

Abstract
Background and Objectives
Pathogenic CHD2 variants are associated with neurodevelopmental disorders and developmental and epileptic encephalopathy. While pediatric CHD2 phenotypes have been readily explored, adult phenotypes are not well understood. We aimed to investigate the phenotypic spectrum of adult patients with CHD2 variants.
Methods
Patients 18 years or older with likely pathogenic or pathogenic (LP/P) CHD2 variants were included. We used standardized tools to evaluate current seizures, medication use, sleep, gastrointestinal symptoms, pain response, gait, social communication disorder, and adaptive behavioral skills of patients.
Results
In this prospective study, 14 unrelated adult patients (age range: 18–45 years, median: 21 years) with LP/P CHD2 variants were described. Eleven novel variants were identified. No genotype-phenotype correlations were identified. 79% of adults still have ongoing seizures. Photosensitivity was present in 64% of adult patients. Autism spectrum disorder was diagnosed in 71% of patients. Only 29% were able to read and understand material at a sixth-grade level or higher. Behavioral issues were reported in 100% of adult patients, and 71% had internalizing features, such as anxiety. Self-injurious behaviors were present in 50%. Only 43% could ambulate independently. Additional characteristics included reflux (36%), constipation (71%), and abnormal pain responsiveness (43%). 1 patient presented with nonepileptic breath-holding spells leading to cyanosis. No patient could perform all basic activities of daily living independently, all the time. Higher seizure severity was associated with worse nonseizure outcomes (p = 0.04).
Discussion
Most adults with CHD2 continue to have seizures, and seizure severity is associated with worse comorbidities such as maladaptive behaviors, gait, gastrointestinal, sleep, and abnormal pain responsiveness. Longevity has not been systematically studied in this group of patients. Here we describe a group of adult patients (up to 45 years of age) and the natural history of this condition. These data may provide prognostic insights for families of pediatric patients and help identify key points to be addressed in future precision trials for patients with CHD2 variants.
Introduction
The CHD2 gene, located at chromosome 15q26.1, encodes chromodomain helicase DNA-binding protein 2 (CHD2), which is an ATP-dependent chromatin-remodeling enzyme. This gene interacts with gene regulatory sequences and histones necessary for early cortical development and synaptic function.1
In humans, pathogenic variants leading to CHD2 loss of function are mainly associated with developmental and epileptic encephalopathy (DEE) (OMIM#615369) but may also manifest as neurodevelopmental disorders (NDDs) without epilepsy.2 In 2009, a 30-month-old girl was described with refractory myoclonic epilepsy, intellectual disability (ID), delayed growth, and a distinctive facial appearance.3 She had a microdeletion of 15q26.1, which encompasses the CHD2 gene. In 2013, the CHD2 gene was specifically identified as a cause of DEE.4
The true prevalence of CHD2-related disorders is not known, in part because of its clinical heterogeneity and limited genetic testing to date, especially in adults. However, de novo CHD2 pathogenic variants were present in an estimated 1% of individuals included in cohorts with various DEEs.4,5 In another report of nearly 10,000 pediatric patients with epilepsy referred for genetic testing using a comprehensive epilepsy panel, 1 in 400 individuals were identified with a CHD2 pathogenic variant.6
Of all patients with CHD2 reported so far, only 27 individuals, reported across 16 publications, are adults older than 18 years2,4,7–21 Given the paucity of information on adult outcomes of patients with CHD2 variants, we performed a literature review of all reported cases of adults with CHD2 variants, both with and without seizures. We then conducted a prospective, cross-sectional study by recruiting and evaluating an international group of adults with DEE and NDDs without seizures. All patients harbored likely pathogenic or pathogenic (LP/P) CHD2 variants. Seizures, sleep and gastrointestinal disturbances, pain responsiveness, walking abilities, social communication skills, daily living abilities, and maladaptive behaviors were assessed. Findings were compared with those of previously reported adults with CHD2 and other adults with DEE to better distinguish the phenotype of adults with CHD2 variants specifically.
Methods
Search Strategy
A systematic online literature search of the PubMed and MEDLINE databases from inception through June 2024 was conducted using the search term “CHD2” (Figure 1). The references cited in the identified publications were searched for additional studies. Inclusion criteria included studies with clinical outcomes of humans. Studies were excluded if they (1) did not involve humans, (2) did not present new or unique data, (3) were non-English studies, (4) were unrelated, and (5) did not provide information on clinical phenotypes. Information that was not reported by the authors was left blank. “N/A” was used when authors explicitly stated that information was not available.
Figure 1. Study Identification Flow Diagram.
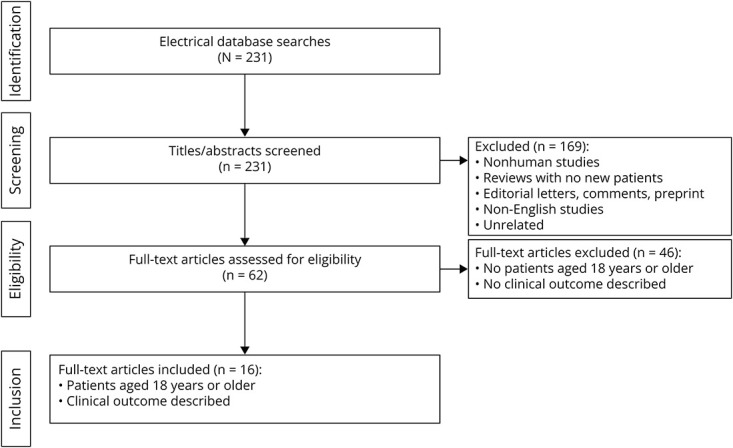
Of 231 searches for “CHD2,” 16 full-text articles featuring patients aged 18 years or older were identified.
Study Population and Data Collection
Between July 2021 and February 2023, patients aged 18 years or older with CHD2 LP/P variants were enrolled in this prospective study. Recruitment was conducted using the author's practices and with the assistance of Coalition to Cure CHD2. Study materials were translated and made available in English and Spanish. Clinical data collection involved the completion of 3 validated assessment tools, described further.
Standard Protocol Approvals, Registrations, and Patient Consents
Institutional approval for the study was granted by the Research Ethics Board at the University Health Network (protocol number 21-5009) based in Toronto, Canada. Informed and written consent was obtained from the substitute decision makers for all patients.
