

Abstract
FLT3 activating mutations are detected in approximately 30 % of newly diagnosed acute myeloid leukemia (AML) cases, most commonly consisting of internal tandem duplication (ITD) mutations in the juxtamembrane region. Recently, several FLT3 inhibitors have demonstrated clinical activity and three are currently approved – midostaurin, quizartinib, and gilteritinib. Midostaurin is a first-generation FLT3 inhibitor with minimal activity as monotherapy. Midostaurin lacks selectivity and is only approved by the USFDA for use in combination with other chemotherapy agents. The second-generation inhibitors quizartinib and gilteritinib display improved specificity and selectivity, and have been approved for use as monotherapy. However, their clinical efficacies are limited in part due to the emergence of drug-resistant FLT3 secondary mutations in the tyrosine kinase domain at positions D835 and F691. Therefore, in order to overcome drug resistance and further improve outcomes, new compounds targeting FLT3-ITD with secondary mutants are urgently needed. In this study, through the structural modification of a reported compound Ling-5e, we identified compound 24 as a FLT3 inhibitor that is equally potent against FLT3-ITD and the clinically relevant mutants FLT3-ITD/D835Y, and FLT3-ITD/F691L. Its inhibitory effects were demonstrated in both cell viability assays and western blots analyses. When tested against cell lines lacking activating mutations in FLT3, no non-specific cytotoxicity effect was observed. Interestingly, molecular docking results showed that compound 24 may adopt different binding conformations with FLT3-F691L compared to FLT3, which may explain its retained activity against FLT3-ITD/F691L. In summary, compound 24 has inhibition potency on FLT3 comparable to gilteritinib, but a more balanced inhibition on FLT3 secondary mutations, especially FLT3-ITD/F691L which is gilteritinib resistant. Compound 24 may serve as a promising lead for the drug development of either primary or relapsed AML with FLT3 secondary mutations.
Keywords: FLT3-ITD, Cancer, AML, Mutation
1. Introduction
Gain-of-function mutations in FMS-like tyrosine kinase 3 (FLT3) are detected in approximately 30 % of newly diagnosed acute myeloid leukemia (AML) cases [1,2]. Because of its high frequency and association with poor prognosis, mutant FLT3 is an attractive target for AML therapy [3].
FLT3 is composed of four domains: the extracellular portion that binds FLT3 ligand, the transmembrane (TM) domain, the juxtamembrane (JM) domain, and a highly conserved intracellular kinase domain (Fig. 1, Panel A). The 3D structure of the FLT3 kinase domain includes a smaller N-terminal lobe, a larger C-terminal lobe and a hinge region that connects the two lobes. The key structures related to kinase activity include the JM domain, which serves an autoinhibitory function, the activation loop (A-loop) and the catalytic loop (Fig. 1, Panel B). The JM domain contains several key tyrosine residues as phosphorylation sites. The A-loop also has 1–3 tyrosine residues. The A-loop is structurally flexible and has two conformations: DFG-in or DFG-out. The DFG motif refers to three highly conserved amino acid residues at the base of the A-loop (Asp829-Phe830-Gly831 in FLT3). In the DFG-in (active) conformation, the A-loop is in an open and extended state, characterized by the Asp829 residue pointing at the ATP-binding pocket and the Phe830 residue pointing to the back cleft. In this conformation, the Asp829 residue interacts with ATP and promotes kinase activation. In the DFG-out (inactive) conformation, the A-loop is in a closed state, precluding binding of ATP and substrates. Thus, the kinase domain adopts an autoinhibited conformation, characterized by the Asp829 residue pointing to the back cleft and the Phe830 residue pointing to the ATP-binding pocket [4,5]. Notably, the DFG-out conformation generates a hydrophobic pocket adjacent to the ATP-binding pocket, behind the “gatekeeper” residue F691 [6].
Fig. 1.
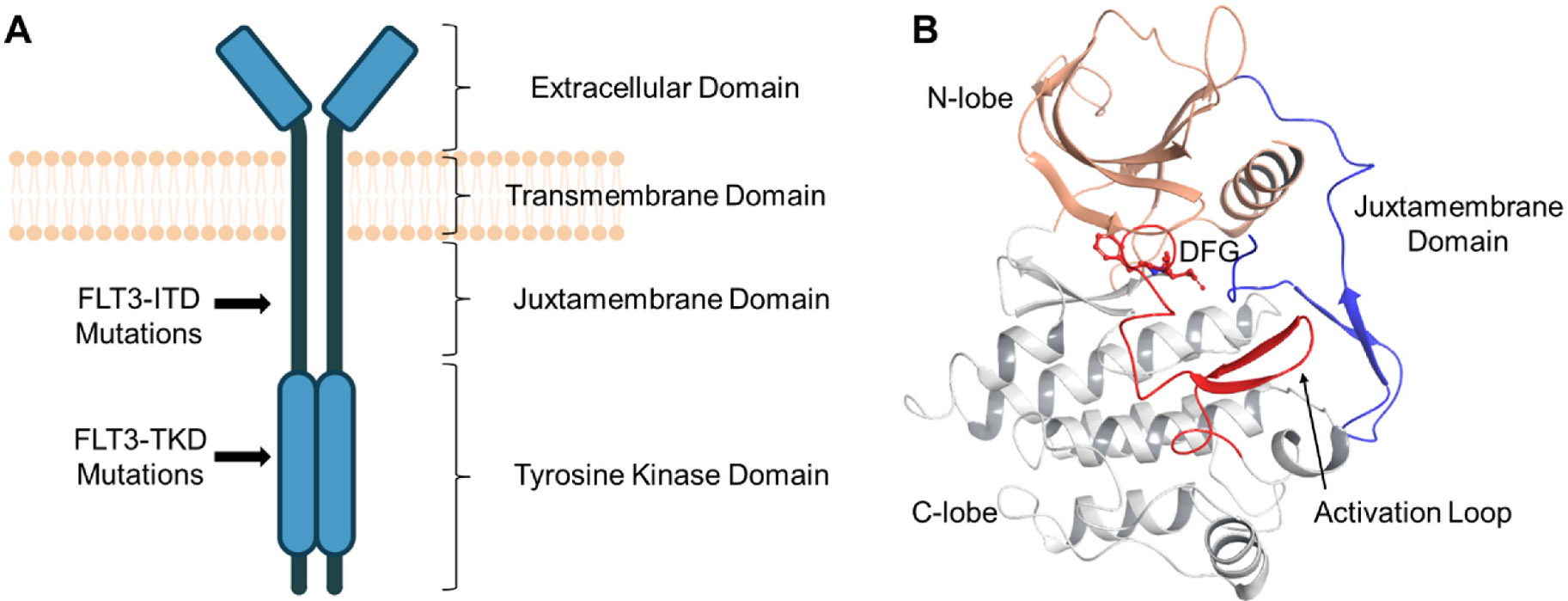
The structure of FLT3. (A) Illustration of FLT3 structure and mutation positions. (B) The crystal structure of FLT3 tyrosine kinase domain (DFG-out conformation, PDB: 1RJB).
The native or “wild type” (WT) FLT3 is kept inactive monometrically. The JM domain binds in a strategic area of FLT3 and interacts with multiple key structural components, thus working as a wedge that stabilizes the inactive state of FLT3 [7]. The A-loop, blocked by the JM domain, and stabilized by multiple residues’ interactions, is kept in the autoinhibited DFG-out conformation (Fig. 1, Panel B). Upon binding to FLT3 ligand (FL), FLT3 dimerizes and brings the two intracellular domains into close proximity. The subsequent transphosphorylation of tyrosine residues at the JM domains forces them to wind back, exposing the A-loops; while the following phosphorylation of A-loops forces them to adopt the DFG-in conformation, promoting the further activation of FLT3 [8]. FLT3 activating mutations have two subtypes: internal tandem duplication at the juxtamembrane domain (“FLT3-ITD”), and point mutations in the activation loop of the tyrosine kinase domain (“FLT3-TKD”) (Fig. 1, Panel A). FLT3-ITD disrupts the inhibitory function of the JM domain, thus promoting FLT3 activation in the absence of FLT3 ligand; FLT3-TKD mutations also enhance unregulated FLT3 activation through stabilization of the active, DFG-in conformation.
Based on their binding mode with the target kinase, inhibitors are categorized as type I or type II. Type I inhibitors bind to the ATP-binding pocket while type II inhibitors bind to both the ATP-binding pocket and the hydrophobic pocket adjacent to the ATP-binding pocket, which is only accessible when the A-loop is in the inactive DFG-out conformation. Two generations of FLT3 inhibitors have been discovered and studied in clinical trials, with three FLT3 inhibitors currently approved for clinical use (Fig. 2, Panel A). Midostaurin is a type I, first-generation FLT3 inhibitor with minimal activity as monotherapy. Midostaurin lacks selectivity and is only approved by the USFDA for use in combination with cytotoxic chemotherapy agents [9-11]. As a type II [12], second-generation FLT3 inhibitor, quizartinib [13-19] has substantially improved specificity and selectivity, and was approved in Japan as monotherapy for AML cases with FLT3-ITD mutation. Quizartinib displays substantial initial clinical efficacy, but resistant mutations rapidly emerge at residues D835 and F691 [20-22]. The D835Y mutation in the activation loop breaks the hydrogen bond between Asp835 and Ser838 which helps stabilize the DFG-out conformation [23]. Because the type II inhibitor quizartinib only binds to a DFG-out conformation, FLT3-D835Y is highly quizartinib-resistant. The F691L mutation at the “gatekeeper” residue is bulky and leads to steric clash with quizartinib, preventing its binding to the hydrophobic pocket behind this position [24-26].
Fig. 2.
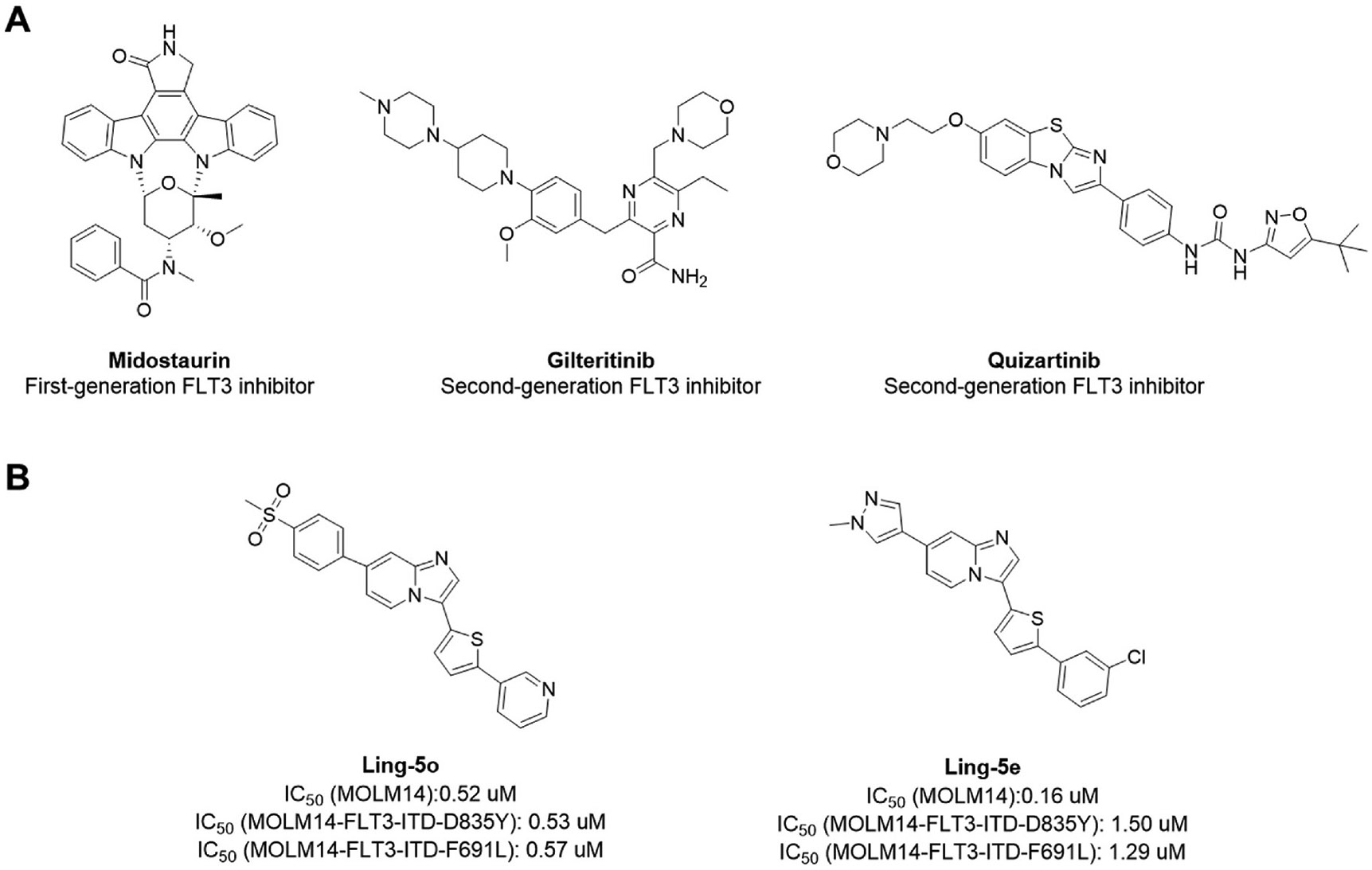
Illustration of FLT3 inhibitors. (A) The structure of midostaurin, gilteritinib, and quizartinib. (B) The structure Ling-5o and Ling-5e as previously reported.
Gilteritinib is a second-generation FLT3 inhibitor that was recently approved by USFDA as monotherapy for the treatment of relapsed or refractory AML with FLT3 mutations [27-29]. As a type I inhibitor, gilteritinib is not impacted by the D835Y mutation. However, its inhibitory effects are diminished by steric clash caused by the F691L mutation [30]. In order to overcome drug resistance, new compounds with balanced inhibition against FLT3-ITD and FLT3-ITD with secondary mutations are needed, especially FLT3-ITD/F691L which is gilteritinib resistant.
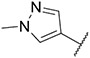
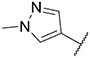
Imidazo [l,2-a]pyridine derivatives were previously identified to potentially have promising FLT3 inhibitory effects [31]. Our studies identified 7-(4-(methylsulfonyl)phenyl)-3-(5-(pyridin-3-yl)thiophen-2-yl)imidazo [1,2-a]pyridine (Ling-5o) as a balanced FLT3-ITD and FLT3-ITD secondary mutants inhibitor (Fig. 2, Panel B) [32]. However, compared to quizartinib and gilteritinib, the inhibitory potency of Ling-5o is low, with sub-micromolar IC50s on MOLM14 cells and MOLM14 cells with drug resistant secondary mutations. Therefore, new FLT3 inhibitors with balanced inhibitory effects on FLT3-ITD and FLT3-ITD secondary mutations, that are more potent than Ling-5o, are needed. Here, we report that through structural modifications, 6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)-N-(4-(methylsulfonyl)phenyl)pyridin-2-amine (24) and its analogues were identified as balanced FLT3-ITD and FLT3-ITD secondary mutants inhibitors with improved potency.
2. Results and discussion
2.1. Discovery
Our discovery strategy is to develop a scaffold with high potency on FLT3 first and then screen derivatives to identify compounds that are equally potent towards FLT3 and its drug-resistant mutants. Our previous studies identified 3-(5-(3-chlorophenyl)thiophen-2-yl)-7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridine (Ling-5e) as a FLT3 inhibitor. Although there is a gap between Ling-5e’s cell-based inhibition of FLT3-ITD and its inhibition of FLT3-ITD/D835Y and FLT3-ITD/F691L, the potency of Ling-5e against FLT3 kinase is much higher than the potency of Ling-5o (Fig. 2, Panel B). Therefore, we chose Ling-5e as our lead compound for further optimization. Our initial aim was to improve the potency of this scaffold against FLT3, and then screen for compounds with a balanced potency against FLT3-ITD, FLT3-ITD/D835Y, and FLT3-ITD/F691L.
From the previously identified binding pose of Ling-5e in FLT3 model, the imidazo [1,2-a]pyridine fragment binds to the hinge region, forming hydrogen bonds with C694 residue. The connective thiophene fragment does not appear to have any crucial interactions with surrounding residues. In addition, the pocket surrounding the thiophen fragment is able to accommodate both five-member rings and six-member rings [31,32]. Finally, the binding of the chlorophenyl fragment and the gatekeeper residue F691 is stabilized by pi-pi interactions; however, this pi-pi interaction is lost when F691 is mutated into leucine (Fig. 3, Panel A). Based on these evaluations, we maintained the imidazo [1,2-a]pyridine structure, which is crucial for hinge binding. Then, we changed the thiophene fragment into six-member aromatic rings. Finally, we switched the chlorophenyl group into various fragments, including non-aromatic six-member rings and phenyl rings with different substitutes. Subsequent screenings of the derivatives led to the discovery of 6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)-N-(4-(methylsulfonyl)phenyl)pyridin-2-amine (24) as an improved structure for FLT3 and FLT3 mutants inhibition (Fig. 3, Panel B).
