

Abstract
Voltage‐gated calcium channels (VGCC) are abundant in the central nervous system and serve a broad spectrum of functions, either directly in cellular excitability or indirectly to regulate Ca2+ homeostasis. Ca2+ ions act as one of the main connections in excitation–transcription coupling, muscle contraction and excitation–exocytosis coupling, including synaptic transmission. In recent years, many genes encoding VGCCs main α or additional auxiliary subunits have been associated with epilepsy. This review sums up the current state of knowledge on disease mechanisms and provides guidance on disease‐specific therapies where applicable.
Keywords: genetic epilepsy, ion channel, precision therapy, voltage gated calcium channel
Voltage‐gated calcium channels are abundant in the central nervous system and serve a broad spectrum of functions. Ca2+ ions act as one of the main connections in excitation–transcription, excitation–contraction and excitation–exocytosis coupling, including synaptic transmission. In recent years, many genes encoding voltage‐gated calcium channel subunits have been associated with epilepsy. This review sums up the current state of knowledge on disease mechanisms and provides guidance on disease‐specific therapies where applicable.
Abbreviations
- ASM
anti‐seizure medication
- CNS
central nervous system
- DEE
developmental and epileptic encephalopathy
- EA2
episodic ataxia type 2
- FHM1
familial hemiplegic migraine type 1
- GOF
gain of function
- KO
knockout
- LOF
loss of function
- o/e
observed/expected
- OMIM
online Mendelian inheritance in man
- pLI
probability of being loss‐of‐function intolerant
- SUDEP
sudden unexpected death in epilepsy patients
- VGCC
voltage‐gated calcium channel
1. INTRODUCTION
Voltage‐gated calcium channels (VGCC) form a class of transmembrane proteins with a main pore forming α‐subunit which is comprised of 4 domains, each containing 6 transmembrane segments. Historically, different channels have been named according to their electrophysiological properties and sensitivity to blockers. A total of 10 α‐subunits of VGCCs can be found in the human genome, forming subfamilies of the high voltage‐activated long‐lasting, L‐type CaV1.X VGCCs, the high and intermediate voltage‐activated CaV2.x VGCC with P/Q‐, N‐, R‐type current conducting channels, or the low‐voltage‐activated, transient‐current (T‐type) CaV3.x channels. CaV1.x and CaV2.x channels assemble into multi‐subunit complexes comprising also β‐, α2δ ‐ and γ‐subunits beside the channel's main α ‐subunit in a 1:1:1:1 ratio (Figure 1) (Takahashi et al., 1987; Müller et al., 2010). T‐type VGCCs do not depend on additional subunits to produce current, but their gating characteristics may be influenced by these auxiliary subunits (Dolphin et al., 1999; Gao et al., 2000; Lacinová & Klugbauer, 2004; Lambert et al., 1997; Leuranguer et al., 1998; see also review by Zamponi et al., 2015). β‐Subunits are intracellular proteins that enhance calcium currents by increasing the membrane expression and facilitating the open state of the channel (Bichet et al., 2000; Williams et al., 1994; Yasuda et al., 2004). α2δ‐Subunits are post‐translationally cleaved and rejoined by a disulphide bond and further promote currents through calcium channels (Hobom et al., 2000; Yasuda et al., 2004). Both β‐ and α2δ‐subunits show distinct expression patterns and do not assemble with specific α‐subunits (Hobom et al., 2000; Ludwig et al., 1997; Yasuda et al., 2004). The association of γ‐subunits with the channel complex is uncertain, with differences between the individual subtypes, whereas their function in the trafficking of AMPA receptors is more clear (see review Chen et al., 2007). The channel complex is embedded in a rich nano‐environment of mostly calcium‐binding proteins serving to directly translate an elevation in intracellular calcium to downstream pathways (Müller et al., 2010). In comparison to other voltage‐gated ion channels, VGCCs take a special role, as the conducted ions render them directly linked to downstream effects beyond their contribution to electrophysiological processes such as exocytosis of neurotransmitters, altered transcription, excitotoxicity and apoptosis (Bucurenciu et al., 2010; Cano‐Abad et al., 2001; Hardingham et al., 1997; Li et al., 2016; Stanika et al., 2012; Stanley, 1993). Furthermore, the expression of VGCCs is not limited to neurons and they can also be found in glia cells (D'Ascenzo et al., 2004).
FIGURE 1.
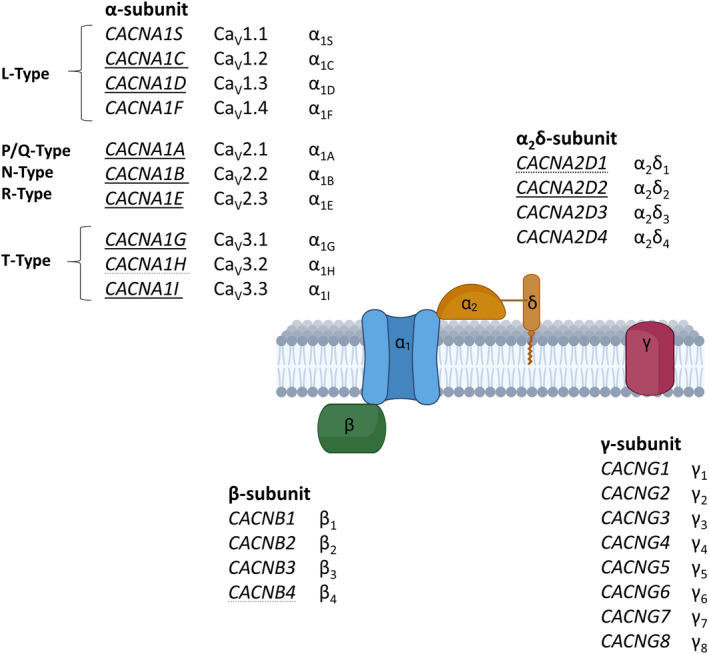
The calcium channel complex. Genes with a clear association with epilepsy are underlined. Genes with disputed disease association are underlined with dotted lines. Created with BioRender.com.
In recent years, all VGCC α‐subunits and a progressing number of auxiliary subunits expressed in the brain have been associated with epilepsy (Table 1). This review will give an overview of the channel family and the associated epileptic and some other phenotypes.
TABLE 1.
Overview of genes, phenotypes and specific therapeutic options for calcium channelopathies.
| Gene | Protein | Phenotype | Specific treatment |
|---|---|---|---|
| CACNA1C | CaV1.2 |
GOF: Timothy syndrome (OMIM #601005) GOF & LOF: Neurodevelopmental disorder with hypotonia, language delay and skeletal defects with or without seizures (OMIM #620029) |
GOF: Verapamil+ (Gershon et al., 2014) Dyhydropyridines? |
| CACNA1D | CaV1.3 |
GOF: Primary aldosteronism, seizures and neurologic abnormalities (OMIM #615474) LOF: Sinoatrial node dysfunction and deafness (OMIM #614896) |
GOF: Verapamil? Dyhydropyridines? |
| CACNA1A | CaV2.1 |
GOF: Migraine, familial hemiplegic, 1 (OMIM #141500), alternating hemiplegia of childhood, DEE42 (OMIM #617106) LOF: Episodic ataxia Type 2 (OMIM #108500), DEE42 with absence seizures (OMIM #617106) CAG‐Expansion: Spinocerebellar ataxia type 6 (OMIM #183086) |
Mixed cohort: TPM+ (Le Roux et al., 2021), LEV+ (Le Roux et al., 2021), LTG+ (Byers et al., 2016; Le Roux et al., 2021), VPA+ (Le Roux et al., 2021) LOF (Episodic ataxia): 4‐AP++ (Muth et al., 2021; Strupp, Kalla, et al., 2011), acetazolamide++ (Muth et al., 2021) |
| CACNA1B | CaV2.2 | Bi‐allelic LOF: Neurodevelopmental disorder with seizures and nonepileptic hyperkinetic movements (OMIM #618497) | |
| CACNA1E | CaV2.3 |
GOF: DEE 69 (OMIM #618285), Developmental delay and regression without epilepsy (Royer‐Bertrand et al., 2021) LOF: DEE 69 (OMIM #618285) |
GOF: TPM+ (Helbig, Lauerer, et al., 2018) |
| CACNA1G | CaV3.1 |
GOF & mixed: Epilepsy, developmental delay, cerebellar atrophy LOF & Mixed: SCA42 (OMIM #616795/#618087) |
GOF: ESL?, ESX?, ZNS? |
| CACNA1I | CaV3.3 |
GOF: Neurodevelopmental disorder with speech impairment and with or without seizures (OMIM #620114) Mixed effects: Familial hemiplegic migraine LOF: Schizophrenia |
GOF: ESX+ (El Ghaleb et al., 2021), ESL?, ZNS? |
| CACNA2D1 | α2δ1 | Bi‐allelic LOF/Monoallelic LOF (debated): Developmental and epileptic encephalopathy 110 (OMIM #620149) | Avoid GPB & PGB? |
| CACNA2D2 | α2δ2 | Bi‐allelic LOF: Cerebellar atrophy with seizures and variable developmental delay (OMIM #618501) | Avoid GPB & PGB? |
Abbreviations: 4‐AP, 4‐aminopyridine; ESL, eslicarbazepine acetate; ESX, ethosuximide; GBP, gabapentin; LEV, levetiracetam; LTG, lamotrigine; PGB, pregabalin; TPM, topiramate; ZNS, Zonisamide; ?, theoretical consideration, based on in vitro data; +, evidence from single case reports or case series; ++, evidence from randomized clinical trials.
2. L‐TYPE CALCIUM CHANNELS (CaV1.x)
L‐type calcium channels are long‐lasting, high‐voltage activated VGCCs. They are widely expressed in the body with subunit‐specific distinct expression patterns. CACNA1S, encoding CaV1.1, is mainly expressed in skeletal muscle and is not expressed in the central nervous system (CNS) to a significant amount (Sinnegger‐Brauns et al., 2009; https://www.proteinatlas.org/ENSG00000081248‐CACNA1S;Uhlén et al., 2015). CACNA1F/CaV1.4 expression is most distinct but not limited to the retina (Doering et al., 2014; Strom et al., 1998). Apart from other tissues, there is some expression in the spinal cord and in the pineal gland (Doering et al., 2014; Hemara‐Wahanui et al., 2005; McRory et al., 2004; Sinnegger‐Brauns et al., 2009). Only CACNA1C/CaV1.2 and CACNA1D/CaV1.3 are significantly expressed in the brain with a variable predominance in different species between the two subunits (Hell et al., 1993; Sinnegger‐Brauns et al., 2009; Splawski et al., 2004). Both CaV1.2 and CaV1.3 are expressed postsynaptically in clusters in dendritic spines and the soma of glutamatergic and GABAergic cells (Di Biase et al., 2008; Hell et al., 1993; Jenkins et al., 2010; Obermair et al., 2004). Although only detected sparsely, there is also evidence of expression in the axon for both subunits (Obermair et al., 2004; Tippens et al., 2008). Both are also expressed in astrocytes, where CaV1.2 channels serve an important role in astrocytic activation and astrogliosis (Cheli et al., 2016; Tippens et al., 2008).
Missense variants in CACNA1C, encoding CaV1.2, had been first established in cardiac arrhythmias, such as Long‐QT syndrome (Boczek et al., 2013; Fukuyama et al., 2014) and Timothy syndrome, a severe multisystem disorder that among other symptoms includes cardiac arrhythmia, syndactyly, developmental delay and occasionally seizures (Boczek et al., 2015; Splawski et al., 2004, 2005). More recently, variants in this gene have been described in patients with a leading neurodevelopmental syndrome consisting of neonatal onset epilepsy, developmental delay, autistic features, hypotonia, orthopaedic abnormalities and ataxia (Bozarth et al., 2018; Rodan et al., 2021). Strikingly, electrophysiological gain‐ (GOF) and loss‐of‐function (LOF) variants as well as variants in which no effect on channel function were described. Truncating variants were only sparsely associated with seizures (Rodan et al., 2021).
Variants in CACNA1D, encoding CaV1.3, have been associated with combined cardiac and inner ear phenotypes. Biallelic LOF variants in this gene have been found in a family with sinuatrial node dysfunction and deafness (Baig et al., 2011). In line with this, KO mice for CACNA1D exhibit hearing loss and bradycardia (Platzer et al., 2000). The evidence for seizure disorders in patients carrying missense variants in CACNA1D is based on a few case reports: Scholl et al. identified two individuals who suffered from aldosterone‐producing adenomas and primary aldosteronism that were associated with somatic and germline CACNA1D missense variants with congenital hyperaldosteronism, arterial hypertonia, intellectual disability and focal to bilateral tonic–clonic seizures. One patient showed a movement disorder with spastic quadriplegia and athetosis. Both patients carried de novo variants showing a GOF in electrophysiological recordings (Scholl et al., 2013). A report in Russia claims to have found a third similar case carrying a missense variant (Semenova et al., 2018). Pingerra et al. describe a patient with developmental delay, autism spectrum disorder and focal epilepsy carrying a missense variant in the S6 segment of domain I in CaV1.3 that results in an electrophysiological mixed effect with a tendency to a GOF of the channel (Pinggera et al., 2017). Another study by Rinné et al. describes a family in which the p.Arg930His variant in CACNA1D cosegregated with a phenotypic variable syndrome of sinus node dysfunction, focal epilepsy and attention deficit hyperactivity disorder (Rinné et al., 2022). However, the interpretation of this variant is intensely debated in genetic databases such as ClinVar, as its frequency is 1:4000 in gnomAD and the functional electrophysiological characterisation produced a heterogenic isoform‐specific GOF or LOF effect. The very same variant was also reported in a patient with primary aldosteronism, seizures and neurological abnormalities in a study on monogenic hypertension in China (Bao et al., 2020). Taken together, there is limited evidence for an epilepsy phenotype in patients carrying missense GOF variants in CACNA1D. Based on the few cases reported in the literature, CACNA1D should be treated as a candidate gene for epilepsy, especially in the context of additional developmental delay.
Even though both genes are expressed in the brain and heart and are associated with cardiac arrhythmia, we could not find evidence in the literature for an involvement of CACNA1C or CACNA1D in sudden unexpected death in epilepsy patients (SUDEP), although CACNA1C variants had been described in cases of sudden unexpected death in youth (Narula et al., 2015). This is not surprising as the currently preferred pathophysiological explanation of SUDEP is a central dysregulation leading to secondary cardiac arrhythmia based on the results of the MORTEMUS study (Ryvlin et al., 2013).
CaV1.2 and CaV1.3 channels differ in their electrophysiological properties (Helton et al., 2005; Koschak et al., 2001; Xu & Lipscombe, 2001). The dissection of the individual function of channels in the brain is difficult and given the overlapping expression patterns and the lack of specific blockers for the subunits can only be obtained by KO models, which on the other hand can imply compensatory mechanisms (Jurkovičová‐Tarabová et al., 2012). CaV1.2 is involved in long‐term potentiation and spatial memory formation in the hippocampus and modulates the spiking behaviour of neurons (Lacinova et al., 2008; Moosmang et al., 2005). Knockout of CaV1.3 enhances excitability in principal neurons but reduces long‐term potentiation in the basal complex of the amygdala (McKinney et al., 2009). Several studies focused on the function of L‐type channels in the nigrostriatal network. Here, both channels can modulate the spiking and pacemaking behaviour of neurons (Guzman et al., 2009; Olson et al., 2005).
Pharmacologically, L‐type calcium channels are targeted by phenylalkylamines, such as verapamil, benzothiazepines, such as diltiazem, dihydropyridines, such as nimodipine and to some extent by flunarizine (Hockerman et al., 2000; Tytgat et al., 1988; Xu & Lipscombe, 2001). In a patient carrying the p.Gly402Ser GOF variant in CACNA1C in a mosaic pattern, suffering from Timothy syndrome with prominent psychiatric disease and without a history of seizures, verapamil has been used as a treatment with some effect on the cardiac but not on the neuropsychiatric phenotype of the patient (Gershon et al., 2014). Diyhydropyridines, which are commonly used drugs to treat high blood pressure, have been proposed as a possible treatment option for GOF variants in CACNA1C (Marcantoni et al., 2020). However, at the time of submitting this article, we could not find any case description in which dihydropyridines were used to treat patients suffering from GOF variants in CACNA1C or CACNA1D. It is noteworthy, that the dihydropyridine nimodipine reduces hippocampal firing in an in vitro model of febrile seizures in wild type and CaV1.3 KO mice and reduces epileptic activity in induced febrile seizures in vivo indicating an important role of CaV1.2 in the generation of febrile seizures (Radzicki et al., 2013). In neurons that were differentiated from induced pluripotent stem cells of patients with Timothy syndrome due to the p.G406R variant in CACNA1C, treatment with nimodipine could rescue the elevated calcium in neurons but not the upregulation of tyrosine hydroxylase, resulting in increased catecholamine production in the patient cell line (Paşca et al., 2011). On the other hand, it is noteworthy that dihydropyridines do not cause CNS side effects in patients treated for arterial hypertonia. Furthermore, vascularly expressed alternatively spliced isoforms are more sensitive to dihydropyridines and their use‐dependent manner of action makes the blockage of L‐type channels by dihydropyridines in the brain less effective (Helton et al., 2005; Welling et al., 1997). Thus, arterial hypotension with an increased risk of syncope would be expected to be a limiting factor in treating patients before a neurological treatment effect is expected. On the contrary, the sensitivity for mutated channels to blocking agents can change in the mutated channel, as can be seen in the p.Val401Leu variant in CACNA1D leading to autism spectrum disorder and epilepsy, in which the channel's affinity for israpidine, another dihydropyridine with an extensively higher affinity to CaV1.3 than CaV1.2, is increased in vitro in comparison to the wild type channel (Koschak et al., 2001; Pinggera et al., 2017). Thus, a drug that would not have an extensive effect in healthy individuals might have an effect in patients carrying GOF variants.
