

Abstract
The long control regions (LCRs) of mucosal epitheliotropic papillomaviruses have similar organizations: a promoter region, an enhancer region, and a highly conserved distribution of E2 DNA binding sites (C. Desaintes and C. Demeret, Semin. Cancer Biol. 7:339–347, 1996). The enhancer of these viruses is epithelial cell specific, as it fails to activate transcription from heterologous promoters in nonepithelial cell types (B. Gloss, H. U. Bernard, K. Seedorf, and G. Klock, EMBO J. 6:3735–3743, 1987). Using the bovine papillomavirus type 4 (BPV-4) LCR and a bovine primary cell system, we have shown previously that a level of epithelial specificity resides in a papillomavirus promoter region. The BPV-4 promoter shows an enhanced response to transcriptional activators in epithelial cells compared with that of fibroblasts (K. W. Vance, M. S. Campo, and I. M. Morgan, J. Biol. Chem. 274:27839–27844, 1999). A chimeric lcr/tk promoter suggests that the upstream BPV-4 promoter region determines the cell-type-selective response of this promoter in fibroblasts and keratinocytes. Promoter deletion analysis identified two novel repressor elements that are, at least in part, responsible for mediating the differential response of this promoter to upstream activators in fibroblasts and keratinocytes. One of these elements, promoter repressor element 2 (PRE-2), is conserved in position and sequence in the related mucosal epitheliotropic papillomaviruses, BPV-3 and BPV-6. PRE-2 functions in cis to repress the basal activity of the simian virus 40 promoter and binds a specific protein complex. We identify the exact nucleotides necessary for binding and correlate loss of binding with loss of transcriptional repression. We also incorporate these mutations into the BPV-4 promoter and demonstrate an enhanced response of the mutated promoter to E2 in fibroblasts. The DNA binding protein in the detected complex is shown to have a molecular mass of approximately 50 kDa. The PRE-2 binding protein represents a novel transcriptional repressor and regulator of papillomavirus transcription.
Human papillomaviruses (HPVs) are a family of small double-stranded DNA viruses with a strict tropism for the epithelial cell type. HPVs are causative agents of squamous cell carcinomas. Over 95% of human cervical cancers contain transcriptionally active DNA of the mucosal epitheliotropic papillomaviruses, such as HPV type 16 (HPV-16) and HPV-18, often integrated into the host genome (41). Papillomaviruses have a closed circular genome that can be divided into three regions: regions encoding the early and late gene products which are separated by a noncoding region of 500 to 1,000 bp called the long control region (LCR). The LCR is the transcriptional control unit of the virus and contains a number of binding sites for transcription factors including virus-encoded E2. One way in which these viruses are restricted to the epithelial cell type is at the transcriptional level. Bovine papillomavirus type 4 (BPV-4) is a mucosal epitheliotropic papillomavirus that infects the upper alimentary canal of cattle. BPV-4 infection causes benign papillomas with a high risk of progressing to carcinoma in cattle feeding on bracken fern (6). Although the overall LCR sequence homology between BPV-4 and HPV-16 and -18 is low, these LCRs have similar organizations: a promoter region, an enhancer region, and a highly conserved distribution of E2 DNA binding sites (10).
The enhancer of these papillomaviruses is epithelial cell specific, as it fails to activate transcription from heterologous promoters in nonepithelial cell types (13). The BPV-4, HPV-16, and HPV-18 enhancers are of similar sizes and positions (25). These epithelial cell-specific enhancers contain numerous binding sites for cellular transcription factors, including AP-1 (7, 32, 38), Oct-1 (15, 27), NF-1 (1, 2, 28), PEF-1 (9, 33), TEF-1 (16), Sp1 (3), YY-1 (31), and the glucocorticoid receptor (24). No one factor that determines the epithelial cell-specific nature of these enhancer elements has been identified. It has been proposed that epithelial specificity is brought about by the cooperative interactions of ubiquitously expressed transcription factors. The mechanism of this activation may involve synergism between DNA-bound factors that are differentially expressed or, alternatively, spliced or modified in a cell-type-dependent manner to establish a pattern of epithelial cell-specific transcriptional regulation.
The promoter regions of BPV-4, HPV-16, and HPV-18 contain the origin of replication, three binding sites for virus-encoded E2, a TATA box, and an initiator element. Binding sites for the cellular factors Sp1 (3), YY-1 (4), CDP/Cut (29), and C/EBP (5) are also commonly found in HPV promoters. Sp1 is a positive regulator and YY-1 is a negative regulator of HPV gene expression. CDP/Cut represses HPV-16 transcription and replication by binding to a conserved element that overlaps the binding site for the viral replication protein E1 (29). Although the BPV-4 promoter does not contain binding sites for Sp1, YY-1, and CDP/Cut, C/EBP family members have been implicated as both positive and negative regulators of transcription from HPV and BPV-4 LCRs (23).
The cellular factors that BPV-4 uses to achieve epithelial cell-specific transcriptional regulation are distinct from those used by the HPVs. However, the conservation of the organization of E2 binding sites between the BPV-4 and HPV-16 and -18 LCRs strongly suggests that the mechanism that E2 uses to regulate transcription from mucosal epitheliotropic LCRs is conserved (26). Immediately upstream of the TATA box are two E2 binding sites separated from each other and the TATA box by 3 or 4 bp. Two additional upstream sites flank the epithelial cell-specific enhancer: one beside the E1 DNA binding site involved in the regulation of viral DNA replication and one a further 300 to 400 bp upstream. This organization of E2 DNA binding sites is not observed in the cutaneous HPV or BPV LCRs.
Previously, we have demonstrated that HPV-16 E2, which functions in a manner identical to that of BPV-4 E2 in our assays, upregulates transcription from the BPV-4 LCR in primary bovine palate keratinocytes (PalK cells) but not in palate fibroblasts (PalF cells) (26). PalK cells are the natural target cell type for transformation by BPV-4. Insertion of multiple E2 sites upstream of the BPV-4 promoter demonstrated that the BPV-4 epithelial cell-specific enhancer is not required for the enhanced activity of E2 in epithelial cells (39). The BPV-4 promoter region itself shows an enhanced epithelial response to activation, not only by E2 but also by several other transcriptional activators. This epithelial preference is not extended to heterologous promoters such as the thymidine kinase (tk) promoter. This was the first time that a level of epithelial specificity has been shown to reside in a papillomavirus promoter region (39).
The 127-bp BPV-4 promoter region used in our previous studies contains the TATA box and has the TATA proximal E2 binding sites mutated to prevent E2 binding, as these sites have previously been shown to mediate downregulation of transcription at high levels of E2 (17, 26). The BPV-4 promoter used does not have the putative initiator element identified in the BPV-4 LCR (8) or contain any potential binding sites for cellular factors commonly found in papillomavirus promoters (18). The results presented here demonstrate that the upstream BPV-4 promoter region contains two novel repressor elements that are, at least in part, responsible for mediating the differential response of this promoter to upstream activators in fibroblasts and keratinocytes. One of these elements, promoter repressor element 2 (PRE-2), is conserved in position and sequence in the related mucosal epitheliotropic papillomaviruses BPV-3 and BPV-6. PRE-2 functions in cis to repress the basal activity of the simian virus (SV40) promoter and binds a specific protein complex. We identify the exact nucleotides necessary for binding and correlate loss of binding with loss of transcriptional repression. Mutation of the PRE-2 site in the BPV-4 promoter results in an elevated response of this promoter to transcriptional activation by E2 in fibroblasts. The DNA binding protein in the detected complex is shown to have a molecular mass of approximately 50 kDa. The results also demonstrate that a minimal BPV-4 TATA-containing promoter is able to support activated transcription by E2 and suggest that the TFIID complexes assembled at the BPV-4 TATA and tk TATA boxes are distinct.