Fig. 3.
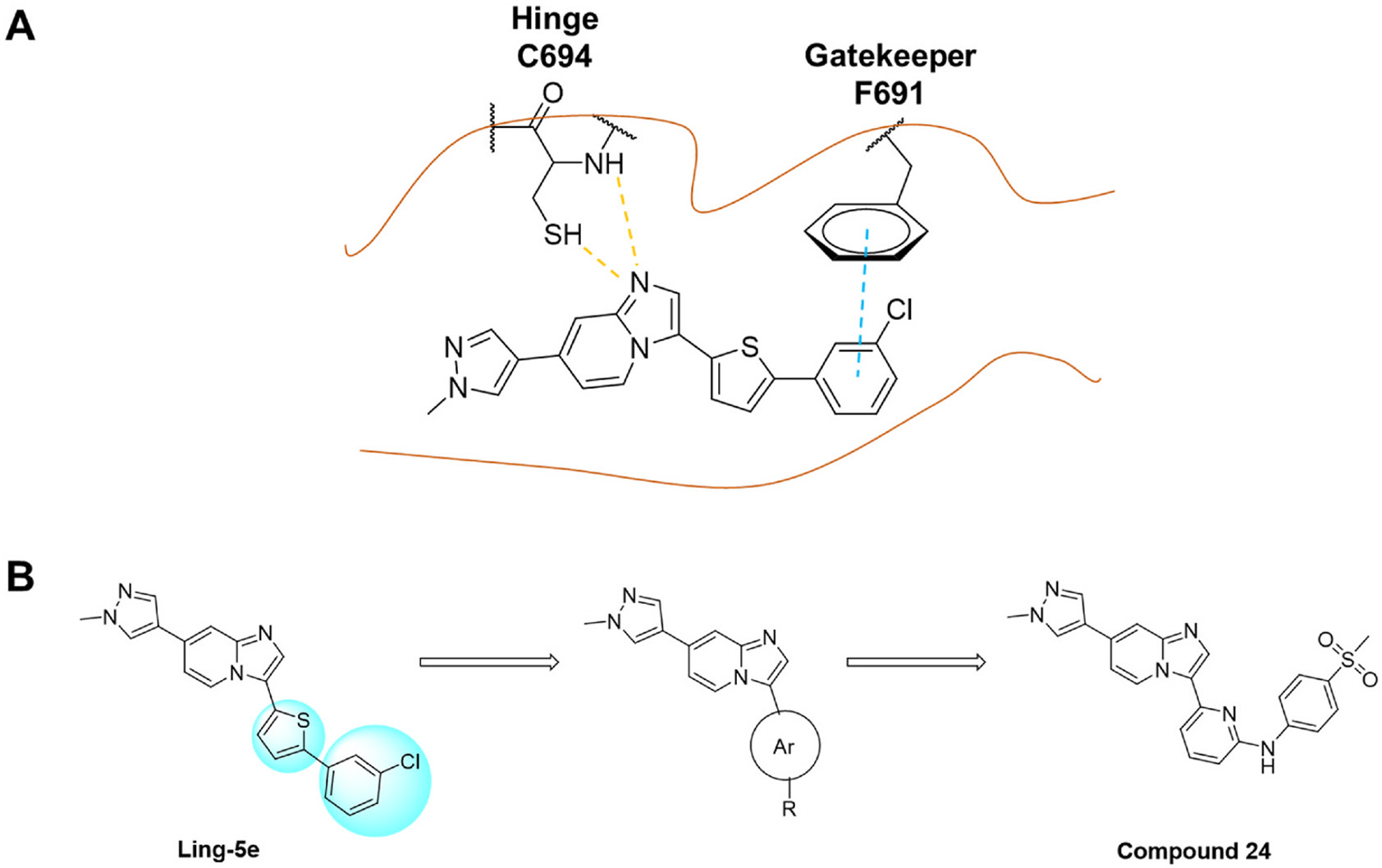
The molecular design rationale from the view of protein-1igand interactions. (A) Binding pose of Ling-5e in FLT3 model. Yellow dashes depict for hydrogen bonds. Blue dash depicts for pi-pi stacking. (B) Structural modification of Ling-5e led to the discovery of compound 24.
2.2. Structure-activity relationship study
MOLM14 is a patient-derived AML cell line with a FLT3-ITD mutation. MOLM14 cells and MOLM14 derivatives sublines that harbor specific secondary mutations were used to verify the compounds’ activity against FLT3 and FLT3 mutants. K562 is another patient-derived leukemia cell line that lacks FLT3 activating mutations that is used as a control cell line to screen for non-specific toxicity. We first compared the structures with different six-member aromatic rings and substitution positions (Table 1). Comparing 2, 3, and 4, we found that compounds with a meta- and para-substitution on the six-member ring may be better than those with an ortho-substitution. The comparison between 1 and 3 also indicates that the effects of pyridine and benzene as the six-member ring are comparable. We then chose pyridine as the six-member aromatic ring and kept exploring the different groups that are substituted at the meta-position (Table 2). The comparison between 5 and 7 indicates that a NH-group instead of a trisubstituted N, that directly connected to the six-member ring, may be beneficial for the potency. Compounds 8–12 show that phenoxy ethyl piperidine series as R2 has very beneficial antiproliferative effects on MOLM14 cells, but not necessarily on MOLM14-F691L cells. Compounds 13–18 show that with alkyl benzene series as R2, the antiproliferative effects of compounds on MOLM14 cells is associated with the molecular size of the alkyl groups. Smaller alkyl groups may be more beneficial than bigger ones. Interestingly, compound 13 has high potency equally on MOLM14 and MOLM14-D835Y cells; its potency margin on MOLM14-F691L cells against MOLM14 cells is also relatively low. However, it also inhibits the proliferation of K562 cells, indicating a low selectivity margin on FLT3 against other kinases. Compounds 19–23 show that methoxy substituted benzenes as R2 positions also have beneficial effects on inhibiting MOLM14 cells. Compounds 24–32 show the effect of an electro withdrawing group substituted phenol ring as R2. Interestingly, 24 is observed to possess potent and balanced inhibitory effects on MOLM14 cells and MOLM14 cells with secondary FLT3 mutations without noticeable inhibition on K562 cells. Several compounds with other groups as R2 were also synthesized and screened. The results are shown as 33–39. Finally, in order to compare methyl pyrazole and methyl sulfonic benzene at R1 position, compounds 40–43 were synthesized. By comparison to their analogues, these compounds were observed to have weaker antiproliferative effects on MOLM14 cells, indicating that methyl pyrazole is better than methyl sulfonyl benzene at R1 position. Interestingly, although the antiproliferative effects of compound 42 is relatively low, it has a low potency margin on MOLM14-F691L cells against MOLM14 cells.
Table 1.
Structure-activity study of the six-member aromatic ring.
| |||||
|---|---|---|---|---|---|
| NO. | Ar | MOLM14 IC50(nM) | MOLM14-FLT3-D835Y IC50(nM) | MOLM14-FLT3-F691L IC50(nM) | K562 IC50(nM) |
| 1 |
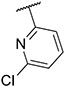
|
28.69 ± 4.56 | 227.8 ± 43.7 | 458.0 ± 34.0 | >1000 |
| 2 |
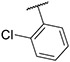
|
397.0 ± 54.9 | >1000 | >1000 | >1000 |
| 3 |
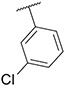
|
29.52 ± 5.54 | 590.6 ± 182.3 | 304.1 ± 96.8 | >1000 |
| 4 |
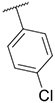
|
16.93 ± 2.52 | 166.5 ± 17.2 | 293.1 ± 72.2 | >1000 |
Table 2.
Structure-activity study of R1 and R2.
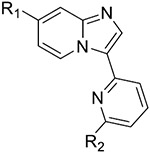
| ||||||
|---|---|---|---|---|---|---|
| NO. | R1 | R2 | MOLM14 IC50(nM) | MOLM14-FLT3-D835Y IC50(nM) | MOLM14-FLT3-F691L IC50(nM) | K562 IC50(nM) |
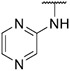
| 5 |
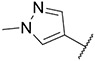
|
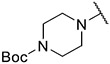
|
51.86 ± 5.39 | 259.3 ± 41.0 | 794.4 ± 164.2 | >1000 |
| 6 |
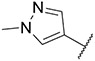
|
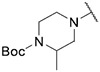
|
100.9 ± 15.2 | 353.1 ± 31.7 | 425.7 ± 134.1 | >1000 |
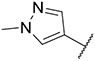
| 7 |
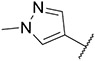
|
|
17.18 ± 1.26 | 18.18 ± 2.48 | 97.46 ± 31.20 | >1000 |
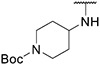
| 8 |
|
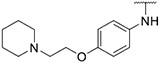
|
0.19 ± 0.08 | 0.77 ± 0.44 | 9.80 ± 1.88 | >1000 |
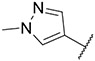
| 9 |
|
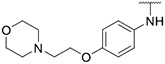
|
1.00 ± 0.22 | 1.83 ± 0.49 | 7.10 ± 1.61 | >1000 |
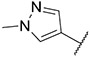
| 10 |
|
|
3.25 ± 0.23 | 1.41 ± 0.32 | 238.5 ± 43.8 | >1000 |
| 11 |
|
|
0.77 ± 0.10 | 2.21 ± 0.57 | 12.47 ± 1.66 | >1000 |
| 12 |
|
|
0.51 ± 0.10 | 0.87 ± 0.19 | 7.42 ± 3.56 | 924 ± 109 |
| 13 |
|
|
5.62 ± 0.45 | 5.67 ± 1.95 | 18.95 ± 3.95 | 219 ± 77 |
| 14 |
|
|
6.76 ± 1.06 | 4.39 ± 0.66 | 90.7 ± 19.7 | >1000 |
| 15 |
|
|
9.03 ± 1.29 | 3.62 ± 0.52 | 294.47 ± 12.09 | 927 ± 88 |
| 16 |
|
|
13.47 ± 2.64 | 8.91 ± 1.71 | 142.1 ± 27.4 | >1000 |
| 17 |
|
|
40.38 ± 6.36 | 41.93 ± 7.91 | >1000 | >1000 |
| 18 |
|
|
153.7 ± 40.8 | 265.4 ± 52.9 | 412.4 ± 101.3 | >1000 |
| 19 |
|
|
2.38 ± 0.31 | 1.65 ± 0.36 | 14.42 ± 1.69 | 274 ± 21 |
| 20 |
|
|
2.49 ± 0.46 | 4.64 ± 1.23 | 13.99 ± 2.35 | 969 ± 181 |
| 21 |
|
|
6.95 ± 0.70 | 27.52 ± 2.34 | 105.2 ± 19.9 | 960 ± 140 |
| 22 |
|
|
6.09 ± 1.04 | 4.91 ± 1.40 | 920 ± 92 | 890 ± 241 |
| 23 |
|
|
1.72 ± 0.31 | 0.87 ± 0.17 | 9.98 ± 2.25 | >1000 |
| 24 |
|
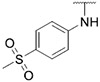
|
4.55 ± 0.39 | 3.16 ± 0.65 | 8.34 ± 1.05 | >1000 |
| 25 |
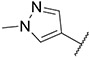
|
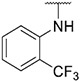
|
0.95 ± 0.04 | 0.31 ± 0.09 | 183.5 ± 35.2 | 262 ± 97 |
| 26 |
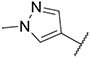
|
|
2.24 ± 0.47 | 3.20 ± 0.47 | 28.85 ± 2.95 | 132 ± 11 |
| 27 |
|
|
33.31 ± 3.11 | 18.87 ± 2.96 | 160.1 ± 44.2 | >1000 |
| 28 |
|
|
19.29 ± 5.41 | 49.86 ± 6.77 | 462.0 ± 145.7 | >1000 |
| 29 |
|
|
6.82 ± 1.00 | 5.19 ± 0.96 | 314.5 ± 61.3 | 923 ± 245 |
| 30 |
|
|
4.03 ± 0.67 | 2.46 ± 0.36 | 258.8 ± 40.0 | 115 ± 21 |
| 31 |
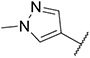
|
|
18.34 ± 2.12 | 47.98 ± 7.30 | 134.9 ± 16.3 | >1000 |
| 32 |
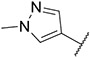
|
|
28.80 ± 3.10 | 23.07 ± 1.25 | 336.8 ± 40.0 | 736 ± 74 |
| 33 |
|
|
1.31 ± 0.29 | 5.90 ± 0.26 | 566.3 ± 108.8 | 119 ± 23 |
| 34 |
|
|
233.1 ± 29.4 | 177.4 ± 57.9 | >1000 | >1000 |
| 35 |
|
|
76.59 ± 9.04 | 76.59 ± 17.91 | 792.3 ± 218.7 | >1000 |
| 36 |
|
|
99.04 ± 4.05 | 121.0 ± 17.0 | >1000 | >1000 |
| 37 |
|
|
3.80 ± 0.73 | 2.59 ± 0.03 | 32.59 ± 10.85 | 210 ± 45 |
| 38 |
|
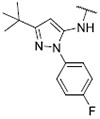
|
18.03 ± 1.91 | 55.98 ± 6.23 | >1000 | >1000 |
| 39 |
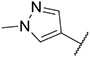
|
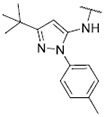
|
161.9 ± 36.7 | 414.3 ± 107.1 | >1000 | >1000 |
| 40 |
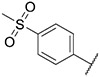
|
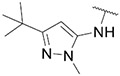
|
4.30 ± 0.55 | 5.32 ± 2.00 | 54.16 ± 16.23 | 688 ± 151 |
| 41 |
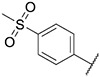
|
|
278.4 ± 52.5 | 613.2 ± 147.2 | >1000 | >1000 |
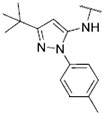
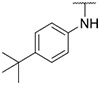
| 42 |
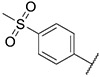
|
|
43.82 ± 8.32 | 43.86 ± 4.88 | 130.1 ± 41.3 | >1000 |
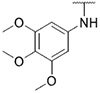
| 43 |
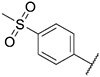
|
|
109.6 ± 19.1 | 203.3 ± 48.3 | >1000 | >1000 |
| Gilteritinib | 5.47 ± 0.25 | 7.83 ± 0.03 | 58.78 ± 0.96 | >1000 | ||
| Vandetanib | 1108 ± 84 | 2805 ± 139 | >20000 | 1535 ± 150 | ||
The screenings resulted in the discovery of several compounds (18, 20, 24, and 42) with balanced inhibition on MOLM14 cells and MOL M14 cells with secondary FLT3 mutations. Meanwhile, they largely lack antiproliferative effects on K562 cells. Interestingly, 6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)-N-(4-(methylsulfonyl)phenyl)pyridin-2-amine (24) was observed to have a potent and balanced inhibition on MOLM14, MOLM14-D835Y, and MOLM14-F691L cells. Its inhibitory effects are more potent than Ling-5o and are more balanced than gilteritinib. The cell antiproliferative effects of compound 24 was verified with cell viability experiments comparing with gilteritinib. From the cell viability curve, compound 24 has comparable inhibition to gilteritinib on MOLM14 cells, but has much more balanced inhibition on MOLM14 cells with secondary FLT3 mutations (Fig. 4).
Fig. 4.
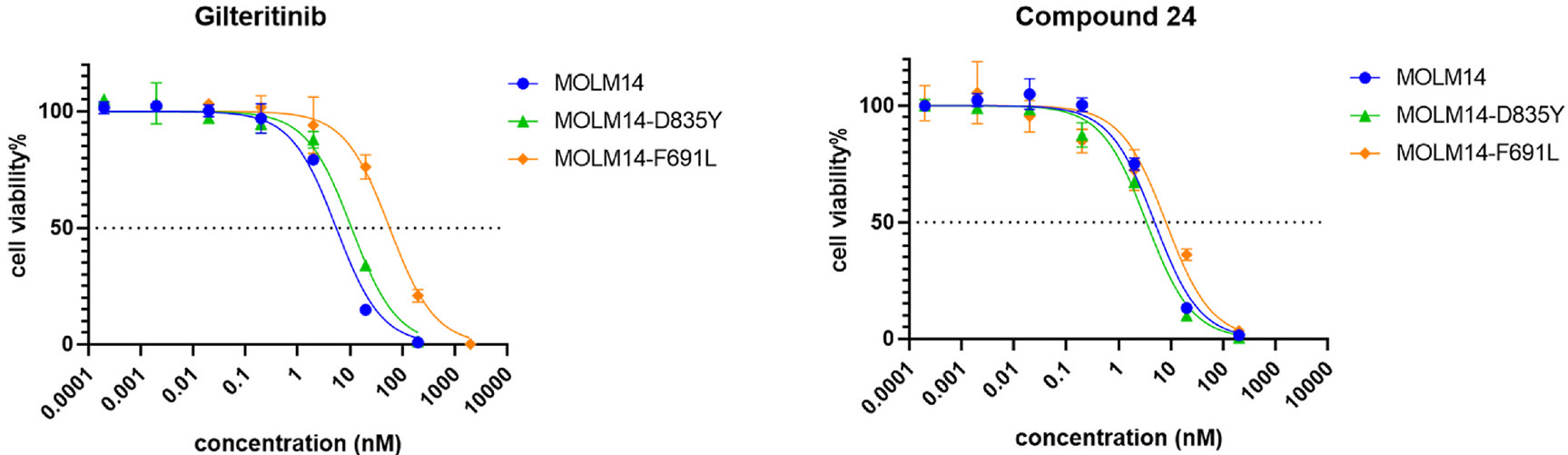
Cell-based viability curve of gilteritinib and Compound 24.
