

Abstract
The chemistry of bidentate ligands with a dppf-like motif, where phosphorus is fully or partially replaced by other pnictogens as donor sites, is summarized and discussed in this comprehensive review, while covering the literature from 1966 to 2024, related to more than 165 original references and discussing more than 75 independent chemical entities (1–41). Besides addressing synthetic, structural, and electrochemical aspects of such compounds, their donor properties and metal coordination behavior is discussed, along with catalytic applications. Based on their electronic and steric situations, trends in the performance of such compounds, either as ligands for catalysis or on their own merits for non-catalytic purposes, have been elucidated. Related topics that could not be covered in this article have been acknowledged by referring to the literature for completeness.
Keywords: pnictogens, ferrocene, dppf-analogs, amine, arsenic, antimony, bismuth, homoditopic ligands, heteroditopic ligands
1. Introduction
By hosting mono- [1], di- [2], tri- [3,4], and multidentate phosphanyl ligand systems [5], ferrocene has played a vital role in complexation and catalysis for almost six decades. Despite the dominance of dppf (i.e., 1,1′-bis(diphenylphosphino)ferrocene or 1,1′-bis(diphenylphosphanyl)ferrocene according to IUPAC) [6] and its slimmer and bulkier counterparts as bidentate ligands [7], related 1,1′-bischalcogen and 1,1′-bispnictogen ligands have further emerged over time [8,9,10]. Unlike other bidentate ligands with alkyl (e.g., 1,2-bis(diphenylphosphino)ethane) and alkenyl (e.g., cis-1,2-bis(diphenylphosphino)ethylene) spacers, ferrocene provides a robust yet flexible backbone, which allows a variety of metal centers to be stabilized by attaining various facile spatial orientations (such as classical chelated; open-bridged; double-bridged; quasi-closed bridged; η1, η1-intrabridged; η1, η1-interbridged; and quasi-closed double-bridged complexes) [7,11,12]. At the same time, the 1,1′-(bisphosphino)ferrocene ligand family shows higher bite angles (βn, Figure 1A) during complexation [13], which further plays an instrumental role in catalysis, usually resulting in higher conversion rates than comparable reactions with their alkyl and alkenyl counterparts [14].
Other than contributing to ligand chemistry and catalysis, pnictogen-substituted ferrocenylene species further constitute a major subsection of ferrocenophanes (FCPs, Figure 1B,C), where the resulting rings feature moderate to high molecular strain [15]. Dihedral angle α is considered the key parameter of molecular deformation, which has been related to ring strain and thermodynamic aspects of ring-opening polymerization (ROP) reactions [16], allowing different compounds to be compared. Following the previously demonstrated trends, the α angle decreases with the increase in the size and number of bridging atoms [15,17,18]. In this respect, the largest α angle is manifested by P-bridged [1]FCPs, for which the values vary narrowly between 26.9 and 27.9°, depending upon the nature of the substituents on phosphorus and in the α-positions of the ferrocene (Figure 1D) [15]. Owing to the bigger atomic size of arsenic, the α angle of arsa [1]FCP (α = 22.9°) is significantly smaller than those of phospha [1]FCPs (Figure 1E) [19].
Besides [1]FCPs, there are a handful of examples reported for pnictogen-bridged [n] FCPs (n = 2, 3) [17,18,20,21,22,23,24], among which the highest dihedral angles were found for a family of B,N-bridged [2]FCPs (α = 22.9–24.2°), where the bridging B=N bonds can further be considered isoelectronic to the C=C bond (Figure 1F) [25]. On the other hand, N,Si and N,Sn [2]FCPs (Figure 1G) showed low to moderate dihedral angles (α = 9.36–15.73°), and therefore, no detectable polymers were observed after ROP reactions [23]. N,P [2]FCP H demonstrates a rare example for mixed pnictogen-bridged [2]FCP, which, upon prolonged standing at room temperature, isomerizes into N,C,P [3]FCP I, accompanied by a decrease in the α angle (from 17.93–18.15° for H to 5.81–9.49° for I), which is a determining factor in transforming [2]FCP to [3]FCP [24]. Upon replacement of P in the ansa-position with a smaller pnictogen-like N, diazacarba [3]FCP with paramagnetic (J, α = 11.80–14.31°) and arylamino- (K, α = 15.57–16.44°) substituents showed considerably higher dihedral angles [26] compared to alkylidene-bridged aminophosphanyl [3]FCP I (α = 5.81–9.49°) [24]. However, none of the compounds D–K have been used for complexation and catalysis, except for photoinduced ROP of D with R = Ph and R’ = H, where the ring opening reaction is believed to proceed via in situ formation of a complex L [15,27].
Figure 1.

Bite angle βn for a chelated complex of dppf (A) [7]; [1]FCP and its dihedral angle α (B) [16]; [2]FCP and its dihedral angle α (C) [23,24]; phospha [1]FCP, where R = Ph, CH(Me)(NMe2) and R’ = NiPr2, Ph, tBu, Cl, etc. (D) [15]; arsa [1]FCP (E) [15]; azabora [2]FCP (F) [25]; aza [2]FCP, where ER’2 = SiMe2, SntBu2, SitBu2, and R = SiMe3 (G) [23]; azaphospha [2]FCP (H) [24]; azacarbaphospha [3]FCP (I) [24]; diazacarba [3]FCP with paramagnetic substituents (J) [26,28,29,30]; arylamino-1,3-diaza [3]ferrocenophanes (K) [26]; intermediate for photoinduced ROP of D (L) [15,27]; 1,1′-diisocyanatoferrocene (M) [31,32,33]; and 1,1′-dipthalimidoferrocene (N) [34,35].
Although several excellent review articles and book chapters discuss syntheses and catalytic features of dppf and its analogs [7,8,11,36,37,38,39,40,41], to the best of our knowledge, there are no reports available that solely concentrate on the recent advancements in their non-phosphanyl counterparts. Unlike several bisphosphanyl-substituted dppf analogs, where detailed computational assessment on the bite angles βn and catalytic activities have frequently been reported [7,42], such data are unavailable for their (N,N), (As,As), (Sb,Sb), (Bi,Bi), or (N,P) counterparts. As they are lacking pronounced chemical applications, 1,1′-diisocyanato- (Figure 1M) and 1,1′-dipthalimidoferrocene (Figure 1N) have not been put into focus here. Compounds M and N have notably been used as starting materials to functionalize ferrocenes with amino and (oxycarbonyl)amino moieties via reduction with other amines (for M, Scheme 1) [31,32,33], phosphines (for M, Scheme 1) [43], and alcohols (for M, Scheme 1) [44,45], and via Gabriel-type synthesis with hydrazine (for N, Scheme 1) [34,35].
Scheme 1.

A few selected reactions with compounds M (A–C) and N (D) [32,35,43,45,46].
For a better overview, the wealth of 1,1′-bispnictogen-substituted dppf analogs has been divided into two major categories: 1,1′-symmetrically and -unsymmetrically substituted systems. 1,1′-Symmetrically substituted compounds are discussed depending on their substituents on ferrocenes, and therefore, have further been classified as 1,1′-diamino- (1–12), 1,1′-diimidazolium- (13–16), 1,1′-diimino- (17–19), 1,1′-diarsanyl- (20–23), 1,1′-distibanyl- (24), and 1,1′-dibismuthanyl ferrocenes (25 and 26). On the other hand, 1,1′-unsymmetrically substituted systems are subdivided into the following three groups: 1,1′-N,P (27–39), 1,1′-arsanylphosphanyl- (40), and 1,1′-arsanylstibanylferrocenes (41). Owing to the sake of simplicity and their low abundance, multiferrocenyl and multidentate ligand systems are kept out of our discussion, and consequently, readers interested in such ligands are referred to specialized articles for further information [47,48].
2. Itemization and Inventory
Although the main text of the current article summarizes 1,1′-pnictogen-disubstituted dppf analogs, their chemical structures, synthetic precursors, respective complexes, and applications have been listed in Table 1 for a better overview. Table 1 further serves the purpose of synoptical documentation, so that the functional details of 1–38 can be found via a quick and easy inspectional survey, without investing much time in comprehensive reading.
Table 1.
Overview of the synthetic access and chemical uses for 1,1′-bispnictogen-substituted dppf analogs.
| Group 15 Elements Substituting 1,1′-Ferrocenes | Precursors for Syntheses | Chemical Applications and Complexes |
|---|---|---|
| 1,1′-Symmetrically substituted systems: 1,1′-diaminoferrocenes | ||
|
C5H4LiNMe2 and FeCl2 (Scheme 2A) [49] | Isolated complexes of 1 are reported with TiCl2 [50]. Compound 1 has frequently been used for the purpose of electrochemical measurements [51,52,53]. |
|
Fc’Br2, NaNPh2, and CuI [54] | Compound 2 has been used for electrochemical measurements [51,53,54]. |
|
Fc’(NH2)2, 3,5-Me2-C6H3-Br, and PdBINAP [55] | No application reported [55]. |
|
Fc’(NH2)2 and (2R,5R)-2,5-hexanediol cyclic sulfate [56] | Used for comprehending N-CpFc electron donation [56]. |
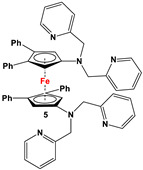
|
C5H4LiN(CH2Py)2 and FeCl2 (Scheme 2B) [57] | Isolated complex of 5 is reported with ZnBr2, Zn(CF3SO3)2, and Co(CF3SO3)2 [58,59,60]. Compound 5 has further been used for synthesizing redox-switchable complexes. |
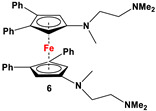
|
C5H4LiNMe(CH2CH2NMe2) and FeCl2 (Scheme 2B) [57] | Compound 6 has been used for electrochemical measurements [58]. |
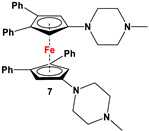
|
C5H4LiN(CH2)4NMe and FeCl2 (Scheme 2B) [57,58] | Compound 7 was used for electrochemical measurements [58]. |
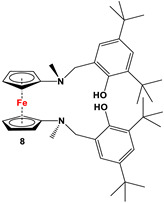
|
Fc’(NH2)2 and 3,5-di-tert-butyl-2-hydroxybenzaldehyde [61]. | Isolated complexes of 8 are reported with Zr(OtBu)2, which was further used in ROP for L-lactide and ε-caprolactone [61]. |
|
Fc’(NH2)2 and aldehydes or silylchlorides or respective ketones (with p-toluene sulfonic acid monohydrate) [62,63,64,65,66]. | Different variations of 9 were used for electrochemical measurements and computational purposes [64], and to act as substituents for carbenes, stannylenes, germylenes [62,63,67], Zr(Bn)2, Mg(THF)2, TiCl2, and TiMe2 [68]. Germylenes with deprotonated 9a, 9d, 9e, and 9h were further explored for oxidation reactions with S, Se, and (PhSe)2 [69]. Isolated complexes of [M(CH2Ar)(THF)] (M = Sc, Y, La, Lu) with 9d and 9f were used for dearomatization and ring-opening reactions [70,71,72,73,74]. |
|
Fc’(NH2)2, PhBr, and Pd2(dba)3 (similar to Scheme 2D) [75]. | Compound 10 was used to synthesize zirconium chelates [76]. |
|
Fc’(NH2)2, respective arylbromides, and Pd2(dba)3 (similar to Scheme 2D) [65,75,77]. | 11b and 11c were used to synthesize N-heterocyclic silylenes [78], germylenes, and stannylenes [79]. Germylenes of 11b and 11c were further explored for oxidation reactions with S, Se, and (PhSe)2 [69]. Isolated complexes of 11c were reported with Al(III) [77]. Isolated complexes of 11d were reported with Zr(NMe2)2 and Zr(Bz)2 [76]. |
|
Fc’(NH2)2, 3,5-Me2-C6H3-Br, and PdBINAP [55]. | No application reported [55]. |
| 1,1′-Symmetrically substituted systems: 1,1′-diimidazoliumferrocenes | ||
|
Fc’I2, imidazole and CuI [80]. | Compound 13 was used to synthesize ferrocene-based redox-responsive receptors [80]. |
|
Fc’(NH2)2 and 2-fluoronitrobenzene [81]. | Isolated complexes of compound 14 were reported with Ir(cod), where “cod” stands for 1,5-cyclooctadiene [81]. |
|
Fc’(N3)2, ethynylpyrene and CuSO4 [82]. | Compound 15 was used to synthesize ion-pair recognition receptors [82]. |
|
M and L-proline (Scheme 2E) [31]. | No application reported [31]. |
| 1,1′-Symmetrically substituted systems: 1,1′-diiminoferrocenes | ||
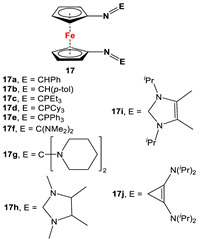
|
Fc’(NH2)2 and respective arylaldehydes [83,84,85,86,87]. | Isolated complex of 17a was reported with PdCl2 and PdMeCl, which have further been used for catalytic purposes [88,89]. Reduced versions of 17 were used for complexation with Zr(Bz)4 [90]. Cationic Ni(II) and Pd(II) complexes are reported with 17c–17i [84,85,86,87]. |
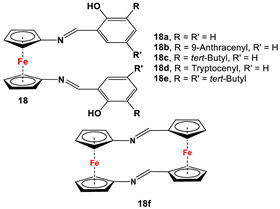
|
Fc’(NH2)2 and respective arylaldehydes (for 18a–18e) [89,91]; Fc’(N3)2, PPh3, and Fc’(CHO)2 (for 18f) [92]. | Isolated complexes for different variations of 18 were reported with Zr(Bz)2 [91], Mg(THF)2 [91], TiCl2 [89], Ti(OiPr)2 [89,93,94], Ce(OtBu)2 [95], In(OtBu) [94,96], Zn [97], Co [97], Zr(OiPr)2 [98], Zr(OtBu)2 [94,98], and Al(OiPr) [94]. Some of these complexes were further used for ethylene, lactone, and lactide polymerizations [89,93,94,95,96,98]. |
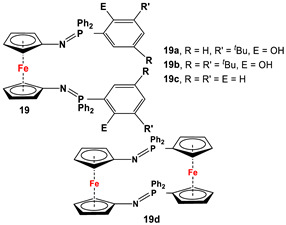
|
Fc’(N3)2 and respective arylphosphine (Scheme 2F) [92,99,100]. | Isolated complexes with 19 are reported with Ce(OtBu)2 [99], Ce(OtBu)THF [99], CeCl(THF) [99], CeI(THF) [99], YCl [99], Y(OtBu) [99], YCl [101], Y(CH2Ph) [101], and Y(CH2SiMe3) [101]. |
| 1,1′-Symmetrically substituted systems: 1,1’-diarsanylferrocenes | ||
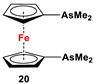
|
Fc’Li2·2/3tmeda and Me2AsCl (Scheme 3A) [2]. | Isolated complexes with compounds 20 are reported with Cr(CO)4 [102], Mo(CO)4 [102], W(CO)4 [102], PdCl2 [103], PdBr2 [103], PtCl2 [103], PtBr2 [103], PtI2 [103], Ni(CO)2 [103], Ni(CO)I2 [103], NiBr2 [104], and [Cu(MeCN)2]BF4 [105]. |
|
1,1′-bis(benzodithiaarsole) ferrocene and excess CyMgCl (Scheme 3E) [106]. | 21 was reported for in situ complexation with Pd2(dba)3, which was further used as an arsa-Buchwald ligand for catalytic purposes [106]. |
|
Fc’Li2·2/3tmeda and Ph2AsCl (Scheme 3A) [2]. | Isolated complexes with compounds 20 are reported with Cr(CO)4 [102], Mo(CO)4 [102], W(CO)4 [102], Ni(CO)2 [103], Ni(CO)I2 [103], (η2-C60)Pt [107], and PdCl2 [108]. |
|
(α-CH2NMe2, β-SiMe3-C5H3)(C5H5)Fe, nBuLi, and Ph2AsCl (Scheme 3C) [109]. | Fungicidal activity of compound 23 for crop plants was examined against fusarium head blight of wheat, early blight of tomato, wilt disease of cotton, ring-rot disease of apple, and brown blotch disease of peanut [109]. |
| 1,1′-Symmetrically substituted systems: 1,1′-distibanylferrocene | ||
|
Fc’Br2, nBuLi, and Ph2SbCl (Scheme 3B) [110]. | Isolated complexes with compounds 24 are reported with AgClO4 [110]. Compound 24 was further oxidized to stiboranes, which was eventually converted to a rare SbOSb [3]FCP [110]. |
| 1,1′-Symmetrically substituted systems: 1,1′-dibismuthanylferrocenes | ||
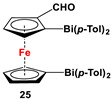
|
(α-CHO-C5H4)(C5H5)Fe, Me2N(CH2)2NMeLi, and nBuLi, and (p-Tol)2BiCl in the next step [111]. | No application reported for 25 [111]. |
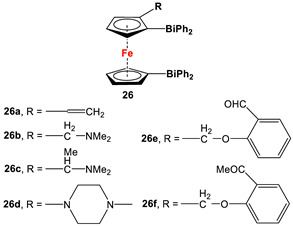
|
(α-CH2NMe2-C5H3)(C5H5)Fe, nBuLi, and Ph2BiCl (Scheme 3D) for 26b, [112] which acted as a precursor for other species. | No application reported for 26 [112]. |
| 1,1′-Unsymmetrically substituted systems: 1,1′-aminophosphanylferrocenes | ||
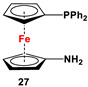
|
Fc’(PPh2·BH3)N3 and DABCO (Scheme 4A) [113]. | No application reported for 27 [113]. |
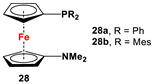
|
Fc’(NMe2)Br, nBuLi, and R2PCl (R = Ph and Mes) [114]. | Isolated complexes with compound 28a were reported with PdCl2, PdCl(SbF6), PPh3(BF4)2, PdPPh2Cp(SbF6)2, [28a·Pd(PPh2)Fc’(NMe2)], and P(p-OMe-C6H4)3(BF4)2 [114]. |
|
Fc’(PPh2·S)N3, Raney Ni, and HCOOAc (Scheme 4B) [113]. | Compound 29 was used to synthesize 32a [113]. |
|
30·S and Raney Ni [115]. | No application reported for 30 [115]. |
|
Fc’(PPh2·BH3)NH2 and Ph2PCH2CO2H·BH3, followed by DABCO [116]. | Isolated complexes with compounds 31 are reported with PdCl2, PdCl(SbF6), and PdCl(SbF6)2 [116]. |
|
29 and BOP/DBU for 32a [113], Fc’(Ph2P·BH3)Br or Fc’(Ph2P·S)Br, and nBuLi and TsN3 for 32b, followed by removal of BH3 or S [113]. | Isolated complexes with compound 32a are reported with AgCl [113], Ag(SbF6) [113], Ag(Me2CO)(SbF6) [113], AuCl [113], (AuCl)2 [113], (AuCN)2(SbF6)2 [113], and (AuCN)2(NTf)2 [113]. Au-complexes of 32a were used for cyclodimerization of enynol [113]. Fischer-type and Mesoionic carbenes were also synthesized from 32a and 32b, respectively [117,118]. |
|
Fc’(PPh2)NH2 and Me2NCH(OMe)2 [119]. | Isolated complexes with compounds 33 are reported with PdCl2, PdCl(BARF) (BARF = B(3,5-(CF3)2C6H3)4), (η6–1-Me, 3-iPr-C6H4)Ru, and (η6-C5Me5)Rh [119]. |
|
Fc’(PPh2)NH2, nBuLi, and respective RNCNR [120,121]. | Isolated complexes with compounds 34 are reported with PdCl2 [120], PdCl(SbF6) [120], PdCl(BF4) [120], PtCl2 [121], PdCl(BARF) [121], PdBr(4-CN-C6H4) [121], and Pd(4-CN-C6H4)SbF6 [121]. Isolated complexes with [34·H]SbF6 were reported with PdCl2 and μ-Pd2Cl4 [120]. |
|
35·PdCl2: Fc’(PPh2)NC, PdCl2(cod), and respective RR’NH [118]. | Isolated complexes with compound 35 are reported with PdCl2, which was further reported with Miyaura borylation [118]. |
|
36·PdCl2: Fc’(PPh2)NC, PdCl2(cod), and respective [Cl(CH2)2NH2Me]Cl [118]. | Isolated complexes with compound 36 are reported with PdCl2, which was further reported with Miyaura borylation [118]. |
|
37·PdCl2: Fc’(PPh2)NC, PdCl2(cod), and (S)-2-(chloromethyl)pyrrolidine [118]. | No application reported for 37 [118]. |
|
38·PdCl2: Fc’(PPh2)NC, PdCl2(cod), and respective (MeO)2CHCH2NHR (R = Me, iPr) [118]. | Isolated complexes with compound 38 are reported with PdCl2, which was further reported with Miyaura borylation [118]. |
|
Fc’(PPh2·BH3)N3, RC≡CH (R = Bz, Mes), and CuSO4·5H2O [122]. | Isolated complexes with compound 39 are reported with MeBF4, PdCl2(MeBF4), AuiPr(MeBF4), and [PdCl2(AuiPr)2(MeBF4)2]1/2 [122]. |
| 1,1’-Unsymmetrically substituted systems: 1,1’-arsanylphosphanylferrocenes | ||
|
[1]PhosphaFCP and nBuLi, followed by Ph2AsCl [123,124]. | No application reported for 37 [123,124,125]. |
| 1,1′-Unsymmetrically substituted systems: 1,1′-phosphanylstibanylferrocene | ||
|
Fc’(PPh2)Br and nBuLi, followed by Ph2SbCl [126]. | Isolated complexes with compound 41 are reported with AuCl, which was further treated with 3,5-di-tert-butyl-o-benzoquinone for further complexation with Sb. Both complexes were eventually used for gold catalysis [127]. |
Scheme 2.