MATERIALS AND METHODS
Plasmid constructs and expression vectors.
Construction of the PV2 6E2 and tk 6E2 reporter constructs has been described previously (39). The lcr/tk hybrid promoter was generated by splicing-by-overlap-extension PCR. The BPV-4 promoter region from nucleotides 184 to 279 and the tk promoter from nucleotides 120 to 199 were PCR amplified. The internal primers used ensured that the primary PCR products had 20-bp overlapping complementary ends. A second PCR using primers annealing at the nonoverlapping ends was performed to generate the hybrid lcr/tk promoter as a BglII-HindIII fragment. This fragment was then cloned into pGL36E2. pGL36E2 contains six E2 DNA binding sites cloned into the BglII site of pGL3. The 80bpTATA, 66bpTATA, 41bpTATA, 19bpTATA, and 3bpTATA BPV-4 promoter deletions were PCR amplified as BglII-HindIII fragments from PV2 6E2. These fragments were then cloned into pGL36E2. PRO4XPRE-2, PRO4XPRE-2(−), and PRO4XPRE-2mt1 were all generated in the same manner as described below. Oligonucleotides were synthesized on an Applied Biosystems model 381A DNA synthesizer and are as follows: PRE-2 upper strand, 5′GATCCGCTAGGTAAGTGTTGTACCTA; PRE-2 lower strand, 5′GATCTAGGTACAACACTTACCTAGCG; PRE-2mt1 upper strand, 5′GATCCGCTAGGTCCTTGTTGTACCTA; and PRE-2mt1 lower strand, 5′GATCTAGGTACAACAAGGACCTAGCG. Oligonucleotides were separated on a 6% polyacrylamide gel, excised, eluted overnight in a minimal volume of elution buffer (0.1% sodium dodecyl sulfate [SDS], 0.5 M ammonium acetate, 10 mM magnesium acetate), and purified by LiCl and ethyl alcohol (EtOH) precipitation Pairs of these oligonucleotides when annealed together generate a binding motif with BamHI/BglII-compatible ends. After end labeling with T4 kinase (Kramel Biotech) and ligation, the double-stranded oligonucleotides were restriction digested with BamHI/BglII. This ensures that only concatemers of binding sites facing in the same orientation are generated, as ligation in the wrong orientation reconstitutes a restriction site. The oligonucleotides were separated on a 6% polyacrylamide gel, and the bands corresponding to four binding sites were excised, eluted, and purified by sodium acetate and EtOH precipitation. The correct numbers of synthetic binding sites were then cloned into the BglII site upstream of the SV40 promoter in the pGL3 luciferase vector. 80bpmt1 was generated by PCR amplifying the corresponding BPV-4 promoter region as a BglII-HindIII fragment from 80bpTATA using a 5′ primer containing the mutation. The amplified fragment was then cloned into pGL36E2. The fidelity of all plasmid constructions was verified using an Applied Biosystems 373A automated sequencer. pCMVHPV16-E2 and pCGVP16-E2 have been described previously (39).
Cell culture.
PalK cells were prepared from fetal biopsy specimens as described previously for human cervical keratinocytes (9). They were cultured on irradiated Swiss 3T3 feeders under conditions described elsewhere (17). The Swiss 3T3 feeders were grown in SLM (Gibco) with 10% fetal calf serum and irradiated with 60 Gy prior to use as feeders. PalF cells were prepared as described previously (19) from the same palate biopsy specimen used to prepare the PalK cells and cultured in Dulbecco modified Eagle medium with 10% fetal calf serum. At least two different sources of these primary cell types were used in the experiments described.
Transfection.
PalK cells were transfected using the Polybrene-dimethyl sulfoxide technique as described elsewhere (20). Briefly, 5 × 105 cells were seeded on a 60-mm-diameter tissue culture dish without feeders. On the following day, the medium was replaced with 10-mg/ml-Polybrene-containing medium and the DNA was added. After 6 h, the DNA-Polybrene-containing medium was removed and a 35% dimethyl sulfoxide solution was added to the cells for 3 min. Following this incubation, the cells were washed two times with phosphate-buffered saline (PBS) and then refed with normal medium. The cells were harvested 44 to 48 h later. PalF cells were transfected using a standard calcium phosphate precipitation technique. Briefly, cells were plated out at 2 × 105 per 60-mm-diameter tissue culture disk. On the following day, a calcium phosphate precipitate containing the DNA was added to the cells and they were incubated overnight. On the following morning, the cells were washed twice with PBS and refed with normal medium. The cells were harvested 28 to 32 h later. All transfection experiments whose results are shown represent the averages of at least three independent experiments carried out in duplicate. Within each experiment, the total amount of DNA transfected into each sample was made constant using empty expression vector.
Transcription assay.
PalK and PalF cells were lysed directly on the tissue culture plates. The medium was removed, and the cells were washed twice with PBS. Three hundred microliters of reporter lysis buffer (Promega) was added to the plate and left for 10 min. The cell lysate was then scraped from the dish and placed in a 1.5-ml centrifuge tube. The lysate was cleared by centrifuging the sample for 10 min and removing the supernatant to a fresh tube. Eighty microliters of the supernatant was then assayed for luciferase activity using the luciferase assay system from Promega with a Tropix TR717 Microplate luminometer. To standardize for cell number, the protein concentration was determined. pGL3CONT (which contains the SV40 promoter and enhancer driving expression of the luciferase gene) was transfected in parallel to confirm efficient transfection. This construct demonstrates high levels of transcriptional activity in both keratinocytes and fibroblasts. All transfections were repeated at least three times in duplicate.
Nuclear extract preparation.
PalK and PalF nuclear extracts were prepared as follows. Approximately 107 cells were washed twice with 5 ml of ice-cold PBS and removed from the tissue culture dish by scraping. The cells were pelleted and resuspended in 1.5 ml of ice-cold PBS. The cells were pelleted again, resuspended in 400 μl of hypotonic lysis buffer (10 mM HEPES-KOH [pH 7.9], 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol [DTT], and 0.2 mM phenylmethylsulfonyl fluoride), and allowed to swell on ice for 10 min. Samples were vortexed for 10 s and pelleted, and the supernatant was removed. The pellet was resuspended in 50 μl of high-salt buffer (20 mM HEPES-KOH [pH 7.9], 25% [vol/vol] glycerol, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, and 0.2 mM phenylmethylsulfonyl fluoride) and incubated on ice for 20 min. The cellular debris was removed by centrifuging. The supernatant was removed, frozen on dry ice, and stored at −70°C.