To our knowledge, activators of the channel are not available for clinical use. The applicability of commonly used activators in vitro, such as BayK8644, is limited due to a dystonic CNS‐mediated effect (Bourson et al., 1989). A targeted therapy for patients carrying LOF variants is thus currently not available (see also review in Zamponi et al., 2015).
3. P/Q‐TYPE CALCIUM CHANNELS (CaV2.1)
CACNA1A encodes for the P/Q‐type channel CaV2.1, which is ubiquitously distributed in the brain with high expression in cerebellar and hippocampal neurons (Ludwig et al., 1997; Schlick et al., 2010; Westenbroek et al., 1995). CaV2.1 channels are expressed in both inhibitory (Althof et al., 2015; Zaitsev et al., 2007) and excitatory cells (Althof et al., 2015).
CAG expansions in this gene lead to spinocerebellar ataxia type 6 (Zhuchenko et al., 1997). Missense and nonsense variants in CACNA1A have been identified as disease‐causing in a spectrum of disorders that are often distinct between GOF and LOF variants and can in both cases imply an epileptic phenotype and in most cases, a cerebellar phenotype: GOF variants have been identified in non‐epileptic and epileptic paroxysmal disorders such as familial hemiplegic migraine type 1 (FHM1) and alternating hemiplegia of childhood as well as in patients suffering from an epileptic encephalopathy with convulsive seizures often with overlapping phenotypes of the three entities (Chan et al., 2008; Ducros et al., 2001; Le Roux et al., 2021; Stam et al., 2009; Zangaladze et al., 2010). Patients with a leading CACNA1A GOF epileptic encephalopathy show often intractable seizures with frequent status epilepticus, developmental delay with autistic features, ataxia and hemiplegic attacks (Le Roux et al., 2021).
The electrophysiological consequences of GOF variants in CACNA1A have mostly been studied in the context of FHM1, in which hemiplegic attacks occur due to a cortical spreading depolarisation, that causes transient neuronal dysfunction (for review see Pietrobon & Moskowitz, 2014). As an extraordinary example, the recurrent p.Ser218Leu missense variant in CACNA1A leads to a characteristic phenotype of FHM1 with unprovoked seizures and brain oedema after minor head trauma with subsequent seizures (Chan et al., 2008; Stam et al., 2009; Zangaladze et al., 2010). Homozygous p.Ser218Leu knock‐in mice carrying this variant show a similar phenotype to humans with recurrent spontaneous epileptic seizures and often die after epileptic seizures, possibly caused by spreading depolarisations to the brainstem (Loonen et al., 2019; van den Maagdenberg et al., 2010). As expected for familial hemiplegic migraine, the threshold for eliciting cortical spreading depression is lower in these mice (van den Maagdenberg et al., 2010). Due to the shift of the activation and inactivation curves to more hyperpolarised potentials, the basal concentration of calcium in neurons is elevated and leads to stronger and more active excitatory synapses and increased short‐term depression (Di Guilmi et al., 2014).
Monoallelic LOF variants in CACNA1A have been associated with an often overlapping disease spectrum of episodic ataxia type 2 (EA2), non‐episodic, chronic ataxia and cerebellar dysfunction, cognitive impairment and ADHD. Whereas the associated epileptic phenotype with predominant absence seizures is often milder compared to the GOF phenotype, cases of severe DEE have been described (Damaj et al., 2015; Imbrici et al., 2004; Jiang et al., 2019; Jouvenceau et al., 2001; Le Roux et al., 2021; Rajakulendran et al., 2010; Stendel et al., 2020). Furthermore, several missense variants that have not been electrophysiologically characterised have been found in patients with congenital ataxia or EA2 who often also exhibit a seizure phenotype (Byers et al., 2016; Gur‐Hartman et al., 2021; Travaglini et al., 2017). The phenotype in patients carrying LOF variants is in many cases strikingly consistent with that of CACNA1A knockout (KO) mice which exhibit a prominent ataxia and dystonia and absence seizures with behavioural arrest and spike–wave discharges in EEG (Jun et al., 1999; Llinas et al., 2007; Song et al., 2004). Other mouse models for EA2, such as tottering and leaner, which carry missense or splice variants resulting in a LOF of CaV2.1, show a comparable phenotype with ataxia, absence and convulsive seizures (Fletcher et al., 1996; Jun et al., 1999; Noebels & Sidman, 1979; Wakamori et al., 1998).
Absence seizures go along with oscillations in the thalamo‐cortical network between cortex, inhibitory neurons in the reticular thalamic nucleus and thalamo‐cortical projection neurons. These oscillations can be triggered both in cortex and in thalamus. They rely on the rebound burst firing mediated by T‐type channels in the thalamus and cortex, although these mechanistic details have been critically discussed recently (McCafferty et al., 2018; for review and discussion see Huguenard, 2019 and Crunelli et al., 2020). The global dysfunction of CaV2.1 function in tottering mice triggers a compensatory upregulation of T‐type currents, but not the mRNA in thalamo‐cortical projection neurons, potentially tipping the balance in the thalamo‐cortical loop towards the generation of absence seizures (Zhang et al., 2002). Interestingly, the selective knockout of CaV2.1 in cortical interneurons is sufficient to generate absence and convulsive seizures without eliciting an ataxic or dystonic phenotype by disrupting the GABA release from parvalbumin positive fast‐spiking interneurons. This is consistent with the impaired feedforward inhibition of thalamic projections to layer IV of the cortex in tottering mice mediated by these interneurons (Rossignol et al., 2013; Sasaki et al., 2006). Likewise, the isolated disruption of CaV2.1 in murine layer VI pyramidal neurons is also sufficient to elicit an absence seizure phenotype with compensatory presynaptically driven upregulation of T‐type currents in thalamic relay and reticular neurons (Bomben et al., 2016). In other cell types, the loss of CaV2.1 is compensated by other calcium channels, especially CaV2.2 channels, partially preserving their function in the release of neurotransmitters (Cao & Tsien, 2005; Jun et al., 1999; Qian & Noebels, 2000; Rossignol et al., 2013). Knockout of CaV2.1 channels in adult mice still elicits an absence seizure phenotype indicating that the underlying pathophysiology stems from these direct electrophysiological changes and is not due to secondary structural defects in network architecture (Miao et al., 2020). Crossbreeding tottering mice with KCNA1 KO mice, which as well show a seizure phenotype, masks the absence seizures in these mice. The same effect could be observed when blocking KV1.1 channels with 4‐aminopyridine (Glasscock et al., 2007).
There are two established treatments in patients with EA2 carrying LOF variants, the carboanhydrase inhibitor acetazolamide and the potassium channel blocker 4‐aminopyridine, with evidence from randomised clinical trials (Muth et al., 2021; Strupp, Kalla, et al., 2011; Strupp, Thurtell, et al., 2011). Topiramate, which is also a partial carboanhydrase inhibitor, showed the best efficacy for treating seizures in a mixed cohort of GOF and LOF patients with CACNA1A‐associated epilepsy, whereas Levetiracetam, Lamotrigine and Valproate have also been reported as effective (Byers et al., 2016; Le Roux et al., 2021). Single cases have been rendered seizure‐free by ACTH or pyridoxine (Du et al., 2017; Le Roux et al., 2021).
4. N‐TYPE CALCIUM CHANNELS (CaV2.2)
N‐type or CaV2.2 channels, encoded by CACNA1B, have been proposed to be potentially involved in genetic generalised epilepsy by the Epi25 Collaborative and the Epi4K Consortium (Epi25 Collaborative, 2021; Epi4K Consortium & Epilepsy Phenome/Genome Project, 2017). In addition, Gorman et al. described three families in which bi‐allelic predicted LOF variants due to protein truncation or nonsense‐mediated decay of CACNA1B led to a developmental and epileptic encephalopathy with a hyperkinetic movement disorder. Epilepsy often began with epileptic spasms and was regularly accompanied by a developmental regression. A majority of the patients died at a young age (Gorman et al., 2019). Contrary to human cases, CaV2.2 knockout mice display neither seizures nor hyperkinetic movement disorder, but the reduced response to sensory stimuli and pain, hyperaggressive behaviour, altered sleep architecture and hyperactive behaviour (Beuckmann et al., 2003; Kim et al., 2009; Kim, Jun, et al., 2001). CaV2.2 is widely expressed in the nervous system including the cortex, hippocampus and cerebellum (Fujita et al., 1993; Jones et al., 1997; Ludwig et al., 1997; Westenbroek et al., 1992). On a subcellular level, CaV2.2 channels are expressed in dendrites, as well as presynaptic nerve terminals where they mediate neurotransmitter release together with CaV2.1 (Hirning et al., 1988; Westenbroek et al., 1992). During development, the role of CaV2.2 in neurotransmitter release is predominantly taken over by CaV2.1 channels, which are located more closely to the synaptic release machinery (Iwasaki et al., 2000; Wu et al., 1999).
Due to the nature of a homozygous loss of CACNA1B as a cause of disease, it is hard to derive a specific therapy for the disease as no functional CaV2.2 channels will be expressed in these patients. In the small cohort of Gorman et al., no anti‐seizure medication did stand out in providing seizure control in the 6 patients described (Gorman et al., 2019). Further research needs to be conducted to get more insights into the clinical course of disease in these patients and successful treatment options.
5. R‐TYPE CALCIUM CHANNELS (CaV2.3)
CACNA1E encodes the R‐type calcium channel CaV2.3, the third presynaptic calcium channel in this subgroup. Missense variants in this gene have been associated with a complex phenotype including largely pharmacoresistant early onset infantile epileptic encephalopathy with severe developmental delay. Helbig et al. reported 30 patients with de novo missense variants. Additional symptoms were congenital contractures with arthrogryposis, perinatal hypotonia and extrapyramidal movement disorders. Three patients with variants predicted to be LOF by nonsense‐mediated decay had a milder phenotype (Helbig, Lauerer, et al., 2018). A second cohort of patients with developmental delay and no epileptic phenotype was described later (Royer‐Bertrand et al., 2021). A wide phenotypic spectrum can be observed in patients with the same variant, implicating other confounding factors, as has been argued before for monogenic epilepsies in general (Campbell et al., 2022). Seizures in CACNA1E encephalopathy are often pharmacoresistant, however, Topiramate, an antiseizure drug (ASM) acting on CaV2.3 channels, has been identified as an effective treatment option in some patients (Helbig, Lauerer, et al., 2018).
Interestingly, most disease‐causing missense variants in CACNA1E cluster are in the distal S6 segment of the channel. This mutational hotspot is common in VGCCs, as can be also observed in Timothy syndrome with the common variants p.Gly402Ser and p.Gly406Arg in CACNA1C, as well as disease‐causing variants in the T‐type channel genes CACNA1G and CACNA1I. These variants in the activation gate of the channels usually result in a GOF effect by facilitating channel activation and prolonging the time course of inactivation (Chemin et al., 2018; El Ghaleb et al., 2021; Helbig, Lauerer, et al., 2018; Splawski et al., 2004, 2005).
CaV2.3 is expressed ubiquitously in the brain in both excitatory and inhibitory cells as well as astrocytes (D'Ascenzo et al., 2004; Ludwig et al., 1997; Parajuli et al., 2012; Soong et al., 1993; Weiergräber et al., 2006, 2008; Williams et al., 1994). On a subcellular level, the channel has been detected pre‐ and postsynaptically (Breustedt et al., 2003; Dietrich et al., 2003; Parajuli et al., 2012). However, caution should be exercised when interpreting studies on the localisation of CaV2.3 as the R‐type current does not fully correspond to CaV2.3 channels and the commonly used regimes of blocking this current with SNX‐482 or a low concentration of Ni+ ions are not fully specific for CaV2.3 channels (Bourinet et al., 2001; Kimm & Bean, 2014; Lee et al., 1999; Tottene et al., 2000). Before the description of CACNA1E as an epilepsy gene in humans, a possible involvement of CaV2.3 in the pathophysiology of absence seizures had been discussed based on experiments in KO mice (Weiergräber et al., 2008; Zaman et al., 2011). KO mice show reduced susceptibility to seizure induction with pentylenetetrazol and kainate, but not with 4‐aminopyridine (Weiergräber et al., 2006, 2007). Furthermore, KO animals showed reduced excitotoxicity in the hippocampus after seizure induction with kainate (Weiergräber et al., 2007). R‐type calcium channels are involved in long‐term potentiation and rhythmic firing of neurons (Breustedt et al., 2003; Dietrich et al., 2003; Zaman et al., 2011). CaV2.3 is also a target of CDKL5, a kinase in which variants lead to a similar although not completely identical phenotype as in CACNA1E‐GOF‐DEE patients. Interestingly, the lack of phosphorylation due to a defect in CDKL5 leads to a GOF in CaV2.3 (Sampedro‐Castañeda et al., 2023).
6. T‐TYPE CALCIUM CHANNELS (CaV3.x)
T‐type VGCCs form low‐voltage activated channels that conduct transient calcium currents which are essential for rebound burst firing in a subunit‐specific manner (Huguenard & Prince, 1992; Klöckner et al., 1999; McRory et al., 2001). T‐type channels have been accounted to be crucial for the generation of absence seizures in the network of GABAergic neurons in the reticular thalamic nucleus, excitatory thalamo‐cortical neurons of the thalamic relay nucleus and the cortex (for review see Crunelli et al., 2020). They are essential in the neuronal switch from tonic to burst firing, which is elicited by Ca2+‐mediated low threshold spikes, an important prerequisite for oscillations in the thalamo‐cortical circuitry between somatosensory cortex and the thalamus (Huguenard & Prince, 1992; Kim, Song, et al., 2001). It was recently proposed by experiments in freely moving animals, that T‐type channels are involved in the absence seizure generation in the cortex and reticular thalamic nucleus, but not in thalamo‐cortical relay neurons (McCafferty et al., 2018). Thus, the distinct expression patterns of the three T‐type channel subunits determine their importance in the generation of absence seizures in this network as is outlined below. Although many studies do not distinguish between the three different T‐type channels, the expression pattern is distinct. In rats, CaV3.1 channels are strongly expressed in the lateral geniculate nucleus, especially in thalamo‐cortical projection neurons, but only weakly in the reticular thalamic nucleus, whereas CaV3.2 is strongly expressed in the reticular thalamic nucleus and not in the corpus geniculatum laterale and intralaminar nuclei of the thalamus. CaV3.3 is expressed throughout the whole thalamus (Broicher et al., 2007, 2008). Whereas CaV3.1 is the predominant T‐type calcium channel in the cortex, hippocampal pyramidal neurons express all three channels (Talley et al., 1999; Zhang et al., 2002).
All three T‐type VGCCs have been associated with epilepsy with a different extent of evidence with either GOF or LOF mechanisms, as outlined in detail below. Besides the well‐described pathophysiological role of T‐type channels in absence seizures, they are targets of licenced anti‐seizure medications, which can help as guidance to preferentially use these drugs in patients carrying GOF variants and avoid them in patients carrying LOF variants. Ethosuximide, a pan‐T‐type channel blocker, is a first‐line treatment for absence seizures (Gomora et al., 2001) and zonisamide, which is used in Japan to treat genetic generalised epilepsies, also blocks all three T‐type channels with a preference for CaV3.2 beside voltage‐gated Na+ channels (Matar et al., 2009; Suzuki et al., 1992). Another combined blocker of Na+ and T‐type Ca2+ channels is eslicarbazepine acetate, which has been shown to prevent epileptogenesis in a mouse model of temporal lobe epilepsy (Doeser et al., 2014). Valproic acid, the first line treatment in generalised genetic epilepsy, shows a minor block of T‐type channels (Kelly et al., 1990; Todorovic & Lingle, 1998). Recently, it was also shown that stiripentol, another drug, mainly used in Dravet syndrome, also acts on T‐type calcium channels beside its GABAergic mechanism of action (Riban et al., 2022). Other drugs blocking T‐type channels include pimozide, flunarizine, phytocannabinoids and amiloride (Santi et al., 2002; Tang et al., 1988; for review see also Mirlohi et al., 2022; Zamponi et al., 2015). Another substance, not approved for the use in humans, is Z944, a specifically designed T‐type channel blocker that also modifies T‐type channel expression and shows promising results in mouse models of temporal lobe epilepsy (Casillas‐Espinosa et al., 2015, 2019; Tringham et al., 2012).
6.1. CACNA1G (CaV3.1)
Missense variants in CACNA1G have been identified in patients with DEE with varying severe electroclinical phenotypes with a wide variety of seizure types. Epilepsy and developmental delay are often accompanied by cerebral and/or cerebellar atrophy and digital and facial dysmorphisms (Berecki et al., 2020; Chemin et al., 2018; Kunii et al., 2020). Functional testing of these missense variants in heterologous expression systems frequently revealed mixed GOF and LOF effects, for example, shifting the voltage dependence of both activation and inactivation to more hyperpolarised potentials (Berecki et al., 2020; Chemin et al., 2018; Kunii et al., 2020). In silico modelling pointed to a resulting GOF in neurons for some of these variants (Chemin et al., 2018). Another study detected missense variants in families with juvenile myoclonic epilepsy that did not change the gating parameters of the channel significantly, and have not been entered as pathogenic into ClinVar by other submitters since then, rendering this association rather questionable (Singh et al., 2007).
On the broader spectrum, missense variants in CACNA1G have also been described in cerebellar phenotypes: LOF variants cause autosomal dominant cerebellar ataxia without seizures (Coutelier et al., 2015), and mixed GOF and LOF variants have been described in spinocerebellar ataxia type 42 without seizures (Morino et al., 2015). Furthermore, patients with microdeletions of the 17q21.33 locus, that involve CACNA1G among other genes, show developmental delay and in some cases cerebral atrophy, whereas an epileptic phenotype has not been reported (Bardai et al., 2016; Harbuz et al., 2013; Jewell et al., 2017; Preiksaitiene et al., 2012). This may suggest that a mono‐allelic LOF in CACNA1G is not sufficient to elicit an epileptic phenotype. However, dominant‐negative effects of missense variants leading to the complete disruption of functional CaV3.1 channels cannot be excluded.