2.3. Cytotoxicity assay and kinase inhibition assay of selected compounds
We selected compounds 18, 20, 24, and 42 for further studies. In addition to K562 cells, we also tested these compounds’ antiproliferative effects on adherent cell lines MDA-MB-231 and HEK-293, which both lack FLT3 activating mutations. There were no profound antiproliferative effects observed in either cell line (Table 3), indicating that these compounds are not cytotoxic [33]. These compounds were also subjected to FLT3 kinase inhibition assays. Their IC50 values on FLT3 kinase correspond with their IC50 values on the MOLM14 cell line (Table 3). This result strongly supports the use of the MOLM14 cell line to assess compounds’ inhibition effects on FLT3 kinase.
Table 3.
Cell antiproliferative effects and FLT3 kinase inhibition of selected compounds.
| NO. | MDA-MB-231 IC50(nM) |
HEK-293 IC50(nM) |
FLT3 kinase IC50(nM) |
|---|---|---|---|
| 18 | >1000 | 993 ± 228 | 288.0 ± 72.9 |
| 20 | >1000 | >1000 | 6.63 ± 0.70 |
| 24 | >1000 | >1000 | 7.94 ± 0.88 |
| 42 | >1000 | >1000 | 94.58 ± 16.82 |
| Gilteritinib | – | – | 10.59 ± 1.42 |
| Vandetanib | – | – | >1000 |
2.4. Western blot
In order to understand the biochemical effects of compound 24 on FLT3 and its downstream targets, we performed Western blot analysis. MOLM14, MOLM14-D835Y, and MOLM14-F691L cell lines were treated for 2 h with 1/10X, 1X, 10X, and 100X of 24’s IC50 on MOLM14 cells (4.5 nM). Compound 24 was observed to have inhibitory effects on the phosphorylation of FLT3 and ERK in MOLM14 cells, with noticeable signal downregulation starting at 1X IC50, and nearly complete signal downregulation at 10X IC50 (Fig. 5, Panel A). Its effects on MOLM14-D835Y and MOLM14-F691L cells are similar to that seen in MOLM14 cells. A trace phospho-ERK signal is observed in MOLM-F691L cells after 10X IC50 treatment with compound 24; however, in general, compound 24 has comparable inhibitory effects on phospho-FLT3 and phospho-ERK in MOLM14 cells and MOLM14 cells with secondary FLT3 mutations (Fig. 5, Panel B and C). The result of Western blot verifies that 24 inhibits the proliferation of MOLM14, MOLM14-D835Y, and MOLM14-F691L by inhibiting their FLT3 phosphorylation and activation of downstream pathways.
Fig. 5.
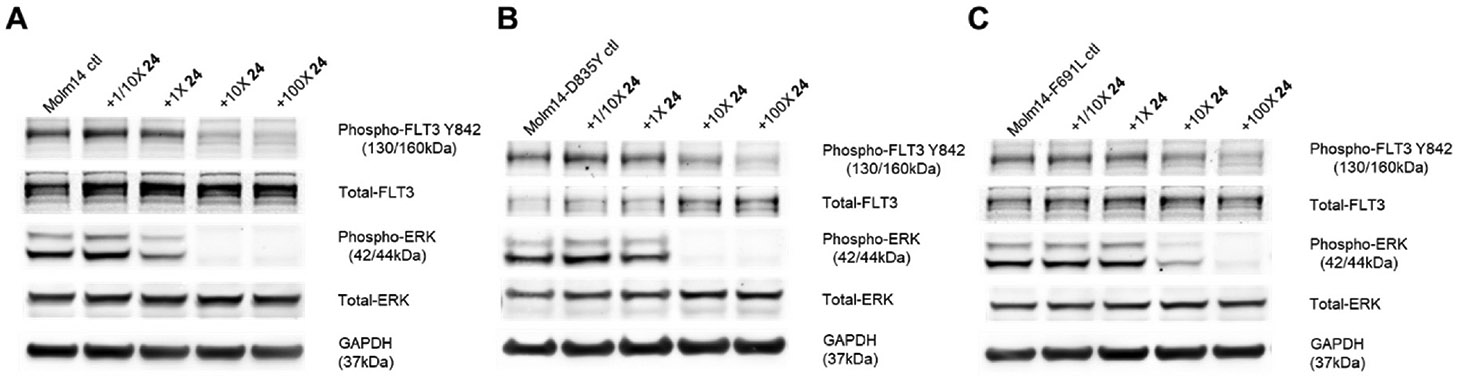
Western blot analysis of MOLM14 (A), MOLM14-D835Y (B), and MOLM14-F691L (C) cells treated with compound 24 for 2 h. DMSO diluent was used as a treatment control in each experiment, and GAPDH as a loading control.
2.5. Molecular docking
The FLT3-D835Y mutation does not induce significant conformational changes of the ATP-binding pocket in the DFG-in conformation of FLT3. Compound 24, being a type I inhibitor that binds to FLT3 in the active conformation, maintains its inhibitory potency on the FLT3-D835Y mutation. Therefore, it is expected that MOLM14-D835Y cells are sensitive to compound 24. Furthermore, in order to understand the structural basis of 24’s maintained inhibitory effects on the FLT3-F691L mutation, we performed computer-aided molecular docking. The results showed that 24 bind to FLT3-WT with its methyl-pyridine warhead directed into the ATP-binding pocket, forming hydrogen bonds with amino acid residues C694 and C695, as well as aromatic - H bonds with L616, C694, C695, and F830 (Figure 6, Panel A). Interestingly, the predicted binding mode of 24 with FLT3-F691L is identical to that with FLT3-WT (re 6, Panel A).). Interestingly, the predicted binding mode of 24 with FLT3-F691L is identical to that with FLT3-WT (Figure 6, Panel B). The F691 residue of FLT3 is known to engage in π–π stacking interactions with the majority of FLT3 inhibitors used clinically [12, 20, 34, 35]. However, the F691L mutation primarily confers resistance through two mechanisms: 1) the disruption of previous interactions with inhibitors and 2) an increase in steric clash on-site. Notably, compound 24 is predicted to lack crucial molecular interactions with F691. Consequently, the F691L mutation is unlikely to substantially reduce the binding affinity between compound 24 and FLT3. Moreover, the clash scores do not exhibit a significant increase between compound 24 and FLT3 with the F691L mutation. In conclusion, the FLT3 F691L mutation neither substantially reduces the binding affinity between FLT3 and compound 24 nor significantly increases steric clash. The results of molecular docking provide insights into the maintained potency of 24 on MOLM14-F691L cells.
Fig. 6.
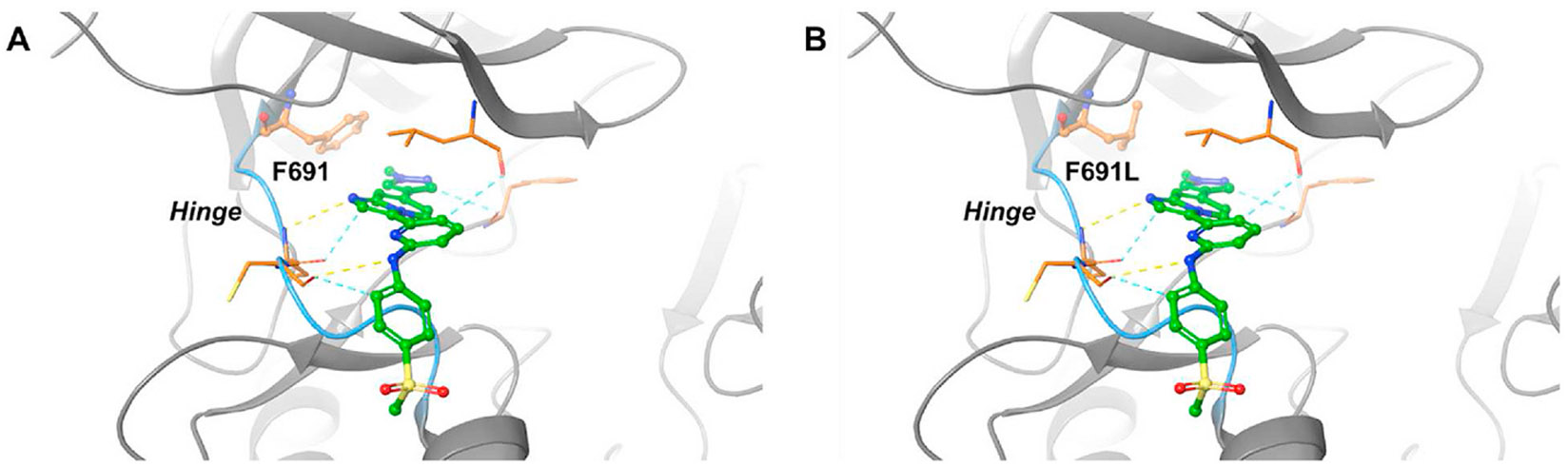
Computer-aided molecular modeling of compound 24 bound to FLT3 (A) and FLT3-F691L (B). The ribbons representing hinges are colored in blue. The hydrogen bonds and Aromatic - H bonds are depicted as dashed lines in yellow and in blue, respectively. The FLT3 crystal structure (in complex with gilteritinib) was applied from PDB: 6JQR.
2.6. Kinase selectivity profiling
To evaluate the kinase selectivity of compound 24, we conducted a site-directed competition binding assay for 97 kinases (Fig. 7). The results revealed that at 4.5 nM concentration, compound 24 displayed interactions (>65 % wash-off) only with FLT3 and TRKA. Notably, simultaneous inhibition of FLT3 and KIT causes profound myelosuppression through a synthetic lethal mechanism, and is also the reason for multiple side effects of FLT3 inhibitors [33-35]. However, compound 24 showed minimal binding affinity with KIT, highlighting its high selectivity for FLT3 over KIT. In summary, compound 24 is a selective FLT3 inhibitor, and is only observed to bind to TRKA as a side kinase target.
Fig. 7.
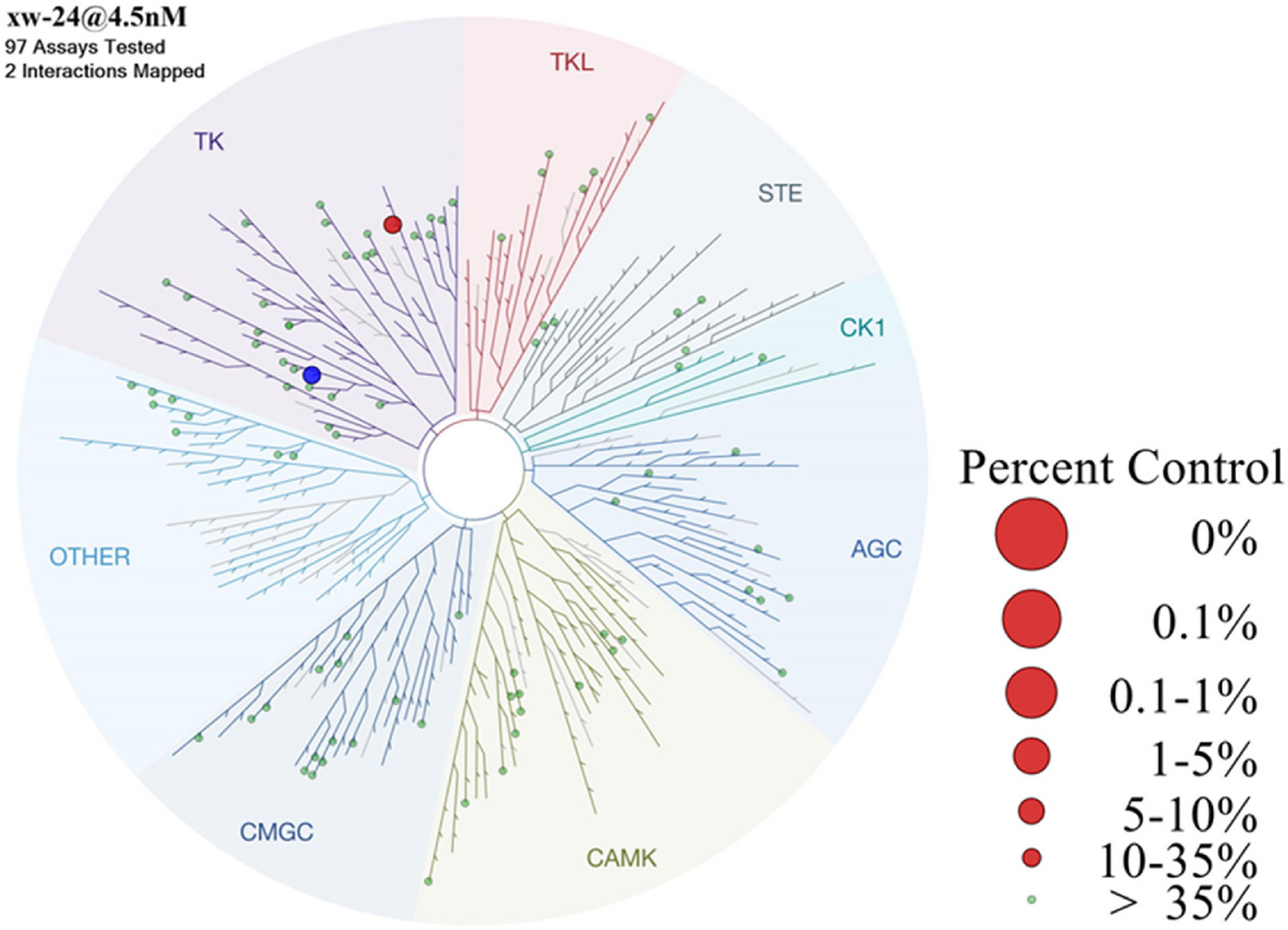
Kinase selectivity profiling of compound 24. FLT3 kinase highlighted in blue.
3. Conclusion
Although three FLT3 inhibitors have been clinically approved, there remains an unmet need for an inhibitor with balanced inhibition of FLT3-ITD and FLT3-ITD secondary mutants, specifically on FLT3-ITD/D835Y and FLT3-ITD/F691L. Through structural modifications of Ling-5e and subsequent screenings, we identified 6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)-N-(4-(methylsulfonyl)phenyl)pyridin-2-amine (compound 24) as a potent FLT3-ITD and FLT3-ITD secondary mutants inhibitor. Its cell-based inhibitory effects are more potent than previously reported Ling-5o. It has comparable antiproliferative effects to gilteritinib on FLT3-ITD expressing cell line MOLM14; however, its inhibitory effects on MOLM14-D835Y and MOLM14-F691L cells is more balanced than gilteritinib. From the results of Western blot analysis, compound 24 inhibits the proliferation of FLT3 mutated cell lines by inhibiting the phosphorylation of FLT3 and its activation of downstream targets. No non-specific cytotoxic effect was observed with compound 24. Interestingly, our molecular docking results indicate that it may have flexible binding modes with FLT3 and FLT3 mutants, with either methyl-pyridine or methyl-sulfonyl-benzene warhead directing inside the ATP-binding pocket; which may explain its balanced inhibitory effects on FLT3 and FLT3 mutants. Finally, the kinase selectivity profiling of compound 24 showed that it is a selective FLT3 inhibitor, with relative lower binding affinity for TRKA and other kinases. In conclusion, compound 24 is a selective FLT3 inhibitor with a balanced inhibition on FLT3-ITD, FLT3-ITD/D835Y, and FLT3-ITD/F691L, and may serve as a promising lead for the drug development of the treatment of both primary and relapsed AML with FLT3 mutations.
Recently, numerous exciting works have been reported in the discovery of FLT3 inhibitors, many of them observed inhibitory effects with their lead compounds on FLT3-ITD/F691L dependent cell lines [36-40]. However, the activities of these FLT3 inhibitors against FLT3-ITD/F691L versus FLT3-ITD are reduced, indicating possibly reduced clinical efficacy in AML patients with FLT3-ITD/F691L secondary mutations. In addition, impressive studies have been reported by the Lu group on the identification of FLT3 inhibitors with equivalent activity against artificially engineered Ba/F3-FLT3-ITD cells and Ba/F3-FLT3-ITD/F691L cells, indicating a balanced inhibition on FLT3-ITD and FLT3-ITD/F691L [41,42]. However, the Ba/F3 cells are mice-derived, and may not reflect the potential activity of these FLT3 inhibitors in AML patients. In contrast, the patient-derived MOLM14 cell line and sublines we employ are more clinically relevant, and may better reflect how FLT3 inhibitors work on AML patients. Therefore, the observation of balanced inhibition of compound 24 on MOLM14 and MOLM14-FLT3-F691L is unique and of vital importance.