Band shift assays.
PRE-2, PRE-2mt1, PRE-2mt2 (see Fig. 5A), and AP-1 oligonucleotides were synthesized on an Applied Biosystems model 381A DNA synthesizer. Single-stranded oligonucleotides were electrophoresed on a 6% polyacrylamide gel, and the bands were excised, eluted overnight in a minimal volume of elution buffer (0.1% SDS, 0.5 M ammonium acetate, 10 mM magnesium acetate), and purified by LiCl and EtOH precipitation. Purified oligonucleotides were annealed using standard methods. Three picomoles of double-stranded PRE-2 was 32P labeled with T4 polynucleotide kinase (Kramel Biotech) and electrophoresed on an 8% polyacrylamide gel, and the band was excised and eluted overnight in 500 μl of distilled H2O.
FIG. 5.

(A) PRE-2 SV40 constructs. Four copies of PRE-2 were inserted into the BglII site, upstream of the SV40 promoter in the pGL3 luciferase vector, in both a positive and a negative orientation. The position and sequence of PRE-2 are conserved among the mucosal epitheliotropic papillomaviruses BPV-4, BPV-3, and BPV-6. (B) PRE-2 strongly represses SV40 promoter activity in an orientation-independent manner in both PalK and PalF cells. PalK and PalF cells were transfected with the indicated amounts of pGL3PRO, PRO4XPRE-2, and PRO4XPRE-2(−). Results are expressed relative to the luciferase activity of pGL3PRO (set arbitrarily at 100%), which contains only the SV40 promoter.
Electrophoretic mobility shift assay reactions were performed as follows. Ten to fifteen micrograms of nuclear extract was added to 3 μg of poly(dI-dC) in a final volume of 30 μl of binding buffer (20 mM HEPES [pH 7.9], 4% Ficoll, 2 mM MgCl2, 40 mM KCl, 0.1 mM EGTA, and 0.5 mM DTT). After 15 min of preincubation at room temperature, approximately 5 fmol of 32P-labeled PRE-2 probe was added. The binding reaction mixture was incubated for a further 15 min at room temperature and then electrophoresed on a 6% polyacrylamide gel. Competition band shifts were performed under the same conditions, except that a 100- or 500-fold excess of unlabeled PRE-2, PRE-2mt1, PRE-2mt2, or AP-1 oligonucleotide was added where indicated. The gel was dried and visualized by autoradiography.
UV cross-linking.
A derivative of the PRE-2 oligonucleotide was synthesized with bromodeoxyuridine (BrdU) in place of thymine to enhance UV-induced protein-DNA cross-linking. 32P-radiolabeled BrdU–PRE-2 was used to probe PalK and PalF nuclear extracts under conditions the same as those described above. After electrophoresis, the gel was UV irradiated (304 nm) for 45 min at 4°C. The bands were visualized using a phosphorimager, excised, soaked in SDS-polyacrylamide gel electrophoresis sample buffer for 15 min at 37°C, and electrophoresed on a 10% SDS gel. The gel was dried and visualized by autoradiography.
RESULTS
Identification of the promoter region responsible for the enhanced epithelial response of the BPV-4 promoter to upstream activators.
The enhanced epithelial response of the BPV-4 promoter may be determined by DNA-bound proximal promoter factors that are either cell type specific, differentially expressed, alternatively spliced, or modified in a cell-type-dependent manner. Also, cell-type-specific components of the basal transcription machinery have been identified previously; for example, TAFII105 is a TATA-binding protein-associated factor highly expressed in B lymphocytes, indicating that the TATA box itself might dictate cell-type-specific transcriptional regulation (11). To identify the BPV-4 promoter region responsible for the differential response of this promoter in keratinocytes and fibroblasts, a chimeric promoter with six E2 DNA binding sites inserted upstream was generated using a PCR-based strategy. The region of the heterologous tk promoter containing the tk TATA box and initiator was exchanged with the corresponding core BPV-4 promoter region to generate the lcr/tk chimeric promoter (Fig. 1). The ability of HPV-16 E2 and VP16-E2, which comprise the DNA binding domain of BPV-1 E2 fused to the acidic transactivation domain of VP16, to upregulate transcription from this construct was assayed in PalK and PalF cells. Figure 2A shows the enhanced epithelial response of the BPV-4 promoter to upstream activators. E2 upregulates transcription by a maximum of 7-fold in PalF cells and 60-fold in PalK cells, while VP16-E2 transactivates the PV2 promoter a maximum of 700-fold in PalF cells and 2,500-fold in PalK cells. The tk promoter shows no such epithelial preference (Fig. 2B). E2 activates transcription from the tk promoter in a cell-type-independent manner. A maximum of approximately 90-fold activation is observed with a 0.1:1 ratio of expression vector to reporter plasmid in both cell types. VP16-E2 activates transcription from the tk promoter preferentially in fibroblasts. A maximum of approximately 2,500-fold activation in PalF cells and 1,000-fold activation in PalK cells is observed. The lcr/tk hybrid promoter retains the enhanced epithelial response of the BPV-4 promoter to activation by E2 and VP16-E2 (Fig. 2C). E2 activates transcription by a maximum of 2.5-fold in PalF cells and 11-fold in PalK cells. VP16-E2 upregulates transcription by a maximum of 130-fold in fibroblasts and 300-fold in keratinocytes. These results demonstrate that the upstream BPV-4 LCR promoter region is involved in mediating the enhanced epithelial response of the BPV-4 promoter to upstream activators. Although the hybrid lcr/tk promoter retains an enhanced epithelial response to upstream activators, the overall levels of response are reduced in comparison with those of the PV2 promoter, suggesting that the BPV-4 TATA box region is more responsive than that of the tk promoter.
FIG. 1.
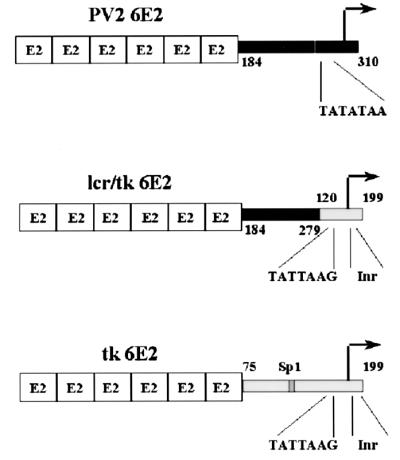
E2-responsive promoter constructs. Promoter regions were PCR amplified as BglII-HindIII fragments and cloned into the pGL3 luciferase vector. Six E2 DNA binding sites were inserted into the BglII site immediately upstream of these promoters. The PV2 6E2 and tk 6E2 constructs have been described previously (39). The lcr/tk hybrid promoter, generated by splicing-by-overlap-extension PCR, contains the BPV-4 upstream promoter region from nucleotides 184 to 279 fused to the tk promoter region from nucleotides 120 to 199. This region of the tk promoter contains the TATA box and initiator element.
FIG. 2.