Severity of Clinical Outcomes
Disease severity was assessed using the modified version of the Severity Assessment22 tool that provided a composite score. 51 items consisting of a Likert scale rating were used to investigate seizures, treatments, gait, pain responsiveness, toileting, reflux, and sleep abnormalities. Ratings of each patient's overall condition and therapy effectiveness were also assessed. Clinical phenotype was considered most severe with a maximum score of 129/129. Seizures were classified using the 2017 International League Against Epilepsy guidelines.23
Social Communication Skills
The presence of communication deficits was screened using the Social Communication Questionnaire (SCQ) (Lifetime Version).24 Total scores were yielded and interpreted in reference to the cutoff score of 15. If patients scored more than the cutoff, it is likely that they are on the autism spectrum, suggesting that a more extensive evaluation should be conducted to confirm the screening findings.
Adaptive Behavioral Skills
Adaptive behavioral abilities were assessed using the Vineland Adaptive Behavior Scale 3 (VABS3).25 The VABS3 assessed abilities in the following domains: communication (receptive, expressive, and written), daily living skills (personal, domestic, and community), socialization (interpersonal relationships, play/leisure, and coping skills), motor ability (fine and gross motor), and maladaptive behaviors. An overall rating was given based on the patient's results compared with those of a norm sample, which is a representative group of patients the same age from across the United States. From each domain, raw scores were extracted and selected because of floor effects found in age-wise comparisons.
Statistical Analyses
Patient characteristics and clinical features were summarized using descriptive statistics. A comparison of assessment scores with variant types (missense, nonsense, deletion, splice acceptor, and frameshift where applicable) and affected functional domains was conducted using the Kruskal-Wallis test. Analyses were conducted using R, and statistical significance was set to <0.05. Statistical values were not reported for nonstatistically significant findings. Figures were created using GraphPad Prism.
Variant Interpretation
The pathogenicity of CHD2 variants was interpreted as per the American College of Medical Genetics and Genomics guidelines.26 The GRCh38 or hg38 build was used. The ClinVar database and GnomAD browser were used to query each of the CHD2 variants.27,28
Data Availability
Deidentified data may be provided on reasonable request.
Results
Cohort Description
Fourteen adult patients (7 women) participated in the study, with a median age of 21 years (range: 18–45 years) (Table 1). Patients were recruited from the following countries: Canada, the United States, and Spain. All patients came from nonconsanguineous families. One patient had a family history of epilepsy. This study presents the oldest patient with a CHD2 variant, who is a 45-year-old woman.
Table 1.
Demographics of 14 Adult Patients With CHD2 Variants and Their Genetic Profiles (n = 14)
| Patient # | Age range (y) | Family history | Exon # | cDNA | Protein consequence | Variant type | Inheritance | ACMG interpretation | Variant previously reported? |
| 1 | 18–25 | No | N/A | 93306085_93529447del | 223.362 kbp | Deletion | De novo | Pathogenic | No |
| 2 | 18–25 | No | 21 | c.2674delC | p.Gln892fs*19 | Frameshift | De novo | Pathogenic | No |
| 3 | 26–30 | No | 19 | c.2397_2398delTG | p.Leu800Aspfs*25 | Frameshift | De novo | Pathogenic | No |
| 4 | 18–25 | No | 19 | c.2423_2424insAT | p.Asn808Lysfs*11 | Frameshift | De novo | Likely pathogenic | No |
| 5 | 18–25 | No | 17 | c.2087delT | p.Phe696Serfs*38 | Frameshift | Parents not tested | Likely pathogenic | No |
| 6 | 30+ | No | 19 | c.2416_2417dupAG | p.Asn808Glyfs*11 | Frameshift | De novo | Pathogenic | No |
| 7 | 18–25 | Yes | 38 | c.4968dup | p.Trp1657Metfs*10 | Frameshift | De novo | Pathogenic | Suls et al. (2013) |
| 8 | 18–25 | No | 32 | c.4133T > G | p.Val1378Gly | Missense | De novo | Likely pathogenic | No |
| 9 | 18–25 | No | 21 | c.2636C > T | p.Ala879Val | Missense | De novo | Pathogenic | No |
| 10 | 18–25 | No | 26 | c.3298G > A | p.Ala1100Thr | Missense | De novo | Likely pathogenic | No |
| 11 | 26–30 | No | 36 | c.4602G > T | p.Trp1534Cys | Missense | De novo | Likely pathogenic | No |
| 12 | 30+ | No | 17 | c.2095C > T | p.Arg699Trp | Missense | De novo | Pathogenic | De Maria et al. (2022) |
| 13 | 18–25 | No | 38 | c.4909C > T | p.Arg1637* | Nonsense | De novo | Pathogenic | Suls et al. (2013); Carvill et al. (2013); Galizia et al. (2015) |
| 14 | 18–25 | No | 8 | c.789dupT | p.Glu264* | Nonsense | De novo | Likely pathogenic | No |
Abbreviations: ASD = autism spectrum disorder; F = female; M = male.
Sex, age, ASD diagnosis, family history of epilepsy, and ethnicity are listed when provided. The zygosity of all variants was heterozygous.
Patients had a spectrum of genetic variants: 2 patients had nonsense, 5 had missense, and 6 had frameshift leading to protein truncation. In addition, 1 patient had a pathogenic 223.362 kb deletion in chromosome 15q26.1, which partially overlaps with the CHD2 gene. Of our cohort, 13 (93%) of 14 variants arose de novo. One patient did not have both parents available for testing, so inheritance is unknown.
11 LP/P variants in our cohort were novel and have not been previously reported (Table 1).
Phenotypic Spectrum
Seizures
Of 14 patients, 7 patients (50%) reported at least 1 nonconvulsive seizure in the past 12 months, 1 (7%) had them monthly, 3 (21%) had them weekly, and 3 (21%) had them daily. 6 (43%) of the 14 patients had at least 1 convulsive seizure in the past 12 months. Specifically, of 14 patients, 3 (21%) had monthly convulsive seizures, 1 (7%) had them weekly, and 2 (14%) had them daily. Disruptive isolated epileptic spasms in the past 12 months were noted in 6 (43%) of the 14 patients, where 3 (21%) had them monthly, 2 (14%) had them weekly, and 1 (7%) had them daily. 1 patient (7%) had prolonged seizures lasting longer than 5 minutes in the previous 12 months. These prolonged seizures happened approximately once a week (Table 2).
