribeys P, Vernachio J, Brown EL. 2004. Inhibition of complement activation by a secreted Staphylococcus aureus protein. J Infect Dis 190:571–579 10.1086/422259. [DOI] [PubMed] [Google Scholar]
- 124.Hammel M, Sfyroera G, Ricklin D, Magotti P, Lambris JD, Geisbrecht BV. 2007. A structural basis for complement inhibition by Staphylococcus aureus. Nat Immunol 8:430–437 10.1038/ni1450. [DOI] [PubMed] [Google Scholar]
- 125.Ko YP, Kuipers A, Freitag CM, Jongerius I, Medina E, van Rooijen WJ, Spaan AN, van Kessel KPM, Höök M, Rooijakkers SHM. 2013. Phagocytosis escape by a Staphylococcus aureus protein that connects complement and coagulation proteins at the bacterial surface. PLoS Pathog 9:e1003816 10.1371/journal.ppat.1003816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kuipers A, Stapels DA, Weerwind LT, Ko YP, Ruyken M, Lee JC, van Kessel KPM, Rooijakkers SHM. 2016. The Staphylococcus aureus polysaccharide capsule and Efb-dependent fibrinogen shield act in concert to protect against phagocytosis. Microbiology 162:1185–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rothfork JM, Dessus-Babus S, Van Wamel WJB, Cheung AL, Gresham HD. 2003. Fibrinogen depletion attenuates Staphyloccocus aureus infection by preventing density-dependent virulence gene up-regulation. J Immunol 171:5389–5395. [DOI] [PubMed] [Google Scholar]
- 128.Hammel M, Sfyroera G, Pyrpassopoulos S, Ricklin D, Ramyar KX, Pop M, Jin Z, Lambris JD, Geisbrecht BV. 2007. Characterization of Ehp, a secreted complement inhibitory protein from Staphylococcus aureus. J Biol Chem 282:30051–30061 10.1074/jbc.M704247200. [DOI] [PubMed] [Google Scholar]
- 129.Jongerius I, Garcia BL, Geisbrecht BV, van Strijp JA, Rooijakkers SH. 2010. Convertase inhibitory properties of staphylococcal extracellular complement-binding protein. J Biol Chem 285:14973–14979 10.1074/jbc.M109.091975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jongerius I, von Köckritz-Blickwede M, Horsburgh MJ, Ruyken M, Nizet V, Rooijakkers SHM. 2012. Staphylococcus aureus virulence is enhanced by secreted factors that block innate immune defenses. J Innate Immun 4:301–311 10.1159/000334604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sharp JA, Echague CG, Hair PS, Ward MD, Nyalwidhe JO, Geoghegan JA, Foster TJ, Cunnion KM. 2012. Staphylococcus aureus surface protein SdrE binds complement regulator factor H as an immune evasion tactic. PLoS One 7:e38407 10.1371/journal.pone.0038407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhang Y, Wu M, Hang T, Wang C, Yang Y, Pan W, Zang J, Zhang M, Zhang X. 2017. Staphylococcus aureus SdrE captures complement factor H’s C-terminus via a novel ‘close, dock, lock and latch’ mechanism for complement evasion. Biochem J 474:1619–1631 10.1042/BCJ20170085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hair PS, Ward MD, Semmes OJ, Foster TJ, Cunnion KM. 2008. Staphylococcus aureus clumping factor A binds to complement regulator factor I and increases factor I cleavage of C3b. J Infect Dis 198:125–133 10.1086/588825. [DOI] [PubMed] [Google Scholar]
- 134.Hair PS, Echague CG, Sholl AM, Watkins JA, Geoghegan JA, Foster TJ, Cunnion KM. 2010. Clumping factor A interaction with complement factor I increases C3b cleavage on the bacterial surface of Staphylococcus aureus and decreases complement-mediated phagocytosis. Infect Immun 78:1717–1727 10.1128/IAI.01065-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Woehl JL, Stapels DAC, Garcia BL, Ramyar KX, Keightley A, Ruyken M, Syriga M, Sfyroera G, Weber AB, Zolkiewski M, Ricklin D, Lambris JD, Rooijakkers SHM, Geisbrecht BV. 2014. The extracellular adherence protein from Staphylococcus aureus inhibits the classical and lectin pathways of complement by blocking formation of the C3 proconvertase. J Immunol 193:6161–6171 10.4049/jimmunol.1401600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rooijakkers SHM, van Wamel WJB, Ruyken M, van Kessel KPM, van Strijp JA. 2005. Anti-opsonic properties of staphylokinase. Microbes Infect 7:476–484 10.1016/j.micinf.