The lcr/tk hybrid promoter retains the enhanced epithelial response of the BPV-4 promoter to upstream activators. PalK and PalF cells were cotransfected with the indicated amounts of pCMV HPV16-E2 and pCGVP16-E2 expression vectors and 1 μg of either the PV2 6E2 (A), tk 6E2 (B), or lcr/tk 6E2 (C) reporter construct. The total amount of DNA transfected was made equal in each case with pCMV or pCG containing no insert. Results are expressed as fold transactivation relative to the luciferase activity in the absence of E2 or VP16-E2 expression vector.
Identification of the DNA elements responsible for the differential response of the BPV-4 promoter to upstream activators in fibroblasts and keratinocytes.
The lcr/tk hybrid promoter result demonstrates that the upstream BPV-4 promoter region contributes to the differential response of this promoter to upstream activators in fibroblasts and keratinocytes. A series of 5′ deletions of the papillomavirus promoter were therefore generated to identify the specific LCR promoter elements that cooperate with E2 to activate transcription preferentially in keratinocytes (Fig. 3B). The ability of HPV-16 E2 to upregulate transcription from the promoter deletions with six E2 binding sites inserted upstream was assayed in PalK and PalF cells (Fig. 4A and Table 1). It should be noted that, in each construct, the E2 DNA binding sites are an extra 6 bp upstream from the TATA box due to the restriction enzyme site used for cloning. Deletion analysis of the papillomavirus promoter identifies two novel repressor elements that are at least in part responsible for mediating the cell-type-selective response of this promoter to activation by E2 (Fig. 4A). Deletion of the region from 19 to 3 bp from the TATA box results in a 3.5-fold increase in transactivation in PalK cells and a 7-fold increase in PalF cells. This region defines PRE-1. The PRE-1 region spans the TATA box proximal E2 binding sites that have been mutated to prevent E2 binding. These sites have previously been shown to mediate downregulation of transcription at high levels of E2. These mutations do not affect the epithelial cell-specific response of the BPV-4 promoter to E2 (39). Deletion from 80 to 66 bp from the TATA box (PRE-2) results in a 3-fold increase in transcriptional activation by E2 in PalK cells and a 6.5-fold increase in PalF cells. Deletion of either of these regions does not have a significant effect on basal promoter activity. The PRE-2 region was chosen for further investigation, as a recently identified repressor element in the HPV-16 LCR has been described in a similar location (30). Also, the PRE-2 element is conserved in other BPV mucosal epitheliotropic papillomavirus promoters, both in location and in sequence (see below). Both of these observations suggest that PRE-2 is an important element in the regulation of the BPV-4 promoter.
FIG. 3.

(A) Sequence of the BPV-4 promoter from nucleotides 184 to 310. The TATA box and potential binding sites for transcription factors as shown by footprinting studies are shown in boldface (28). The TATA box proximal E2 binding sites are underlined. Mutations preventing E2 binding have been introduced into these sites, as these have been shown to mediate downregulation of transcription at high levels of E2. Promoter deletions are indicated by arrows. (B) E2-responsive promoter deletion constructs. A series of 5′ deletions of the BPV-4 promoter were PCR amplified as BglII-HindIII fragments. These fragments were cloned into pGL36E2, which contains six E2 DNA binding sites inserted into the BglII site of pGL3.
FIG. 4.

(A) Transcriptional activation of the BPV-4 promoter deletions by HPV-16 E2. PalK and PalF cells were cotransfected with 1 μg of reporter plasmid and 0.1 μg of pCMV HPV-16 E2 expression vector. This ratio has previously been shown to be optimal for maximal activation of the LCR promoter by E2. Results are expressed as fold activation relative to the luciferase activity of each reporter in the absence of E2. (B) Transcriptional activation of the minimal BPV-4 TATA-containing promoter. PalK and PalF cells were cotransfected with 1 μg of the 3bpTATA construct, which contains neither an initiator element nor a binding site for an upstream factor, and the indicated amount of either the pCMVHPV16E2 or the pCGVP16-E2 expression vector. pCMV or pCG was used to make the total amount of DNA transfected equal in all cases. Results are expressed as fold transactivation over the luciferase activity in the absence of activator. pGL3CONT, which contains the SV40 promoter and enhancer driving expression of the luciferase gene, was transfected in parallel in all cases to confirm efficient transfection. This construct demonstrates high levels of transcriptional activity in both keratinocytes and fibroblasts.
TABLE 1.
Response of BPV-4 promoter deletion mutants to transcriptional upregulation by HPV-16 E2 (Fig. 4A)
| Promoter deletion | PalF
|
PalK
|
||
|---|---|---|---|---|
| Fold TAa | SE | Fold TAa | SE | |
| PV2 6E2 | 5.06 | 0.97 | 36.06 | 2.97 |
| 80bpTATA | 3.54 | 0.82 | 23.88 | 1.44 |
| 66bpTATA | 23.09 | 4.92 | 68.83 | 9.06 |
| 41bpTATA | 20.86 | 3.30 | 122.40 | 24.15 |
| 19bpTATA | 40.52 | 6.85 | 157.68 | 12.77 |
| 3bpTATA | 286.22 | 12.01 | 566.55 | 50.27 |
Fold TA, transcriptional activation by HPV-16 E2 over background.
A core tk promoter must contain at least two elements to be able to respond to E2, and these elements, the TATA box, the initiator element, and a binding site for an upstream promoter factor, are interchangeable (14). In contrast to a minimal tk TATA box-containing promoter, E2 efficiently activates a minimal BPV-4 TATA promoter that contains neither an initiator element nor a binding site for an upstream factor. E2 upregulates transcription from the 3bpTATA construct approximately 560-fold in keratinocytes and 280-fold in fibroblasts (Fig. 4A). This minimal promoter contains only 32 bp of the BPV-4 promoter sequence. This result suggests that the TFIID complexes assembled at the BPV-4 and tk TATA boxes are distinct.
Deletion analysis also demonstrates that the eight- to ninefold-enhanced epithelial response of the BPV-4 promoter to activation by E2, compared with fibroblasts, has been reduced to only twofold with a minimal BPV-4 TATA-containing promoter (Fig. 4A). This is in agreement with the result observed with the lcr/tk chimeric promoter, suggesting that the upstream BPV-4 promoter region contributes to the enhanced epithelial response of this promoter to upstream activators. To extend this observation, we determined the levels of activation of the 3bpTATA construct over a range of HPV-16 E2 concentrations (Fig. 4B). Lower levels of E2 activate transcription from the 3bpTATA construct to similar levels in PalK and PalF cells. At these levels of E2, the activation of the PV2 promoter is eight- to ninefold enhanced in epithelial cells. However, high levels of E2 (1 μg as shown in Fig. 4B) downregulate transcription from the 3bpTATA construct in fibroblasts while activation remains elevated in keratinocytes. Previously, we have shown that E2 is expressed to similar levels in both PalK and PalF cells (39). It seems probable that the downregulation of transcription in fibroblasts at elevated levels of E2 is due to a squelching mechanism. In keratinocytes, it is possible that E2, when expressed at elevated levels, can use additional coactivators to activate transcription from the BPV-4 TATA box or that the same coactivator is used in both cell types but overexpressed in keratinocytes compared with fibroblasts. VP16-E2 also activates transcription from PV2 fivefold better in epithelial cells than in fibroblasts (39). We therefore used a range of VP16-E2 concentrations to activate the 3bpTATA construct to determine whether the upstream BPV-4 promoter region plays a role in the cell-type-selective response of this promoter to VP16-E2. The results of these experiments are shown in Fig. 4B and confirm a role for the promoter repressor elements in mediating the enhanced epithelial response of the BPV-4 promoter to upstream activators. Over a range of concentrations, VP16-E2 upregulates transcription from the 3bpTATA construct more efficiently in fibroblasts than in epithelial cells. Clearly, this is the reverse of the situation with the PV2 promoter, where the response is four- to fivefold enhanced in epithelial cells.