Table 2.
Summary of Seizure and Photosensitivity Findings for 14 Adult Patients With CHD2 Variants in the Past 12 Months
| Seizure types | % of patients (n = 14) | Frequency in the past 12 mo | |||
| At least once | Monthly | Weekly | Daily | ||
| Nonconvulsive seizures, n (%) | 7/14 (50) | 7/14 (50) | 1/14 (7) | 3/14 (21) | 3/14 (21) |
| Convulsive seizures, n (%) | 6/14 (43) | 6/14 (43) | 3/14 (21) | 1/14 (7) | 2/14 (14) |
| Disruptive isolated spasms, n (%) | 6/14 (43) | 6/14 (43) | 3/14 (21) | 2/14 (14) | 1/14 (7) |
| Prolonged seizures (>5 mins), n (%) | 1/14 (7) | — | — | 1/14 (7) | — |
| Photosensitive epilepsy, n (%) | 9/14 (64) | — | — | — | — |
| PPR response on EEG, n (%) | 3/14 (21) | — | — | — | — |
One patient (1/14, 7%) did not have any seizures ever.
Photosensitive epilepsy was reported in 9 (64%) of 14 patients. One patient had strobe light–induced seizures. Avoidance of sunlight was noted in 2 (14%) of 14 adults. Four patients (29%) showed sensitivity to flashing lights. Three (21%) of 14 patients demonstrated photoparoxysmal responses on EEG. Of these 3 patients with PPR, 2 also had clinical photosensitivity (Table 2).
Caregivers reported a median of at least 5 lifetime antiseizure medications (ASMs) (range: 0–5+ ASMs) and a median current usage of 2 ASMs (range: 0–5+ ASMs). Only 2 (14%) of 14 patients have tried the ketogenic diet. Trial of vagus nerve stimulation (VNS) was reported in 2 (14%) of 14 patients, and deep brain stimulation (DBS) was reported in 2 (14%) of 14 patients. Currently, patients #7 and #14 have implanted VNS and patient #9 has an active DBS device.
Walking Abilities and Scoliosis
All patients were able to walk at least 15–50 feet outside the home (Figure 2), but only 6 (43%) of 14 patients could walk independently with no assistance, including the abilities to run, climb on level and uneven terrain, and climb stairs without difficulty or assistance. Of the 8 patients (57%) who could not ambulate independently, 5 walked with assistance and 3 used wheelchairs most of the time.
Figure 2. Walking Abilities of Adults With CHD2.
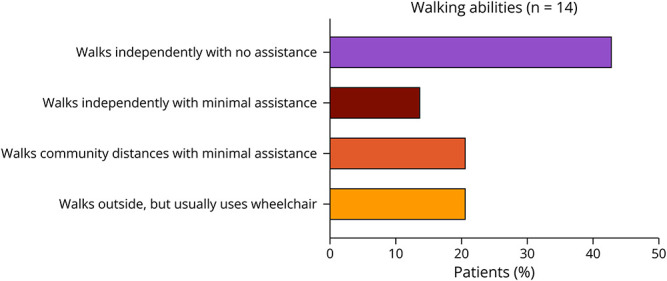
Percentage prevalence of a range of walking abilities is provided (n = 14).
Eight (57%) of 14 patients did not have scoliosis, and 6 (43%) of 14 patients dealt with mild scoliosis not requiring treatment. Leg deformities were reported in 2 patients, as well as gigantism in 1 man.
Social Communication Disorders and Autism Spectrum Disorders
In our cohort, 11 (79%) of 14 patients scored above the threshold on the SCQ. Ten of these 11 patients had a previous formal diagnosis of ASD given by a physician or health care provider. Specific ASD-like features were consistent across patients, including ritual-like behaviors (14/14, 100%), special interests with unusual intensity (10/14, 71%), and odd phrases or echolalia (9/14, 64%).
Adaptive Behaviors
All patients in this cohort had lower scores compared with normative data. All patients (100%) were able to feed themselves with a fork and spoon. Twelve patients (86%) were cooperative in personal activities such as undressing, dressing, and washing of the hands and face. Thirteen (93%) of the 14 patients could dress themselves, such as putting on clothes from the front and pulling up pants with elastic waistbands. 8 patients (57%) could bathe themselves. Twelve patients (86%) were able to sometimes sit unsupported for at least 10 minutes, and all patients were able to stand unsupported for at least 1 minute.
Regarding language abilities, all patients could understand at least 50 words. Thirteen (93%) of 14 patients could say one-word requests and could speak at least 50 words. All patients could recognize their own written name. Thirteen (93%) of 14 could recognize at least 10 alphabet letters while 11 (79%) of 14 could read at least 10 words. Four (29%) of 14 patients were able to read and understand material at a sixth-grade level or higher.
Maladaptive Behaviors
All patients exhibited maladaptive behavioral concerns. Various internalizing behaviors were reported, such as being overly needy/dependent (10/14, 71%), anxiety (10/14, 71%), and irritability (10/14, 71%). Avoidant behavior (8/14, 57%) and fearfulness of common objects or situations (10/14, 71%) were also present. Externalizing features included stubbornness (10/14, 71%), temper tantrums (7/14, 50%), and physical aggression (7/14, 50%). Compulsive behaviors (8/14, 57%) and overfixation on certain topics (10/14, 71%) were also observed. Delusional beliefs were present in 2 (14%) of 14 patients, and self-injurious behaviors were reported in 50%.
Comorbidities
Summary graphs of gastrointestinal clinical features in CHD2 adults are shown in Figure 3. In the 12 months before enrolling, swallowing was normal in 13 (93%) of 14 patients, but 1 patient required more than 30 minutes to eat a meal. Reflux was uncontrolled in 2 (14%) of 14 patients but was controlled in 3 (21%) and absent in 9 (64%) of 14 patients. Ten (71%) of 14 patients dealt with constipation issues while 3 of these patients had uncontrolled constipation. One woman was diagnosed with celiac disease at 19 years.
Figure 3. Summary Graphs of Gastrointestinal Clinical Features in Adults With CHD2.