4. Experimental section
4.1. Chemistry
All the starting materials were obtained from commercial suppliers and used without further purification. Thermo Finnigan LCQ Deca with Thermo Surveyor LCMS System at variable wavelengths of 254 nm and 214 nm was used to monitor the reaction and test the purity of the compounds. The purity of all the final compounds is >95 %. The water-methanol gradient buffered with 0.1 % formic acid was used as the mobile phase for the HPLC system. NMR spectra was completed on a Varian 400 MHz instrument. The 1H NMR spectra and 13C spectra were recorded at 400 MHz and 101 MHz, respectively. All final compounds were purified using Silica gel (0.035e0.070 mm, 60 Å) flash chromatography, unless otherwise noted. Microwave assisted reactions were completed in sealed vessels using a Biotage Initiator microwave synthesizer (Biotage, Uppsala, Sweden).
4.1.1. The general synthetic scheme
Scheme 1 depicts the general synthetic route of compounds 1–43. The synthesis of intermediate I, II and compound 1 was described in another report. 4-chloro-2-amino pyridine and 2-chloroacetaldehyde were subjected to a condensation, a Suzuki reaction, and a palladium catalyzed Heck type coupling to yield compound 1. Further Buchwald-Hartwig cross coupling reactions of compound 1 with respective nonaromatic-amines or aromatic-amines afforded compound 5–39 in 30–40 % yields, respectively. Compounds 2–4 were synthesized from II by the same palladium catalyzed Heck type coupling as 1 with a higher temperature and longer reaction time, because of lower reactivity of the benzene ring than pyridine. Compounds 40–43 were afforded from III by the same Buchwald-Hartwig cross coupling as reaction d.
Scheme 1.
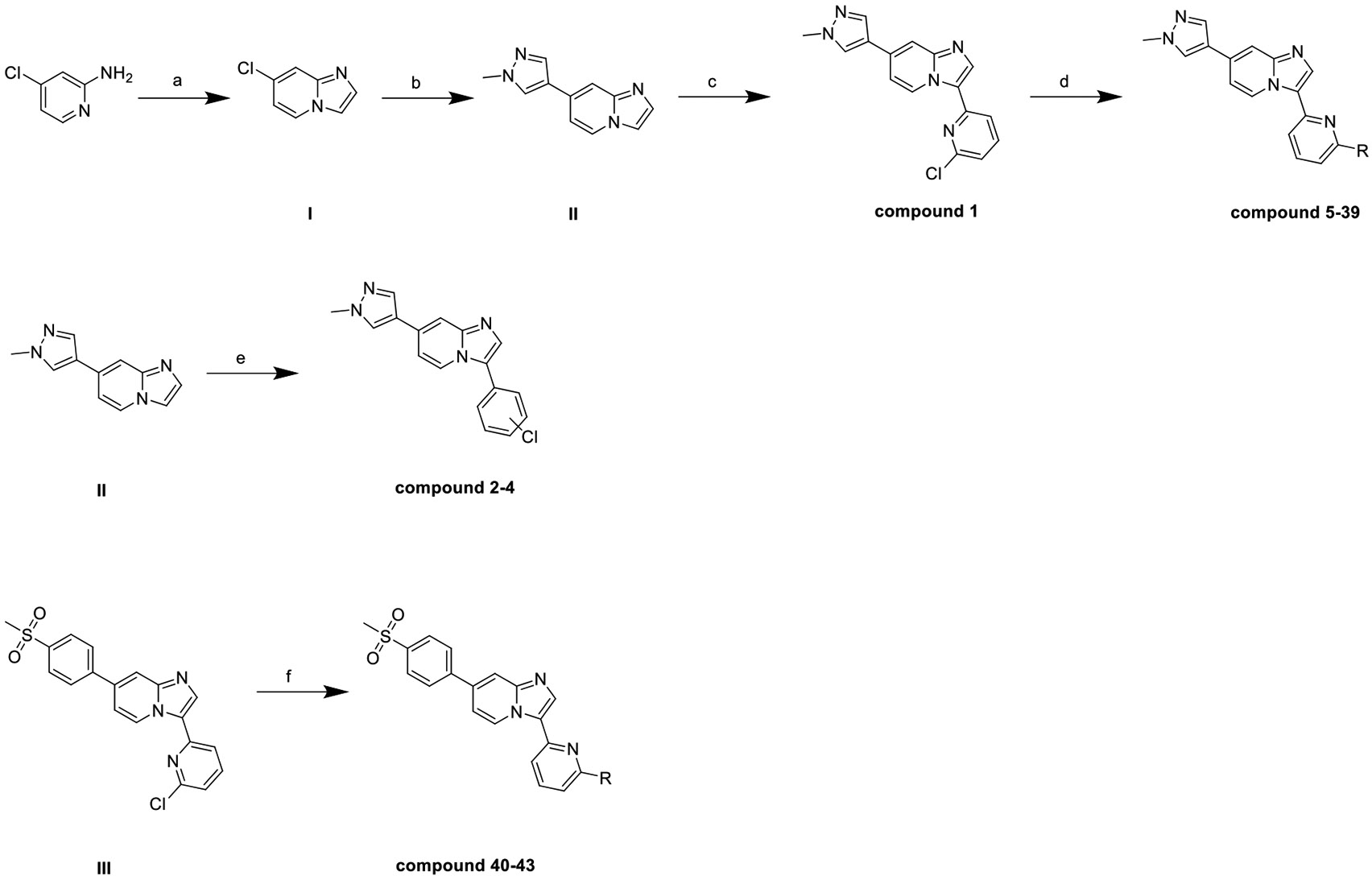
A general synthetic scheme. Reagents and conditions: (a) 2-chloroacetaldehyde, n-Butanol, 130 °C, 12 h; (b) 1-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole, Pd2 (dba)3, PCy3, CH3COOK, DMF/H2O (4:1), 130 °C, 1 h, M/W; (c) 2,6-dichloropyridine, Pd(PPh3)4, Pd(OAc)2, K2CO3, 1,4-Dioxane/H2O (10:1), 100 °C, 14 h; (d) respective amines, Pd(PPh3)4, Cs2CO3, 1,4-Dioxane, 100 °C, 14 h; (e) respective dichlorobenzene, Pd(PPh3)4, Pd(OAc)2, K2CO3, 1,4-Dioxane/H2O (10:1), 120 °C, 24 h; (f) respective amines, Pd(PPh3)4, Cs2CO3, 1,4-Dioxane, 100 °C, 14 h.
4.1.2. Synthesis of 3-(2-chlorophenyl)-7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridine, 2
Compound II (1.50 g, 7.57 mmol), 1,2-dichlorobenzene (3.34 g, 22.70 mmol), potassium carbonate (2.61 g, 18.92 mmol) were dissolved into 20 mL mixed solvent (Dioxane:H2O = 10:1). The resulting reaction mixture was degassed with nitrogen for 5–10 min. After that, Pd(PPh3)4 (0.87 g, 757 μmol) and Pd(OAc)2 (85 mg, 378 μmol) were added. The resulting mixture was stirred at 120 °C under N2 atmosphere for 24 h (sealed bottle). After completion of reaction, the reaction mixture was filtered and concentrated in vacuo. Flash chromatography (MeOH/DCM 0–7 %) yielded compound 2 as a yellow solid (0.71 g, 30.39 %). MS m/z [M + 1] 309.2. 1H NMR (400 MHz, cdcl3) δ 7.83 (d, J = 0.7 Hz, 1H), 7.81 (dd, J = 7.2, 0.8 Hz, 1H), 7.73 (dd, J = 1.7, 0.9 Hz, 1H), 7.71 (s, 1H), 7.68 (s, 1H), 7.60–7.55 (m, 1H), 7.50–7.46 (m, 1H), 7.43–7.39 (m, 2H), 6.94 (dd, J = 7.2, 1.7 Hz, 1H), 3.98 (s, 3H). 13C NMR (101 MHz, cdcl3) δ 146.58, 137.05, 134.70, 134.33, 132.85, 130.39, 130.37, 129.54, 128.28, 127.52, 127.38, 124.63, 122.88, 121.40, 112.23, 111.39, 39.43.
4.1.3. Synthesis of 3-(3-chlorophenyl)-7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridine, 3
Compound II (1.00 g, 5.04 mmol), 1,3-dichlorobenzene (2.22 g, 15.13 mmol), potassium carbonate (1.39 g, 10.09 mmol) were dissolved into 15 mL mixed solvent (Dioxane:H2O = 10:1). The resulting reaction mixture was degassed with nitrogen for 5–10 min. After that, Pd(PPh3)4 (0.58 g, 504 μmol) and Pd(OAc)2 (57 mg, 252 μmol) were added. The resulting mixture was stirred at 120 °C under N2 atmosphere for 24 h (sealed bottle). After completion of reaction, the reaction mixture was filtered and concentrated in vacuo. Flash chromatography (MeOH/DCM 0–7 %) yielded compound 3 as a yellow solid (0.52 g, 33.38 %). MS m/z [M + 1] 309.2. 1H NMR (400 MHz, cdcl3) δ 8.34–8.27 (m, 1H), 7.83 (d, J = 1.8 Hz, 1H), 7.71 (d, J = 2.0 Hz, 2H), 7.67 (q, J = 2.3 Hz, 1H), 7.57–7.55 (m, 1H), 7.49–7.43 (m, 2H), 7.42–7.36 (m, 1H), 7.01–6.90 (m, 1H), 3.98 (s, 3H). 13C NMR (101 MHz, cdcl3) δ 147.13, 137.02, 133.62, 132.17, 130.69, 129.40, 128.57, 128.21, 128.01, 127.72, 127.56, 125.91, 123.43, 121.18, 112.43, 112.08, 39.44.
4.1.4. Synthesis of 3-(4-chlorophenyl)-7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridine, 4
Compound II (1.50 g, 7.57 mmol), 1,4-dichlorobenzene (3.34 g, 22.70 mmol), potassium carbonate (2.61 g, 18.92 mmol) were dissolved into 20 mL mixed solvent (Dioxane:H2O = 10:1). The resulting reaction mixture was degassed with nitrogen for 5–10 min. After that, Pd(PPh3)4 (0.87 g, 757 μmol) and Pd(OAc)2 (85 mg, 378 μmol) were added. The resulting mixture was stirred at 120 °C under N2 atmosphere for 24 h (sealed bottle). After completion of reaction, the reaction mixture was filtered and concentrated in vacuo. Flash chromatography (MeOH/DCM 0–7 %) yielded compound 2 as a yellow solid (0.73 g, 31.24 %). MS m/z [M + 1] 309.2. 1H NMR (400 MHz, cdcl3) δ 8.34–8.22 (m, 1H), 7.83 (s, 1H), 7.71 (s, 2H), 7.66 (d, J = 4.4 Hz, 1H), 7.59–7.51 (m, 2H), 7.49 (s, 2H), 6.95 (td, J = 7.5, 1.8 Hz, 1H), 3.98 (s, 3H). 13C NMR (101 MHz, cdcl3) δ 147.01, 137.00, 134.12, 134.06, 133.34, 129.67 (2C), 129.40 (2C), 129.17, 128.23, 128.01, 127.52, 127.49, 123.35, 121.22, 112.44, 111.98, 39.44.
4.1.5. Synthesis of tert-butyl 4-(6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)pyridin-2-yl)piperazine-1-carboxylate, compound 5
Compound 1 (100 mg, 322.83 μmol), the respective amine (645.67 μmol, 2 equiv), cesium carbonate (157.78 mg, 434.25 μmol) were dissolved into 5 mL Dioxane. The resulting reaction mixture was degassed with nitrogen for 5–10 min. After that, Pd(PPh3)4 (37.31 mg, 32.28 μmol) was added. The resulting mixture was stirred at 100 °C under N2 atmosphere overnight (sealed bottle). After completion of reaction, the reaction mixture was filtered and concentrated in vacuo. Silica flash chromatography (MeOH/DCM 0–7 %) followed by C18 flash chromatography (MeOH/H2O 10–80 %) yielded compound 2 as a yellow solid (54 mg, 36.40 %). MS m/z [M + 1] 460.1. 1H NMR (400 MHz, dmso) δ 9.65 (d, J = 7.4 Hz, 1H), 8.35 (s, 1H), 8.21 (s, 1H), 8.08 (s, 1H), 7.88 (s, 1H), 7.65 (t, J = 8.0 Hz, 1H), 7.39 (d, J = 7.6 Hz, 1H), 7.26 (d, J = 7.6 Hz, 1H), 6.76 (d, J = 8.5 Hz, 1H), 3.89 (s, 3H), 3.65–3.55 (m, 4H), 3.55–3.45 (m, 4H), 1.44 (s, 9H). 13C NMR (101 MHz, cdcl3) δ 158.84, 154.86, 148.57, 147.80, 138.42, 137.10, 135.22, 129.86, 127.64, 127.38, 124.31, 121.31, 111.93, 111.60, 110.64, 104.81, 80.15 (2C), 45.55 (2C), 39.25, 28.51 (3C).
4.1.6. Synthesis of tert-butyl 2-methyl-4-(6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)pyridin-2-yl)piperazine-1-carboxylate, compound 6
Compound 6 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a yellow compound. MS m/z [M + 1] 474.1. 1H NMR (400 MHz, dmso) δ 9.67 (d, J = 7.4 Hz, 1H), 8.36 (s, 1H), 8.21 (s, 1H), 8.08 (s, 1H), 7.88 (s, 1H), 7.65–7.61 (m, 1H), 7.40 (d, J = 7.5 Hz, 1H), 7.22 (d, J = 7.5 Hz, 1H), 6.73 (d, J = 8.6 Hz, 1H), 4.27 (s, 1H), 4.18–4.07 (m, 2H), 3.89 (s, 3H), 3.84 (s, 1H), 3.23 (d, J = 10.4 Hz, 2H), 3.06–2.98 (m, 1H), 1.44 (s, 9H), 1.18 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, cdcl3) δ 159.14, 154.89, 148.61, 147.85, 138.36, 137.09, 135.17, 132.27, 132.17, 129.88, 128.69, 128.57, 127.57, 127.52, 124.44, 121.41, 111.96, 111.57, 110.17, 104.25, 80.06, 49.45, 45.36, 39.41, 28.62 (3C), 16.53.
4.1.7. Synthesis of tert-butyl 4-((6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)pyridin-2-yl)amino)pipendine-1-carboxylate, compound 7
Compound 7 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a white compound. MS m/z [M + 1] 474.1. 1H NMR (400 MHz, cdcl3) δ 9.77 (dd, J = 7.3, 0.8 Hz, 1H), 8.03 (s, 1H), 7.85 (d, J = 0.7 Hz, 1H), 7.74 (s, 1H), 7.71 (d, J = 1.0 Hz, 1H), 7.49–7.43 (m, 1H), 7.03 (d, J = 7.6 Hz, 1H), 6.98 (dd, J = 7.3, 1.8 Hz, 1H), 6.27 (d, J = 8.3 Hz, 1H), 4.51 (d, J = 7.6 Hz, 1H), 4.17 (dd, J = 33.6, 8.2 Hz, 2H), 3.97 (s, 3H), 3.91 (ddd, J = 14.0, 10.5, 5.5 Hz, 1H), 2.99 (t, J = 11.8 Hz, 2H), 2.13 (d, J = 10.7 Hz, 2H), 1.47 (s, 9H). 13C NMR (101 MHz, cdcl3) δ 157.05, 154.96, 148.81, 147.65, 138.09, 137.07, 134.71, 130.02, 127.75, 124.10, 121.29, 111.78, 111.43, 109.63, 104.94, 79.90, 50.85, 49.13 (2C), 39.39, 32.54 (2C), 29.82, 28.58 (3C).
4.1.8. Synthesis of 6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)-N-(4-(2-(piperidin-1-yl)ethoxy)phenyl)pyridin-2-amine, compound 8
Compound 8 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a white compound. MS m/z [M + 1] 494.3. 1H NMR (400 MHz, dmso) δ 9.71 (d, J = 7.3 Hz, 1H), 8.88 (s, 1H), (s, 1H),8.36 (s, 1H), 8.22 (s, 1H), 8.08 (s, 1H), 7.86 (s, 1H), 7.59 (t, J = 7.9 Hz, 1H), 7.41 (d, J = 8.9 Hz, 2H), 7.26 (d, J = 7.5 Hz, 1H), 7.14 (dd, J = 7.3, 1.7 Hz, 1H), 6.97 (d, J = 8.9 Hz, 2H), 6.61 (d, J = 8.3 Hz, 1H), 4.08 (t, J = 6.1 Hz, 2H), 3.89 (s, 3H), 2.66 (t, J = 6.0 Hz, 2H), 2.48–2.37 (m, 4H), 1.49 (dt, J = 10.9, 5.5 Hz, 4H), 1.38 (dd, J = 10.9, 5.5 Hz, 2H). 13C NMR (101 MHz, dmso) δ 155.90, 153.84, 147.69, 147.13, 138.04, 136.69, 135.22, 134.07, 129.69, 128.83, 128.01, 123.36, 122.10 (2C), 120.07, 114.80 (2C), 110.99, 110.55, 109.86, 106.28, 65.84, 57.51, 54.45 (2C), 38.79, 25.58 (2C), 23.94.