The papillomavirus PRE-2 can repress the basal activity of a strong heterologous promoter.
The PRE-2 motif is conserved in position and sequence in the related mucosal epitheliotropic papillomaviruses BPV-3 and BPV-6 (Fig. 5A). Also, a YY-1-independent silencer in roughly the same position as PRE-2 in the HPV-16 LCR has recently been identified (30). Two functional types of repressor elements exist: negative regulatory elements (NREs) that are promoter specific and silencer elements that are able to repress heterologous promoter activity out of context of the native promoter. To further characterize the functional properties of PRE-2, oligomers corresponding to this sequence were multimerized upstream of the SV40 promoter in the pGL3 luciferase vector in both a positive and a negative orientation (Fig. 5A). The ability of these elements to direct repression of SV40 promoter activity was assayed in both PalK and PalF cells. Figure 5B shows that, over a range of concentrations, four copies of PRE-2 repress SV40 promoter activity approximately 40 to 80% in PalK cells and 60 to 80% in PalF cells. The orientation of PRE-2 had no effect on the extent of repression in both cell types. These results demonstrate that PRE-2 can repress the basal activity of a strong promiscuous constitutive promoter and is therefore a silencer.
PRE-2 binds a specific protein complex in both PalK and PalF cells.
In vitro footprinting shows that PRE-2 contains a DNA binding site for a potential transcription factor (18). Extensive database searches revealed that PRE-2 does not contain any known transcription factor binding sites. As functional analysis has identified PRE-2 as a transcriptional silencer, we performed band shift assays to determine whether PRE-2 could bind a nuclear protein. A double-stranded radiolabeled oligonucleotide containing the 20-bp PRE-2 motif detected a single protein complex with both PalK and PalF nuclear extracts (Fig. 6). Binding to PRE-2 was confirmed to be specific, as it was competed by a 100-fold excess of nonradioactive self oligonucleotide but not by the unrelated AP-1 oligonucleotide.
FIG. 6.

PRE-2 binds a specific protein complex in both PalK and PalF cells. A single radiolabeled PRE-2 motif was used to probe PalK and PalF nuclear extracts in a band shift assay. One-hundredfold excesses of unlabeled PRE-2 and AP-1 oligonucleotides were used as indicated to assess the specificity of binding.
Functional characterization of the PRE-2 binding complex.
To identify the exact nucleotides necessary for in vitro PRE-2–nuclear protein interaction, we generated two double-stranded oligonucleotides each containing a single PRE-2 motif with a 3-bp substitution in the footprint (Fig. 7A). The mutant PRE-2 oligonucleotides were tested for competition using the band shift assay. Figure 7B demonstrates that binding of the detected protein complex is not competed by either of the two mutant sequences, showing these mutated residues to be important for sequence-specific binding in both PalK and PalF cells. In Fig. 7B, there is an apparent enhanced binding in PalK cells to PRE-2. However, this is due to the PalK cell gel being exposed slightly longer (as can be seen from the residual signals in the wells and the increased intensity of the free oligonucleotide). Also, the minor band just below the specific major band was not always observed and may represent a breakdown or modification product of the major band. To determine the functional consequences of loss of binding, four copies of PRE-2mt1 were inserted upstream of the SV40 promoter in the pGL3PRO reporter vector, generating PRO4Xmt1. The ability of this construct to relieve PRE-2-mediated repression of SV40 promoter activity was assayed in both PalK and PalF cells. Figure 8 shows that loss of factor binding correlates with loss of transcriptional repression. Over a range of concentrations, the activity of the PRO4Xmt1 construct is similar to that of the SV40 promoter alone in both PalF and PalK cells. To confirm a role for PRE-2 in regulation of transcription from the BPV-4 promoter, the mutations in PRE-2 that lost repressor function and failed to bind the cellular protein were incorporated into the 80bpTATA promoter construct, creating 80bpmt1. 80bpTATA has properties similar to those of the PV2 construct (Fig. 4A and Table 1). In fibroblasts, mutation of PRE-2 results in a threefold increase in the response of the papillomavirus promoter to E2 (Fig. 9). This indicates that PRE-2 acts as a repressor element in the context of the BPV-4 promoter. Also, in keratinocytes mutation of PRE-2 does not increase the response of the promoter to E2 as dramatically as is observed in the fibroblasts, indicating that PRE-2 is involved in the cell-type-specific response of this promoter to activation by E2. It should be noted that mutation of the PRE-2 element does not increase the response of the promoter to the levels observed with 66bpTATA (Fig. 4A and Table 1). This may be due to other factors that we cannot detect in our assays interacting with the DNA in this region, or perhaps the factor binding PRE-2 has residual binding capacity for the mutated PRE-2 site and can therefore still repress the response to E2. However, it is clear that PRE-2 is involved in regulating the transcriptional response of the papillomavirus promoter to E2.
FIG. 7.

(A) Sequences of PRE-2 mutants. Footprinting studies demonstrate that PRE-2 contains a potential binding site for a transcription factor (underlined). Two double-stranded oligonucleotides, each containing a single PRE-2 motif with a 3-bp substitution in this footprint, were generated and tested for competition in the band shift assay. (B) PRE-2mt1 and PRE-2mt2 do not compete for binding to the detected protein complex. A single radiolabeled PRE-2 motif was used in a band shift assay to probe PalK and PalF nuclear extracts. Cold competitors were added as indicated at either a 100-fold (+) or a 500-fold (∗) molar excess.
FIG. 8.

Loss of complex binding to PRE-2 correlates with loss of transcriptional repression. Four copies of mt1 were inserted into the BglII site, upstream of the SV40 promoter in the pGL3PRO luciferase vector, generating PRO4Xmt1. PalK and PalF cells were transfected with the indicated amounts of pGL3PRO, PRO4XPRE-2, and PRO4Xmt1. Results are expressed relative to the luciferase activity of pGL3PRO (set arbitrarily at 100%), which contains only the SV40 promoter.
FIG. 9.