Cacna1g KO mice are less prone to the induction of spike‐wave discharges by the application of γ‐butyrolactone and baclofen but not to the induction of cortically induced absence seizures with bicuculine or convulsive seizures with 4‐aminopyridine (Kim, Song, et al., 2001). The knockout of Cana1g in mice abolishes rebound bursts in thalamo‐cortical projection neurons within the ventrobasal complex which were deemed to be essential in generating spike–wave discharges in absence seizures (Kim, Song, et al., 2001). More recent studies could show that selective blockage of T‐type currents in this region of the thalamus, in which CaV3.1 is primarily expressed, does not abolish absence seizures (Crunelli et al., 2020; McCafferty et al., 2018). On the contrary, crossbreeding Cacna1g KO mice with Cacna1a KO mice abolishes the absence seizures that are usually observed upon Cacna1a KO, indicating the importance of CaV3.1 T‐type currents in the generation of absence seizures (Jun et al., 1999; Llinas et al., 2007; Song et al., 2004). Similarly, KO of Cacna1g reduces the phenotypic severity in mouse models of Scn1a and Scn2a encephalopathies and kainate‐induced hippocampal seizures (Calhoun et al., 2016, 2017; Kim, 2015).
Taken together, there are hints for a GOF mechanism of CaV3.1 in CACNA1G variant carriers with reported epilepsy, and that a LOF is protective against absence seizures, although the mechanistic details remain uncertain. However, the heterogenicity of data may complicate the interpretation of functional effects derived from heterologous expression systems for the clinical geneticist and should be followed by in vivo or in silico verification.
6.2. CACNA1H (CaV3.2)
CACNA1H has been proposed as an epilepsy gene first in 2003 when a study in the Chinese Han population found inherited but not co‐segregating missense variants in the gene in patients with childhood absence epilepsy (Chen et al., 2003). Various other reports have observed inherited heterozygous or compound heterozygous missense variants in CACNA1H in patients with an extensive variability of phenotypes including the whole phenotypic spectrum of genetic generalised epilepsy as well as febrile seizures, epilepsy with myoclonic–atonic seizures and temporal lobe epilepsy, which co‐segregated with an epilepsy phenotype only in few families (Becker et al., 2017; Chourasia et al., 2019; Heron et al., 2007; Liang et al., 2006; Vitko, 2005). Many of the detected missense variants alter the gating mechanisms of CaV3.2 channels and lead to either GOF or LOF in the channel in vitro with a tendency of more GOF variants with mostly small effect sizes, but also functional effects on neuronal firing (Becker et al., 2017; Eckle et al., 2014; Heron et al., 2007; Khosravani et al., 2005; Peloquin et al., 2006; Vitko, 2005). In contrast, missense variants in CACNA1H have repeatedly not been identified as enriched in other large cohorts of patients with epilepsy (Chioza et al., 2006; Epi4K Consortium et al., 2013; Epi4K Consortium & Epilepsy Phenome/Genome Project, 2017; Heyne et al., 2019) and the existence of a monogenetic syndrome caused by missense variants in this gene is now strongly debated, leading to the suggestion to exclude the gene from gene panels in clinical testing (Calhoun et al., 2020; Helbig, Riggs, et al., 2018).
Interestingly, a homozygous missense variant leading to an amino acid substitution in the linker between domains III and IV of CaV3.2 has been described to co‐segregate with seizure count and seizure activity in the Genetic Absence Epilepsy in Rats from Strasbourg (GAERS) model (Powell et al., 2009). However, even though the variant showed a splice‐variant specific GOF with accelerated recovery from inactivation when heterologously expressed, animals not carrying this variant still showed seizures, indicating a polygenic basis of disease in this model (Powell et al., 2009). This is underlined by follow‐up experiments of the same group showing that the introduction of this variant into congenic animals derived from a non‐epileptic control strain by crossbreeding could not elicit a seizure phenotype but modified the seizure phenotype in GAERS animals facilitating absence seizures (Casillas‐Espinosa et al., 2023). Epileptogenesis in the hippocampus is largely prevented in Cacna1h KO mice, which do not show neuronal cell loss after the induction of status epilepticus with pilocarpine and are less likely to develop chronic epilepsy (Becker et al., 2008). This fits the population statistics in humans that indicate that CACNA1H is opposing to other voltage‐gated ion channels tolerant to LOF (Calhoun et al., 2020). Also, an additional KO of Cacna1h did not alter the seizure susceptibility and survival in a crossbred Scn1a KO mouse line to a clinically significant amount (Calhoun et al., 2020).
Taken together, there is currently no clear evidence that missense variants in CACNA1H contribute to epilepsy in affected individuals, but experiments in GAERS suggest that a homozygous GOF variant facilitates absence seizures in this model. Therefore, the current state of research cannot fully exclude CACNA1H to be a candidate gene to contribute to the polygenic burden in generalised genetic epilepsies, especially with missense variants that lead to an electrophysiological GOF in the channel. However, a monogenetic cause of disease should not be proclaimed (Becker et al., 2017; Calhoun et al., 2020; Powell et al., 2009). In addition, the KO of CACNA1H and inhibitors of this channel can prevent epileptogenesis in the hippocampus so that blockers of CaV3.2 channels may be a promising anti‐epileptogenic future therapeutic option (Becker et al., 2008; Doeser et al., 2014).
6.3. CACNA1I (CaV3.3)
Recently, four different de novo missense variants in CACNA1I, which encodes the CaV3.3 channel have been described in patients with a neurodevelopmental syndrome of varying severity and epilepsy. In three patients, de novo variants led to a severe phenotype with global developmental delay, hypotonia, cortical blindness and epilepsy during the first 2 years of life. Another variant co‐segregated in a family with cognitive impairment of affected individuals, and one individual of which developed seizures with 58 years of age. However, in this patient, no MRI but only a CT scan was obtained to rule out other causes of structural epilepsy, which renders an association to the genetic findings questionable. This p.I860M variant showed the weakest electrophysiological alterations of all variants tested, but was affecting the same amino acid as the p.I860N variant identified in more severe cases. All variants induced a GOF by shifting the activation curve and the window current to more hyperpolarised potentials, prolonging inactivation, and in some variants also deactivation. The shift in window current leads to an increase in firing and to a slow oscillation mode when expressed in chromaffin cells. Consistent with a GOF effect, in one severely affected individual, ethosuximide improved seizure control (El Ghaleb et al., 2021). Beyond epilepsy, variants in CACNA1I with a mixed effect in heterologous expression systems have been identified in familial hemiplegic migraine, whereas LOF variants were detected in schizophrenia (Andrade et al., 2016; Maksemous et al., 2022).
CACNA1I thus appears to be a promising epilepsy gene in individuals carrying de novo GOF variants given the remarkable work by El Ghaleb et al. (2021). Further case descriptions are necessary to establish reproducibility for this gene in epilepsy.
7. AUXILIARY SUBUNITS
7.1. CACNB4
CACNB4 encodes the β4 subunit of the calcium channel complex. The first description of CACNB4 variants in epilepsy in humans was in 1999, when a group of researchers screened patients with familial epilepsy and ataxia for variants in this gene. This was influenced by the mouse model lethargic, which carries a bi‐allelic LOF Cacnb4 variant and exhibits an absence seizure phenotype and ataxia (Burgess et al., 1997). The authors identified a missense and a nonsense variant, leading to an early stop codon that co‐segregated in the three affected families with disease (Escayg et al., 2000). However, the identified p.Cys104Phe missense variant did not lead to changes in electrophysiological recordings and has meanwhile been found in gnomAD in 117 cases, whereas the p.Arg482* variant leading to a distal stop codon has not been annotated in ClinVar for a second time but in gnomAD in one healthy individual. Another study identified a heterozygous frameshift variant in CACNB4 in 3 individuals with epilepsy, predicted to lead to nonsense‐mediated decay. However, this study lacks further clinical data and information on segregation for the participants, indicating questionable results (Naseer et al., 2022). Protein‐truncating variants in CACNB4 are not enriched in epilepsy cases in the Epi25 cohort and the gnomAD constraint metrics indicate that the gene is not intolerant to loss of function with a probability of being loss‐of‐function intolerant (pLI) score of 0.03, and the observed/expected (oe/e) constraint score of 0.28 (confidence interval 0.17–0.49) (Chen et al., 2022; Epi25 Collaborative et al., 2023; Lek et al., 2016). This renders disease association unlikely for all variants described.
De Bagneaux et al. suggested the homozygous p.Leu125Pro missense variant in CACNB4 to be disease‐causing in two siblings born of consanguineous parents with severe developmental delay, hypotonia, athetoid–dystonic movements, cerebellar atrophy and focal tonic seizures. The mutated protein failed to be trafficked in presynaptic terminals and axon hillocks of transfected hippocampal neurons and activity‐dependent nuclear targeting was inhibited and led to a LOF with decreased current density and altered inactivation kinetics when co‐expressed with CaV2.1 in a heterologous expression system. Furthermore, the mutant proteins did not bind to other protein interaction partners, such as the serin‐threoine kinase TNIK in contrast to the wild‐type protein (Coste de Bagneaux et al., 2020).
Before the description of biallelic variants, CACNB4 had been flagged by the ClinGen consortium to be a disputed epilepsy gene as there was a lack of replication after the initial report by Helbig, Riggs, et al. (2018). Furthermore, ultrarare variants in this gene were not enriched in a cohort of patients with neurodevelopmental disorders with epilepsy (Heyne et al., 2019).
Taken together, there is very limited evidence for a monogenetic epilepsy syndrome involving CACNB4 given the lack of reproducibility and the few cases reported, especially with mono‐allelic variants. The overlap of severe phenotypes in the bi‐allelic LOF of CACNB4 in mouse and humans points towards a possible recessively inherited disease but should be backed up by additional case descriptions.
7.2. CACNA2D1
CACNA2D1 encodes the α2δ1 subunit of VGCCs. Recently, Dahimene et al. reported two patients with biallelic CACNA2D1 variants resulting in biallelic LOF of the α2δ1 subunit that led to a DEE phenotype with cortical visual impairment, severe developmental delay, hypotonia, a movement disorder with spasticity, choreiform movements and orofacial dyskinesia, facial dysmorphism, cerebral atrophy with an emphasis on corpus callosum and an epilepsy syndrome with generalised absence or hemiclonic seizures. CACNA2D1 variants in these patients were either predicted to be LOF by nonsense‐mediated decay due to an early frameshift variant or failed to fulfil their physiological function of enhancing calcium currents and channel expression when co‐expressed with CaV2.2 (Dahimene et al., 2022).
A mono‐allelic predicted LOF variant has been reported in a patient with infantile epileptic spasms (formerly known as West syndrome) without developmental delay (Hino‐Fukuyo et al., 2015). Patients with a microdeletion of the 7q21.11 locus involving the CACNA2D1 gene exhibit developmental delay or reduced cognitive functions and in many but not all cases epilepsy. However, this recurrently deleted sequence contains several candidate genes expressed in CNS, so a clear association with CACNA2D1 remains unclear (Mazzaschi et al., 2013; Mefford et al., 2011; Siddique et al., 2017; Vergult et al., 2015). Another patient with treatment‐resistant epilepsy, intellectual disability and polymicrogyria carried a balanced reciprocal translocation 46,X,t(X;7)(p10;q21.2) potentially disrupting the CACNA2D1 gene (Vergult et al., 2015). Furthermore, a monoallelic intronic variant predicted to result in a splicing defect was reported in a patient with craniofacial dysmorphism, language delay and epilepsy (Valentino et al., 2021). However, Dahimene et al. raise a serious doubt regarding the pathogenicity of monoallelic LOF variants with regard to epilepsy as the previously reported patients show phenotypic heterogenicity. They furthermore argue that gnomAD and LOVD list various likely LOF variants. Contradicting the authors' theory is that the expected frequency of LOF variants in CACNA2D1 in gnomAD is lower than expected, resulting in a probability of being loss‐of‐function intolerant (pLI) score of 1 and the observed/expected (oe/e) constraint score of 0.15 (confidence interval 0.09–0.24) (Chen et al., 2022; Lek et al., 2016). Also, phenotypic heterogeneity is not uncommon in channelopathies even within patients carrying the same variants (Martins Custodio et al., 2023; Royer‐Bertrand et al., 2021). On the other hand, Cacna2d1 KO mice exhibit a mild cardiac phenotype and sensory deficits whereas a seizure phenotype has not been reported (Fuller‐Bicer et al., 2009; Patel et al., 2013).
Further supporting the pathogenicity of an LOF of CACNA2D1, two patients, seropositive for antibodies in serum and CSF directed against the α2δ1 subunit exhibited autoimmune encephalitis with psychiatric alteration and seizures. Interestingly, in vitro electrophysiological recordings led to the conclusion that these antibodies result in a loss of protein function downstream of enhancing calcium currents, reducing the frequency of miniature excitatory and inhibitory post‐synaptic currents in an autaptic hippocampal neuronal culture model, but not reducing calcium currents at the soma (Lee et al., 2021).
The α2δ1 subunit has a primary expression pattern in cortex and hippocampus (Hobom et al., 2000). A LOF in this protein is thus expected to result in a functional loss in calcium currents in these cells, potentially mimicking the pathomechanisms of epileptic encephalopathy with a LOF in co‐expressed VGCC α‐subunits, most likely CaV2.1 and CaV2.3 (Hobom et al., 2000). A compensatory upregulation of other α2δ‐subunits could not be observed in Cacna2d1 KO mice (Fuller‐Bicer et al., 2009; Patel et al., 2013). Beyond their function as calcium channel subunits, α2δ‐subunits are essential for establishing the physiological architecture of glutamatergic synapses via their function as thrombospondin receptors and interact with NMDA receptors (Chen et al., 2018; Eroglu et al., 2009; Schöpf et al., 2021).
Taken together, a LOF in CACNA2D1 might lead to an epileptic phenotype although the evidence that a monogenetic LOF is sufficient to lead to a clinical phenotype remains open. Interestingly, a mouse model overexpressing Cacna2d1 also shows a seizure phenotype with the behavioural arrest that responds to the T‐type calcium channel blocker ethosuximide, making GOF missense variants interesting candidates for causing epilepsy (Faria et al., 2017).
α2δ‐subunits are targeted by gabapentin and pregabalin, anti‐seizure medications that nowadays are more often used to treat neuropathic pain (Bian et al., 2006; Gee et al., 1996). Even though no clinical evidence is present, we suggest therefore to avoid these drugs in patients carrying LOF variants in genes encoding α2δ‐subunits as the additional blockage of these proteins may result in clinical worsening as can be seen in other channelopathies resulting from an LOF such as SCN1A‐associated Dravet syndrome and SCN2A LOF‐DEE (Brunklaus et al., 2012; Wolff et al., 2017).
7.3. CACNA2D2
CACNA2D2 encodes for the auxiliary subunit α2δ2, which enhances divalent cation currents through VGCCs (Brodbeck et al., 2002; Hobom et al., 2000). Long before the discovery of disease association in man, spontaneous variants in Cacna2d2 had been associated with epilepsy and ataxia in mutated mouse lines: Truncating variants in Cacna2d2 have been found in the absence seizure mouse models ducky and ducky2j (Barclay et al., 2001; Brodbeck et al., 2002). In the entla mouse line, a 38 kb duplication in Cacna2d2 results in an electrophysiological loss of function of the protein (Brill et al., 2004). In all three mouse lines, homozygous animals exhibit ataxia, paroxysmal dyskinesia and seizures with spike–wave activity in EEG recordings, which is consistent with the phenotype in homozygous knockout animals (Barclay et al., 2001; Brill et al., 2004; Ivanov et al., 2004). Homozygous loss of Cacna2d2 results in a reduced current of CaV2.1 and other VGCCs, which explains the similar phenotype to CACNA1A LOF models such as tottering (Brodbeck et al., 2002; Fletcher et al., 1996; Jun et al., 1999; Noebels & Sidman, 1979; Wakamori et al., 1998). In addition, Cacna2d2 dysfunction can alter neuronal morphology, as mice homozygous for the truncating ducky allele show reduced dendritic arbours of Purkinje cells (Brodbeck et al., 2002).
Consistent with the phenotypic similarities in the mouse models, the expression of α2δ2 is widely overlapping with the expression of CaV2.1 and CaV2.3 with a hotspot in the Purkinje cell layer of the cerebellum, which corresponds with the cerebellar phenotype in mice and humans (Barclay et al., 2001; Brodbeck et al., 2002; Butler et al., 2018; Edvardson et al., 2013; Hobom et al., 2000; Pippucci et al., 2013; Punetha et al., 2019; Valence et al., 2019). α2δ2 is furthermore expressed in the thalamus, hypothalamus, the olfactory bulb, the habenulae, the superior and inferior colliculus, the striatum and the septal nuclei with a minor expression in the cortex. There is opposing data on the expression in the hippocampus (Barclay et al., 2001; Hobom et al., 2000).
Years after the first description of these mouse phenotypes, homozygous and compound‐heterozygous predicted LOF or missense variants have been described in patients with a DEE phenotype with multifocal tonic, atonic, tonic–clonic and absence seizures refractory to anti‐seizure medications. Patients regularly showed dyskinesia, cerebellar atrophy, ataxia and atypical eye movements consistent with the murine phenotype (Butler et al., 2018; Edvardson et al., 2013; Pippucci et al., 2013; Punetha et al., 2019). The monogenetic origin of the disease was debated in the first place, as the first two reported families additionally carried rare homozygous variants in the CELSR3 gene (Edvardson et al., 2013; Pippucci et al., 2013) and in the third family a common heterozygous variant in CELSR3 was present (Butler et al., 2018). Later on, additional patients with homozygous variants in CACNA2D1, but not CELSR3, with similar (Punetha et al., 2019) and milder phenotypes, namely congenital ataxia with one febrile seizure, have been described (Valence et al., 2019). It is noteworthy that missense variants in CELSR3, which encodes the cadherin egf lag seven‐pass g‐type receptor 3, a protein involved in axonal guidance, have also been associated with febrile seizures in a Chinese cohort (Li et al., 2022).