Synthetic routes to selected 1,1′-N,N-ferrocenes (A–F) [31,49,55,56,58,99].
Scheme 3.

Synthetic routes to 1,1′-diarsanyl- (A,C,E), 1,1′-distibanyl- (B), and 1,1′-dibismuthanylferrocenes (D) [2,109,110].
Scheme 4.

Synthetic routes for 1,1′-N,P-ferrocenes (A–D), where DABCO, BOP, and DBU stand for 1,4-diazabicyclo [2.2.2]octane, (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate, and 1,8-diazabicyclo(5.4.0)undec-7-ene, respectively [113,114,119,122].
3. Synthetic Aspects
The earliest example of 1,1′-symmetrically substituted diaminoferrocene was synthesized via a “modified fly-trap methodology”, where a general entry of cyclopentadienylamine is provided by the reaction of C5H5Li with the hydroxylamine derivative of Me2N-OSO2Me. The resulting C5H5NMe2 was then deprotonated and in situ reacted with FeCl2 to obtain title compound 1 (Scheme 2A) [49]. This methodology has further been extended to synthesize 3,4,3′,4′-tetraphenyl-substituted 1,1′-diaminoferrocene (5–7), where a family of 3,4-diphenyl-substituted cyclopentadienylamine has initially been used for deprotonation and subsequent salt metathesis reactions with FeCl2 (Scheme 2B) [57,58,59,60]. While the above-mentioned “modified fly-trap methodology” is restricted to Fc′(NMe2)2 (1) and a few Ph4-substituted 1,1′-diaminoferrocenes 5–7 [57,58,59,60], the majority of other diamines were typically synthesized using a series of well-established synthetic methodologies, starting from Fc′(NH2)2 (selected examples in Scheme 2C,D) [55,56,61,81,91], which is synthesized either via catalytic hydrogenation of Fc′(N3)2 [128] or via two-step Gabriel-type synthesis, starting from Fc’Br2 or Fc’I2 [35]. Other case-specific synthetic strategies have occasionally been employed to access 16 and 19, involving condensation and Staudinger reactions, starting from M and Fc′(N3)2, respectively (Scheme 2E,F) [31,99].
Dimethyl- and diphenyl-substituted 1,1′-diarsanyl- and 1,1′-distibanylferrocenes 20, 22, and 24 were synthesized by salt metathesis reactions of tmeda-stabilized (i.e., Fc’Li2·2/3tmeda) [129] or in situ-synthesized Fc’Li2 with Me2AsCl (Scheme 3A) [2], Ph2AsCl (Scheme 3A) [2], and Ph2SbCl (Scheme 3B) [110], respectively. A similar methodology has further been applied for plana r-enantiomeric versions of 1,1′-distibanyl- and dibismuthanylferrocenes 23 and 26b, where -CH2NMe2 units guided the corresponding lithiation to α-Cp positions (Scheme 3C,D) [109,112,130,131,132]. Compound 26b was further treated with a series of common organic reagents, giving rise to a family of 1,1′-dibismuthanylferrocenes with different pendant difunctional substituents (selected example of 26e in Scheme 3D) [112]. On the other hand, 1,1′-bis(dicyclohexylarsanyl)ferrocene (21) was synthesized via a CuI-catalyzed reaction of 1,1′-bis(dithiaarsole)ferrocene with an excess amount of cyclohexylmagnesium chloride (Scheme 3E) [106].
The main precursors for all previously reported N,P-substituted ferrocene ligands (such as 27 and Fc′(PPh2)N3) have been synthesized, starting from protected ferrocenyl phosphanes to avoid unwanted Staudinger reactions. For example, when Fc′(Ph2PBH3)Br was selectively lithiated and subsequently reacted with TsN3, Fc′(Ph2PBH3)N3 was obtained. In the next step, Fc′(Ph2PBH3)N3 was further reduced and deprotected to obtain title compound 27 (Scheme 4A) [113]. Alternatively, thionation has been used to protect the P-functionality. To this end, selective lithiation was first performed on Fc′(Ph2P = S)Br, followed by salt metathesis reaction with TsN3. The resulting Fc′(Ph2P = S)N3 was either reduced selectively at the P-functionality to obtain Fc′(PPh2)N3 (Scheme 4A), or the P and N-functionalities simultaneously transformed to title compound 32a (Scheme 4B) [113]. Starting from 27 and Fc′(PPh2)N3, a family of 1,1′-azaphospha ferrocenylene ligands (such as 29 [113], 30 [115], 31 [116], 32a [113], 33 [119], 35 [118], and 36 [122], has been accessible using a series of well-established synthetic methodologies, as outlined in Scheme 4A,C. On the other hand, the syntheses of ligands 28 was based upon the successful and large-scale preparation of an unsymmetrically substituted 1,1′-aminobromoferrocene Fc′(NMe2)Br (Scheme 4D), which was first lithiated and subsequently reacted with R2PCl (where R = Ph and Mes) to obtain target compounds 28a and 28b [114]. Here, it is to be noted that in order to synthesize 28, N (i.e., NMe2) was first introduced at ferrocene, followed by P (i.e., PR2), whereas an opposite synthetic order was followed for Fc′(PPh2)N3, 27, and 32a (compare Scheme 4A,B,D) [113].
1,1′-Arsanylphosphanylferrocenes have been synthesized via ring-opening reactions of phospha [1]FCPs, where PhLi has reportedly been used as a ring-opening agent. The resulting anionic species were further in situ reacted with Ph2AsCl to selectively synthesize 40b (Scheme 5A) [123,124,125]. On the other hand, the only example of 1,1′-phosphanylstibanylferrocene was synthesized in a modular approach, where 1,1′-dibromoferrocene was first selectively lithiated and subsequently reacted with Ph2PCl. The resulting Fc′(PPh2)Br was further lithiated and in situ reacted with Ph2AsCl to synthesize mixed compound 41 (Scheme 5B) [126].
Scheme 5.

Synthetic routes for 1,1′-arsanylphosphanyl- (A) and 1,1′-arsanylstibanylferrocenes (B) [113,114,119,122,123,124,125,126].
4. Complexation Motifs of Pnictogen-Substituted 1,1′-Ferrocenes
The steric situation of the phosphanyl units in dppf analogs is one of the key features of these compounds, allowing for a wide variation of complexation modes [7]. By contrast, the pnictogen-substituted non-phosphanyl species show only a handful examples for open-bridged (entries 1–10, Table S1, ESI; Figure 2A), quasi-closed-bridged (entry 11, Table S1, ESI; Figure 2B), double-bridged (entries 12 and 13, Table S1, ESI; Figure 2C), lower- (entries 14–16, Table S1, ESI; Figure 2D), and higher-order η1, η1-interbridged (entry 17, Table S1, ESI; Figure 2E) complexes. However, 1,1′-bisimino- (17a and 17i), diarsanyl- (20 and 22), distibanyl- (24), and phosphanyliminoferrocenes (33 and 34a) predominantly show chelation as their preferred mode of complexation (entries 23–54; Figure 2F), which are further compared with similar complexes from dppf analogs (entries 18–22), and a rare example of double chelation for 17j (Entry 55; Figure 2G) in Table S1 (ESI). When secondary amines (9–11) and substituted imines with proximal hydroxyl groups (18 and 19) were deprotonated and in situ reacted with metal halides, cyclic (entries 56 and 57, Table S1, ESI; Figure 2H), multidentate chelated (entries 58–81, Table S1, ESI; Figure 2I,J), and higher-order species with intermolecular N-M-N bridges (entries 82 and 83, Table S1, ESI; Figure 2K) were obtained. 1,1′-Diaminoferrocenes with secondary amines (9 and 11) have notably been found to be useful in stabilizing carbenes, silylenes, germylenes, and stannylenes, which are further showcased as entries 84–120 in Table S1 (ESI) (Figure 2L). In order to present a complete picture to the readers, Table S1 (ESI) has further been equipped with compounds (entries 121–127; Figure 2M,N), obtained by oxidation of 1,1′-distibanylferrocene 24, featuring a rare family of SbOSb [3]FCPs (entries 124–127; Figure 2N).
Figure 2.

Simplified illustrations demonstrating the binding modes for pnictogen-disubstituted 1,1′-ferrocenes: open- (A), quasi-closed- (B), and double-bridged complex (C); lower (D) and higher aggregated η1, η1-interconnected complex (E); chelated (F) and double-chelated complex (G); cyclic ((H), E’ = FcNR−) and multidentate chelated species ((I,J), O’ = PhO−, E’ = FcNR−); higher-aggregated species with transition metal bridges ((K), E’ = PhO−); tetrylene-bridged species ((L), E = carbene, silylene, stannylene, and germylene), compounds obtained via oxidation of 24 ((M), where X = F and Cl, n = 2 and 4; and (N), where X = F, Cl, ONO2, and OClO3). Note For the sake of simplicity, top views of the complexes are depicted for (E,K).
Each complexation mode is exemplified with examples, which are arranged following the order of Table 1. Complexes from each ligand were then arranged by increasing atomic number of the corresponding complexation partners in Table S1 (ESI). As the complexes with deprotonated ligands (entries 58–81, Table S1, ESI) exhibit E-M distances in an acceptable range of polar coordination bonds and tetrylene-bridged cyclic species (entries 84–120, Table S1, ESI) structurally behave similar to [3]FCPs, the corresponding dihedral angles (α) are recorded for them in Table S1 (ESI). In order to complete the data-set, dihedral (α) angles are listed for all chelating complexes and cyclic species, in contrast to open- (entries 1–10, Table S1, ESI; Figure 2A) and η1, η1-interconnecting complexes (entries 14–16, Table S1, ESI; Figure 2D,E). No meaningful bite angles (βn) have been defined for open- (entries 1–10; Figure 2A), quasi- (entry 11; Figure 2B), double-bridged (entries 12 and 13; Figure 2C), lower- (entries 14–16; Figure 2D), or higher-order η1, η1-interconnected complexes (entry 17; Figure 2E) in Table S1, ESI. For acyclic coordination such as for 24(F)2, 24(F)4, and 24(Cl)4 (entries 121–123; Figure 2M), and higher-order compounds with intermolecular N-M-N bridges (entries 82 and 83, Table S1, ESI; Figure 2N), α and βn are not listed. Similarly, Fischer-type carbenes are not included in this table, as they do not feature any direct bonding connectivity between transition metal and a donor atom directly attached to the ferrocene.
After methodically arranging the entire wealth of complexes and compounds derived from ligands 1–41, our next aim was to compare bite angles (βn) for bisphosphanyl and bisarsanyl ligands. In order to do so, Pd(II) complexes of dppf and its analogs (dppf·Pd(η2-C60), dppf·PdCl2, and Fc′(PMes2)(PtBu2) PdCl2 in entries 18, 19, and 22, respectively, in Table S1, ESI) were first structurally compared with 24·PdCl2 (entry 39, Table S1, ESI), where the βns (complex type Figure 2F) were found following the trend of dppf·Pd(η2-C60), dppf·PdCl2, and Fc′(PMes2)(PtBu2)·PdCl2 > 24·PdCl2. This trend possibly resulted from the long Sb-Pd bonds (2.5020(5) Å), which push the bridging PdCl2 moiety away from the ferrocenylene unit, decreasing the βn compared to dppf and its analogs (where P-Pd = 2.262(4)-2.286(3) Å; case Figure 3A). This argument holds true for a similar comparison between dppf·Pt(η2-C60) and 24·PtCl2 (entries 20 and 43, Table S1, ESI), where the latter showed higher βn compared to the former. However, on comparison between 24·PtCl2 and dppf·PtCl2 (entries 43 and 21, Table S1, ESI), an opposite trend could also be found, where, despite longer Sb-Pt bonds (2.5007(5) Å) and a smaller α angle (1.0°), the former complex showed a higher value of βn (96.49(1)°, complex type Figure 2F) than the latter (P-Pt = 2.266(5) Å, α = 5.0°, and βn = 91.6(2)°). However, a close inspection of their structural features reveals larger twist angle in 24·PtCl2 (32.6°), making βn higher than that of dppf·PtCl2 (tilt angle 30.7°, case Figure 3B).
Figure 3.