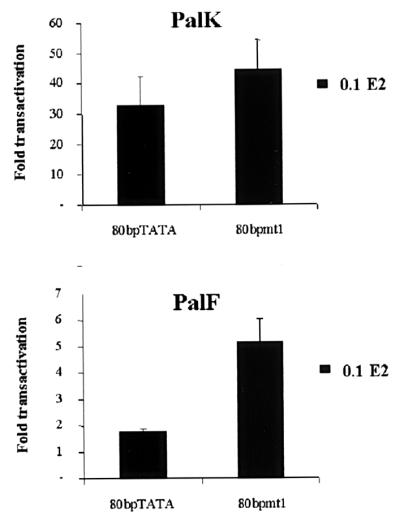
Mutation of PRE-2 results in an elevated transcriptional response of the BPV-4 promoter. PRE-2mt1 was introduced into the 80bpTATA promoter construct, generating 80bpmt1. One microgram of 80bpTATA and 80bpmt1 reporter constructs was cotransfected into PalK and PalF cells with 0.1 μg of pCMVHPV16-E2 expression vector. pCMV vector was added so that an equivalent amount of DNA was used in each transfection. Results are expressed as fold transactivation relative to the luciferase activity of each reporter in the absence of E2.
PRE-2 specifically binds a 50-kDa cellular protein.
UV cross-linking was performed to determine the molecular mass of the active DNA binding form of the detected protein complex. A single radiolabeled BrdU-substituted PRE-2 motif was used to probe PalK and PalF nuclear extracts in a band shift assay. No difference in binding complexes was observed between the BrdU and non-BrdU oligonucleotides. The gel was UV irradiated (304 nm) to cross-link the protein to the DNA, the bands of interest were excised, and the protein complexes were resolved by SDS–10% polyacrylamide gel electrophoresis. A nonlabeled BrdU PRE-2 motif and the unrelated AP-1 oligonucleotide were used to assess the specificity of binding. Figure 10 shows that BrdU PRE-2 binds a major species of approximately 50 kDa in both PalK and PalF nuclear extracts. Binding of this factor was confirmed to be specific, as it was competed by excess nonlabeled BrdU PRE-2 but was not competed by AP-1 in either cell type.
FIG. 10.

Molecular weight determination of the PRE-2 binding factor. PalK and PalF nuclear extracts were probed with a radiolabeled BrdU-substituted PRE-2 motif in a band shift assay. After UV exposure, the PRE-2 cross-linked protein complex was electrophoresed on an SDS–10% polyacrylamide gel. Competition band shift reactions with either 100-fold nonlabeled BrdU or 100-fold nonlabeled AP-1 were performed as indicated to assess the specificity of binding. Numbers at left are molecular masses in kilodaltons.
DISCUSSION
Previously, we have demonstrated that the BPV-4 promoter shows an enhanced epithelial response to activation, not only by E2 but also by VP16-E2 and VP16-LexA (39). In this study, we demonstrate that the upstream BPV-4 promoter region contributes to the enhanced epithelial response of this promoter to upstream activators. This is clear from Fig. 2, where swapping of the tk and LCR upstream elements demonstrates that the upstream promoter region of the BPV-4 LCR dictates a cell-type-specific response. Previously, we have shown that VP16-E2 activates transcription from the BPV-4 promoter construct PV2 6E2 fivefold better in keratinocytes (39). However, when the upstream region is removed VP16-E2 activates transcription from the BPV-4 LCR TATA box better in fibroblasts (Fig. 4B). This conclusively demonstrates that the upstream promoter region of the BPV-4 LCR contributes toward the cell-type-specific response of this promoter. The BPV-4 promoter contains a novel silencer element, PRE-2, that represses the transcriptional response to the E2 protein and may be one component involved in mediating the cell-type-selective response of the BPV-4 promoter to upstream activators. PRE-2 functions as an autonomous cis-acting element to repress the basal activity of the promiscuous SV40 promoter in a cell-type-independent manner. Database searches reveal that PRE-2 shows no homology to any of the published transcription factor binding sites. The PRE-2 motif is conserved in position and sequence in the related mucosal epitheliotropic papillomaviruses BPV-3 and BPV-6, suggesting functional significance. Band shift assays show that PRE-2 binds a specific protein complex in both PalK and PalF cells. PRE-2 mutants that do not compete for binding in band shift assays do not repress transcription when multimerized upstream of the SV40 promoter. Incorporation of these mutations into the BPV-4 promoter region results in an elevation of the transcriptional response to E2 (Fig. 9). This elevation is more marked in fibroblasts, suggesting that PRE-2 may be involved in mediating the cell-type-specific response of the BPV-4 promoter. UV cross-linking demonstrates that the PRE-2 binding form of the protein complex has a molecular mass of approximately 50 kDa. None of the transcriptional repressors known to bind papillomavirus promoters, for example, YY-1 (4), Sp3 (3), C/EBPβ (5), and CDP/Cut (29), are this size. These results demonstrate that the PRE-2 binding protein is a novel transcriptional repressor and regulator of mucosal epitheliotropic papillomavirus transcription.
E2 requires the cooperation of at least one additional DNA binding proximal promoter factor to activate a minimal tk TATA box promoter (14). The results presented here demonstrate that a minimal BPV-4 TATA promoter containing neither an initiator element nor a binding site for an upstream factor is sufficient to support activated transcription by E2. This result suggests that the TFIID complexes assembled at the BPV-4 and tk TATA boxes are distinct. The HPV-16 enhancer-promoter is virtually inactive in normal human diploid fibroblasts but active in human fibroblasts with a deletion in the short arm of chromosome 11 (del-11 cells). Mutation of the HPV-16 TATAAAA box to the SV40 TATTTAT sequence reduces the activity of the HPV-16 enhancer-promoter in del-11 cells. DNA-protein complexes formed with an HPV-16 promoter fragment are quantitatively different in del-11 and diploid fibroblasts. This difference disappears upon mutation of the HPV-16 TATA to the SV40 TATA sequence, indicating specificity of the HPV-16 TATA box sequence (34). When these data are considered together with our results, it seems possible that the mucosal epitheliotropic papillomavirus promoters may be recognized by a distinct TFIID complex. This possibility is currently under investigation.
In general, transcriptional repressors can work either passively to antagonize activator function or actively to target components of the general transcription machinery in such a way as to decrease the frequency of transcriptional initiation (21). For example, E2 can repress transcription of the HPV-16 promoter by a passive mechanism displacing Sp1 from a proximal promoter element (37), while the unliganded thyroid hormone receptor α actively represses transcription by targeting TATA-binding protein (12). The protein interacting with the papillomavirus promoter repressor element PRE-2 does not simply displace the binding of a positively acting factor but directs an active repression mechanism, as PRE-2 can repress SV40 promoter activity. Active repressors interact to regulate multiple steps in gene expression from chromatin unfolding and transcription initiation to processing of the message. PRE-2 clearly does not act in a cell-selective manner when placed out of context of the BPV-4 promoter, as it represses transcription from the promiscuous SV40 promoter in both fibroblasts and keratinocytes. Also, the protein binding to this element can be detected in both cell types. This is in common with other papillomavirus transcriptional control elements that, when taken out of the context of papillomavirus LCRs, behave in a cell-type-independent manner. We propose that cooperative interactions among upstream bound activators, non-DNA-bound cofactors, repressor molecules bound to the PRE motifs, and the basal machinery assembled at the BPV-4 TATA box all determine the differential response of the BPV-4 promoter in keratinocytes and fibroblasts.