Taken together, bi‐allelic LOF variants in CACNA2D2 seem to be sufficient to explain the phenotype of the reported patients given the concordance with the murine phenotypes in terms of ataxia and absence seizures. The experimental in vitro evidence of a reduction of calcium channel currents by the co‐expression of the pathological p.L1040P CACNA2D2 allele found in patients with CaV1.2 and CaV2.2 in Xenopus laevis oocytes and the reduction of calcium currents in Purkinje cells in the ducky and ducky2j mouse models supports this conclusion, with the constraint that not all missense variants found in humans have been functionally characterised (Brodbeck et al., 2002; Donato et al., 2006; Edvardson et al., 2013). Nevertheless, it cannot be fully excluded that variants in CELSR3 serve as modifying factors of disease, given its role in axonal guidance and neuronal development. Regarding a targeted therapy, we propose to avoid drugs that act on α2δ‐subunits in these patients, as with LOF variants in CACNA2D1, although to our knowledge no clinical evidence is present for this suggestion.
8. SUMMARY
All voltage‐gated calcium channel α‐subunits and some of their auxiliary subunits have been associated with epilepsy in the past with often strikingly overlapping phenotypes in murine models. While the disease association has been reproduced in some of the genes, over time some of these associations have been loosened, such as for CACNA1H and CACNB4. Further research is needed to extend our knowledge on pathophysiology in all VGCC‐associated syndromes to derive specific therapies for the often severely disabled patients.
AUTHOR CONTRIBUTIONS
Robert Johannes Lauerer‐Braun: Conceptualization; data curation; writing – original draft. Holger Lerche: Conceptualization; supervision; writing – original draft.
FUNDING INFORMATION
The study was funded by the Bundesministerium für Bildung und Forschung, Treat‐ION, grant # 01GM2210A and the Deutsche Forschungsgemeinschaft, FOR‐2715, grants Le1030/16‐2 and Le1030/23‐1 and the Else Kröner Fresenius Stiftung through the EKFS Kolleg Precise.net.
CONFLICT OF INTEREST STATEMENT
The authors have no conflict of interest to disclose.
PEER REVIEW
The peer review history for this article is available at https://www.webofscience.com/api/gateway/wos/peer‐review/10.1111/jnc.15983.
ACKNOWLEDGMENTS
Open Access funding enabled and organized by Projekt DEAL.
Lauerer, R. J. , & Lerche, H. (2024). Voltage‐gated calcium channels in genetic epilepsies. Journal of Neurochemistry, 168, 3853–3871. 10.1111/jnc.15983
This Review is part of the Special issue “Ion Channels and Genetic Epilepsy”.
DATA AVAILABILITY STATEMENT
Data sharing does not apply to this article as no datasets were generated or analysed during the current study.
REFERENCES
- Althof, D. , Baehrens, D. , Watanabe, M. , Suzuki, N. , Fakler, B. , & Kulik, A. (2015). Inhibitory and excitatory axon terminals share a common nano‐architecture of their Cav2.1 (P/Q‐type) Ca(2+) channels. Frontiers in Cellular Neuroscience, 9, 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade, A. , Hope, J. , Allen, A. , Yorgan, V. , Lipscombe, D. , & Pan, J. Q. (2016). A rare schizophrenia risk variant of CACNA1I disrupts Ca(V)3.3 channel activity. Scientific Reports, 6, 34233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baig, S. M. , Koschak, A. , Lieb, A. , Gebhart, M. , Dafinger, C. , Nürnberg, G. , Ali, A. , Ahmad, I. , Sinnegger‐Brauns, M. J. , Brandt, N. , Engel, J. , Mangoni, M. E. , Farooq, M. , Khan, H. U. , Nürnberg, P. , Striessnig, J. , & Bolz, H. J. (2011). Loss of Ca(v)1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nature Neuroscience, 14, 77–84. [DOI] [PubMed] [Google Scholar]
- Bao, M. , Li, P. , Li, Q. , Chen, H. , Zhong, Y. , Li, S. , Jin, L. , Wang, W. , Chen, Z. , Zhong, J. , Geng, B. , Fan, Y. , Yang, X. , & Cai, J. (2020). Genetic screening for monogenic hypertension in hypertensive individuals in a clinical setting. Journal of Medical Genetics, 57, 571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barclay, J. , Balaguero, N. , Mione, M. , Ackerman, S. L. , Letts, V. A. , Brodbeck, J. , Canti, C. , Meir, A. , Page, K. M. , Kusumi, K. , Perez‐Reyes, E. , Lander, E. S. , Frankel, W. N. , Gardiner, R. M. , Dolphin, A. C. , & Rees, M. (2001). Ducky mouse phenotype of epilepsy and ataxia is associated with mutations in the Cacna2d2 gene and decreased calcium channel current in cerebellar Purkinje cells. The Journal of Neuroscience, 21, 6095–6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardai, G. , Lemyre, E. , Moffatt, P. , Palomo, T. , Glorieux, F. H. , Tung, J. , Ward, L. , & Rauch, F. (2016). Osteogenesis Imperfecta type I caused by COL1A1 deletions. Calcified Tissue International, 98, 76–84. [DOI] [PubMed] [Google Scholar]
- Becker, A. J. , Pitsch, J. , Sochivko, D. , Opitz, T. , Staniek, M. , Chen, C. C. , Campbell, K. P. , Schoch, S. , Yaari, Y. , & Beck, H. (2008). Transcriptional upregulation of Cav3.2 mediates epileptogenesis in the pilocarpine model of epilepsy. The Journal of Neuroscience, 28, 13341–13353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, F. , Reid, C. A. , Hallmann, K. , Tae, H. S. , Phillips, A. M. , Teodorescu, G. , Weber, Y. G. , Kleefuss‐Lie, A. , Elger, C. , Perez‐Reyes, E. , Petrou, S. , Kunz, W. S. , Lerche, H. , & Maljevic, S. (2017). Functional variants in HCN4 and CACNA1H may contribute to genetic generalized epilepsy. Epilepsia Open, 2, 334–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berecki, G. , Helbig, K. L. , Ware, T. L. , Grinton, B. , Skraban, C. M. , Marsh, E. D. , Berkovic, S. F. , & Petrou, S. (2020). Novel missense CACNA1G mutations associated with infantile‐onset developmental and epileptic encephalopathy. International Journal of Molecular Sciences, 21, 6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuckmann, C. T. , Sinton, C. M. , Miyamoto, N. , Ino, M. , & Yanagisawa, M. (2003). N‐type calcium channel α1B subunit (CaV2.2) knock‐out mice display hyperactivity and vigilance state differences. The Journal of Neuroscience, 23, 6793–6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian, F. , Li, Z. , Offord, J. , Davis, M. D. , McCormick, J. , Taylor, C. P. , & Walker, L. C. (2006). Calcium channel alpha2‐delta type 1 subunit is the major binding protein for pregabalin in neocortex, hippocampus, amygdala, and spinal cord: An ex vivo autoradiographic study in alpha2‐delta type 1 genetically modified mice. Brain Research, 1075, 68–80. [DOI] [PubMed] [Google Scholar]
- Bichet, D. , Cornet, V. , Geib, S. , Carlier, E. , Volsen, S. , Hoshi, T. , Mori, Y. , & De Waard, M. (2000). The I‐II loop of the Ca2+ channel α1 subunit contains an endoplasmic reticulum retention signal antagonized by the β subunit. Neuron, 25, 177–190. [DOI] [PubMed] [Google Scholar]
- Boczek, N. J. , Best, J. M. , Tester, D. J. , Giudicessi, J. R. , Middha, S. , Evans, J. M. , Kamp, T. J. , & Ackerman, M. J. (2013). Exome sequencing and systems biology converge to identify novel mutations in the L‐type calcium channel, CACNA1C, linked to autosomal dominant long QT syndrome. Circulation. Cardiovascular Genetics, 6, 279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boczek, N. J. , Miller, E. M. , Ye, D. , Nesterenko, V. V. , Tester, D. J. , Antzelevitch, C. , Czosek, R. J. , Ackerman, M. J. , & Ware, S. M. (2015). Novel Timothy syndrome mutation leading to increase in CACNA1C window current. Heart Rhythm, 12, 211–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomben, V. C. , Aiba, I. , Qian, J. , Mark, M. D. , Herlitze, S. , & Noebels, J. L. (2016). Isolated P/Q calcium channel deletion in layer VI corticothalamic neurons generates absence epilepsy. The Journal of Neuroscience, 36, 405–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourinet, E. , Stotz, S. C. , Spaetgens, R. L. , Dayanithi, G. , Lemos, J. , Nargeot, J. , & Zamponi, G. W. (2001). Interaction of SNX482 with domains III and IV inhibits activation gating of α1E (CaV2.3) calcium channels. Biophysical Journal, 81, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourson, A. , Moser, P. C. , Gower, A. J. , & Mir, A. K. (1989). Central and peripheral effects of the dihydropyridine calcium channel activator BAY K 8644 in the rat. European Journal of Pharmacology, 160, 339–347. [DOI] [PubMed] [Google Scholar]
- Bozarth, X. , Dines, J. N. , Cong, Q. , Mirzaa, G. M. , Foss, K. , Lawrence Merritt, J., 2nd , Thies, J. , Mefford, H. C. , & Novotny, E. (2018). Expanding clinical phenotype in CACNA1C related disorders: From neonatal onset severe epileptic encephalopathy to late‐onset epilepsy. American Journal of Medical Genetics. Part A, 176, 2733–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breustedt, J. , Vogt, K. E. , Miller, R. J. , Nicoll, R. A. , & Schmitz, D. (2003). Alpha1E‐containing Ca2+ channels are involved in synaptic plasticity. Proceedings of the National Academy of Sciences of the United States of America, 100, 12450–12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brill, J. , Klocke, R. , Paul, D. , Boison, D. , Gouder, N. , Klugbauer, N. , Hofmann, F. , Becker, C. M. , & Becker, K. (2004). Entla, a novel epileptic and ataxic Cacna2d2 mutant of the mouse. The Journal of Biological Chemistry, 279, 7322–7330. [DOI] [PubMed] [Google Scholar]
- Brodbeck, J. , Davies, A. , Courtney, J. M. , Meir, A. , Balaguero, N. , Canti, C. , Moss, F. J. , Page, K. M. , Pratt, W. S. , Hunt, S. P. , Barclay, J. , Rees, M. , & Dolphin, A. C. (2002). The ducky mutation in Cacna2d2 results in altered Purkinje cell morphology and is associated with the expression of a truncated alpha 2 delta‐2 protein with abnormal function. The Journal of Biological Chemistry, 277, 7684–7693. [DOI] [PubMed] [Google Scholar]
- Broicher, T. , Kanyshkova, T. , Landgraf, P. , Rankovic, V. , Meuth, P. , Meuth, S. G. , Pape, H. C. , & Budde, T. (2007). Specific expression of low‐voltage‐activated calcium channel isoforms and splice variants in thalamic local circuit interneurons. Molecular and Cellular Neurosciences, 36, 132–145. [DOI] [PubMed] [Google Scholar]
- Broicher, T. , Kanyshkova, T. , Meuth, P. , Pape, H.‐C. , & Budde, T. (2008). Correlation of T‐channel coding gene expression, IT, and the low threshold Ca2+ spike in the thalamus of a rat model of absence epilepsy. Molecular and Cellular Neuroscience, 39, 384–399. [DOI] [PubMed] [Google Scholar]
- Brunklaus, A. , Ellis, R. , Reavey, E. , Forbes, G. H. , & Zuberi, S. M. (2012). Prognostic, clinical and demographic features in SCN1A mutation‐positive Dravet syndrome. Brain, 135, 2329–2336. [DOI] [PubMed] [Google Scholar]
- Bucurenciu, I. , Bischofberger, J. , & Jonas, P. (2010). A small number of open Ca2+ channels trigger transmitter release at a central GABAergic synapse. Nature Neuroscience, 13, 19–21. [DOI] [PubMed] [Google Scholar]
- Burgess, D. L. , Jones, J. M. , Meisler, M. H. , & Noebels, J. L. (1997). Mutation of the Ca2+ channel β subunit gene Cchb4 is associated with ataxia and seizures in the lethargic (lh) mouse. Cell, 88, 385–392. [DOI] [PubMed] [Google Scholar]
- Butler, K. M. , Holt, P. J. , Milla, S. S. , da Silva, C. , Alexander, J. J. , & Escayg, A. (2018). Epileptic encephalopathy and cerebellar atrophy resulting from compound heterozygous CACNA2D2 variants. Case Reports in Genetics, 2018, 6308283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers, H. M. , Beatty, C. W. , Hahn, S. H. , & Gospe, S. M., Jr. (2016). Dramatic response after lamotrigine in a patient with epileptic encephalopathy and a De NovoCACNA1A variant. Pediatric Neurology, 60, 79–82. [DOI] [PubMed] [Google Scholar]
- Calhoun, J. D. , Hawkins, N. A. , Zachwieja, N. J. , & Kearney, J. A. (2016). Cacna1g is a genetic modifier of epilepsy caused by mutation of voltage‐gated sodium channel Scn2a. Epilepsia, 57, e103–e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun, J. D. , Hawkins, N. A. , Zachwieja, N. J. , & Kearney, J. A. (2017). Cacna1g is a genetic modifier of epilepsy in a mouse model of Dravet syndrome. Epilepsia, 58, e111–e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun, J. D. , Huffman, A. M. , Bellinski, I. , Kinsley, L. , Bachman, E. , Gerard, E. , Kearney, J. A. , & Carvill, G. L. (2020). CACNA1H variants are not a cause of monogenic epilepsy. Human Mutation, 41, 1138–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, C. , Leu, C. , Feng, Y. A. , Wolking, S. , Moreau, C. , Ellis, C. , Ganesan, S. , Martins, H. , Oliver, K. , Boothman, I. , Benson, K. , Molloy, A. , Brody, L. , Epi4K Collaborative , Genomics England Research Consortium , Michaud, J. L. , Hamdan, F. F. , Minassian, B. A. , Lerche, H. , … Epi25 Collaborative . (2022). The role of common genetic variation in presumed monogenic epilepsies. eBioMedicine, 81, 104098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano‐Abad, M. a. F. , Villarroya, M. , García, A. G. , Gabilan, N. H. , & López, M. G. (2001). Calcium entry through L‐type calcium channels causes mitochondrial disruption and chromaffin cell death. Journal of Biological Chemistry, 276, 39695–39704. [DOI] [PubMed] [Google Scholar]
- Cao, Y. Q. , & Tsien, R. W. (2005). Effects of familial hemiplegic migraine type 1 mutations on neuronal P/Q‐type Ca2+ channel activity and inhibitory synaptic transmission. Proceedings of the National Academy of Sciences of the United States of America, 102, 2590–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casillas‐Espinosa, P. M. , Hicks, A. , Jeffreys, A. , Snutch, T. P. , O'Brien, T. J. , & Powell, K. L. (2015). Z944, a novel selective T‐type Calcium Channel antagonist delays the progression of seizures in the amygdala kindling model. PLoS One, 10, e0130012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casillas‐Espinosa, P. M. , Lin, R. , Li, R. , Nandakumar, N. M. , Dawson, G. , Braine, E. L. , Martin, B. , Powell, K. L. , & O'Brien, T. J. (2023). Effects of the T‐type calcium channel Ca(V)3.2 R1584P mutation on absence seizure susceptibility in GAERS and NEC congenic rats models. Neurobiology of Disease, 184, 106217. [DOI] [PubMed] [Google Scholar]
- Casillas‐Espinosa, P. M. , Shultz, S. R. , Braine, E. L. , Jones, N. C. , Snutch, T. P. , Powell, K. L. , & O'Brien, T. J. (2019). Disease‐modifying effects of a novel T‐type calcium channel antagonist, Z944, in a model of temporal lobe epilepsy. Progress in Neurobiology, 182, 101677. [DOI] [PubMed] [Google Scholar]
- Chan, Y. C. , Burgunder, J. M. , Wilder‐Smith, E. , Chew, S. E. , Lam‐Mok‐Sing, K. M. , Sharma, V. , & Ong, B. K. (2008). Electroencephalographic changes and seizures in familial hemiplegic migraine patients with the CACNA1A gene S218L mutation. Journal of Clinical Neuroscience, 15, 891–894. [DOI] [PubMed] [Google Scholar]
- Cheli, V. T. , Santiago González, D. A. , Smith, J. , Spreuer, V. , Murphy, G. G. , & Paez, P. M. (2016). L‐type voltage‐operated calcium channels contribute to astrocyte activation in vitro. Glia, 64, 1396–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemin, J. , Siquier‐Pernet, K. , Nicouleau, M. , Barcia, G. , Ahmad, A. , Medina‐Cano, D. , Hanein, S. , Altin, N. , Hubert, L. , Bole‐Feysot, C. , Fourage, C. , Nitschké, P. , Thevenon, J. , Rio, M. , Blanc, P. , vidal, C. , Bahi‐Buisson, N. , Desguerre, I. , Munnich, A. , … Cantagrel, V. (2018). De novo mutation screening in childhood‐onset cerebellar atrophy identifies gain‐of‐function mutations in the CACNA1G calcium channel gene. Brain, 141, 1998–2013. [DOI] [PubMed] [Google Scholar]