Simplified illustrations for the dependence of βn on the lengths of E-M bonds (case (A)), Sb-Pd > P-Pd, βndppf·PdCl2 > βn24·PdCl2) and Cp/Cp twist angles τ (case (B)), τ 24·PdCl2 > τ dppf·PdCl2, βn24·PdCl2 > βndppf·PdCl2). Note Dihedral angle α is not being considered while drawing for case (A), and the Cp rings were consequently drawn as parallel to each other.
When comparing complexes of a ligand scaffold with different substituents, bite angles (βn) increase with enhanced steric interactions. For example, βns for complexes of 17a, 24, and 34a showed the following trends: 17a·PdClMe > 17a·PdCl2 (entries 23 and 24, Table S1, ESI), 24·[Ru(η6-1-Me,3-iPr-C6H4)Cl][PF6] > 24·[Ru(η5-C5Me5)Cl] (entries 36 and 37, Table S1, ESI), and 34a·PdBr(p-CN-C6H4) > [34a·Pd(acac)](SbF6) (entries 49 and 52, Table S1, ESI). On the other hand, when the PdCl2 complex of sterically bulky ligand 17i was compared with that of its slimmer counterpart 17a, βns showed the expected trend, i.e., 17i·PdCl2 > 17a·PdCl2 (entries 23 and 32, Table S1, ESI). Chelate complexes, such as 24·PdCl2 (entry 39, Table S1, ESI), showed a larger βn value than their counterparts with a shared metal cation, such as (24)2·(μ-Pd)(SbF6) (entry 40, Table S1, ESI). This is likely due to the steric interactions between two adjacent ligand molecules of 24, which is further supported by the elongation of the Sb-Pd distances from 24·PdCl2 (2.5020(5) Å) to (24)2·(μ-Pd)(SbF6) (2.6142(4) Å). Despite having a pool of complexes with different metal ions, intraspecific comparisons did not deliver any clear trends for complexes with 17i (entries 25–32, Table S1, ESI) and 24 (entries 36–45, Table S1, ESI).
Considering the effect of secondary ligands, Zr-complexes of 11d feature higher steric congestion in (11d-2H)Zr(CH2Ph)2 than in (11d-2H)Zr(NMe2)2, based on the larger βn angle (complex type H, Figure 2) in the former (112.94(11) Å, entry 57, Table S1, ESI) than in the latter (104.68(15) Å, entry 56, Table S1, ESI). Similarly, by comparison of multidentate chelated species from 18e, (18e-2H)Zr(OiPr)2 (βn = 98.26°, entry 72, Table S1, ESI) showed higher βn (complex type I, Figure 2) than that of (18e-2H)Zr(OnPr)2 (βn = 96.93°, Entry 71, Table S1, ESI). However, despite increased steric bulk and decreased α angle, βn surprisingly decreased on moving from (18e-2H)Zr(OiPr)2 (α = 5.2°, βn = 98.26°, entry 72, Table S1, ESI) to (18e-2H)Zr(OtBu)2 (α = 3.1°, βn = 85.90°, entry 73, Table S1, ESI), which is speculatively due to elongation of OPh-Zr distance in the latter (from 2.031 Å to 2.120(1) Å for (18e-2H)Zr(OiPr)2 and (18e-2H)Zr(OtBu)2, respectively).
Tetrylene-bridged 1,1′-diamino-ferrocenes exhibit an easily comprehendible relation between dihedral angles (α) and corresponding bridging elements, where, α varies in a substantially wider range for carba- (α = 15.4–18.3°), sila- (α = 6.2–16.4°), germa- (α = 5.5–10.2°), and stanna-bridged compounds (α = 1.9–5.6°), listed in entries 84–120 (Table S1, ESI). Similar to [n]FCPs, the dihedral angles (α) increase and decrease with the size of the bridging element. For example, when tetrylene-bridged species derived from 9 and 11 were compared, the following trends were observed for α angles: (9a-2H)Ge, (9a-2H)Ge(SePh)2 > (9a-2H)Sn (entries 84–86, Table S1, ESI); (9b-2H)C, [(9b-2H)CH][BF4], (9b-2H)[C-RhCl(cod)] > (9b-2H)Ge > (9b-2H)2Sn (entries 87–91, Table S1, ESI); [(9d-2H)CH][BF4], (9d-2H)C > (9d-2H)Ge, [(9d-2H)Ge(μ-S)]2, [(9d-2H)Ge(μ-Se)]2, (9d-2H)(Ge3OCl2) (entries 92–97, Table S1, ESI); (9e-2H)Ge, (9e-2H)Ge(SePh)2 > (9e-2H)Sn (entries 98–100, Table S1, ESI); (11b-2H)Si(SePh)2, (11b-2H)Ge, (11b-2H)Ge(SePh)2, [(11b-2H)Ge(μ-Se)]2, [(11b-2H)Ge]2·(μ-Mo(CO)4) > (11b-2H)Sn (entries 104–109, Table S1, ESI); (11c-2H)Si > (11c-2H)Ge (entries 110 and 118, Table S1, ESI); and (11c-2H)Si(SePh)2 > (11c-2H)Ge(SePh)2·1/2C6H6 (entries 113 and 120, Table S1, ESI), where “≈” (almost equal to) and “>” (greater than) were used to indicate trends for a given parameter (i.e., α).
In the next step, the chelated complexes with Fe→Pd and Fe→Ni interactions are discussed, and Table S2 (ESI) summarizes all related species for ligands 17, 28, 31, and 34. Their molecular parameters (such as avg. Cipso,Cp-E bond lengths, Ni/Pd-Fe distances, and tilt and bite angles) will further be compared with the corresponding values for similar complexes with P,P-substituted dppf analogs (entries 1–3, 26, and 27, Table S2, ESI). When analyzing and discussing the lengths of Fe-Pd distances for cationic Pd(II)-complexes with Fe-Pd interactions, it is observed that the differences in Cipso,Cp-P or Cipso,Cp-N bond lengths majorly affect the respective Pd-Fe distances and interactions for related P,P-, N,N-, or P,N-analogs of dppf. Shorter Pd-Fe distances are observed for ferrocene-based N,N ligand scaffolds (Pd-Fe = 2.6297(4)-2.7954(5) Å for complexes with 17c and 17e–17i; entries 4–16, Table S2, ESI) compared with their P,P counterparts (Pd-Fe = 2.7974(10)-3.0014(4) Å for complexes with dppf and Fc’(PMes2)(PtBu2); entries 1–3, Table S2, ESI), whereas for related mixed P,N scaffolds (Pd-Fe = 2.7384(18)-2.8349(11) Å for complexes with 28a, 31, 34a, 34b, and 34c; entries 17–25, Table S2, ESI), intermediate Pd-Fe distances can be seen. Similar trends could also be observed for complexes with Fe-Ni interactions, where complexes with N,N-substituted dppf analogs show shorter Ni-Fe distances (Ni-Fe = 2.6268(4)-2.8244(6) Å for complexes with 17e and 17i; entries 28–30, Table S2, ESI) than those of corresponding P,P-substituted counterparts (Ni-Fe = 3.498 Å for complexes with (C5H4PiPr2)Fe; entries 26 and 27, Table S2, ESI). However, the weakest Fe→M interactions and, consequently, the longest Fe-M bond distances could be observed for Sc (III), Y (III), La (III), and Lu (III) compounds, listed in entries 31–34 (Table S2, ESI), where, despite having short Cipso,Cp-E bond lengths (1.366(8)-1.401(7) Å), the Fe-M bond distance varies between 3.158(2) and 3.3857(8) Å. It is also to be noted that ferrocene moieties in these complexes are tilted in the opposite direction of the E-Pd-E’ or E-Ni-E’ bridges, and the Pd-Fe or Ni-Fe distances are not a consequence of either steric repulsion or any sort of geometric distortions alone in the related molecules (Figure 4). As per Pietschnig and co-workers, intermetallic distances in these complexes result from a compromise between minimized steric repulsions, rotational distortions, and secondary interactions of the ligand systems in the solid state [114].
Figure 4.

Molecular parameters of Pd(II)/Ni(II) complexes of dppf analogs with Fe→Pd/Ni interactions with tilt (α, (A)) and bite (βn, (B)) angles.
DFT calculations performed on these complexes were further able to verify their intrinsic structural features and trends, which further shed light on the nature of the Fe→Pd bonding interactions. In case of [17i·Pd(NCMe)][BF4]2 and [17i·NiPh][BPh4] (entries 16 and 28, Table S2, ESI), Tamm and co-workers demonstrated the possible existence of second minima on the potential energy surface, where the Pd-Fe distance is significantly longer [87,133]. Eventually, Pietschnig and coworkers further increased the distances between the Pd and Fe centers in case of model systems A–C, where second minima were found around ~3.78 Å (A’), ~4.01 Å (B’), and ~4.29 Å (C’), and the Pd atom adopted a slightly distorted T-shaped geometry, which in turn complies with earlier knowledge (Figure 5) [133]. In contrast to A and B, in the case of C, the T-shaped second minima (C’) were more stable by 8.1 kcal mol−1 (ΔEisomer-scan, Figure 5). Pietschnig and co-workers came to an additional conclusion, where the introduction of bulky substituents at the donor atoms prevent the formation of the T-shaped isomer, which can further prevent dimerization via the formation of an intermolecular Pd2Cl2 bridging unit. Therefore, once the chlorine substituent on Pd was replaced with more bulky phosphanes in [28a·Pd(PPh2C5H5)][SbF6]2, [28a·Pd(PPh3)][BF4]2, [28a·Pd(PPh2)Fc′(NMe2)][BF4]2, and [28a·PdP(p-OMe-C6H4)3][BF4]2, due to steric factors, the stability of the T-shaped molecular geometry at Pd centers substantially decreased compared to [28a·PdCl][SbF6]2 [114].
Figure 5.

Potential energy surface scan for the elongation of the Fe-Pd distances in the model compounds containing (N,N’-), (N,P-), and (P,P’-) donating centers. Reproduced from Ref. [114] with permission from the Chinese Chemical Society (CCS), Peking University (PKU), and the Royal Society of Chemistry.
5. Electronic Situation in Pnictogen-Disubstituted 1,1′-Ferrocenes and Their Complexes
The electrochemistry of ferrocenyl amines have been studied extensively for decades. As per Britton and Herberhold et al., the correlation of oxidation potentials for ferrocenyl moieties are in good agreement with Taft’s constants (σop) rather than Hammet’s constants (σ), which indicates predominant resonance effects of N atoms over to their inductive effects [51]. Such efficient N-to-Cp electron donations result in electron-rich Fe centers, which show considerably low redox potentials for 1,1′-N,N-substituted species (such as 1, 2, 4–7, 9b–9e, 11b, and 19; entries 5–16, 19, and 20, Table S3, ESI) and 1,1′-P,N-substituted species (33 and 34a; entries 26 and 27, Table S3, ESI), with respect to those of ferrocene and dppf (entries 1 and 2, Table S3, ESI). N-to-Cp extended electronic conjugations can further be supported by shortening N-CCp bonds (1.377(2) Å for 4) and planar N atoms [56]. For 1,1′-diiminoferrocenes with CHAr substituents (17a and 18e), despite having planar N centers (N-CCp = 1.397(4) Å for 18b, isostructural with 18e) [89], their lone pair is partially conjugated with phenyl groups, which further decreases N-to-Cp electron donation and consequently increases the values of E0 (entries 17 and 18, Table S3, ESI). On the other hand, due to featuring non-planar N atoms, N-to-Cp electron donations are not fully supported for 28a and 28b, resulting in a substantial increase in E0 (entries 23 and 24, Table S3, ESI). Owing to a substantial energy difference between 2p and 5p orbitals, the lone pairs of Sb are not conjugated with the Cp rings, resulting in tetrahedral Sb moieties and relatively high EO values (entry 21, Table S3, ESI). It is here noteworthy that Fe→Pd interactions in 34a·PdCl(SbF6) and [28a·Pd(PPh2)Fc′(NMe2)][BF4]2 are fairly strong and, consequently, the Fe atom is sparingly available for reversible oxidation (entries 36 and 46, Table S3, ESI).
As the coordination complexes are formed by donating lone pairs of electrons from N to corresponding metal ions, N-to-Cp electron donations become no longer possible, and as a consequence of such restricted conjugation, their E0 values increase from 5 to 5·Zn(CF3SO3)2, 5·[Zn(CF3SO3)2]2, 5Chelate·Zn(CF3SO3)2, 5·Co(CF3SO3)2, 5·[Co(CF3SO3)2]2, and 5Chelate·Co(CF3SO3)2 (entries 9, 28–33, Table S3, ESI); from 17a to 17a·PdMeCl and [17a·PdMeCl]BAF (entries 17, 35 and 36, Table S3, ESI); from 28a to [28achelate]28a·Pd(BF4)2 (entries 23 and 40, Table S3, ESI); from 32a to 32a·AuCl, [32a·AuCl]2, and [32a·μ-Au]2X2 (X = SbF6, NTf2) (entries 25, 41–43, Table S3, ESI); from 33 to 33·PdCl2 (entries 26 and 44, Table S3, ESI); and from 34a to 34a·PdCl2, 34a·PdCl(SbF6), and 34a·PdCl(SbF6) (entries 27, 45, and 46, Table S3, ESI). Although P and Sb centers in dppf and 24 do not have lone pairs suitable for P/Sb-to-Cp donations, a negative inductive effect has been considered upon complexation for dppf·PdCl2, Fc′(PMes2)(PtBu2)·PdCl2, 24·PdCl2, and 24·[Pd(η2-maleic anhydride)], accompanied by increasing E0 values for dppf and 24 versus their respective complexes (entries 3, 4, 37, and 38, Table S3, ESI). For compounds with covalently bonded metal bridges and tetrylenes, the increase in E0 values is accompanied by the formation of N-E (where E = metal atoms and tetrylenes) bonds, forcing the N atoms from planar configuration to tetrahedral, and restricting N-to-Cp extended electronic conjugations. For example, E0 values increased from 9b to [9b-2H]CH[BF4], [9b-2H]C, [9b-2H][C-RhCl(CO)2], and [9b-2H]Ge (entries 12, 50–53, Table S3, ESI); from 9c to [9c-2H]Ge (entries 13 and 54, Table S3, ESI); from 9d to [9d-2H]C (entries 14 and 55, Table S3, ESI; from 9e to [9e-2H]Ge (entries 15 and 56, Table S3, ESI); from 11b to [11b-2H]Ge (entries 16 and 57, Table S3, ESI); from 18e to (18e-2H)AlOiPr, (18e-2H)Zn, (18e-2H)Co, (18e-2H)Y(OtBu)THF, (18e-2H)Ce(OtBu)2, and (18e-2H)Ce(OtBu)THF (entries 18, 61–66, Table S3, ESI); from 19a to (19a-2H)Y(OtBu), (19a-2H)Ce(OtBu)THF, and (19a-2H)Ce(OtBu)2 (entries 19, 67–69, Table S3, ESI); and from 19b to (19b-2H)YCl and (19b-2H)Y(CH2Ph) (entries 20, 70, and 71, Table S3, ESI).
6. Applications
The multitude of applied aspects has been sorted into two major divisions: redox-active sensoric materials, where no catalytic reactivity is involved, and catalytic reactions, where the respective ligands have first been used to synthesize isolable or in situ prepared metal complexes, which have been used for various catalytic reactions. On the basis of applications, the non-catalytic reactions were further classified into the following sub-topics: redox-responsive molecular switches, ion recognition receptors, mesoionic and Fischer-type carbenes, dearomatization reactions of N-heterocycles, and the exploration of oxidation reactions on germylenes. On the other hand, the catalytic reactions are categorized under the following sub-headings: ring-opening polymerization of lactides and cyclic esters, Pd-catalyzed cross-coupling reactions, and Au-catalyzed annellation reactions.
6.1. Redox-Active Sensoric Materials
6.1.1. Redox-Responsive Molecular Switches
The most interesting aspect for non-catalytic applications is that of molecular switches, where a characteristic property of a molecule can reversibly be switched on or off by changing the oxidation state of the organometallic scaffold, which is coupled to a macrocyclic ligand. In molecular switches, the coordination of a metal cation is destabilized upon oxidation of the redox-active unit, and can further be restabilized upon reduction of the same. The concept of molecular switches on N-containing dppf analogs were first introduced by Plenio et al. [59], where a molecular switch was coupled with a redox-responsive ligand 5. The interaction of a redox-responsive chelating aminoferrocene 5, a redox-switchable oxaferrocene cryptand (Fc′Crypt), with Zn2+ and Na+ is shown in Scheme 6. The addition of two equivalents of Zn(CF3SO3)2 to an equimolecular mixture of Fc′Crypt·NaCF3SO3 and 5+PF6− in acetonitrile led to the complex 5+·2Zn2+, which is a strong oxidant and capable of oxidizing Fc′Crypt·Na+ quantitatively. The resulting Fc′Crypt+·Na+ subsequently displayed a drastically decreased affinity for Na+, resulting in quantitative removal of Na+ from Fc′Crypt+·Na+. In order to obtain the free molecule of 5, a strong ligand cyclam was added to the reaction mixture, which was capable of removing Zn2+ ions irreversibly (Scheme 6). Free aminoferrocene ligand 5 further acted as a reducing agent to reduce Fc′Crypt + to Fc′Crypt, which finally regained its ability to bind Na+. The reactions shown in Scheme 6 could further be monitored by UV/Vis spectroscopy, where the absorption spectra of 5, 5+, 5·2Zn2+, and 5+·2Zn2+ displayed very distinctive signals [59].
Scheme 6.

Electron-transfer-mediated regulation of the Na+ concentration by Zn2+ ions [59,60].
In order to determine whether the reactions depicted in Scheme 6 actually took place and to further determine whether these reactions are kinetically feasible within a given time frame, 1H NMR and UV/Vis titration were performed. In both experiments, a solution of one equivalent of Zn(CF3SO3)2 in CH3CN was added stepwise to a mixture of 5+PF6− and Fc′Crypt·Na+ in CH3CN, followed by one equivalent of cyclam. Although the UV/Vis experiment is ideal for observing species associated with 5 and 5+, due to the negligible extinction coefficients, it is not suitable for the detection of species derived from Fc′Crypt. To cover this gap, a 1H NMR titration experiment was performed, where CD3CN was used as solvent, and one equivalent of Zn2+ salt was found to be sufficient to initiate the reaction sequence, as shown in Scheme 6 [60].
6.1.2. Ion Recognition Receptor
Ferrocene–triazole and imidazole derivatives have found potential applications in the fields of electrochemical detection and sensing via host–guest chemistry [134]. N centers act as a Lewis bases and bind cations via inter- or intramolecular coordination, whereas anions are recognized through a complimentary C–H···anion or N-H···anion hydrogen bond formation for triazoles and imidazoles, respectively. Upon recognizing the cations, anions, or ion pairs, the resulting in situ-formed LM+, LA−, or LM+A− complexes exhibit an easily detectable change in the redox potential of the ferrocene/ferrocenium redox couple, accompanied by perturbation of the emission signal in the emission spectrum [135]. Although such heteroditopic receptors for ion pair recognition involving organic triazoles are common [136,137], systems with ferrocene backbones are very rare [135,138,139]. Here it is noteworthy that Otón, Tárraga, and Molina et al. introduced an unsymmetrically substituted ferrocenylene triazole with sensing properties for unusual ion pair recognition [82]. In order to synthesize the sensor molecule, compound 15 was reacted with 2-quinaldoyl chloride to obtain species 42, where one half of the ferrocene unit is linked to a pyrene through a 1,2,3-triazole and the other half is substituted by a quinoline ring, linked through amide linkage (Scheme 7).
Scheme 7.

