

Abstract
Background: Metabolic cancers are defined by metabolic reprogramming. Although this reprograming drives rapid tumour growth and invasion, it also reveals specific metabolic vulnerabilities that can be therapeutically exploited in cancer therapy. A novel form of programmed cell death, known as disulfidptosis, was identified last year; tumour cells with high SLC7A11 expression undergo disulfidptosis when deprived of glucose. Natural products have attracted increasing attention and have shown potential to treat metabolic cancers through diverse mechanisms. Methods: We systematically searched electronic databases involving PubMed, Web of Science, Gooale Scholar. To ensue comprehensive exploration, keywords including metabolic reprogramming, metabolic cancer, disulfidptosis, natural products and some other words were employed. Results: In this review, we focus on the shared characteristics and metabolic vulnerabilities of metabolic cancers. Additionally, we discuss the molecular mechanisms underlying disulfidptosis and highlight key regulatory genes. Furthermore, we predict bioactive natural products that target disulfidptosis-related genes, offering new perspectives for anticancer strategies through the modulation of disulfidptosis. Conclusions: By summarizing current research progress, this review mainly analyzed the potential mechanisms of natural products in the treatment of metabolic cancer.
Keywords: metabolic reprogramming, metabolic cancer, disulfidptosis, glucose starvation, natural products
1. Introduction
Metabolic cancer is defined by the metabolic reprogramming within tumour cells, recognized as a critical hallmark of cancer development and maintenance [1,2,3]. Cancer cells, in contrast to normal cells, exhibit abnormal metabolic behaviours, such as excessive reliance on glycolysis despite sufficient oxygen availability, a phenomenon known as the Warburg effect [1,4]. Additionally, abnormalities in lipid metabolism and glutamine metabolism have been noted across various types of cancer [5]. These metabolic alterations not only support the rapid proliferation of tumour cells but also enhance tumour invasiveness and drug resistance by modulating the tumour microenvironment [6].
Despite the growing understanding of cancer metabolism, therapeutic strategies based on these insights remain significantly challenged. Currently, drugs targeting glycolysis and lipid metabolism are undergoing clinical trials [7]. However, due to the high metabolic heterogeneity of cancer cells, the efficacy of these drugs is often limited. Moreover, many metabolic targets also play essential roles in normal cells, resulting in substantial side effects that limit the broader application of these therapies [8]. The limitations of existing therapeutic approaches underscore the need for exploring novel targets and strategies. Recently, a research team led by Professor Boyi Gan at the MD Anderson Cancer Center identified and characterized a novel form of cell death, distinct from apoptosis, necroptosis, pyroptosis, ferroptosis, cuproptosis and autophagy-dependent cell death, termed “disulfidptosis” [9]. This discovery provides a new therapeutic avenue for cancer treatment. The study found that under glucose starvation, excessive consumption of reduced coenzyme II (NADPH) in tumour cells leads to abnormal accumulation of disulfides such as cystine, inducing disulfide stress, which triggers actin cytoskeleton contraction and cell death. Furthermore, cystine uptake is primarily mediated by the solute carrier family member SLC7A11 on the cell membrane. High expression of SLC7A11 significantly accelerates disulfidptosis in tumour cells under glucose starvation. This research suggests that targeting tumour cell glucose metabolism pathways could provide a basis for developing new cancer treatment models.
In recent years, natural products have attracted significant attention due to their di-verse biological activities and relatively low toxicity [10,11]. This review aims to explore the potentially bioactive natural products in targeting disulfidptosis. As the role of disulfidptosis in metabolic cancer becomes increasingly recognized, natural products may offer unique therapeutic advantages in targeting this emerging pathway. By summarizing current research progress, this review primarily examines the mechanisms through which natural products could treat metabolic cancer.
1.1. Characteristics of Metabolic Cancer
Unlike normal cells, metabolic tumours are characterized by metabolic reprogramming, which affects several key metabolic pathways to meet the demands of rapid tumour cell proliferation and division [5,8,12]. Figure 1 shows the main metabolic pathways including glucose, fat and amino acid metabolism in normal physiological states.
Figure 1.

Metabolic pathways in normal cells. (UDPG: Uridine diphosphate glucose; UGP: Uridine diphosphate glucose -Pyrophosphorylase; PFK-1: Phosphofructokinase-1; FBPase: Fructose-1,6-bisphosphatase; NADPH: Nicotinamide Adenine Dinucleotide Phosphate Hydrogen; Co-A: Coenzyme A).
1.2. Glucose Metabolism Reprogramming
The abnormal glucose metabolism observed in tumour cells is called the “Warburg effect” [13,14]. This describes the preference of cancer cells for glycolysis over mitochondrial oxidative phosphorylation, even in the presence of oxygen. The Warburg effect not only provides sufficient energy for the rapid proliferation of tumour cells, but also provides intermediate products for the biosynthesis required for their growth, such as synthetic raw materials for amino acids and lipids. Anticancer treatments targeting glucose metabolism, such as those inhibiting key glycolytic enzymes, represent a major focus of current research [12].
1.3. Lipid Metabolism Reprogramming
As an important component of metabolic pathways, lipid metabolism in tumour cells has garnered growing attention. Compared with normal cells, tumour cells frequently display enhanced fatty acid synthesis and lipid storage capacity [15]. This not only supports the synthesis of cell membranes but also supplies essential precursors for signalling molecule synthesis. Abnormal lipid metabolism has also been reported to be closely associated with tumour aggressiveness and drug resistance [16,17]. Certain tumours, such as prostate cancer and renal cell carcinoma, are particularly dependent on alterations in lipid metabolism to sustain their growth. Studies have found that inhibiting lipid metabolism pathways can effectively suppress the progression of these tumours, offering new avenues for targeted therapy in metabolic tumours [12,18].
1.4. Amino Acid Metabolism Reprogramming
Amino acid metabolism is also significantly altered in metabolic cancers. Tumour cells exhibit an excessive dependence on specific amino acids, such as glutamine and arginine [19]. These amino acids not only provide an important source of energy but are also involved in many cell biological processes, such as resistance to oxidative stress and nucleic acid synthesis. As a major nitrogen source and energy supply, glutamine not only plays a key role in the nitrogen metabolism of tumour cells, but also promotes energy metabolism by converting it into α-ketoglutarate and entering the tricarboxylic acid cycle. Studies have found that inhibiting glutamine metabolism can significantly reduce the proliferation of tumour cells and induce their apoptosis [20]. Therefore, targeting amino acid metabolism has become an important anticancer strategy.
2. Metabolic Weaknesses
The metabolic reprogramming process acts as a “double-edged sword” in metabolic tumours. On the one hand, it serves as a pathway for energy acquisition to support tumour growth and survival. On the other hand, this reprogramming exposes a range of metabolic vulnerabilities, which form the theoretical foundation for targeted therapies against metabolic tumours.
2.1. Overdependence of Metabolic Pathways
Tumour cells exhibit an overreliance on glycolysis and specific amino acid metabolic pathways, positioning these as potential therapeutic targets. As one of the essential amino acids in the body, arginine is integral in protein synthesis in the body and is also involved in the synthesis of urea, ornithine, nitric oxide and other substances [21]. Many tumours, including MYC gene-driven small cell lung cancer, exhibit increased dependence on arginine [22]. When arginine is depleted, tumour cell viability is markedly reduced, making these cancer cells particularly sensitive to arginine deprivation therapies, such as arginine deiminase (ADI). Drugs targeting these key metabolic pathways can disrupt the energy supply and biosynthesis of cancer cells, thereby inhibiting tumour growth [23].