Information on (A) constipation, (B) toileting, and (C) reflux are provided (n = 14). No statistically significant differences between variant types or functional domains were found.
Breathing was normal in 12 (86%) of 14 patients, but 1 patient had occasional breath-holding or hyperventilating spells. Episodes of cyanosis due to abnormal breathing not associated with seizures were reported in 1 patient.
Of 14 patients, 11 (79%) had normal toileting in the past 12 months, 2 (14%) required timed toileting, and 1 patient wore diapers only. Eight (57%) of 14 patients exhibited normal reactions to pain, 2 (14%) of 14 patients showed delayed reactions to minor pain, and 4 (29%) of 14 patients exhibited delayed pain responsiveness to major pain.
Summary graphs of other clinical features in CHD2 adults are shown in Figure 4. Sleep in the past 12 months was normal for 10 (71%) of 14 patients. Of the remaining patients, 3 aroused once per week and 1 patient aroused more than once per week. Disruptive daytime sleepiness varied in severity across 12 (86%) of 14 patients. Only 2 (14%) of 14 patients had no issues. While it was rare in 5 (36%) of 14 patients, 7 (50%) of 14 patients had disruptive daytime sleepiness ranging from 2 to 6 days per week. Two of these patients struggled with disruptive, constant daytime sleepiness throughout day, every day.
Figure 4. Summary Graphs of Other Clinical Features in Adults With CHD2.

Information on (A) sleep disturbances, (B) disruptive daytime sleepiness, and (C) pain responsiveness are provided (n = 14). No statistically significant differences between variant types or functional domains were found.
When comparing the severity scores of nonseizure outcomes with seizure severity scores, we found a statistically significant linear correlation (p = 0.04) (Figure 5). Increasing seizure severity was associated with worse nonseizure outcomes, as presented in Figures 2 and 3.
Figure 5. Seizure Severity Scores vs Nonseizure Outcome Severity Score.
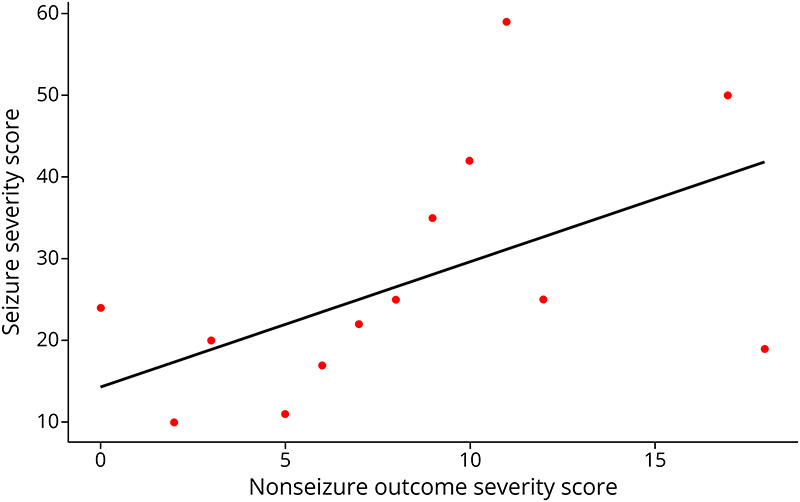
s (p = 0.04) Increasing seizure severity was associated with increasing severity of nonseizure outcomes reported in Figures 2 and 3.
Longevity
This study features the oldest CHD2 patient currently presented in detail in the literature. The patient is a 45-year-old woman carrying a de novo pathogenic missense variant (p.Arg669Trp) in exon 15 of unknown inheritance. The patient does not have epilepsy and has never had any known seizures. She does not have photosensitivity. She was diagnosed with “pervasive development disorder with autistic overtones” at 28 years. The patient walks independently without assistance and does not have scoliosis. She lives in a care home with 2 other intellectually disabled women. She works with the support of a job coach, performing assorted tasks for the care home she lives in and earning minimum wage.
However, the patient demonstrates aggressive behaviors, exhibiting aggression toward peers and family members, and has also harmed pets. As such, she cannot be left alone with her peers. ASD-like traits are also readily seen in the patient, including ritualistic behaviors, special interests, and fixations. She exhibits behaviors such as hoarding and overeating. In addition, the patient has delusions surrounding fictional characters, which she claims are “real and live in another realm.”
Discussion
This study features the molecular and clinical information of 14 adults with CHD2-related NDD. This is the largest study looking at the adult phenotype of this condition, which so far, has been mainly described in children. Our cohort's median age was 21 years (n = 14, range: 18–45 years). Drug-resistant seizures were seen in 79% of patients, adaptive behaviors were low in 100% compared with individuals of their age, and 57% of patients required some form of assistance ambulating. Aggression was observed in half of the adults, and 71% of them had ASD. However, this is possibly an underestimation because 1 patient who scored above the threshold on the SCQ has not yet been formally tested to rule out ASD. No patient was completely independent for activities of daily living (ADLs). In addition, several patients had different degrees of difficulties in other areas such as abnormal sleep, daytime sleepiness, abnormal pain responsivity, reflux, constipation, and toileting issues.
Previously, only 24 adults (median: 26 years, range: 18–65 years) with CHD2 variants have been reported across 16 different studies that focused mainly on children. The oldest patient (65-year-old) previously described had a concurrent paracentric inversion of chromosome 16 (inv(16)(q22.3q24.1)).21 We reviewed all those previously published adult cases here (eTable 1) and compared them with our patients.
No phenotype-genotype correlations could be extrapolated in this study, and 11 of 14 patients had novel P/LP variants in CHD2. Both the pathogenic p.Trp1657Metfs*10 PTV and p.Arg699Trp variants have been reported once.2,9 The p.Arg1637* pathogenic variant has been reported in 2 other patients.4,29 The p.Trp1657Metfs*10 variant has been seen in a 24-year-old woman with normal development before seizure onset at 2 years of age.2 Seizure types included eyelid myoclonia with absences (EMAs), myoclonic seizures, and GTCS. Seizures were pharmacoresistant and often cluster. She has a lifetime medication usage of 8 ASMs and continues to have drug-resistant seizures. Our patient with the same p.Trp1657Metfs*10 variant is an 18-year-old man with the most severe phenotype across our cohort. He has daily convulsive and weekly nonconvulsive seizures. Prolonged seizures lasting longer than 5 minutes requiring rescue medications occurred more than 5 times in the previous 12 months. In addition, the patient has weekly isolated epileptic spasms and myoclonic jerks that tend to cluster. Like the patient previously reported,2 our patient has pharmacoresistant epilepsy despite being on 4 ASMs and having an implanted VNS.
