

Abstract
Autoimmune diseases comprise a large group of conditions characterized by a complex pathogenesis and significant heterogeneity in their clinical manifestations. Advances in sequencing technology have revealed that in addition to genetic susceptibility, various epigenetic mechanisms including DNA methylation and histone modification play critical roles in disease development. The emerging field of epigenetics has provided new perspectives on the pathogenesis and development of autoimmune diseases. Aberrant epigenetic modifications can be used as biomarkers for disease diagnosis and prognosis. Exploration of human epigenetic profiles revealed that patients with autoimmune diseases exhibit markedly altered DNA methylation profiles compared with healthy individuals. Targeted cutting-edge epigenetic therapies are emerging. For example, DNA methylation inhibitors can rectify methylation dysregulation and relieve patients. Histone deacetylase inhibitors such as vorinostat can affect chromatin accessibility and further regulate gene expression, and have been used in treating hematological malignancies. Epigenetic therapies have opened new avenues for the precise treatment of autoimmune diseases and offer new opportunities for improved therapeutic outcomes. Our review can aid in comprehensively elucidation of the mechanisms of autoimmune diseases and development of new targeted therapies that ultimately benefit patients with these conditions.
Keywords: autoimmune disease, epigenetic, systemic lupus erythematosus, DNA methylation, histone modification, RNA modification
1. Introduction
Autoimmune diseases represent a diverse group of conditions characterized by an imbalanced immune system that abnormally attacks the cells and tissues of the body, loss of immune tolerance to autoantigens, and impaired self-nonself discrimination. This dysfunction leads to excessive proliferation and activation of autoreactive T- or B-cells, resulting in the unintentional targeting of normal tissues (1). The establishment of immune tolerance involves several different mechanisms and interactions, resulting in disease-specific heterogeneity in autoimmune diseases (2). The clinical symptoms of autoimmune diseases are highly heterogeneous and related to the specific site of involvement ( Table 1 ); accordingly, they can be categorized into organ-specific and systemic autoimmune diseases (42). However, most of these diseases have similar manifestations, such as the tissue inflammatory response, which is caused by local immune cell infiltration and inflammatory factors (43). In certain diseases, the occurrence of lesions and damage can be triggered by autoantibodies secreted by plasma cells or directly mediated by pathogenic T-cells. Therefore, most autoimmune diseases are hypothesized to have a shared etiology and pathogenesis (44). For instance, genetic factors exert substantial influence on the pathogenesis of autoimmune diseases, and genome-wide association studies (GWAS) are powerful tools for studying genetic susceptibility to disease. However, the technique is limited to analysis at the genetic level, and interactions between genes and downstream factors are not adequately considered; thus, the pathogenesis and regression of autoimmune diseases cannot be comprehensively revealed (45).
Table 1.
Specific antibodies, clinical manifestations, and treatment of autoimmune diseases.
| Disease | Autoantibody | Treatment | References | |
|---|---|---|---|---|
| Rheumatic autoimmune diseases | Systemic lupus erythematosus (SLE) | Anti-nuclear antibodies (ANAs): anti-double-stranded DNA antibody, antiphospholipid antibody, anti-Sm antibody | Nonsteroidal anti-inflammatory drugs (NSAIDs), Glucocorticoids, Hydroxychloroquine, Belimumab, Rituximab, Anifrolumab | (3, 4) |
| Rheumatoid arthritis (RA) | Rheumatoid factor (RF), anti-citrullinated protein antibodies (ACPAs) | Disease-modifying anti-rheumatic drugs (DMARDs): methotrexate, hydroxychloroquine; JAK inhibitors: Tofacitinib, Baricitinib; TNF inhibitors: Adalimumab, Infliximab; Anti-IL-6: Tocilizumab, Sarilumab | (5, 6) | |