2.2. Accumulation of Metabolites
Metabolic reprogramming results in abnormal metabolite accumulation. When metabolic intermediates, such as lactate or lipid intermediates, accumulate, they can induce cytotoxic effects and alter the tumour microenvironment [24,25]. For example, some tumours accumulate intermediate metabolites such as α-ketoglutarate following the inactivation of mutated metabolic enzymes like iso-citrate dehydrogenase (IDH) [26]. These metabolites not only disrupt the intracellular metabolic balance but also inhibit the function of key enzymes, creating an immunosuppressive and pro-tumourigenic environment that promotes tumour progression. These metabolites can serve as biomarkers or therapeutic targets in cancer therapy. For instance, elevated lactate levels are associated with increased tumour aggressiveness and resistance to therapy, making lactate metabolism an attractive target for novel anticancer strategies [25,27]. Additionally, cancer cells often exhibit abnormal metabolic processes, including increased copper uptake. Studies have found that modulating copper homeostasis can selectively induce cancer cell death without affecting normal cells. For example, copper ionophores, such as dithiocarbamates, have shown potential in inducing copper-mediated cell death, and are particularly promising for cancers characterized by high copper accumulation, such as liver cancer [28]. In addition, copper can induce cell death via targeting lipoylated Tricarboxylic Acid cycle (TCA cycle) proteins, a new type of cell death regarded as “cuproptosis” that is distinguishable from established programmed cell death (e.g., apoptosis, pyroptosis, and ferroptosis) and provides novel insights into metabolic cancer therapy [29].
2.3. Abnormal Expression of Metabolic Enzymes
The expression of metabolic enzymes in tumour cells is frequently reprogrammed to meet their rapid proliferation demands [30]. As a key enzyme in the glycolysis pathway, normal cells primarily express pyruvate kinase M1 (PKM1), while pyruvate kinase M2 (PKM2) is highly expressed in tumour cells. PKM2 supports the glycolytic and biosynthetic requirements of cancer cells by favouring glycolysis over complete glucose oxidation for energy production [31]. By inhibiting the activity of these enzymes, the metabolic balance of tumour cells can be disturbed, thereby affecting their growth and survival [32,33].
As research on metabolic tumours advances, an increasing number of metabolic vulnerabilities are being identified and exploited for targeted therapies. Future anticancer strategies should not only inhibit the proliferation of tumour cells, but also disrupt their metabolic balance so that they cannot adapt to treatment pressure by reprogramming metabolic pathways. For instance, drugs that combine targeting glycolysis and glutamine metabolism are expected to show greater potential in treating refractory cancers [34]. By targeting the specific metabolic needs of tumour cells, we can effectively kill cancer cells without damaging normal cells, reduce the side effects of treatment, and improve patient survival rates.
3. Unique Cell Death in SLC7A11 High-Expression Cells
Professor Boyi Gan’s research team identified a novel type of cell death, which they termed “disulfidptosis”. This form of cell death is linked to the vulnerability of the cytoskeleton under disulfide bond stress. The research primarily investigates how cells with high SLC7A11 expression undergo this type of cell death during glucose starvation, driven by disulfide bond stress.
Initially, the researchers tested whether known cell death inhibitors could prevent the death of SLC7A11 high-expression UMRC6, H460, A549, and 786-O cells under glucose starvation. The findings showed that inhibitors of ferroptosis, such as ferrostatin-1 and deferoxamine, or apoptosis inhibitors, like Z-VAD-fmk, were not effective in stopping cell death. But inhibitors of disulfide bond stress, like dithiothreitol, β-mercaptoethanol, and T-CEP, were able to prevent cell death completely. This means that in cells with high SLC7A11 expression, the cell death caused by glucose starvation is closely tied to disulfide bond stress.
Further research showed that SLC7A11 high-expression cells quickly trigger a kind of cell death during glucose starvation that is different from apoptosis and ferroptosis. This death does not involve adenosine triphosphate (ATP ) loss or the formation of cysteine crystals. The researchers named this new type of cell death “disulfidptosis”.
Using a technique called stable isotope labelling by amino acids in cell culture (SILAC), the team analyzed how disulfide bond proteins change in SLC7A11 high-expression cells during glucose starvation. The results revealed that a large number of proteins formed disulfide bonds, especially proteins related to the cytoskeleton. Gene ontology analysis showed that proteins involved in the structure of the cytoskeleton and cell adhesion tend to form disulfide bonds when glucose is lacking. These proteins include FLNA/B, MYH9, TLN1, and ACTB, which have several cysteine residues in their peptides that form disulfide bonds.
The study demonstrated that F-actin cytoskeleton disintegration occurs in SLC7A11 high-expression cells under glucose starvation, accompanied by significant disulfide bond formation. This disulfide bond stress affects the function and stability of cytoskeletal proteins, leading to cell death. Functional studies further revealed that inhibiting the WAVE regulatory complex (WRC), which promotes cytoskeleton polymerization, significantly reduces disulfidptosis. Conversely, activating the Rac signalling pathway promotes this form of cell death. Figure 2 illustrates the detailed molecular mechanism of disulfidptosis.
Figure 2.

Molecular mechanisms of disulfidptosis. The study found that under glucose starvation, excessive consumption of reduced coenzyme II (NADPH) in tumour cells leads to abnormal accumulation of disulfides such as cystine, inducing disulfide stress, which triggers actin cytoskeleton contraction and cell death. Furthermore, cystine uptake is primarily mediated by the solute carrier family member SLC7A11 on the cell membrane. High expression of SLC7A11 significantly accelerates disulfidptosis in tumour cells under glucose starvation. (Glu: glucose; PPP: Pentose Phosphate Pathway; R5P: Ribose-5-phosphate; WRC: WAVE Regulatory Complex) (Figure created using Figdraw).
The study also showed that glucose transport inhibitors could induce disulfidptosis in SLC7A11 high-expression cancer cells, thus suppressing tumour growth. This finding provides a new potential strategy for cancer therapy, suggesting that targeting disulfidptosis in SLC7A11 high-expression cells could be valuable in future clinical applications.
By studying cancer cells with high SLC7A11 expression under glucose starvation using whole-genome CRISPR/Cas9 screening, the authors uncovered the vulnerability of the cytoskeleton to disulfide bond stress, highlighting the critical role of NADPH in maintaining the intracellular reductive environment, and identifying key genes likely to function in disulfidptosis (Table 1). This discovery not only deepens our understanding of cellular stress responses and death mechanisms but also offers a new perspective on how cancer cells adapt to nutrient deprivation. The identification of disulfidptosis opens up new avenues for exploring potential targets in cancer therapy.
Table 1.