The p.Arg1637* variant has been reported in 2 patients.27,28,30 Detailed seizure description of one of these cases showed evolution from early-onset absence epilepsy to idiopathic photosensitive occipital epilepsy.28 In our study, a 19-year-old woman carrying an p. Arg1637* variant has epilepsy, ID, and ASD diagnoses. She has daily nonconvulsive seizures and monthly convulsive seizures. She has isolated epileptic spasms that cluster and occur weekly. She required rescue medications twice in the past 12 months and 2 emergency department visits in the past 12 months because of her seizures.
The 45-year-old patient in this study shares a variant (p.Arg699Trp) previously reported in 2 patients with epilepsy.9 One 21-year-old man exhibited a more severe phenotype featuring DEE and severe ID. This patient was nonverbal, had scoliosis, and demonstrated aggressive behavior. The other patient with the same variant was a 2-year-old girl with a milder clinical presentation with pharmacoresponsive epilepsy, language delay, and attention deficits. Her development was normal with no scoliosis. The exact cause of such variation in the phenotype cannot be confirmed. They are likely multifactorial, including gene-environment interactions and polygenic burden of autism, ID, and epilepsy.30-32 Furthermore, it is unclear how the 2-year-old patient will evolve and whether she will eventually develop pharmacoresistant epilepsy.
Previous investigations of CHD2 patients have noted that seizure onset typically occurs between 6 months and 4 years; however, disease onset may occur as early as 3 months27-29,33 Although 23 (85%) of 27 patients previously described had seizures starting mainly in the first few years of life, it is unclear how many of them still had seizures in adulthood. As such, our findings of drug-resistant seizures in 79% of all adults in this cohort are a significant alert that this symptom needs life-long management.
Clinical photosensitivity is a common feature of CHD2-related conditions, especially EMA, and such seizures can even be self-induced.7,9,29,30,33,34 A review of patients with CHD2 has found that photosensitivity (either clinical or electrographic) has an overall prevalence of 46%.9 Of interest, photosensitivity was slightly more prevalent in our adult population (64%). Despite the strong association between photosensitivity and CHD2 variants, the specific cause is unknown. Knockdown of chd2 in zebrafish results in larvae with stunted growth, seizure-like behavior, and photosensitivity, which align with findings in humans.2,29
In our adult cohort, gait abnormalities requiring walking assistance were seen in over half of the patients (57%). This is a very high proportion of gait abnormalities in a group of mainly “young adults.” Of the 27 adults with CHD2 previously described in 16 different studies, “gait problems” were seen in 26%. The methodology in the different studies may account for the difference in the proportion of adults with gait abnormalities, as in this study we used the Severity Assessment Tool20 in all patients. Gait abnormalities have also been documented in adults with other DEEs. For example, only 21% of patients with STXBP1-DEE35 could walk independently on all surfaces, and no adults with SYNGAP1-DEE36 were able to walk independently (Table 3).
Table 3.
Comparison of CHD2 Adults With Those of Other Studies Systematically Evaluating Nonseizure Outcomes and Adaptive Behaviors in Adults With Other DEEs (SYNGAP1-DEE and STXBP1-DEE)
| CHD2 (this study) | SYNGAP1 (Rong et al., 2023) | STXBP1 (Stamberger et al., 2021) | |
| No. of patients | 14 | 14 | Varied (26–28) |
| Demographic | Adults (18+ y) | Adults (18+ y) | Adults (18+ y) |
| Nonseizure outcomes, % | |||
| Abnormal swallowing | 7 | 36 | |
| Constipation | 71 | 71 | |
| Abnormal toileting | 21 | 71 | 71 |
| Abnormal pain responsiveness | 43 | 100 | |
| Sleep disturbances | 29 | 86 | |
| Disruptive daytime sleepiness | 86 | 79 | |
| Walking abilities, % | |||
| Independent, all surfaces | 43 | 0 | 21 |
| Independent, level surfaces | 0 | 14 | 29 |
| With assistance (crutches, walkers) | 36 | 50 | 11 |
| Usually wheelchair/no regular walking | 21 | 29 | 7 |
| Nonambulatory | 0 | 7 | 32 |
| Verbal abilities, % | |||
| Single words or more | 100 | 100 | 29 |
| Nonverbal | 0 | 0 | 71 |
| Independence for daily living skills, % | |||
| Washing independence | 57 | 14 | 0 |
| Dressing independence | 93 | 64 | 0 |
| Toileting independence | 64 | 21 | 7 |
| Feeding independence | 100 | 64 | 4 |
Abbreviation: DEE = developmental and epileptic encephalopathy.
Features not evaluated were left blank.
Unfortunately, lower mobility has been associated with a decreased ability to complete activities of daily living in older adult patients with ID.37 Finally, scoliosis was observed in 43% of adults in this cohort, which is increased compared with 15% of current adults with CHD2 in the literature.8,14,15
Social communication deficits were an important finding in our adult cohort, recapitulating CHD2 as a high-risk gene associated with ASD, with or without seizures.38 In a literature review of 102 patients with CHD2, ASD and autistic features had a prevalence of 56%.9 Of interest, among the 27 adults identified in our literature review, only 8 (30%) of them had ASD.7-10,17-19 We found a much higher proportion of diagnosed ASD in our adult population (71%). This significantly higher prevalence could be attributed to potential underdiagnoses, as seen with one of our patients who scored above the SCQ threshold but has never been formally tested for ASD.