| Sjögren’s syndrome (SS) | ANAs; anti-ribonucleoprotein complexes Ro/SSA and La/SSB antibodies; RF | Glucocorticoids, hydroxychloroquine, anti BAFF antibody belimumab | (7, 8) | |
| Antiphospholipid syndrome (APS) | Anti-phospholipid antibody, anti-β2 glycoprotein-I antibody, anticardiolipin antibodies | Anti-coagulants, Vitamin K antagonists (VKAs), Aspirin, Hydroxychloroquine | (9, 10) | |
| Systemic sclerosis (SSc) | ANAs, anti-topoisomerase I (Scl-70), anti-nucleolar antibody, anti-RNA polymerase I/III antibody | Mycophenolate, cyclophosphamide, Tocilizumab, Rituximab | (11, 12) | |
| Central nervous system | Multiple sclerosis (MS) | Intrathecal oligoclonal IgM antibodies against myelin lipids, preferentially phosphatidylcholine; anti-Sperm-associated antigen 16 (SPAG16) antibodies | Ocrelizumab, interferons, glatiramer acetate, dimethyl fumarate | (13, 14) |
| Guillain–Barré syndrome (GBS) subtype: acute motor axonal neuropathy (AMAN) | Anti-ganglioside antibodies: anti-GM1a, GM1b, GD1a antibodies | Intravenous immunoglobulin (IVIg) and plasma exchange | (15, 16) | |
| Autoimmune limbic encephalitis (ALE) | Anti-leucine-rich glioma inactivated 1 antibody, anti-γ-aminobutyric acid B receptor antibody, anti-Hu antibody | Intravenous immunoglobulin (IVIG), plasma exchange (PLEX), rituximab | (17, 18) | |
| Anti-NMDA receptor encephalitis | Anti-GluN1 subunit of the NMDA receptor antibody | Steroids, intravenous immunoglobulins (IVIG), or plasma exchange (PLEX) | (19) | |
| Endocrine system | Type 1 diabetes mellitus | Antibodies against specific β-cell proteins, including insulin, glutamate decarboxylase, islet antigen 2, zinc transporter 8, and tetraspanin-7 | Insulin, pramlintide, metformin, glucagon-like peptide-1 receptor agonists | (20, 21) |
| Autoimmune Addison disease | Anti-adrenal cortex autoantibodies (ACAs): antibodies directed against steroid 21-hydroxylase | Corticosteroid replacement | (22) | |
| Hematologic system | Autoimmune hemolytic anemia (AIHA) | Anti-RBC antigens antibodies: Warm AIHA IgG; cold AIHA IgM; Mixed-type AIHA (mAIHA) IgG, IgM; Paroxysmal cold hemoglobinuria (PCH) IgG |
Glucocorticoids, splenectomy, anti-CD20 mAb | (23) |
| Skin | Vitiligo | Anti-epidermal melanocytes antibody | UV Treatments, Topical corticosteroids, Calcineurin inhibitors | (24, 25) |
| Pemphigus | Anti-Desmoglein 1/3 autoantibody | Corticosteroids, immunosuppressant drugs | (26) | |
| Epidermolysis bullosa acquisita (EBA) | Anti- collagen VII antibody | Neutrophil targeting therapies, conventional Immunosuppressives, intravenous immunoglobulin and rituximab | (27, 28) | |
| Digestive system | Primary biliary cirrhosis (PBC) | Anti-mitochondrial antibodies (AMAs), Anti-Sp100 and anti-gp210 antibodies | Ursodeoxycholic acid (UDCA) | (29, 30) |
| Coeliac disease | Anti-transglutaminase 2 (TG2) and anti-endomysial (EMA) antibodies | A gluten-free diet, administration of probiotics. | (31, 32) | |
| Crohn’s disease | Antimicrobial antibodies: anti-Saccharomyces cerevisiae antibody (ASCA) | 5-aminosalicylate, corticosteroids, anti-TNF: Infliximab, adalimumab | (33, 34) | |
| Autoimmune gastritis (AIG) | Anti-parietal cell antibody, anti-H+/K+ ATPase antibody, anti-intrinsic factor antibody | Intramuscular cobalamin, Iron and vitamin B12 supplementation | (35, 36) | |
| Muscle | Myasthenia gravis (MG) | Anti-acetylcholine receptor (AChR) antibody; Anti-muscle-specific kinase antibody | Pyridostigmine, immunosuppressive medication | (37, 38) |
| Lambert-Eaton myasthenic syndrome (LEMS) | Anti-P/Q-type Voltage-gated calcium channel (VGCC) antibody | 3,4-diaminopyridine potassium channel blocker |
(39) | |
| Thyroid gland | Graves’ diseases (GD) | Anti-thyrotropin receptor antibody (TRAb), anti-thyroid stimulating antibody (TSAb) | Methimazole, propylthiouracil, thyroidectomy | (40) |