Modification of chromatin structure represents another level of regulation of gene expression. The chromatin organization of the HPV-16 and -18 genomes suggests important regulatory roles of nucleosomes during the viral life cycle (35, 36). Two nucleosomes are precisely positioned on the HPV-16 LCR: one overlaps the center of the viral enhancer, while a second nucleosome overlaps the origin of replication and the promoter region. The HPV-18 LCR shows specific assembly of a nucleosome over the replication origin and the proximal promoter, positioned about 90 bp upstream of the homologous region of the HPV-16 LCR. E1, the major papillomavirus replication factor, has been shown to bind hSNF5, a component of the SWI-SNF complex (22). SWI-SNF is an ATP-dependent chromatin-remodeling complex. A number of transcriptional repressors have been shown to recruit histone deacetylase activity as part of multiprotein complexes. Deacetylation of histone tails promotes nucleosome assembly, thereby inhibiting the ability of transcription factors to gain access to the DNA. Trichostatin A, a specific inhibitor of histone deacetylase activity, upregulates the HPV-11 LCR promoter in undifferentiated primary human keratinocytes. Deacetylase recruitment is independent of the C/EBPβ binding sites located in the HPV-11 promoter region (40). C/EBPβ has also been implicated as a negative regulator of BPV-4 transcription (23). Recently, a novel YY-1-independent silencer located in approximately the same position as PRE-2 in the HPV-16 LCR has been identified (30). The differentiation-specific factor CDP/Cut binds to this element and represses HPV-16 transcription by a histone-mediated mechanism (29). It therefore seems possible that PRE-2 functions by recruiting deacetylases to the BPV-4 promoter.
The results presented here further our understanding of the mechanisms underlying not only mucosal epitheliotropic papillomavirus transcriptional control but also cell-type-specific transcription in general. The PRE-2 binding protein appears to represent a novel transcriptional repressor and regulator of papillomavirus transcription. Future work will focus on the cloning of the gene encoding PRE-2. Also, the mechanism of repression will be elucidated, whether this is through direct interaction with the basal machinery, via a corepressor, or by recruitment of histone deacetylase activity.
ACKNOWLEDGMENTS
We thank David Gillespie (Beatson Institute) for useful comments on the manuscript and for the AP-1 oligonucleotide.
K.W.V. is the recipient of a UK Medical Research Council Studentship. M.S.C. is supported by a Life Fellowship from the Cancer Research Campaign.
REFERENCES
- 1.Apt D, Chong T, Liu Y, Bernard H U. Nuclear factor I and epithelial cell-specific transcription of human papillomavirus type 16. J Virol. 1993;67:4455–4463. doi: 10.1128/jvi.67.8.4455-4463.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Apt D, Liu Y, Bernard H U. Cloning and functional analysis of spliced isoforms of human nuclear factor I-X: interference with transcriptional activation by NFI/CTF in a cell-type specific manner. Nucleic Acids Res. 1994;22:3825–3833. doi: 10.1093/nar/22.19.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Apt D, Watts R M, Suske G, Bernard H U. High Sp1-Sp3 ratios in epithelial cells during epithelial differentiation and cellular transformation correlate with the activation of the HPV-16 promoter. Virology. 1996;224:281–291. doi: 10.1006/viro.1996.0530. [DOI] [PubMed] [Google Scholar]
- 4.Bauknecht T, Angel P, Royer H, zur Hausen H. Identification of a negative regulatory domain in the human papillomavirus type 18 promoter: interaction with the transcriptional repressor YY1. EMBO J. 1992;11:4607–4617. doi: 10.1002/j.1460-2075.1992.tb05563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bauknecht T, Shi Y. Overexpression of C/EBPB represses human papillomavirus type 18 upstream regulatory region activity in HeLa cells by interfering with the binding of TATA-binding protein. J Virol. 1998;72:2113–2124. doi: 10.1128/jvi.72.3.2113-2124.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campo S M, O'Neil B W, Barron R J, Jarrett W F H. Experimental reproduction of the papilloma-carcinoma complex of the alimentary canal in cattle. Carcinogenesis. 1994;15:1597–1601. doi: 10.1093/carcin/15.8.1597. [DOI] [PubMed] [Google Scholar]
- 7.Chong T, Apt D, Gloss B, Isa M, Bernard H U. The enhancer of human papillomavirus type 16: binding sites for the ubiquitous transcription factors oct-1, NFA, TEF-2, NF1, and AP-1 participate in epithelial cell-specific transcription. J Virol. 1991;65:5933–5943. doi: 10.1128/jvi.65.11.5933-5943.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Connolly J A, Morgan I M, Jackson M E, Campo M S. The BPV-4 co-carcinogen quercetin induces cell cycle arrest and up-regulates transcription from the LCR of BPV-4. Oncogene. 1998;16:2739–2746. doi: 10.1038/sj.onc.1201796. [DOI] [PubMed] [Google Scholar]
- 9.Cuthill S, Sibbet G J, Campo M S. Characterization of a nuclear factor, papilloma enhancer binding factor-1, that binds the long control region of human papillomavirus type 16 and contributes to enhancer activity. Mol Carcinog. 1993;8:96–104. doi: 10.1002/mc.2940080206. [DOI] [PubMed] [Google Scholar]
- 10.Desaintes C, Demeret C. Control of papillomavirus DNA replication and transcription. Semin Cancer Biol. 1996;7:339–347. doi: 10.1006/scbi.1996.0043. [DOI] [PubMed] [Google Scholar]
- 11.Dikstein R, Zhou S, Tjian R. Human TAFII105 is a cell type specific TFIID subunit related to hTAFII130. Cell. 1996;87:137–146. doi: 10.1016/s0092-8674(00)81330-6. [DOI] [PubMed] [Google Scholar]
- 12.Fondell J D, Brunel F, Hisatake K, Roeder R G. Unliganded thyroid hormone receptor α can target TATA-binding protein for transcriptional repression. Mol Cell Biol. 1996;16:281–287. doi: 10.1128/mcb.16.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gloss B, Bernard H U, Seedorf K, Klock G. The upstream regulatory region of the human papilloma virus-16 contains an E2 protein-independent enhancer which is specific for cervical carcinoma cells and regulated by glucocorticoid hormones. EMBO J. 1987;6:3735–3743. doi: 10.1002/j.1460-2075.1987.tb02708.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ham J, Dostatni N, Arnos F, Yaniv M. Several different upstream promoter elements can potentiate transactivation by the BPV-1 E2 protein. EMBO J. 1991;10:2931–2940. doi: 10.1002/j.1460-2075.1991.tb07843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoppe-Seyler F, Butz K, zur Hausen H. Repression of the human papillomavirus type 18 enhancer by the cellular transcription factor Oct-1. J Virol. 1991;65:877–883. doi: 10.1128/jvi.65.10.5613-5618.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishiji T, Lace M J, Parkkinen S, Anderson R D, Haugen T H, Cripe T P, Xiao J H, Davidson I, Chambon P, Turek L P. Transcriptional enhancer factor (TEF)-1 and its cell-specific co-activator activate human papillomavirus-16 E6 and E7 oncogene transcription in keratinocytes and cervical carcinoma cells. EMBO J. 1992;11:2271–2281. doi: 10.1002/j.1460-2075.1992.tb05286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jackson E M, Campo M S. Both viral E2 protein and the cellular factor PEBP2 regulate transcription via E2 consensus sites within the bovine papillomavirus type 4 long control region. J Virol. 1995;69:6038–6046. doi: 10.1128/jvi.69.10.6038-6046.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jackson E M, Campo M S. Positive and negative E2-independent regulatory elements in the long control region of bovine papillomaviruses type 4. J Gen Virol. 1991;72:877–883. doi: 10.1099/0022-1317-72-4-877. [DOI] [PubMed] [Google Scholar]