Disulfidptosis-related genes and functions [35].
| Category | Gene | Gene Function | Expression in Disulfidptosis |
|---|---|---|---|
| Amino Acid/Nutrient Transport | SLC7A11 | Responsible for amino acid (e.g., cystine) transport, involved in antioxidant stress response | Upregulated |
| SLC3A2 | Forms a heterodimer with SLC7A11, cooperatively transports amino acids | Upregulated | |
| GLUT1 | Primarily responsible for transmembrane glucose transport, major carrier for glucose uptake | Downregulated | |
| GLUT3 | High glucose affinity, primarily expressed in neurons, involved in glucose uptake | Downregulated | |
| Cytoskeleton and Signal Transduction | NCKAP1 | A member of the WAVE complex, which helps with changing the cell’s structure and is linked to cell movement | Upregulated |
| CYFIP1 | A member of the WAVE complex, which helps with changing the cell’s structure and is linked to cell movement | Upregulated | |
| WAVE2 | A member of the WAVE complex, which helps with changing the cell’s structure and is linked to cell movement | Upregulated | |
| ABI2 | A member of the WAVE complex, which helps with changing the cell’s structure and is linked to cell movement | Upregulated | |
| HSPC300 | A member of the WAVE complex, which helps with changing the cell’s structure and is linked to cell movement | Upregulated | |
| RAC1 | Small GTPase, regulates the cytoskeleton, plays a key role in cell migration and adhesion | Upregulated | |
| Endoplasmic reticulum stress response | RPN1 | Involved in post-translational modification and degradation of proteins, key protein in ER glycosylation | Upregulated |
| ATF4 | Transcription factor, plays a role in cellular stress response, regulates gene expression | Upregulated | |
| Mitochondrial Function and Energy Metabolism | NUBPL | Mitochondrial assembly factor that is involved in putting together mitochondrial respiratory chain complex I | Downregulated |
| NDUFA11 | A part of mitochondrial respiratory chain complex I, which is involved in moving electrons and making ATP. | Downregulated | |
| LRPPRC | Regulates mitochondrial gene expression, influences oxidative phosphorylation process | Downregulated | |
| OXSM | Involved in fatty acid oxidation, part of the mitochondrial β-oxidation pathway | Downregulated | |
| NDUFS1 | A main part of mitochondrial respiratory chain complex I, which is involved in oxidative phosphorylation | Downregulated | |
| Glucose Metabolism | GYS1 | Glycogen synthase, involved in glycogen synthesis, affects energy storage | Downregulated |
| G6PD | Responsible for the first step of the pentose phosphate pathway, produces NADPH for reductive biosynthesis | Downregulated | |
| 6-PGD | Involved in the pentose phosphate pathway | Downregulated | |
| TALDO1 | Involved in the pentose phosphate pathway, generates nucleotides and NADPH | Downregulated | |
| TKT | Key enzyme in the pentose phosphate pathway, involved in carbon skeleton rearrangement | Downregulated | |
| Cell Division | PRC1 | Regulates the cell division process, particularly playing a role in cytokinesis during the final stages of cell division | Downregulated |
4. Application Prospect of Natural Products in Metabolic Therapy Targeting Disulfidptosis
Natural products are naturally occurring compounds primarily derived from plants, microorganisms, animals, and marine organisms. These compounds are renowned for their structural diversity and wide-ranging biological activities, making them a significant resource for drug development [10,11]. Over the past 20 years, research on natural products has advanced significantly, particularly in the areas of anticancer [36], anti-inflammatory [37], and antiviral therapies [38]. Recently, natural products have shown great promise in inducing programmed cell death pathways, especially ferroptosis. By modulating intracellular iron metabolism and lipid peroxidation, natural products can effectively trigger ferroptosis. For example, epigallocatechin gallate (EGCG), a compound found in green tea, can induce ferroptosis by elevating intracellular reactive oxygen species (ROS) levels and promoting lipid peroxidation [39]. Other compounds, such as curcumin [40] and quercetin [41], have been found to enhance lipid peroxidation and induce ferroptosis by inhibiting glutathione peroxidase 4 (GPX4). In addition to ferroptosis, compounds like resveratrol, curcumin, and quercetin exhibit pro-oxidative effects in the presence of copper. These compounds can amplify copper-mediated reactive oxygen species (ROS) generation, leading to DNA damage and ultimately causing cancer cell death. Although copper overload can be toxic, chelation therapy can be strategically employed for the treatment of cancers with hypercupremic states, striking a careful balance between therapeutic efficacy and toxicity [42]. The ability of these natural products to enhance ROS generation in the presence of copper presents a novel therapeutic approach, particularly for cancers characterized by elevated copper levels, thus improving treatment selectivity and effectiveness [43].
As potential drug candidates, natural products targeting ferroptosis and cuproptosis provide promising avenues for the treatment of metabolic cancers. This raises the prospect of targeting disulfidptosis for therapeutic intervention. Future research could focus on screening natural products that are effective against tumours sensitive to disulfidptosis or using them to overcome tumour resistance [44] to chemotherapy by inducing disulfidptosis. Although there are no current reports of natural products directly affecting disulfidptosis, a deeper understanding of its molecular mechanisms, combined with in-sights from natural product research, could help identify natural compounds with therapeutic potential (Table 2).
Table 2.
Potentially bioactive natural products that may target disulfidptosis.
| Natural Product | Gene | Structure | Origin | Function | Reference |
|---|---|---|---|---|---|
| Curculigoside | SLC7A11 |
|
C. orchioide | Downregulating the SLC7A11/GPX4 signalling pathway promotes ferroptosis and improves Alzheimer’s disease | [45] |
| Glabridin | SLC3A2/SLC7A11 |
|
Licorice | Enhancing SLC3A2/SLC7A11 expression in diabetic nephropathy rats inhibits ferroptosis | [46] |
| Ginsenoside Rh2 | IRF1 |
|
Ginseng | Upregulating IRF1 expression inhibits SLC7A11, enhances ferroptosis, reduces liver inflammation, and suppresses liver fibrosis | [47] |
| Rehmannioside A | NRF2 |
|
Rehmannia glutinosa Libosch | Activating the Nrf2 and SLC7A11/GPX4 signalling pathways inhibits ferroptosis and improves cognitive dysfunction after cerebral ischemia | [48] |
| Kaempferol | NRF2 |
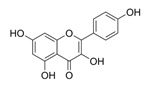
|
Green tea, Broccoli, Delphinium | Activating the Nrf2 and SLC7A11/GPX4 signalling pathways inhibits ferroptosis in OGD/R-treated neurons | [49] |
| Lycium barbarum polysaccharide | NRF2 | Consisted of several monosaccharides (galactose, rhamnose, arabinose, glucose, mannose, xylose) and proteins | L. barbarum | Activating NRF2 promotes the expression of downstream targets such as HO-1 and SLC7A11, reducing ferroptosis | [50] |
| Genistein | GLUT1 |
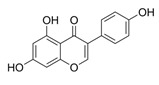
|
Soya | Binds to GLUT1 and inhibits GLUT1 transport | [51] |
| Epigallocatechol Gallate | GLUT1, GLUT3 |
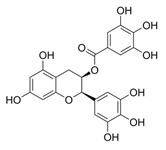
|
Green tea | Binds to GLUT1/3 and inhibits GLUT1/3 transport efficiency | [52] |
| Diallyl disulfide | RAC1 |
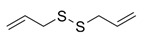
|
Garlic | Downregulates the TGF-β1/Rac1 pathway in gastric cancer, inhibits tumour invasiveness | [53] |
| Kuwanon H | ATF4 |
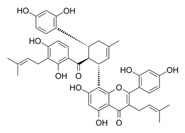
|
Morus alba L. | Upregulates ATF4, induces endoplasmic reticulum stress, leading to apoptosis and autophagy | [54] |
| Agrimol B | NDUFS1 |
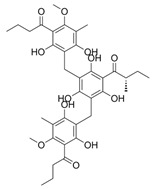
|
Agrimonia pilosa Ledeb | Downregulates NDUFS1, and increases mROS, inducing autophagy arrest | [55] |
| R001 | G6PD |
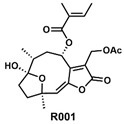
|
Vernonia cinerea plant | Inhibits G6PD and TrxR1 functions, leading to limited GSH synthesis and promoting oxidative stress | [56,57,58] |
4.1. Amino Acid and Nutrients Transport
SLC7A11 and SLC3A2 form a heterodimer, and together constitute the cystine/glutamate antiporter system, transporting cystine into the cell in exchange for glutamate [59]. This transporter plays a critical role in maintaining intracellular cystine levels and facilitating the synthesis of glutathione, which is the cell’s primary antioxidant [60]. In cancer cells, the overexpression of SLC7A11 promotes cystine uptake, thereby increasing resistance to oxidative stress and ferroptosis [61]. However, under glucose-starved conditions, excessive cystine uptake can lead to intracellular disulfide accumulation, triggering disulfidptosis. By upregulating the expression of SLC7A11 and SLC3A2, the cystine burden inside the cell increases, thereby promoting disulfidptosis in metabolically stressed cancer cells. Glabridin [46] has been reported to directly upregulate SLC3A2/SLC7A11, suppressing ferroptosis in diabetic nephropathy rat models. Unlike direct modulation of SLC7A11, many studies aim to regulate its expression by targeting upstream pathways, with NRF2 being the most notable molecular target. Rehmannioside A [48], Kaempferol [49], and Lycium barbarum polysaccharides [50] have all been shown to activate the Nrf2/SLC7A11/GPX4 pathway, mitigating ferroptosis.