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 137.Langley R, Wines B, Willoughby N, Basu I, Proft T, Fraser JD. 2005. The staphylococcal superantigen-like protein 7 binds IgA and complement C5 and inhibits IgA-Fc alpha RI binding and serum killing of bacteria. J Immunol 174:2926–2933 10.4049/jimmunol.174.5.2926. [DOI] [PubMed] [Google Scholar]
- 138.Laursen NS, Gordon N, Hermans S, Lorenz N, Jackson N, Wines B, Spillner E, Christensen JB, Jensen M, Fredslund F, Bjerre M, Sottrup-Jensen L, Fraser JD, Andersen GR. 2010. Structural basis for inhibition of complement C5 by the SSL7 protein from Staphylococcus aureus. Proc Natl Acad Sci U S A 107:3681–3686 10.1073/pnas.0910565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bestebroer J, Aerts PC, Rooijakkers SHM, Pandey MK, Köhl J, van Strijp JA, de Haas CJ. 2010. Functional basis for complement evasion by staphylococcal superantigen-like 7. Cell Microbiol 12:1506–1516 10.1111/j.1462-5822.2010.01486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kang M, Ko YP, Liang X, Ross CL, Liu Q, Murray BE, Höök M. 2013. Collagen-binding microbial surface components recognizing adhesive matrix molecule (MSCRAMM) of Gram-positive bacteria inhibit complement activation via the classical pathway. J Biol Chem 288:20520–20531 10.1074/jbc.M113.454462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. 2004. Neutrophil extracellular traps kill bacteria. Science 303:1532–1535 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 142.Metzler KD, Goosmann C, Lubojemska A, Zychlinsky A, Papayannopoulos V. 2014. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Reports 8:883–896 10.1016/j.celrep.2014.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.von Köckritz-Blickwede M, Nizet V. 2009. Innate immunity turned inside-out: antimicrobial defense by phagocyte extracellular traps. J Mol Med (Berl) 87:775–783 10.1007/s00109-009-0481-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P. 2012. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe 12:324–333 10.1016/j.chom.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 145.Berends ETM, Horswill AR, Haste NM, Monestier M, Nizet V, von Köckritz-Blickwede M. 2010. Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J Innate Immun 2:576–586 10.1159/000319909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Thammavongsa V, Missiakas DM, Schneewind O. 2013. Staphylococcus aureus degrades neutrophil extracellular traps to promote immune cell death. Science 342:863–866 10.1126/science.1242255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Manda-Handzlik A, Demkow U. 2015. Neutrophils: the role of oxidative and nitrosative stress in health and disease. Adv Exp Med Biol 857:51–60 10.1007/5584_2015_117. [DOI] [PubMed] [Google Scholar]
- 148.Borregaard N, Cowland JB. 1997. Granules of the human neutrophilic polymorphonuclear leukocyte. Blood 89:3503–3521. [PubMed] [Google Scholar]
- 149.Egesten A, Breton-Gorius J, Guichard J, Gullberg U, Olsson I. 1994. The heterogeneity of azurophil granules in neutrophil promyelocytes: immunogold localization of myeloperoxidase, cathepsin G, elastase, proteinase 3, and bactericidal/permeability increasing protein. Blood 83:2985–2994. [PubMed] [Google Scholar]
- 150.Levy O. 2004. Antimicrobial proteins and peptides: anti-infective molecules of mammalian leukocytes. J Leukoc Biol 76:909–925 10.1189/jlb.0604320. [DOI] [PubMed] [Google Scholar]