4.1.9. Synthesis of 6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)-N-(4-(2-morpholinoethoxy)phenyl)pyridin-2-amine, compound 9
Compound 9 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a white compound. MS m/z [M + 1] 496.3. 1H NMR (400 MHz, dmso) δ 9.71 (d, J = 7.3 Hz, 1H), 8.89 (s, 1H), 8.36 (s, 1H), 8.22 (s, 1H), 8.08 (s, 1H), 7.86 (s, 1H), 7.59 (t, J = 7.9 Hz, 1H), 7.42 (d, J = 8.9 Hz, 2H), 7.26 (d, J = 7.6 Hz, 1H), 7.14 (dd, J = 7.3, 1.6 Hz, 1H), 6.98 (d, J = 8.9 Hz, 2H), 6.62 (d, J = 8.3 Hz, lH), 4.10 (dd, J = 10.8, 5.4 Hz, 2H), 3.89 (s, 3H), 3.65–3.50 (m, 4H), 2.71 (t, J = 5.8 Hz, 2H). 13C NMR (101 MHz, dmso) δ 155.89, 153.77, 147.68, 147.13, 138.03, 136.68, 135.21, 134.14, 129.67, 128.80, 128.00, 123.36, 122.09 (2C), 120.07, 114.81 (2C), 110.98, 110.55, 109.87, 106.28, 66.16 (2C), 65.59, 57.14, 53.66 (2C), 38.78.
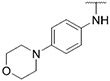
4.1.10. Synthesis of 6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)-N-(4-morpholinophenyl)pyridin-2-amine, compound 10
Compound 10 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a yellow compound. MS m/z [M + 1] 452.3. 1H NMR (400 MHz, cdcl3) δ 9.70 (d, J = 7.3 Hz, 1H), 8.06 (s, 1H), 7.83 (s, 1H), 7.69 (s, 2H), 7.50 (t, J = 7.9 Hz, 1H), 7.34–7.27 (m, 2H), 7.13 (d, J = 7.6 Hz, 1H), 6.99–6.93 (m, 2H), 6.90 (dd, J = 7.3, 1.7 Hz, 1H), 6.55 (d, J = 8.2 Hz, 1H), 6.50 (s, 1H), 3.97 (s, 3H), 3.93–3.87 (m, 4H), 3.21–3.14 (m, 4H). 13C NMR (101 MHz, cdcl3) δ 156.57, 149.01, 148.22, 147.92, 138.33, 137.06, 135.17, 132.96, 129.93, 128.37, 127.55, 124.22 (2C), 123.90, 121.46, 116.80 (2C), 111.77, 111.40, 111.02, 104.81, 67.09 (2C), 50.03 (2C), 39.40, 29.83.
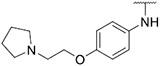
4.1.11. Synthesis of 6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)-N-(4-(2-(pyrrolidin-1-yl)ethoxy)phenyl)pyridin-2-amine, compound 11
Compound 11 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a white compound. MS m/z [M + 1] 480.2. 1H NMR (400 MHz, cdcl3) δ 9.70 (dd, J = 7.3, 0.8 Hz, 1H), 8.07 (s, 1H), 7.84 (d, J = 0.7 Hz, 1H), 7.72 (s, 1H), 7.70 (dd, J = 1.8, 0.8 Hz, 1H), 7.54–7.48 (m, 1H), 7.34–7.27 (m, 2H), 7.14 (d, J = 7.6 Hz, 1H), 6.99–6.93 (m, 2H), 6.91 (dd, J = 7.3, 1.9 Hz, 1H), 6.53 (d, J = 8.2 Hz, 1H), 6.43 (s, 1H), 4.15 (t, J = 5.9 Hz, 2H), 3.98 (s, 3H), 2.95 (t, J = 5.9 Hz, 2H), 2.72–2.61 (m, 4H), 1.89–1.78 (m, 4H). 13C NMR (101 MHz, cdcl3) δ 156.54, 155.77, 149.03, 147.98, 138.35, 137.10, 135.27, 133.42, 129.98, 128.39, 127.63, 124.55 (2C), 123.89, 121.47, 116.54, 115.45 (2C), 111.82, 111.44, 111.06, 104.87, 67.56, 55.31, 54.90 (2C), 39.40, 29.85, 23.66 (2C).
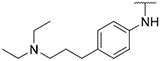
4.1.12. Synthesis of N-(4-(2-(diethylamino)ethoxy)phenyl)-6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)pyridin-2-amine, compound 12
Compound 12 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a yellow compound. MS m/z [M + 1] 482.2. 1H NMR (400 MHz, dmso) δ 9.71 (d, J = 7.3 Hz, 1H), 8.88 (s, 1H), 8.35 (s, 1H), 8.22 (s, 1H), 8.07 (s, 1H), 7.86 (s, 1H), 7.62–7.57 (m, 1H), 7.41 (d, J = 8.9 Hz, 2H), 7.26 (d, J = 7.6 Hz, 1H), 7.13 (dd, J = 7.4, 1.8 Hz, 1H), 6.96 (d, J = 8.9 Hz, 2H), 6.61 (d, J = 8.2 Hz, 1H), 4.03 (t, J = 6.2 Hz, 2H), 3.89 (s, 3H), 2.79 (t, J = 6.2 Hz, 2H), 2.56 (q, J = 7.1 Hz, 4H), 0.98 (t, J = 7.1 Hz, 6H). 13C NMR (101 MHz, cdcl3) δ 156.53, 155.69, 148.91, 147.87, 138.25, 137.01, 135.12, 133.35, 129.89, 128.35, 127.55, 124.40 (2C), 123.86, 121.36, 115.27 (2C), 111.68, 111.33, 110.89, 104.83, 67.08, 51.96, 47.96 (2C), 39.31, 11.96 (2C).
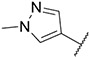
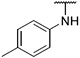
4.1.13. Synthesis of 6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)-N-(p-tolyl)pyridin-2-amine, compound 13
Compound 13 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a green compound. MS m/z [M + 1] 381.3. 1H NMR (400 MHz, cdcl3) δ 9.73 (d, J = 7.3 Hz, 1H), 8.08 (s, 1H), 7.85 (s, 1H), 7.72 (s, 1H), 7.70 (d, J = 0.9 Hz, 1H), 7.54 (t, J = 7.9 Hz, 1H), 7.29 (d, J = 8.3 Hz, 2H), 7.23–7.15 (m, 3H), 6.93 (dd, J = 7.3, 1.8 Hz, 1H), 6.65 (d, J = 8.2 Hz, 1H), 6.52 (s, 1H), 3.98 (s, 3H), 2.38 (s, 3H). 13C NMR (101 MHz, cdcl3) δ 155.83, 149.04, 138.38, 137.77, 137.11, 135.30, 133.12, 129.97, 129.94 (2C), 128.37, 127.58, 121.83 (2C), 121.46, 111.83, 111.43, 105.29, 39.44, 21.05.
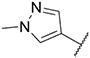
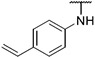
4.1.14. Synthesis of 6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)-N-(4-vinylphenyl)pyridin-2-amine, compound 14
Compound 14 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a yellow compound. MS m/z [M + 1] 393.2. 1H NMR (400 MHz, cdcl3) δ 9.70 (d, J = 7.3 Hz, 1H), 8.09 (s, 1H), 7.85 (s, 1H), 7.71 (s, 2H), 7.57 (t, J = 7.9 Hz, 1H), 7.40 (dd, J = 20.7, 8.5 Hz, 4H), 7.21 (d, J = 7.7 Hz, 1H), 6.92 (d, J = 7.3 Hz, 1H), 6.78–6.68 (m, 2H), 6.65 (s, 1H), 5.71 (d, J = 17.6 Hz, 1H), 5.21 (d, J = 10.8 Hz, 1H), 3.98 (s, 3H). 13C NMR (101 MHz, cdcl3) δ 155.04, 149.00, 148.04, 140.14, 138.45, 137.11, 136.38, 135.37, 132.50, 130.07, 128.37, 127.61, 127.22 (2C), 123.84, 121.39, 120.76 (2C), 112.47, 111.93, 111.79, 111.50, 106.07, 39.44.
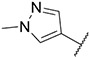
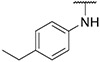
4.1.15. Synthesis of N-(4-ethylphenyl)-6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)pyridin-2-amine, compound 15
Compound 15 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a grey compound. MS m/z [M + 1] 395.3. 1H NMR (400 MHz, cdcl3) δ 9.73 (dd, J = 7.3, 0.9 Hz, 1H), 8.08 (s, 1H), 7.85 (d, J = 0.7 Hz, 1H), 7.72 (s, 1H), 7.71 (dd, J = 1.8, 0.9 Hz, 1H), 7.57–7.52 (m, 1H), 7.33–7.28 (m, 2H), 7.22 (d, J = 8.8 Hz, 1H), 7.18 (dd, J = 7.6, 0.6 Hz, 1H), 6.93 (dd, J = 7.3, 1.9 Hz, 1H), 6.66 (d, J = 8.2 Hz, 1H), 6.46 (s, 1H), 3.99 (s, 3H), 2.68 (q, J = 7.5 Hz, 2H), 1.29 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, cdcl3) δ 158.02, 155.84, 149.06, 148.00, 139.68, 138.40, 137.94, 137.11, 135.32, 129.97, 128.76 (2C), 128.43, 127.57, 122.02 (2C), 121.48, 111.84, 111.44, 110.17, 105.31, 39.46, 28.49, 16.00.
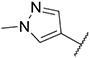
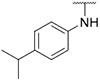
4.1.16. Synthesis of N-(4-isopropylphenyl)-6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)pyridin-2-amine, compound 16
Compound 16 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a white compound. MS m/z [M + 1] 409.3. 1H NMR (400 MHz, dmso) δ 9.75–9.70 (m, 1H), 9.01 (s, 1H), 8.34 (s, 1H), 8.23 (s, 1H), 8.06 (d, J = 0.6 Hz, 1H), 7.87 (d, J = 0.9 Hz, 1H), 7.63 (d, J = 7.7 Hz, 1H), 7.43 (d, J = 8.5 Hz, 2H), 7.29 (d, J = 7.5 Hz, 1H), 7.24 (d, J = 8.5 Hz, 2H), 7.11 (dd, J = 7.3, 1.9 Hz, 1H), 6.70 (d, J = 8.2 Hz, 1H), 3.90 (s, 3H), 2.91 (dt, J = 13.8, 7.0 Hz, 1H), 1.24 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, dmso) δ 155.56, 147.69, 147.16, 141.77, 138.80, 138.14, 136.65, 135.28, 131.52, 131.42, 129.69, 128.81, 128.77, 128.69, 128.07, 126.54 (2C), 123.38, 120.18 (2C), 120.07, 110.93, 110.59, 110.30, 106.69, 38.83, 32.83, 24.12 (2C).
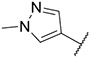
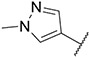
4.1.17. Synthesis of N-(4-(tert-butyl)phenyl)-6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)pyridin-2-amine, compound 17
Compound 17 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a orange compound. MS m/z [M + 1] 423.3. 1H NMR (400 MHz, dmso) δ 9.74 (d, J = 7.3 Hz, 1H), 9.01 (s, 1H), 8.32 (s, 1H), 8.23 (s, 1H), 8.04 (s, 1H), 7.87 (s, 1H), 7.62 (t, J = 7.9 Hz, 1H), 7.40 (dd, J = 20.2, 8.7 Hz, 4H), 7.30 (d, J = 7.6 Hz, 1H), 7.09 (dd, J = 7.3, 1.6 Hz, 1H), 6.70 (d, J = 8.3 Hz, 1H), 3.90 (s, 3H), 1.32 (s, 9H). 13C NMR (101 MHz, dmso) δ 155.56, 147.70, 147.14, 144.04, 138.44, 138.14, 136.58, 135.26, 129.65, 128.69, 128.10, 125.41 (2C), 123.36, 120.07, 119.95 (2C), 110.88, 110.60, 110.29, 106.59, 33.98, 31.33 (3C).
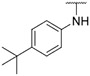
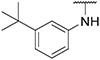
4.1.18. Synthesis of N-(3-(tert-butyl)phenyl)-6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)pyridin-2-amine, compound 18
Compound 18 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a white compound. MS m/z [M + 1] 423.3. 1H NMR (400 MHz, cdcl3) δ 9.73 (dd, J = 7.3, 0.7 Hz, 1H), 8.08 (s, 1H), 7.83 (s, 1H), 7.71–7.69 (m, 1H), 7.69 (s, 1H), 7.55 (t, J = 7.9 Hz, 1H), 7.33–7.28 (m, 3H), 7.18 (d, J = 7.6 Hz, 1H), 7.16–7.13 (m, 1H), 6.91 (dd, J = 7.3, 1.8 Hz, 1H), 6.74–6.68 (m, 2H), 3.96 (s, 3H), 1.33 (s, 9H). 13C NMR (101 MHz, cdcl3) δ 155.67, 152.76, 149.10, 147.96, 140.16, 138.40, 137.08, 135.22, 130.02, 128.98, 128.34, 127.57, 123.88, 121.43, 120.52, 118.52, 111.78, 111.60, 111.53, 105.46, 39.39, 34.90, 31.47 (3C), 29.83.
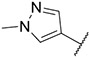
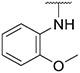
4.1.19. Synthesis of N-(2-methoxyphenyl)-6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)pyridin-2-amine, compound 19
Compound 19 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a grey compound. MS m/z [M + 1] 397.2. 1H NMR (400 MHz, cdcl3) δ 9.71 (d, J = 7.3 Hz, 1H), 8.04 (s, 1H), 8.00 (d, J = 7.5 Hz, 1H), 7.83 (s, 1H), 7.68 (s, 2H), 7.53 (t, J = 7.9 Hz, 1H), 7.16 (d, J = 7.6 Hz, 1H), 7.05–6.92 (m, 4H), 6.89 (d, J = 7.3 Hz, 1H), 6.67 (d, J = 8.2 Hz, 1H), 3.94 (s, 3H), 3.91 (s, 3H). 13C NMR (101 MHz, cdcl3) δ 154.98, 149.15, 148.55, 138.26, 137.13, 135.08, 130.03, 129.98, 128.45, 127.72, 127.43, 124.08, 122.15, 121.34, 120.71, 119.77, 111.81, 111.47, 111.37, 110.55, 107.15, 55.86, 39.39.
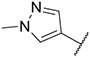
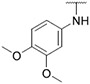
4.1.20. Synthesis of N-(3,4-dimethoxyphenyl)-6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)pyridin-2-amine, compound 20
Compound 20 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a brown compound. MS m/z [M + 1] 427.2. 1H NMR (400 MHz, dmso) δ 9.73 (d, J = 7.4 Hz, 1H), 8.92 (s, 1H), 8.35 (s, 1H), 8.22 (s, 1H), 8.08 (s, 1H), 7.87 (s, 1H), 7.60 (t, J = 7.9 Hz, 1H), 7.27 (d, J = 7.5 Hz, 1H), 7.12 (dd, J = 5.2, 2.0 Hz, 2H), 7.04 (dd, J = 8.6, 2.3 Hz, 1H), 6.96 (d, J = 8.6 Hz, 1H), 6.65 (d, J = 8.2 Hz, 1H), 3.89 (s, 3H), 3.77 (s, 3H), 3.68 (s, 3H). 13C NMR (101 MHz, dmso) δ 155.76, 149.01, 147.69, 147.15, 144.11, 138.21, 136.75, 135.24, 134.66, 129.67, 128.79, 127.98, 123.41, 120.04, 112.57, 112.26, 110.91, 110.45, 110.15, 106.52, 105.61, 56.06, 55.45, 39.52, 38.68.
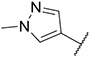
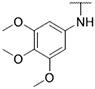
4.1.21. Synthesis of 6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)-N-(3,4,5-trimethoxyphenyl)pyridin-2-amine, compound 21
Compound 21 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a yellow compound. MS m/z [M + 1] 457.2. 1H NMR (400 MHz, dmso) δ 9.70 (d, J = 7.4 Hz, 1H), 9.06 (s, 1H), 8.34 (s, 1H), 8.23 (s, 1H), 8.07 (s, 1H), 7.88 (s, 1H), 7.65 (t, J = 8.0 Hz, 1H), 7.31 (d, J = 7.5 Hz, 1H), 7.14 (dd, J = 7.3, 1.6 Hz, 1H), 6.85 (s, 2H), 6.73 (d, J = 8.2 Hz, 1H), 3.89 (s, 3H), 3.68 (s, 6H), 3.66 (s, 3H). 13C NMR (101 MHz, cdcl3) δ 155.66, 153.49 (2C), 148.47, 147.62, 138.14, 136.85, 136.73, 134.66, 133.74, 132.04, 131.94, 129.99, 128.59, 128.47, 128.30, 127.58, 123.84, 120.92, 111.30, 111.13, 106.16, 98.98 (2C), 61.02, 56.04 (2C), 39.10.
