

ABSTRACT
Streptococcus pyogenes (i.e., the group A Streptococcus) is a human-restricted and versatile bacterial pathogen that produces an impressive arsenal of both surface-expressed and secreted virulence factors. Although surface-expressed virulence factors are clearly vital for colonization, establishing infection, and the development of disease, the secreted virulence factors are likely the major mediators of tissue damage and toxicity seen during active infection. The collective exotoxin arsenal of S. pyogenes is rivaled by few bacterial pathogens and includes extracellular enzymes, membrane active proteins, and a variety of toxins that specifically target both the innate and adaptive arms of the immune system, including the superantigens; however, despite their role in S. pyogenes disease, each of these virulence factors has likely evolved with humans in the context of asymptomatic colonization and transmission. In this article, we focus on the biology of the true secreted exotoxins of the group A Streptococcus, as well as their roles in the pathogenesis of human disease.
Streptococcus pyogenes (i.e., the group A Streptococcus) is a human-restricted and versatile bacterial pathogen that produces an impressive arsenal of both surface-expressed and secreted virulence factors. S. pyogenes exists primarily as an asymptomatic colonizer of the skin and mucous membranes of the nasopharynx, and despite being universally sensitive to β-lactam antibiotics in vitro, this bacterium continues to generate significant morbidity and mortality on a global scale. Human diseases induced by S. pyogenes usually occur as relatively uncomplicated manifestations such as pharyngitis and skin infections, but it may also cause more problematic diseases including erysipelas and scarlet fever. In addition, S. pyogenes can cause devastating invasive diseases including puerperal sepsis, bacteremia, necrotizing fasciitis, and streptococcal toxic shock syndrome (TSS). This bacterium is further recognized as a very important cause of postinfection sequelae including acute rheumatic fever and rheumatic heart disease, acute glomerulonephritis, and potentially, pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections. Although surface-expressed virulence factors are clearly vital for colonization, establishment of infection, and the development of disease, the secreted virulence factors are likely the major mediators of tissue damage and toxicity seen during active infection. The collective exotoxin arsenal of S. pyogenes is rivaled by few bacterial pathogens and includes extracellular enzymes, membrane active proteins, and a variety of toxins that specifically target both the innate and adaptive arms of the immune system, including the superantigens; however, despite their role in S. pyogenes disease, each of these virulence factors has likely evolved with humans in the context of asymptomatic colonization and transmission. In this article, we will focus on the biology of the true secreted exotoxins of the group A Streptococcus, as well as their roles in the pathogenesis of human disease.
THE CYTOLYTIC TOXINS OF S. PYOGENES
β-hemolytic activity on blood agar is a hallmark feature of the group A Streptococcus, and this phenotype has been used clinically for over a century. The streptolysin S (SLS) exotoxin is a small, oxygen stable hemolysin that is responsible for the characteristic β-hemolysis of S. pyogenes (1). SLS is a cytolytic toxin that has features of the ribosomally synthesized and posttranslationally modified lantibiotic bacteriocins from other Gram-positive bacteria; however, lantibiotic bacteriocins mostly function as antibacterials without cytotoxic activity for eukaryotic cells (2). Production of SLS is encoded by the SLS-associated gene (sag) cluster, which contains genes encoding the structural SLS protein (sagA) as well as enzymes responsible for the posttranslational modification and transport of the toxin and, potentially, an immunity function (sagB through sagI) (3). Furthermore, the sag locus is conserved among group A Streptococcus strains regardless of M protein serotype (3). SLS has been shown to target and disrupt the function of multiple host cells, including erythrocytes, macrophages, neutrophils, and keratinocytes (2, 4–6). The exact mechanism of action of SLS is not completely clear, although it is thought to act by forming a pore in host cell membranes (7). However, a recent study has demonstrated that SLS lyses erythrocytes by disrupting the function of anion transporters to cause an influx of chloride anions (Cl–). The influx of Cl– is followed by an influx of water, resulting in colloidal-osmotic rupture of erythrocytes (8). Furthermore, SLS has been shown to work in conjunction with the host protease calpain to facilitate the destruction of intracellular junction proteins, promoting paracellular invasion of the bacteria across the epithelial barrier (9). To examine the role of SLS in invasive models of disease, infection of an S. pyogenes strain deleted for SLS function was tested in a murine model of necrotizing soft tissue infection, and SLS was determined to be essential for pathogenesis (3). A similar study using transposon mutagenesis created a mutant deficient in SLS production that remained normal with respect to other exoprotein expression and was also shown to be markedly less virulent in a mouse model of subcutaneous infection than in the isogenic wild-type strain (10). Therefore, SLS likely contributes to S. pyogenes pathogenesis through a combination of inhibiting phagocyte function and damaging epithelial barriers.
Streptolysin O (SLO) is a well-characterized exotoxin that functions as a thiol-activated cytolysin and is secreted from nearly all group A Streptococcus isolates (11). The slo gene encodes SLO monomers that oligomerize on eukaryotic membranes in a cholesterol-dependent manner, resulting in the formation of a large pore. This produces significant damage and ultimately results in host cell apoptosis (12). The SLO toxin is active in a reduced state and is rapidly inactivated in the presence of oxygen; thus, activity is not thought to be visible on routine blood agar plates (13). Previous studies have shown that the host targets of SLO include erythrocytes, macrophages, neutrophils, and keratinocytes (14–17). SLO has also been shown to inhibit neutrophil degranulation at sublytic concentrations (16). In addition to SLO, the slo gene resides in an operon that additionally encodes S. pyogenes NADase (SPN). The NADase functions by hydrolyzing cellular NAD+ and by depleting cellular ATP to decrease the host energy sources (18). Multiple studies have concluded that the entry of SPN into host cells is dependent on SLO pore formation (19, 20), and SPN and SLO in conjunction increase epithelial cell apoptosis, compared to SLO alone (21). Using immortalized human keratinocytes, strains lacking SLO were attenuated for intracellular survival compared to the wild-type strain (20). Furthermore, in a murine model of necrotizing soft tissue infection, S. pyogenes strains lacking SLO were deficient in causing myositis, bacteremia, and soft tissue infection (22). Another study demonstrated lower infectivity of SPN mutant strains in models of necrotizing fasciitis, although the deletion of SLO led to an inability to cause disease (22). Therefore, SLO likely promotes the pathogenesis of S. pyogenes in invasive skin and soft tissue infection through the destruction of phagocytes, as well as the cytolysin-mediated translocation of SPN into target cells.
Independent studies investigating the role of both SLO and SLS in murine models of necrotizing fasciitis and systemic infection each demonstrated that both cytolytic toxins significantly contribute to the development of murine necrotizing fasciitis (23, 24). Although SLO and SLS have been extensively studied and shown to be important contributors to the pathogenesis of invasive S. pyogenes infections, their roles during colonization and acute nasopharyngeal infection have yet to be defined.
STREPTOCOCCAL SUPERANTIGENS
S. pyogenes is recognized as one of the few bacterial pathogens that produces superantigen exotoxins. Streptococcal superantigens are ribosomally synthesized, relatively low molecular mass (∼22 to 28 kDa) proteins that contain classical signal peptides that are cleaved after secretion to release the mature toxin. Superantigens function by activating T cells and are among the most potent known activators of T cells. The term “superantigen” was first used to describe the massive primary T cell response to these bacterial toxins (25). However, the S. pyogenes superantigens are also historically known as the erythrogenic toxins or scarlet fever toxins, due to their role in causing a red rash in the context of scarlet fever (26–28). In fact, the characteristic rash seen in scarlet fever is likely due to an amplified hypersensitivity reaction resulting from superantigen activity (28). Since the appearance of a rash was the defining feature of the “erythrogenic” toxins, Watson (27) first proposed the name “streptococcal pyrogenic exotoxin” (Spe) owing to the fever-producing ability, because he initially believed that the Spes belonged to a separate family of toxins. We now know that these represent the same group that belongs to a larger group of structurally conserved exotoxins that are also produced by Staphylococcus aureus (29–32) and some Lancefield group C and G β-hemolytic streptococci (33–37). There are now at least 14 characterized and genetically distinct S. pyogenes superantigens, and many of them are encoded within lysogenic bacteriophage, or putative bacteriophage, elements. Therefore, different strains often encode different repertoires of typically between 3 and 6 distinct superantigen genes (38–42). Although nomenclatural inconsistencies exist in the literature, a recent comprehensive review of the streptococcal superantigens has clarified many of these issues (43). Using this updated nomenclature, the currently known repertoire includes streptococcal pyrogenic exotoxin (Spe) types A, C, G, H, I, J, K, L, M, N, O, and P, as well as the streptococcal superantigen and streptococcal mitogenic exotoxin Z (SmeZ) (Fig. 1A). The SpeN, SpeO, and SpeP superantigens were first identified in Staphylococcus equi subsp. zooepidemicus (originally defined as SzeN, SzeF, and SzeP, respectively) (36), but to our knowledge, these particular superantigens have not been identified in S. pyogenes. Additionally, S. equi subsp. equi can encode SeeH, SeeI, SeeL, and SeeM, which are highly orthologous to SpeH, SpeI, SpeL, and SpeM, respectively (37). Although most of the streptococcal superantigens are encoded on bacteriophage elements, SpeG and SmeZ are exceptions in that they appear to be encoded within the core chromosome (35, 38–42). SpeJ is also not found in association with bacteriophage genes (40), yet it is not present in the many sequenced strains. SmeZ, in particular, is known to have many allelic variants, and most of the sequence changes are located on the surface of the toxin and rarely found within the predicted receptor binding domains. This indicates that SmeZ is likely under significant immunological pressure to alter antigenic characteristics as a possible immune evasion strategy (35). Despite the fairly weak primary amino acid sequence homology (Fig. 1B), crystal structures determined for these toxins share the “generic” superantigen fold (Fig. 1C) (44). Each structure includes an N-terminal α-helix which leads into a β-barrel domain, also known as the oligosaccharide/oligonucleotide binding fold (45). A central α-helix then joins this domain to a β-sheet structure known as the β-grasp domain. Despite the clear similarities to the overall fold, there are also important differences in how these toxins engage their host receptors.
FIGURE 1.
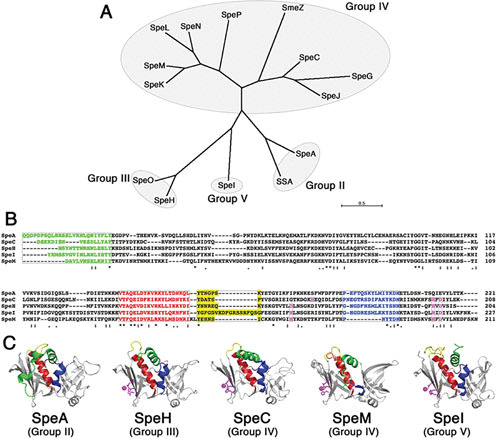
Phylogenetic relationships and structural conservation of the streptococcal superantigens. (A) Unrooted neighbor-joining tree showing phylogenetic relationships of known streptococcal superantigens. The unrooted tree was based on the alignment of amino acid sequences using CLUSTAL W (166) and constructed using MEGA7 (167). The groups indicate a prior classification scheme for the superantigen family (32). (B) Amino acid alignment of five representative streptococcal superantigens. The colors designate distinct domains in the superantigen structure, including the N-terminal α-helix (green), the central α-helix (red), the α3-β8 loop that is unique to the group V superantigens (168), and a C-terminal α-helix that is lacking in a subgroup of group IV. Residues involved in the coordination of a zinc atom important for binding to the MHC class II β-chain are colored magenta. (C) Crystal structures of representative streptococcal superantigens are colored as in panel B.