- 151.Turner J, Cho Y, Dinh NN, Waring AJ, Lehrer RI. 1998. Activities of LL-37, a cathelin-associated antimicrobial peptide of human neutrophils. Antimicrob Agents Chemother 42:2206–2214 10.1128/AAC.42.9.2206. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Noore J, Noore A, Li B. 2013. Cationic antimicrobial peptide LL-37 is effective against both extra- and intracellular Staphylococcus aureus. Antimicrob Agents Chemother 57:1283–1290 10.1128/AAC.01650-12. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wimley WC, Selsted ME, White SH. 1994. Interactions between human defensins and lipid bilayers: evidence for formation of multimeric pores. Protein Sci 3:1362–1373 10.1002/pro.5560030902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Pham CTN. 2006. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol 6:541–550 10.1038/nri1841. [PubMed] [DOI] [PubMed] [Google Scholar]
- 155.Reeves EP, Lu H, Jacobs HL, Messina CGM, Bolsover S, Gabella G, Potma EO, Warley A, Roes J, Segal AW. 2002. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature 416:291–297 10.1038/416291a. [PubMed] [DOI] [PubMed] [Google Scholar]
- 156.Corbin BD, Seeley EH, Raab A, Feldmann J, Miller MR, Torres VJ, Anderson KL, Dattilo BM, Dunman PM, Gerads R, Caprioli RM, Nacken W, Chazin WJ, Skaar EP, Skaar EP. 2008. Metal chelation and inhibition of bacterial growth in tissue abscesses. Science 319:962–965 10.1126/science.1152449. [PubMed] [DOI] [PubMed] [Google Scholar]
- 157.Stríz I, Trebichavský I. 2004. Calprotectin: a pleiotropic molecule in acute and chronic inflammation. Physiol Res 53:245–253. [PubMed] [PubMed] [Google Scholar]
- 158.Schindler M, Assaf Y, Sharon N, Chipman DM. 1977. Mechanism of lysozyme catalysis: role of ground-state strain in subsite D in hen egg-white and human lysozymes. Biochemistry 16:423–431 10.1021/bi00622a013. [PubMed] [DOI] [PubMed] [Google Scholar]
- 159.Selsted ME, Martinez RJ. 1978. Lysozyme: primary bactericidin in human plasma serum active against Bacillus subtilis. Infect Immun 20:782–791. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Babior BM, Lambeth JD, Nauseef W. 2002. The neutrophil NADPH oxidase. Arch Biochem Biophys 397:342–344 10.1006/abbi.2001.2642. [PubMed] [DOI] [PubMed] [Google Scholar]
- 161.Kettle AJ, Anderson RF, Hampton MB, Winterbourn CC. 2007. Reactions of superoxide with myeloperoxidase. Biochemistry 46:4888–4897 10.1021/bi602587k. [PubMed] [DOI] [PubMed] [Google Scholar]
- 162.Klebanoff SJ, Kettle AJ, Rosen H, Winterbourn CC, Nauseef WM. 2013. Myeloperoxidase: a front-line defender against phagocytosed microorganisms. J Leukoc Biol 93:185–198 10.1189/jlb.0712349. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Klebanoff SJ. 2005. Myeloperoxidase: friend and foe. J Leukoc Biol 77:598–625 10.1189/jlb.1204697. [PubMed] [DOI] [PubMed] [Google Scholar]
- 164.Klebanoff SJ, Hamon CB. 1972. Role of myeloperoxidase-mediated antimicrobial systems in intact leukocytes. J Reticuloendothel Soc 12:170–196. [PubMed] [PubMed] [Google Scholar]
- 165.Aratani Y, Kura F, Watanabe H, Akagawa H, Takano Y, Suzuki K, Maeda N, Koyama H. 2000. Differential host susceptibility to pulmonary infections with bacteria and fungi in mice deficient in myeloperoxidase. J Infect Dis 182:1276–1279 10.1086/315843. [PubMed] [DOI] [PubMed] [Google Scholar]
- 166.Jensen MS, Bainton DF. 1973. Temporal changes in pH within the phagocytic vacuole of the polymorphonuclear neutrophilic leukocyte. J Cell Biol 56:379–388 10.1083/jcb.56.2.379. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Dri P, Presani G, Perticarari S, Albèri L, Prodan M, Decleva E. 2002. Measurement of phagosomal pH of normal and CGD-like human neutrophils by dual fluorescence flow cytometry. Cytometry 48:159–166 10.1002/cyto.10123. [PubMed] [DOI] [PubMed] [Google Scholar]