Synthesis of heteroditopic receptor 42 and its ion recognition (shown as blue dashed bonds) properties with Pb2+ and HP2O73− (1:1) [82].
Signal response of the emission for 42 in the presence of several anions (such as F−, Cl−, Br−, AcO−, NO3−, HSO4−, H2PO4−, and HP2O73− as TBA+ salts) was also studied, with only HP2O73− anion causing a small but significant change in the fluorescence spectrum. During the course of the titration, an isoemissive point at λ = 425 nm was conserved, which indicates the existence of an equilibrium between species 42 and complex [42·HP2O73−]. The cross-selectivity of 42 was further tested with several cations (such as Li+, Na+, K+, Ca2+, Mg2+, Ni2+, Zn2+, Cd2+, Pb2+, Cu2+, and Hg2+), where only Pb2+ (ΔE1/2 = 75 mV) and Hg2+ (ΔE1/2 = 155 mV) displaying considerable perturbation in the oxidation wave, with a moderate amount of the cation (10 equivalents), while others required higher amounts (100 equivalents) or showed no changes at all. Upon testing the change in fluorescence spectra with the above-mentioned cations, it was revealed that only Hg2+ caused variations in the emission properties of receptor 42, where a progressive decrease in the monomer emission intensity of about 85% (from ΦF = 0.071 to ΦF = 0.012) with the addition of 60 equivalents of Hg2+ was observed. When the ion pair recognition capability of 42 was studied via UV/Vis spectroscopy for Pb2+ and HP2O73− (1:1), a remarkable red shift in color was observed, where 42, [42·Pb2+], [42·HP2O73−], and [42·(Pb2+)(HP2O73−)] displayed the visible colors of yellow, red, yellow, and green, respectively. In order to have a further insight into the structure of the resulting [42·(Pb2+)(HP2O73−)] anion, theoretical calculations were used, and Pb···Ntriazole, Pb···OHP2O7, Pb···Oimide, Pb-Nquinoline, and OHP2O7···Hpyrene connectivities were observed in the optimized structure (see [42·(Pb2+)(HP2O73−)] in Scheme 7).
The metal recognition properties of the imino-bridged [2.2]ferrocenophanes 18f and 19d were also evaluated by cyclic voltammetry, and a reversible electrochemical response was observed for Zn2+ complexation/decomplexation of 18f [92]. Species 18f further underwent altered oxidation in the presence of Cu2+ and Hg2+ cations, in contrast to Li+, Na+, K+, Mg2+, Ca2+, Cd2+, and Ni2+, for which no significant change in the corresponding electrochemical processes was found. Monitoring the recognition property of 18f with UV/Vis spectroscopy, no observable changes were noticed upon addition of Li+, Na+, K+, Mg2+, Ca2+, Cd2+, and Ni2+, whereas significant changes in the absorption bands were observed upon addition of Cu2+, Hg2+, and Zn2+. With increasing amounts of Zn2+ added to 18f, the low-energy (LE) metal-to-ligand transition band (MLCT) at λ = 491 nm gradually disappeared and a new band at λ = 600 nm progressively appeared, accompanied by a visible transformation in color from red to deep green. The presence of an isosbestic point at λ = 505 nm indicates a clean interconversion between the uncomplexed 18f and complexed species 18f·Zn2+. In sharp contrast to 18f, species 19d exhibited electrochemical responses only in the presence of Li+, accompanied by a red shift in the absorption signal from λ = 480 nm to λ = 669 nm and a clear isosbestic point located at λ = 614 nm. The exceptionally selective complexation behavior of 19d can be explained by size selectivity, where, based on SCXRD of [19d·Li]B(C6H5)4, a small cavity is formed by two N atoms and Fe of bisiminophosphoranylferrocene, providing space up to the size of Li+ only.
Jin and Liu et al. introduced a ferrocene-based receptor for the chloride anion 43, (Scheme 8) which was prepared from compound 13 by stepwise reaction with MeI and NH4PF6. The addition of a sub-equivalent amount of Cl− to receptor 43 caused a significant potential shift of ΔE1/2 = 310 mV, where the second oxidation wave overlapped with the oxidation wave of Cl− in the CV curve. Upon addition of Cl−, the square wave voltammogram (SWV) curve of 43 showed a gradual anodic shift for the 43·Cl− complex, along with an increase in peak current. Upon addition of one equivalent of chloride, a clear two-wave potential was observed in SWV experiments, with a separation of ca. 160 mV from each other. Upon addition of more than two equivalents of Cl−, only the peak corresponding to the oxidation of the 43·Cl− complex was observed on the SWV curve, which implies a strong complexation between 43 and Cl−. When more than three equivalents of Cl− were added to 43, an additional peak for Cl− oxidation (at ca. 0.7 V) was noticed [80].
Scheme 8.

Synthesis of ferrocenylene bisimidazolium salts acting as Cl− recognition receptor [80].
6.1.3. Mesoionic and Fischer-Type Carbenes
Mesoionic carbenes (MICs) are a type of a stable yet fairly reactive carbenoid intermediate that, despite being related to N-heterocyclic carbenes (NHCs), are an abnormal variant of the latter and therefore are sometimes referred to as remote N-heterocyclic carbenes [140]. MICs were notably introduced in the ferrocene system by Sarkar et al., where the resulting ferrocenyl heteromultimetallic iridium(I) and gold(I) complexes were used to demonstrate redox-switchable catalysis to synthesize oxazoline, furan, and phenols [141,142]. Unsymmetrically substituted ferrocenylene-based MICs were introduced by Štěpnička et al., where species 32b·BH3 was first reacted with benzyl- and mesityl-substituted acetylene to obtain triazole 39 (Scheme 9A) [122]. After deprotonation with [Me3O][BF4] and subsequent removal of BH3, active carbenoids 44 was obtained, which was then reacted with transition-metal precursors to synthesize chelated and η1, η1-interbridged complexes 45 and 46, respectively (Scheme 9B,C). When the Au-complex of 39a (47 with R = Bn) was further reacted with [PdCl2(MeCN)2], heterobimetallic gold and palladium complex 48 resulted (Scheme 9D) [122].
Scheme 9.

Mesoionic carbenes 39 (A) and their complexation with Pd(II) (B,C) and Au(II) (D), where IPr stands for 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene (inset) [122].
Fischer-type carbenes are in their corresponding singlet states, often featuring an empty and accessible pz orbital on the carbene C atom, which is capable of accepting π-back donation from the coordinating low-valent, late-transition elements [143]. The stability of such carbenes is further attained by the π-donor substituents in conjugation of the carbene atoms (e.g., alkoxy and alkylated amino groups). Although the concept of Fischer-type carbene complexes is well explored, with almost all transition metals and several organic moieties as backbones to host the molecule, Štěpnička and co-workers have recently reported synthesis and complexation for a unsymmetrically substituted ferrocenylene scaffold, where a second coordination from PPh2 unit helps to stabilize the metal cation (Pd2+) [117]. In order to synthesize these complexes, when 32a was separately reacted with (cod)PdClMe, [(RR’)Pd(μ-Cl)]2 (where R = Me and R’ = PPh3, (RR’) = 2-(dimethylamino)methylphenyl, and (RR’) = η3-C3H5), (η3-C3H5)Pd(PPh3)Cl), simultaneous coordination of the Ph2P with Pd2+ and insertion of the isocyanide groups into the Pd–C bonds were observed (species 49–53, Scheme 10).
Scheme 10.

Fischer-type carbene complexes (49–53) with Pd(II), synthesized from unsymmetrically substituted compound 32b [117].
6.1.4. Dearomatization Reactions of N-Heterocycles
Benzyl complexes of group 3 elements [(9f-2H)M(CH2Ar)(THF)] (where Ar = 3,5-Me2C6H3 and M = Sc, Y, La, and Lu), supported by a ferrocene diamide ligand 9f, are reactive toward aromatic N-heterocycles via the coupling or breaking of C-N bonds (Scheme 11) [70,71,72,73,74]. For example, when a toluene solution of [(9f-2H)Sc(CH2Ar)(THF)] (where Ar = 3,5-Me2C6H3) was heated with 2-phenylpyridine at 70 °C, the first step of the reaction was commenced by the THF displacement and coordination of 2-phenylpyridine to form 54, followed by ortho-metalation of the pyridine ring and simultaneous removal of mesitylene to produce the THF adduct 55 [70]. Upon prolonged heating at 70 °C in toluene solution, species 55 converted into C-C-coupled product 56, where among the two pyridine rings, one was dearomatized (Scheme 11A). When species [(9f-2H)Sc(CH2Ar)(THF)] (where Ar = 3,5-Me2C6H3) was reacted with 1-methylimidazole, displacement of THF was followed by the formation of imidazole-coordinated intermediate 57, which underwent simultaneous removal of mesitylene to produce C-H activation product 58 (Scheme 11B). In the next step, C-C coupling occurred between two neighboring imidazole units, followed by the dearomatization of one imidazole ring to yield intermediate 59, which very rapidly arranged itself to produce final product 60 (Scheme 11B). When 55 was synthesized, isolated, and subsequently reacted with 8-methylisoquinoline, corresponding C-C-coupled product 61 with a dearomatized isoquinoline unit was obtained (Scheme 11C) [72]. On the other hand, upon reaction of isoquinoline or 2,2′-bipyridine with [(9f-2H)M(CH2Ar)(THF)] (where M = Sc, Y, La, or Lu, and Ar = 3,5-Me2C6H3), alkyl migration of the benzyl ligand onto the pyridine ring was facilitated, accompanied by the dearomatization of the corresponding N-heterocycle, to yield 62 and 63 (Scheme 11D,E) [73,74]. When a toluene solution of [(9d-2H)Lu(CH2Ar)(THF)2] was heated at 70 °C separately with 1-methylimidazole or isoquinoline, corresponding products similar to 60 and 62 were obtained (with R = adamantyl), respectively [74].
Scheme 11.

Proposed mechanistic details for dearomatization and ring-opening reactions (A–E) using [(9f-2H)M(CH2Ar)(THF)] (M = Sc, Y, La, Lu), where Ar, R, and iqn stand for 3,5-Me2C6H3, SiMe2tBu, and isoquinoline, respectively [70,71,72,73,74].
6.1.5. Exploration of Oxidation Reactions of Germylenes
Siemeling and co-workers recently explored the oxidization reaction on their flagship germylenes, prepared from 1,1′-diaza ferrocenes (9a, 9b, 9d, 9e, 9h, 11b, and 11c), where species (9a-2H)Ge, (9d-2H)Ge, (9e-2H)Ge, (9h-2H)Ge, (11b-2H)Ge, and (11c-2H)Ge were separately treated with elemental sulfur (S8), elemental selenium (red Se), and PhSe-SePh to obtain the oxidized products, such as [(9a-2H)Ge(SePh)2], [(9d-2H)Ge(μ-S)]2, [(9d-2H)Ge(μ-Se)]2, [(9e-2H)Ge(SePh)2], [(9h-2H)Ge(μ-S)]2, [(9h-2H)Ge(μ-Se)]2, [(9h-2H)Ge(SePh)2], [(11b-2H)Ge(μ-Se)]2, and [(11c-2H)Ge(μ-Se)]2 (Scheme 12). Unprecedentedly short intramolecular CH···Se distances were observed in SCXRD-analyzed structures of [(9a-2H)Ge(SePh)2] and [(9d-2H)Ge(μ-Se)]2 (Scheme 12) [69].
Scheme 12.

Oxidative addition reactions of germylenes and intramolecular CH···Se linkages (shown as blue dashed bonds) for [(9a-2H)Ge(SePh)2] and [(9d-2H)Ge(μ-Se)]2 [69].
6.2. Catalytic Reactions
6.2.1. Ring-Opening Polymerization (ROP) of Lactides and Cyclic Esters
In order to evaluate the catalytic activity for the ROP, (18e-2H)Ce(OtBu)2 was reacted at 70 °C with L-lactide and ε-caprolactone (Scheme 13A,B) [95]. The reaction with 100 equiv. of ε-caprolactone took 4 h to reach 80% conversion, whereas a similar reaction with 100 equiv. of L-lactide required only 20 min. Although ROP of ε-caprolactone is generally more facile than that of L-lactide, the extraordinarily high reactivity of L-lactide is not fully understood. Here, it is to be noted that the isotactic polymer was formed exclusively without epimerization of the stereogenic centers (Scheme 13A). By comparison, the simple alkoxide Ce(OtBu)4(THF)2 was found to be more active than (18e-2H)Ce(OtBu)2 for L-lactide polymerization (Scheme 13A). When (18e-2H)Y(OtBu)(THF) was used for L-lactide polymerization (Scheme 13A), it was observed that the Y-complex was more active than (18e-2H)Ce(OtBu)2, with the ROP occurring at room temperature within minutes for the Y-counterpart. Molecular weight analyses of the polymers resulting from ROPs showed that the polymers from (18e-2H)Y(OtBu)(THF) had lower poly-dispersity indices (PDIs) than those from (18e-2H)Ce(OtBu)2. The Mulliken charges, calculated by DFT on (18e-2H)Y(OtBu)(THF) and (18e-2H)Ce(OtBu)2, indicated that the Y-center in (18e-2H)Y(OtBu)(THF) is more electrophilic than the Ce-center in (18e-2H)Ce(OtBu)2, making yttrium more reactive than cerium toward L-lactide [95]. However, unprecedentedly high reactivity towards the polymerization of L-lactide, ε-caprolactone, trimethylene carbonate, and δ-valerolactone was further achieved by ROP reaction with complex (18e-2H)In(OtBu) at room temperature (Scheme 13) [96].
Scheme 13.

Polymerization of L-lactide (A), ε-caprolactone (B), trimethylene carbonate (C), and δ-valerolactone (D) [95,96].
In order to explore the geometric change during lactide ring-opening polymerization, Diaconescu and co-workers used (18e-2H)Zr(OnPr)2, (18e-2H)Zr(OiPr)2, and (18e-2H)Zr(OtBu)2 as precatalysts, where (18e-2H)Zr(OtBu)2 showed no activity but both (18e-2H)Zr(OnPr)2 and (18e-2H)Zr(OiPr)2 enabled yields in the range of 60–70% at 100 °C over a reaction time of 24 h [98]. 1H NMR experiments of the reaction mixtures indicated that the corresponding reactions with (18e-2H)Zr(OnPr)2 and (18e-2H)Zr(OiPr)2 proceeded with a geometric change from cis-β to trans within 2 h upon heating at 100 °C. A similar geometric change was not observed with (18e-2H)Zr(OtBu)2 even after 24 h heating at 100 °C, and consequently, catalysis did not occur for the latter. Here, it is noteworthy that (18e-2H)Zr(OnPr)2, (18e-2H)Zr(OiPr)2, and (18e-2H)Zr(OtBu)2 contain cis-β and trans isomers in ratios of 71:29, 84:16, and 95:5 at room temperature [98]. 1H NMR experiments of the reaction mixtures further indicated that the polymerization majorly propagates after the previously mentioned change in geometry (i.e., cis-β to trans, Scheme 14). This observation further complies with the previously reported reactivity of salen-TiCl2 complexes [144,145,146].
Scheme 14.

Proposed reactions of (18e-2H)Zr(OR)2 (where R = nPr and iPr) and lactide [98].
6.2.2. Redox-Switchable Catalysis
The activities of group 4 metals (Zr and Ti) for ROP of L-lactide and ε-caprolactone were further explored with redox-moderated precatalyst 8·Zr(OtBu)2, where oxidized and reduced forms of the corresponding metal complex affected the rate of polymerization [61]. When 8·Zr(OtBu)2 was heated at 100 °C in the presence of 100 equiv. of L-lactide, 90% conversion could be achieved within 2 h. On the other hand, with the oxidized version of 8·Zr(OtBu)2 as a catalyst (i.e., [8·Zr(OtBu)2]BARF, synthesized via oxidation of 8·Zr(OtBu)2 with AcFcBARF, where AcFc = Fc(COCH3)), <5% conversion was observed under the same reaction conditions as before. However, the activity toward ε-caprolactone showed the opposite trend, where [8·Zr(OtBu)2]BARF and 8·Zr(OtBu)2 exhibited 98% and <5% conversion, respectively, with 100 equiv. of starting material at 25 °C over 24 h. In situ conversion between the oxidized and reduced forms of 8·Zr(OtBu)2 was further examined regarding their catalytic implications, where AcFcBARF was added to the reaction mixture at 43% conversion of L-lactide to polylactide (Figure 6A). Owing to the oxidation of 8·Zr(OtBu)2 to [8·Zr(OtBu)2]BARF, the polymerization halted and resumed at the previous rate upon reduction with Co(η5-Cp)2 (Figure 6A). Similarly, when Co(η5-Cp)2 was added to the reaction mixture for the polymerization of ε-caprolactone with [8·Zr(OtBu)2]BARF, the conversion halted until further oxidation (via the in situ addition of AcFcBARF) was performed (Figure 6B). Upon analyses with gel-permeation chromatography (GPC), the resulting polymers showed narrow molecular weight distribution with PDIs in the range of 1.1 to 1.2 [61].
Figure 6.

Plot of catalytic conversion vs. time (min) for the polymerization of (A) L-lactide with 8·Zr(OtBu)2, and (B) ε-caprolactone with [8·Zr(OtBu)2]BARF, where AcFcBARF (AcFc = Fc(COCH3)) and Co(η5-Cp)2 were used as the oxidant and reductant, respectively. Adapted with permission from Ref [61]. Copyright 2014 American Chemical Society.
By using these redox switches, Diaconescu and coworkers also demonstrated the successful syntheses of AB- and BA-type diblock and ABA- and ABC-type triblock copolymers [147]. For example, L-lactide was first polymerized in the presence of 8·Zr(OtBu)2, followed by in situ oxidation of 8·Zr(OtBu)2 with AcFcBARF and the addition of cyclohexene oxide to obtain diblock co-polymer [L-lactide]a-[cyclohexene oxide]b. Co(η5-Cp)2 was then added to the resulting reaction mixture, followed by the addition of β-butyrolactone to obtain ABC-type triblock copolymer [L-lactide]a-[cyclohexene oxide]b-[β-butyrolactone]c. When the mechanistic study for block-dependent copolymerization of cyclohexene oxide and lactide was performed for ring-opening polymerization, it was found that the reaction is thermodynamically unfavorable for lactide alone with [8·Zr(OtBu)2]BARF [61,147,148]. However, this reaction becomes thermodynamically favorable for lactide after the polymerization of cyclohexene oxide with [8·Zr(OtBu)2]BARF, where the initiation (or ring-opening) of lactide is thermodynamically favorable but the propagation is not. The propagation step for the polymerization of lactide is only possible after the polymerization of cyclohexene oxide [148].
The same group further reported an electrochemically controlled synthesis of multiblock copolymers, where the redox state of the precatalyst (8-2H)·Zr(OtBu)2 was electrochemically altered with a glassy carbon electrode, which resulted in a change in the catalytic selectivity of the catalyst [149]. For example, a sequential addition of L-lactide to a solution of TPANTf2 (75 mM; TPANTf2 = tetrapropylammonium bistriflimide) and 1,2-difluorobenzene (1.5 mL) of (8-2H)·Zr(OtBu)2, followed by electrochemical oxidation and addition of cyclohexene oxide, yielded AB-type diblock copolymer [L-lactide]a-[cyclohexene oxide]b. When the resulting reaction mixture was further electrochemically reduced and L-lactide monomer was added, ABA-type triblock copolymer [L-lactide]a-[cyclohexene oxide]b-[L-lactide]c resulted. The electrochemically controlled redox reaction of (8-2H)·Zr(OtBu)2 ⇌ (8-2H)+·Zr(OtBu)2, along with their corresponding bulk electrolysis potentials (vs. Ag/Ag+ pseudoreference electrode), are demonstrated in Figure 7.
Figure 7.