Superantigens function by causing excessive T cell activation through simultaneous engagement of both the variable region of the T cell antigen receptor (TCR) β-chain (referred to as “Vβ”) on T cells and to different regions of major histocompatibility complex (MHC) class II molecules on antigen-presenting cells (44, 46). Thus, superantigens are remarkable in that they have evolved to target two critical and extremely diverse receptors of adaptive immunity. In superantigen-mediated T cell activation, the toxin binds directly to MHC class II molecules, and this occurs without processing by the antigen-presenting cell. Superantigens do not undergo major modifications after release from the cell or major structural alterations upon binding to their ligands. The interaction with the TCR is dependent on binding to different Vβ regions, and this occurs, generally, away from the regions of the TCR that are critical for peptide-specific recognition. Due to the unconventional contacts created by these interactions, superantigen-mediated T cell activation is also not MHC restricted, and furthermore, both CD4+ and CD8+ T cells are stimulated in a Vβ-specific manner. Thus, superantigens can “force” an excessive primary response that is not seen with conventional peptide antigens. In the most severe case, this excessive activation of T cells results in the massive release of proinflammatory cytokines from both T cells and antigen-presenting cells (47) that can result in a serious and potentially lethal disease defined as TSS (32).
Cocrystallization structures have been determined for SpeA and SpeC in complex with the mouse Vβ8.1 and human Vβ2.1 chains, respectively (48). Earlier cocrystal structures exist for the staphylococcal superantigens staphylococcal enterotoxin B (SEB) and staphylococcal enterotoxin C (SEC) in complex with the mouse Vβ8.1 chain (49, 50). Although highly homologous superantigens, such as SEB, SEC, and SpeA, may have similar architectures for their Vβ chain interactions, the β-chain-SpeC complex involved a significantly larger contact surface, a different orientation, and contacts with the complementarity determining region (CDR) 3 loop, whereas SEB, SEC, and SpeA had no intermolecular contacts with this loop (48). Nevertheless, it appears that the critical component for functional recognition of the TCR is the CDR 2 loop of the β-chain (51).
The interaction of superantigens with MHC class II can also show considerable diversity, because superantigens have two independent binding domains. One binding face occurs through the N-terminal oligosaccharide/oligonucleotide binding fold domain and is of relatively low affinity (∼10−5 M). Although somewhat controversial (52–55), it appears likely that most, if not all, streptococcal superantigens contain this low-affinity MHC binding domain. A second, zinc-dependent MHC binding face occurs on the β-grasp domain and is of relatively high affinity (∼10−7). The zinc ion, shown in magenta in Fig. 1C, is coordinated through three conserved residues, whereas the fourth ligand is from a conserved residue on the MHC β-chain.
Figure 2 presents the complexes for a conventional TCR-MHC class II interaction, as well as how SpeA (which lacks a high-affinity MHC class II binding domain) likely engages TCR Vβ and MHC class II and SpeC, which crosslinks MHC class II (56). The SpeA model is analogous to the standard “wedge” model of superantigen-mediated T cell activation (49), whereas the SpeC model is analogous to the proposed model for staphylococcal enterotoxin A (57, 58). In these models, the mechanism by which superantigens subvert the normal T cell activation process is apparent, whereby the antigenic peptide is displaced from the TC, and the specificity of the reaction is governed by superantigen recognition of the Vβ CDR2 loop (51).
FIGURE 2.
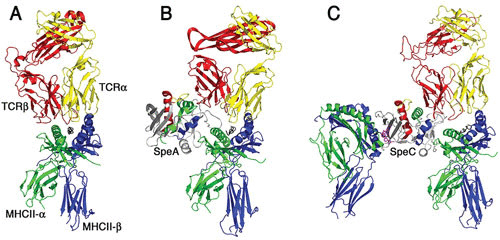
Models of T cell activation complexes for streptococcal superantigens. Ribbon diagrams demonstrating typical antigen-mediated T cell activation (A) and modeled T cell activation complexes for SpeA (B) and SpeC (C). The cocrystal structures of SpeA and SpeC in complex with their respective TCR β-chains (48) and of SpeC in complex with the MHC class II through the zinc-dependent high-affinity binding domain have been determined (169). SpeC also activates T cells in a mode similar to the staphylococcal enterotoxin A model (58) where SpeC engages MHC class II α-chain through a generic low-affinity binding domain (170) and engages the MHC class II β-chain through a zinc-dependent, high-affinity binding domain (169). The binding architecture for the generic low-affinity MHC class II binding to SpeA and SpeC is modeled using the staphylococcal enterotoxin B-MHC class II cocrystal structure (171). Note the presence of the zinc ion (magenta) coordinated in the high-affinity binding site for SpeC and that SpeA lacks this zinc site. The TCR α-chain (shown in gray) for both the SpeA and SpeC diagrams is modeled for clarity by superimposition of the α/β TCR shown on the left of the respective TCR β-chains for both superantigens. The figure was generated using Pymol.
Since early in the past century, the incidence of serious streptococcal disease greatly decreased, likely owing to the widespread use of antibiotics (59). However, it was not until the late 1980s that descriptions of well-defined cases led to the recognition of a severely toxic streptococcal superantigen-mediated disease called streptococcal TSS (60, 61). Indeed, since the mid-1980s there has been a sustained resurgence in the frequency and severity of streptococcal TSS and other invasive diseases caused by group A streptococci (62–64). This led to the hypotheses that there was an emergence of an excessively virulent strain or strains or, alternatively, that there had been major changes in host susceptibility to these infections. Indeed, many of the contemporary invasive infections have been attributed to the M1T1 clone (62), which is now recognized to have acquired an SpeA2- and DNAse-encoding bacteriophage, as well as a horizontally acquired region encoding the SLO and SPN, likely from an M12 serotype (65). These acquisitions increase the virulence of this clone (66). Over the past decade, there has also been a dramatic resurgence in scarlet fever in mainland China and Hong Kong, South Korea, the United Kingdom, and Germany (67–70). Although it is not yet entirely clear, these large-scale outbreaks are likely linked to the acquisition of superantigen-producing bacteriophage elements in S. pyogenes (71–73).
Multiple lines of experimental evidence indicate that the streptococcal superantigens are significant virulence factors for severe streptococcal disease. The prototypical streptococcal superantigens are SpeA and SpeC, and these two toxins have been recognized as the likely causative agents in most cases of streptococcal TSS (61, 62, 74). Indeed, SpeA and SpeC are produced by many S. pyogenes strains isolated from patients with streptococcal TSS (29, 74–78). Due to the association of SpeA and SpeC with streptococcal TSS, two toxoid vaccines have been generated for these toxins that, when used in the rabbit model of TSS, provided complete protection to lethal quantities of the respective superantigen (53, 79). There are at least six naturally occurring alleles of speA (80, 81), and serotype M1 and M3 strains expressing the speA2 and speA3 alleles caused the majority of streptococcal TSS in the 1980s (81). Of the streptococcal superantigens that have been tested, the toxins share the properties of inducing fever (pyrogenicity) and lethal shock in the rabbit model. For example, recombinant SpeA, SpeC, and SpeJ can induce symptoms of streptococcal TSS without necrotizing fasciitis/myositis in this model (53, 79, 82), and SpeA administered to rabbits as a vaccine also conferred significant protection from TSS with necrotizing fasciitis/myositis after challenge with viable M1 and M3 streptococci (64). Circulating superantigens have also been found in patients with streptococcal TSS (83), and the lack of humoral immunity to the superantigens is a risk factor for the development of streptococcal TSS (84, 85).
Although significant immunological research with bacterial superantigens has been conducted in mouse models, conventional mice are no longer considered appropriate models to evaluate the biological effects of these toxins. Lethal effects are not seen at very high relative doses without the coadministration of a liver-damaging agent, whereas the rabbit model is susceptible to fairly low doses when administered continuously (86). It has further been demonstrated that HLA-DQ transgenic mice were more susceptible to SpeA-induced lethality whether administered as a pure toxin or during infection with SpeA+ S. pyogenes, whereas nontransgenic mice did not show an obvious effect (87, 88). Consistent with this, it has been established that in humans, MHC class II allelic variation contributes to the differences in severity seen for invasive streptococcal infections (89). This is understood to be due to differential cytokine production triggered by streptococcal superantigens, and MHC class II α-chain polymorphisms affect superantigen responses (90, 91). Recently, using infection models with transgenic mice expressing human MHC class II alleles, we demonstrated that the SpeA superantigen is a critical bacterial factor that is necessary to induce an acute nasopharyngeal infection (92). Using this model, SpeA was shown to activate specific Vβ-subsets in vivo (93), and strikingly, functional Vβ-specific T cell subsets were required for the infection phenotype (94). Furthermore, humoral immunity to the SpeA superantigen dramatically inhibited infection by S. pyogenes (92, 94). We believe that these findings support the further development of toxoid superantigens as vaccines to target the transmission and colonization by S. pyogenes.
STREPTOCOCCAL PYROGENIC EXOTOXIN B
SpeB is highly conserved in virtually all S. pyogenes isolates (95, 96), although despite its original name as a streptococcal pyrogenic exotoxin, SpeB is known to function as a broad-spectrum protease and is secreted as a 40-kDa zymogen. The pro-domain is auto-catalytically cleaved to generate an active 28-kDa proteinase (97, 98). Additionally, an SpeB inhibitor located downstream of speB is cotranscribed with the protease. The inhibitor, termed Spi, has sequence and predicted structural homology with the pro-domain of SpeB (99). The inhibitor is resistant to cleavage by active SpeB, making it an effective inhibitor of protease activity within the bacterial cytoplasm (99). Although SpeB is highly conserved across most S. pyogenes strains, its expression may vary depending on the type of infection (100). For example, when analyzing isolates from patients with pharyngitis, impetigo, invasive disease, or acute rheumatic fever, 40% of isolates from patients with acute rheumatic fever produced SpeB, compared to only 5.5% of isolates from impetigo patients (100). Therefore, the variability in SpeB expression suggests that SpeB is more important for certain forms of disease progression.