- 168.Cech P, Lehrer RI. 1984. Phagolysosomal pH of human neutrophils. Blood 63:88–95. [PubMed] [PubMed] [Google Scholar]
- 169.Levine AP, Duchen MR, de Villiers S, Rich PR, Segal AW. 2015. Alkalinity of neutrophil phagocytic vacuoles is modulated by HVCN1 and has consequences for myeloperoxidase activity. PLoS One 10:e0125906 10.1371/journal.pone.0125906. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kettle AJ, Winterbourn CC. 2001. A kinetic analysis of the catalase activity of myeloperoxidase. Biochemistry 40:10204–10212 10.1021/bi010940b. [PubMed] [DOI] [PubMed] [Google Scholar]
- 171.Levine AP, Segal AW. 2016. The NADPH oxidase and microbial killing by neutrophils, with a particular emphasis on the proposed antimicrobial role of myeloperoxidase within the phagocytic vacuole. Microbiol Spectr 4:MCHD-0018-2015. 10.1128/microbiolspec.MCHD-0018-2015. [DOI] [PubMed] [Google Scholar]
- 172.de Jong NWM, Ramyar KX, Guerra FE, Nijland R, Fevre C, Voyich JM, McCarthy AJ, Garcia BL, van Kessel KPM, van Strijp JAG, Geisbrecht BV, Haas PA. 2017. Immune evasion by a staphylococcal inhibitor of myeloperoxidase. Proc Natl Acad Sci U S A 114:9439–9444 10.1073/pnas.1707032114. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Liu GY, Essex A, Buchanan JT, Datta V, Hoffman HM, Bastian JF, Fierer J, Nizet V. 2005. Staphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. J Exp Med 202:209–215 10.1084/jem.20050846. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Liu C-I, Liu GY, Song Y, Yin F, Hensler ME, Jeng W-Y, Nizet V, Wang AH-J, Oldfield E. 2008. A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence. Science 319:1391–1394 10.1126/science.1153018. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Song Y, Liu CI, Lin FY, No JH, Hensler M, Liu YL, Jeng WY, Low J, Liu GY, Nizet V, Wang AHJ, Oldfield E. 2009. Inhibition of staphyloxanthin virulence factor biosynthesis in Staphylococcus aureus: in vitro, in vivo, and crystallographic results. J Med Chem 52:3869–3880 10.1021/jm9001764. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Karavolos MH, Horsburgh MJ, Ingham E, Foster SJ. 2003. Role and regulation of the superoxide dismutases of Staphylococcus aureus. Microbiology 149:2749–2758 10.1099/mic.0.26353-0. [PubMed] [DOI] [PubMed] [Google Scholar]
- 177.Valderas MW, Hart ME. 2001. Identification and characterization of a second superoxide dismutase gene (sodM) from Staphylococcus aureus. J Bacteriol 183:3399–3407 10.1128/JB.183.11.3399-3407.2001. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Mandell GL. 1975. Catalase, superoxide dismutase, and virulence of Staphylococcus aureus. In vitro and in vivo studies with emphasis on staphylococcal-leukocyte interaction. J Clin Invest 55:561–566 10.1172/JCI107963. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Horsburgh MJ, Clements MO, Crossley H, Ingham E, Foster SJ. 2001. PerR controls oxidative stress resistance and iron storage proteins and is required for virulence in Staphylococcus aureus. Infect Immun 69:3744–3754 10.1128/IAI.69.6.3744-3754.2001. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Cosgrove K, Coutts G, Jonsson I-M, Tarkowski A, Kokai-Kun JF, Mond JJ, Foster SJ. 2007. Catalase (KatA) and alkyl hydroperoxide reductase (AhpC) have compensatory roles in peroxide stress resistance and are required for survival, persistence, and nasal colonization in Staphylococcus aureus. J Bacteriol 189:1025–1035 10.1128/JB.01524-06. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Guerra FE, Addison CB, de Jong NWM, Azzolino J, Pallister KB, van Strijp JAG, Voyich JM. 2016. Staphylococcus aureus SaeR/S-regulated factors reduce human neutrophil reactive oxygen species production. J Leukoc Biol 100:1005–1010 10.1189/jlb.4VMAB0316-100RR. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Richardson AR, Dunman PM, Fang FC. 2006. The nitrosative stress response of Staphylococcus aureus is required for resistance to innate immunity. Mol Microbiol 61:927–939 10.1111/j.1365-2958.2006.05290.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 183.Richardson AR, Libby SJ, Fang FC. 2008. A nitric oxide-inducible lactate dehydrogenase enables Staphylococcus aureus to resist innate immunity. Science 319:1672–1676 10.1126/science.1155207. [PubMed] [DOI] [PubMed] [Google Scholar]
- 184.Peschel A, Otto M, Jack RW, Kalbacher H, Jung G, Götz F. 1999. Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J Biol Chem 274:8405–8410 10.1074/jbc.274.13.8405. [PubMed] [DOI] [PubMed] [Google Scholar]
- 185.Collins LV, Kristian SA, Weidenmaier C, Faigle M, Van Kessel KP, Van Strijp JA, Götz F, Neumeister B, Peschel A. 2002. Staphylococcus aureus strains lacking D-alanine modifications of teichoic acids are highly susceptible to human neutrophil killing and are virulence attenuated in mice. J Infect Dis 186:214–219 10.1086/341454. [PubMed] [DOI] [PubMed] [Google Scholar]
- 186.Peschel A, Jack RW, Otto M, Collins LV, Staubitz P, Nicholson G, Kalbacher H, Nieuwenhuizen WF, Jung G, Tarkowski A, van Kessel KPM, van Strijp JAG, Peschel A, Jack RW, Otto M, Collins LV, Staubitz P, Nicholson G, Kalbacher H, Nieuwenhuizen WF, Jung G, Tarkowski A, van Kessel KPM, van Strijp JG. 2001. Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with l-lysine. J Exp Med 193:1067–1076 10.1084/jem.193.9.1067. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Oku Y, Kurokawa K, Ichihashi N, Sekimizu K. 2004. Characterization of the Staphylococcus aureus mprF gene, involved in lysinylation of phosphatidylglycerol. Microbiology 150:45–51 10.1099/mic.0.26706-0. [PubMed] [DOI] [PubMed] [Google Scholar]
- 188.Sieprawska-Lupa M, Mydel P, Krawczyk K, Wójcik K, Puklo M, Lupa B, Suder P, Silberring J, Reed M, Pohl J, Shafer W, McAleese F, Foster T, Travis J, Potempa J. 2004. Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob Agents Chemother 48:4673–4679 10.1128/AAC.48.12.4673-4679.2004. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Bera A, Herbert S, Jakob A, Vollmer W, Götz F. 2005. Why are pathogenic staphylococci so lysozyme resistant? The peptidoglycan O-acetyltransferase OatA is the major determinant for lysozyme resistance of Staphylococcus aureus. Mol Microbiol 55:778–787 10.1111/j.1365-2958.2004.04446.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 190.Herbert S, Bera A, Nerz C, Kraus D, Peschel A, Goerke C, Meehl M, Cheung A, Götz F. 2007. Molecular basis of resistance to muramidase and cationic antimicrobial peptide activity of lysozyme in staphylococci. PLoS Pathog 3:e102 10.1371/journal.ppat.0030102. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Stapels DA, Ramyar KX, Bischoff M, von Köckritz-Blickwede M, Milder FJ, Ruyken M, Eisenbeis J, McWhorter WJ, Herrmann M, van Kessel KP, Geisbrecht BV, Rooijakkers SH. 2014. Staphylococcus aureus secretes a unique class of neutrophil serine protease inhibitors. Proc Natl Acad Sci U S A 111:13187–13192 10.1073/pnas.1407616111. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Stapels DC, Woehl JL, Milder FJ, Tromp AT, van Batenburg A, de Graaf WC, Broll SC, White NM, Rooijakkers SHM, Geisbrecht BV. 2017. Evidence for multiple modes of neutrophil serine protease recognition by the EAP family of staphylococcal innate immune evasion proteins. Protein Sci 27:509–522. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Stapels DA, Kuipers A, von Köckritz-Blickwede M, Ruyken M, Tromp AT, Horsburgh MJ, de Haas CJ, van Strijp JA, van Kessel KP, Rooijakkers SH. 2016. Staphylococcus aureus protects its immune-evasion proteins against degradation by neutrophil serine proteases. Cell Microbiol 18:536–545 10.1111/cmi.12528. [PubMed] [DOI] [PubMed] [Google Scholar]