On the other hand, glucose transporters GLUT1 and GLUT3 facilitate glucose entry into cells [62]. Inhibiting GLUT1/GLUT3 induces glucose starvation, increasing NADPH consumption and promoting disulfide bond formation, which in turn leads to disulfidptosis in cells with high SLC7A11 expression. Both Genistein [63] and Epigallocatechol Gallate [39] have been reported to bind to GLUT, reducing its transport efficiency and limiting glucose uptake into cells.
4.2. Cytoskeleton and Signal Transduction
NCKAP1, CYFIP1, WAVE2, ABI2, and HSPC300 encode components of the WAVE regulatory complex (WRC) [64], which plays a critical role in actin polymerization and cytoskeletal rearrangement. RAC1, a small GTPase, efficiently activates WRC [65], driving the Arp2/3 complex to regulate the actin cytoskeleton, promote lamellipodia formation and participating in cell migration and morphology maintenance. In disulfidptosis, upregulation of WRC-related genes and RAC1 leads to actin polymerization and disulfide bond crosslinking within the actin cytoskeleton, which is a key event causing cytoskeletal collapse and cell death. Enhancing the expression of WRC-related proteins or persistently activating RAC1 could promote disulfidptosis in metabolic tumours. Diallyl disulfide [53] derived from garlic has been shown to inhibit tumour invasiveness in gastric cancer by downregulating the TGF-β1/Rac1 pathway. Whether Diallyl disulfide plays a role in disulfidptosis remains unknown.
4.3. ER Stress Response
RPN1 is a subunit of the 26S proteasome, located in the 19S regulatory particle (RP) cap of the proteasome. It is a component of the endoplasmic reticulum-associated degradation (ERAD) system, which is responsible for degrading misfolded or accumulated proteins within the endoplasmic reticulum [66]. Additionally, RPN1 is part of the oligosaccharyl transferase (OST) complex and is critical for the N-glycosylation of glycoproteins, affecting protein folding and post-translational modification [67]. When disulfide bonds accumulate excessively within the cell, RPN1 helps mitigate ER stress by regulating the degradation of misfolded proteins. Silencing or inhibiting RPN1 exacerbates disulfide bond and misfolded protein accumulation, thus promoting disulfidptosis. ATF4, a key transcription factor in the cellular stress response, is essential for regulating endoplasmic reticulum stress, oxidative stress, and amino acid metabolism [68]. ATF4 controls the expression of various antioxidant genes, such as glutathione reductase (GSR) and glutathione peroxidase (GSH-Px), and is involved in the metabolism of glutamine and cysteine, which enables cells to counteract oxidative stress. During disulfidptosis, ATF4 is upregulated, enhancing the cells’ antioxidant defences against disulfide-induced death. Continuous upregulation of ATF4 expression may exacerbate endoplasmic reticulum stress under glucose starvation, thereby promoting disulfidptosis. Kuwanon H [54] has been shown to induce ER stress, leading to apoptosis and autophagy via ATF4 upregulation, and may also play a role in disulfidptosis.
4.4. Mitochondrial Function and Energy Metabolism
NUBPL, NDUFA11, and NDUFS1 are involved in encoding mitochondrial respiratory chain complex I [69,70,71], which plays a crucial role in oxidative phosphorylation and ATP production. LRPPRC regulates mitochondrial gene expression and affects energy metabolism [72]. Downregulating LRPPRC can inhibit tumour growth, invasion, and metastasis. OXSM participates in mitochondrial fatty acid synthesis [73]. Defects in mitochondrial function can increase oxidative stress and lead to energy depletion. Under glucose deprivation conditions, inhibiting these genes can heighten the susceptibility of tumour cells to disulfidptosis. Agrimol B [55] can induce autophagy reduction by downregulating NDUFS1 in liver cancer cells, leading to speculation that it could similarly influence disulfidptosis.
4.5. Glucose Metabolism
GYS1 is one of the key enzymes involved in glycogen synthesis, regulating the balance between glycogen production and breakdown [74,75]. In disulfidptosis, GYS1 expression is downregulated due to glucose starvation and reduced intake. Silencing GYS1 can further heighten cell sensitivity to glucose deprivation by reducing glycogen accumulation, inhibiting the glycolytic pathway, and promoting disulfidptosis. G6PD, 6-PGD, TALDO1, and TKT are key players in the pentose phosphate pathway (PPP) [76], which is crucial for maintaining NADPH levels, regulating redox balance, and resisting oxidative stress. Inhibiting this pathway depletes NADPH, exacerbates abnormal disulfide bond accumulation, and promotes disulfidptosis. R001 [56,57,58] is an unnamed natural compound extracted from the Vernonia cinerea plant. It has been shown to inhibit G6PD and TrxR1 functions in triple-negative breast cancer, limiting GSH synthesis and promoting oxidative stress. The inhibitory effect of R001 on G6PD suggests its potential application in promoting disulfidptosis.
4.6. Cell Division
PRC1 regulates cell division, ensuring correct differentiation and function during development [77]. Abnormal cell division can result in chromosomal instability [78]. Based on PRC1’s genetic functions, it is speculated that PRC1 may not directly participate in disulfide-induced cell death but rather plays an indirect role. It is possible that PRC1 influences the cellular decision to enter an oxidative stress state or proceed directly to disulfide-induced death by acting on cell cycle check-points [79]. Further research is required to confirm PRC1’s role in disulfide-induced cell death.
5. Prospects and Challenges
As mentioned above, therapies targeting disulfidptosis show great potential for treating metabolic tumours, especially when combined with natural product interventions. However, the development and application of natural products often face numerous challenges, with low bioavailability being one of the primary limitations for some of these compounds [80,81,82], which significantly hinders their clinical application. Many natural products, such as curcumin [83], have relatively low solubility due to their hydrophobic structures, limiting their absorption and bioavailability in the body. Additionally, some structurally complex natural products administered orally are poorly absorbed through the gastrointestinal tract, with only a small portion reaching systemic circulation [84]. Even if absorbed into the bloodstream, some natural products are often rapidly metabolized or eliminated due to enzymatic activity, limiting their ability to maintain effective concentrations in the blood [85,86]; their short half-life further limits their therapeutic potential.