Electrochemically moderated one-pot polymerization with 8·Zr(OtBu)2 [149].
Polymerization of L-lactide was further performed at 90 °C with (18e-2H)Ti(OiPr)2 and its oxidized version, [(18e-2H)Ti(OiPr)2]BARF (synthesized via oxidation of (18e-2H)Ti(OiPr)2 with AcFcBARF), and the catalytic conversion was plotted against time, with (18e-2H)Ti(OiPr)2 showing an extraordinarily low catalytic conversion rate (red markers in Figure 8A) [93]. This observation is opposite to the previously reported catalytic trend, where electron deficient complexes showed substantially lower conversion rates than their corresponding electron-rich counterparts [61,150]. To examine the redox-switching ability, (18e-2H)Ti(OiPr)2 was reacted with 100 equiv. of L-lactide, where the oxidation state of the catalyst was in situ modulated via the addition of AcFcBARF and Co(η5-Cp)2 as the oxidant and reductant, respectively. As shown in Figure 8B, the catalytic activity of (18e-2H)Ti(OiPr)2 was substantially low until the complex is in situ oxidized with AcFcBARF, but subsided after reaching ca. 4–6%. When the oxidized catalyst was further in situ reduced upon addition of Co(η5-Cp)2, the catalyst surprisingly began to perform at a greater rate until up to ca. 40% conversion [93]. Further in situ oxidation with AcFcBARF halted the catalytic activity, which was followed by restoration of the same upon addition of Co(η5-Cp)2. As the trend in the in situ redox-switchable catalysis (Figure 8B) is different than that found in Figure 8A, the in situ oxidation of (18e-2H)Ti(OiPr)2 was performed with AcFcBARF in the presence of excess L-lactide, which showed a halted reactivity at ca. 4–6% catalytic conversion. Upon subsequent in situ reduction of the resulting oxidized species with Co(η5-Cp)2 in the presence of excess L-lactide, a dramatic increase in polymeric activity could be observed, with the conversation reaching up to ca. 80% over 5 h. As an outcome of the previous observations, Long and coworkers concluded that the catalytic activities of (18e-2H)Ti(OiPr)2 and [(18e-2H)Ti(OiPr)2]BARF not only depend on the oxidation states of the metal ions in the respective precatalysts, but also considerably depend on the chemical species present during their catalytic reactions.
Figure 8.

Plot for polymerization (%) of L-lactide with reaction time (h), (A) where red and blue markers have been used for catalysis with (18e-2H)Ti(OiPr)2 and [(18e-2H)Ti(OiPr)2]BARF, respectively, (B) and in situ redox-switching with (18e-2H)Ti(OiPr)2 as starting precatalyst. Adapted with permission from Ref. [93]. Copyright 2015 American Chemical Society.
Iso-propoxide complexes of aluminum, supported by 18e (i.e., (18e-2H)AlOiPr and [(18e-2H)AlOiPr][BARF]), were further used to examine the redox switchability for the ring-opening polymerization of L-lactide, ɛ-caprolactone, δ-valerolactone, β-butyrolactone, trimethylene carbonate, and cyclohexene oxide, where only the non-oxidized compound (i.e., (18e-2H)AlOiPr) was found to be active for L-lactide, β-butyrolactone, and trimethylene carbonate [94]. Although 64% and 98% conversion were observed after 24 h at 100 °C (catalyst:monomer, 1:100), for L-lactide and β-butyrolactone, respectively, a quantitative conversion was observed for trimethylene carbonate (catalyst:monomer, 1:100) even after 2.5 h at room temperature. In the case of ɛ-caprolactone and δ-valerolactone (catalyst:monomer, 1:100), no difference in the activity of oxidized and reduced forms of the catalyst could be observed, as in both cases the quantitative transformation could be achieved within 2 h at room temperature. On the other hand, when similar ring-opening polymerization was investigated for cyclohexene oxide separately, with reduced and oxidized forms of the above-mentioned catalyst (catalyst:monomer, 1:100), only the oxidized form of the catalyst (i.e., [(18e-2H)AlOiPr][BARF]) was found to be active [94]. By using the selectivity for the catalytic reactions of the above-mentioned monomers, the syntheses of AB block copolymers were attempted with L-lactide and cyclohexene oxide. In order to do so, polymerization of L-lactide was first performed with catalyst (18e-2H)AlOiPr, followed by the addition of AcFcBARF and cyclohexene oxide to stop the L-lactide polymerization and initiate the corresponding polymerization of cyclohexene oxide to obtain [L-lactide]a-[cyclohexene oxide]b-OiPr (Scheme 15A). The reverse diblock co-polymer [cyclohexene oxide]b-[L-lactide]a-OiPr could further be synthesized via the following steps: initial polymerization of cyclohexene oxide with [(18e-2H)AlOiPr][BARF], followed by the addition of Co(η5-Cp)2, along with L-lactide (Scheme 15B). Triblock ABA co-polymer [L-lactide]a-[cyclohexene oxide]b-[L-lactide]c-OiPr was synthesized from [L-lactide]a-[cyclohexene oxide]b-OiPr via subsequent in situ reduction of [(18e-2H)AlOiPr][BARF] with Co(η5-Cp)2, followed by the addition of L-lactide monomers to the reaction mixture (Scheme 15A) [94]. Similarly, when trimethylene carbonate was added at 100 °C after the formation of [cyclohexene oxide]b-[L-lactide]a-OiPr, the triblock co-polymer [trimethylene carbonate]c-[cyclohexene oxide]b-[L-lactide]a-OiPr was obtained (Scheme 15B) [94].
Scheme 15.

ABA- (A) and ABC-type (B) triblock polymers, synthesized with redox-switchable catalyst (18e-2H)AlOiPr [94].
Markovnikov hydroalkoxylation of unactivated olefins with cobalt complexes of salen-ligands accompanied by silane and N-fluoropyridinium salt was primarily reported by Hiroya and co-workers [151]. Inspired by the work of Hiroya et al. [151], Diaconescu and coworkers optimized the catalytic activity of [(18e-2H)Co] towards hydroalkoxylation of olefins in presence of siloxane TMDSO (TMDSO = HMe2Si-O-SiMe2H) and electrophilic fluorinating agent NFPBF4 (where NFPBF4 = N-fluoro-2,4,6-trimethylpyridiniumtetrafluoroborate) in CH2Cl2 (Scheme 16) [97]. Although this catalytic system was effective (yield ca. 99%) for many different varieties of styrene derivatives (Scheme 16), little to no activity was observed for alkyl or norbornyl derivatives. Moreover, when the tetrameric Co- and monomeric Zn-complexes were used in place of [(18e-2H)Co], little and no catalytic conversion were observed for [(18e-2H)Co]4 and [(18e-2H)Zn], respectively. In situ oxidation of [(18e-2H)Co] by addition of AcFcBARF halted the catalytic reaction, which further resumed to the previous rate upon in situ reduction with Co(η5-Cp)2 (Scheme 16).
Scheme 16.

Catalytic reaction by [(18e-2H)Co] and plot of conversion versus time of the redox switch (inset). Adapted with permission from Ref. [97]. Copyright 2016 American Chemical Society.
6.2.3. Pd(II)-Catalyzed Cross-Coupling Reactions
Phosphine ligands have been employed for Pd(II)-catalyzed Suzuki cross-coupling of haloarenes with arylboronic acid for decades [152]. Being interested in developing a new generation of non-poisonous, environment-friendly, water-based, and phosphine-free catalysts of high efficiency, Hor and coworkers investigated the catalytic efficiency of the Pd(II)-complex of 1,1′-diiminoferrocene 17a for such cross-coupling reactions [88]. As the products were water-insoluble, their easy separation and isolation from the crude reaction mixture provided an additional advantage for this catalyst (Scheme 17A). Although 17a·PdCl2 has successfully catalyzed cross-coupling reactions between aryl bromides/iodides and aryl boronic acids in non-homogenous aqueous reaction conditions, it failed to display any catalytic activity for Cl-substituted starting materials (Scheme 17A). The choices of base (Scheme 17B), catalytic load, and recoverability have further been investigated for 17a·PdCl2 by Hor et al [88].
Scheme 17.

Suzuki cross-coupling reactions with catalysts 17a·PdCl2 (A), 24·PdCl2 (B), and 24·[Pd(η2-maleic anhydride)] (C) [88,110].
In order to compare the catalytic activity of 1,1′-distibanylferrocene 24 with dppf, Štěpnička et al. used 24·PdCl2 and 24·[Pd(η2-maleic anhydride)] as catalysts for Suzuki cross-coupling reactions [110]. However, the yields for reactions with Pd(II)-complexes of 24 (i.e., 24·PdCl2 and 24·[Pd(η2-maleicanhydride)]) were rather small compared to those from respective complexes of dppf (i.e., dppf·PdCl2 and dppf·[Pd(η2-maleic anhydride)], Scheme 17C).
In order to explore the Miyaura borylation reaction with Pd(II)-complexes of 1,1′-aminophosphanylferrocene carbene ligands, 64–68 were first synthesized from 32a via reaction with PdCl2(COD) and primary or secondary amines or ammonium salt (Scheme 18A) [118]. Precatalysts 64–66 and 68 were then reacted with 4-bromotoluene and bis(pinacolato)diborane to synthesize the corresponding boronic esters (Scheme 18B) [118]. A series of optimization experiments with different solvents and bases revealed iPrOH and KOAc to be suitable for these reactions. When 64–66 and 68 were further used as precatalysts for the Miyaura borylation reaction of 4-bromotoluene, 65b and 66 showed the maximum catalytic activity and selectivity, resulting in ca. 98% yield of boronic ester and 0% yield of homocoupled product 4,4′-dimethylbiphenyl (based on NMR spectra measured from the reaction mixture, Scheme 18B) [118]. When the most synthetically accessible complex, 66, was used as catalyst for reactions with several other aryl bromides, the lowest coupling yields could be observed for mesityl bromide (Scheme 18C). Nonetheless, the reactions with other bromides resulted in decent to excellent yields, varying in a range of 66–97% (Scheme 18C).
Scheme 18.

Syntheses (A) and Miyaura borylation reactions (B,C) with precatalysts 64–66 and 68 [118]. The yields of 4,4′-dimethylbiphenyl are given in parentheses (B).
6.2.4. Au(I)-Catalyzed Annellation Reactions
Being inspired by the catalytic properties of gold(I) complexes of Fc′(PPh2)(CN) [153], complexes Fc′(Ph2P·AuCl)NC (i.e., 32a·AuCl); Fc’(Ph2P·AuCl)(NC·AuCl) (i.e., 32a·(AuCl)2)); and η1, η1-interbridged complexes [Fc′(Ph2P·Au)NC]2[SbF6]2 (i.e., [32a·μ-Au]2[SbF6]2) and [Fc′(Ph2P·Au)NC]2[NTF2]2 (i.e., [32a·μ-Au]2[NTF2]2) were used as catalysts for cycloisomerization reaction of enynol by Štěpnička et al. [113]. Owing to very strong Au-CN bonds in the dimeric complexes, formation of the catalytically active mono-gold species was suppressed, and consequently, no substantial yields of 2,3-dimethylfuran were observed for [32a·μ-Au]2[SbF6]2 or [32a·μ-Au]2[NTF2]2 (Scheme 19). Although mono-gold species 32a·AuCl was found to be ineffective for catalysis, di-gold species 32a·(AuCl)2 demonstrated the highest catalytic yield of 75–89% after 3 h (Scheme 19).
Scheme 19.

Au-catalyzed cycloisomerization of enynol [113].
Au(I)-complexes derived from ligand 41 (i.e., 69 and 70, following reactions depicted in Scheme 20A) have further been used for in situ AgNTf2-activated cyclization of N-propargylbenzamide to produce 4,5-dihydro-5-methylene-2-phenyloxazole (Scheme 20B) [126]. Although the yield obtained with phosphine complex 69 was very high (ca. 97% NMR yield), the initial acceleration of the reaction, followed by catalyst decomposition, was observed for the analogous reaction with complex 70. Moreover, complexes 69 and 70 have further been used for Au-catalyzed oxidative [2 + 2 + 1] cyclization of ethynylbenzene with acetonitrile (Scheme 20C), with 69 producing a higher yield (37%) than 70 (27%). As the previous records demonstrated, the outcome of such catalytic reactions is dependent on the N-oxide [154]. Štěpnička and co-workers have further reported the catalytic yields using several different N-oxides with complex 69, where the substituted (with 4-Me, 4-OMe, and 4-NO2) pyridine N-oxides produced substantially lower yields (3–23%) than the unsubstituted one (37%). When sterically demanding N-oxides (such as 2,4-Me2-pyridine N-oxide, 1,5-Me2-pyridine N-oxide, 8-methylquinoline N-oxide, and 2-methylquinoline N-oxide) were compared in this reaction, the highest yield was obtained with 8-methylquinoline N-oxide (73%), whereas the other sterically encumbered species produced yields in the range of 11–28% for the reaction demonstrated in Scheme 20C [126].
Scheme 20.

Syntheses (A) and AgNTf2-activated cyclization of N-propargylbenzamide (B,C) by complexes 69 and 70 [126].
Owing to their inherent carbophilic nature, Au(I) complexes have frequently been used as catalysts for various functionalization reactions with C=C and C≡C bonds [155]. In order to test their carbophilic nature for annellation reactions of 4-fluoro-N-propargylbenzamide, four Au(I) complexes of phosphanylstibanyl ligands (i.e., 69 and 71–73) were synthesized as outlined in Scheme 21A,B [127]. While complexes 69, 71, and 73 were almost ineffective for catalysis, an onset of product formation could be observed right after the addition of 72 (Scheme 21C). This observation directly supports the effectiveness and increased carbophilicity of the AuCl center in 72, where the Sb(V) center potentially engaged with the Au-Cl bond. On the other hand, as the ferrocenylene system freely rotated around the η5-Cpcenter—Fe—η5-Cpcenter axis, species 73 attained a pre-organized orientation prior catalysis, which minimized the scope for the formation of AuCl→Sb(V) linkage. As a result, despite containing an Sb(V) center, species 73 showed only a modicum of catalytic activity (Scheme 21C). In order to explain the above catalytic results, Gabbaï and coworkers computed the structure of the presumed adduct 74, formed between precatalyst 72 and the alkenyl substrate. The computationally optimized structure of adduct 74 displayed coordination of the alkyne to the Au center, with a simultaneous formation of an AuCl→Sb(V) linkage (shown by the blue dashed bond in the inset of Scheme 21), with the Au center being more exposed and consequently more active towards electrophilic addition.
Scheme 21.