| Hashimoto’s thyroiditis | Anti-thyroperoxidase antibody (TPOAb), anti-thyroglobulin antibody (TgAb) | Levothyroxine, glucocorticoids | (41) |
Although most autoimmune diseases are inextricably linked to genetic factors, recent advance in epigenetics has greatly enriched our understanding of their pathogenesis, and provided a comprehensive review of the pathogenesis of the mechanisms involved. Epigenetics elucidates the unconventional changes in gene expression that ultimately lead to differences in phenotypes without altering DNA sequences (46). The core of the epigenetic regulatory mechanism is a variety of reversible covalent modifications of nucleic acids and histones in the presence of multiple chemical modifying enzymes, such as DNA/RNA methylation and histone acetylation ( Figure 1 , Table 2 ). Meanwhile, these modifications can induce sophisticated crosstalk for regulating gene expression (49). In addition, the mechanisms of epigenetic regulation also include chromatin remodeling and non-coding RNAs, the former uses chromatin remodeling complexes to regulate the denseness or looseness of chromatin structure, thereby affecting the binding of transcription factors to control gene expression (58). Although non-coding RNAs are unable to translate proteins, they have an essential role in epigenetics by coordinating DNA methylation, chromatin structure remodeling, and histone chemical modification (59).
Figure 1.

An overview of epigenetic mechanisms. The basic unit of chromatin is the nucleosome. Two molecules of H2A, H2B, H3, and H4 form a histone octamer, and DNA molecules are coiled around the histone octamer to form the nucleosome’s core particles. DNA methylation is mediated by the enzyme DNA methyltransferase (DNMT), which transfers methyl groups to adenine (A), guanine (G), or cytosine (C) by using S-adenosyl methionine (SAM) as a substrate. Histone modifications include acetylation, methylation, phosphorylation and so on, and they mainly occur on lysine (K), arginine (R) and serine (S). RNA modifications are similar to DNA modifications, including m1A, m6A and many other types. Chemical structures are prepared using ChemDraw.
Table 2.
Comparison of DNA, RNA, and histone modifications.
| Writer | Eraser | Reader | Types of modification | Site of modification | Biological effect | References | |
|---|---|---|---|---|---|---|---|
| DNA modification | DNMT1, DNMT3A and DNMT3B | TET1/2/3 | MBD, MeCP2 | 5mC, 6mA, 7mG | Genome DNA, mitochondrial DNA | DNA methylation at gene promoter is associated with transcriptional silencing and it mediates chromatin inaccessibility | (47–50) |
| RNA modification | METTL3, METTL14, WTAP, KIAA1492 | FTO, ALKBH5 | YTHDF1/2/3, YTHDC1/2, IGF2BP1/2/3, HNRNP protein family | m6A, m5C, m7G, Pseudouridine | messenger RNA, ribosomal RNA, transfer RNA, non-coding RNA | m6A regulates various post-transcriptional aspects of the mRNA lifecycle, including RNA degradation, processing, splicing, nuclear export, mRNA translation, and also regulates gene expression | (49, 51, 52) |
| Histone modification | HMT, HAT | HDM, HDAC | Chromodomain, PHD fingers, bromodomain proteins | Methylation, Acetylation, Phosphorylation, Ubiquitination | H2A, H2B, H3, H4 | Transcriptional activation: H3K4me3, H3K4ac. Transcriptional repression: H3K27me3. |
(53–57) |
DNMT, DNA methyltransferase; TET, Ten-eleven translocation; MBD, methyl-CpG-binding domain; MeCP, methyl-CpG binding protein; HDAC, histone deacetylase; METTL, Methyltransferase like; WTAP, Wilms tumor 1-associating protein; FTO, fat-mass and obesity-associated protein; ALKBH, AlkB homologue; YTHDF, YTH domain family; YTHDC, YTH domain-containing protein; IGF2BP, Insulin-like growth factor-2 mRNA-binding proteins; HNRNP, heterogeneous nuclear ribonucleoprotein; HMT, histone methyltransferase; HAT, histone acetyltransferases; HDM, histone demethylase; HDAC, histone deacetylase; PHD, plant homeodomain.