Nanotechnology offers an effective strategy to overcome the challenges in drug delivery [80,81]. Using nanotechnology to encapsulate natural products can significantly improve their water solubility, stability and bioavailability. For example, nano-modification of curcumin is an effective strategy to address its low solubility, greatly enhancing its bioavailability and targeted delivery efficacy. Delivering resveratrol through biomimetic nano-systems not only improves its efficacy in treating colorectal cancer, but also addresses the challenge of low bioavailability of natural products [87]. In addition, solid lipid nanoparticles (SLNs), as a new type of nano-drug delivery system, can effectively extend the half-life of drugs in vivo and significantly enhance oral bioavailability [84]. SLNs significantly improve the solubility and bioavailability of dihydroartemisinin (DHA), greatly improving its anticancer effect in vivo [88]. Although the current nano-drug delivery system has shown great potential in the laboratory stage, it still faces many challenges in clinical application, especially in terms of biocompatibility, toxicity evaluation, and controlled drug release [89].
As a newly proposed form of programmed cell death, disulfidptosis currently has limited experimental research. Although studies have revealed some mechanisms underlying disulfidptosis, including the association between intracellular disulfide overaccumulation and redox imbalance, our understanding of its key molecules and regulatory pathways remains incomplete [3,9,35,90,91,92,93,94]. For instance, tumour cells originating from different primary tissues may exhibit variations in their sensitivity to disulfidptosis due to differences in tissue-specific functions and energy metabolism, such as glucose and lipid metabolism. Current research has primarily focused on cell types with high SLC7A11 expression, raising the question of whether disulfidptosis occurs in cells with low SLC7A11 expression. And if so, how does it differ from the characteristics of those in high-expressing cells? Moreover, while most studies focus on disulfidptosis as a distinct programmed cell death pathway, potential links between disulfidptosis and ferroptosis, which also involves SLC7A11, remain to be explored. The possibility of interactions or synergistic effects between disulfidptosis and other forms of regulated cell death, such as apoptosis and autophagy, also warrants further investigation. Additionally, the regulatory influence of the tumour microenvironment on disulfidptosis remains poorly understood. Developing in vivo strategies to induce glucose deprivation that can selectively target tumour cells while sparing normal tissues requires further research and optimization. Future studies should focus on elucidating these mechanisms and developing strategies for the precise targeting of tumour cells, and clinical trials based on disulfidptosis regulation are also necessary for targeting treatment.
In summary, although targeting disulfidptosis with natural products shows promise for the treatment of metabolic tumours, challenges remain in optimizing bioavailability, understanding the underlying mechanisms, and achieving clinical translation. As research into the molecular mechanisms of disulfidptosis progresses and the development of new drugs advances, this strategy could offer novel and more effective therapeutic approaches for precision oncology.
Author Contributions
Conceptualization, X.L. and P.L.; writing—original draft preparation, X.L. and P.L.; figure drawing, Y.Z. and S.T.; writing—review and editing, P.L., J.X. and L.Y.; supervision, P.L., M.S. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding Statement
This study was supported by National Natural Science Foundation of China (82370583), Innovation Capability Support Program of Shaanxi Province (2023KJXX-033), Key Research and Development Program of Shaanxi Province (2022-LL-JB-30), Natural Science Basic Research Program of Shaanxi Province (2024JC-YBQN-0091), “The Young Talent Support Plan” of Xi’an Jiaotong University (RCYJ0013), Medical “Base-Clinic” Integrated Innovation Project of Xi’an Jiaotong University (YXJLRH2022058), the Fundamental Research Funds for the Central University (xzy012023120 and xzd012024061), Science and Technology Program of Xi’an City (23YXYJ0125), Shaanxi Fundamental Science Research Project for Chemistry and Biology (23JHQ036), Natural Science Foundation of Shaanxi Province (2020JQ-541) and Research Foundation of the Second Affiliated Hospital of Xi’an Jiaotong University (YJ(QN)202301).
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Pavlova N.N., Thompson C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martinez-Outschoorn U.E., Peiris-Pagés M., Pestell R.G., Sotgia F., Lisanti M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017;14:11–31. doi: 10.1038/nrclinonc.2016.60. [DOI] [PubMed] [Google Scholar]
- 3.Zheng P., Zhou C., Ding Y., Duan S. Disulfidptosis: A new target for metabolic cancer therapy. J. Exp. Clin. Cancer Res. CR. 2023;42:103. doi: 10.1186/s13046-023-02675-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vander Heiden M.G., DeBerardinis R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell. 2017;168:657–669. doi: 10.1016/j.cell.2016.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Faubert B., Solmonson A., DeBerardinis R.J. Metabolic reprogramming and cancer progression. Science. 2020;368:eaaw5473. doi: 10.1126/science.aaw5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pavlova N.N., Zhu J., Thompson C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022;34:355–377. doi: 10.1016/j.cmet.2022.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tufail M., Jiang C.-H., Li N. Altered metabolism in cancer: Insights into energy pathways and therapeutic targets. Mol. Cancer. 2024;23:203. doi: 10.1186/s12943-024-02119-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ward P.S., Thompson C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu X., Nie L., Zhang Y., Yan Y., Wang C., Colic M., Olszewski K., Horbath A., Chen X., Lei G., et al. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat. Cell Biol. 2023;25:404–414. doi: 10.1038/s41556-023-01091-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newman D.J., Cragg G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016;79:629–661. doi: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- 11.Newman D.J., Cragg G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020;83:770–803. doi: 10.1021/acs.jnatprod.9b01285. [DOI] [PubMed] [Google Scholar]