- Chen, J. , Li, L. , Chen, S. R. , Chen, H. , Xie, J. D. , Sirrieh, R. E. , MacLean, D. M. , Zhang, Y. , Zhou, M. H. , Jayaraman, V. , & Pan, H. L. (2018). The alpha2delta‐1‐NMDA receptor complex is critically involved in neuropathic pain development and gabapentin therapeutic actions. Cell Reports, 22, 2307–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, R.‐S. , Deng, T.‐C. , Garcia, T. , Sellers, Z. M. , & Best, P. M. (2007). Calcium channel γ subunits: A functionally diverse protein family. Cell Biochemistry and Biophysics, 47, 178–186. [DOI] [PubMed] [Google Scholar]
- Chen, S. , Francioli, L. C. , Goodrich, J. K. , Collins, R. L. , Wang, Q. , Alföldi, J. , Watts, N. A. , Vittal, C. , Gauthier, L. D. , Poterba, T. , Wilson, M. W. , Tarasova, Y. , Phu, W. , Yohannes, M. T. , Koenig, Z. , Farjoun, Y. , Banks, E. , Donnelly, S. , Gabriel, S. , … Karczewski, K. J. (2022). A genome‐wide mutational constraint map quantified from variation in 76,156 human genomes. bioRxiv, 2022.2003.2020.485034. [Google Scholar]
- Chen, Y. , Lu, J. , Pan, H. , Zhang, Y. , Wu, H. , Xu, K. , Liu, X. , Jiang, Y. , Bao, X. , Yao, Z. , Ding, K. , Lo, W. H. Y. , Qiang, B. , Chan, P. , Shen, Y. , & Wu, X. (2003). Association between genetic variation of CACNA1H and childhood absence epilepsy. Annals of Neurology, 54, 239–243. [DOI] [PubMed] [Google Scholar]
- Chioza, B. , Everett, K. , Aschauer, H. , Brouwer, O. , Callenbach, P. , Covanis, A. , Dulac, O. , Durner, M. , Eeg‐Olofsson, O. , Feucht, M. , Friis, M. , Heils, A. , Kjeldsen, M. , Larsson, K. , Lehesjoki, A. E. , Nabbout, R. , Olsson, I. , Sander, T. , Sirén, A. , … Gardiner, R. M. (2006). Evaluation of CACNA1H in European patients with childhood absence epilepsy. Epilepsy Research, 69, 177–181. [DOI] [PubMed] [Google Scholar]
- Chourasia, N. , Ossó‐Rivera, H. , Ghosh, A. , Von Allmen, G. , & Koenig, M. K. (2019). Expanding the phenotypic Spectrum of CACNA1H mutations. Pediatric Neurology, 93, 50–55. [DOI] [PubMed] [Google Scholar]
- Epi25 Collaborative , Chen, S. , Neale, B. M. , & Berkovic, S. F. (2023). Shared and distinct ultra‐rare genetic risk for diverse epilepsies: A whole‐exome sequencing study of 54,423 individuals across multiple genetic ancestries. medRxiv, 2023.2002.2022.23286310. [Google Scholar]
- Coste de Bagneaux, P. , von Elsner, L. , Bierhals, T. , Campiglio, M. , Johannsen, J. , Obermair, G. J. , Hempel, M. , Flucher, B. E. , & Kutsche, K. (2020). A homozygous missense variant in CACNB4 encoding the auxiliary calcium channel beta4 subunit causes a severe neurodevelopmental disorder and impairs channel and non‐channel functions. PLoS Genetics, 16, e1008625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutelier, M. , Blesneac, I. , Monteil, A. , Monin, M. L. , Ando, K. , Mundwiller, E. , Brusco, A. , le Ber, I. , Anheim, M. , Castrioto, A. , Duyckaerts, C. , Brice, A. , Durr, A. , Lory, P. , & Stevanin, G. (2015). A recurrent mutation in CACNA1G alters Cav3.1 T‐type calcium‐channel conduction and causes autosomal‐dominant cerebellar ataxia. American Journal of Human Genetics, 97, 726–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunelli, V. , Lorincz, M. L. , McCafferty, C. , Lambert, R. C. , Leresche, N. , Di Giovanni, G. , & David, F. (2020). Clinical and experimental insight into pathophysiology, comorbidity and therapy of absence seizures. Brain, 143, 2341–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahimene, S. , von Elsner, L. , Holling, T. , Mattas, L. S. , Pickard, J. , Lessel, D. , Pilch, K. S. , Kadurin, I. , Pratt, W. S. , Zhulin, I. B. , Dai, H. , Hempel, M. , Ruzhnikov, M. R. Z. , Kutsche, K. , & Dolphin, A. C. (2022). Biallelic CACNA2D1 loss‐of‐function variants cause early‐onset developmental epileptic encephalopathy. Brain, 145, 2721–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damaj, L. , Lupien‐Meilleur, A. , Lortie, A. , Riou, É. , Ospina, L. H. , Gagnon, L. , Vanasse, C. , & Rossignol, E. (2015). CACNA1A haploinsufficiency causes cognitive impairment, autism and epileptic encephalopathy with mild cerebellar symptoms. European Journal of Human Genetics, 23, 1505–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Ascenzo, M. , Vairano, M. , Andreassi, C. , Navarra, P. , Azzena, G. B. , & Grassi, C. (2004). Electrophysiological and molecular evidence of L‐(Cav1), N‐ (Cav2.2), and R‐ (Cav2.3) type Ca2+ channels in rat cortical astrocytes. Glia, 45, 354–363. [DOI] [PubMed] [Google Scholar]
- Di Biase, V. , Obermair, G. J. , Szabo, Z. , Altier, C. , Sanguesa, J. , Bourinet, E. , & Flucher, B. E. (2008). Stable membrane expression of postsynaptic CaV1.2 calcium channel clusters is independent of interactions with AKAP79/150 and PDZ proteins. The Journal of Neuroscience, 28, 13845–13855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Guilmi, M. N. , Wang, T. , Inchauspe, C. G. , Forsythe, I. D. , Ferrari, M. D. , van den Maagdenberg, A. M. , Borst, J. G. , & Uchitel, O. D. (2014). Synaptic gain‐of‐function effects of mutant Cav2.1 channels in a mouse model of familial hemiplegic migraine are due to increased basal [Ca2+]i. The Journal of Neuroscience, 34, 7047–7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich, D. , Kirschstein, T. , Kukley, M. , Pereverzev, A. , von der Brelie, C. , Schneider, T. , & Beck, H. (2003). Functional specialization of presynaptic Cav2.3 Ca2+ channels. Neuron, 39, 483–496. [DOI] [PubMed] [Google Scholar]
- Doering, C. J. , Peloquin, J. B. , & McRory, J. E. (2014). The CaV1.4 calcium channel: More than meets the eye. Channels, 1, 4–11. [PubMed] [Google Scholar]
- Doeser, A. , Dickhof, G. , Reitze, M. , Uebachs, M. , Schaub, C. , Pires, N. M. , Bonifácio, M. J. , Soares‐da‐Silva, P. , & Beck, H. (2014). Targeting pharmacoresistant epilepsy and epileptogenesis with a dual‐purpose antiepileptic drug. Brain, 138, 371–387. [DOI] [PubMed] [Google Scholar]
- Dolphin, A. C. , Wyatt, C. N. , Richards, J. , Beattie, R. E. , Craig, P. , Lee, J. H. , Cribbs, L. L. , Volsen, S. G. , & Perez‐Reyes, E. (1999). The effect of alpha2‐delta and other accessory subunits on expression and properties of the calcium channel alpha1G. The Journal of Physiology, 519(Pt 1), 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato, R. , Page, K. M. , Koch, D. , Nieto‐Rostro, M. , Foucault, I. , Davies, A. , Wilkinson, T. , Rees, M. , Edwards, F. A. , & Dolphin, A. C. (2006). The ducky(2J) mutation in Cacna2d2 results in reduced spontaneous Purkinje cell activity and altered gene expression. The Journal of Neuroscience, 26, 12576–12586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, X. , Chen, Y. , Zhao, Y. , Luo, W. , Cen, Z. , & Hao, W. (2017). Dramatic response to pyridoxine in a girl with absence epilepsy with ataxia caused by a de novo CACNA1A mutation. Seizure, 45, 189–191. [DOI] [PubMed] [Google Scholar]
- Ducros, A. , Denier, C. , Joutel, A. , Cecillon, M. , Lescoat, C. , Vahedi, K. , Darcel, F. , Vicaut, E. , Bousser, M. G. , & Tournier‐Lasserve, E. (2001). The clinical spectrum of familial hemiplegic migraine associated with mutations in a neuronal calcium channel. New England Journal of Medicine, 345, 17–24. [DOI] [PubMed] [Google Scholar]
- Eckle, V. S. , Shcheglovitov, A. , Vitko, I. , Dey, D. , Yap, C. C. , Winckler, B. , & Perez‐Reyes, E. (2014). Mechanisms by which a CACNA1H mutation in epilepsy patients increases seizure susceptibility. The Journal of Physiology, 592, 795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvardson, S. , Oz, S. , Abulhijaa, F. A. , Taher, F. B. , Shaag, A. , Zenvirt, S. , Dascal, N. , & Elpeleg, O. (2013). Early infantile epileptic encephalopathy associated with a high voltage gated calcium channelopathy. Journal of Medical Genetics, 50, 118–123. [DOI] [PubMed] [Google Scholar]
- El Ghaleb, Y. , Schneeberger, P. E. , Fernández‐Quintero, M. L. , Geisler, S. M. , Pelizzari, S. , Polstra, A. M. , van Hagen, J. M. , Denecke, J. , Campiglio, M. , Liedl, K. R. , Stevens, C. A. , Person, R. E. , Rentas, S. , Marsh, E. D. , Conlin, L. K. , Tuluc, P. , Kutsche, K. , & Flucher, B. E. (2021). CACNA1I gain‐of‐function mutations differentially affect channel gating and cause neurodevelopmental disorders. Brain, 144, 2092–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epi4K Consortium and Epilepsy Phenome/Genome Project . (2017). Ultra‐rare genetic variation in common epilepsies: a case‐control sequencing study. Lancet Neurology, 16, 135–143. [DOI] [PubMed] [Google Scholar]
- Epi25 Collaborative . (2021). Sub‐genic intolerance, ClinVar, and the epilepsies: A whole‐exome sequencing study of 29,165 individuals. American Journal of Human Genetics, 108, 965–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epi4K Consortium , Epilepsy Phenome/Genome Project , Allen, A. S. , Berkovic, S. F. , Cossette, P. , Delanty, N. , Dlugos, D. , Eichler, E. E. , Epstein, M. P. , Glauser, T. , Goldstein, D. B. , Han, Y. , Heinzen, E. L. , Hitomi, Y. , Howell, K. B. , Johnson, M. R. , Kuzniecky, R. , Lowenstein, D. H. , Lu, Y.‐F. , … Winawer, M. R. (2013). De novo mutations in epileptic encephalopathies. Nature, 501, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu, C. , Allen, N. J. , Susman, M. W. , O'Rourke, N. A. , Park, C. Y. , Özkan, E. , Chakraborty, C. , Mulinyawe, S. B. , Annis, D. S. , Huberman, A. D. , Green, E. M. , Lawler, J. , Dolmetsch, R. , Garcia, K. C. , Smith, S. J. , Luo, Z. D. , Rosenthal, A. , Mosher, D. F. , & Barres, B. A. (2009). Gabapentin receptor alpha2delta‐1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell, 139, 380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escayg, A. , De Waard, M. , Lee, D. D. , Bichet, D. , Wolf, P. , Mayer, T. , Johnston, J. , Baloh, R. , Sander, T. , & Meisler, M. H. (2000). Coding and noncoding variation of the human calcium‐channel beta4‐subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. American Journal of Human Genetics, 66, 1531–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria, L. C. , Gu, F. , Parada, I. , Barres, B. , Luo, Z. D. , & Prince, D. A. (2017). Epileptiform activity and behavioral arrests in mice overexpressing the calcium channel subunit alpha2delta‐1. Neurobiology of Disease, 102, 70–80. [DOI] [PubMed] [Google Scholar]
- Fletcher, C. F. , Lutz, C. M. , OSullivan, T. N. , Shaughnessy, J. D. , Hawkes, R. , Frankel, W. N. , Copeland, N. G. , & Jenkins, N. A. (1996). Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell, 87, 607–617. [DOI] [PubMed] [Google Scholar]
- Fujita, Y. , Mynlieff, M. , Dirksen, R. T. , Kim, M. S. , Niidome, T. , Nakai, J. , Friedrich, T. , Iwabe, N. , Miyata, T. , Furuichi, T. , Furutama, D. , Mikoshiba, K. , Mori, Y. , & Beam, K. G. (1993). Primary structure and functional expression of the omega‐conotoxin‐sensitive N‐type calcium channel from rabbit brain. Neuron, 10, 585–598. [DOI] [PubMed] [Google Scholar]
- Fukuyama, M. , Wang, Q. , Kato, K. , Ohno, S. , Ding, W. G. , Toyoda, F. , Itoh, H. , Kimura, H. , Makiyama, T. , Ito, M. , Matsuura, H. , & Horie, M. (2014). Long QT syndrome type 8: Novel CACNA1C mutations causing QT prolongation and variant phenotypes. Europace, 16, 1828–1837. [DOI] [PubMed] [Google Scholar]
- Fuller‐Bicer, G. A. , Varadi, G. , Koch, S. E. , Ishii, M. , Bodi, I. , Kadeer, N. , Muth, J. N. , Mikala, G. , Petrashevskaya, N. N. , Jordan, M. A. , Zhang, S. P. , Qin, N. , Flores, C. M. , Isaacsohn, I. , Varadi, M. , Mori, Y. , Jones, W. K. , & Schwartz, A. (2009). Targeted disruption of the voltage‐dependent calcium channel alpha2/delta‐1‐subunit. American Journal of Physiology. Heart and Circulatory Physiology, 297, H117–H124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, B. , Sekido, Y. , Maximov, A. , Saad, M. , Forgacs, E. , Latif, F. , Wei, M. H. , Lerman, M. , Lee, J. H. , Perez‐Reyes, E. , Bezprozvanny, I. , & Minna, J. D. (2000). Functional properties of a new voltage‐dependent calcium channel alpha(2)delta auxiliary subunit gene (CACNA2D2). The Journal of Biological Chemistry, 275, 12237–12242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee, N. S. , Brown, J. P. , Dissanayake, V. U. K. , Offord, J. , Thurlow, R. , & Woodruff, G. N. (1996). The novel anticonvulsant drug, gabapentin (Neurontin), binds to the α2δ subunit of a calcium channel. Journal of Biological Chemistry, 271, 5768–5776. [DOI] [PubMed] [Google Scholar]
- Gershon, E. S. , Grennan, K. , Busnello, J. , Badner, J. A. , Ovsiew, F. , Memon, S. , Alliey‐Rodriguez, N. , Cooper, J. , Romanos, B. , & Liu, C. (2014). A rare mutation of CACNA1C in a patient with bipolar disorder, and decreased gene expression associated with a bipolar‐associated common SNP of CACNA1C in brain. Molecular Psychiatry, 19, 890–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasscock, E. , Qian, J. , Yoo, J. W. , & Noebels, J. L. (2007). Masking epilepsy by combining two epilepsy genes. Nature Neuroscience, 10, 1554–1558. [DOI] [PubMed] [Google Scholar]
- Gomora, J. C. , Daud, A. N. , Weiergräber, M. , & Perez‐Reyes, E. (2001). Block of cloned human T‐type calcium channels by succinimide antiepileptic drugs. Molecular Pharmacology, 60, 1121–1132. [PubMed] [Google Scholar]
- Gorman, K. M. , Meyer, E. , Grozeva, D. , Spinelli, E. , McTague, A. , Sanchis‐Juan, A. , Carss, K. J. , Bryant, E. , Reich, A. , Schneider, A. L. , Pressler, R. M. , Simpson, M. A. , Debelle, G. D. , Wassmer, E. , Morton, J. , Sieciechowicz, D. , Jan‐Kamsteeg, E. , Paciorkowski, A. R. , King, M. D. , … Yu, P. (2019). Bi‐allelic loss‐of‐function CACNA1B mutations in progressive epilepsy‐dyskinesia. American Journal of Human Genetics, 104, 948–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur‐Hartman, T. , Berkowitz, O. , Yosovich, K. , Roubertie, A. , Zanni, G. , Macaya, A. , Heimer, G. , Dueñas, B. P. , Sival, D. A. , Pode‐Shakked, B. , López‐Laso, E. , Humbertclaude, V. , Riant, F. , Bosco, L. , Cayron, L. B. , Nissenkorn, A. , Nicita, F. , Bertini, E. , Hassin, S. , … Blumkin, L. (2021). Clinical phenotypes of infantile onset CACNA1A‐related disorder. European Journal of Paediatric Neurology, 30, 144–154. [DOI] [PubMed] [Google Scholar]
- Guzman, J. N. , Sanchez‐Padilla, J. , Chan, C. S. , & Surmeier, D. J. (2009). Robust pacemaking in substantia nigra dopaminergic neurons. The Journal of Neuroscience, 29, 11011–11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbuz, R. , Bilan, F. , Couet, D. , Charraud, V. , Kitzis, A. , & Gilbert‐Dussardier, B. (2013). Osteogenesis imperfecta, tricho‐dento‐osseous syndrome and intellectual disability: A familial case with 17q21.33‐q22 (COL1A1 and DLX3) deletion and 7q32.3‐q33 duplication resulting from a reciprocal interchromosomal insertion. American Journal of Medical Genetics. Part A, 161A, 2504–2511. [DOI] [PubMed] [Google Scholar]
- Hardingham, G. E. , Chawla, S. , Johnson, C. M. , & Bading, H. (1997). Distinct functions of nuclear and cytoplasmic calcium in the control of gene expression. Nature, 385, 260–265. [DOI] [PubMed] [Google Scholar]