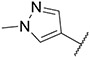
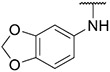
4.1.22. Synthesis of N-(benzo[d][1,3]dioxol-5-yl)-6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)pyridin-2-amine, compound 22
Compound 22 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a yellow compound. MS m/z [M + 1] 411.2. 1H NMR (400 MHz, cdcl3) δ 9.83 (d, J = 7.3 Hz, 1H), 8.02 (s, 1H), 7.85 (s, 1H), 7.72 (s, 1H), 7.71–7.67 (m, 1H), 7.50–7.45 (m, 1H), 7.29–7.21 (m, 1H), 7.06 (d, J = 7.6 Hz, 1H), 7.02 (dd, J = 7.3, 1.5 Hz, 1H), 6.92 (t, J = 7.8 Hz, 2H), 6.38 (d, J = 8.2 Hz, 1H), 4.92 (t, J = 6.2 Hz, 1H), 4.75 (d, J = 6.2 Hz, 2H), 3.97 (s, 3H). 13C NMR (101 MHz, cdcl3) δ 160.53, 157.50, 148.92, 147.86, 138.17, 137.10, 135.08, 129.89, 129.51, 128.15, 127.56, 124.12, 121.53, 111.77, 111.52, 111.41, 110.29, 104.55, 50.88, 39.38, 34.32, 29.84.
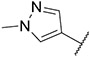
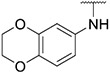
4.1.23. Synthesis of N-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)pyridin-2-amine, compound 23
Compound 23 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a white compound. MS m/z [M + 1] 425.2. 1H NMR (400 MHz, dmso) δ 9.76 (d, J = 7.3 Hz, 1H), 8.90 (s, 1H), 8.36 (s, 1H), 8.23 (s, 1H), 8.09 (s, 1H), 7.87 (d, J = 0.8 Hz, 1H), 7.60 (t, J = 7.9 Hz, 1H), 7.28 (d, J = 7.6 Hz, 1H), 7.15 (dd, J = 7.4, 1.4 Hz, 1H), 7.09 (d, J = 2.4 Hz, 1H), 6.95 (dd, J = 8.5, 2.3 Hz, 1H), 6.86 (d, J = 8.6 Hz, 1H), 6.62 (d, J = 8.3 Hz, 1H), 4.27 (dd, J = 10.5, 5.2 Hz, 4H), 3.90 (s, 3H). 13C NMR (101 MHz, dmso) δ 155.55, 147.60, 147.13, 143.26, 138.41, 138.07, 136.69, 135.19, 134.68, 129.76, 128.81, 128.02, 123.37, 120.05, 116.87, 113.47, 111.07, 110.48, 110.06, 109.26, 106.61, 64.21, 63.98, 38.81.
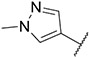
4.1.24. Synthesis of 6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)-N-(4-(methylsulfonyl)phenyl)pyridin-2-amine, compound 24
Compound 24 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a yellow compound. MS m/z [M + 1] 445.2. 1H NMR (400 MHz, dmso) δ 9.77 (s, 1H), 9.57 (d, J = 7.3 Hz, 1H), 8.36 (s, 1H), 8.27 (s, 1H), 8.10 (s, 1H), 7.92 (s, 1H), 7.86–7.75 (m, 4H), 7.48 (d, J = 7.7 Hz, 1H), 7.24 (d, J = 6.5 Hz, 1H), 6.87 (d, J = 8.1 Hz, 1H), 3.90 (s, 3H), 3.19 (s, 3H). 13C NMR (101 MHz, dmso) δ 153.96, 147.62, 147.36, 146.09, 138.71, 136.74, 135.70, 131.58, 130.00, 128.85, 128.34 (2C), 127.41, 123.30, 120.00, 117.47 (2C), 112.75, 111.32, 110.66, 108.88, 44.06, 38.81.
4.1.25. Synthesis of 6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)-N-(2-(trifluoromethyl)phenyl)pyridin-2-amine, compound 25
Compound 25 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a black compound. MS m/z [M + 1] 435.1. 1H NMR (400 MHz, cdcl3) δ 9.62 (dd, J = 7.3, 0.8 Hz, 1H), 8.11 (s, 1H), 7.93 (d, J = 8.2 Hz, 1H), 7.85 (d, J = 0.7 Hz, 1H), 7.75–7.67 (m, 3H), 7.61 (t, J = 7.9 Hz, 1H), 7.55 (t, J = 7.3 Hz, 1H), 7.30 (dd, J = 7.7, 0.5 Hz, 1H), 7.21 (t, J = 7.7 Hz, 1H), 6.90 (dd, J = 7.3, 1.8 Hz, 1H), 6.71 (s, 1H), 6.68 (d, J = 8.1 Hz, 1H), 3.99 (s, 3H). 13C NMR (101 MHz, cdcl3) δ 154.64, 149.11, 148.15, 138.65, 137.12, 135.63, 132.56, 130.16, 128.35, 127.62, 126.94, 123.97, 122.99, 121.38, 112.70, 111.86, 111.57, 106.69, 104.50, 39.45.
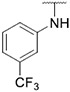
4.1.26. Synthesis of 6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)-N-(3-(trifluoromethyl)phenyl)pyridin-2-amine, compound 26
Compound 26 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a yellow compound. MS m/z [M + 1] 435.2. 1H NMR (400 MHz, dmso) δ 9.54 (d, J = 7.0 Hz, 2H), 8.35 (s, 1H), 8.25 (s, 1H), 8.07 (s, 1H), 8.00 (s, 1H), 7.90 (s, 1H), 7.82 (d, J = 7.7 Hz, 1H), 7.73 (t, J = 7.7 Hz, 1H), 7.57 (t, J = 8.0 Hz, 1H), 7.42 (d, J = 7.6 Hz, 1H), 7.28 (d, J = 8.2 Hz, 1H), 7.14 (d, J = 7.4 Hz, 1H), 6.78 (d, J = 8.2 Hz, 1H), 3.89 (s, 3H). 13C NMR (101 MHz, dmso) δ 154.47, 147.45, 147.29, 142.08, 138.57, 136.64, 135.53, 129.91, 129.85, 128.79, 127.30, 123.37, 122.19, 120.01, 117.02, 116.98, 114.31, 114.27, 111.92, 111.23, 110.58, 108.27, 38.78.
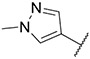
4.1.27. Synthesis of 6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)-N-(4-(trifluoromethyl)phenyl)pyridin-2-amine, compound 27
Compound 27 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a grey compound. MS m/z [M + 1] 435.2. 1H NMR (400 MHz, cdcl3) δ 9.63 (dd, J = 7.3, 0.5 Hz, 1H), 8.11 (s, 1H), 7.86 (s, 1H), 7.76–7.71 (m, 2H), 7.64 (t, J = 8.0 Hz, 1H), 7.60 (d, J = 8.7 Hz, 2H), 7.54 (d, J = 8.6 Hz, 2H), 7.30 (d, J = 7.6 Hz, 1H), 6.93 (dd, J = 7.3, 1.8 Hz, 1H), 6.78–6.70 (m, 2H), 3.99 (s, 3H). 13C NMR (101 MHz, cdcl3) δ 154.02, 149.02, 143.82, 138.70, 137.12, 135.67, 130.29, 128.15, 127.65, 126.60, 126.56, 121.29, 119.22 (2C), 113.01, 111.90, 111.62, 107.06, 39.46.
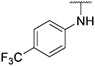
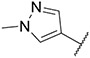
4.1.28. Synthesis of 6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)-N-(4-(trifluoromethoxy)phenyl)pyridin-2-amine, compound 28
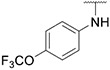
Compound 28 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a white compound. MS m/z [M + 1] 451.3. 1H NMR (400 MHz, cdcl3) δ 9.63 (d, J = 7.3 Hz, 1H),8.08 (s, 1H), 7.84 (s, 1H), 7.72 (s, 1H), 7.70 (s, 1H), 7.58 (t, J = 7.9 Hz, 1H), 7.45 (d, J = 8.8 Hz, 2H), 7.23 (d, J = 7.7 Hz, 2H), 6.88 (d, J = 7.3 Hz, 1H), 6.73 (s, 1H), 6.65 (d, J = 8.2 Hz, 1H), 3.98 (s, 3H). 13C NMR (101 MHz, dmso) δ 154.79, 151.31, 147.53, 142.11, 140.53, 138.43, 136.66, 135.38, 129.89, 128.82, 127.66, 123.35, 121.79, 120.27, 120.02, 111.39, 111.08, 110.59, 107.65, 38.80.
4.1.29. Synthesis of 6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)-N-(4-nitrophenyl)pyridin-2-amine, compound 29
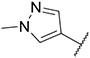
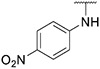
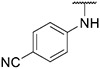
Compound 29 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a orange compound. MS m/z [M + 1] 412.1. 1H NMR (400 MHz, cdcl3) δ 9.59 (d, J = 7.3 Hz, 1H), 8.22 (d, J = 9.2 Hz, 2H), 8.12 (s, 1H), 7.86 (s, 1H), 7.74 (s, 2H), 7.69 (t, J = 7.9 Hz, 1H), 7.58 (d, J = 9.2 Hz, 2H), 7.38 (d, J = 7.7 Hz, 1H), 7.03–6.94 (m, 2H), 6.77 (d, J = 8.1 Hz, 1H), 3.98 (s, 3H). 13C NMR (101 MHz, dmso) δ 153.34, 148.04, 147.73, 147.49, 139.63, 138.95, 136.82, 135.93, 130.18, 128.99, 127.37, 125.52 (2C), 123.19, 120.01, 116.80 (2C), 113.60, 111.51, 110.63, 109.62, 38.83.
4.1.30. Synthesis of N-(6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)pyridin-2-yl)pyrazin-2-amine, compound 30
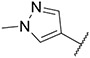
Compound 30 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a yellow compound. MS m/z [M + 1] 369.3. 1H NMR (400 MHz, dmso) δ 10.17 (s, 1H), 10.02 (d, J = 7.3 Hz, 1H), 8.71 (s, 1H), 8.40 (s, 1H), 8.33 (s, 1H), 8.32 (dd, J = 2.3, 1.1 Hz, 1H), 8.15–8.09 (m, 2H), 7.91 (s, 1H), 7.84–7.75 (m, 2H), 7.56 (d, J = 7.3 Hz, 1H), 7.30 (dd, J = 7.3, 1.6 Hz, 1H), 3.91 (s, 3H). 13C NMR (101 MHz, dmso) δ 152.43, 150.97, 148.14, 147.42, 141.44, 138.69, 136.77, 135.72, 135.62, 135.41, 130.14, 128.93, 128.39, 122.81, 120.02, 113.26, 111.39, 110.48, 108.66, 38.82.
4.1.31. Synthesis of 4-((6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)pyridin-2-yl)amino)benzonitrile, compound 31
Compound 31 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a yellow compound. MS m/z [M + 1] 392.3. 1H NMR (400 MHz, dmso) δ 9.96 (s, 1H), 9.69 (d, J = 7.4 Hz, 1H), 8.71 (s, 1H), 8.59 (s, 1H), 8.27 (s, 1H), 8.06 (s, 1H), 7.92–7.84 (m, 1H), (s, 3H), 7.71 (dd, J = 7.4, 1.4 Hz, 1H), 7.55 (d, J = 7.5 Hz, 1H), 7.00 (d, J = 8.3 Hz, 1H), 3.93 (s, 3H). 13C NMR (101 MHz, dmso) δ 172.06, 154.01, 145.37, 145.19, 139.25, 137.76, 133.41 (2C), 130.53, 128.91, 124.35, 119.71, 118.70, 117.92 (2C), 114.37, 114.11, 111.30, 106.02, 102.00, 39.02.
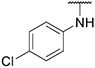
4.1.32. Synthesis of N-(4-chlorophenyl)-6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)pyridin-2-amine, compound 32
Compound 32 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a white compound. MS m/z [M + 1] 401.3. 1H NMR (400 MHz, cdcl3) δ 9.64 (d, J = 7.3 Hz, 1H), 8.10 (d, J = 4.6 Hz, 1H), 7.86 (s, 1H), 7.73 (d, J = 3.2 Hz, 2H), 7.58 (t, J = 7.9 Hz, 1H), 7.40–7.31 (m, 3H), 7.23 (d, J = 7.7 Hz, 1H), 6.93 (dd, J = 7.3, 1.6 Hz, 1H), 6.64 (d, J = 8.2 Hz, 1H), 6.53 (s, 1H), 3.99 (s, 3H). 13C NMR (101 MHz, dmso) δ 154.74, 147.55, 147.24, 140.24, 138.36, 136.75, 135.45, 129.88, 128.89, 128.64 (2C), 127.55, 124.43, 123.37, 120.32 (2C), 120.02, 111.33, 111.20, 110.60, 107.76, 38.78.
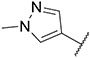
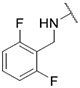
4.1.33. Synthesis of N-(2,6-difluorobenzyl)-6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)pyridin-2-amine, compound 33
Compound 33 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a white compound. MS m/z [M + 1] 417.2. 1H NMR (400 MHz, dmso) δ 9.73 (d, J = 7.5 Hz, 1H), 8.96 (s, 1H), 8.36 (s, 1H), 8.23 (s, 1H), 8.09 (s, 1H), 7.87 (s, 1H), 7.61 (t, J = 7.8 Hz, 1H), 7.29 (d, J = 7.3 Hz, 1H), 7.16 (s, 1H), 7.14 (s, 1H), 6.94 (s, 2H), 6.63 (d, J = 8.3 Hz, 1H), 6.04 (s, 2H), 3.90 (s, 3H). 13C NMR (101 MHz, dmso) δ 155.60, 147.60, 147.37, 147.17, 142.10, 138.11, 136.69, 135.42, 135.28, 129.71, 128.82, 127.95, 123.33, 120.02, 112.96, 110.99, 110.53, 110.13, 108.14, 106.61, 102.72, 100.89, 38.78.
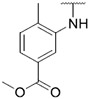
4.1.34. Synthesis of methyl 4-methyl-3-((6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)pyridin-2-yl)amino)benzoate, compound 34
Compound 34 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a yellow compound. MS m/z [M + 1] 439.2. 1H NMR (400 MHz, dmso) δ 9.40 (d, J = 7.3 Hz, 1H),8.59 (s, 1H), 8.29 (d, J = 7.9 Hz, 2H), 8.17 (d, J = 1.5 Hz, 1H), 8.02 (s, 1H), 7.83 (s, 1H), 7.73 (dd, J = 7.9, 1.6 Hz, 1H), 7.65 (t, J = 7.9 Hz, 1H), 7.48 (d, J = 7.9 Hz, 1H), 7.36 (d, J = 7.6 Hz, 1H), 6.80 (dd, J = 7.4, 1.6 Hz, 1H), 6.71 (d, J = 8.2 Hz, 1H), 3.89 (s, 3H), 3.72 (s, 3H), 2.34 (s, 3H). 13C NMR (101 MHz, cdcl3) δ 174.77, 167.05, 155.79, 147.20, 138.90, 138.44, 137.28, 136.90, 133.75, 132.08, 131.05, 128.81, 128.32, 127.75, 125.43, 124.82, 123.59, 120.96, 111.43, 110.87, 106.02, 52.10, 39.23, 21.61, 18.53.
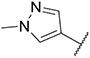
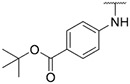
4.1.35. Synthesis of tert-butyl 4-((6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)pyridin-2-yl)amino)benzoate, compound 35
Compound 35 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a yellow compound. MS m/z [M + 1] 467.0. 1H NMR (400 MHz, cdcl3) δ 9.69 (dd, J = 7.3, 0.6 Hz, 1H), 8.11 (s, 1H), 7.99 (d, J = 8.7 Hz, 2H), 7.86 (d, J = 0.7 Hz, 1H), 7.74 (s, 1H), 7.73 (d, J = 0.9 Hz, 1H), 7.63 (t, J = 7.9 Hz, 1H), 7.46 (d, J = 8.8 Hz, 2H), 7.30 (d, J = 7.4 Hz, 1H), 6.98 (dd, J = 7.3, 1.8 Hz, 1H), 6.80 (s, 1H), 6.77 (d, J = 8.1 Hz, 1H), 3.99 (s, 3H), 1.61 (s, 9H). 13C NMR (101 MHz, cdcl3) δ 165.72, 153.93, 149.09, 148.18, 144.57, 138.62, 137.12, 135.64, 131.08 (2C), 130.28, 128.19, 127.70, 125.50, 123.73, 121.33, 118.15 (2C), 113.00, 111.88, 111.73, 107.20, 80.81, 39.44, 28.46 (3C).