Syntheses (A,B) and catalytic reactions with precatalysts 69, 71–73 (C), and the computationally optimized structure of adduct 74 for cycloisomerization of 4-fluro-N-propargylbenzamide with precatalyst 72 [127].
7. Perspectives
In contrast to readily and commercially available dppf and its bulky bisphosphanyl ferrocene analogs (e.g., 1,1′-bis(di-tert-butylphosphanyl)ferrocene), their non-phosphanyl counterparts are relatively unexplored. Nevertheless, the so-far reported investigations indicate their application potential for redox-responsive molecular switches, ligands, and ion-recognition receptors. Although a new variety of redox-switchable catalysts for ring-opening polymerization of cyclic lactides and lactones could be developed from diamino-substituted ferrocenylenes, the catalytic activities for their distibanyl and mixed phosphanylstibanyl counterparts fall short compared to dppf for cross-coupling reactions. One of the key features for diamino-substituted ferrocenylenes is their ability to host (hetero)carbenes, which allow for the exploration of ferrocene-bridged N-heterocyclic systems with low-coordinate group 14 elements in the ansa-bridge. Intermetallic interactions (Fe→M) have further been explored within the framework of the ferrocenylene scaffolds, featuring extreme Fe→M distances with mixed N and P donor sites, based on experimental and computational evidence. The readily accessible mesoionic and Fischer-type carbenes further highlight the relevance of the mixed P,N-substituted ferrocenylene scaffold. Overall, pnictogen-substituted non-phosphanyl ferrocenes have found their major chemical impact in non-catalytic sectors, but their catalytic potential is just starting to emerge, and consequently, provides the scope for future investigation and development.
Acknowledgments
S.D. thanks Vellore Institute of Technology, Chennai Campus, India, for providing “VIT RGEMS SEED GRANT” for carrying out this research work.
Supplementary Materials
The following supporting information (SI) can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29225283/s1, Table S1: Survey of SCXRD characterized complexes and compounds from 1,1’-bispnictogen substituted dppf-analogs, Table S2: Molecular parameters of cationic Ni(II), Pd(II), Sc(III), Lu(III) complexes of dppf and its 1,1’-diamino-, selected 1,1’-bisphosphanyl- and 1,1’-aminophosphanylferrocenes with Fe→Pd bonding interactions, Table S3: Survey of oxidation potentials (EO) for 1,1’-bispnictogen substituted dppf analogs and their complexes with potentials converted to the (C5Me5)2Fe/(C5Me5)2Fe+ scale. Other than the so-far cited references, a few additional reports have further been cited in the SI file [156,157,158,159,160,161,162,163,164,165,166].
Conflicts of Interest
The authors declare no conflicts of interest.
Funding Statement
This research was carried out with the support of “VIT RGEMS SEED GRANT” (Sanction Order No. PH1/2024/SG2024001) Vellore Institute of Technology, Chennai Campus, India, and the German Science Fund (DFG).
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Sollott G.P., Mertwoy H.E., Portnoy S., Snead J.L. Unsymmetrical Tertiary Phosphines of Ferrocene by Friedel-Crafts Reactions. I. Ferrocenylphenylphosphines. J. Org. Chem. 1963;28:1090–1092. doi: 10.1021/jo01039a055. [DOI] [Google Scholar]
- 2.Bishop J.J., Davison A., Katcher M.L., Lichtenberg D.W., Merrill R.E., Smart J.C. Symmetrically disubstituted ferrocenes. J. Organomet. Chem. 1971;27:241–249. doi: 10.1016/S0022-328X(00)80571-9. [DOI] [Google Scholar]
- 3.Butler I.R., Coles S.J., Fontani M., Hursthouse M.B., Lewis E., Malik K.L.M.A., Meunier M., Zanello P. Diferrocenyltriphosphines 2. Reversible phosphine deligation in the chemistry of diferrocenyltriphosphine Ru(II) dichloride complexes with nitriles and pyridines: Towards a pH-switchable catalyst? J. Organomet. Chem. 2001;637–639:538–548. doi: 10.1016/S0022-328X(01)01135-4. [DOI] [Google Scholar]
- 4.Mizuta T., Aotani T., Imamura Y., Kubo K., Miyoshi K. Structure and properties of the macrocyclic tridentate ferrocenylphosphine ligand (-PhPC5H4FeC5H4-)3. Organometallics. 2008;27:2457–2463. doi: 10.1021/om800057w. [DOI] [Google Scholar]
- 5.Mizuta T., Inami Y., Kubo K., Miyoshi K. Synthesis of binuclear complexes bound in an enlarged tetraphosphamacrocycle: Two diphosphine metal units linked in front-to-front style. Inorg. Chem. 2009;48:7534–7536. doi: 10.1021/ic9013224. [DOI] [PubMed] [Google Scholar]
- 6.Favre H.A., Powell W.H. Nomenclature of Organic Chemistry: IUPAC Recommendations and Preferred Names. The Royal Society of Chemistry; London, UK: 2013. [Google Scholar]
- 7.Dey S., Pietschnig R. Chemistry of sterically demanding dppf-analogs. Coord. Chem. Rev. 2021;437:213850. doi: 10.1016/j.ccr.2021.213850. [DOI] [Google Scholar]
- 8.Siemeling U. Other Symmetric 1,1′-Bidentate Ferrocene Ligands. In: Štěpnička P., editor. Ferrocenes: Ligands, Materials and Biomolecules. John Wiley & Sons; Hoboken, NJ, USA: 2008. pp. 141–176. [Google Scholar]
- 9.Stepnicka P. Forever young: The first seventy years of ferrocene. Dalton Trans. 2022;51:8085–8102. doi: 10.1039/D2DT00903J. [DOI] [PubMed] [Google Scholar]
- 10.Štěpnička P. Ferrocene Chemistry. Eur. J. Inorg. Chem. 2022;2022:e202200388. doi: 10.1002/ejic.202200388. [DOI] [Google Scholar]
- 11.Gan K.-S., Hor T.S.A. 1,1′-Bis(diphenylphosphino)ferrocene—Coordination Chemistry, Organic Syntheses, and Catalysis. In: Togni A., Hayashi T., editors. Ferrocenes: Homogeneous Catalysis, Organic Synthesis, Materials Science. John Wiley & Sons; Hoboken, NJ, USA: 2007. pp. 3–104. [Google Scholar]
- 12.Dey S., Roesler F., Höfler M.V., Bruhn C., Gutmann T., Pietschnig R. Synthesis, Structure and Cu-Phenylacetylide Coordination of an Unsymmetrically Substituted Bulky dppf-Analog. Eur. J. Inorg. Chem. 2021;2022:e202100939. doi: 10.1002/ejic.202100939. [DOI] [Google Scholar]
- 13.Tolman C.A. Steric effects of phosphorus ligands in organometallic chemistry and homogeneous catalysis. Chem. Rev. 2002;77:313–348. doi: 10.1021/cr60307a002. [DOI] [Google Scholar]
- 14.Dierkes P., van Leeuwen P.W.N.M. The bite angle makes the difference: A practical ligand parameter for diphosphine ligands. J. Chem. Soc. Dalton Trans. 1999:1519–1529. doi: 10.1039/a807799a. [DOI] [Google Scholar]
- 15.Herbert D.E., Mayer U.F., Manners I. Strained metallocenophanes and related organometallic rings containing π-hydrocarbon ligands and transition-metal centers. Angew. Chem. Int. Ed. 2007;46:5060–5081. doi: 10.1002/anie.200604409. [DOI] [PubMed] [Google Scholar]
- 16.Green J.C. Bent metallocenes revisited. Chem. Soc. Rev. 1998;27:263–272. doi: 10.1039/a827263z. [DOI] [Google Scholar]
- 17.Dey S., Szathmari B., Franz R., Bruhn C., Kelemen Z., Pietschnig R. Controlled Ring Opening of a Tetracyclic Tetraphosphane with Twofold Metallocene Bridging. Chem. Eur. J. 2024;30:e202400194. doi: 10.1002/chem.202400194. [DOI] [PubMed] [Google Scholar]
- 18.Dey S., Kargin D., Höfler M.V., Szathmári B., Bruhn C., Gutmann T., Kelemen Z., Pietschnig R. Oligo- and polymerization of phospha [2]ferrocenophanes to one dimensional phosphorus chains with ferrocenylene handles. Polymer. 2022;242:124589. doi: 10.1016/j.polymer.2022.124589. [DOI] [Google Scholar]
- 19.Butler I.R., Cullen W.R., Einstein F.W.B., Rettig S.J., Willis A.J. Synthesis of some ring-substituted [1]ferrocenophanes and the structure of four representative examples. Organometallics. 1983;2:128–135. doi: 10.1021/om00073a024. [DOI] [Google Scholar]
- 20.Abramovitch R.A., Azogu C.I., Sutherland R.G. A novel bridged ferrocene derivative. J. Chem. Soc. D Chem. Commun. 1969:1439–1440. doi: 10.1039/c29690001439. [DOI] [Google Scholar]
- 21.Abramovitch R.A., Azogu C.I., Sutherland R.G. The thermolysis and photolysis of ferrocenylsulphonyl azide. Evidence for a metal-nitrene complex in the thermolysis. Tetrahedron Lett. 1971;12:1637–1640. doi: 10.1016/S0040-4039(01)87421-9. [DOI] [Google Scholar]
- 22.Abramovitch R.A., Atwood J.L., Good M.L., Lampert B.A. Crystal structure and Moessbauer spectrum of [2]-ferrocenophanethiazine 1,1-dioxide. Inorg. Chem. 1975;14:3085–3089. doi: 10.1021/ic50154a045. [DOI] [Google Scholar]
- 23.Dey S., Quail J.W., Müller J. [2]Ferrocenophanes with Nitrogen in Bridging Positions. Organometallics. 2015;34:3039–3046. doi: 10.1021/acs.organomet.5b00340. [DOI] [Google Scholar]
- 24.Dey S., Sun W., Muller J. [n]Ferrocenophanes (n = 2, 3) with Nitrogen and Phosphorus in Bridging Positions. Inorg. Chem. 2016;55:3630–3639. doi: 10.1021/acs.inorgchem.6b00170. [DOI] [PubMed] [Google Scholar]
- 25.Bhattacharjee H., Dey S., Zhu J., Sun W., Muller J. Strained azabora[2]ferrocenophanes. Chem. Commun. 2018;54:5562–5565. doi: 10.1039/C8CC02965B. [DOI] [PubMed] [Google Scholar]
- 26.Gurskaya L., Bagryanskaya I., Amosov E., Kazantsev M., Politanskaya L., Zaytseva E., Bagryanskaya E., Chernonosov A., Tretyakov E. 1,3-Diaza[3]ferrocenophanes functionalized with a nitronyl nitroxide group. Tetrahedron. 2018;74:1942–1950. doi: 10.1016/j.tet.2018.02.062. [DOI] [Google Scholar]
- 27.Mizuta T., Yamasaki T., Miyoshi K. Syntheses and X-Ray Crystal Structures of Platinum(II) Complexes Bearing Two and Three Phosphorus-Bridged [1]Ferrocenophanes, cis-[PtCl2(fcpp)2] and [PtCl(fcpp)3]BF4. Chem. Lett. 2000;29:924–925. doi: 10.1246/cl.2000.924. [DOI] [Google Scholar]
- 28.Zayakin I.A., Syroeshkin M.A., Shangin P.G., Korlyukov A.A., Dmitriev A.A., Gritsan N.P., Tretyakov E.V. Binuclear complex of the organogold derivative of nitronyl nitroxide with 1,1′-bis(diphenylphosphino)ferrocene: Synthesis, structure, and properties. Russ. Chem. Bull. 2024;73:1216–1228. doi: 10.1007/s11172-024-4238-2. [DOI] [Google Scholar]
- 29.Bagryanskaya I., Fedin M., Gorbunov D., Gritsan N., Gurskaya L., Kazantsev M., Polienko Y., Stass D., Tretyakov E. A nitroxide diradical containing a ferrocen-1,1′-diyl-substituted 1,3-diazetidine-2,4-diimine coupler. Tetrahedron Lett. 2017;58:478–481. doi: 10.1016/j.tetlet.2016.12.068. [DOI] [Google Scholar]
- 30.Gurskaya L.Y., Polienko Y.F., Rybalova T.V., Gritsan N.P., Dmitriev A.A., Kazantsev M.S., Zaytseva E.V., Parkhomenko D.A., Beregovaya I.V., Zakabluk G.A., et al. Multispin Systems with a Rigid Ferrocene-1,1′-diyl-Substituted 1,3-Diazetidine-2,4-diimine Coupler: A General Approach. Eur. J. Org. Chem. 2022;2022:e202101234. doi: 10.1002/ejoc.202101234. [DOI] [Google Scholar]
- 31.Talisic L., Snelgrove C., Wijesundara M., Pilkington M., Metallinos C. Stereoselective Synthesis of 1,1′,2,2′-Tetrasubstituted Ferrocenes by Double Lithiation of a 1,1′-Disubstituted Ferrocenyl Pyrroloimidazolone. Eur. J. Inorg. Chem. 2023;26:e202200694. doi: 10.1002/ejic.202200694. [DOI] [Google Scholar]
- 32.Dascalu M., Balan M., Shova S., Racles C., Cazacu M. Design and synthesis of the first ferrocenylsiloxane urea: Structure and properties. Polyhedron. 2015;102:583–592. doi: 10.1016/j.poly.2015.11.013. [DOI] [Google Scholar]
- 33.Károlyi B.I., Bősze S., Orbán E., Sohár P., Drahos L., Gál E., Csámpai A. Acylated mono-, bis- and tris- Cinchona-Based Amines Containing Ferrocene or Organic Residues: Synthesis, Structure and In Vitro Antitumor Activity on Selected Human Cancer Cell Lines. Molecules. 2012;17:2316–2329. doi: 10.3390/molecules17032316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sato M., Ebine S., Tiedemann S. Condensation of Halobenzenes and Haloferrocenes with Phthalimide in the Presence of Copper(I) Oxide; A Simplified Gabriel Reaction. Synthesis. 1981;1981:472–473. doi: 10.1055/s-1981-29492. [DOI] [Google Scholar]
- 35.Abdulmalic M.A., Rüffer T. A New Facile Two-Step Synthetic Procedure of 1,1′-Diaminoferrocene. Bull. Chem. Soc. Jpn. 2013;86:724–728. doi: 10.1246/bcsj.20130031. [DOI] [Google Scholar]
- 36.Colacot T.J., Parisel S. Synthesis, Coordination Chemistry and Catalytic Use of dppf Analogs. In: Štěpnička P., editor. Ferrocenes: Ligands, Materials and Biomolecules. John Wiley & Sons; Hoboken, NJ, USA: 2008. pp. 117–140. [Google Scholar]
- 37.Štěpnička P. 1′-Functionalised Ferrocene Phosphines: Synthesis, Coordination Chemistry and Catalytic Applications. In: Štěpnička P., editor. Ferrocenes: Ligands, Materials and Biomolecules. John Wiley & Sons; Hoboken, NJ, USA: 2008. pp. 177–204. [Google Scholar]
- 38.Štěpnička P., Lamač M. Synthesis and Catalytic Use of Planar Chiral and Polydentate Ferrocene Donors. In: Štěpnička P., editor. Ferrocenes: Ligands, Materials and Biomolecules. John Wiley & Sons; Hoboken, NJ, USA: 2008. pp. 237–277. [Google Scholar]
- 39.Blaser H.-U., Chen W., Camponovo F., Togni A. Chiral 1,2-Disubstituted Ferrocene Diphosphines for Asymmetric Catalysis. In: Štěpnička P., editor. Ferrocenes: Ligands, Materials and Biomolecules. John Wiley & Sons; Hoboken, NJ, USA: 2008. pp. 205–235. [Google Scholar]
- 40.Fihri A., Meunier P., Hierso J.-C. Performances of symmetrical achiral ferrocenylphosphine ligands in palladium-catalyzed cross-coupling reactions: A review of syntheses, catalytic applications and structural properties. Coord. Chem. Rev. 2007;251:2017–2055. doi: 10.1016/j.ccr.2007.03.020. [DOI] [Google Scholar]
- 41.Dey S., Buzsaki D., Bruhn C., Kelemen Z., Pietschnig R. Bulky 1,1′-bisphosphanoferrocenes and their coordination behaviour towards Cu(i) Dalton Trans. 2020;49:6668–6681. doi: 10.1039/D0DT00941E. [DOI] [PubMed] [Google Scholar]
- 42.Dey S., Dettling L., Gál D., Bruhn C., Kelemen Z., Müller C., Pietschnig R. Tuning the Sterics: Rh-Catalyzed Hydroformylation Reactions with Ferrocene Based Diphosphorus Ligands. Soc. Sci. Res. Netw. 2024:preprint. doi: 10.2139/ssrn.4972596. [DOI] [Google Scholar]
- 43.Ito M., Iseki M., Itazaki M., Nakazawa H. Tetrahedral cage complex with planar vertices: Selective synthesis of Pt4L6 cage complexes involving hydrogen bonds driven by halide binding. Chem. Commun. 2016;52:7205–7208. doi: 10.1039/C6CC01448H. [DOI] [PubMed] [Google Scholar]
- 44.Dascalu M., Musteata V.E., Vacareanu L., Racles C., Cazacu M. Synthesis and characterization of metal-containing poly(siloxane-urethane) crosslinked structures derived from siloxane diols and ferrocene diisocyanate. RSC Adv. 2015;5:99193–99200. doi: 10.1039/C5RA15290A. [DOI] [Google Scholar]
- 45.Petrov A.R., Jess K., Freytag M., Jones P.G., Tamm M. Large-Scale Preparation of 1,1′-Ferrocenedicarboxylic Acid, a Key Compound for the Synthesis of 1,1′-Disubstituted Ferrocene Derivatives. Organometallics. 2013;32:5946–5954. doi: 10.1021/om4004972. [DOI] [Google Scholar]
- 46.Bildstein B., Malaun M., Kopacka H., Wurst K., Mitterböck M., Ongania K.-H., Opromolla G., Zanello P. N,N′-Diferrocenyl-N-heterocyclic Carbenes and Their Derivatives. Organometallics. 1999;18:4325–4336. doi: 10.1021/om990377h. [DOI] [Google Scholar]
- 47.Otón F., Espinosa A., Tárraga A., Molina P. Synthesis of Multifunctional Aza-Substituted Ruthenocene Derivatives Displaying Charge-Transfer Transitions and Selective Zn(II) Ions Sensing Properties. Organometallics. 2007;26:6234–6242. doi: 10.1021/om700757m. [DOI] [Google Scholar]
- 48.You S.-L., Hou X.-L., Dai L.-X., Zhu X.-Z. Highly Efficient Ligands for Palladium-Catalyzed Asymmetric Alkylation of Ketone Enolates. Org. Lett. 2001;3:149–151. doi: 10.1021/ol0067033. [DOI] [PubMed] [Google Scholar]
- 49.Bernheim M., Boche G. Electrophilic Amination of Cyclopentadienyllithium Compounds. Angew. Chem. Int. Ed. 1980;19:1010–1011. doi: 10.1002/anie.198010101. [DOI] [Google Scholar]