- Helbig, I. , Riggs, E. R. , Barry, C. A. , Klein, K. M. , Dyment, D. , Thaxton, C. , Sadikovic, B. , Sands, T. T. , Wagnon, J. L. , Liaquat, K. , Cilio, M. R. , Mirzaa, G. , Park, K. , Axeen, E. , Butler, E. , Bardakjian, T. M. , Striano, P. , Poduri, A. , Siegert, R. K. , … Mefford, H. C. (2018). The ClinGen Epilepsy gene curation expert panel‐bridging the divide between clinical domain knowledge and formal gene curation criteria. Human Mutation, 39, 1476–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbig, K. L. , Lauerer, R. J. , Bahr, J. C. , Souza, I. A. , Myers, C. T. , Uysal, B. , Schwarz, N. , Gandini, M. A. , Huang, S. , Keren, B. , Mignot, C. , Afenjar, A. , Billette de Villemeur, T. , Héron, D. , Nava, C. , Valence, S. , Buratti, J. , Fagerberg, C. R. , Soerensen, K. P. , … Wiener, J. (2018). De novo pathogenic variants in CACNA1E cause developmental and epileptic encephalopathy with contractures, macrocephaly, and dyskinesias. American Journal of Human Genetics, 103, 666–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell, J. W. , Westenbroek, R. E. , Warner, C. , Ahlijanian, M. K. , Prystay, W. , Gilbert, M. M. , Snutch, T. P. , & Catterall, W. A. (1993). Identification and differential subcellular localization of the neuronal class C and class D L‐type calcium channel alpha 1 subunits. Journal of Cell Biology, 123, 949–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helton, T. D. , Xu, W. , & Lipscombe, D. (2005). Neuronal L‐type calcium channels open quickly and are inhibited slowly. The Journal of Neuroscience, 25, 10247–10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemara‐Wahanui, A. , Berjukow, S. , Hope, C. I. , Dearden, P. K. , Wu, S. B. , Wilson‐Wheeler, J. , Sharp, D. M. , Lundon‐Treweek, P. , Clover, G. M. , Hoda, J. C. , Striessnig, J. , Marksteiner, R. , Hering, S. , & Maw, M. A. (2005). A CACNA1F mutation identified in an X‐linked retinal disorder shifts the voltage dependence of Cav1.4 channel activation. Proceedings of the National Academy of Sciences of the United States of America, 102, 7553–7558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heron, S. E. , Khosravani, H. , Varela, D. , Bladen, C. , Williams, T. C. , Newman, M. R. , Scheffer, I. E. , Berkovic, S. F. , Mulley, J. C. , & Zamponi, G. W. (2007). Extended spectrum of idiopathic generalized epilepsies associated with CACNA1H functional variants. Annals of Neurology, 62, 560–568. [DOI] [PubMed] [Google Scholar]
- Heyne, H. O. , Artomov, M. , Battke, F. , Bianchini, C. , Smith, D. R. , Liebmann, N. , Tadigotla, V. , Stanley, C. M. , Lal, D. , Rehm, H. , Lerche, H. , Daly, M. J. , Helbig, I. , Biskup, S. , Weber, Y. G. , & Lemke, J. R. (2019). Targeted gene sequencing in 6994 individuals with neurodevelopmental disorder with epilepsy. Genetics in Medicine, 21, 2496–2503. [DOI] [PubMed] [Google Scholar]
- Hino‐Fukuyo, N. , Kikuchi, A. , Arai‐Ichinoi, N. , Niihori, T. , Sato, R. , Suzuki, T. , Kudo, H. , Sato, Y. , Nakayama, T. , Kakisaka, Y. , Kubota, Y. , Kobayashi, T. , Funayama, R. , Nakayama, K. , Uematsu, M. , Aoki, Y. , Haginoya, K. , & Kure, S. (2015). Genomic analysis identifies candidate pathogenic variants in 9 of 18 patients with unexplained West syndrome. Human Genetics, 134, 649–658. [DOI] [PubMed] [Google Scholar]
- Hirning, L. D. , Fox, A. P. , McCleskey, E. W. , Olivera, B. M. , Thayer, S. A. , Miller, R. J. , & Tsien, R. W. (1988). Dominant role of N‐type Ca2+ channels in evoked release of norepinephrine from sympathetic neurons. Science, 239, 57–61. [DOI] [PubMed] [Google Scholar]
- Hobom, M. , Dai, S. , Marais, E. , Lacinova, L. , Hofmann, F. , & Klugbauer, N. (2000). Neuronal distribution and functional characterization of the calcium channel alpha2delta‐2 subunit. The European Journal of Neuroscience, 12, 1217–1226. [DOI] [PubMed] [Google Scholar]
- Hockerman, G. H. , Dilmac, N. , Scheuer, T. , & Catterall, W. A. (2000). Molecular determinants of diltiazem block in domains IIIS6 and IVS6 of L‐type Ca2+ channels. Molecular Pharmacology, 58, 1264–1270. [DOI] [PubMed] [Google Scholar]
- Huguenard, J. (2019). Current controversy: Spikes, bursts, and synchrony in generalized absence Epilepsy: Unresolved questions regarding thalamocortical synchrony in absence Epilepsy. Epilepsy Currents, 19, 105–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard, J. , & Prince, D. (1992). A novel T‐type current underlies prolonged Ca(2+)‐dependent burst firing in GABAergic neurons of rat thalamic reticular nucleus. The Journal of Neuroscience, 12, 3804–3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbrici, P. , Jaffe, S. L. , Eunson, L. H. , Davies, N. P. , Herd, C. , Robertson, R. , Kullmann, D. M. , & Hanna, M. G. (2004). Dysfunction of the brain calcium channel CaV2.1 in absence epilepsy and episodic ataxia. Brain, 127, 2682–2692. [DOI] [PubMed] [Google Scholar]
- Ivanov, S. V. , Ward, J. M. , Tessarollo, L. , McAreavey, D. , Sachdev, V. , Fananapazir, L. , Banks, M. K. , Morris, N. , Djurickovic, D. , Devor‐Henneman, D. E. , Wei, M. H. , Alvord, G. W. , Gao, B. , Richardson, J. A. , Minna, J. D. , Rogawski, M. A. , & Lerman, M. I. (2004). Cerebellar ataxia, seizures, premature death, and cardiac abnormalities in mice with targeted disruption of the Cacna2d2 gene. The American Journal of Pathology, 165, 1007–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki, S. , Momiyama, A. , Uchitel, O. D. , & Takahashi, T. (2000). Developmental changes in calcium channel types mediating central synaptic transmission. The Journal of Neuroscience, 20, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins, M. A. , Christel, C. J. , Jiao, Y. , Abiria, S. , Kim, K. Y. , Usachev, Y. M. , Obermair, G. J. , Colbran, R. J. , & Lee, A. (2010). Ca2+−dependent facilitation of Cav1.3 Ca2+ channels by densin and Ca2+/calmodulin‐dependent protein kinase II. The Journal of Neuroscience, 30, 5125–5135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewell, R. , Sarkar, A. , Jones, R. , Wilkinson, A. , Martin, K. , Arundel, P. , & Balasubramanian, M. (2017). Atypical osteogenesis imperfecta caused by a 17q21.33 deletion involving COL1A1. Clinical Dysmorphology, 26, 228–230. [DOI] [PubMed] [Google Scholar]
- Jiang, X. , Raju, P. K. , D'Avanzo, N. , Lachance, M. , Pepin, J. , Dubeau, F. , Mitchell, W. G. , Bello‐Espinosa, L. E. , Pierson, T. M. , Minassian, B. A. , Lacaille, J. C. , & Rossignol, E. (2019). Both gain‐of‐function and loss‐of‐function de novo CACNA1A mutations cause severe developmental epileptic encephalopathies in the spectrum of Lennox‐Gastaut syndrome. Epilepsia, 60, 1881–1894. [DOI] [PubMed] [Google Scholar]
- Jones, O. T. , Bernstein, G. M. , Jones, E. J. , Jugloff, D. G. , Law, M. , Wong, W. , & Mills, L. R. (1997). N‐type calcium channels in the developing rat hippocampus: Subunit, complex, and regional expression. The Journal of Neuroscience, 17, 6152–6164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvenceau, A. , Eunson, L. H. , Spauschus, A. , Ramesh, V. , Zuberi, S. M. , Kullmann, D. M. , & Hanna, M. G. (2001). Human epilepsy associated with dysfunction of the brain P/Q‐type calcium channel. Lancet, 358, 801–807. [DOI] [PubMed] [Google Scholar]
- Jun, K. , Piedras‐Rentería, E. S. , Smith, S. M. , Wheeler, D. B. , Lee, S. B. , Lee, T. G. , Chin, H. , Adams, M. E. , Scheller, R. H. , Tsien, R. W. , & Shin, H. S. (1999). Ablation of P/Q‐type Ca(2+) channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the alpha(1A)‐subunit. Proceedings of the National Academy of Sciences of the United States of America, 96, 15245–15250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkovičová‐Tarabová, B. , Griesemer, D. , Pirone, A. , Sinnegger‐Brauns, M. J. , Striessnig, J. , & Friauf, E. (2012). Repertoire of high voltage‐activated Ca2+ channels in the lateral superior olive: Functional analysis in wild‐type, Cav1.3−/−, and Cav1.2DHP−/− mice. Journal of Neurophysiology, 108, 365–379. [DOI] [PubMed] [Google Scholar]
- Kelly, K. M. , Gross, R. A. , & Macdonald, R. L. (1990). Valproic acid selectively reduces the low‐threshold (T) calcium current in rat nodose neurons. Neuroscience Letters, 116, 233–238. [DOI] [PubMed] [Google Scholar]
- Khosravani, H. , Bladen, C. , Parker, D. B. , Snutch, T. P. , McRory, J. E. , & Zamponi, G. W. (2005). Effects of Cav3.2 channel mutations linked to idiopathic generalized epilepsy. Annals of Neurology, 57, 745–749. [DOI] [PubMed] [Google Scholar]
- Kim, C. , Jeon, D. , Kim, Y. H. , Lee, C. J. , Kim, H. , & Shin, H. S. (2009). Deletion of N‐type Ca(2+) channel Ca(v)2.2 results in hyperaggressive behaviors in mice. The Journal of Biological Chemistry, 284, 2738–2745. [DOI] [PubMed] [Google Scholar]
- Kim, C. , Jun, K. , Lee, T. , Kim, S. S. , McEnery, M. W. , Chin, H. , Kim, H. L. , Park, J. M. , Kim, D. K. , Jung, S. J. , Kim, J. , & Shin, H. S. (2001). Altered nociceptive response in mice deficient in the alpha(1B) subunit of the voltage‐dependent calcium channel. Molecular and Cellular Neurosciences, 18, 235–245. [DOI] [PubMed] [Google Scholar]
- Kim, C.‐H. (2015). Cav3.1 T‐type calcium channel modulates the epileptogenicity of hippocampal seizures in the kainic acid‐induced temporal lobe epilepsy model. Brain Research, 1622, 204–216. [DOI] [PubMed] [Google Scholar]
- Kim, D. , Song, I. , Keum, S. , Lee, T. , Jeong, M.‐J. , Kim, S.‐S. , McEnery, M. W. , & Shin, H.‐S. (2001). Lack of the burst firing of Thalamocortical relay neurons and resistance to absence seizures in mice lacking α1G T‐type Ca2+ channels. Neuron, 31, 35–45. [DOI] [PubMed] [Google Scholar]
- Kimm, T. , & Bean, B. P. (2014). Inhibition of A‐type potassium current by the peptide toxin SNX‐482. The Journal of Neuroscience, 34, 9182–9189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klöckner, U. , Lee, J.‐H. , Cribbs, L. L. , Daud, A. , Hescheler, J. , Pereverzev, A. , Perez‐Reyes, E. , & Schneider, T. (1999). Comparison of the Ca2+ currents induced by expression of three cloned α1 subunits, α1G, α1H and α1I, of low‐voltage‐activated T‐type Ca2+ channels. European Journal of Neuroscience, 11, 4171–4178. [DOI] [PubMed] [Google Scholar]
- Koschak, A. , Reimer, D. , Huber, I. , Grabner, M. , Glossmann, H. , Engel, J. , & Striessnig, J. (2001). α1D (Cav1.3) subunits can form L‐type Ca2+ channels activating at negative voltages. Journal of Biological Chemistry, 276, 22100–22106. [DOI] [PubMed] [Google Scholar]
- Kunii, M. , Doi, H. , Hashiguchi, S. , Matsuishi, T. , Sakai, Y. , Iai, M. , Okubo, M. , Nakamura, H. , Takahashi, K. , Katsumoto, A. , Tada, M. , Takeuchi, H. , Ishikawa, T. , Miyake, N. , Saitsu, H. , Matsumoto, N. , & Tanaka, F. (2020). De novo CACNA1G variants in developmental delay and early‐onset epileptic encephalopathies. Journal of the Neurological Sciences, 416, 117047. [DOI] [PubMed] [Google Scholar]
- Lacinová, L. , & Klugbauer, N. (2004). Modulation of gating currents of the Ca(v)3.1 calcium channel by alpha 2 delta 2 and gamma 5 subunits. Archives of Biochemistry and Biophysics, 425, 207–213. [DOI] [PubMed] [Google Scholar]
- Lacinova, L. , Moosmang, S. , Langwieser, N. , Hofmann, F. , & Kleppisch, T. (2008). Cav1.2 calcium channels modulate the spiking pattern of hippocampal pyramidal cells. Life Sciences, 82, 41–49. [DOI] [PubMed] [Google Scholar]
- Lambert, R. C. , Maulet, Y. , Mouton, J. , Beattie, R. , Volsen, S. , De Waard, M. , & Feltz, A. (1997). T‐type Ca2+ current properties are not modified by Ca2+ channel β subunit depletion in nodosus ganglion neurons. The Journal of Neuroscience, 17, 6621–6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Roux, M. , Barth, M. , Gueden, S. , Desbordes de Cepoy, P. , Aeby, A. , Vilain, C. , Hirsch, E. , de Saint Martin, A. , Portes, V. D. , Lesca, G. , Riquet, A. , Chaton, L. , Villeneuve, N. , Villard, L. , Cances, C. , Valton, L. , Renaldo, F. , Vermersch, A. I. , Altuzarra, C. , … Van Bogaert, P. (2021). CACNA1A‐associated epilepsy: Electroclinical findings and treatment response on seizures in 18 patients. European Journal of Paediatric Neurology, 33, 75–85. [DOI] [PubMed] [Google Scholar]
- Lee, J.‐H. , Gomora, J. C. , Cribbs, L. L. , & Perez‐Reyes, E. (1999). Nickel block of three cloned T‐type calcium channels: Low concentrations selectively block α1H. Biophysical Journal, 77, 3034–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S. T. , Lee, B. J. , Bae, J. Y. , Kim, Y. S. , Han, D. H. , Shin, H. S. , Kim, S. , Park, D. K. , Seo, S. W. , Chu, K. , Lee, S. K. , & Ho, W. K. (2021). Ca(V) alpha2delta autoimmune encephalitis: A novel antibody and its characteristics. Annals of Neurology, 89, 740–752. [DOI] [PubMed] [Google Scholar]
- Lek, M. , Karczewski, K. J. , Minikel, E. V. , Samocha, K. E. , Banks, E. , Fennell, T. , O'Donnell‐Luria, A. H. , Ware, J. S. , Hill, A. J. , Cummings, B. B. , Tukiainen, T. , Birnbaum, D. P. , Kosmicki, J. A. , Duncan, L. E. , Estrada, K. , Zhao, F. , Zou, J. , Pierce‐Hoffman, E. , Berghout, J. , … Exome Aggregation Consortium . (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuranguer, V. , Bourinet, E. , Lory, P. , & Nargeot, J. (1998). Antisense depletion of β‐subunits fails to affect T‐type calcium channels properties in a neuroblastoma cell line. Neuropharmacology, 37, 701–708. [DOI] [PubMed] [Google Scholar]
- Li, B. , Tadross, M. R. , & Tsien, R. W. (2016). Sequential ionic and conformational signaling by calcium channels drives neuronal gene expression. Science, 351, 863–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Lin, S. M. , Qiao, J. D. , Liu, X. R. , Wang, J. , Jiang, M. , Zhang, J. , Zhong, M. , Chen, X. Q. , Zhu, J. , He, N. , Su, T. , Shi, Y. W. , Yi, Y. H. , Liao, W. P. , & for the China Epilepsy Gene 1.0 Project . (2022). CELSR3 variants are associated with febrile seizures and epilepsy with antecedent febrile seizures. CNS Neuroscience & Therapeutics, 28, 382–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, J. , Zhang, Y. , Wang, J. , Pan, H. , Wu, H. , Xu, K. , Liu, X. , Jiang, Y. , Shen, Y. , & Wu, X. (2006). New variants in the CACNA1H gene identified in childhood absence epilepsy. Neuroscience Letters, 406, 27–32. [DOI] [PubMed] [Google Scholar]
- Llinas, R. R. , Choi, S. , Urbano, F. J. , & Shin, H. S. (2007). Gamma‐band deficiency and abnormal thalamocortical activity in P/Q‐type channel mutant mice. Proceedings of the National Academy of Sciences of the United States of America, 104, 17819–17824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loonen, I. C. M. , Jansen, N. A. , Cain, S. M. , Schenke, M. , Voskuyl, R. A. , Yung, A. C. , Bohnet, B. , Kozlowski, P. , Thijs, R. D. , Ferrari, M. D. , Snutch, T. P. , van den Maagdenberg, A. M. J. M. , & Tolner, E. A. (2019). Brainstem spreading depolarization and cortical dynamics during fatal seizures in Cacna1a S218L mice. Brain, 142, 412–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig, A. , Flockerzi, V. , & Hofmann, F. (1997). Regional expression and cellular localization of the α1 and β subunit of high voltage‐activated calcium channels in rat brain. The Journal of Neuroscience, 17, 1339–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksemous, N. , Blayney, C. D. , Sutherland, H. G. , Smith, R. A. , Lea, R. A. , Tran, K. N. , Ibrahim, O. , McArthur, J. R. , Haupt, L. M. , Cader, M. Z. , Finol‐Urdaneta, R. K. , Adams, D. J. , & Griffiths, L. R. (2022). Investigation of CACNA1I Cav3.3 dysfunction in hemiplegic migraine. Frontiers in Molecular Neuroscience, 15, 892820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcantoni, A. , Calorio, C. , Hidisoglu, E. , Chiantia, G. , & Carbone, E. (2020). Cav1.2 channelopathies causing autism: New hallmarks on Timothy syndrome. Pflügers Archiv, 472, 775–789. [DOI] [PubMed] [Google Scholar]
- Martins Custodio, H. , Clayton, L. M. , Bellampalli, R. , Pagni, S. , Silvennoinen, K. , Caswell, R. , Genomics England Research Consortium , Ambrose, J. C. , Arumugam, P. , Bevers, R. , Bleda, M. , Boardman‐Pretty, F. , Boustred, C. R. , Brittain, H. , Brown, M. A. , Caulfield, M. J. , Chan, G. C. , Giess, A. , Griffin, J. N. , … Sisodiya, S. M. (2023). Widespread genomic influences on phenotype in Dravet syndrome, a 'monogenic' condition. Brain, 146, 3885–3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matar, N. , Jin, W. , Wrubel, H. , Hescheler, J. , Schneider, T. , & Weiergräber, M. (2009). Zonisamide block of cloned human T‐type voltage‐gated calcium channels. Epilepsy Research, 83, 224–234. [DOI] [PubMed] [Google Scholar]