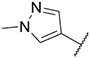
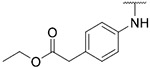
4.1.36. Synthesis of ethyl 2-(4-((6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)pyridin-2-yl)amino)phenyl)acetate, compound 36
Compound 36 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a white compound. MS m/z [M + 1] 453.2. 1H NMR (400 MHz, cdcl3) δ 9.68 (dd, J = 7.3, 0.8 Hz, 1H), 8.08 (s, 1H), 7.88 (d, J = 0.7 Hz, 1H), 7.76 (s, 1H), 7.69 (dd, J = 1.8, 0.8 Hz, 1H), 7.57–7.52 (m, 1H), 7.39–7.27 (m, 4H), 7.19 (dd, J = 7.6, 0.5 Hz, 1H), 6.97 (dd, J = 7.3, 1.9 Hz, 1H), 6.68 (s, 1H), 6.64 (d, J = 7.8 Hz, 1H), 4.19 (q, J = 7.1 Hz, 2H), 3.96 (s, 3H), 3.63 (s, 2H), 1.28 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, cdcl3) δ 172.02, 155.41, 148.93, 148.00, 139.46, 138.37, 137.15, 135.18, 130.17 (2C), 128.97, 128.59, 127.76, 123.79, 121.80 (2C), 121.43, 111.54, 105.86, 61.04, 40.89, 39.35, 29.83, 14.38.
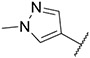
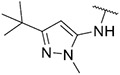
4.1.37. Synthesis of N-(3-(tert-butyl)-1-methyl-1H-pyrazol-5-yl)-6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)pyridin-2-amine, compound 37
Compound 37 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a white compound. MS m/z [M + 1] 427.2. 1H NMR (400 MHz, cdcl3) δ 9.41 (dd, J = 7.3, 0.8 Hz, 1H), 8.09 (d, J = 2.0 Hz, 1H), 7.82 (d, J = 0.7 Hz, 1H), 7.71–7.65 (m, 2H), 7.56 (t, J = 7.9 Hz, 1H), 7.22 (dd, J = 7.7, 0.5 Hz, 1H), 6.93 (dd, J = 7.3, 1.9 Hz, 1H), 6.45–6.40 (m, 1H), 6.29 (s, 1H), 6.04 (s, 1H), 3.96 (s, 3H), 3.73 (s, 3H), 1.36 (s, 9H). 13C NMR (101 MHz, cdcl3) δ 161.15, 156.04, 149.22, 148.12, 138.74, 138.47, 137.08, 135.37, 130.23, 128.27, 127.56, 121.33, 111.84, 111.64, 111.58, 104.70, 97.34, 39.40, 35.33, 32.48, 30.75 (3C).
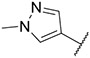
4.1.38. Synthesis of N-(3-(tert-butyl)-1-(4-fluorophenyl)-1H-pyrazol-5-yl)-6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)pyridin-2-amine, compound 38
Compound 38 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a yellow compound. MS m/z [M + 1] 507.3. 1H NMR (400 MHz, cdcl3) δ 9.58 (d, J = 7.3 Hz, 1H), 8.09 (s, 1H), 7.83 (s, 1H), 7.75 (s, 1H), 7.72 (s, 1H), 7.62–7.53 (m, 3H), 7.13–7.06 (m, 2H), 7.01–6.97 (m, 1H), 6.56 (d, J = 8.1 Hz, 1H), 6.31 (s, 1H), 6.13 (s, 1H), 3.98 (s, 3H), 1.40 (s, 9H). 13C NMR (101 MHz, cdcl3) δ 162.72, 162.48, 160.26, 155.27, 149.02, 147.92, 138.80, 138.60, 136.91, 135.18, 130.12, 128.27, 127.47, 126.07, 125.98, 123.41, 121.16, 116.14, 115.92, 111.85, 111.65, 111.48, 105.32, 97.64, 39.28, 32.63, 30.53 (3C).
4.1.39. Synthesis of N-(3-(tert-butyl)-1-(p-tolyl)-1H-pyrazol-5-yl)-6-(7-(1-methyl-1H-pyrazol-4-yl)imidazo [1,2-a]pyridin-3-yl)pyridin-2-amine, compound 39
Compound 39 was synthesized according to the procedure outlined in section 4.1.5 and was isolated as a white compound. MS m/z [M + 1] 503.3. 1H NMR (400 MHz, cdcl3) δ 9.61 (d, J = 7.3 Hz, 1H), 8.07 (s, 1H), 7.81 (s, 1H), 7.69 (s, 1H), 7.67 (s, 1H), 7.57 (t, J = 7.9 Hz, 1H), 7.47 (s, 1H), 7.45 (s, 1H), 7.25–7.18 (m, 3H), 6.94 (dd, J = 7.3, 0.9 Hz, 1H), 6.57 (d, J = 8.2 Hz, 1H), 6.34 (s, 1H), 6.32 (s, 1H), 3.97 (s, 3H), 2.35 (s, 3H), 1.40 (s, 9H). 13C NMR (101 MHz, cdcl3) δ 162.26, 154.91, 149.17, 148.09, 138.67, 138.45, 137.42, 137.07 (2C), 136.42, 135.45, 130.17, 130.02, 128.32, 127.53, 124.39 (2C), 123.57, 121.38, 112.16, 111.77, 111.74, 105.44, 96.51, 39.42, 32.68, 30.63 (3C), 21.21.
4.1.40. Synthesis of N-(3-(tert-butyl)-1-methyl-1H-pyrazol-5-yl)-6-(7-(4-(methylsulfonyl)phenyl)imidazo [1,2-a]pyridin-3-yl)pyridin-2-amine, compound 40
Compound III (124 mg, 322.83 μmol), 3-(tert-butyl)-1-methyl-1H-pyrazol-5-amine (99 mg, 645.67 μmol, 2 equiv), cesium carbonate (157.78 mg, 434.25 μmol) were dissolved into 5 mL Dioxane. The resulting reaction mixture was degassed with nitrogen for 5–10 min. After that, Pd(PPh3)4 (37.31 mg, 32.28 μmol) was added. The resulting mixture was stirred at 100 °C under N2 atmosphere overnight (sealed bottle). After completion of reaction, the reaction mixture was filtered and concentrated in vacuo. Silica flash chromatography (MeOH/DCM 0–7 %) followed by C18 flash chromatography (MeOH/H2O 10–80 %) yielded compound 2 as a green solid (58 mg, 35.86 %). MS m/z [M + 1] 501.2. 1H NMR (400 MHz, cdcl3) δ 9.54 (d, J = 7.4 Hz, 1H), 8.19 (s, 1H), 8.08–8.01 (m, 2H), 7.90 (d, J = 1.9 Hz, 1H), 7.83 (dq, J = 8.3, 2.1 Hz, 2H), 7.60 (t, J = 7.9 Hz, 1H), 7.30–7.25 (m, 1H), 7.08 (dt, J = 7.0, 1.8 Hz, 1H), 6.48 (d, J = 8.2 Hz, 1H), 6.26 (s, 1H), 6.05 (s, 1H), 3.73 (s, 3H), 3.11 (s, 3H), 1.36 (s, 9H). 13C NMR (101 MHz, cdcl3) δ 161.19, 156.11, 148.84, 147.53, 144.14, 140.08, 138.90, 138.33, 136.06, 135.67, 128.62, 128.37 (2C), 127.71 (2C), 123.80, 115.46, 112.16, 111.91, 105.33, 97.43, 44.73, 35.37, 32.50, 30.77 (3C).
4.1.41. Synthesis of N-(3-(tert-butyl)-1-(p-tolyl)-1H-pyrazol-5-yl)-6-(7-(4-(methylsulfonyl)phenyl)imidazo [1,2-a]pyridin-3-yl)pyridin-2-amine, compound 41
Compound 41 was synthesized according to the procedure outlined in section 4.1.40 and was isolated as a green compound. MS m/z [M + 1] 577.3. 1H NMR (400 MHz, cdcl3) δ 9.75 (dd, J = 7.3, 0.5 Hz, 1H), 8.18 (s, 1H), 8.08–8.00 (m, 2H), 7.92 (d, J = 1.1 Hz, 1H), 7.87–7.81 (m, 2H), 7.61 (t, J = 7.9 Hz, 1H), 7.46 (d, J = 8.4 Hz, 2H), 7.27 (d, J = 7.6 Hz, 1H), 7.21 (d, J = 8.1 Hz, 2H), 7.11 (dd, J = 7.4, 2.0 Hz, 1H), 6.61 (d, J = 8.2 Hz, 1H), 6.33 (s, 1H), 6.29 (s, 1H), 3.11 (s, 3H), 2.35 (s, 3H), 1.40 (s, 9H). 13C NMR (101 MHz, cdcl3) δ 162.29, 155.02, 148.81, 147.52, 144.20, 140.14, 138.84, 138.29, 137.49, 136.41, 136.16, 135.66, 130.05 (2C), 128.66, 128.41 (2C), 127.72 (2C), 124.36 (2C), 115.58, 112.51, 112.11, 106.03, 96.68, 44.74, 32.71, 30.65 (3C), 21.22.
4.1.42. Synthesis of 6-(7-(4-(methylsulfonyl)phenyl)imidazo [1,2-a]pyridin-3-yl)-N-(3,4,5-trimethoxyphenyl)pyridin-2-amine, compound 42
Compound 42 was synthesized according to the procedure outlined in section 4.1.40 and was isolated as a yellow compound. MS m/z [M + 1] 531.1. 1H NMR (400 MHz, cdcl3) δ 9.86 (d, J = 7.4 Hz, 1H), 8.18 (s, 1H), 8.03 (dd, J = 8.5, 1.7 Hz, 2H), 7.91 (d, J = 1.1 Hz, 1H), 7.82 (dd, J = 8.5, 1.8 Hz, 2H), 7.59 (t, J = 7.9 Hz, 1H), 7.24 (dd, J = 10.9, 4.3 Hz, 1H), 7.07 (dd, J = 7.4, 1.9 Hz, 1H), 6.74–6.58 (m, 4H), 3.87 (s, 3H), 3.81 (s, 6H), 3.10 (s, 3H). 13C NMR (101 MHz, cdcl3) δ 155.86, 153.82 (2C), 148.60, 147.38, 144.11, 140.09, 138.62, 136.36, 135.95, 135.61, 134.58, 128.71, 128.36 (2C), 127.76 (2C), 124.30, 115.58, 111.84, 106.32, 99.85 (2C), 61.21, 56.30 (2C), 44.74, 29.82.
4.1.43. Synthesis of N-(4-(tert-butyl)phenyl)-6-(7-(4-(methylsulfonyl)phenyl)imidazo [1,2-a]pyridin-3-yl)pyridin-2-amine, compound 43
Compound 43 was synthesized according to the procedure outlined in section 4.1.40 and was isolated as a yellow compound. MS m/z [M + 1] 497.2. 1H NMR (400 MHz, cdcl3) δ 9.85 (dd, J = 7.4, 0.8 Hz, 1H), 8.17 (s, 1H), 8.08–8.02 (m, 2H), 7.92 (dd, J = 1.9, 0.8 Hz, 1H), 7.87–7.80 (m, 2H), 7.59–7.54 (m, 1H), 7.45–7.38 (m, 2H), 7.37–7.30 (m, 2H), 7.21 (d, J = 7.2 Hz, 1H), 7.04 (dd, J = 7.4, 2.0 Hz, 1H), 6.71 (d, J = 8.1 Hz, 1H), 6.63 (s, 1H), 3.11 (s, 3H), 1.37 (s, 9H). 13C NMR (101 MHz, cdcl3) δ 155.96, 148.60, 147.35, 146.70, 144.28, 140.04, 138.49, 137.63, 135.89, 135.45, 128.84, 128.37 (2C), 127.70 (2C), 126.20 (2C), 124.37, 121.77 (2C), 115.52, 111.69, 111.67, 105.99, 44.73, 34.56, 31.62 (3C).
4.2. Cell cultures and viability assay
MOLM14, MOLM14-D835Y and MOLM14-F691L cell lines were obtained from the laboratory of Dr. Shah in University of California San Francisco and cultured in RPMI (Gibco, USA) containing 10 % fetal bovine serum (FBS) (Gibco, USA). All media contained 100 units/mL penicillin (Gibco, USA), and 100 μg/mL streptomycin (Gibco, USA). Other cell lines were cultured in appropriate media according to provider’s instructions. Cells were incubated at 37 °C in a humidified atmosphere of 5 % CO2.
For the viability assays, cells in logarithmic phase were seeded into 96-well culture plates at densities of (5000) cells per well. Then, after 24 h of incubation, cells were treated in triplicate with various concentrations of compounds for 72 h in final volumes of 200 μL. Upon end point, 20 μL Resazurin solution (Biotium, USA) was added to each well, and the cells were incubated for an additional 4–6 h. Fluorescence values at a wavelength of 590 nM were taken on a spectrophotometer (BioTek, USA). GI50 values were calculated using percentage of growth versus vehicle (DMSO) treated control. The data were finally fitted in GraphPad Prism V9.0 software to obtain IC50 (half-1ife) values using [inhibitor] vs. normalized response (variable slope) model. Each compound was tested at least three times.
4.3. FLT3 kinase activity assay
Kinase assays was conducted using the bioluminescent ADP-Glo™ kinase assay (Promega, USA), following the manufacturer’s instructions. Assay was performed with the test compounds at 8-point half log dilutions (1 μM to 0.316 nM). Luminescence signal was measured on a spectrophotometer (BioTek, USA), and IC50 values reported are based on the dose-response curve fitted in GraphPad Prism V9.0 using [inhibitor] vs. normalized response model. Each compound was tested at least three times.
4.4. Western blot
MOLM14, MOLM14-D835Y and MOLM14-F691L cells, at 5 × 105 cells/ml, were exposed to compound 24 for 2 h at 37° at the indicated doses. Total cell lysates were resolved by SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. The membranes were then probed with primary antibodies purchased from Cell Signaling (Danvers, MA, USA) [pFLT3 Y842 (#4577), FLT3 (#3462), pERK (#4370), ERK (#9107) and GAPDH (#5174)] followed by secondary antibodies purchased from Licor (Lincoln, NE, USA). Immunoblots were then visualized by a Biorad ChemiDoc MP Imaging System (Hercules, CA, USA).
4.5. Molecular docking
The molecular docking studies were completed using Maestro 11.8 (released 2018-4). The ligand was prepared by the LigPrep tool. The crystal structure of FLT3 bound to gilteritinib (PDB ID: 6JQR) was used as template. The structure of FLT3-F691L was generated by 3D builder tool – mutate residue. The protein preparation was done in Maestro using the protein preparation wizard tool. A grid box around the ATP binding site was created. The ligands were docked in this grid using Ligand Docking. The results were visualized and analyzed with the Maestro suite.
4.6. Kinase selectivity profiling
Compound 24 was dissolved in DMSO to required concentrations and sent to Eurofins DiscoverX Corporation located in San Diego, CA USA for KINOMEscan™ Profiling Service.
The kinases were tagged with DNA for qPCR detection. Streptavidin-coated magnetic beads were treated with biotinylated small molecule ligands for 30 min at room temperature to generate affinity resins for kinase assays. The liganded beads were blocked with excess biotin and washed with blocking buffer (SeaBlock (Pierce), 1 % BSA, 0.05 % Tween 20, 1 mM DTT) to remove unbound ligand and to reduce non-specific phage binding. Binding reactions were assembled by combining kinases, liganded affinity beads, and test compounds in 1x binding buffer (20 % SeaBlock, 0.17x PBS, 0.05 % Tween 20, 6 mM DTT). Test compounds were prepared as 100x stocks in 100 % DMSO and directly diluted into the assay. All reactions were performed in polypropylene 384-well plates in a final volume of 0.02 mL. The assay plates were incubated at room temperature with shaking for 1 h and the affinity beads were washed with wash buffer (1x PBS, 0.05 % Tween 20). The beads were then re-suspended in elution buffer (1x PBS, 0.05 % Tween 20, 0.5 μM non-biotinylated affinity ligand) and incubated at room temperature with shaking for 30 min. The kinase concentration in the eluates was measured by qPCR.
The TREEspot™ Interaction Maps was generated online based on the testing results.
Acknowledgements
For completion of this work, H. L. & N. S. were supported by the grants (NIHR01 CA249282). H. L. was also supported by Helen Adams & Arkansas Research Alliance Endowment.
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Submission declaration and verification
We wish to confirm that the work described in the article has not been published previously (except in the form of an abstract, a published lecture or academic thesis), that it is not under consideration for publication elsewhere, that its publication is approved by all authors and tacitly or explicitly by the responsible authorities where the work was carried out, and that, if accepted, it will not be published elsewhere in the same form, in English or in any other language, including electronically without the written consent of the copyright-holder.
We confirm that the manuscript has been read and approved by all named authors and that there are no other persons who satisfied the criteria for authorship but are not listed. We further confirm that the order of authors listed in the manuscript has been approved by all of us.