- 50.Stahl K.-P., Boche G., Massa W. 1,1′-Bis(N,N-dimethylamino)ferrocene,1,1′-bis(N,N-dimethylamino) cobaltocenium hexafluorophosphate and 1,1′-bis(N,N-dimethylamino)titanocene dichloride. Crystal structure of 1,1′-bis(N,N-dimethylamino)titanocene dichloride. J. Organomet. Chem. 1984;277:113–125. doi: 10.1016/0022-328X(84)80686-5. [DOI] [Google Scholar]
- 51.Britton W.E., Kashyap R., El-Hashash M., El-Kady M., Herberhold M. The anomalous electrochemistry of the ferrocenylamines. Organometallics. 1986;5:1029–1031. doi: 10.1021/om00136a033. [DOI] [Google Scholar]
- 52.Cerveau G., Chuit C., Colomer E., Corriu R.J.P., Reye C. Ferrocenyl compounds containing two hypervalent silicon species. Electrochemical studies. Organometallics. 1990;9:2415–2417. doi: 10.1021/om00159a002. [DOI] [Google Scholar]
- 53.Lu S., Strelets V.V., Ryan M.F., Pietro W.J., Lever A.B.P. Electrochemical Parametrization in Sandwich Complexes of the First Row Transition Metals. Inorg. Chem. 1996;35:1013–1023. doi: 10.1021/ic950620e. [DOI] [PubMed] [Google Scholar]
- 54.Herberhold M., Ellinger M., Kremnitz W. Ferrocenylamine. J. Organomet. Chem. 1983;241:227–240. doi: 10.1016/S0022-328X(00)98509-7. [DOI] [Google Scholar]
- 55.Lee J.A., Williams B.N., Ogilby K.R., Miller K.L., Diaconescu P.L. Synthesis of symmetrically and unsymmetrically 3,5-dimethylbenzyl-substituted 1,1′-ferrocene diamines. J. Organomet. Chem. 2011;696:4090–4094. doi: 10.1016/j.jorganchem.2011.06.043. [DOI] [Google Scholar]
- 56.Blank N.F., Glueck D.S., Zakharov L.N., Rheingold A.L., Saybolt M.D., Ghent B.L., Nataro C. Synthesis, Structure, and Electrochemistry of an Electron-Rich Chiral Diaminoferrocene, (S,S)-Bis(2,5-dimethylpyrrolidinyl)ferrocene. Organometallics. 2005;24:5184–5187. doi: 10.1021/om050367i. [DOI] [Google Scholar]
- 57.Plenio H., Burth D. Aminocyclopentadienes, Aminoferrocenes, and Aminocobaltocenes. Angew. Chem. Int. Ed. 1995;34:800–803. doi: 10.1002/anie.199508001. [DOI] [Google Scholar]
- 58.Plenio H., Burth D. Aminoferrocenes and Aminocobaltocenes as Redox-Active Chelating Ligands: Syntheses, Structures, and Coordination Chemistry1. Organometallics. 1996;15:4054–4062. doi: 10.1021/om9602771. [DOI] [Google Scholar]
- 59.Plenio H., Aberle C. An Artificial Regulatory System with Coupled Molecular Switches. Angew. Chem. Int. Ed. 1998;37:1397–1399. doi: 10.1002/(SICI)1521-3773(19980605)37:10<1397::AID-ANIE1397>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 60.Plenio H., Aberle C. Coupled Molecular Switches: A Redox-Responsive Ligand and the Redox-Switched Complexation of Metal Ions. Chem. Eur. J. 2001;7:4438–4446. doi: 10.1002/1521-3765(20011015)7:20<4438::AID-CHEM4438>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 61.Wang X., Thevenon A., Brosmer J.L., Yu I., Khan S.I., Mehrkhodavandi P., Diaconescu P.L. Redox Control of Group 4 Metal Ring-Opening Polymerization Activity toward l-Lactide and ε-Caprolactone. J. Am. Chem. Soc. 2014;136:11264–11267. doi: 10.1021/ja505883u. [DOI] [PubMed] [Google Scholar]
- 62.Siemeling U., Färber C., Bruhn C. A stable crystalline N-heterocyclic carbene with a 1,1′-ferrocenediyl backbone. Chem. Commun. 2009:98–100. doi: 10.1039/B813809E. [DOI] [PubMed] [Google Scholar]
- 63.Volk J., Bicho B.A.C., Bruhn C., Siemeling U. N-Heterocyclic germylenes and stannylenes of the type [Fe{(η5-C5H4)NR}2E] with bulky alkyl substituents. Z. Naturforsch. B. 2017;72:785–794. doi: 10.1515/znb-2017-0085. [DOI] [Google Scholar]
- 64.Duhović S., Diaconescu P.L. An experimental and computational study of 1,1′-ferrocene diamines. Polyhedron. 2013;52:377–388. doi: 10.1016/j.poly.2012.08.063. [DOI] [Google Scholar]
- 65.Shafir A., Arnold J. Zirconium complexes incorporating diaryldiamidoferrocene ligands: Generation of cationic derivatives and polymerization activity towards ethylene and 1-hexene. Inorg. Chim. Acta. 2003;345:216–220. doi: 10.1016/S0020-1693(02)01300-2. [DOI] [Google Scholar]
- 66.Siemeling U., Färber C., Leibold M., Bruhn C., Mücke P., Winter R.F., Sarkar B., von Hopffgarten M., Frenking G. Six-Membered N-Heterocyclic Carbenes with a 1,1′-Ferrocenediyl Backbone: Bulky Ligands with Strong Electron-Donor Capacity and Unusual Non-Innocent Character. Eur. J. Inorg. Chem. 2009;2009:4607–4612. doi: 10.1002/ejic.200900863. [DOI] [Google Scholar]
- 67.Oetzel J., Weyer N., Bruhn C., Leibold M., Gerke B., Pöttgen R., Maier M., Winter R.F., Holthausen M.C., Siemeling U. Redox-Active N-Heterocyclic Germylenes and Stannylenes with a Ferrocene-1,1′-diyl Backbone. Chem. Eur. J. 2017;23:1187–1199. doi: 10.1002/chem.201605074. [DOI] [PubMed] [Google Scholar]
- 68.Shafir A., Power M.P., Whitener G.D., Arnold J. Silylated 1,1′-Diaminoferrocene: Ti and Zr Complexes of a New Chelating Diamide Ligand. Organometallics. 2001;20:1365–1369. doi: 10.1021/om0008979. [DOI] [Google Scholar]
- 69.Weyer N., Guthardt R., Correia Bicho B.A., Oetzel J., Bruhn C., Siemeling U. Stable N-Heterocyclic Germylenes of the Type [Fe{(η5-C5H4)NR}2Ge] and Their Oxidation Reactions with Sulfur, Selenium, and Diphenyl Diselenide. Z. Anorg. Allg. Chem. 2019;645:188–197. doi: 10.1002/zaac.201800449. [DOI] [Google Scholar]
- 70.Carver C.T., Diaconescu P.L. Ring-Opening Reactions of Aromatic N-Heterocycles by Scandium and Yttrium Alkyl Complexes. J. Am. Chem. Soc. 2008;130:7558–7559. doi: 10.1021/ja802728k. [DOI] [PubMed] [Google Scholar]
- 71.Carver C.T., Monreal M.J., Diaconescu P.L. Scandium Alkyl Complexes Supported by a Ferrocene Diamide Ligand. Organometallics. 2008;27:363–370. doi: 10.1021/om7007277. [DOI] [Google Scholar]
- 72.Carver C.T., Benitez D., Miller K.L., Williams B.N., Tkatchouk E., Goddard W.A., III, Diaconescu P.L. Reactions of Group III Biheterocyclic Complexes. J. Am. Chem. Soc. 2009;131:10269–10278. doi: 10.1021/ja902794w. [DOI] [PubMed] [Google Scholar]
- 73.Miller K.L., Williams B.N., Benitez D., Carver C.T., Ogilby K.R., Tkatchouk E., Goddard W.A., III, Diaconescu P.L. Dearomatization Reactions of N-Heterocycles Mediated by Group 3 Complexes. J. Am. Chem. Soc. 2010;132:342–355. doi: 10.1021/ja908489p. [DOI] [PubMed] [Google Scholar]
- 74.Wong A.W., Miller K.L., Diaconescu P.L. Reactions of aromatic N-heterocycles with a lutetium benzyl complex supported by a ferrocene-diamide ligand. Dalton Trans. 2010;39:6726–6731. doi: 10.1039/c001927e. [DOI] [PubMed] [Google Scholar]
- 75.Siemeling U., Auch T.C., Kuhnert O., Malaun M., Kopacka H., Bildstein B. 1, 1′-Di(arylamino)ferrocenes. A New Family of Privileged [N, N] Ligands with Tunable Steric Control for Applications in Homogeneous Organometallic Catalysis and Coordination Chemistry. Z. Anorg. Allg. Chem. 2003;629:1334–1336. doi: 10.1002/zaac.200300080. [DOI] [Google Scholar]
- 76.Siemeling U., Auch T.-C., Tomm S., Fink H., Bruhn C., Neumann B., Stammler H.-G. Zirconium Chelates of a Bulky Ferrocene-Based Diamido Ligand. Organometallics. 2007;26:1112–1115. doi: 10.1021/om061063e. [DOI] [Google Scholar]
- 77.Correia Bicho B.A., Bruhn C., Guthardt R., Weyer N., Siemeling U. [Fe{(η5-C5H4)N(SiMe2tBu)}2Al]2: A Stable Redox-Functionalized Dialumane(4) Z. Anorg. Allg. Chem. 2018;644:1329–1336. doi: 10.1002/zaac.201800250. [DOI] [Google Scholar]
- 78.Weyer N., Heinz M., Schweizer J.I., Bruhn C., Holthausen M.C., Siemeling U. A Stable N-Heterocyclic Silylene with a 1,1′-Ferrocenediyl Backbone. Angew. Chem. Int. Ed. 2021;60:2624–2628. doi: 10.1002/anie.202011691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Walz F., Moos E., Garnier D., Köppe R., Anson C.E., Breher F. A Redox-Switchable Germylene and its Ligating Properties in Selected Transition Metal Complexes. Chem. Eur. J. 2017;23:1173–1186. doi: 10.1002/chem.201605073. [DOI] [PubMed] [Google Scholar]
- 80.Kong D., Weng T., He W., Liu B., Jin S., Hao X., Liu S. Synthesis, characterization, and electrochemical properties of ferrocenylimidazolium. J. Organomet. Chem. 2013;727:19–27. doi: 10.1016/j.jorganchem.2012.12.028. [DOI] [Google Scholar]
- 81.Varnado C.D., Jr., Lynch V.M., Bielawski C.W. 1,1′-Bis(N-benzimidazolylidene)ferrocene: Synthesis and study of a novel ditopic ligand and its transition metal complexes. Dalton Trans. 2009:7253–7261. doi: 10.1039/b908696j. [DOI] [PubMed] [Google Scholar]
- 82.Otón F., González M.D.C., Espinosa A., Ramírez de Arellano C., Tárraga A., Molina P. Ion Pair Recognition Receptor Based on an Unsymmetrically 1,1′-Disubstituted Ferrocene–Triazole Derivative. J. Org. Chem. 2012;77:10083–10092. doi: 10.1021/jo301570u. [DOI] [PubMed] [Google Scholar]
- 83.Gibson V.C., Long N.J., Marshall E.L., Oxford P.J., White A.J.P., Williams D.J. The synthesis and metal coordination chemistry of new 1,1′-N-substituted ferrocenediyl ligands derived from 1,1′-diaminoferrocene. J. Chem. Soc. Dalton Trans. 2001:1162–1164. doi: 10.1039/b101795k. [DOI] [Google Scholar]
- 84.Abubekerov M., Khan S.I., Diaconescu P.L. Ferrocene-bis(phosphinimine) Nickel(II) and Palladium(II) Alkyl Complexes: Influence of the Fe–M (M = Ni and Pd) Interaction on Redox Activity and Olefin Coordination. Organometallics. 2017;36:4394–4402. doi: 10.1021/acs.organomet.7b00626. [DOI] [Google Scholar]
- 85.Metallinos C., Tremblay D., Barrett F.B., Taylor N.J. 1,1′-Bis(phosphoranylidenamino)ferrocene palladium(II) complexes: An unusual case of dative Fe→Pd bonding. J. Organomet. Chem. 2006;691:2044–2047. doi: 10.1016/j.jorganchem.2006.01.012. [DOI] [Google Scholar]
- 86.Klapp L.R.R., Bruhn C., Leibold M., Siemeling U. Ferrocene-Based Bis(guanidines): Superbases for Tridentate N, Fe, N-Coordination. Organometallics. 2013;32:5862–5872. doi: 10.1021/om400454v. [DOI] [Google Scholar]
- 87.Jess K., Baabe D., Bannenberg T., Brandhorst K., Freytag M., Jones P.G., Tamm M. Ni–Fe and Pd–Fe Interactions in Nickel(II) and Palladium(II) Complexes of a Ferrocene-Bridged Bis(imidazolin-2-imine) Ligand. Inorg. Chem. 2015;54:12032–12045. doi: 10.1021/acs.inorgchem.5b02457. [DOI] [PubMed] [Google Scholar]
- 88.Weng Z., Koh L.L., Hor T.S.A. Suzuki cross-coupling in aqueous media catalyzed by a 1,1′-N-substituted ferrocenediyl Pd(II) complex. J. Organomet. Chem. 2004;689:18–24. doi: 10.1016/j.jorganchem.2003.09.028. [DOI] [Google Scholar]
- 89.Gibson V.C., Gregson C.K.A., Halliwell C.M., Long N.J., Oxford P.J., White A.J.P., Williams D.J. The synthesis, coordination chemistry and ethylene polymerisation activity of ferrocenediyl nitrogen-substituted ligands and their metal complexes. J. Organomet. Chem. 2005;690:6271–6283. doi: 10.1016/j.jorganchem.2005.05.051. [DOI] [Google Scholar]
- 90.Shafir A., Fiedler D., Arnold J. Highly diastereoselective reduction of ferrocene bis-imines with methyllithium and the formation of C2-symmetric Zr complexes. Chem. Commun. 2003:2598–2599. doi: 10.1039/b308360h. [DOI] [PubMed] [Google Scholar]
- 91.Shafir A., Fiedler D., Arnold J. Formation of 1:1 complexes of ferrocene-containing salen ligands with Mg, Ti and Zr. J. Chem. Soc. Dalton Trans. 2002:555–560. doi: 10.1039/b107066p. [DOI] [Google Scholar]
- 92.Otón F., Ratera I., Espinosa A., Wurtz K., Parella T., Tárraga A., Veciana J., Molina P. Selective Metal-Cation Recognition by [2.2]Ferrocenophanes: The Cases of Zinc- and Lithium-Sensing. Chem. Eur. J. 2010;16:1532–1542. doi: 10.1002/chem.200901421. [DOI] [PubMed] [Google Scholar]
- 93.Brown L.A., Rhinehart J.L., Long B.K. Effects of Ferrocenyl Proximity and Monomer Presence during Oxidation for the Redox-Switchable Polymerization of l-Lactide. ACS Catal. 2015;5:6057–6060. doi: 10.1021/acscatal.5b01434. [DOI] [Google Scholar]
- 94.Lai A., Hern Z.C., Diaconescu P.L. Switchable Ring-Opening Polymerization by a Ferrocene Supported Aluminum Complex. ChemCatChem. 2019;11:4210–4218. doi: 10.1002/cctc.201900747. [DOI] [Google Scholar]
- 95.Broderick E.M., Diaconescu P.L. Cerium(IV) Catalysts for the Ring-Opening Polymerization of Lactide. Inorg. Chem. 2009;48:4701–4706. doi: 10.1021/ic802047u. [DOI] [PubMed] [Google Scholar]
- 96.Quan S.M., Diaconescu P.L. High activity of an indium alkoxide complex toward ring opening polymerization of cyclic esters. Chem. Commun. 2015;51:9643–9646. doi: 10.1039/C5CC01312G. [DOI] [PubMed] [Google Scholar]
- 97.Shepard S.M., Diaconescu P.L. Redox-Switchable Hydroelementation of a Cobalt Complex Supported by a Ferrocene-Based Ligand. Organometallics. 2016;35:2446–2453. doi: 10.1021/acs.organomet.6b00317. [DOI] [Google Scholar]
- 98.Dai R., Lai A., Alexandrova A.N., Diaconescu P.L. Geometry Change in a Series of Zirconium Compounds during Lactide Ring-Opening Polymerization. Organometallics. 2018;37:4040–4047. doi: 10.1021/acs.organomet.8b00620. [DOI] [Google Scholar]
- 99.Broderick E.M., Thuy-Boun P.S., Guo N., Vogel C.S., Sutter J., Miller J.T., Meyer K., Diaconescu P.L. Synthesis and Characterization of Cerium and Yttrium Alkoxide Complexes Supported by Ferrocene-Based Chelating Ligands. Inorg. Chem. 2011;50:2870–2877. doi: 10.1021/ic102076g. [DOI] [PubMed] [Google Scholar]
- 100.Tárraga A., Otón F., Espinosa A., Desamparados Velasco M., Molina P., Evans D.J. Synthesis and properties of a new class of nitrogen-rich multinuclear[m.n] ferrocenophanes. Chem. Commun. 2004:458–459. doi: 10.1039/B313797J. [DOI] [PubMed] [Google Scholar]
- 101.Brosmer J.L., Diaconescu P.L. Yttrium-Alkyl Complexes Supported by a Ferrocene-Based Phosphinimine Ligand. Organometallics. 2015;34:2567–2572. doi: 10.1021/om501227t. [DOI] [Google Scholar]
- 102.Davison A., Bishop J.J. Symmetrically disubstituted ferrocenes. II. Complexes of ferrocene-1,1′-bis(dimethylarsine) and ferrocene-1,1′-bis(diphenylarsine) with Group VI carbonyls. Inorg. Chem. 1971;10:826–831. doi: 10.1021/ic50098a032. [DOI] [Google Scholar]
- 103.Davison A., Bishop J.J. Symmetrically disubstituted ferrocenes. III. Complexes of ferrocene-1,1′-bis(dimethylarsine) and ferrocene-1,1′-bis(diphenylarsine) with the Group VIII metals. Inorg. Chem. 1971;10:832–837. doi: 10.1021/ic50098a033. [DOI] [Google Scholar]
- 104.Fitzpatrick M.G., Hanton L.R., Simpson J. Conformationally flexible arsine ligands: Two forms of bis [1,1′-bis (dimethylarsino) ferrocene] dibromonickel (II) Inorg. Chim. Acta. 1996;244:131–136. doi: 10.1016/0020-1693(95)04763-8. [DOI] [Google Scholar]
- 105.Fitzpatrick M.G., Hanton L.R., Henderson W., Kneebone P.E., Levy E.G., McCaffrey L.J., McMorran D.A. Application of electrospray mass spectrometry to the characterisation of tertiary arsine ligands. Inorg. Chim. Acta. 1998;281:101–110. doi: 10.1016/S0020-1693(98)00152-2. [DOI] [Google Scholar]
- 106.Sumida A., Imoto H., Naka K. Synthetic Strategy for AB2-Type Arsines via Bidentate Dithiolate Leaving Groups. Inorg. Chem. 2022;61:17419–17426. doi: 10.1021/acs.inorgchem.2c01974. [DOI] [PubMed] [Google Scholar]
- 107.Song L.-C., Wang G.-F., Liu P.-C., Hu Q.-M. Synthetic and Structural Studies on Transition Metal Fullerene Complexes Containing Phosphorus and Arsenic Ligands: Crystal and Molecular Structures of (η2-C60)M(dppf) (dppf = 1,1′-Bis(diphenylphosphino)ferrocene; M = Pt, Pd), (η2-C60)Pt(AsPh3)2, (η2-C60)Pt(dpaf) (dpaf = 1,1′-Bis(diphenylarsino)ferrocene), and (η2-C70)Pt(dpaf) Organometallics. 2003;22:4593–4598. [Google Scholar]
- 108.Warnick E.P., Dupuis R.J., Piro N.A., Scott Kassel W., Nataro C. Compounds containing weak, non-covalent interactions to the metal in the backbone of 1,1′-bis(phosphino)metallocene ligands. Polyhedron. 2016;114:156–164. doi: 10.1016/j.poly.2015.11.025. [DOI] [Google Scholar]