- 12.Phan L.M., Yeung S.C., Lee M.H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014;11:1–19. doi: 10.7497/j.issn.2095-3941.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vander Heiden M.G., Cantley L.C., Thompson C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liberti M.V., Locasale J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016;41:211–218. doi: 10.1016/j.tibs.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirschey M.D., DeBerardinis R.J., Diehl A.M.E., Drew J.E., Frezza C., Green M.F., Jones L.W., Ko Y.H., Le A., Lea M.A., et al. Dysregulated metabolism contributes to oncogenesis. Semin. Cancer Biol. 2015;35((Suppl.)):S129–S150. doi: 10.1016/j.semcancer.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Menendez J.A., Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer. 2007;7:763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- 17.Cheng C., Geng F., Cheng X., Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. 2018;38:27. doi: 10.1186/s40880-018-0301-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van der Mijn J.C., Panka D.J., Geissler A.K., Verheul H.M., Mier J.W. Novel drugs that target the metabolic reprogramming in renal cell cancer. Cancer Metab. 2016;4:14. doi: 10.1186/s40170-016-0154-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen J., Cui L., Lu S., Xu S. Amino acid metabolism in tumour biology and therapy. Cell Death Dis. 2024;15:42. doi: 10.1038/s41419-024-06435-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin J., Byun J.-K., Choi Y.-K., Park K.-G. Targeting glutamine metabolism as a therapeutic strategy for cancer. Exp. Mol. Med. 2023;55:706–715. doi: 10.1038/s12276-023-00971-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia-Bermudez J., Williams R.T., Guarecuco R., Birsoy K. Targeting extracellular nutrient dependencies of cancer cells. Mol. Metab. 2020;33:67–82. doi: 10.1016/j.molmet.2019.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chalishazar M.D., Wait S.J., Huang F., Ireland A.S., Mukhopadhyay A., Lee Y., Schuman S.S., Guthrie M.R., Berrett K.C., Vahrenkamp J.M., et al. MYC-Driven Small-Cell Lung Cancer is Metabolically Distinct and Vulnerable to Arginine Depletion. Clin. Cancer Res. 2019;25:5107–5121. doi: 10.1158/1078-0432.CCR-18-4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gatenby R.A., Gillies R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 24.Yoshida G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. CR. 2015;34:111. doi: 10.1186/s13046-015-0221-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen A.-N., Luo Y., Yang Y.-H., Fu J.-T., Geng X.-M., Shi J.-P., Yang J. Lactylation, a Novel Metabolic Reprogramming Code: Current Status and Prospects. Front. Immunol. 2021;12:688910. doi: 10.3389/fimmu.2021.688910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang C., Moore L.M., Li X., Yung W.K.A., Zhang W. IDH1/2 mutations target a key hallmark of cancer by deregulating cellular metabolism in glioma. Neuro Oncol. 2013;15:1114–1126. doi: 10.1093/neuonc/not087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Z.-H., Peng W.-B., Zhang P., Yang X.-P., Zhou Q. Lactate in the tumour microenvironment: From immune modulation to therapy. EBioMedicine. 2021;73:103627. doi: 10.1016/j.ebiom.2021.103627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang D., Kroemer G., Kang R. Targeting cuproplasia and cuproptosis in cancer. Nat. Rev. Clin. Oncol. 2024;21:370–388. doi: 10.1038/s41571-024-00876-0. [DOI] [PubMed] [Google Scholar]
- 29.Tsvetkov P., Coy S., Petrova B., Dreishpoon M., Verma A., Abdusamad M., Rossen J., Joesch-Cohen L., Humeidi R., Spangler R.D., et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375:1254–1261. doi: 10.1126/science.abf0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cairns R.A., Harris I.S., Mak T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 31.Zhu S., Guo Y., Zhang X., Liu H., Yin M., Chen X., Peng C. Pyruvate kinase M2 (PKM2) in cancer and cancer therapeutics. Cancer Lett. 2021;503:240–248. doi: 10.1016/j.canlet.2020.11.018. [DOI] [PubMed] [Google Scholar]
- 32.Yu X., Li S. Non-metabolic functions of glycolytic enzymes in tumourigenesis. Oncogene. 2017;36:2629–2636. doi: 10.1038/onc.2016.410. [DOI] [PubMed] [Google Scholar]
- 33.You M., Xie Z., Zhang N., Zhang Y., Xiao D., Liu S., Zhuang W., Li L., Tao Y. Signaling pathways in cancer metabolism: Mechanisms and therapeutic targets. Signal Transduct. Target. Ther. 2023;8:196. doi: 10.1038/s41392-023-01442-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Porporato P.E. Understanding cachexia as a cancer metabolism syndrome. Oncogenesis. 2016;5:e200. doi: 10.1038/oncsis.2016.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao D., Meng Y., Dian Y., Zhou Q., Sun Y., Le J., Zeng F., Chen X., He Y., Deng G. Molecular landmarks of tumour disulfidptosis across cancer types to promote disulfidptosis-target therapy. Redox Biol. 2023;68:102966. doi: 10.1016/j.redox.2023.102966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cragg G.M., Pezzuto J.M. Natural Products as a Vital Source for the Discovery of Cancer Chemotherapeutic and Chemopreventive Agents. Med. Princ. Pract. 2016;25((Suppl. 2)):41–59. doi: 10.1159/000443404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo Y., Peng X., Liu F., Zhang Q., Ding L., Li G., Qiu F. Potential of natural products in inflammation: Biological activities, structure-activity relationships, and mechanistic targets. Arch. Pharm. Res. 2024;47:377–409. doi: 10.1007/s12272-024-01496-z. [DOI] [PubMed] [Google Scholar]
- 38.Chen J., Zhao Y., Cheng J., Wang H., Pan S., Liu Y. The Antiviral Potential of Perilla frutescens: Advances and Perspectives. Molecules. 2024;29:3328. doi: 10.3390/molecules29143328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang C., Wu A., Tan L., Tang D., Chen W., Lai X., Gu K., Chen J., Chen D., Tang Q. Epigallocatechin-3-Gallate Alleviates Liver Oxidative Damage Caused by Iron Overload in Mice through Inhibiting Ferroptosis. Nutrients. 2023;15:1993. doi: 10.3390/nu15081993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang X., Ding H., Liang M., Chen X., Yan Y., Wan N., Chen Q., Zhang J., Cao J. Curcumin induces ferroptosis in non-small-cell lung cancer via activating autophagy. Thorac. Cancer. 2021;12:1219–1230. doi: 10.1111/1759-7714.13904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ding L., Dang S., Sun M., Zhou D., Sun Y., Li E., Peng S., Li J., Li G. Quercetin induces ferroptosis in gastric cancer cells by targeting SLC1A5 and regulating the p-Camk2/p-DRP1 and NRF2/GPX4 Axes. Free Radic. Biol. Med. 2024;213:150–163. doi: 10.1016/j.freeradbiomed.2024.01.002. [DOI] [PubMed] [Google Scholar]
- 42.Farhan M., Rizvi A. Understanding the Prooxidant Action of Plant Polyphenols in the Cellular Microenvironment of Malignant Cells: Role of Copper and Therapeutic Implications. Front. Pharmacol. 2022;13:929853. doi: 10.3389/fphar.2022.929853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ge E.J., Bush A.I., Casini A., Cobine P.A., Cross J.R., DeNicola G.M., Dou Q.P., Franz K.J., Gohil V.M., Gupta S., et al. Connecting copper and cancer: From transition metal signalling to metalloplasia. Nat. Rev. Cancer. 2022;22:102–113. doi: 10.1038/s41568-021-00417-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y., Wu X., Ren Z., Li Y., Zou W., Chen J., Wang H. Overcoming cancer chemotherapy resistance by the induction of ferroptosis. Drug Resist. Updat. 2023;66:100916. doi: 10.1016/j.drup.2022.100916. [DOI] [PubMed] [Google Scholar]