- 19.Jaggar R T, Pennie W D, Smith K T, Jackson M E, Campo M S. Co-operation between bovine papillomavirus type 4 and ras in the morphological transformation of primary bovine fibroblasts. J Gen Virol. 1990;71:3041–3046. doi: 10.1099/0022-1317-71-12-3041. [DOI] [PubMed] [Google Scholar]
- 20.Jiang C-K, Connolly D, Blumenberg M. Comparison of methods for transfection of human epidermal keratinocytes. J Investig Dermatol. 1991;97:969–973. doi: 10.1111/1523-1747.ep12491889. [DOI] [PubMed] [Google Scholar]
- 21.Johnson A D. The price of repression. Cell. 1995;81:655–658. doi: 10.1016/0092-8674(95)90524-3. [DOI] [PubMed] [Google Scholar]
- 22.Lee D, Sohn H, Kalpane G V, Choe J. Interaction of E1 and hSNF5 proteins stimulates replication of human papillomavirus DNA. Nature. 1999;399:487–491. doi: 10.1038/20966. [DOI] [PubMed] [Google Scholar]
- 23.McCaffery R E, Jackson M E. An element binding a C/EBP-related transcription factor contributes to negative regulation of the bovine papillomavirus type 4 long control region. J Gen Virol. 1994;75:3047–3056. doi: 10.1099/0022-1317-75-11-3047. [DOI] [PubMed] [Google Scholar]
- 24.Mittal R, Tsutsami K, Pater A, Pater M M. Human papillomavirus type 16 expression in cervical keratinocytes: role of progesterone and glucocorticoid hormones. Obstet Gynecol. 1993;81:5–12. [PubMed] [Google Scholar]
- 25.Morgan I M, Grindlay G J, Campo M S. The bovine papillomavirus type 4 long control region contains an epithelial specific enhancer. J Gen Virol. 1999;80:23–27. doi: 10.1099/0022-1317-80-1-23. [DOI] [PubMed] [Google Scholar]
- 26.Morgan I M, Grindlay G J, Campo M S. Epithelial specific transcriptional regulation of the bovine papillomavirus 4 promoter by E2. J Gen Virol. 1998;79:501–508. doi: 10.1099/0022-1317-79-3-501. [DOI] [PubMed] [Google Scholar]
- 27.Morris P J, Dent C L, Ring C J, Latchman D S. The octamer binding site in the HPV-16 regulatory region produces opposite effects on gene expression in cervical and non-cervical cells. Nucleic Acids Res. 1993;21:1019–1023. doi: 10.1093/nar/21.4.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O'Connor M, Bernard H U. Oct-1 activates the epithelial-specific enhancer of human papillomavirus type 16 via a synergistic interaction with NFI at a conserved composite regulatory element. Virology. 1995;207:77–88. doi: 10.1006/viro.1995.1053. [DOI] [PubMed] [Google Scholar]
- 29.O'Connor M J, Stunkel W, Koh C H, Zimmermann H, Bernard H U. The differentiation-specific factor CDP/Cut represses transcription and replication of human papillomaviruses through a conserved silencing element. J Virol. 2000;74:401–410. [PMC free article] [PubMed] [Google Scholar]
- 30.O'Connor M J, Stunkel W, Zimmermann H, Koh C H, Bernard H U. A novel YY1-independent silencer represses the activity of the human papillomavirus type 16 enhancer. J Virol. 1998;72:10083–10092. doi: 10.1128/jvi.72.12.10083-10092.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O'Connor M J, Tan S H, Tan C H, Bernard H U. YY1 represses human papillomavirus type 16 transcription by quenching AP-1 activity. J Virol. 1996;70:6529–6539. doi: 10.1128/jvi.70.10.6529-6539.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Offord E A, Beard P. A member of the activator protein 1 family found in keratinocytes but not in fibroblasts required for transcription from a human papillomavirus type 18 promoter. Virology. 1990;64:4792–4798. doi: 10.1128/jvi.64.10.4792-4798.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sibbet G J, Cuthill S, Campo M S. The enhancer in the long control region of human papillomavirus type 16 is up-regulated by PEF-1 and down-regulated by Oct-1. J Virol. 1995;69:4006–4011. doi: 10.1128/jvi.69.7.4006-4011.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smits P H M, Smits H L, Minnaar R P, ter Schegget J. Regulation of human papillomavirus type 16 (HPV-16) transcription by loci on the short arm of chromosome 11 is mediated by the TATAAAA motif of the HPV-16 promoter. J Gen Virol. 1993;74:121–124. doi: 10.1099/0022-1317-74-1-121. [DOI] [PubMed] [Google Scholar]
- 35.Stunkel W, Bernard H U. The chromatin structure of the long control region of human papillomavirus type 16 represses viral oncoprotein expression. J Virol. 1999;73:1918–1930. doi: 10.1128/jvi.73.3.1918-1930.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stunkel W, Huang Z, Tan S H, O'Connor M J, Bernard H U. Nuclear matrix attachment regions of human papillomavirus type 16 repress or activate the E6 promoter, depending on the physical state of the viral DNA. J Virol. 2000;74:2489–2501. doi: 10.1128/jvi.74.6.2489-2501.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tan S H, Gloss B, Bernard H U. During negative regulation of the human papillomavirus-16 E6 promoter, the viral E2 protein can displace Sp1 from a proximal promoter element. Nucleic Acids Res. 1992;20:251–256. doi: 10.1093/nar/20.2.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thierry F, Spyrou G, Yaniv M, Howley P. Two AP1 sites binding JunB are essential for human papillomavirus type 18 transcription in keratinocytes. J Virol. 1992;66:3740–3748. doi: 10.1128/jvi.66.6.3740-3748.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vance K W, Campo M S, Morgan I M. An enhanced epithelial response of a papillomavirus promoter to transcriptional activators. J Biol Chem. 1999;274:27839–27844. doi: 10.1074/jbc.274.39.27839. [DOI] [PubMed] [Google Scholar]
- 40.Zhao W, Noya F, Chen W Y, Townes T M, Chow L T, Broker T R. Trichostatin A up-regulates human papillomavirus type 11 upstream regulatory region-E6 promoter activity in undifferentiated primary human keratinocytes. J Virol. 1999;73:5026–5033. doi: 10.1128/jvi.73.6.5026-5033.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.zur Hausen H. Human papillomaviruses in the pathogenesis of anogenital cancer. Virology. 1991;184:9–13. doi: 10.1016/0042-6822(91)90816-t. [DOI] [PubMed] [Google Scholar]