A review of patients with CHD2 (across pediatric and adult cases) found that behavioral issues were noted in 42% of patients in the literature.9 However, no systematic evaluation of maladaptive behaviors has been previously published in patients with CHD2. Of interest, using the VABS3, 100% of our adults with CHD2 showed maladaptive behavioral issues. The discrepancy may be attributed to differences in methodology or the fact that maladaptive behaviors are more common (or more easily diagnosed) in adults. Specific internalizing maladaptive behaviors such as anxiety and being overly needy have not been systematically evaluated in patients with CHD2. We found that those symptoms were present in 71% of our adults studied. When compared with descriptions in 3 (11%) of 27 adults with CHD2 (with or without seizures) in the literature,2,4,7-16 this finding offers new insights into the adult CHD2 phenotype. Finally, aggression and oppositional behavior have been reported in 14 (52%) of 27 adults even in nonepileptic phenotypes,9,17 which falls in line with the 50% prevalence in our cohort.
Verbal ability has been reported in 11 (41%) of 27 adults in the literature, and 100% demonstrated language and speech deficits.7-10,13,14,17,22 While the exact definitions of verbal ability were not specified in previous works, all adults in this study demonstrated language abilities of at least 50 words. None of our CHD2 adults was nonverbal, although most had a very limited vocabulary of 50 words or less. Verbal skills in adults with CHD2 were similar to those seen in adults with SYNGAP1 and much better than those seen in adults with STXBP1 where 71% of adults were nonverbal (Table 3).35,36
Delusions were observed in 2 adults in this study. Psychotic episodes and delusions not associated with seizures have been noted in 4 (15%) of 27 patients in the literature, where all patients were adults.7,10,14
Basic activities of daily living refer to the ability of a person to manage his/her own physical needs, including eating, transferring, or ambulating; personal hygiene; dressing; and toileting. ADLs have not been systematically evaluated in adults nor pediatric populations with P/LP CHD2. In this study, we evaluated adult independency to complete ADLs. No adult patients with CHD2 were completely independent for all their ADLs. However, they overall did better than adults with SYNGAP1 and STXBP1-DEEs (Table 3).
Various clinical features such as sleep dysfunction, toileting accidents, and abnormal pain processing are also seen in our CHD2 adults.39 These abnormalities are also seen in other patients with DEEs and are compared in Table 3.
This study details the oldest patient with CHD2 reported so far. This is a 45-year-old patient, who has never had seizures but has problematic aggressive behaviors. She has a de novo c.2095C > T CHD2 missense variant. The lifespan of patients with CHD2-associated conditions is currently unknown. The previously oldest patient in the literature was a 35-year-old woman reported in 2015. That patient had a de novo 2.4 Mb deletion involving CHD2 and had a mild phenotype, described only as “disadvantaged.” She also had epilepsy and exhibited ASD-like traits and aggression.7
Limitations of this study include its cross-sectional design. Furthermore, despite being large for such rare conditions, the sample size is still small. This might be in part due to the somewhat recency of CHD2 being linked to NDDs and DEEs and the fact that adult neurologists generally do not update genetic testing previously performed in childhood.40,41
This study presented the spectrum of clinical outcomes of adults specifically with CHD2-associated disorders and the oldest patient (45-year-old woman) currently described in the literature. Specifically, 79% of adults still have pharmacoresistant seizures. Both externalizing and internalizing behavioral issues were identified as prominent in adults with CHD2. While 57% of patients required some form of assistance walking, all adults with CHD2 were ambulatory. Overall, adult patients with CHD2 are not independent for ADLs and dependent on their caregivers. These data will be relevant when evaluating the results of precision drug trials on seizures and nonseizure comorbidities. Further longitudinal studies using standardized scales will help build a more comprehensive landscape of adult CHD2 phenotypes.
Acknowledgment
The authors thank all patients and their families and the Coalition to Cure CHD2 for participating in this study.
Glossary
- ADLs
activities of daily living
- ASD
autism spectrum disorder
- DBS
deep brain stimulation
- DEE
developmental and epileptic encephalopathy
- EMA
eyelid myoclonia with absence
- ID
intellectual disability
- NDD
neurodevelopmental disorder
- SCQ
Social Communication Questionnaire
- VABS3
Vineland Adaptive Behavior Scale 3
- VNS
vagus nerve stimulation
Appendix. Authors
| Name | Location | Contribution |
| Marlene Rong, MSc | Institute of Medical Science, University of Toronto, Adult Genetic Epilepsy (AGE) Program, Krembil Neurosciences Institute, Toronto Western Hospital, University Health Network, Canada | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data |
| Quratulain Zulfiqar Ali, MD | Adult Genetic Epilepsy (AGE) Program, Krembil Neurosciences Institute, Toronto Western Hospital, University Health Network, Canada | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data |
| Angel Aledo-Serrano, MD | Epilepsy Unit, Vithas Clinical Neuroscience Institute, Vithas Madrid University Hospitals, Faculty of Experimental Sciences, Francisco de Vitoria University, Madrid, Spain | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data |
| Allan Bayat, MD, PhD | Department of Drug Design and Pharmacology, University of Copenhagen, Department for Genetics and Personalized Medicine, Danish Epilepsy Centre, Dianalund, Institute for Regional Health Services, University of Southern Denmark, Odense | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data |
| Orrin Devinsky, MD, PhD | NYU Langone Epilepsy Center | Drafting/revision of the manuscript for content, including medical writing for content |
| Farah Qaiser | Adult Genetic Epilepsy (AGE) Program, Krembil Neurosciences Institute, Toronto Western Hospital, University Health Network, Canada | Drafting/revision of the manuscript for content, including medical writing for content; analysis or interpretation of data |
| Ilakkiah Chandran, BSc | Adult Genetic Epilepsy (AGE) Program, Krembil Neurosciences Institute, Toronto Western Hospital, University Health Network, Canada | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data |
| Anum Ali | Adult Genetic Epilepsy (AGE) Program, Krembil Neurosciences Institute, Toronto Western Hospital, University Health Network, Canada | Analysis or interpretation of data |
| Alfonso Fasano, MD, PhD | Division of Neurology, Department of Medicine, University of Toronto, Krembil Brain Institute, Edmond J. Safra Program in Parkinson's Disease, Morton and Gloria Shulman Movement Disorders Clinic, Toronto Western Hospital, UHN, ON, Canada | Drafting/revision of the manuscript for content, including medical writing for content; study concept or design |
| Anne S. Bassett, MD | Clinical Genetics Research Program, Centre for Addiction and Mental Health, The Dalglish Family 22q Clinic, Toronto General Hospital, University Health Network, Department of Psychiatry, University of Toronto, Toronto Congenital Cardiac Centre for Adults, Division of Cardiology, Department of Medicine, and Department of Psychiatry, University Health Network, Toronto General Hospital Research Institute and Campbell Family Mental Health Research Institute, ON, Canada | Study concept or design |
| Danielle M. Andrade, MD | Division of Neurology, Department of Medicine, University of Toronto, Krembil Brain Institute, ON, Adult Genetic Epilepsy (AGE) Program, Krembil Neurosciences Institute, Toronto Western Hospital, University Health Network, Canada | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data |
Study Funding
M. Rong received unrestricted educational funding from Biocodex. A. Bayat is funded by a BRIDGE—Translational Excellence Programme grant funded by the Novo Nordisk Foundation, grant agreement number: NNF20SA0064340. A.S. Bassett holds the Dalglish Chair in 22q11.2 Deletion Syndrome at the University Health Network and University of Toronto.