- 194.Jin T, Bokarewa M, Foster T, Mitchell J, Higgins J, Tarkowski A. 2004. Staphylococcus aureus resists human defensins by production of staphylokinase, a novel bacterial evasion mechanism. J Immunol 172:1169–1176 10.4049/jimmunol.172.2.1169. [PubMed] [DOI] [PubMed] [Google Scholar]
- 195.Wilke GA, Bubeck Wardenburg J. 2010. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus α-hemolysin-mediated cellular injury. Proc Natl Acad Sci U S A 107:13473–13478 10.1073/pnas.1001815107. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Powers ME, Kim HK, Wang Y, Bubeck Wardenburg J. 2012. ADAM10 mediates vascular injury induced by Staphylococcus aureus α-hemolysin. J Infect Dis 206:352–356 10.1093/infdis/jis192. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Inoshima I, Inoshima N, Wilke GA, Powers ME, Frank KM, Wang Y, Bubeck Wardenburg J. 2011. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat Med 17:1310–1314 10.1038/nm.2451. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Valeva A, Walev I, Pinkernell M, Walker B, Bayley H, Palmer M, Bhakdi S. 1997. Transmembrane β-barrel of staphylococcal α-toxin forms in sensitive but not in resistant cells. Proc Natl Acad Sci U S A 94:11607–11611 10.1073/pnas.94.21.11607. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Jayasinghe L, Bayley H. 2005. The leukocidin pore: evidence for an octamer with four LukF subunits and four LukS subunits alternating around a central axis. Protein Sci 14:2550–2561 10.1110/ps.051648505. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Koop G, Vrieling M, Storisteanu DML, Lok LSC, Monie T, van Wigcheren G, Raisen C, Ba X, Gleadall N, Hadjirin N, Timmerman AJ, Wagenaar JA, Klunder HM, Fitzgerald JR, Zadoks R, Paterson GK, Torres C, Waller AS, Loeffler A, Loncaric I, Hoet AE, Bergström K, De Martino L, Pomba C, de Lencastre H, Ben Slama K, Gharsa H, Richardson EJ, Chilvers ER, de Haas C, van Kessel K, van Strijp JAG, Harrison EM, Holmes MA. 2017. Identification of LukPQ, a novel, equid-adapted leukocidin of Staphylococcus aureus. Sci Rep 7:40660 10.1038/srep40660. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Vrieling M, Koymans KJ, Heesterbeek DA, Aerts PC, Rutten VP, de Haas CJ, van Kessel KP, Koets AP, Nijland R, van Strijp JA. 2015. Bovine Staphylococcus aureus secretes the leukocidin LukMF’ to kill migrating neutrophils through CCR1. MBio 6:e00335 10.1128/mBio.00335-15. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Alonzo F III, Torres VJ. 2014. The bicomponent pore-forming leucocidins of Staphylococcus aureus. Microbiol Mol Biol Rev 78:199–230 10.1128/MMBR.00055-13. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Spaan AN, van Strijp JAG, Torres VJ. 2017. Leukocidins: staphylococcal bi-component pore-forming toxins find their receptors. Nat Rev Microbiol 15:435–447 10.1038/nrmicro.2017.27. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Spaan AN, Reyes-Robles T, Badiou C, Cochet S, Boguslawski KM, Yoong P, Day CJ, de Haas CJC, van Kessel KPM, Vandenesch F, Jennings MP, Le Van Kim C, Colin Y, van Strijp JA, Henry T, Torres VJ. 2015. Staphylococcus aureus targets the Duffy antigen receptor for chemokines (DARC) to lyse erythrocytes. Cell Host Microbe 18:363–370 10.1016/j.chom.2015.08.001. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.DuMont AL, Yoong P, Surewaard BGJ, Benson MA, Nijland R, van Strijp JA, Torres VJ. 2013. Staphylococcus aureus elaborates leukocidin AB to mediate escape from within human neutrophils. Infect Immun 81:1830–1841 10.1128/IAI.00095-13. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Naimi TS, LeDell KH, Como-Sabetti K, Borchardt SM, Boxrud DJ, Etienne J, Johnson SK, Vandenesch F, Fridkin S, O’Boyle C, Danila RN, Lynfield R. 2003. Comparison of community- and health care-associated methicillin-resistant Staphylococcus aureus infection. JAMA 290:2976–2984 10.1001/jama.290.22.2976. [PubMed] [DOI] [PubMed] [Google Scholar]
- 207.Wang R, Braughton KR, Kretschmer D, Bach T-HL, Queck SY, Li M, Kennedy AD, Dorward DW, Klebanoff SJ, Peschel A, DeLeo FR, Otto M. 2007. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med 13:1510–1514 10.1038/nm1656. [PubMed] [DOI] [PubMed] [Google Scholar]
- 208.Surewaard BGJ, Nijland R, Spaan AN, Kruijtzer JA, de Haas CJ, van Strijp JA. 2012. Inactivation of staphylococcal phenol soluble modulins by serum lipoprotein particles. PLoS Pathog 8:e1002606 10.1371/journal.ppat.1002606. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Surewaard BGJ, de Haas CJC, Vervoort F, Rigby KM, DeLeo FR, Otto M, van Strijp JA, Nijland R. 2013. Staphylococcal alpha-phenol soluble modulins contribute to neutrophil lysis after phagocytosis. Cell Microbiol 15:1427–1437 10.1111/cmi.12130. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Lina G, Bohach GA, Nair SP, Hiramatsu K, Jouvin-Marche E, Mariuzza R, International Nomenclature Committee for Staphylococcal Superantigens. 2004. Standard nomenclature for the superantigens expressed by Staphylococcus. J Infect Dis 189:2334–2336 10.1086/420852. [PubMed] [DOI] [PubMed] [Google Scholar]
- 211.Fraser JD, Proft T. 2008. The bacterial superantigen and superantigen-like proteins. Immunol Rev 225:226–243 10.1111/j.1600-065X.2008.00681.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 212.Finlay BB, McFadden G. 2006. Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell 124:767–782 10.1016/j.cell.2006.01.034. [PubMed] [DOI] [PubMed] [Google Scholar]
- 213.Verkaik NJ, Lebon A, de Vogel CP, Hooijkaas H, Verbrugh HA, Jaddoe VW, Hofman A, Moll HA, van Belkum A, van Wamel WJ. 2010. Induction of antibodies by Staphylococcus aureus nasal colonization in young children. Clin Microbiol Infect 16:1312–1317 10.1111/j.1469-0691.2009.03073.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 214.Schmidt F, Meyer T, Sundaramoorthy N, Michalik S, Surmann K, Depke M, Dhople V, Gesell Salazar M, Holtappels G, Zhang N, Bröker BM, Bachert C, Völker U. 2017. Characterization of human and Staphylococcus aureus proteins in respiratory mucosa by in vivo- and immunoproteomics. J Proteomics 155:31–39 10.1016/j.jprot.2017.01.008. [PubMed] [DOI] [PubMed] [Google Scholar]
- 215.Lo H, Tang CM, Exley RM. 2009. Mechanisms of avoidance of host immunity by Neisseria meningitidis and its effect on vaccine development. Lancet Infect Dis 9:418–427 10.1016/S1473-3099(09)70132-X. [DOI] [PubMed] [Google Scholar]
- 216.Stollerman GH, Dale JB. 2008. The importance of the group a Streptococcus capsule in the pathogenesis of human infections: a historical perspective. Clin Infect Dis 46:1038–1045 10.1086/529194. [PubMed] [DOI] [PubMed] [Google Scholar]
- 217.Pozzi C, Olaniyi R, Liljeroos L, Galgani I, Rappuoli R, Bagnoli F. 2016. Vaccines for Staphylococcus aureus and target populations. Curr Top Microbiol Immunol 6:23–27. [DOI] [PubMed] [Google Scholar]