SpeB is considered an important virulence factor most likely due to the immunomodulating effects of its protease activity (101, 102). SpeB has broad proteolytic activity against a number of human proteins, including interleukin-1β (102), immunoglobulins (101), fibrinogen (103), fibronectin (104), kininogens (105), and a metalloprotease (106). Although SpeB has been shown to cleave certain immunoglobulins, a study investigating the cleavage of IgG under physiologic conditions found that IgG is only cleaved in a reduced form, whereas physiologic IgG is active in an oxidized form (107). Therefore, SpeB likely does not contribute to S. pyogenes pathogenesis through the cleavage of IgG. The innate host immune system is also weakened through proteolytic destruction of complement factors such as C3 and the membrane attack complex (108, 109). SpeB also has the capacity to cleave many human chemokines (110), further disrupting the immune response. In addition to the many immune modulating factors degraded by SpeB, the cysteine protease also degrades tight-junction proteins such as E-cadherin and desmogleins, facilitating the paracellular invasion of the bacteria across the epithelial barrier (111, 112). Therefore, the broad spectrum of SpeB targets likely ameliorates the virulence of S. pyogenes by perturbing the host immune system and compromising the epithelial barrier.
Given the broad spectrum of SpeB, S. pyogenes proteins can also be proteolyzed (113). SpeB can directly degrade multiple superantigens, such as SmeZ, which affects the ability of the superantigen to induce proliferation of human lymphocytes (114, 115). In addition, the cleavage of protein F1, an S. pyogenes cell wall-attached fibronectin-binding protein, decreased the fibronectin-dependent internalization of S. pyogenes into human cells, suggesting that SpeB also plays a role in the internalization process (116). Likewise, SpeB-deficient mutants were shown to have enhanced in vitro internalization into human epithelial and endothelial cells (117). Therefore, an inactive SpeB may be favored later during infection to promote the dissemination of the bacteria (116). Furthermore, earlier work identified a phase shift in an invasive M1T1 strain in vivo favoring a nonfunctional cysteine protease to conserve the function of the multiple virulence factors normally degraded by SpeB (118, 119). This phase shift is linked to inactivating mutations within the two-component CovRS sensor kinase, which results in a hypervirulent phenotype in the serotype M1 background (120). An inverse relationship has also been identified between SpeB expression and the severity of invasive group A streptococci infections, further suggesting that S. pyogenes may need to differentially regulate SpeB expression, depending on the site of infection and events occurring during infection (121).
In vivo studies supporting the role of SpeB as a virulence factor have not always been congruent. For example, an S. pyogenes strain with decreased SpeB expression exhibited a decreased capacity to cause murine necrotizing fasciitis, and complementation of SpeB restored the wild-type phenotype (122). Alternatively, isogenic gene replacement strains and a mouse model of invasive soft tissue infection showed that speB mutants had no apparent effect on the ability of group A streptococci to cause local tissue injury and invasive infection (123). Overall, the diversity of SpeB targets may contribute to the protease being both a virulence factor and a form of virulence regulation through the degradation of bacterial proteins.
DNases OF S. PYOGENES
The DNases of S. pyogenes constitute an additional mechanism that contributes to the pathogenicity of the bacterium. Through the analysis of all known S. pyogenes genomes, eight DNases have been identified (124): spnA, spdB, sda1, sda2, spd1, spd3, spd4, and sdn. Of the known DNases, spnA and spdB are chromosomally encoded and are found across all S. pyogenes isolates (125, 126). The six other DNases are prophage associated, and therefore they are only found in certain S. pyogenes strains (124). Furthermore, the DNase spnA is the only known cell wall-anchored DNase of S. pyogenes. (124, 126).
DNases contribute to the pathogenesis of S. pyogenes by facilitating innate immune evasion of the pathogen. During infection, neutrophils release antibacterial granule proteins and chromatin to create neutrophil extracellular traps (NETs) (127). NETs bind and trap bacteria and degrade bacterial virulence factors, ultimately resulting in bacterial death (127). The secreted DNase Sda1 and the cell wall-anchored DNase SpnA are both able to degrade the chromatin backbone of NETs, allowing the bacteria to avoid the degradation of their virulence factors and promote bacterial survival (128–130). S. pyogenes DNases further contribute to pathogenesis through the degradation of bacterial DNA (131). By degrading CpG-rich bacterial DNA, the TLR9-mediated immune response is weakened due to the degradation of the TLR9 activating signal (131). In a model of mouse necrotizing fasciitis, Sda1 was shown to suppress TLR9-dependent tumor necrosis factor-α and tumor necrosis factor-α induction, promoting evasion of the innate immune system (131). Additionally, a murine model of skin and soft tissue and a cynomolgus macaque model of pharyngitis infection showed that isogenic mutants of S. pyogenes lacking DNase production were less virulent than the wild-type strain, highlighting their importance in pathogenesis (129). The ability of DNases to degrade NETs and bacterial DNA, along with the decreased virulence of DNase-deficient mutants in vivo, emphasize the importance of S. pyogenes DNases as extracellular virulence factors.
STREPTOCOCCAL INHIBITOR OF COMPLEMENT
The streptococcal inhibitor of complement (SIC) is a 31-kDa protein secreted by certain S. pyogenes strains, most notably M1 serotypes (132). SIC is highly variable, with large numbers of variants arising in vivo during epidemic spread of the organism (133, 134). Such high variation suggests that the phenomenon offers selective advantage to the microbe. Most of the variants are single amino acid changes, insertions, or deletions. Other M types may make related proteins, some of which have minimal effect on the complement system (135). SIC was originally identified as a protein that inhibits the membrane attack complex of the complement system, with its mechanism being the inhibition of C567 insertion into the membrane (136). Furthermore, SIC is rapidly internalized by neutrophils and binds specifically to ezrin and moesin, proteins that link the actin cytoskeleton to the host cell surface, interfering with polymorphonuclear opsonophagocytosis and intracellular killing (136). SIC also directly inhibits other components of the innate immune system, including lysozyme, secretory leukocyte proteinase inhibitor, human defensins, and cathelicidin LL-37, all of which can be toxic to S. pyogenes (137–140). Additionally, SIC inhibits the release of bradykinin, a potent vasodilator, which could function to decrease inflammation at the site of infection, allowing the bacteria to persist (141). More recently, SIC also inhibited the bactericidal activity of histones, which are found in NETs (142).
In vivo studies suggest that SIC increases streptococcal virulence. Mice that were inoculated intranasally with M1 SIC-negative streptococci had a lower incidence of throat colonization than mice inoculated with the wild-type M1 strain (143). SIC-negative strains also exhibited a decrease in survival in macrophages (137). Therefore, SIC likely promotes bacterial survival and dissemination by allowing the organisms to avoid innate immune defenses in the extracellular environment.
IgG-TARGETING ENZYMES OF S. PYOGENES
S. pyogenes has evolved multiple mechanisms to target IgG antibodies, a major effector molecule of the humoral immune system. The immunoglobulin G-degrading enzyme (IdeS, also known as Mac) is a protease that removes the Fc region of IgG antibodies and can thus inhibit opsonophagocytosis of S. pyogenes (144, 145). This virulence factor is specific for IgG and does not target IgM, IgE, or IgD antibodies (144). However, in vivo studies using a mouse invasive infection model failed to demonstrate a contribution of IdeS to virulence (146). Endoglycosidase S (EndoS) is an ∼108-kDa secreted enzyme that hydrolyzes the β-1,4 linkage between the first two N-acetylglucosamine residues on the glycan linked to Asn297 within the CH2 domain of the IgG Fc region (147). Deglycosylation of IgG through this mechanism impairs antibody effector functions, including binding to the Fc-gamma receptor and complement activation (148). Nevertheless, similar to IdeS, deletion of the gene encoding EndoS in the M1 5448 background had no obvious phenotype in an invasive intraperitoneal mouse infection model (149). In both cases, the lack of an in vivo phenotype may reflect a more subtle role for these enzymes that is not apparent through acute, invasive infections. In addition to EndoS and IdeS, which show high levels of specificity for IgG, the broad-spectrum SpeB cysteine protease is also capable of cleaving IgG in the hinge region, similar to protease papain (147); however, the in vivo relevance of this has been questioned (107).
STREPTOKINASE
Streptokinase is a single-chain 414-amino acid protein that is able to activate the host protein plasminogen (150). Streptokinase forms a complex with the inactive zymogen plasminogen, or a trimolecular complex with plasminogen and fibrinogen, to generate the active serine protease plasmin (151, 152). These activator complexes bind to the bacterial surface, allowing the bacteria to acquire additional protease activity. Plasmin is responsible for degrading fibrin clots, connective tissue, extracellular matrix components, and adhesion proteins (153). Therefore, the improper activation of plasmin could result in widespread tissue damage and dissemination of the bacteria (154, 155). Transgenic mice expressing human plasminogen had an increased mortality rate compared to mice without the transgene. This susceptibility was directly related to streptokinase expression, highlighting the importance of streptokinase in S. pyogenes virulence (154). Furthermore, increased levels of bacteria-bound plasmin correlated with a decrease in C3b deposition and a decrease in C3b-mediated neutrophil killing (156). Additionally, bacteria-associated plasmin has been shown to prevent histone-mediated killing by NETs (157). In addition to activating plasminogen, streptokinase has also been shown to activate the contact system, resulting in the release of bradykinin (158). The release of bradykinin triggers vascular leakage, which could further promote the dissemination of the bacteria (159, 160). The release of streptokinase by S. pyogenes ultimately promotes the dissemination of the bacteria by facilitating the conversion of plasminogen to plasmin.
FUTURE PERSPECTIVES
The increased incidence of severe invasive group A streptococcal diseases, and the recent resurgence of scarlet fever, still remain incompletely explained; however, the streptococcal superantigens are clearly implicated in both illnesses and thus are key virulence factors driving the evolution of S. pyogenes pathogenesis. Indeed, the majority of genetic diversity in different strains and serotypes of S. pyogenes occurs due to the presence or absence of large mobile genetic elements, including lysogenic bacteriophage and integrative conjugative elements (161, 162). In particular, many of the superantigens, as well as DNases, and some other select toxins are encoded within bacteriophage elements, and this provides S. pyogenes with an extra level of adaptability. The apparent excessive redundancy of these toxins has yet to be explained, although we believe this allows S. pyogenes to avoid neutralizing antisuperantigen antibodies and may also provide additional means to efficiently target polymorphic MHC class II molecules from different populations.
Although most research on streptococcal exotoxins has focused on their role in severe streptococcal diseases, the established niche for S. pyogenes is a state of colonization in the throat or on the skin of humans. Also, multiple S. pyogenes virulence factors do not operate efficiently in mouse models (92, 154, 163–165). To understand the basic biology of this organism, which is not related to severe and invasive disease, better models will be necessary to evaluate specific virulence factors, therapies, and vaccines. Thus, most, if not all, of these remarkable exotoxins that can alter normal immune system function, damage tissue, and promote disease have each likely evolved in the context of streptococcal persistence and transmission. We hope a clearer understanding will lead to further rationales to design vaccines capable of targeting the colonization state of S. pyogenes.
ACKNOWLEDGMENTS
We acknowledge support from the University of Iowa and the Canadian Institutes of Health Research (CIHR) operating grant MOP-142137 and the Discovery Grant program from the Natural Sciences and Engineering Council of Canada.