With the advancement of detection technologies over the past few decades, knowledge and research on epigenetic mechanisms have progressively deepened, and an increasing body of evidence has shown that epigenetic mechanisms positively regulate the growth and aging of people and are closely linked to a wide variety of diseases, especially autoimmune diseases (60). Epigenetic modifications are highly dynamic systems that constantly change in response to environmental factors and organism development (61). Notably, epigenetic modifications are reversible processes mediated by a variety of enzymes, exhibiting specificity to certain cells and diseases, consistent with the fact that autoimmune diseases share similar pathogenesis but present with distinct clinical manifestations. Therefore, epigenetic modifications are promising biomarkers for disease diagnosis (62). The reversal of aberrant modifications through the use of targeted drugs has enormous potential for treating autoimmune diseases. Epigenetic drug research is an emerging field in the treatment of diseases at the level of gene regulation that opens a new path for precision medicine (63). Globally, the incidence of autoimmune diseases persists at elevated levels coupled with the absence of efficient therapeutic interventions with minimum side effects. Conventional nonsteroidal anti-inflammatory drugs (NSAIDs) can only alleviate the symptoms, while long-term use of immunosuppressive drugs compromises the immune function of patients (64). Therefore, it is essential to explore new research directions to comprehensively understand the mechanisms of autoimmune diseases and develop new targeted therapies, ultimately benefiting patients with these conditions.
In this review, we summarize the major mechanisms of autoimmune diseases and epigenetics, emphasizing the close association of epigenetic modifications with autoimmune diseases and providing novel perspectives on leveraging epigenetics for the diagnosis and treatment of diseases.
2. The mechanisms of autoimmune disease
2.1. Potential pathogenesis of autoimmune diseases
Many years ago, it was observed that autoimmune diseases had a tendency for family inheritance, with their pathogenesis primarily driven by genetic factors. GWAS provides a powerful tool for investigating disease susceptibility genetically, but this technique is limited to genetic analysis and is unable to comprehensively reveal the pathogenesis of autoimmune diseases (45). A deeper exploration of disease mechanisms has highlighted the indispensability of environmental and epigenetic factors in autoimmune disease development. Epigenetics has emerged as a prominent research topic in this field, not only bridging the gap between environmental and genetic factors but also acting as an independent factor that induces or exacerbates autoimmune diseases (65). However, the current accumulated knowledge is still unable to fully explain the heterogeneity of the disease; therefore, finding new research directions is necessary to comprehensively sort out their mechanisms.
Among the genetically driven autoimmune diseases, immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome is a classic example of a monogenic autoimmune disease in which the mutated forkhead box P3 (FOXP3) gene disrupts the transcription of Treg signature genes, such as interleukin 2 receptor subunit alpha (IL-2RA) and cytotoxic T-lymphocyte associated protein 4 (CTLA4), thus affecting the development and function of Tregs and ultimately resulting in several autoimmune disorders, such as type 1 diabetes mellitus (T1D) and severe bowel disease (66). Mutations in the AIRE gene impede the expression of the tissue-specific antigen by medullary thymic epithelial cells (mTECs), resulting in the compromised clearance of autoreactive T-cells or induction of Treg production, which in turn triggers a severe multiorgan damaging autoimmune disease, namely autoimmune polyendocrine syndrome type 1 (APS-1) (67). In addition, various environmental risk factors, including drugs, viral infections, and ultraviolet radiation, can influence the pathogenesis of autoimmune diseases by affecting epigenetic modifications. One hypothesis regarding the mechanism by which air pollution contributes to autoimmune diseases is that it can enhance systemic inflammatory and autoimmune responses through a variety of mechanisms (68). For instance, various infectious factors, particularly viruses, contribute to systemic lupus erythematosus (SLE) pathogenesis through multiple mechanisms. One such mechanism is molecular mimicry, which refers to the use of exogenous antigens that resemble autoantigens in their sequence or structure, thereby activating autoreactive T- or B-cells to produce autoantibodies, resulting in tissue and organ damage. A typical example is the crossreactivity between Epstein-Barr virus (EBV) nuclear antigens and host autoantigens (69). Moreover, CD8+ T-cells of patients with SLE exhibit an impaired immune response to EBV, leading to a significant increase in the EBV viral load in patients, where the latent viruses cause persistent infections and constantly stimulate the immune system to continuously produce antibodies and circulating immune complexes, ultimately exacerbating the disease (70).