- 218.Bagnoli F, Bertholet S, Grandi G. 2012. Inferring reasons for the failure of Staphylococcus aureus vaccines in clinical trials. Front Cell Infect Microbiol 2:16 10.3389/fcimb.2012.00016. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Skurnik D, Kropec A, Roux D, Theilacker C, Huebner J, Pier GB. 2012. Natural antibodies in normal human serum inhibit Staphylococcus aureus capsular polysaccharide vaccine efficacy. Clin Infect Dis 55:1188–1197 10.1093/cid/cis624. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Fowler VG, Allen KB, Moreira ED, Moustafa M, Isgro F, Boucher HW, Corey GR, Carmeli Y, Betts R, Hartzel JS, Chan ISF, McNeely TB, Kartsonis NA, Guris D, Onorato MT, Smugar SS, DiNubile MJ, Sobanjo-ter Meulen A. 2013. Effect of an investigational vaccine for preventing Staphylococcus aureus infections after cardiothoracic surgery: a randomized trial. JAMA 309:1368–1378 10.1001/jama.2013.3010. [PubMed] [DOI] [PubMed] [Google Scholar]
- 221.Alonzo F III, Kozhaya L, Rawlings SA, Reyes-Robles T, DuMont AL, Myszka DG, Landau NR, Unutmaz D, Torres VJ. 2013. CCR5 is a receptor for Staphylococcus aureus leukotoxin ED. Nature 493:51–55 10.1038/nature11724. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Hua L, Hilliard JJ, Shi Y, Tkaczyk C, Cheng LI, Yu X, Datta V, Ren S, Feng H, Zinsou R, Keller A, O’Day T, Du Q, Cheng L, Damschroder M, Robbie G, Suzich J, Stover CK, Sellman BR. 2014. Assessment of an anti-alpha-toxin monoclonal antibody for prevention and treatment of Staphylococcus aureus-induced pneumonia. Antimicrob Agents Chemother 58:1108–1117 10.1128/AAC.02190-13. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Sause WE, Buckley PT, Strohl WR, Lynch AS, Torres VJ. 2016. Antibody-based biologics and their promise to combat Staphylococcus aureus infections. Trends Pharmacol Sci 37:231–241 10.1016/j.tips.2015.11.008. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Hoekstra H, Romero Pastrana F, Bonarius HPJ, van Kessel KPM, Elsinga GS, Kooi N, Groen H, van Dijl JM, Buist G. 2017. A human monoclonal antibody that specifically binds and inhibits the staphylococcal complement inhibitor protein SCIN. Virulence 9:70–82. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.François B, Barraud O, Jafri HS. 2017. Antibody-based therapy to combat Staphylococcus aureus infections. Clin Microbiol Infect 23:219–221 10.1016/j.cmi.2017.02.035. [PubMed] [DOI] [PubMed] [Google Scholar]
- 226.Koymans KJ, Vrieling M, Gorham RD, van Strijp JAG. 2016. Staphylococcal immune evasion proteins: structure, function, and host adaptation. Curr Top Microbiol Immunol 6:23–27. [DOI] [PubMed] [Google Scholar]
- 227.Du C, Xie X. 2012. G protein-coupled receptors as therapeutic targets for multiple sclerosis. Cell Res 22:1108–1128 10.1038/cr.2012.87. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Ricklin D, Lambris JD. 2013. Complement in immune and inflammatory disorders: pathophysiological mechanisms. J Immunol 190:3831–3838 10.4049/jimmunol.1203487. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Liu Y, Yin H, Zhao M, Lu Q. 2014. TLR2 and TLR4 in autoimmune diseases: a comprehensive review. Clin Rev Allergy Immunol 47:136–147 10.1007/s12016-013-8402-y. [PubMed] [DOI] [PubMed] [Google Scholar]
- 230.Summers BJ, Garcia BL, Woehl JL, Ramyar KX, Yao X, Geisbrecht BV. 2015. Identification of peptidic inhibitors of the alternative complement pathway based on Staphylococcus aureus SCIN proteins. Mol Immunol 67(2 Pt B):193–205 10.1016/j.molimm.2015.05.012. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Boer JC, Domanska UM, Timmer-Bosscha H, Boer IGJ, de Haas CJC, Joseph JV, Kruyt FA, de Vries EG, den Dunnen WF, van Strijp JA, Walenkamp AM. 2013. Inhibition of formyl peptide receptor in high-grade astrocytoma by CHemotaxis Inhibitory Protein of S. aureus. Br J Cancer 108:587–596 10.1038/bjc.2012.603. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]