REFERENCES
- 1.Nizet V, Beall B, Bast DJ, Datta V, Kilburn L, Low DE, De Azavedo JC. 2000. Genetic locus for streptolysin S production by group A streptococcus. Infect Immun 68:4245–4254 10.1128/IAI.68.7.4245-4254.2000. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Molloy EM, Cotter PD, Hill C, Mitchell DA, Ross RP. 2011. Streptolysin S-like virulence factors: the continuing sagA. Nat Rev Microbiol 9:670–681 10.1038/nrmicro2624. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Datta V, Myskowski SM, Kwinn LA, Chiem DN, Varki N, Kansal RG, Kotb M, Nizet V. 2005. Mutational analysis of the group A streptococcal operon encoding streptolysin S and its virulence role in invasive infection. Mol Microbiol 56:681–695 10.1111/j.1365-2958.2005.04583.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 4.Goldmann O, Sastalla I, Wos-Oxley M, Rohde M, Medina E. 2009. Streptococcus pyogenes induces oncosis in macrophages through the activation of an inflammatory programmed cell death pathway. Cell Microbiol 11:138–155 10.1111/j.1462-5822.2008.01245.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 5.Miyoshi-Akiyama T, Takamatsu D, Koyanagi M, Zhao J, Imanishi K, Uchiyama T. 2005. Cytocidal effect of Streptococcus pyogenes on mouse neutrophils in vivo and the critical role of streptolysin S. J Infect Dis 192:107–116 10.1086/430617. [PubMed] [DOI] [PubMed] [Google Scholar]
- 6.Flaherty RA, Puricelli JM, Higashi DL, Park CJ, Lee SW. 2015. Streptolysin S promotes programmed cell death and enhances inflammatory signaling in epithelial keratinocytes during group A Streptococcus infection. Infect Immun 83:4118–4133 10.1128/IAI.00611-15. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carr A, Sledjeski DD, Podbielski A, Boyle MDP, Kreikemeyer B. 2001. Similarities between complement-mediated and streptolysin S-mediated hemolysis. J Biol Chem 276:41790–41796 10.1074/jbc.M107401200. [PubMed] [DOI] [PubMed] [Google Scholar]
- 8.Higashi DL, Biais N, Donahue DL, Mayfield JA, Tessier CR, Rodriguez K, Ashfeld BL, Luchetti J, Ploplis VA, Castellino FJ, Lee SW. 2016. Activation of band 3 mediates group A Streptococcus streptolysin S-based beta-haemolysis. Nat Microbiol 1:15004 10.1038/nmicrobiol.2015.4. [PubMed] [DOI] [PubMed] [Google Scholar]
- 9.Sumitomo T, Nakata M, Higashino M, Jin Y, Terao Y, Fujinaga Y, Kawabata S. 2011. Streptolysin S contributes to group A streptococcal translocation across an epithelial barrier. J Biol Chem 286:2750–2761 10.1074/jbc.M110.171504. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Betschel SD, Borgia SM, Barg NL, Low DE, De Azavedo JCS. 1998. Reduced virulence of group A streptococcal Tn916 mutants that do not produce streptolysin S. Infect Immun 66:1671–1679. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kehoe MA, Miller L, Walker JA, Boulnois GJ. 1987. Nucleotide sequence of the streptolysin O (SLO) gene: structural homologies between SLO and other membrane-damaging, thiol-activated toxins. Infect Immun 55:3228–3232. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tweten RK, Hotze EM, Wade KR. 2015. The unique molecular choreography of giant pore formation by the cholesterol-dependent cytolysins of Gram-positive bacteria. Annu Rev Microbiol 69:323–340 10.1146/annurev-micro-091014-104233. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhakdi S, Tranum-Jensen J, Sziegoleit A. 1985. Mechanism of membrane damage by streptolysin-O. Infect Immun 47:52–60. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shewell LK, Harvey RM, Higgins MA, Day CJ, Hartley-Tassell LE, Chen AY, Gillen CM, James DBA, Alonzo F III, Torres VJ, Walker MJ, Paton AW, Paton JC, Jennings MP. 2014. The cholesterol-dependent cytolysins pneumolysin and streptolysin O require binding to red blood cell glycans for hemolytic activity. Proc Natl Acad Sci U S A 111:E5312–E5320 10.1073/pnas.1412703111. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Timmer AM, Timmer JC, Pence MA, Hsu LC, Ghochani M, Frey TG, Karin M, Salvesen GS, Nizet V. 2009. Streptolysin O promotes group A Streptococcus immune evasion by accelerated macrophage apoptosis. J Biol Chem 284:862–871 10.1074/jbc.M804632200. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uchiyama S, Döhrmann S, Timmer AM, Dixit N, Ghochani M, Bhandari T, Timmer JC, Sprague K, Bubeck-Wardenburg J, Simon SI, Nizet V. 2015. Streptolysin O rapidly impairs neutrophil oxidative burst and antibacterial responses to group A Streptococcus. Front Immunol 6:581 10.3389/fimmu.2015.00581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruiz N, Wang B, Pentland A, Caparon M. 1998. Streptolysin O and adherence synergistically modulate proinflammatory responses of keratinocytes to group A streptococci. Mol Microbiol 27:337–346 10.1046/j.1365-2958.1998.00681.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 18.Michos A, Gryllos I, Håkansson A, Srivastava A, Kokkotou E, Wessels MR. 2006. Enhancement of streptolysin O activity and intrinsic cytotoxic effects of the group A streptococcal toxin, NAD-glycohydrolase. J Biol Chem 281:8216–8223 10.1074/jbc.M511674200. [PubMed] [DOI] [PubMed] [Google Scholar]
- 19.Madden JC, Ruiz N, Caparon M. 2001. Cytolysin-mediated translocation (CMT): a functional equivalent of type III secretion in Gram-positive bacteria. Cell 104:143–152 10.1016/S0092-8674(01)00198-2. [DOI] [PubMed] [Google Scholar]
- 20.Sharma O, O’Seaghdha M, Velarde JJ, Wessels MR. 2016. NAD+-glycohydrolase promotes intracellular survival of group A Streptococcus. PLoS Pathog 12:e1005468 10.1371/journal.ppat.1005468. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bricker AL, Cywes C, Ashbaugh CD, Wessels MR. 2002. NAD+-glycohydrolase acts as an intracellular toxin to enhance the extracellular survival of group A streptococci. Mol Microbiol 44:257–269 10.1046/j.1365-2958.2002.02876.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 22.Zhu L, Olsen RJ, Lee JD, Porter AR, DeLeo FR, Musser JM. 2017. Contribution of Secreted NADase and streptolysin O to the pathogenesis of epidemic serotype M1 Streptococcus pyogenes infections. Am J Pathol 187:605–613 10.1016/j.ajpath.2016.11.003. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sierig G, Cywes C, Wessels MR, Ashbaugh CD. 2003. Cytotoxic effects of streptolysin O and streptolysin S enhance the virulence of poorly encapsulated group A streptococci. Infect Immun 71:446–455 10.1128/IAI.71.1.446-455.2003. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fontaine MC, Lee JJ, Kehoe MA. 2003. Combined contributions of streptolysin O and streptolysin S to virulence of serotype M5 Streptococcus pyogenes strain Manfredo. Infect Immun 71:3857–3865 10.1128/IAI.71.7.3857-3865.2003. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.White J, Herman A, Pullen AM, Kubo R, Kappler JW, Marrack P. 1989. The V beta-specific superantigen staphylococcal enterotoxin B: stimulation of mature T cells and clonal deletion in neonatal mice. Cell 56:27–35 10.1016/0092-8674(89)90980-X. [DOI] [PubMed] [Google Scholar]
- 26.Dick GF, Dick GH. 1924. A skin test for susceptibility to scarlet fever. JAMA 82:256–266 10.1001/jama.1924.02650300011003. [DOI] [Google Scholar]
- 27.Watson DW. 1960. Host-parasite factors in group A streptococcal infections. Pyrogenic and other effects of immunologic distinct exotoxins related to scarlet fever toxins. J Exp Med 111:255–284 10.1084/jem.111.2.255. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schlievert PM, Bettin KM, Watson DW. 1979. Reinterpretation of the Dick test: role of group A streptococcal pyrogenic exotoxin. Infect Immun 26:467–472. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bohach GA, Fast DJ, Nelson RD, Schlievert PM. 1990. Staphylococcal and streptococcal pyrogenic toxins involved in toxic shock syndrome and related illnesses. Crit Rev Microbiol 17:251–272 10.3109/10408419009105728. [PubMed] [DOI] [PubMed] [Google Scholar]
- 30.Kotb M. 1995. Bacterial pyrogenic exotoxins as superantigens. Clin Microbiol Rev 8:411–426 10.1128/CMR.8.3.411. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marrack P, Kappler J. 1990. The staphylococcal enterotoxins and their relatives. Science 248:1066 10.1126/science.248.4959.1066-b. [PubMed] [DOI] [PubMed] [Google Scholar]
- 32.McCormick JK, Yarwood JM, Schlievert PM. 2001. Toxic shock syndrome and bacterial superantigens: an update. Annu Rev Microbiol 55:77–104 10.1146/annurev.micro.55.1.77. [PubMed] [DOI] [PubMed] [Google Scholar]
- 33.Assimacopoulos AP, Stoehr JA, Schlievert PM. 1997. Mitogenic factors from group G streptococci associated with scarlet fever and streptococcal toxic shock syndrome. Adv Exp Med Biol 418:109–114 10.1007/978-1-4899-1825-3_27. [PubMed] [DOI] [PubMed] [Google Scholar]
- 34.Miyoshi-Akiyama T, Zhao J, Kato H, Kikuchi K, Totsuka K, Kataoka Y, Katsumi M, Uchiyama T. 2003. Streptococcus dysgalactiae-derived mitogen (SDM), a novel bacterial superantigen: characterization of its biological activity and predicted tertiary structure. Mol Microbiol 47:1589–1599 10.1046/j.1365-2958.2003.03411.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 35.Proft T, Moffatt SL, Weller KD, Paterson A, Martin D, Fraser JD. 2000. The streptococcal superantigen SMEZ exhibits wide allelic variation, mosaic structure, and significant antigenic variation. J Exp Med 191:1765–1776 10.1084/jem.191.10.1765. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paillot R, Darby AC, Robinson C, Wright NL, Steward KF, Anderson E, Webb K, Holden MTG, Efstratiou A, Broughton K, Jolley KA, Priestnall SL, Marotti Campi MC, Hughes MA, Radford A, Erles K, Waller AS. 2010. Identification of three novel superantigen-encoding genes in Streptococcus equi subsp. zooepidemicus, szeF, szeN, and szeP. Infect Immun 78:4817–4827 10.1128/IAI.00751-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paillot R, Robinson C, Steward K, Wright N, Jourdan T, Butcher N, Heather Z, Waller AS. 2010. Contribution of each of four superantigens to Streptococcus equi-induced mitogenicity, gamma interferon synthesis, and immunity. Infect Immun 78:1728–1739 10.1128/IAI.01079-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Banks DJ, Porcella SF, Barbian KD, Beres SB, Philips LE, Voyich JM, DeLeo FR, Martin JM, Somerville GA, Musser JM. 2004. Progress toward characterization of the group A Streptococcus metagenome: complete genome sequence of a macrolide-resistant serotype M6 strain. J Infect Dis 190:727–738 10.1086/422697. [PubMed] [DOI] [PubMed] [Google Scholar]