- 109.Du H., Park K.C., Wang F., Wang S., Liu Q., Zhang S., Huang Y., Shi S. Synthesis and Characterization of Homoannularly Disubstituted and Heteroannularly Trisubstituted Ferrocene Derivatives by Arsenic and Silicon or by Arsenic and Tin. Organometallics. 2007;26:6219–6224. doi: 10.1021/om7004468. [DOI] [Google Scholar]
- 110.Schulz J., Antala J., Cisarova I., Stepnicka P. Beyond phosphorus: Synthesis, reactivity, coordination behaviour and catalytic properties of 1,1′-bis(diphenylstibino)ferrocene. Dalton Trans. 2023;52:1198–1211. doi: 10.1039/D2DT03770J. [DOI] [PubMed] [Google Scholar]
- 111.Murafuji T., Mutoh T., Sugihara Y. Synthesis of (formylferrocenyl)bismuthanes. A way to control the stereochemistry at the chiral bismuth centre using hypervalent bond formation and planar chirality of ferrocene. Chem. Commun. 1996:1693–1694. doi: 10.1039/cc9960001693. [DOI] [Google Scholar]
- 112.Villamizar C C.P., Anzaldo B., Sharma P., González R., Toscano A., Peña M.A., Sharma M., Gutierrez R. 1,1′-2-Trisubstituted ferrocenyl dibismuthines containing N/O donor pendant arm. J. Organomet. Chem. 2020;930:121593. doi: 10.1016/j.jorganchem.2020.121593. [DOI] [Google Scholar]
- 113.Skoch K., Cisarova I., Schulz J., Siemeling U., Stepnicka P. Synthesis and characterization of 1′-(diphenylphosphino)-1-isocyanoferrocene, an organometallic ligand combining two different soft donor moieties, and its Group 11 metal complexes. Dalton Trans. 2017;46:10339–10354. doi: 10.1039/C7DT02336G. [DOI] [PubMed] [Google Scholar]
- 114.Dey S., Roesler F., Bruhn C., Kelemen Z., Pietschnig R. Tailoring the Fe→Pd interaction in cationic Pd(ii) complexes via structural variation of the ligand scaffold of sterically demanding dppf-analogs and their P,N-counterparts. Inorg. Chem. Front. 2023;10:3828–3843. doi: 10.1039/D3QI00576C. [DOI] [Google Scholar]
- 115.Ritte M., Bruhn C., Siemeling U. Synthesis and Crystal Structures of the P,N-Substituted Ferrocenes [Fe{η5-C5H4–P(S)Ph2}(η5-C5H4–NHCH2tBu)] and [Fe(η5-C5H4–PPh2)(η5-C5H4–NHCH2tBu)] Z. Naturforsch. B. 2014;69:906–912. doi: 10.5560/znb.2014-4132. [DOI] [Google Scholar]
- 116.Navrátil M., Císařová I., Štěpnička P. Synthesis and coordination of hybrid phosphinoferrocenes with extended donor pendants. Dalton Trans. 2022;51:14618–14629. doi: 10.1039/D2DT02514K. [DOI] [PubMed] [Google Scholar]
- 117.Skoch K., Cisarova I., Uhlik F., Stepnicka P. Comparing the reactivity of isomeric phosphinoferrocene nitrile and isocyanide in Pd(ii) complexes: Synthesis of simple coordination compounds vs. preparation of P-chelated insertion products and Fischer-type carbenes. Dalton Trans. 2018;47:16082–16101. doi: 10.1039/C8DT03564D. [DOI] [PubMed] [Google Scholar]
- 118.Škoch K., Schulz J., Císařová I., Štěpnička P. Pd(II) Complexes with Chelating Phosphinoferrocene Diaminocarbene Ligands: Synthesis, Characterization, and Catalytic Use in Pd-Catalyzed Borylation of Aryl Bromides. Organometallics. 2019;38:3060–3073. doi: 10.1021/acs.organomet.9b00398. [DOI] [Google Scholar]
- 119.Binnani C., Leitner Z., Císařová I., Štěpnička P. Synthesis and Coordination Behavior of a Hybrid Phosphanylferrocene Amidine Ligand. Eur. J. Inorg. Chem. 2024;27:e202300644. doi: 10.1002/ejic.202300644. [DOI] [Google Scholar]
- 120.Bárta O., Gyepes R., Císařová I., Alemayehu A., Štěpnička P. Synthesis and study of Fe→Pd interactions in unsymmetric Pd(ii) complexes with phosphinoferrocene guanidine ligands. Dalton Trans. 2020;49:4225–4229. doi: 10.1039/D0DT00812E. [DOI] [PubMed] [Google Scholar]
- 121.Bárta O., Císařová I., Štěpnička P. The protonation state governs the coordination of phosphinoferrocene guanidines. Dalton Trans. 2021;50:14662–14671. doi: 10.1039/D1DT02884G. [DOI] [PubMed] [Google Scholar]
- 122.Škoch K., Vosáhlo P., Císařová I., Štěpnička P. Synthesis and characterisation of Pd(ii) and Au(i) complexes with mesoionic carbene ligands bearing phosphinoferrocene substituents and isomeric carbene moieites. Dalton Trans. 2020;49:1011–1021. doi: 10.1039/C9DT04521J. [DOI] [PubMed] [Google Scholar]
- 123.Withers H.P., Seyferth D., Fellmann J.D., Garrou P.E., Martin S. Phosphorus- and arsenic-bridged [1]ferrocenophanes. 2. Synthesis of poly((1,1′-ferrocenediyl)phenylphosphine) oligomers and polymers. Organometallics. 1982;1:1283–1288. doi: 10.1021/om00070a005. [DOI] [Google Scholar]
- 124.Butler I.R., Cullen W.R. The synthesis of α-N,N-dimethyl-1′-diphenylphosphinoferrocenylethylamine and related ligands. Can. J. Chem. 1983;61:147–153. doi: 10.1139/v83-026. [DOI] [Google Scholar]
- 125.Butler I.R., Cullen W.R. Lithiophosphinoferrocenes. A route to polyphosphines and ring-substituted ferrocenophanes. Organometallics. 1986;5:2537–2542. doi: 10.1021/om00143a023. [DOI] [Google Scholar]
- 126.Schulz J., Antala J., Rezazgui D., Cisarova I., Stepnicka P. Synthesis, Structure, Reactivity, and Intramolecular Donor-Acceptor Interactions in a Phosphinoferrocene Stibine and Its Corresponding Phosphine Chalcogenides and Stiboranes. Inorg. Chem. 2023;62:14028–14043. doi: 10.1021/acs.inorgchem.3c02075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhou B., Bedajna S., Gabbaï F.P. Pnictogen bonding at the service of gold catalysis: The case of a phosphinostiborane gold complex. Chem. Commun. 2024;60:192–195. doi: 10.1039/D3CC04942F. [DOI] [PubMed] [Google Scholar]
- 128.Shafir A., Power M.P., Whitener G.D., Arnold J. Synthesis, Structure, and Properties of 1,1′-Diamino- and 1,1′-Diazidoferrocene. Organometallics. 2000;19:3978–3982. doi: 10.1021/om0004085. [DOI] [Google Scholar]
- 129.Butler I.R., Cullen W.R., Ni J., Rettig S.J. The structure of the 3:2 adduct of 1,1′-dilithioferrocene with tetramethylethylenediamine. Organometallics. 1985;4:2196–2201. doi: 10.1021/om00131a023. [DOI] [Google Scholar]
- 130.Boston G.M.R., Philipp H.M., Butenschön H. Fluorosulfonylferrocene, (Trifluoromethylsulfonyl)ferrocene and New Ferrocenyl Sulfonates: Directed ortho Lithiation and New Anionic Thia-Fries Rearrangements at Ferrocene. Eur. J. Inorg. Chem. 2021;2021:4903–4914. doi: 10.1002/ejic.202100785. [DOI] [Google Scholar]
- 131.Sadeh S., Cao M.P.T., Quail J.W., Zhu J., Müller J. Enantiopure Phospha[1]ferrocenophanes: Textbook Examples of Through-Space Nuclear Spin–Spin Coupling. Chem. Eur. J. 2018;24:8298–8301. doi: 10.1002/chem.201801139. [DOI] [PubMed] [Google Scholar]
- 132.Sadeh S., Schatte G., Müller J. A Flexible Approach to Strained Sandwich Compounds: Chiral [1]Ferrocenophanes with Boron, Gallium, Silicon, and Tin in Bridging Positions. Chem. Eur. J. 2013;19:13408–13417. doi: 10.1002/chem.201301991. [DOI] [PubMed] [Google Scholar]
- 133.Ringenberg M.R. Beyond Common Metal–Metal Bonds, κ3-Bis(donor)ferrocenyl → Transition-Metal Interactions. Chem. Eur. J. 2019;25:2396–2406. doi: 10.1002/chem.201803150. [DOI] [PubMed] [Google Scholar]
- 134.Ganesh V., Sudhir V.S., Kundu T., Chandrasekaran S. 10 Years of Click Chemistry: Synthesis and Applications of Ferrocene-Derived Triazoles. Chem. Asian J. 2011;6:2670–2694. doi: 10.1002/asia.201100408. [DOI] [PubMed] [Google Scholar]
- 135.Alfonso M., Espinosa A., Tárraga A., Molina P. A Simple but Effective Dual Redox and Fluorescent Ion Pair Receptor Based on a Ferrocene–Imidazopyrene Dyad. Org. Lett. 2011;13:2078–2081. doi: 10.1021/ol2004935. [DOI] [PubMed] [Google Scholar]
- 136.Chang K.-C., Su I.-H., Wang Y.-Y., Chung W.-S. A Bifunctional Chromogenic Calix[4]arene Chemosensor for Both Cations and Anions: A Potential Ca2+ and F− Switched INHIBIT Logic Gate with a YES Logic Function. Eur. J. Org. Chem. 2010;2010:4700–4704. doi: 10.1002/ejoc.201000581. [DOI] [Google Scholar]
- 137.Picot S.C., Mullaney B.R., Beer P.D. Ion-Pair Recognition by a Heteroditopic Triazole-Containing Receptor. Chem. Eur. J. 2012;18:6230–6237. doi: 10.1002/chem.201200251. [DOI] [PubMed] [Google Scholar]
- 138.Alfonso M., Tárraga A., Molina P. Ferrocene-Based Heteroditopic Receptors Displaying High Selectivity toward Lead and Mercury Metal Cations through Different Channels. J. Org. Chem. 2011;76:939–947. doi: 10.1021/jo102243e. [DOI] [PubMed] [Google Scholar]
- 139.Alfonso M., Espinosa A., Tárraga A., Molina P. Multichannel HSO4− recognition promoted by a bound cation within a ferrocene-based ion pair receptor. Chem. Commun. 2012;48:6848–6850. doi: 10.1039/c2cc32215c. [DOI] [PubMed] [Google Scholar]
- 140.Vivancos Á., Segarra C., Albrecht M. Mesoionic and Related Less Heteroatom-Stabilized N-Heterocyclic Carbene Complexes: Synthesis, Catalysis, and Other Applications. Chem. Rev. 2018;118:9493–9586. doi: 10.1021/acs.chemrev.8b00148. [DOI] [PubMed] [Google Scholar]
- 141.Hettmanczyk L., Suntrup L., Klenk S., Hoyer C., Sarkar B. Heteromultimetallic Complexes with Redox-Active Mesoionic Carbenes: Control of Donor Properties and Redox-Induced Catalysis. Chem. Eur. J. 2017;23:576–585. doi: 10.1002/chem.201604615. [DOI] [PubMed] [Google Scholar]
- 142.Klenk S., Rupf S., Suntrup L., van der Meer M., Sarkar B. The Power of Ferrocene, Mesoionic Carbenes, and Gold: Redox-Switchable Catalysis. Organometallics. 2017;36:2026–2035. doi: 10.1021/acs.organomet.7b00270. [DOI] [Google Scholar]
- 143.Dötz K.H. Carbene Complexes in Organic Synthesis [New Synthetic Methods (47)] Angew. Chem. Int. Ed. 1984;23:587–608. doi: 10.1002/anie.198405871. [DOI] [Google Scholar]
- 144.Chen H., White P.S., Gagné M.R. Synthesis and Reactivity of Titanium(IV)–Salen Complexes Containing Oxygen and Chloride Ligands. Organometallics. 1998;17:5358–5366. doi: 10.1021/om980414p. [DOI] [Google Scholar]
- 145.Clarkson G.J., Gibson V.C., Goh P.K.Y., Hammond M.L., Knight P.D., Scott P., Smit T.M., White A.J.P., Williams D.J. Group 4 catalysts for ethene polymerization containing tetradentate salicylaldiminato ligands. Dalton Trans. 2006:5484–5491. doi: 10.1039/b610555f. [DOI] [PubMed] [Google Scholar]
- 146.Gregson C.K.A., Blackmore I.J., Gibson V.C., Long N.J., Marshall E.L., White A.J.P. Titanium–salen complexes as initiators for the ring opening polymerisation of rac-lactide. Dalton Trans. 2006:3134–3140. doi: 10.1039/B518266B. [DOI] [PubMed] [Google Scholar]
- 147.Quan S.M., Wang X., Zhang R., Diaconescu P.L. Redox Switchable Copolymerization of Cyclic Esters and Epoxides by a Zirconium Complex. Macromolecules. 2016;49:6768–6778. doi: 10.1021/acs.macromol.6b00997. [DOI] [Google Scholar]
- 148.Quan S.M., Wei J., Diaconescu P.L. Mechanistic Studies of Redox-Switchable Copolymerization of Lactide and Cyclohexene Oxide by a Zirconium Complex. Organometallics. 2017;36:4451–4457. doi: 10.1021/acs.organomet.7b00672. [DOI] [Google Scholar]
- 149.Hern Z.C., Quan S.M., Dai R., Lai A., Wang Y., Liu C., Diaconescu P.L. ABC and ABAB Block Copolymers by Electrochemically Controlled Ring-Opening Polymerization. J. Am. Chem. Soc. 2021;143:19802–19808. doi: 10.1021/jacs.1c08648. [DOI] [PubMed] [Google Scholar]
- 150.Gregson C.K.A., Gibson V.C., Long N.J., Marshall E.L., Oxford P.J., White A.J.P. Redox Control within Single-Site Polymerization Catalysts. J. Am. Chem. Soc. 2006;128:7410–7411. doi: 10.1021/ja061398n. [DOI] [PubMed] [Google Scholar]
- 151.Shigehisa H., Aoki T., Yamaguchi S., Shimizu N., Hiroya K. Hydroalkoxylation of Unactivated Olefins with Carbon Radicals and Carbocation Species as Key Intermediates. J. Am. Chem. Soc. 2013;135:10306–10309. doi: 10.1021/ja405219f. [DOI] [PubMed] [Google Scholar]
- 152.Yin J., Rainka M.P., Zhang X.-X., Buchwald S.L. A Highly Active Suzuki Catalyst for the Synthesis of Sterically Hindered Biaryls: Novel Ligand Coordination. J. Am. Chem. Soc. 2002;124:1162–1163. doi: 10.1021/ja017082r. [DOI] [PubMed] [Google Scholar]
- 153.Škoch K., Císařová I., Štěpnička P. Synthesis and Catalytic Use of Gold(I) Complexes Containing a Hemilabile Phosphanylferrocene Nitrile Donor. Chem. Eur. J. 2015;21:15998–16004. doi: 10.1002/chem.201502968. [DOI] [PubMed] [Google Scholar]
- 154.He W., Li C., Zhang L. An Efficient [2 + 2 + 1] Synthesis of 2,5-Disubstituted Oxazoles via Gold-Catalyzed Intermolecular Alkyne Oxidation. J. Am. Chem. Soc. 2011;133:8482–8485. doi: 10.1021/ja2029188. [DOI] [PubMed] [Google Scholar]
- 155.Bhoyare V.W., Tathe A.G., Das A., Chintawar C.C., Patil N.T. The interplay of carbophilic activation and Au(i)/Au(iii) catalysis: An emerging technique for 1,2-difunctionalization of C–C multiple bonds. Chem. Soc. Rev. 2021;50:10422–10450. doi: 10.1039/D0CS00700E. [DOI] [PubMed] [Google Scholar]
- 156.de Lima G.M., Filgueiras C.A.L., Giotto M.T.S., Mascarenhas Y.P. Tin, palladium and platinum derivatives of 1,1′-bis(diphenylphosphine)ferrocene. Crystal and molecular structures of 1,1′- bis-(diphenylphosphine)ferrocenedichloropalladium(II) and of 1,1′-bis(diphenylphosphine)ferrocenedichloroplatinum(II) Transit. Met. Chem. 1995;20:380–383. doi: 10.1007/BF00139135. [DOI] [Google Scholar]
- 157.Jess K., Baabe D., Freytag M., Jones P.G., Tamm M. Transition-Metal Complexes with Ferrocene-Bridged Bis(imidazolin-2-imine) and Bis(diaminocyclopropenimine) Ligands. Eur. J. Inorg. Chem. 2017;2017:412–423. doi: 10.1002/ejic.201600841. [DOI] [Google Scholar]
- 158.Khramov D.M., Rosen E.L., Lynch V.M., Bielawski C.W. Diaminocarbene[3]ferrocenophanes and Their Transition-Metal Complexes. Angew. Chem. Int. Ed. 2008;47:2267–2270. doi: 10.1002/anie.200704978. [DOI] [PubMed] [Google Scholar]
- 159.Macrae C.F., Edgington P.R., McCabe P., Pidcock E., Shields G.P., Taylor R., Towler M., van de Streek J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006;39:453–457. doi: 10.1107/S002188980600731X. [DOI] [Google Scholar]
- 160.Macrae C.F., Bruno I.J., Chisholm J.A., Edgington P.R., McCabe P., Pidcock E., Rodriguez-Monge L., Taylor R., van de Streek J., Wood P.A. Mercury CSD 2.0– new features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008;41:466–470. doi: 10.1107/S0021889807067908. [DOI] [Google Scholar]
- 161.Gramigna K.M., Oria J.V., Mandell C.L., Tiedemann M.A., Dougherty W.G., Piro N.A., Kassel W.S., Chan B.C., Diaconescu P.L., Nataro C. Palladium(II) and Platinum(II) Compounds of 1,1′-Bis(phosphino)metallocene (M = Fe, Ru) Ligands with Metal–Metal Interactions. Organometallics. 2013;32:5966–5979. doi: 10.1021/om400494t. [DOI] [Google Scholar]
- 162.Cabrera K.D., Rowland A.T., Szarko J.M., Diaconescu P.L., Bezpalko M.W., Kassel W.S., Nataro C. Monodentate phosphine substitution in [Pd(κ3-dppf)(PR3)][BF4]2 (dppf = 1,1′-bis(diphenylphosphino)ferrocene) compounds. Dalton Trans. 2017;46:5702–5710. doi: 10.1039/C6DT04921D. [DOI] [PubMed] [Google Scholar]
- 163.Jover J., Miloserdov F.M., Benet-Buchholz J., Grushin V.V., Maseras F. On the Feasibility of Nickel-Catalyzed Trifluoromethylation of Aryl Halides. Organometallics. 2014;33:6531–6543. doi: 10.1021/om5008743. [DOI] [Google Scholar]
- 164.Blass B.L., Sánchez R.H., Decker V.A., Robinson M.J., Piro N.A., Kassel W.S., Diaconescu P.L., Nataro C. Structural, Computational, and Spectroscopic Investigation of [Pd(κ3-1,1′-bis(di-tert-butylphosphino)ferrocenediyl)X]+ (X = Cl, Br, I) Compounds. Organometallics. 2016;35:462–470. doi: 10.1021/acs.organomet.5b00889. [DOI] [Google Scholar]
- 165.Elschenbroich C. Organometallics. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim, Germany: 2006. [Google Scholar]
- 166.Noviandri I., Brown K.N., Fleming D.S., Gulyas P.T., Lay P.A., Masters A.F., Phillips L. The decamethylferrocenium/decamethylferrocene redox couple: A superior redox standard to the ferrocenium/ferrocene redox couple for studying solvent effects on the thermodynamics of electron transfer. J. Phys. Chem. B. 1999;103:6713–6722. doi: 10.1021/jp991381+. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.