- 45.Gong Y., Wang Y., Li Y., Weng F., Chen T., He L. Curculigoside, a traditional Chinese medicine monomer, ameliorates oxidative stress in Alzheimer’s disease mouse model via suppressing ferroptosis. Phytother. Res. 2024;38:2462–2481. doi: 10.1002/ptr.8152. [DOI] [PubMed] [Google Scholar]
- 46.Tan H., Chen J., Li Y., Li Y., Zhong Y., Li G., Liu L., Li Y. Glabridin, a bioactive component of licorice, ameliorates diabetic nephropathy by regulating ferroptosis and the VEGF/Akt/ERK pathways. Mol. Med. 2022;28:58. doi: 10.1186/s10020-022-00481-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lang Z., Yu S., Hu Y., Tao Q., Zhang J., Wang H., Zheng L., Yu Z., Zheng J. Ginsenoside Rh2 promotes hepatic stellate cell ferroptosis and inactivation via regulation of IRF1-inhibited SLC7A11. Phytomedicine. 2023;118:154950. doi: 10.1016/j.phymed.2023.154950. [DOI] [PubMed] [Google Scholar]
- 48.Fu C., Wu Y., Liu S., Luo C., Lu Y., Liu M., Wang L., Zhang Y., Liu X. Rehmannioside A improves cognitive impairment and alleviates ferroptosis via activating PI3K/AKT/Nrf2 and SLC7A11/GPX4 signaling pathway after ischemia. J. Ethnopharmacol. 2022;289:115021. doi: 10.1016/j.jep.2022.115021. [DOI] [PubMed] [Google Scholar]
- 49.Yuan Y., Zhai Y., Chen J., Xu X., Wang H. Kaempferol Ameliorates Oxygen-Glucose Deprivation/Reoxygenation-Induced Neuronal Ferroptosis by Activating Nrf2/SLC7A11/GPX4 Axis. Biomolecules. 2021;11:923. doi: 10.3390/biom11070923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiang S.-J., Xiao X., Li J., Mu Y. Lycium barbarum polysaccharide-glycoprotein ameliorates ionizing radiation-induced epithelial injury by regulating oxidative stress and ferroptosis via the Nrf2 pathway. Free Radic. Biol. Med. 2023;204:84–94. doi: 10.1016/j.freeradbiomed.2023.04.020. [DOI] [PubMed] [Google Scholar]
- 51.Guelfi G., Pasquariello R., Anipchenko P., Capaccia C., Pennarossa G., Brevini T.A.L., Gandolfi F., Zerani M., Maranesi M. The Role of Genistein in Mammalian Reproduction. Molecules. 2023;28:7436. doi: 10.3390/molecules28217436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ojelabi O.A., Lloyd K.P., De Zutter J.K., Carruthers A. Red wine and green tea flavonoids are cis-allosteric activators and competitive inhibitors of glucose transporter 1 (GLUT1)-mediated sugar uptake. J. Biol. Chem. 2018;293:19823–19834. doi: 10.1074/jbc.RA118.002326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Su B., Su J., Zeng Y., Ding E., Liu F., Tan T., Xia H., Wu Y.-H., Zeng X., Ling H., et al. Diallyl disulfide inhibits TGF-β1-induced upregulation of Rac1 and β-catenin in epithelial-mesenchymal transition and tumour growth of gastric cancer. Oncol. Rep. 2018;39:2797–2806. doi: 10.3892/or.2018.6345. [DOI] [PubMed] [Google Scholar]
- 54.Hu X., Pan G., Luo J., Gao X., Mu Y., Wang Z., Hu X., Li C., Abbas M.N., Zhang K., et al. Kuwanon H Inhibits Melanoma Growth Through Cytotoxic Endoplasmic Reticulum Stress and Impaired Autophagy Flux. J. Agric. Food Chem. 2023;71:13768–13782. doi: 10.1021/acs.jafc.3c02257. [DOI] [PubMed] [Google Scholar]
- 55.Dong L., Luo L., Wang Z., Lian S., Wang M., Wu X., Fan J., Zeng Y., Li S., Lv S., et al. Targeted degradation of NDUFS1 by agrimol B promotes mitochondrial ROS accumulation and cytotoxic autophagy arrest in hepatocellular carcinoma. Free Radic. Biol. Med. 2024;220:111–124. doi: 10.1016/j.freeradbiomed.2024.04.242. [DOI] [PubMed] [Google Scholar]
- 56.Brotherton-Pleiss C., Yue P., Zhu Y., Nakamura K., Chen W., Fu W., Kubota C., Chen J., Alonso-Valenteen F., Mikhael S., et al. Discovery of Novel Azetidine Amides as Potent Small-Molecule STAT3 Inhibitors. J. Med. Chem. 2021;64:695–710. doi: 10.1021/acs.jmedchem.0c01705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu Y., Yue P., Dickinson C.F., Yang J.K., Datanagan K., Zhai N., Zhang Y., Miklossy G., Lopez-Tapia F., Tius M.A., et al. Natural product preferentially targets redox and metabolic adaptations and aberrantly active STAT3 to inhibit breast tumour growth in vivo. Cell Death Dis. 2022;13:1022. doi: 10.1038/s41419-022-05477-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yue P., Zhu Y., Brotherton-Pleiss C., Fu W., Verma N., Chen J., Nakamura K., Chen W., Chen Y., Alonso-Valenteen F., et al. Novel potent azetidine-based compounds irreversibly inhibit Stat3 activation and induce antitumour response against human breast tumour growth in vivo. Cancer Lett. 2022;534:215613. doi: 10.1016/j.canlet.2022.215613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu J., Xia X., Huang P. xCT: A Critical Molecule That Links Cancer Metabolism to Redox Signaling. Mol. Ther. 2020;28:2358–2366. doi: 10.1016/j.ymthe.2020.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Diaz-Vivancos P., de Simone A., Kiddle G., Foyer C.H. Glutathione—Linking cell proliferation to oxidative stress. Free Radic. Biol. Med. 2015;89:1154–1164. doi: 10.1016/j.freeradbiomed.2015.09.023. [DOI] [PubMed] [Google Scholar]
- 61.Yu D., Wang Q., Zhang Q., Cai M., Liu S., Zhang W. Molecular mechanisms of ferroptosis and its antitumour applications in natural products. Acta Biochim. Biophys. Sin. 2023;55:1337–1347. doi: 10.3724/abbs.2023120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Holman G.D. Structure, function and regulation of mammalian glucose transporters of the SLC2 family. Pflugers Arch. 2020;472:1155–1175. doi: 10.1007/s00424-020-02411-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vera J.C., Reyes A.M., Cárcamo J.G., Velásquez F.V., Rivas C.I., Zhang R.H., Strobel P., Iribarren R., Scher H.I., Slebe J.C. Genistein is a natural inhibitor of hexose and dehydroascorbic acid transport through the glucose transporter, GLUT1. J. Biol. Chem. 1996;271:8719–8724. doi: 10.1074/jbc.271.15.8719. [DOI] [PubMed] [Google Scholar]
- 64.Han K.A., Ko J. Orchestration of synaptic functions by WAVE regulatory complex-mediated actin reorganization. Exp. Mol. Med. 2023;55:1065–1075. doi: 10.1038/s12276-023-01004-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ding B., Yang S., Schaks M., Liu Y., Brown A.J., Rottner K., Chowdhury S., Chen B. Structures reveal a key mechanism of WAVE regulatory complex activation by Rac1 GTPase. Nat. Commun. 2022;13:5444. doi: 10.1038/s41467-022-33174-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jiang T.-X., Zhao M., Qiu X.-B. Substrate receptors of proteasomes. Biol. Rev. Camb. Philos. Soc. 2018;93:1765–1777. doi: 10.1111/brv.12419. [DOI] [PubMed] [Google Scholar]