- 39.Beres SB, Sylva GL, Barbian KD, Lei B, Hoff JS, Mammarella ND, Liu MY, Smoot JC, Porcella SF, Parkins LD, Campbell DS, Smith TM, McCormick JK, Leung DY, Schlievert PM, Musser JM. 2002. Genome sequence of a serotype M3 strain of group A Streptococcus: phage-encoded toxins, the high-virulence phenotype, and clone emergence. Proc Natl Acad Sci U S A 99:10078–10083 10.1073/pnas.152298499. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferretti JJ, McShan WM, Ajdic D, Savic DJ, Savic G, Lyon K, Primeaux C, Sezate S, Suvorov AN, Kenton S, Lai HS, Lin SP, Qian Y, Jia HG, Najar FZ, Ren Q, Zhu H, Song L, White J, Yuan X, Clifton SW, Roe BA, McLaughlin R. 2001. Complete genome sequence of an M1 strain of Streptococcus pyogenes. Proc Natl Acad Sci U S A 98:4658–4663 10.1073/pnas.071559398. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakagawa I, Kurokawa K, Yamashita A, Nakata M, Tomiyasu Y, Okahashi N, Kawabata S, Yamazaki K, Shiba T, Yasunaga T, Hayashi H, Hattori M, Hamada S. 2003. Genome sequence of an M3 strain of Streptococcus pyogenes reveals a large-scale genomic rearrangement in invasive strains and new insights into phage evolution. Genome Res 13:1042–1055 10.1101/gr.1096703. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smoot JC, Barbian KD, Van Gompel JJ, Smoot LM, Chaussee MS, Sylva GL, Sturdevant DE, Ricklefs SM, Porcella SF, Parkins LD, Beres SB, Campbell DS, Smith TM, Zhang Q, Kapur V, Daly JA, Veasy LG, Musser JM. 2002. Genome sequence and comparative microarray analysis of serotype M18 group A Streptococcus strains associated with acute rheumatic fever outbreaks. Proc Natl Acad Sci U S A 99:4668–4673 10.1073/pnas.062526099. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Commons RJ, Smeesters PR, Proft T, Fraser JD, Robins-Browne R, Curtis N. 2014. Streptococcal superantigens: categorization and clinical associations. Trends Mol Med 20:48–62 10.1016/j.molmed.2013.10.004. [PubMed] [DOI] [PubMed] [Google Scholar]
- 44.Li H, Llera A, Malchiodi EL, Mariuzza RA. 1999. The structural basis of T cell activation by superantigens. Annu Rev Immunol 17:435–466 10.1146/annurev.immunol.17.1.435. [PubMed] [DOI] [PubMed] [Google Scholar]
- 45.Mitchell DT, Levitt DG, Schlievert PM, Ohlendorf DH. 2000. Structural evidence for the evolution of pyrogenic toxin superantigens. J Mol Evol 51:520–531 10.1007/s002390010116. [PubMed] [DOI] [PubMed] [Google Scholar]
- 46.Sundberg EJ, Andersen PS, Schlievert PM, Karjalainen K, Mariuzza RA. 2003. Structural, energetic, and functional analysis of a protein-protein interface at distinct stages of affinity maturation. Structure 11:1151–1161 10.1016/S0969-2126(03)00187-4. [DOI] [PubMed] [Google Scholar]
- 47.Bueno C, Criado G, McCormick JKJK, Madrenas J. 2007. T cell signalling induced by bacterial superantigens. Chem Immunol Allergy 93:161–180 10.1159/000100894. [PubMed] [DOI] [PubMed] [Google Scholar]
- 48.Sundberg EJ, Li H, Llera AS, McCormick JK, Tormo J, Schlievert PM, Karjalainen K, Mariuzza RA. 2002. Structures of two streptococcal superantigens bound to TCR beta chains reveal diversity in the architecture of T cell signaling complexes. Structure 10:687–699 10.1016/S0969-2126(02)00759-1. [DOI] [PubMed] [Google Scholar]
- 49.Li H, Llera A, Tsuchiya D, Leder L, Ysern X, Schlievert PM, Karjalainen K, Mariuzza RA. 1998. Three-dimensional structure of the complex between a T cell receptor beta chain and the superantigen staphylococcal enterotoxin B. Immunity 9:807–816 10.1016/S1074-7613(00)80646-9. [DOI] [PubMed] [Google Scholar]
- 50.Fields BA, Malchiodi EL, Li H, Ysern X, Stauffacher CV, Schlievert PM, Karjalainen K, Mariuzza RA. 1996. Crystal structure of a T-cell receptor β-chain complexed with a superantigen. Nature 384:188–192 10.1038/384188a0. [PubMed] [DOI] [PubMed] [Google Scholar]
- 51.Nur-ur Rahman AK, Bonsor DA, Herfst CA, Pollard F, Peirce M, Wyatt AW, Kasper KJ, Madrenas J, Sundberg EJ, McCormick JK. 2011. The T cell receptor β-chain second complementarity determining region loop (CDR2β) governs T cell activation and Vβ specificity by bacterial superantigens. J Biol Chem 11:286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li PL, Tiedemann RE, Moffat SL, Fraser JD. 1997. The superantigen streptococcal pyrogenic exotoxin C (SPE-C) exhibits a novel mode of action. J Exp Med 186:375–383 10.1084/jem.186.3.375. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McCormick JK, Tripp TJ, Olmsted SB, Matsuka YV, Gahr PJ, Ohlendorf DH, Schlievert PM. 2000. Development of streptococcal pyrogenic exotoxin C vaccine toxoids that are protective in the rabbit model of toxic shock syndrome. J Immunol 165:2306–2312 10.4049/jimmunol.165.4.2306. [PubMed] [DOI] [PubMed] [Google Scholar]
- 54.Swietnicki W, Barnie AM, Dyas BK, Ulrich RG. 2003. Zinc binding and dimerization of Streptococcus pyogenes pyrogenic exotoxin C are not essential for T-cell stimulation. J Biol Chem 278:9885–9895 10.1074/jbc.M206957200. [PubMed] [DOI] [PubMed] [Google Scholar]
- 55.Tripp TJ, McCormick JK, Webb JM, Schlievert PM. 2003. The zinc-dependent major histocompatibility complex class II binding site of streptococcal pyrogenic exotoxin C is critical for maximal superantigen function and toxic activity. Infect Immun 71:1548–1550 10.1128/IAI.71.3.1548-1550.2003. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kasper KJ, Xi W, Rahman AK, Nooh MM, Kotb M, Sundberg EJ, Madrenas J, McCormick JK. 2008. Molecular requirements for MHC class II α-chain engagement and allelic discrimination by the bacterial superantigen streptococcal pyrogenic exotoxin C. J Immunol 181:3384–3392 10.4049/jimmunol.181.5.3384. [PubMed] [DOI] [PubMed] [Google Scholar]
- 57.Li Y, Li H, Dimasi N, McCormick JKJK, Martin R, Schuck P, Schlievert PMPM, Mariuzza RA. 2001. Crystal structure of a superantigen bound to the high-affinity, zinc-dependent site on MHC class II. Immunity 14:93–104 10.1016/S1074-7613(01)00092-9. [PubMed] [DOI] [PubMed] [Google Scholar]
- 58.Petersson K, Thunnissen M, Forsberg G, Walse B. 2002. Crystal structure of a SEA variant in complex with MHC class II reveals the ability of SEA to crosslink MHC molecules. Structure 10:1619–1626 10.1016/S0969-2126(02)00895-X. [PubMed] [DOI] [PubMed] [Google Scholar]
- 59.Massell BF, Chute CG, Walker AM, Kurland GS. 1988. Penicillin and the marked decrease in morbidity and mortality from rheumatic fever in the United States. N Engl J Med 318:280–286 10.1056/NEJM198802043180504. [PubMed] [DOI] [PubMed] [Google Scholar]
- 60.Cone LA, Woodard DR, Schlievert PM, Tomory GS. 1987. Clinical and bacteriologic observations of a toxic shock-like syndrome due to Streptococcus pyogenes. N Engl J Med 317:146–149 10.1056/NEJM198707163170305. [PubMed] [DOI] [PubMed] [Google Scholar]
- 61.Stevens DL, Tanner MH, Winship J, Swarts R, Ries KM, Schlievert PM, Kaplan E. 1989. Severe group A streptococcal infections associated with a toxic shock-like syndrome and scarlet fever toxin A. N Engl J Med 321:1–7 10.1056/NEJM198907063210101. [PubMed] [DOI] [PubMed] [Google Scholar]
- 62.Cleary PP, Kaplan EL, Handley JP, Wlazlo A, Kim MH, Hauser AR, Schlievert PM. 1992. Clonal basis for resurgence of serious Streptococcus pyogenes disease in the 1980s. Lancet 339:518–521 10.1016/0140-6736(92)90339-5. [DOI] [PubMed] [Google Scholar]
- 63.Musser JM, Kapur V, Szeto J, Pan X, Swanson DS, Martin DR. 1995. Genetic diversity and relationships among Streptococcus pyogenes strains expressing serotype M1 protein: recent intercontinental spread of a subclone causing episodes of invasive disease. Infect Immun 63:994–1003. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schlievert PM, Assimacopoulos AP, Cleary PP. 1996. Severe invasive group A streptococcal disease: clinical description and mechanisms of pathogenesis. J Lab Clin Med 127:13–22 10.1016/S0022-2143(96)90161-4. [DOI] [PubMed] [Google Scholar]
- 65.Sumby P, Porcella SF, Madrigal AG, Barbian KD, Virtaneva K, Ricklefs SM, Sturdevant DE, Graham MR, Vuopio-Varkila J, Hoe NP, Musser JM. 2005. Evolutionary origin and emergence of a highly successful clone of serotype M1 group A Streptococcus involved multiple horizontal gene transfer events. J Infect Dis 192:771–782 10.1086/432514. [PubMed] [DOI] [PubMed] [Google Scholar]
- 66.Maamary PG, Ben Zakour NL, Cole JN, Hollands A, Aziz RK, Barnett TC, Cork AJ, Henningham A, Sanderson-Smith M, McArthur JD, Venturini C, Gillen CM, Kirk JK, Johnson DR, Taylor WL, Kaplan EL, Kotb M, Nizet V, Beatson SA, Walker MJ, Zakour B. 2012. Tracing the evolutionary history of the pandemic group A streptococcal M1T1 clone. FASEB J 26:4675–4684 10.1096/fj.12-212142. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu Y, Chan TC, Yap LW, Luo Y, Xu W, Qin S, Zhao N, Yu Z, Geng X, Liu SL. 2018. Resurgence of scarlet fever in China: a 13-year population-based surveillance study. Lancet Infect Dis 18:903–912 10.1016/S1473-3099(18)30231-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lamagni T, Guy R, Chand M, Henderson KL, Chalker V, Lewis J, Saliba V, Elliot AJ, Smith GE, Rushton S, Sheridan EA, Ramsay M, Johnson AP. 2018. Resurgence of scarlet fever in England, 2014-16: a population-based surveillance study. Lancet Infect Dis 18:180–187 10.1016/S1473-3099(17)30693-X. [PubMed] [DOI] [PubMed] [Google Scholar]