- Mazzaschi, R. L. , Ashton, F. , Aftimos, S. , George, A. M. , & Love, D. R. (2013). Implications of a Chr7q21.11 microdeletion and the role of the PCLO gene in developmental delay. Sultan Qaboos University Medical Journal, 13, 306–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCafferty, C. , David, F. , Venzi, M. , Lőrincz, M. L. , Delicata, F. , Atherton, Z. , Recchia, G. , Orban, G. , Lambert, R. C. , di Giovanni, G. , Leresche, N. , & Crunelli, V. (2018). Cortical drive and thalamic feed‐forward inhibition control thalamic output synchrony during absence seizures. Nature Neuroscience, 21, 744–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney, B. C. , Sze, W. , Lee, B. , & Murphy, G. G. (2009). Impaired long‐term potentiation and enhanced neuronal excitability in the amygdala of Ca(V)1.3 knockout mice. Neurobiology of Learning and Memory, 92, 519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRory, J. E. , Hamid, J. , Doering, C. J. , Garcia, E. , Parker, R. , Hamming, K. , Chen, L. , Hildebrand, M. , Beedle, A. M. , Feldcamp, L. , Zamponi, G. W. , & Snutch, T. P. (2004). The CACNA1F gene encodes an L‐type calcium channel with unique biophysical properties and tissue distribution. The Journal of Neuroscience, 24, 1707–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRory, J. E. , Santi, C. M. , Hamming, K. S. , Mezeyova, J. , Sutton, K. G. , Baillie, D. L. , Stea, A. , & Snutch, T. P. (2001). Molecular and functional characterization of a family of rat brain T‐type calcium channels. The Journal of Biological Chemistry, 276, 3999–4011. [DOI] [PubMed] [Google Scholar]
- Mefford, H. C. , Yendle, S. C. , Hsu, C. , Cook, J. , Geraghty, E. , McMahon, J. M. , Eeg‐Olofsson, O. , Sadleir, L. G. , Gill, D. , Ben‐Zeev, B. , Lerman‐Sagie, T. , Mackay, M. , Freeman, J. L. , Andermann, E. , Pelakanos, J. T. , Andrews, I. , Wallace, G. , Eichler, E. E. , Berkovic, S. F. , & Scheffer, I. E. (2011). Rare copy number variants are an important cause of epileptic encephalopathies. Annals of Neurology, 70, 974–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao, Q. L. , Herlitze, S. , Mark, M. D. , & Noebels, J. L. (2020). Adult loss of Cacna1a in mice recapitulates childhood absence epilepsy by distinct thalamic bursting mechanisms. Brain, 143, 161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirlohi, S. , Bladen, C. , Santiago, M. J. , Arnold, J. C. , McGregor, I. , & Connor, M. (2022). Inhibition of human recombinant T‐type calcium channels by phytocannabinoids in vitro. British Journal of Pharmacology, 179, 4031–4043. [DOI] [PubMed] [Google Scholar]
- Moosmang, S. , Haider, N. , Klugbauer, N. , Adelsberger, H. , Langwieser, N. , Müller, J. , Stiess, M. , Marais, E. , Schulla, V. , Lacinova, L. , Goebbels, S. , Nave, K. A. , Storm, D. R. , Hofmann, F. , & Kleppisch, T. (2005). Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor‐independent synaptic plasticity and spatial memory. The Journal of Neuroscience, 25, 9883–9892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morino, H. , Matsuda, Y. , Muguruma, K. , Miyamoto, R. , Ohsawa, R. , Ohtake, T. , Otobe, R. , Watanabe, M. , Maruyama, H. , Hashimoto, K. , & Kawakami, H. (2015). A mutation in the low voltage‐gated calcium channel CACNA1G alters the physiological properties of the channel, causing spinocerebellar ataxia. Molecular Brain, 8, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, C. S. , Haupt, A. , Bildl, W. , Schindler, J. , Knaus, H. G. , Meissner, M. , Rammner, B. , Striessnig, J. , Flockerzi, V. , Fakler, B. , & Schulte, U. (2010). Quantitative proteomics of the Cav2 channel nano‐environments in the mammalian brain. Proceedings of the National Academy of Sciences of the United States of America, 107, 14950–14957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muth, C. , Teufel, J. , Schöls, L. , Synofzik, M. , Franke, C. , Timmann, D. , Mansmann, U. , & Strupp, M. (2021). Fampridine and acetazolamide in EA2 and related familial EA: A prospective randomized placebo‐controlled trial. Neurology Clinical Practice, 11, e438–e446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narula, N. , Tester, D. J. , Paulmichl, A. , Maleszewski, J. J. , & Ackerman, M. J. (2015). Post‐mortem whole exome sequencing with gene‐specific analysis for autopsy‐negative sudden unexplained death in the young: A case series. Pediatric Cardiology, 36, 768–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naseer, M. I. , Abdulkareem, A. A. , Rasool, M. , Algahtani, H. , Muthaffar, O. Y. , & Pushparaj, P. N. (2022). Whole‐exome sequencing identifies novel SCN1A and CACNB4 genes mutations in the cohort of Saudi patients with epilepsy. Frontiers in Pediatrics, 10, 919996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noebels, J. L. , & Sidman, R. L. (1979). Inherited Epilepsy: Spike‐wave and focal motor seizures in the mutant mouse tottering. Science, 204, 1334–1336. [DOI] [PubMed] [Google Scholar]
- Obermair, G. J. , Szabo, Z. , Bourinet, E. , & Flucher, B. E. (2004). Differential targeting of the L‐type Ca2+ channel alpha 1C (CaV1.2) to synaptic and extrasynaptic compartments in hippocampal neurons. The European Journal of Neuroscience, 19, 2109–2122. [DOI] [PubMed] [Google Scholar]
- Olson, P. A. , Tkatch, T. , Hernandez‐Lopez, S. , Ulrich, S. , Ilijic, E. , Mugnaini, E. , Zhang, H. , Bezprozvanny, I. , & Surmeier, D. J. (2005). G‐protein‐coupled receptor modulation of striatal CaV1.3 L‐type Ca2+ channels is dependent on a Shank‐binding domain. The Journal of Neuroscience, 25, 1050–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parajuli, L. K. , Nakajima, C. , Kulik, A. , Matsui, K. , Schneider, T. , Shigemoto, R. , & Fukazawa, Y. (2012). Quantitative regional and ultrastructural localization of the Cav2.3 subunit of R‐type calcium channel in mouse brain. The Journal of Neuroscience, 32, 13555–13567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paşca, S. P. , Portmann, T. , Voineagu, I. , Yazawa, M. , Shcheglovitov, A. , Paşca, A. M. , Cord, B. , Palmer, T. D. , Chikahisa, S. , Nishino, S. , Bernstein, J. A. , Hallmayer, J. , Geschwind, D. H. , & Dolmetsch, R. E. (2011). Using iPSC‐derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nature Medicine, 17, 1657–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, R. , Bauer, C. S. , Nieto‐Rostro, M. , Margas, W. , Ferron, L. , Chaggar, K. , Crews, K. , Ramirez, J. D. , Bennett, D. L. H. , Schwartz, A. , Dickenson, A. H. , & Dolphin, A. C. (2013). alpha2delta‐1 gene deletion affects somatosensory neuron function and delays mechanical hypersensitivity in response to peripheral nerve damage. The Journal of Neuroscience, 33, 16412–16426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peloquin, J. B. , Khosravani, H. , Barr, W. , Bladen, C. , Evans, R. , Mezeyova, J. , Parker, D. , Snutch, T. P. , McRory, J. E. , & Zamponi, G. W. (2006). Functional analysis of Cav3.2 T‐type calcium channel mutations linked to childhood absence epilepsy. Epilepsia, 47, 655–658. [DOI] [PubMed] [Google Scholar]
- Pietrobon, D. , & Moskowitz, M. A. (2014). Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nature Reviews. Neuroscience, 15, 379–393. [DOI] [PubMed] [Google Scholar]
- Pinggera, A. , Mackenroth, L. , Rump, A. , Schallner, J. , Beleggia, F. , Wollnik, B. , & Striessnig, J. (2017). New gain‐of‐function mutation shows CACNA1D as recurrently mutated gene in autism spectrum disorders and epilepsy. Human Molecular Genetics, 26, 2923–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pippucci, T. , Parmeggiani, A. , Palombo, F. , Maresca, A. , Angius, A. , Crisponi, L. , Cucca, F. , Liguori, R. , Valentino, M. L. , Seri, M. , & Carelli, V. (2013). A novel null homozygous mutation confirms CACNA2D2 as a gene mutated in epileptic encephalopathy. PLoS One, 8, e82154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platzer, J. , Engel, J. , Schrott‐Fischer, A. , Stephan, K. , Bova, S. , Chen, H. , Zheng, H. , & Striessnig, J. (2000). Congenital deafness and sinoatrial node dysfunction in mice lacking class D L‐type Ca2+ channels. Cell, 102, 89–97. [DOI] [PubMed] [Google Scholar]
- Powell, K. L. , Cain, S. M. , Ng, C. , Sirdesai, S. , David, L. S. , Kyi, M. , Garcia, E. , Tyson, J. R. , Reid, C. A. , Bahlo, M. , Foote, S. J. , Snutch, T. P. , & O'Brien, T. J. (2009). A Cav3.2 T‐type calcium channel point mutation has splice‐variant‐specific effects on function and segregates with seizure expression in a polygenic rat model of absence epilepsy. Journal of Neuroscience, 29, 371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preiksaitiene, E. , Mannik, K. , Dirse, V. , Utkus, A. , Ciuladaite, Z. , Kasnauskiene, J. , Kurg, A. , & Kucinskas, V. (2012). A novel de novo 1.8 Mb microdeletion of 17q21.33 associated with intellectual disability and dysmorphic features. European Journal of Medical Genetics, 55, 656–659. [DOI] [PubMed] [Google Scholar]
- Punetha, J. , Karaca, E. , Gezdirici, A. , Lamont, R. E. , Pehlivan, D. , Marafi, D. , Appendino, J. P. , Hunter, J. V. , Akdemir, Z. C. , Fatih, J. M. , Jhangiani, S. N. , Gibbs, R. A. , Innes, A. M. , Posey, J. E. , & Lupski, J. R. (2019). Biallelic CACNA2D2 variants in epileptic encephalopathy and cerebellar atrophy. Annals of Clinical Translational Neurology, 6, 1395–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian, J. , & Noebels, J. L. (2000). Presynaptic Ca(2+) influx at a mouse central synapse with Ca(2+) channel subunit mutations. The Journal of Neuroscience, 20, 163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radzicki, D. , Yau, H. J. , Pollema‐Mays, S. L. , Mlsna, L. , Cho, K. , Koh, S. , & Martina, M. (2013). Temperature‐sensitive Cav1.2 calcium channels support intrinsic firing of pyramidal neurons and provide a target for the treatment of febrile seizures. The Journal of Neuroscience, 33, 9920–9931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajakulendran, S. , Graves, T. D. , Labrum, R. W. , Kotzadimitriou, D. , Eunson, L. , Davis, M. B. , Davies, R. , Wood, N. W. , Kullmann, D. M. , Hanna, M. G. , & Schorge, S. (2010). Genetic and functional characterisation of the P/Q calcium channel in episodic ataxia with epilepsy. The Journal of Physiology, 588, 1905–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riban, V. , Heulard, I. , Reversat, L. , Si Hocine, H. , & Verleye, M. (2022). Stiripentol inhibits spike‐and‐wave discharges in animal models of absence seizures: A new mechanism of action involving T‐type calcium channels. Epilepsia, 63, 1200–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinné, S. , Stallmeyer, B. , Pinggera, A. , Netter, M. F. , Matschke, L. A. , Dittmann, S. , Kirchhefer, U. , Neudorf, U. , Opp, J. , Striessnig, J. , Decher, N. , & Schulze‐Bahr, E. (2022). Whole exome sequencing identifies a heterozygous variant in the Cav1.3 gene CACNA1D associated with familial sinus node dysfunction and focal idiopathic epilepsy. International Journal of Molecular Sciences, 23, 14215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodan, L. H. , Spillmann, R. C. , Kurata, H. T. , Lamothe, S. M. , Maghera, J. , Jamra, R. A. , Alkelai, A. , Antonarakis, S. E. , Atallah, I. , Bar‐Yosef, O. , Bilan, F. , Bjorgo, K. , Blanc, X. , van Bogaert, P. , Bolkier, Y. , Burrage, L. C. , Christ, B. U. , Granadillo, J. L. , Dickson, P. , … Shashi, V. (2021). Phenotypic expansion of CACNA1C‐associated disorders to include isolated neurological manifestations. Genetics in Medicine, 23, 1922–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol, E. , Kruglikov, I. , van den Maagdenberg, A. M. J. M. , Rudy, B. , & Fishell, G. (2013). CaV2.1 ablation in cortical interneurons selectively impairs fast‐spiking basket cells and causes generalized seizures. Annals of Neurology, 74, 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer‐Bertrand, B. , Jequier Gygax, M. , Cisarova, K. , Rosenfeld, J. A. , Bassetti, J. A. , Moldovan, O. , O'Heir, E. , Burrage, L. C. , Allen, J. , Emrick, L. T. , Eastman, E. , Kumps, C. , Abbas, S. , van Winckel, G. , Undiagnosed Diseases Network , Chabane, N. , Zackai, E. H. , Lebon, S. , Keena, B. , … Superti‐Furga, A. (2021). De novo variants in CACNA1E found in patients with intellectual disability, developmental regression and social cognition deficit but no seizures. Molecular Autism, 12, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryvlin, P. , Nashef, L. , Lhatoo, S. D. , Bateman, L. M. , Bird, J. , Bleasel, A. , Boon, P. , Crespel, A. , Dworetzky, B. A. , Høgenhaven, H. , Lerche, H. , Maillard, L. , Malter, M. P. , Marchal, C. , Murthy, J. M. K. , Nitsche, M. , Pataraia, E. , Rabben, T. , Rheims, S. , … Tomson, T. (2013). Incidence and mechanisms of cardiorespiratory arrests in epilepsy monitoring units (MORTEMUS): A retrospective study. Lancet Neurology, 12, 966–977. [DOI] [PubMed] [Google Scholar]
- Sampedro‐Castañeda, M. , Baltussen, L. L. , Lopes, A. T. , Qiu, Y. , Sirvio, L. , Mihaylov, S. R. , Claxton, S. , Richardson, J. C. , Lignani, G. , & Ultanir, S. (2023). Epilepsy‐linked kinase CDKL5 phosphorylates voltage‐gated calcium channel Cav2.3, altering inactivation kinetics and neuronal excitability. bioRxiv, 2022.2011.2024.517538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santi, C. M. , Cayabyab, F. S. , Sutton, K. G. , McRory, J. E. , Mezeyova, J. , Hamming, K. S. , Parker, D. , Stea, A. , & Snutch, T. P. (2002). Differential inhibition of T‐type calcium channels by neuroleptics. The Journal of Neuroscience, 22, 396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki, S. , Huda, K. , Inoue, T. , Miyata, M. , & Imoto, K. (2006). Impaired feedforward inhibition of the thalamocortical projection in epileptic Ca2+ channel mutant mice, tottering. The Journal of Neuroscience, 26, 3056–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlick, B. , Flucher, B. E. , & Obermair, G. J. (2010). Voltage‐activated calcium channel expression profiles in mouse brain and cultured hippocampal neurons. Neuroscience, 167, 786–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl, U. I. , Goh, G. , Stölting, G. , de Oliveira, R. C. , Choi, M. , Overton, J. D. , Fonseca, A. L. , Korah, R. , Starker, L. F. , Kunstman, J. W. , Prasad, M. L. , Hartung, E. A. , Mauras, N. , Benson, M. R. , Brady, T. , Shapiro, J. R. , Loring, E. , Nelson‐Williams, C. , Libutti, S. K. , … Lifton, R. P. (2013). Somatic and germline CACNA1D calcium channel mutations in aldosterone‐producing adenomas and primary aldosteronism. Nature Genetics, 45, 1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöpf, C. L. , Ablinger, C. , Geisler, S. M. , Stanika, R. I. , Campiglio, M. , Kaufmann, W. A. , Nimmervoll, B. , Schlick, B. , Brockhaus, J. , Missler, M. , Shigemoto, R. , & Obermair, G. J. (2021). Presynaptic α(2)δ subunits are key organizers of glutamatergic synapses. Proceedings of the National Academy of Sciences of the United States of America, 118, e1920827118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova, N. A. , Ryzhkova, O. R. , Strokova, T. V. , & Taran, N. N. (2018). The third case report a patient with primary aldosteronism, seizures, and neurologic abnormalities (PASNA) syndrome de novo variant mutations in the CACNA1D gene. Zhurnal Nevrologii i Psikhiatrii Imeni S.S. Korsakova, 118, 49–52. [DOI] [PubMed] [Google Scholar]