Data availability
Data will be made available on request.
References
- [1].Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ Jr.,Laird PW, Baty JD, Fulton LL, Fulton R, Heath SE, Kalicki-Veizer J, Kandoth C, Klco JM, Koboldt DC, Kanchi KL, Kulkarni S, Lamprecht TL, Larson DE, Lin L, Lu C, McLellan MD, McMichael JF, Payton J, Schmidt H, Spencer DH, Tomasson MH, Wallis JW, Wartman LD, Watson MA, Welch J, Wendl MC, Ally A, Balasundaram M, Birol I, Butterfield Y, Chiu R, Chu A, Chuah E, Chun HJ, Corbett R, Dhalla N, Guin R, He A, Hirst C, Hirst M, Holt RA, Jones S, Karsan A, Lee D, Li HI, Marra MA, Mayo M, Moore RA, Mungall K, Parker J, Pleasance E, Plettner P, Schein J, Stoll D, Swanson L, Tam A, Thiessen N, Varhol R, Wye N, Zhao Y, Gabriel S, Getz G, Sougnez C, Zou L, Leiserson MD, Vandin F, Wu HT, Applebaum F, Baylin SB, Akbani R, Broom BM, Chen K, Motter TC, Nguyen K, Weinstein JN, Zhang N, Ferguson ML, Adams C, Black A, Bowen J, Gastier-Foster J, Grossman T, Lichtenberg T, Wise L, Davidsen T, Demchok JA, Shaw KR, Sheth M, Sofia HJ, Yang L, Downing JR, Eley G, Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia, N. Engl. J. Med 368 (2013) 2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kihara R, Nagata Y, Kiyoi H, Kato T, Yamamoto E, Suzuki K, Chen F, Asou N, Ohtake S, Miyawaki S, Miyazaki Y, Sakura T, Ozawa Y, Usui N, Kanamori H, Kiguchi T, Imai K, Uike N, Kimura F, Kitamura K, Nakaseko C, Onizuka M, Takeshita A, Ishida F, Suzushima H, Kato Y, Miwa H, Shiraishi Y, Chiba K, Tanaka H, Miyano S, Ogawa S, Naoe T, Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients, Leukemia 28 (2014) 1586–1595. [DOI] [PubMed] [Google Scholar]
- [3].Dohner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, Dombret H, Ebert BL, Fenaux P, Larson RA, Levine RL, Lo-Coco F, Naoe T, Niederwieser D, Ossenkoppele GJ, Sanz M, Sierra J, Tallman MS, Tien HF, Wei AH, Lowenberg B, Bloomfield CD, Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel, Blood 129 (2017) 424–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Huse M, Kuriyan J, The conformational plasticity of protein kinases, Cell 109 (2002) 275–282. [DOI] [PubMed] [Google Scholar]
- [5].Vijayan RS, He P, Modi V, Duong-Ly KC, Ma H, Peterson JR, Dunbrack RL Jr., Levy RM, Conformational analysis of the DFG-out kinase motif and biochemical profiling of structurally validated type II inhibitors, J. Med. Chem 58 (2015) 466–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Liao JJ, Molecular recognition of protein kinase binding pockets for design of potent and selective kinase inhibitors, J. Med. Chem 50 (2007) 409–424. [DOI] [PubMed] [Google Scholar]
- [7].Griffith J, Black J, Faerman C, Swenson L, Wynn M, Lu F, Lippke J, Saxena K, The structural basis for autoinhibition of FLT3 by the juxtamembrane domain, Mol. Cell 13 (2004) 169–178. [DOI] [PubMed] [Google Scholar]
- [8].Grafone T, Palmisano M, Nicci C, Storti S, An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: biology and treatment, Onco Rev. 6 (2012) e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Fischer T, Stone RM, Deangelo DJ, Galinsky I, Estey E, Lanza C, Fox E, Ehninger G, Feldman EJ, Schiller GJ, Klimek VM, Nimer SD, Gilliland DG, Dutreix C, Huntsman-Labed A, Virkus J, Giles FJ, Phase IIB trial of oral Midostaurin (PKC412), the FMS-1ike tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3, J. Clin. Oncol 28 (2010) 4339–4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Stone RM, DeAngelo DJ, Klimek V, Galinsky I, Estey E, Nimer SD, Grandin W, Lebwohl D, Wang Y, Cohen P, Fox EA, Neuberg D, Clark J, Gilliland DG, Griffin JD, Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412, Blood 105 (2005) 54–60. [DOI] [PubMed] [Google Scholar]
- [11].Weisberg E, Boulton C, Kelly LM, Manley P, Fabbro D, Meyer T, Gilliland DG, Griffin JD, Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412, Cancer Cell 1 (2002) 433–443. [DOI] [PubMed] [Google Scholar]
- [12].Zorn JA, Wang Q, Fujimura E, Barros T, Kuriyan J, Crystal structure of the FLT3 kinase domain bound to the inhibitor Quizartinib (AC220), PLoS One 10 (2015), e0121177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zarrinkar PP, Gunawardane RN, Cramer MD, Gardner MF, Brigham D, Belli B, Karaman MW, Pratz KW, Pallares G, Chao Q, Sprankle KG, Patel HK, Levis M, Armstrong RC, James J, Bhagwat SS, AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML), Blood 114 (2009) 2984–2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chao Q, Sprankle KG, Grotzfeld RM, Lai AG, Carter TA, Velasco AM, Gunawardane RN, Cramer MD, Gardner MF, James J, Zarrinkar PP, Patel HK, Bhagwat SS, Identification of N-(5-tert-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea dihydrochloride (AC220), a uniquely potent, selective, and efficacious FMS-1ike tyrosine kinase-3 (FLT3) inhibitor, J. Med. Chem 52 (2009) 7808–7816. [DOI] [PubMed] [Google Scholar]
- [15].Cortes J, Perl AE, Döhner H, Kantarjian H, Martinelli G, Kovacsovics T, Rousselot P, Steffen B, Dombret H, Estey E, Strickland S, Altman JK, Baldus CD, Burnett A, Krämer A, Russell N, Shah NP, Smith CC, Wang ES, Ifrah N, Gammon G, Trone D, Lazzaretto D, Levis M, Quizartinib, an FLT3 inhibitor, as monotherapy in patients with relapsed or refractory acute myeloid leukaemia: an open-1abel, multicentre, single-arm, phase 2 trial, Lancet Oncol. 19 (2018) 889–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cortes JE, Khaled S, Martinelli G, Perl AE, Ganguly S, Russell N, Krämer A, Dombret G, Hogge D, Jonas BA, Leung AY-H, Mehta P, Montesinos P, Radsak M, Sica S, Arunachalam M, Holmes M, Kobayashi K, Namuyinga R, Ge N, Yver A, Zhang Y, Levis MJ, Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-R): a multicentre, randomised, controlled, open-1abel, phase 3 trial, Lancet Oncol. 20 (2019) 984–997. [DOI] [PubMed] [Google Scholar]
- [17].Cortes JE, Tallman MS, Schiller GJ, Trone D, Gammon G, Goldberg SL, Perl AD, Marie JP, Martinelli G, Kantarjian HM, Levis MJ, Phase 2b study of 2 dosing regimens of quizartinib monotherapy in FLT3-ITD-mutated, relapsed or refractory AML, Blood 132 (2018) 598–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Levis MJ, Perl AE, Dombret H, Döhner H, Steffen B.r., Rousselot P, Martinelli G, Estey EH, Burnett AK, Gammon G, Trone D, Leo E, Cortes JE, Final results of a phase 2 open-1abel, monotherapy efficacy and safety study of quizartinib (AC220) in patients with FLT3-ITD positive or negative relapsed/refractory acute myeloid leukemia after second-1ine chemotherapy or hematopoietic stem cell transplantation, Blood 120 (2012), 673–673. [Google Scholar]
- [19].Patel HK, Grotzfeld RM, Lai AG, Mehta SA, Milanov ZV, Chao Q, Sprankle KG, Carter TA, Velasco AM, Fabian MA, James J, Treiber DK, Lockhart DJ, Zarrinkar PP, Bhagwat SS, Arylcarboxyamino-substituted diaryl ureas as potent and selective FLT3 inhibitors, Bioorg. Med. Chem. Lett 19 (2009) 5182–5185. [DOI] [PubMed] [Google Scholar]
- [20].Smith CC, Wang Q, Chin CS, Salerno S, Damon LE, Levis MJ, Perl AE, Travers KJ, Wang S, Hunt JP, Zarrinkar PP, Schadt EE, Kasarskis A, Kuriyan J, Shah NP, Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia, Nature 485 (2012) 260–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Daver N, Cortes J, Ravandi F, Patel KP, Burger JA, Konopleva M, Kantarjian H, Secondary mutations as mediators of resistance to targeted therapy in leukemia, Blood 125 (2015) 3236–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhao Z, Wu H, Wang L, Liu Y, Knapp S, Liu Q, Gray NS, Exploration of type II binding mode: a privileged approach for kinase inhibitor focused drug discovery? ACS Chem. Biol 9 (2014) 1230–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Smith CC, Lin K, Stecula A, Sali A, Shah NP, FLT3 D835 mutations confer differential resistance to type II FLT3 inhibitors, Leukemia 29 (2015) 2390–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Short NJ, Rytting ME, Cortes JE, Acute myeloid leukaemia, Lancet 392 (2018) 593–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nagel G, Weber D, Fromm E, Erhardt S, Lubbert M, Fiedler W, Kindler T, Krauter J, Brossart P, Kundgen A, Salih HR, Westermann J, Wulf G, Hertenstein B, Wattad M, Gotze K, Kraemer D, Heinicke T, Girschikofsky M, Derigs HG, Horst HA, Rudolph C, Heuser M, Gohring G, Teleanu V, Bullinger L, Thol F, Gaidzik VI, Paschka P, Dohner K, Ganser A, Dohner H, Schlenk RF, A.M.L.S.G. German-Austrian, Epidemiological, genetic, and clinical characterization by age of newly diagnosed acute myeloid leukemia based on an academic population-based registry study (AMLSG BiO), Ann. Hematol 96 (2017) 1993–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Smith CC, Zhang C, Lin KC, Lasater EA, Zhang Y, Massi E, Damon LE, Pendleton M, Bashir A, Sebra R, Perl A, Kasarskis A, Shellooe R, Tsang G, Carias G, Powell B, Burton EA, Matusow B, Zhang J, Spevak W, Ibrahim PN, Le MH, Hsu HH, Habets G, West BL, Bollag G, Shah NP, Characterizing and overriding the structural mechanism of the quizartinib-resistant FLT3 "gatekeeper" F691L mutation with PLX3397, Cancer Discov. 5 (2015) 668–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Perl AE, Martinelli G, Cortes JE, Neubauer A, Berman E, Paolini S, Montesinos P, Baer MR, Larson RA, Ustun C, Fabbiano F, Erba HP, Di Stasi A, Stuart R, Olin R, Kasner M, Ciceri F, Chou WC, Podoltsev N, Recher C, Yokoyama H, Hosono N, Yoon SS, Lee JH, Pardee T, Fathi AT, Liu C, Hasabou N, Liu X, Bahceci E, Levis MJ, Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML, N. Engl. J. Med 381 (2019) 1728–1740. [DOI] [PubMed] [Google Scholar]
- [28].Perl AE, Altman JK, Cortes J, Smith C, Litzow M, Baer MR, Claxton D, Erba HP, Gill S, Goldberg S, Jurcic JG, Larson RA, Liu C, Ritchie E, Schiller G, Spira AI, Strickland SA, Tibes R, Ustun C, Wang ES, Stuart R, Röllig C, Neubauer A, Martinelli G, Bahceci E, Levis M, Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-1abel, phase 1–2 study, Lancet Oncol. 18 (2017) 1061–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mori M, Kaneko N, Ueno Y, Yamada M, Tanaka R, Saito R, Shimada I, Mori K, Kuromitsu S, Gilteritinib, a FLT3/AXL inhibitor, shows antileukemic activity in mouse models of FLT3 mutated acute myeloid leukemia, Invest. N. Drugs 35 (2017) 556–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zhou S, Yang B, Xu Y, Gu A, Peng J, Fu J, Understanding gilteritinib resistance to FLT3-F691L mutation through an integrated computational strategy, J. Mol. Model 28 (2022) 247. [DOI] [PubMed] [Google Scholar]
- [31].Frett B, McConnell N, Smith CC, Wang Y, Shah NP, Li HY, Computer aided drug discovery of highly ligand efficient, low molecular weight imidazopyridine analogs as FLT3 inhibitors, Eur. J. Med. Chem 94 (2015) 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhang L, Lakkaniga NR, Bharate JB, McConnell N, Wang X, Kharbanda A, Leung YK, Frett B, Shah NP, Li HY, Discovery of imidazo[1,2-a]pyridine-thiophene derivatives as FLT3 and FLT3 mutants inhibitors for acute myeloid leukemia through structure-based optimization of an NEK2 inhibitor, Eur. J. Med. Chem 225 (2021), 113776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Warkentin AA, Lopez MS, Lasater EA, Lin K, He BL, Leung AY, Smith CC, Shah NP, Shokat KM, Overcoming myelosuppression due to synthetic lethal toxicity for FLT3-targeted acute myeloid leukemia therapy, Elife 3 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Galanis A, Levis M, Inhibition of c-Kit by tyrosine kinase inhibitors, Haematologica 100 (2015) e77–e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yamaura T, Nakatani T, Uda K, Ogura H, Shin W, Kurokawa N, Saito K, Fujikawa N, Date T, Takasaki M, Terada D, Hirai A, Akashi A, Chen F, Adachi Y, Ishikawa Y, Hayakawa F, Hagiwara S, Naoe T, Kiyoi H, A novel irreversible FLT3 inhibitor, FF-10101, shows excellent efficacy against AML cells with FLT3 mutations, Blood 131 (2018) 426–438. [DOI] [PubMed] [Google Scholar]
- [36].Tong L, Wang P, Li X, Dong X, Hu X, Wang C, Liu T, Li J, Zhou Y, Identification of 2-Aminopyrimidine derivatives as FLT3 kinase inhibitors with high selectivity over c-KIT, J. Med. Chem 65 (2022) 3229–3248. [DOI] [PubMed] [Google Scholar]
- [37].Wang J, Pan X, Song Y, Liu J, Ma F, Wang P, Liu Y, Zhao L, Kang D, Hu L, Discovery of a potent and selective FLT3 inhibitor (Z)-N-(5-((5-Fluoro-2-oxoindolin-3-ylidene)methyl)-4-methyl-1H-pyrrol-3-yl)-3-(pyrrolidin-1-yl) propanamide with improved drug-1ike properties and superior efficacy in FLT3-ITD-positive acute myeloid leukemia, J. Med. Chem 64 (2021) 4870–4890. [DOI] [PubMed] [Google Scholar]
- [38].Cho H, Shin I, Yoon H, Jeon E, Lee J, Kim Y, Ryu S, Song C, Kwon NH, Moon Y, Kim S, Kim ND, Choi HG, Sim T, Identification of thieno[3,2-d] pyrimidine derivatives as dual inhibitors of focal Adhesion kinase and FMS-like tyrosine kinase 3, J. Med. Chem 64 (2021) 11934–11957. [DOI] [PubMed] [Google Scholar]
- [39].Zhi Y, Wang Z, Yao C, Li B, Heng H, Cai J, Xiang L, Wang Y, Lu T, Lu S, Design and synthesis of 4-(heterocyclic substituted amino)-1H-Pyrazole-3-Carboxamide derivatives and their potent activity against acute myeloid leukemia (AML), Int. J. Mol. Sci 20 (2019) 5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Heng H, Wang Z, Li H, Huang Y, Lan Q, Guo X, Zhang L, Zhi Y, Cai J, Qin T, Xiang L, Wang S, Chen Y, Lu T, Lu S, Combining structure- and property-based optimization to identify selective FLT3-ITD inhibitors with good antitumor efficacy in AML cell inoculated mouse xenograft model, Eur. J. Med. Chem 176 (2019) 248–267. [DOI] [PubMed] [Google Scholar]
- [41].Zhi Y, Li H, Yang P, Jin Q, Yao C, Li B, Ling J, Guo H, Li T, Jin J, Wang Y, Chen Y, Lu T, Lu S, Rational design of 4-((6-phenoxypyrimidin-4-yl)amino)-N-(4-(piperazin-1-yl)phenyl)-1H-pyrazole-3-carboxamide (LT-540-717) as orally bioavailable FLT3 inhibitor, Eur. J. Med. Chem 256 (2023) 115448. [DOI] [PubMed] [Google Scholar]
- [42].Wang Z, Cai J, Ren J, Chen Y, Wu Y, Cheng J, Jia K, Huang F, Cheng Z, Sheng T, Song S, Heng H, Zhu Y, Tang W, Li H, Lu T, Chen Y, Lu S, Discovery of a potent FLT3 inhibitor (LT-850-166) with the capacity of overcoming a variety of FLT3 mutations, J. Med. Chem 64 (2021) 14664–14701. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available on request.