Disclosure
M. Rong received an unrestricted education grant from Biocodex. Q. Zulfiqar Ali reports no disclosures relevant to the manuscript. A. Aledo‐Serrano received educational, scientific and consulting fees from UCB, Biocodex, Eisai, Angelini, Jazz pharma. A. Bayat reports no disclosures relevant to the manuscript. O. Devinsky receives grant support from NINDS, NIMH, MURI, CDC and NSF. He has equity and/or compensation from the following companies: Ajna Biosciences, Tilray, Receptor Life Sciences, Hitch Biosciences, Tevard Biosciences, Regel Biosciences, Script Biosciences, Actio Biosciences, Empatica, SilverSpike, and California Cannabis Enterprises. He has received consulting fees from Zogenix, Ultragenyx, BridgeBio, GeneMedicine and Marinus. He holds patents for the use of cannabidiol in treating neurologic disorders which are owned by GW Pharmaceuticals for which he waived financial interests. He holds other patents in molecular biology. He is the managing partner of the PhiFund Ventures. F. Qaiser, I. Chandran, A. Ali, A. Fasano, and A.S. Bassett report no disclosures relevant to the manuscript. D.M. Andrade receives grant support from Ontario Brain Institute, McLaughlin Foundation, UHN Foundation, Dravet Syndrome Foundation, SYNGAP1 Research Fund. She also received consulting fees from UCB, Biocodex, Paladin, Jazz, Stoke, and Eisai. Finally, she receives royalties from UpToDate. Go to Neurology.org/NG for full disclosures.
References
- 1.Liu JC, Ferreira CG, Yusufzai T. Human CHD2 is a chromatin assembly ATPase regulated by its chromo- and DNA-binding domains. J Biol Chem. 2015;290(1):25-34. doi: 10.1074/jbc.M114.609156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suls A, Jaehn JA, Kecskés A, et al. De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am J Hum Genet. 2013;93(5):967-975. doi: 10.1016/j.ajhg.2013.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Veredice C, Bianco F, Contaldo I, et al. Early onset myoclonic epilepsy and 15q26 microdeletion: observation of the first case. Epilepsia. 2009;50(7):1810-1815. doi: 10.1111/j.1528-1167.2009.02078.x [DOI] [PubMed] [Google Scholar]
- 4.Carvill GL, Heavin SB, Yendle SC, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet. 2013;45(7):825-830. doi: 10.1038/ng.2646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Epi4K Consortium, Epilepsy Phenome/Genome Project, Allen AS, et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501(7466):217-221. doi: 10.1038/nature12439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Truty R, Patil N, Sankar R, et al. Possible precision medicine implications from genetic testing using combined detection of sequence and intragenic copy number variants in a large cohort with childhood epilepsy. Epilepsia Open. 2019;4(3):397-408. doi: 10.1002/epi4.12348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomas RH, Zhang LM, Carvill GL, et al. CHD2 myoclonic encephalopathy is frequently associated with self-induced seizures. Neurology. 2015;84(9):951-958. doi: 10.1212/WNL.0000000000001305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lebrun N, Parent P, Gendras J, Billuart P, Poirier K, Bienvenu T. Autism spectrum disorder recurrence, resulting of germline mosaicism for a CHD2 gene missense variant. Clin Genet. 2017;92(6):669-670. doi: 10.1111/cge.13073 [DOI] [PubMed] [Google Scholar]
- 9.De Maria B, Balestrini S, Mei D, et al. Expanding the genetic and phenotypic spectrum of CHD2‐related disease: from early neurodevelopmental disorders to adult‐onset epilepsy. Am J Med Genet A. 2022;188(2):522-533. doi: 10.1002/ajmg.a.62548 [DOI] [PubMed] [Google Scholar]
- 10.Bernardo P, Galletta D, Iasevoli F, et al. CHD2 mutations: only epilepsy? Description of cognitive and behavioral profile in a case with a new mutation. Seizure. 2017;51:186-189. doi: 10.1016/j.seizure.2017.09.001 [DOI] [PubMed] [Google Scholar]
- 11.Chen J, Zhang J, Liu A, et al. CHD2 ‐related epilepsy: novel mutations and new phenotypes. Dev Med Child Neurol. 2020;62(5):647-653. doi: 10.1111/dmcn.14367 [DOI] [PubMed] [Google Scholar]
- 12.Zhou P, He N, Zhang J‐W, et al. Novel mutations and phenotypes of epilepsy‐associated genes in epileptic encephalopathies. Genes Brain Behav. 2018;17(8):e12456. doi: 10.1111/gbb.12456 [DOI] [PubMed] [Google Scholar]
- 13.Petersen AK, Streff H, Tokita M, Bostwick BL. The first reported case of an inherited pathogenic CHD2 variant in a clinically affected mother and daughter. Am J Med Genet A. 2018;176(7):1667-1669. doi: 10.1002/ajmg.a.38835 [DOI] [PubMed] [Google Scholar]
- 14.Courage C, Houge G, Gallati S, Schjelderup J, Rieubland C. 15q26.1 microdeletion encompassing only CHD2 and RGMA in two adults with moderate intellectual disability, epilepsy and truncal obesity. Eur J Med Genet. 2014;57(9):520-523. doi: 10.1016/j.ejmg.2014.06.003 [DOI] [PubMed] [Google Scholar]
- 15.Borlot F, de Almeida BI, Combe SL, Andrade DM, Filloux FM, Myers KA. Clinical utility of multigene panel testing in adults with epilepsy and intellectual disability. Epilepsia. 2019;60(8):1661-1669. doi: 10.1111/epi.16273 [DOI] [PubMed] [Google Scholar]