- 69.Park DW, Kim SH, Park JW, Kim MJ, Cho SJ, Park HJ, Jung SH, Seo MH, Lee YS, Kim BH, Min H, Lee SY, Ha DR, Kim ES, Hong Y, Chung JK. 2017. Incidence and characteristics of scarlet fever, South Korea, 2008-2015. Emerg Infect Dis 23:658–661 10.3201/eid2304.160773. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brockmann SO, Eichner L, Eichner M. 2018. Constantly high incidence of scarlet fever in Germany. Lancet Infect Dis 18:499–500 10.1016/S1473-3099(18)30210-X. [DOI] [PubMed] [Google Scholar]
- 71.Davies MR, Holden MT, Coupland P, Chen JHK, Venturini C, Barnett TC, Zakour NL, Tse H, Dougan G, Yuen K-Y, Walker MJ. 2015. Emergence of scarlet fever Streptococcus pyogenes emm12 clones in Hong Kong is associated with toxin acquisition and multidrug resistance. Nat Genet 47:84–87 10.1038/ng.3147. [PubMed] [DOI] [PubMed] [Google Scholar]
- 72.Ben Zakour NL, Davies MR, You Y, Chen JHK, Forde BM, Stanton-Cook M, Yang R, Cui Y, Barnett TC, Venturini C, Ong CL, Tse H, Dougan G, Zhang J, Yuen K-Y, Beatson SA, Walker MJ. 2015. Transfer of scarlet fever-associated elements into the group A Streptococcus M1T1 clone. Sci Rep 5:15877 10.1038/srep15877. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Turner CE, Pyzio M, Song B, Lamagni T, Meltzer M, Chow JY, Efstratiou A, Curtis S, Sriskandan S. 2016. Scarlet fever upsurge in England and molecular-genetic analysis in North-West London, 2014. Emerg Infect Dis 22:1075–1078 10.3201/eid2206.151726. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Musser JM, Hauser AR, Kim MH, Schlievert PM, Nelson K, Selander RK. 1991. Streptococcus pyogenes causing toxic-shock-like syndrome and other invasive diseases: clonal diversity and pyrogenic exotoxin expression. Proc Natl Acad Sci U S A 88:2668–2672 10.1073/pnas.88.7.2668. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cockerill FR III, MacDonald KL, Thompson RL, Roberson F, Kohner PC, Besser-Wiek J, Manahan JM, Musser JM, Schlievert PM, Talbot J, Frankfort B, Steckelberg JM, Wilson WR, Osterholm MT. 1997. An outbreak of invasive group A streptococcal disease associated with high carriage rates of the invasive clone among school-aged children. JAMA 277:38–43 10.1001/jama.1997.03540250046030. [PubMed] [DOI] [PubMed] [Google Scholar]
- 76.Demers B, Simor AE, Vellend H, Schlievert PM, Byrne S, Jamieson F, Walmsley S, Low DE. 1993. Severe invasive group A streptococcal infections in Ontario, Canada: 1987-1991. Clin Infect Dis 16:792–800, discussion 801–802 10.1093/clind/16.6.792. [PubMed] [DOI] [PubMed] [Google Scholar]
- 77.Hauser AR, Stevens DL, Kaplan EL, Schlievert PM. 1991. Molecular analysis of pyrogenic exotoxins from Streptococcus pyogenes isolates associated with toxic shock-like syndrome. J Clin Microbiol 29:1562–1567. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Holm SE, Norrby A, Bergholm AM, Norgren M. 1992. Aspects of pathogenesis of serious group A streptococcal infections in Sweden, 1988-1989. J Infect Dis 166:31–37 10.1093/infdis/166.1.31. [PubMed] [DOI] [PubMed] [Google Scholar]
- 79.Roggiani M, Stoehr JA, Olmsted SB, Matsuka YV, Pillai S, Ohlendorf DH, Schlievert PM. 2000. Toxoids of streptococcal pyrogenic exotoxin A are protective in rabbit models of streptococcal toxic shock syndrome. Infect Immun 68:5011–5017 10.1128/IAI.68.9.5011-5017.2000. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bessen DE, Izzo MW, Fiorentino TR, Caringal RM, Hollingshead SK, Beall B. 1999. Genetic linkage of exotoxin alleles and emm gene markers for tissue tropism in group A streptococci. J Infect Dis 179:627–636 10.1086/314631. [PubMed] [DOI] [PubMed] [Google Scholar]
- 81.Nelson K, Schlievert PM, Selander RK, Musser JM. 1991. Characterization and clonal distribution of four alleles of the speA gene encoding pyrogenic exotoxin A (scarlet fever toxin) in Streptococcus pyogenes. J Exp Med 174:1271–1274 10.1084/jem.174.5.1271. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McCormick JK, Pragman AA, Stolpa JC, Leung DYM, Schlievert PM. 2001. Functional characterization of streptococcal pyrogenic exotoxin J, a novel superantigen. Infect Immun 69:1381–1388 10.1128/IAI.69.3.1381-1388.2001. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sriskandan S, Moyes D, Cohen J. 1996. Detection of circulating bacterial superantigen and lymphotoxin-alpha in patients with streptococcal toxic-shock syndrome. Lancet 348:1315–1316 10.1016/S0140-6736(05)65800-X. [DOI] [PubMed] [Google Scholar]
- 84.Basma H, Norrby-Teglund A, Guedez Y, McGeer A, Low DE, El-Ahmedy O, Schwartz B, Kotb M. 1999. Risk factors in the pathogenesis of invasive group A streptococcal infections: role of protective humoral immunity. Infect Immun 67:1871–1877. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Eriksson BK, Andersson J, Holm SE, Norgren M. 1999. Invasive group A streptococcal infections: T1M1 isolates expressing pyrogenic exotoxins A and B in combination with selective lack of toxin-neutralizing antibodies are associated with increased risk of streptococcal toxic shock syndrome. J Infect Dis 180:410–418 10.1086/314872. [PubMed] [DOI] [PubMed] [Google Scholar]
- 86.Parsonnet J, Gillis ZA, Richter AG, Pier GB. 1987. A rabbit model of toxic shock syndrome that uses a constant, subcutaneous infusion of toxic shock syndrome toxin 1. Infect Immun 55:1070–1076. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sriskandan S, Unnikrishnan M, Krausz T, Cohen J. 1999. Molecular analysis of the role of streptococcal pyrogenic exotoxin A (SPEA) in invasive soft-tissue infection resulting from Streptococcus pyogenes. Mol Microbiol 33:778–790 10.1046/j.1365-2958.1999.01525.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 88.Unnikrishnan M, Cohen J, Sriskandan S. 2001. Complementation of a speA negative Streptococcus pyogenes with speA: effects on virulence and production of streptococcal pyrogenic exotoxin A. Microb Pathog 31:109–114 10.1006/mpat.2001.0453. [PubMed] [DOI] [PubMed] [Google Scholar]
- 89.Kotb M, Norrby-Teglund A, McGeer A, El-Sherbini H, Dorak MT, Khurshid A, Green K, Peeples J, Wade J, Thomson G, Schwartz B, Low DE. 2002. An immunogenetic and molecular basis for differences in outcomes of invasive group A streptococcal infections. Nat Med 8:1398–1404 10.1038/nm1202-800. [PubMed] [DOI] [PubMed] [Google Scholar]
- 90.Norrby-Teglund A, Nepom GT, Kotb M. 2002. Differential presentation of group A streptococcal superantigens by HLA class II DQ and DR alleles. Eur J Immunol 32:2570–2577 . [DOI] [PubMed] [Google Scholar]
- 91.Llewelyn M, Sriskandan S, Peakman M, Ambrozak DR, Douek DC, Kwok WW, Cohen J, Altmann DM. 2004. HLA class II polymorphisms determine responses to bacterial superantigens. J Immunol 172:1719–1726 10.4049/jimmunol.172.3.1719. [PubMed] [DOI] [PubMed] [Google Scholar]
- 92.Kasper KJ, Zeppa JJ, Wakabayashi AT, Xu SX, Mazzuca DM, Welch I, Baroja ML, Kotb M, Cairns E, Cleary PP, Haeryfar SMM, McCormick JK. 2014. Bacterial superantigens promote acute nasopharyngeal infection by Streptococcus pyogenes in a human MHC class II-dependent manner. PLoS Pathog 10:e1004155 10.1371/journal.ppat.1004155. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zeppa JJ, Wakabayashi AT, Kasper KJ, Xu SX, Haeryfar SMM, McCormick JK. 2016. Nasopharyngeal infection of mice with Streptococcus pyogenes and in vivo detection of superantigen activity. Methods Mol Biol 1396:95–107 10.1007/978-1-4939-3344-0_8. [PubMed] [DOI] [PubMed] [Google Scholar]
- 94.Zeppa JJ, Kasper KJ, Mohorovic I, Mazzuca DM, Haeryfar SMM, McCormick JK. 2017. Nasopharyngeal infection by Streptococcus pyogenes requires superantigen-responsive Vβ-specific T cells. Proc Natl Acad Sci U S A 114:10226–10231 10.1073/pnas.1700858114. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chaussee MS, Phillips ER, Ferretti JJ. 1997. Temporal production of streptococcal erythrogenic toxin B (streptococcal cysteine proteinase) in response to nutrient depletion. Infect Immun 65:1956–1959. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yu CE, Ferretti JJ. 1991. Frequency of the erythrogenic toxin B and C genes (speB and speC) among clinical isolates of group A streptococci. Infect Immun 59:211–215. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bustin M, Lin MC, Stein WH, Moore S. 1970. Activity of the reduced zymogen of streptococcal proteinase. J Biol Chem 245:846–849. [PubMed] [PubMed] [Google Scholar]
- 98.Doran JD, Nomizu M, Takebe S, Ménard R, Griffith D, Ziomek E. 1999. Autocatalytic processing of the streptococcal cysteine protease zymogen: processing mechanism and characterization of the autoproteolytic cleavage sites. Eur J Biochem 263:145–151 10.1046/j.1432-1327.1999.00473.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 99.Kagawa TF, O’Toole PW, Cooney JC. 2005. SpeB-Spi: a novel protease-inhibitor pair from Streptococcus pyogenes. Mol Microbiol 57:650–666 10.1111/j.1365-2958.2005.04708.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 100.Ly AT, Noto JP, Walwyn OL, Tanz RR, Shulman ST, Kabat W, Bessen DE. 2017. Differences in SpeB protease activity among group A streptococci associated with superficial, invasive, and autoimmune disease. PLoS One 12:e0177784 10.1371/journal.pone.0177784. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Collin M, Olsén A. 2003. Extracellular enzymes with immunomodulating activities: variations on a theme in Streptococcus pyogenes. Infect Immun 71:2983–2992 10.1128/IAI.71.6.2983-2992.2003. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kapur V, Majesky MW, Li LL, Black RA, Musser JM. 1993. Cleavage of interleukin 1 beta (IL-1 beta) precursor to produce active IL-1 beta by a conserved extracellular cysteine protease from Streptococcus pyogenes. Proc Natl Acad Sci U S A 90:7676–7680 10.1073/pnas.90.16.7676. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Matsuka YV, Pillai S, Gubba S, Musser JM, Olmsted SB. 1999. Fibrinogen cleavage by the Streptococcus pyogenes extracellular cysteine protease and generation of antibodies that inhibit enzyme proteolytic activity. Infect Immun 67:4326–4333. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kapur V, Topouzis S, Majesky MW, Li L-L, Hamrick MR, Hamill RJ, Patti JM, Musser JM. 1993. A conserved Streptococcus pyogenes extracellular cysteine protease cleaves human fibronectin and degrades vitronectin. Microb Pathog 15:327–346 10.1006/mpat.1993.1083. [PubMed] [DOI] [PubMed] [Google Scholar]