- Siddique, A. , Willoughby, J. , DDD Study , & McNeill, A. (2017). A 7q21.11 microdeletion presenting with apparent intellectual disability without epilepsy. American Journal of Medical Genetics. Part A, 173, 1128–1130. [DOI] [PubMed] [Google Scholar]
- Singh, B. , Monteil, A. , Bidaud, I. , Sugimoto, Y. , Suzuki, T. , Hamano, S. I. , Oguni, H. , Osawa, M. , Alonso, M. E. , Delgado‐Escueta, A. V. , Inoue, Y. , Yasui‐Furukori, N. , Kaneko, S. , Lory, P. , & Yamakawa, K. (2007). Mutational analysis of CACNA1G in idiopathic generalized epilepsy. Mutation in brief #962. Online. Human Mutation, 28, 524–525. [DOI] [PubMed] [Google Scholar]
- Sinnegger‐Brauns, M. J. , Huber, I. G. , Koschak, A. , Wild, C. , Obermair, G. J. , Einzinger, U. , Hoda, J. C. , Sartori, S. B. , & Striessnig, J. (2009). Expression and 1,4‐dihydropyridine‐binding properties of brain L‐type calcium channel isoforms. Molecular Pharmacology, 75, 407–414. [DOI] [PubMed] [Google Scholar]
- Song, I. , Kim, D. , Choi, S. , Sun, M. , Kim, Y. , & Shin, H. S. (2004). Role of the alpha1G T‐type calcium channel in spontaneous absence seizures in mutant mice. The Journal of Neuroscience, 24, 5249–5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soong, T. W. , Stea, A. , Hodson, C. D. , Dubel, S. J. , Vincent, S. R. , & Snutch, T. P. (1993). Structure and functional expression of a member of the low voltage‐activated calcium channel family. Science, 260, 1133–1136. [DOI] [PubMed] [Google Scholar]
- Splawski, I. , Timothy, K. W. , Decher, N. , Kumar, P. , Sachse, F. B. , Beggs, A. H. , Sanguinetti, M. C. , & Keating, M. T. (2005). Severe arrhythmia disorder caused by cardiac L‐type calcium channel mutations. Proceedings of the National Academy of Sciences of the United States of America, 102, 8089–8096. discussion 8086–8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski, I. , Timothy, K. W. , Sharpe, L. M. , Decher, N. , Kumar, P. , Bloise, R. , Napolitano, C. , Schwartz, P. J. , Joseph, R. M. , Condouris, K. , Tager‐Flusberg, H. , Priori, S. G. , Sanguinetti, M. C. , & Keating, M. T. (2004). Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell, 119, 19–31. [DOI] [PubMed] [Google Scholar]
- Stam, A. H. , Luijckx, G.‐J. , Poll‐Thé, B. T. , Ginjaar, I. B. , Frants, R. R. , Haan, J. , Ferrari, M. D. , Terwindt, G. M. , & van den Maagdenberg, A. M. J. M. (2009). Early seizures and cerebral oedema after trivial head trauma associated with the CACNA1A S218L mutation. Journal of Neurology, Neurosurgery and Psychiatry, 80, 1125–1129. [DOI] [PubMed] [Google Scholar]
- Stanika, R. I. , Villanueva, I. , Kazanina, G. , Andrews, S. B. , & Pivovarova, N. B. (2012). Comparative impact of voltage‐gated calcium channels and NMDA receptors on mitochondria‐mediated neuronal injury. The Journal of Neuroscience, 32, 6642–6650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley, E. F. (1993). Single calcium channels and acetylcholine release at a presynaptic nerve terminal. Neuron, 11, 1007–1011. [DOI] [PubMed] [Google Scholar]
- Stendel, C. , D'Adamo, M. C. , Wiessner, M. , Dusl, M. , Cenciarini, M. , Belia, S. , Nematian‐Ardestani, E. , Bauer, P. , Senderek, J. , Klopstock, T. , & Pessia, M. (2020). Association of a novel splice site mutation in P/Q‐type calcium channels with childhood epilepsy and late‐onset slowly progressive non‐episodic cerebellar ataxia. International Journal of Molecular Sciences, 21, 3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom, T. M. , Nyakatura, G. , Apfelstedt‐Sylla, E. , Hellebrand, H. , Lorenz, B. , Weber, B. H. F. , Wutz, K. , Gutwillinger, N. , Rüther, K. , Drescher, B. , Sauer, C. , Zrenner, E. , Meitinger, T. , Rosenthal, A. , & Meindl, A. (1998). An L‐type calcium‐channel gene mutated in incomplete X‐linked congenital stationary night blindness. Nature Genetics, 19, 260–263. [DOI] [PubMed] [Google Scholar]
- Strupp, M. , Kalla, R. , Claassen, J. , Adrion, C. , Mansmann, U. , Klopstock, T. , Freilinger, T. , Neugebauer, H. , Spiegel, R. , Dichgans, M. , Lehmann‐Horn, F. , Jurkat‐Rott, K. , Brandt, T. , Jen, J. C. , & Jahn, K. (2011). A randomized trial of 4‐aminopyridine in EA2 and related familial episodic ataxias. Neurology, 77, 269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strupp, M. , Thurtell, M. J. , Shaikh, A. G. , Brandt, T. , Zee, D. S. , & Leigh, R. J. (2011). Pharmacotherapy of vestibular and ocular motor disorders, including nystagmus. Journal of Neurology, 258, 1207–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, S. , Kawakami, K. , Nishimura, S. , Watanabe, Y. , Yagi, K. , Scino, M. , & Miyamoto, K. (1992). Zonisamide blocks T‐type calcium channel in cultured neurons of rat cerebral cortex. Epilepsy Research, 12, 21–27. [DOI] [PubMed] [Google Scholar]
- Takahashi, M. , Seagar, M. J. , Jones, J. F. , Reber, B. F. , & Catterall, W. A. (1987). Subunit structure of dihydropyridine‐sensitive calcium channels from skeletal muscle. Proceedings of the National Academy of Sciences, 84, 5478–5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talley, E. M. , Cribbs, L. L. , Lee, J. H. , Daud, A. , Perez‐Reyes, E. , & Bayliss, D. A. (1999). Differential distribution of three members of a gene family encoding low voltage‐activated (T‐type) calcium channels. The Journal of Neuroscience, 19, 1895–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, C. M. , Presser, F. , & Morad, M. (1988). Amiloride selectively blocks the low threshold (T) calcium channel. Science, 240, 213–215. [DOI] [PubMed] [Google Scholar]
- Tippens, A. L. , Pare, J. F. , Langwieser, N. , Moosmang, S. , Milner, T. A. , Smith, Y. , & Lee, A. (2008). Ultrastructural evidence for pre‐ and postsynaptic localization of Cav1.2 L‐type Ca2+ channels in the rat hippocampus. The Journal of Comparative Neurology, 506, 569–583. [DOI] [PubMed] [Google Scholar]
- Todorovic, S. M. , & Lingle, C. J. (1998). Pharmacological properties of T‐type Ca2+ current in adult rat sensory neurons: Effects of anticonvulsant and anesthetic agents. Journal of Neurophysiology, 79, 240–252. [DOI] [PubMed] [Google Scholar]
- Tottene, A. , Volsen, S. , & Pietrobon, D. (2000). α1E subunits form the pore of three cerebellar R‐type calcium channels with different pharmacological and permeation properties. The Journal of Neuroscience, 20, 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travaglini, L. , Nardella, M. , Bellacchio, E. , D'Amico, A. , Capuano, A. , Frusciante, R. , di Capua, M. , Cusmai, R. , Barresi, S. , Morlino, S. , Fernández‐Fernández, J. M. , Trivisano, M. , Specchio, N. , Valeriani, M. , Vigevano, F. , Bertini, E. , & Zanni, G. (2017). Missense mutations of CACNA1A are a frequent cause of autosomal dominant nonprogressive congenital ataxia. European Journal of Paediatric Neurology, 21, 450–456. [DOI] [PubMed] [Google Scholar]
- Tringham, E. , Powell, K. L. , Cain, S. M. , Kuplast, K. , Mezeyova, J. , Weerapura, M. , Eduljee, C. , Jiang, X. , Smith, P. , Morrison, J. L. , Jones, N. C. , Braine, E. , Rind, G. , Fee‐Maki, M. , Parker, D. , Pajouhesh, H. , Parmar, M. , O'Brien, T. J. , & Snutch, T. P. (2012). T‐type calcium channel blockers that attenuate thalamic burst firing and suppress absence seizures. Science Translational Medicine, 4, 121ra119. [DOI] [PubMed] [Google Scholar]
- Tytgat, J. , Vereecke, J. , & Carmeliet, E. (1988). Differential effects of verapamil and flunarizine on cardiac L‐type and T‐type Ca channels. Naunyn‐Schmiedeberg's Archives of Pharmacology, 337, 690–692. [DOI] [PubMed] [Google Scholar]
- Uhlén, M. , Fagerberg, L. , Hallström, B. M. , Lindskog, C. , Oksvold, P. , Mardinoglu, A. , Sivertsson, Å. , Kampf, C. , Sjöstedt, E. , Asplund, A. , Olsson, I. M. , Edlund, K. , Lundberg, E. , Navani, S. , Szigyarto, C. A. K. , Odeberg, J. , Djureinovic, D. , Takanen, J. O. , Hober, S. , … Pontén, F. (2015). Tissue‐based map of the human proteome. Science, 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Valence, S. , Cochet, E. , Rougeot, C. , Garel, C. , Chantot‐Bastaraud, S. , Lainey, E. , Afenjar, A. , Barthez, M. A. , Bednarek, N. , Doummar, D. , Faivre, L. , Goizet, C. , Haye, D. , Heron, B. , Kemlin, I. , Lacombe, D. , Milh, M. , Moutard, M. L. , Riant, F. , … Burglen, L. (2019). Exome sequencing in congenital ataxia identifies two new candidate genes and highlights a pathophysiological link between some congenital ataxias and early infantile epileptic encephalopathies. Genetics in Medicine, 21, 553–563. [DOI] [PubMed] [Google Scholar]
- Valentino, F. , Bruno, L. P. , Doddato, G. , Giliberti, A. , Tita, R. , Resciniti, S. , Fallerini, C. , Bruttini, M. , Lo Rizzo, C. , Mencarelli, M. A. , Mari, F. , Pinto, A. M. , Fava, F. , Baldassarri, M. , Fabbiani, A. , Lamacchia, V. , Benetti, E. , Zguro, K. , Furini, S. , … Ariani, F. (2021). Exome sequencing in 200 intellectual disability/autistic patients: New candidates and atypical presentations. Brain Sciences, 11, 936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Maagdenberg, A. M. J. M. , Pizzorusso, T. , Kaja, S. , Terpolilli, N. , Shapovalova, M. , Hoebeek, F. E. , Barrett, C. F. , Gherardini, L. , van de Ven, R. C. G. , Todorov, B. , Broos, L. A. M. , Tottene, A. , Gao, Z. , Fodor, M. , de Zeeuw, C. I. , Frants, R. R. , Plesnila, N. , Plomp, J. J. , Pietrobon, D. , & Ferrari, M. D. (2010). High cortical spreading depression susceptibility and migraine‐associated symptoms in Cav2.1 S218L mice. Annals of Neurology, 67, 85–98. [DOI] [PubMed] [Google Scholar]
- Vergult, S. , Dheedene, A. , Meurs, A. , Faes, F. , Isidor, B. , Janssens, S. , Gautier, A. , Le Caignec, C. , & Menten, B. (2015). Genomic aberrations of the CACNA2D1 gene in three patients with epilepsy and intellectual disability. European Journal of Human Genetics, 23, 628–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitko, I. (2005). Functional characterization and neuronal modeling of the effects of childhood absence epilepsy variants of CACNA1H, a T‐type calcium channel. Journal of Neuroscience, 25, 4844–4855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakamori, M. , Yamazaki, K. , Matsunodaira, H. , Teramoto, T. , Tanaka, I. , Niidome, T. , Sawada, K. , Nishizawa, Y. , Sekiguchi, N. , Mori, E. , Mori, Y. , & Imoto, K. (1998). Single tottering mutations responsible for the neuropathic phenotype of the P‐type calcium channel. The Journal of Biological Chemistry, 273, 34857–34867. [DOI] [PubMed] [Google Scholar]
- Weiergräber, M. , Henry, M. , Ho, M. S. P. , Struck, H. , Hescheler, J. , & Schneider, T. (2008). Altered thalamocortical rhythmicity in Cav2.3‐deficient mice. Molecular and Cellular Neuroscience, 39, 605–618. [DOI] [PubMed] [Google Scholar]
- Weiergräber, M. , Henry, M. , Krieger, A. , Kamp, M. , Radhakrishnan, K. , Hescheler, J. , & Schneider, T. (2006). Altered seizure susceptibility in mice lacking the Cav2.3 E‐type Ca2+ channel. Epilepsia, 47, 839–850. [DOI] [PubMed] [Google Scholar]
- Weiergräber, M. , Henry, M. , Radhakrishnan, K. , Hescheler, J. , & Schneider, T. (2007). Hippocampal seizure resistance and reduced neuronal excitotoxicity in mice lacking the Cav2.3 E/R‐type voltage‐gated calcium channel. Journal of Neurophysiology, 97, 3660–3669. [DOI] [PubMed] [Google Scholar]
- Welling, A. , Ludwig, A. , Zimmer, S. , Klugbauer, N. , Flockerzi, V. , & Hofmann, F. (1997). Alternatively spliced IS6 segments of the α1C gene determine the tissue‐specific Dihydropyridine sensitivity of cardiac and vascular smooth muscle L‐type Ca2+ channels. Circulation Research, 81, 526–532. [DOI] [PubMed] [Google Scholar]
- Westenbroek, R. E. , Hell, J. W. , Warner, C. , Dubel, S. J. , Snutch, T. P. , & Catterall, W. A. (1992). Biochemical properties and subcellular distribution of an N‐type calcium channel alpha 1 subunit. Neuron, 9, 1099–1115. [DOI] [PubMed] [Google Scholar]
- Westenbroek, R. E. , Sakurai, T. , Elliott, E. M. , Hell, J. W. , Starr, T. V. B. , Snutch, T. P. , & Catterall, W. A. (1995). Immunochemical identification and subcellular‐distribution of the alpha(1a) subunits of brain calcium channels. Journal of Neuroscience, 15, 6403–6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, M. E. , Marubio, L. M. , Deal, C. R. , Hans, M. , Brust, P. F. , Philipson, L. H. , Miller, R. J. , Johnson, E. C. , Harpold, M. M. , & Ellis, S. B. (1994). Structure and functional characterization of neuronal alpha 1E calcium channel subtypes. Journal of Biological Chemistry, 269, 22347–22357. [PubMed] [Google Scholar]
- Wolff, M. , Johannesen, K. M. , Hedrich, U. B. S. , Masnada, S. , Rubboli, G. , Gardella, E. , Lesca, G. , Ville, D. , Milh, M. , Villard, L. , Afenjar, A. , Chantot‐Bastaraud, S. , Mignot, C. , Lardennois, C. , Nava, C. , Schwarz, N. , Gérard, M. , Perrin, L. , Doummar, D. , … Møller, R. S. (2017). Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A‐related disorders. Brain, 140, 1316–1336. [DOI] [PubMed] [Google Scholar]
- Wu, L. G. , Westenbroek, R. E. , Borst, J. G. , Catterall, W. A. , & Sakmann, B. (1999). Calcium channel types with distinct presynaptic localization couple differentially to transmitter release in single calyx‐type synapses. The Journal of Neuroscience, 19, 726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, W. , & Lipscombe, D. (2001). Neuronal CaV1.3α1 L‐type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. The Journal of Neuroscience, 21, 5944–5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda, T. , Chen, L. , Barr, W. , McRory, J. E. , Lewis, R. J. , Adams, D. J. , & Zamponi, G. W. (2004). Auxiliary subunit regulation of high‐voltage activated calcium channels expressed in mammalian cells. The European Journal of Neuroscience, 20, 1–13. [DOI] [PubMed] [Google Scholar]
- Zaitsev, A. V. , Povysheva, N. V. , Lewis, D. A. , & Krimer, L. S. (2007). P/Q‐type, but not N‐type, calcium channels mediate GABA release from fast‐spiking interneurons to pyramidal cells in rat prefrontal cortex. Journal of Neurophysiology, 97, 3567–3573. [DOI] [PubMed] [Google Scholar]
- Zaman, T. , Lee, K. , Park, C. , Paydar, A. , Choi, J. H. , Cheong, E. , Lee, C. J. , & Shin, H.‐S. (2011). CaV2.3 channels are critical for oscillatory burst discharges in the reticular thalamus and absence epilepsy. Neuron, 70, 95–108. [DOI] [PubMed] [Google Scholar]
- Zamponi, G. W. , Striessnig, J. , Koschak, A. , & Dolphin, A. C. (2015). The physiology, pathology, and pharmacology of voltage‐gated calcium channels and their future therapeutic potential. Pharmacological Reviews, 67, 821–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zangaladze, A. , Asadi‐Pooya, A. A. , Ashkenazi, A. , & Sperling, M. R. (2010). Sporadic hemiplegic migraine and epilepsy associated with CACNA1A gene mutation. Epilepsy & Behavior, 17, 293–295. [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Mori, M. , Burgess, D. L. , & Noebels, J. L. (2002). Mutations in high‐voltage‐activated calcium channel genes stimulate low‐voltage‐activated currents in mouse thalamic relay neurons. Journal of Neuroscience, 22, 6362–6371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuchenko, O. , Bailey, J. , Bonnen, P. , Ashizawa, T. , Stockton, D. W. , Amos, C. , Dobyns, W. B. , Subramony, S. H. , Zoghbi, H. Y. , & Lee, C. C. (1997). Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the α1A‐voltage‐dependent calcium channel. Nature Genetics, 15, 62–69. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing does not apply to this article as no datasets were generated or analysed during the current study.