- 16.Mullen SA, Carvill GL, Bellows S, et al. Copy number variants are frequent in genetic generalized epilepsy with intellectual disability. Neurology. 2013;81(17):1507-1514. doi: 10.1212/WNL.0b013e3182a95829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blackburn PR, Sullivan AE, Gerassimou AG, et al. Functional analysis of the SIM1 variant p.G715V in 2 patients with obesity. J Clin Endocrinol Metab. 2020;105(1):355-361. doi: 10.1210/clinem/dgz192 [DOI] [PubMed] [Google Scholar]
- 18.Lund C, Brodtkorb E, Røsby O, Rødningen OK, Selmer KK. Copy number variants in adult patients with Lennox-Gastaut syndrome features. Epilepsy Res. 2013;105(1–2):110-117. doi: 10.1016/j.eplepsyres.2013.01.009 [DOI] [PubMed] [Google Scholar]
- 19.Lund C, Brodtkorb E, Øye AM, Røsby O, Selmer KK. CHD2 mutations in Lennox-Gastaut syndrome. Epilepsy Behav. 2014;33:18-21. doi: 10.1016/j.yebeh.2014.02.005 [DOI] [PubMed] [Google Scholar]
- 20.Saldaris J, Weisenberg J, Pestana-Knight E, et al. Content validation of clinician-reported items for a severity measure for CDKL5 deficiency disorder. J Child Neurol. 2021;36(11):998-1006. doi: 10.1177/08830738211019576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Angelopoulou E, Theodosiou A, Papaevripidou I, et al. CHD2 pathogenic nonsense variant in a three-generation family with variable phenotype and a paracentric inversion 16: case report. Heliyon. 2023;9(12):e22987. doi: 10.1016/j.heliyon.2023.e22987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cogliati F, Straniero L, Rimoldi V, et al. Low-grade parental gonosomal mosaicism in CHD2 siblings with Smith-Magenis-like syndrome. Am J Med Genet B Neuropsychiatr Genet. 2024;195(6):e32976. doi: 10.1002/ajmg.b.32976 [DOI] [PubMed] [Google Scholar]
- 23.Fisher RS, Cross JH, French JA, et al. Operational classification of seizure types by the international League against epilepsy: position paper of the ILAE commission for classification and terminology. Epilepsia. 2017;58(4):522-530. doi: 10.1111/epi.13670 [DOI] [PubMed] [Google Scholar]
- 24.Rutter M, Bailey A, Lord C, et al. Social Communication Questionnaire, 2003: Western Psychoical Services; 2003. [Google Scholar]
- 25.Sparrow SS, Cicchetti DV, Saulnier CA. Vineland Adaptive Behavior Scales, Third Edition (Vineland‐3). NCS Pearson; 2016. [Google Scholar]
- 26.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062-D1067. doi: 10.1093/nar/gkx1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434-443. doi: 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Galizia EC, Myers CT, Leu C, et al. CHD2 variants are a risk factor for photosensitivity in epilepsy. Brain. 2015;138(pt 5):1198-1207. doi: 10.1093/brain/awv052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cardoso AR, Lopes-Marques M, Silva RM, et al. Essential genetic findings in neurodevelopmental disorders. Hum Genomics. 2019;13(1):31. doi: 10.1186/s40246-019-0216-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trost B, Thiruvahindrapuram B, Chan AJS, et al. Genomic architecture of autism from comprehensive whole-genome sequence annotation. Cell. 2022;185(23):4409-4427.e18. doi: 10.1016/j.cell.2022.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.International Schizophrenia Consortium, Purcell SM, Wray NR, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748-752. doi: 10.1038/nature08185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trivisano M, Striano P, Sartorelli J, et al. CHD2 mutations are a rare cause of generalized epilepsy with myoclonic-atonic seizures. Epilepsy Behav. 2015;51:53-56. doi: 10.1016/j.yebeh.2015.06.029 [DOI] [PubMed] [Google Scholar]
- 34.Smith KM, Wirrell EC, Andrade DM, et al. Clinical presentation and evaluation of epilepsy with eyelid myoclonia: results of an international expert consensus panel. Epilepsia. 2023;64(9):2330-2341. doi: 10.1111/epi.17683 [DOI] [PubMed] [Google Scholar]
- 35.Stamberger H, Crosiers D, Balagura G, et al. Natural history study of STXBP1-developmental and epileptic encephalopathy into adulthood. Neurology. 2022;99(3):e221-e233. doi: 10.1212/WNL.0000000000200715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rong M, Benke T, Zulfiqar Ali Q, et al. Adult phenotype of SYNGAP1 -DEE. Neurol Genet. 2023;9(6):e200105. doi: 10.1212/NXG.0000000000200105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hilgenkamp TIM, van Wijck R, Evenhuis HM. (Instrumental) activities of daily living in older adults with intellectual disabilities. Res Dev Disabil. 2011;32(5):1977-1987. doi: 10.1016/j.ridd.2011.04.003 [DOI] [PubMed] [Google Scholar]
- 38.O'Roak BJ, Stessman HA, Boyle EA, et al. Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat Commun. 2014;5(1):5595. doi: 10.1038/ncomms6595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scheffer IE, Liao J. Deciphering the concepts behind “Epileptic encephalopathy” and “Developmental and epileptic encephalopathy”. Eur J Paediatr Neurol. 2020;24:11-14. doi: 10.1016/j.ejpn.2019.12.023 [DOI] [PubMed] [Google Scholar]
- 40.Andrade DM, Bassett AS, Bercovici E, et al. Epilepsy: transition from pediatric to adult care. Recommendations of the Ontario epilepsy implementation task force. Epilepsia. 2017;58(9):1502-1517. doi: 10.1111/epi.13832 [DOI] [PubMed] [Google Scholar]
- 41.Aledo-Serrano A, García-Morales I, Toledano R, et al. Diagnostic gap in genetic epilepsies: a matter of age. Epilepsy Behav. 2020;111:107266. doi: 10.1016/j.yebeh.2020.107266 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Deidentified data may be provided on reasonable request.