- 105.Herwald H, Collin M, Müller-Esterl W, Björck L. 1996. Streptococcal cysteine proteinase releases kinins: a virulence mechanism. J Exp Med 184:665–673 10.1084/jem.184.2.665. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Burns EH Jr, Marciel AM, Musser JM. 1996. Activation of a 66-kilodalton human endothelial cell matrix metalloprotease by Streptococcus pyogenes extracellular cysteine protease. Infect Immun 64:4744–4750. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Persson H, Vindebro R, von Pawel-Rammingen U. 2013. The streptococcal cysteine protease SpeB is not a natural immunoglobulin-cleaving enzyme. Infect Immun 81:2236–2241 10.1128/IAI.00168-13. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kuo CF, Lin YS, Chuang WJ, Wu JJ, Tsao N. 2008. Degradation of complement 3 by streptococcal pyrogenic exotoxin B inhibits complement activation and neutrophil opsonophagocytosis. Infect Immun 76:1163–1169 10.1128/IAI.01116-07. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Honda-Ogawa M, Ogawa T, Terao Y, Sumitomo T, Nakata M, Ikebe K, Maeda Y, Kawabata S. 2013. Cysteine proteinase from Streptococcus pyogenes enables evasion of innate immunity via degradation of complement factors. J Biol Chem 288:15854–15864 10.1074/jbc.M113.469106. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Egesten A, Olin AI, Linge HM, Yadav M, Mörgelin M, Karlsson A, Collin M. 2009. SpeB of Streptococcus pyogenes differentially modulates antibacterial and receptor activating properties of human chemokines. PLoS One 4:e4769 10.1371/journal.pone.0004769. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sumitomo T, Nakata M, Higashino M, Terao Y, Kawabata S. 2013. Group A streptococcal cysteine protease cleaves epithelial junctions and contributes to bacterial translocation. J Biol Chem 288:13317–13324 10.1074/jbc.M113.459875. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sumitomo T, Mori Y, Nakamura Y, Honda-Ogawa M, Nakagawa S, Yamaguchi M, Matsue H, Terao Y, Nakata M, Kawabata S. 2018. Streptococcal cysteine protease-mediated cleavage of desmogleins is involved in the pathogenesis of cutaneous infection. Front Cell Infect Microbiol 8:10 10.3389/fcimb.2018.00010. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Berge A, Björck L. 1995. Streptococcal cysteine proteinase releases biologically active fragments of streptococcal surface proteins. J Biol Chem 270:9862–9867 10.1074/jbc.270.17.9862. [PubMed] [DOI] [PubMed] [Google Scholar]
- 114.Kansal RG, Nizet V, Jeng A, Chuang WJ, Kotb M. 2003. Selective modulation of superantigen-induced responses by streptococcal cysteine protease. J Infect Dis 187:398–407 10.1086/368022. [PubMed] [DOI] [PubMed] [Google Scholar]
- 115.Nooh MM, Aziz RK, Kotb M, Eroshkin A, Chuang WJ, Proft T, Kansal R. 2006. Streptococcal mitogenic exotoxin, SmeZ, is the most susceptible M1T1 streptococcal superantigen to degradation by the streptococcal cysteine protease, SpeB. J Biol Chem 281:35281–35288 10.1074/jbc.M605544200. [DOI] [PubMed] [Google Scholar]
- 116.Nyberg P, Rasmussen M, Von Pawel-Rammingen U, Björck L. 2004. SpeB modulates fibronectin-dependent internalization of Streptococcus pyogenes by efficient proteolysis of cell-wall-anchored protein F1. Microbiology 150:1559–1569 10.1099/mic.0.27076-0. [PubMed] [DOI] [PubMed] [Google Scholar]
- 117.Lukomski S, Burns EH Jr, Wyde PR, Podbielski A, Rurangirwa J, Moore-Poveda DK, Musser JM. 1998. Genetic inactivation of an extracellular cysteine protease (SpeB) expressed by Streptococcus pyogenes decreases resistance to phagocytosis and dissemination to organs. Infect Immun 66:771–776. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kansal RG, McGeer A, Low DE, Norrby-Teglund A, Kotb M. 2000. Inverse relation between disease severity and expression of the streptococcal cysteine protease, SpeB, among clonal M1T1 isolates recovered from invasive group A streptococcal infection cases. Infect Immun 68:6362–6369 10.1128/IAI.68.11.6362-6369.2000. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Aziz RK, Pabst MJ, Jeng A, Kansal R, Low DE, Nizet V, Kotb M. 2004. Invasive M1T1 group A Streptococcus undergoes a phase-shift in vivo to prevent proteolytic degradation of multiple virulence factors by SpeB. Mol Microbiol 51:123–134 10.1046/j.1365-2958.2003.03797.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 120.Sumby P, Whitney AR, Graviss EA, DeLeo FR, Musser JM. 2006. Genome-wide analysis of group a streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog 2:e5 10.1371/journal.ppat.0020005. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rasmussen M, Björck L. 2002. Proteolysis and its regulation at the surface of Streptococcus pyogenes. Mol Microbiol 43:537–544 10.1046/j.1365-2958.2002.02766.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 122.Olsen RJ, Sitkiewicz I, Ayeras AA, Gonulal VE, Cantu C, Beres SB, Green NM, Lei B, Humbird T, Greaver J, Chang E, Ragasa WP, Montgomery CA, Cartwright J Jr, McGeer A, Low DE, Whitney AR, Cagle PT, Blasdel TL, DeLeo FR, Musser JM. 2010. Decreased necrotizing fasciitis capacity caused by a single nucleotide mutation that alters a multiple gene virulence axis. Proc Natl Acad Sci U S A 107:888–893 10.1073/pnas.0911811107. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ashbaugh CD, Wessels MR. 2001. Absence of a cysteine protease effect on bacterial virulence in two murine models of human invasive group A streptococcal infection. Infect Immun 69:6683–6688 10.1128/IAI.69.11.6683-6686.2001. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Remmington A, Turner CE. 2018. The DNases of pathogenic Lancefield streptococci. Microbiology 164:242–250 10.1099/mic.0.000612. [PubMed] [DOI] [PubMed] [Google Scholar]
- 125.Matsumoto M, Sakae K, Hashikawa S, Torii K, Hasegawa T, Horii T, Endo M, Okuno R, Murayama S, Hirasawa K, Suzuki R, Isobe J, Tanaka D, Katsukawa C, Tamaru A, Tomita M, Ogata K, Ikebe T, Watanabe H, Ohta M, Working Group for Group A Streptococci in Japan. 2005. Close correlation of streptococcal DNase B (sdaB) alleles with emm genotypes in Streptococcus pyogenes. Microbiol Immunol 49:925–929 10.1111/j.1348-0421.2005.tb03684.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 126.Hasegawa T, Minami M, Okamoto A, Tatsuno I, Isaka M, Ohta M. 2010. Characterization of a virulence-associated and cell-wall-located DNase of Streptococcus pyogenes. Microbiology 156:184–190 10.1099/mic.0.031955-0. [PubMed] [DOI] [PubMed] [Google Scholar]
- 127.Brinkmann V. 2004. Neutrophil extracellular traps kill bacteria. Science 303:1532–1535. [PubMed] [DOI] [PubMed] [Google Scholar]
- 128.Chang A, Khemlani A, Kang H, Proft T. 2011. Functional analysis of Streptococcus pyogenes nuclease A (SpnA), a novel group A streptococcal virulence factor. Mol Microbiol 79:1629–1642 10.1111/j.1365-2958.2011.07550.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 129.Sumby P, Barbian KD, Gardner DJ, Whitney AR, Welty DM, Long RD, Bailey JR, Parnell MJ, Hoe NP, Adams GG, Deleo FR, Musser JM. 2005. Extracellular deoxyribonuclease made by group A Streptococcus assists pathogenesis by enhancing evasion of the innate immune response. Proc Natl Acad Sci U S A 102:1679–1684 10.1073/pnas.0406641102. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Buchanan JT, Simpson AJ, Aziz RK, Liu GY, Kristian SA, Kotb M, Feramisco J, Nizet V. 2006. DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr Biol 16:396–400 10.1016/j.cub.2005.12.039. [PubMed] [DOI] [PubMed] [Google Scholar]
- 131.Uchiyama S, Andreoni F, Schuepbach RA, Nizet V, Zinkernagel AS. 2012. DNase Sda1 allows invasive M1T1 group A Streptococcus to prevent TLR9-dependent recognition. PLoS Pathog 8:e1002736 10.1371/journal.ppat.1002736. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Akesson P, Sjöholm AG, Björck L. 1996. Protein SIC, a novel extracellular protein of Streptococcus pyogenes interfering with complement function. J Biol Chem 271:1081–1088 10.1074/jbc.271.2.1081. [PubMed] [DOI] [PubMed] [Google Scholar]
- 133.Hoe NP, Kordari P, Cole R, Liu M, Palzkill T, Huang W, McLellan D, Adams GJ, Hu M, Vuopio-Varkila J, Cate TR, Pichichero ME, Edwards KM, Eskola J, Low DE, Musser JM. 2000. Human immune response to streptococcal inhibitor of complement, a serotype M1 group A Streptococcus extracellular protein involved in epidemics. J Infect Dis 182:1425–1436 10.1086/315882. [PubMed] [DOI] [PubMed] [Google Scholar]
- 134.Hoe NP, Nakashima K, Lukomski S, Grigsby D, Liu M, Kordari P, Dou SJ, Pan X, Vuopio-Varkila J, Salmelinna S, McGeer A, Low DE, Schwartz B, Schuchat A, Naidich S, De Lorenzo D, Fu YX, Musser JM. 1999. Rapid selection of complement-inhibiting protein variants in group A Streptococcus epidemic waves. Nat Med 5:924–929 10.1038/11369. [PubMed] [DOI] [PubMed] [Google Scholar]