- 67.Ding J., Xu J., Deng Q., Ma W., Zhang R., He X., Liu S., Zhang L. Knockdown of Oligosaccharyltransferase Subunit Ribophorin 1 Induces Endoplasmic-Reticulum-Stress-Dependent Cell Apoptosis in Breast Cancer. Front. Oncol. 2021;11:722624. doi: 10.3389/fonc.2021.722624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wortel I.M.N., van der Meer L.T., Kilberg M.S., van Leeuwen F.N. Surviving Stress: Modulation of ATF4-Mediated Stress Responses in Normal and Malignant Cells. Trends Endocrinol. Metab. 2017;28:794–806. doi: 10.1016/j.tem.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kevelam S.H., Rodenburg R.J., Wolf N.I., Ferreira P., Lunsing R.J., Nijtmans L.G., Mitchell A., Arroyo H.A., Rating D., Vanderver A., et al. NUBPL mutations in patients with complex I deficiency and a distinct MRI pattern. Neurology. 2013;80:1577–1583. doi: 10.1212/WNL.0b013e31828f1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rhein V.F., Carroll J., Ding S., Fearnley I.M., Walker J.E. NDUFAF5 Hydroxylates NDUFS7 at an Early Stage in the Assembly of Human Complex I. J. Biol. Chem. 2016;291:14851–14860. doi: 10.1074/jbc.M116.734970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sheftel A.D., Stehling O., Pierik A.J., Netz D.J.A., Kerscher S., Elsässer H.-P., Wittig I., Balk J., Brandt U., Lill R. Human ind1, an iron-sulfur cluster assembly factor for respiratory complex I. Mol. Cell Biol. 2009;29:6059–6073. doi: 10.1128/MCB.00817-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cui J., Wang L., Ren X., Zhang Y., Zhang H. LRPPRC: A Multifunctional Protein Involved in Energy Metabolism and Human Disease. Front. Physiol. 2019;10:595. doi: 10.3389/fphys.2019.00595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Duan Y., Yu J., Chen M., Lu Q., Ning F., Gan X., Liu H., Ye Y., Lu S., Lash G.E. Knockdown of heat shock protein family D member 1 (HSPD1) promotes proliferation and migration of ovarian cancer cells via disrupting the stability of mitochondrial 3-oxoacyl-ACP synthase (OXSM) J. Ovarian Res. 2023;16:81. doi: 10.1186/s13048-023-01156-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fastman N.M., Liu Y., Ramanan V., Merritt H., Ambing E., DePaoli-Roach A.A., Roach P.J., Hurley T.D., Mellem K.T., Ullman J.C., et al. The structural mechanism of human glycogen synthesis by the GYS1-GYG1 complex. Cell Rep. 2022;40:111041. doi: 10.1016/j.celrep.2022.111041. [DOI] [PubMed] [Google Scholar]
- 75.Xirouchaki C.E., Mangiafico S.P., Bate K., Ruan Z., Huang A.M., Tedjosiswoyo B.W., Lamont B., Pong W., Favaloro J., Blair A.R., et al. Impaired glucose metabolism and exercise capacity with muscle-specific glycogen synthase 1 (gys1) deletion in adult mice. Mol. Metab. 2016;5:221–232. doi: 10.1016/j.molmet.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.TeSlaa T., Ralser M., Fan J., Rabinowitz J.D. The pentose phosphate pathway in health and disease. Nat. Metab. 2023;5:1275–1289. doi: 10.1038/s42255-023-00863-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bordet G., Couillault C., Soulavie F., Filippopoulou K., Bertrand V. PRC1 chromatin factors strengthen the consistency of neuronal cell fate specification and maintenance in C. elegans. PLoS Genet. 2022;18:e1010209. doi: 10.1371/journal.pgen.1010209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li J., Dallmayer M., Kirchner T., Musa J., Grünewald T.G.P. PRC1: Linking Cytokinesis, Chromosomal Instability, and Cancer Evolution. Trends Cancer. 2018;4:59–73. doi: 10.1016/j.trecan.2017.11.002. [DOI] [PubMed] [Google Scholar]
- 79.Piunti A., Rossi A., Cerutti A., Albert M., Jammula S., Scelfo A., Cedrone L., Fragola G., Olsson L., Koseki H., et al. Polycomb proteins control proliferation and transformation independently of cell cycle checkpoints by regulating DNA replication. Nat. Commun. 2014;5:3649. doi: 10.1038/ncomms4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Razavi M.S., Ahmadi F., Ebrahimnejad P., Akbarzadeh A., Farokhrou M., Nokhodchi A. Harnessing Nanotechnology for Optimized Herbal Cancer Treatment: A Comprehensive Review of Nanoscale Drug Delivery Systems. Pharm. Sci. 2024;30:153–186. doi: 10.34172/PS.2023.27. [DOI] [Google Scholar]
- 81.Harakeh S., Saber S.H., Al-Raddadi R., Al-Jaouni S., Tashkandi H., Qari M., Qari Y., Akefe I.O., Abd Elmageed Z.Y., Haque S. Novel 3′-diindolylmethane nanoformulation induces apoptosis, and reduces migration and angiogenesis in liver cancer cells. J. King Saud. Univ.-Sci. 2023;35:102864. doi: 10.1016/j.jksus.2023.102864. [DOI] [Google Scholar]
- 82.Wang Z., Li Q., An Q., Gong L., Yang S., Zhang B., Su B., Yang D., Zhang L., Lu Y. Optimized solubility and bioavailability of genistein based on cocrystal engineering. Nat. Prod. Bioprospecting. 2023;13:30. doi: 10.1007/s13659-023-00397-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhao C., Zhou X., Cao Z., Ye L., Cao Y., Pan J. Curcumin and analogues against head and neck cancer: From drug delivery to molecular mechanisms. Phytomedicine. 2023;119:154986. doi: 10.1016/j.phymed.2023.154986. [DOI] [PubMed] [Google Scholar]
- 84.Lin C.-H., Chen C.-H., Lin Z.-C., Fang J.-Y. Recent advances in oral delivery of drugs and bioactive natural products using solid lipid nanoparticles as the carriers. J. Food Drug Anal. 2017;25:219–234. doi: 10.1016/j.jfda.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rahman H.S., Othman H.H., Hammadi N.I., Yeap S.K., Amin K.M., Abdul Samad N., Alitheen N.B. Novel drug delivery systems for loading of natural plant extracts and their biomedical applications. Int. J. Nanomed. 2020;15:2439–2483. doi: 10.2147/IJN.S227805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hajialyani M., Tewari D., Sobarzo-Sánchez E., Nabavi S.M., Farzaei M.H., Abdollahi M. Natural product-based nanomedicines for wound healing purposes: Therapeutic targets and drug delivery systems. Int. J. Nanomed. 2018;13:5023–5043. doi: 10.2147/IJN.S174072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang Z., Ji Y., Hu N., Yu Q., Zhang X., Li J., Wu F., Xu H., Tang Q., Li X. Ferroptosis-induced anticancer effect of resveratrol with a biomimetic nano-delivery system in colorectal cancer treatment. Asian J. Pharm. Sci. 2022;17:751–766. doi: 10.1016/j.ajps.2022.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wong K.H., Yang D., Chen S., He C., Chen M. Development of nanoscale drug delivery systems of dihydroartemisinin for cancer therapy: A review. Asian J. Pharm. Sci. 2022;17:475–490. doi: 10.1016/j.ajps.2022.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Watkins R., Wu L., Zhang C., Davis R.M., Xu B. Natural product-based nanomedicine: Recent advances and issues. Int. J. Nanomed. 2015;10:6055–6074. doi: 10.2147/IJN.S92162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Machesky L.M. Deadly actin collapse by disulfidptosis. Nat. Cell Biol. 2023;25:375–376. doi: 10.1038/s41556-023-01100-4. [DOI] [PubMed] [Google Scholar]
- 91.Gu Q., An Y., Xu M., Huang X., Chen X., Li X., Shan H., Zhang M. Disulfidptosis, A Novel Cell Death Pathway: Molecular Landscape and Therapeutic Implications. Aging Dis. 2024 doi: 10.14336/AD.2024.0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang X., Zheng C., Yao H., Guo Y., Wang Y., He G., Fu S., Deng X. Disulfidptosis: Six Riddles Necessitating Solutions. Int. J. Biol. Sci. 2024;20:1042–1044. doi: 10.7150/ijbs.90606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xiao F., Li H.-L., Yang B., Che H., Xu F., Li G., Zhou C.-H., Wang S. Disulfidptosis: A new type of cell death. Apoptosis. 2024;29:1309–1329. doi: 10.1007/s10495-024-01989-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu X., Zhuang L., Gan B. Disulfidptosis: Disulfide stress-induced cell death. Trends Cell Biol. 2023;34:327–333. doi: 10.1016/j.tcb.2023.07.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.