- 135.Binks M, Sriprakash KS. 2004. Characterization of a complement-binding protein, DRS, from strains of Streptococcus pyogenes containing the emm12 and emm55 genes. Infect Immun 72:3981–3986 10.1128/IAI.72.7.3981-3986.2004. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Fernie-King BA, Seilly DJ, Willers C, Würzner R, Davies A, Lachmann PJ. 2001. Streptococcal inhibitor of complement (SIC) inhibits the membrane attack complex by preventing uptake of C567 onto cell membranes. Immunology 103:390–398 10.1046/j.1365-2567.2001.01249.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Pence MA, Rooijakkers SHM, Cogen AL, Cole JN, Hollands A, Gallo RL, Nizet V. 2010. Streptococcal inhibitor of complement promotes innate immune resistance phenotypes of invasive M1T1 group A Streptococcus. J Innate Immun 2:587–595 10.1159/000317672. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fernie-King BA, Seilly DJ, Davies A, Lachmann PJ. 2002. Streptococcal inhibitor of complement inhibits two additional components of the mucosal innate immune system: secretory leukocyte proteinase inhibitor and lysozyme. Infect Immun 70:4908–4916 10.1128/IAI.70.9.4908-4916.2002. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fernie-King BA, Seilly DJ, Lachmann PJ. 2004. The interaction of streptococcal inhibitor of complement (SIC) and its proteolytic fragments with the human beta defensins. Immunology 111:444–452 10.1111/j.0019-2805.2004.01837.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Frick I-M, Akesson P, Rasmussen M, Schmidtchen A, Björck L. 2003. SIC, a secreted protein of Streptococcus pyogenes that inactivates antibacterial peptides. J Biol Chem 278:16561–16566 10.1074/jbc.M301995200. [PubMed] [DOI] [PubMed] [Google Scholar]
- 141.Akesson P, Herwald H, Rasmussen M, Håkansson K, Abrahamson M, Hasan AAK, Schmaier AH, Müller-Esterl W, Björck L. 2010. Streptococcal inhibitor of complement-mediated lysis (SIC): an anti-inflammatory virulence determinant. Microbiology 156:3660–3668 10.1099/mic.0.039578-0. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Westman J, Chakrakodi B, Snäll J, Mörgelin M, Bruun Madsen M, Hyldegaard O, Neumann A, Frick I-M, Norrby-Teglund A, Björck L, Herwald H. 2018. Protein SIC secreted from Streptococcus pyogenes forms complexes with extracellular histones that boost cytokine production. Front Immunol 9:236 10.3389/fimmu.2018.00236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lukomski S, Hoe NP, Abdi I, Rurangirwa J, Kordari P, Liu M, Dou SJ, Adams GG, Musser JM. 2000. Nonpolar inactivation of the hypervariable streptococcal inhibitor of complement gene (sic) in serotype M1 Streptococcus pyogenes significantly decreases mouse mucosal colonization. Infect Immun 68:535–542 10.1128/IAI.68.2.535-542.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.von Pawel-Rammingen U, Johansson BP, Björck L. 2002. IdeS, a novel streptococcal cysteine proteinase with unique specificity for immunoglobulin G. EMBO J 21:1607–1615 10.1093/emboj/21.7.1607. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lei B, DeLeo FR, Hoe NP, Graham MR, Mackie SM, Cole RL, Liu M, Hill HR, Low DE, Federle MJ, Scott JR, Musser JM. 2001. Evasion of human innate and acquired immunity by a bacterial homolog of CD11b that inhibits opsonophagocytosis. Nat Med 7:1298–1305 10.1038/nm1201-1298. [PubMed] [DOI] [PubMed] [Google Scholar]
- 146.Okumura CYM, Anderson EL, Döhrmann S, Tran DN, Olson J, von Pawel-Rammingen U, Nizet V. 2013. IgG protease Mac/IdeS is not essential for phagocyte resistance or mouse virulence of M1T1 group A Streptococcus. MBio 4:e00499-13 10.1128/mBio.00499-13. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Collin M, Olsén A. 2001. EndoS, a novel secreted protein from Streptococcus pyogenes with endoglycosidase activity on human IgG. EMBO J 20:3046–3055 10.1093/emboj/20.12.3046. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Collin M, Svensson MD, Sjöholm AG, Jensenius JC, Sjöbring U, Olsén A. 2002. EndoS and SpeB from Streptococcus pyogenes inhibit immunoglobulin-mediated opsonophagocytosis. Infect Immun 70:6646–6651 10.1128/IAI.70.12.6646-6651.2002. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sjögren J, Okumura CY, Collin M, Nizet V, Hollands A. 2011. Study of the IgG endoglycosidase EndoS in group A streptococcal phagocyte resistance and virulence. BMC Microbiol 11:120 10.1186/1471-2180-11-120. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wang X, Lin X, Loy JA, Tang J, Zhang XC. 1998. Crystal structure of the catalytic domain of human plasmin complexed with streptokinase. Science 281:1662–1665 10.1126/science.281.5383.1662. [PubMed] [DOI] [PubMed] [Google Scholar]
- 151.Parry MA, Zhang XC, Bode I. 2000. Molecular mechanisms of plasminogen activation: bacterial cofactors provide clues. Trends Biochem Sci 25:53–59 10.1016/S0968-0004(99)01521-2. [DOI] [PubMed] [Google Scholar]
- 152.Walker MJ, McArthur JD, McKay F, Ranson M. 2005. Is plasminogen deployed as a Streptococcus pyogenes virulence factor? Trends Microbiol 13:308–313 10.1016/j.tim.2005.05.006. [PubMed] [DOI] [PubMed] [Google Scholar]
- 153.Plow EF, Herren T, Redlitz A, Miles LA, Hoover-Plow JL. 1995. The cell biology of the plasminogen system. FASEB J 9:939–945 10.1096/fasebj.9.10.7615163. [PubMed] [DOI] [PubMed] [Google Scholar]
- 154.Sun H, Ringdahl U, Homeister JW, Fay WP, Engleberg NC, Yang AY, Rozek LS, Wang X, Sjöbring U, Ginsburg D. 2004. Plasminogen is a critical host pathogenicity factor for group A streptococcal infection. Science 305:1283–1286 10.1126/science.1101245. [PubMed] [DOI] [PubMed] [Google Scholar]
- 155.Sanderson-Smith ML, De Oliveira DMP, Ranson M, McArthur JD. 2012. Bacterial plasminogen receptors: mediators of a multifaceted relationship. J Biomed Biotechnol 2012:272148 10.1155/2012/272148. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ly D, Taylor JM, Tsatsaronis JA, Monteleone MM, Skora AS, Donald CA, Maddocks T, Nizet V, West NP, Ranson M, Walker MJ, McArthur JD, Sanderson-Smith ML. 2014. Plasmin(ogen) acquisition by group A Streptococcus protects against C3b-mediated neutrophil killing. J Innate Immun 6:240–250 10.1159/000353754. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Nitzsche R, Köhler J, Kreikemeyer B, Oehmcke-Hecht S. 2016. Streptococcus pyogenes escapes killing from extracellular histones through plasminogen binding and activation by streptokinase. J Innate Immun 8:589–600 10.1159/000448039. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Nitzsche R, Rosenheinrich M, Kreikemeyer B, Oehmcke-Hecht S. 2015. Streptococcus pyogenes triggers activation of the human contact system by streptokinase. Infect Immun 83:3035–3042 10.1128/IAI.00180-15. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Olsen RJ, Shelburne SA, Musser JM. 2009. Molecular mechanisms underlying group A streptococcal pathogenesis. Cell Microbiol 11:1–12 10.1111/j.1462-5822.2008.01225.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 160.Frick I-M, Björck L, Herwald H. 2007. The dual role of the contact system in bacterial infectious disease. Thromb Haemost 98:497–502 10.1160/TH07-01-0051. [PubMed] [DOI] [PubMed] [Google Scholar]
- 161.Banks DJ, Beres SB, Musser JM. 2002. The fundamental contribution of phages to GAS evolution, genome diversification and strain emergence. Trends Microbiol 10:515–521 10.1016/S0966-842X(02)02461-7. [DOI] [PubMed] [Google Scholar]
- 162.Beres SB, Musser JM. 2007. Contribution of exogenous genetic elements to the group A Streptococcus metagenome. PLoS One 2:e800 10.1371/journal.pone.0000800. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Accardo P, Sánchez-Corral P, Criado O, García E, Rodríguez de Córdoba S. 1996. Binding of human complement component C4b-binding protein (C4BP) to Streptococcus pyogenes involves the C4b-binding site. J Immunol 157:4935–4939. [PubMed] [PubMed] [Google Scholar]
- 164.Gustafsson MCU, Lannergård J, Nilsson OR, Kristensen BM, Olsen JE, Harris CL, Ufret-Vincenty RL, Stålhammar-Carlemalm M, Lindahl G. 2013. Factor H binds to the hypervariable region of many Streptococcus pyogenes M proteins but does not promote phagocytosis resistance or acute virulence. PLoS Pathog 9:e1003323 10.1371/journal.ppat.1003323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ermert D, Shaughnessy J, Joeris T, Kaplan J, Pang CJ, Kurt-Jones EA, Rice PA, Ram S, Blom AM. 2015. Virulence of group A streptococci is enhanced by human complement inhibitors. PLoS Pathog 11:e1005043 10.1371/journal.ppat.1005043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kumar S, Stecher G, Tamura K. 2016. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Brouillard J-NP, Günther S, Varma AK, Gryski I, Herfst CA, Rahman AKMN, Leung DYM, Schlievert PM, Madrenas J, Sundberg EJ, McCormick JK. 2007. Crystal structure of the streptococcal superantigen SpeI and functional role of a novel loop domain in T cell activation by group V superantigens. J Mol Biol 367:925–934 10.1016/j.jmb.2007.01.024. [DOI] [PubMed] [Google Scholar]
- 169.Li Y, Li H, Dimasi N, McCormick JK, Martin R, Schuck P, Schlievert PM, Mariuzza RA. 2001. Crystal structure of a superantigen bound to the high-affinity, zinc-dependent site on MHC class II. Immunity 14:93–104 10.1016/S1074-7613(01)00092-9. [DOI] [PubMed] [Google Scholar]
- 170.Kasper KJ, Xi W, Rahman AKMN-U, Nooh MM, Kotb M, Sundberg EJ, Madrenas J, McCormick JK. 2008. Molecular requirements for MHC class II alpha-chain engagement and allelic discrimination by the bacterial superantigen streptococcal pyrogenic exotoxin C. J Immunol 181:3384–3392 10.4049/jimmunol.181.5.3384. [DOI] [PubMed] [Google Scholar]
- 171.Jardetzky TS, Brown JH, Gorga JC, Stern LJ, Urban RG, Chi YI, Stauffacher C, Strominger JL, Wiley DC. 1994. Three-dimensional structure of a human class II histocompatibility molecule complexed with superantigen. Nature 368:711–718 10.1038/368711a0. [DOI] [PubMed] [Google Scholar]