Numerous relevant factors and pathological mechanisms are believed to contribute to the onset of autoimmune diseases; however, no specific theory can perfectly explain the underlying mechanisms. Therefore, comprehensively elucidating the genetic, environmental, and epigenetic factors related to autoimmune diseases is imperative. Notably, the interaction among these factors in mediating disease development is particularly important for elucidating their pathogenesis. This knowledge will enable the identification of risk factors and formulation of preventive interventions targeting susceptible populations, ultimately reducing the disease burden.
2.2. Dysfunctional immune cells in autoimmune diseases
Autoimmune diseases arise from a malfunctioning immune system and disrupted immune tolerance mechanisms, resulting in an immune response in which autoreactive lymphocytes target self-antigens and cause damage to the body (1). Under normal circumstances, host immune homeostasis is maintained by both the innate and adaptive immune systems, thus preventing the occurrence of autoimmune diseases (71). However, in patients with autoimmune diseases, the immune system exhibits abnormalities such as heightened activation of immune cells, alterations in their function and phenotype, and aberrant expression of various immune molecules, including complement, antibodies, and cytokines. This pathological state results in an inflammatory response and tissue damage, ultimately contributing to the progression of autoimmune diseases ( Figure 2 ) (72).
Figure 2.

Epigenetic modification of immune cells in autoimmune diseases. Gene transcription in immune cells in autoimmune diseases is affected by epigenetic mechanisms such as histone modification and DNA methylation. In Treg cell from experimental autoimmune encephalomyelitis (EAE), DNMT inhibitor 5-Aza can promote Foxp3 transcription, and inhibit IL-17A transcription, thus ameliorating CNS inflammatory responses and decreasing proinflammatory cytokines. HDAC inhibitor can increase histone acetylation modification and promote IFN-γ transcription in T cell in T1D. (Figure created by Figdraw, ID: TUIUU9ee14).
Among the various immune cells, T- and B-lymphocytes are the most prominent mediators of autoimmunity that contribute to tissue damage, with aberrant alterations in their number and function being key mechanisms driving autoimmune diseases (73, 74). Autoreactive B-cells not only cause tissue and organ damage and exacerbate inflammatory responses through the secretion of autoantibodies and inflammatory factors but also further aggravate disease severity by presenting antigens and expressing costimulatory molecules to activated T-cells (75). B10 cells suppress immune responses by secreting interleukin 10 (IL-10); however, their number and function are dysregulated in rheumatoid arthritis (RA), as tumor necrosis factor-alpha (TNF-α) downregulates IL-10 and upregulates IFN-γ and IL-17A, inducing a proinflammatory B10 cell phenotype, leading to disease deterioration (76). CD4+CD25+forkhead box P3 (Foxp3)+ Tregs have potent immunosuppressive functions and contribute to immune tolerance maintenance and the control of autoimmune disease development. Abnormalities in the number and function of Tregs have been observed in many types of autoimmune disorders (77). Most studies demonstrated that the proportion of Tregs in SLE patients decreased with the increase in disease activity (78, 79); however, some studies have found that the number of Tregs in organ-specific autoimmune diseases increases, potentially depending on the clinical disease activity, further highlighting the great heterogeneity of these diseases (80).
In addition to T- and B-lymphocytes, several other types of immune cells are involved in autoimmune disease development. Neutrophils kill extracellular bacteria by producing neutrophil extracellular traps (NETs), which are composed of DNA, histones, myeloperoxidase (MPO), and other components that play important roles in innate immune responses (81). However, recent evidence has suggested that neutrophils are linked to autoimmune disease pathogenesis because the substances present in NETs can serve as autoantigens and trigger autoimmune reactions; for instance, IL-8 production and exposure to citrullinated autoantigens was shown to exacerbate joint damage by enhancing the inflammatory response of RA synovial fibroblasts (82). Similarly, M1 macrophages in the synovium of patients with RA can release chemokines to promote the recruitment of more inflammatory cells and production of more inflammatory factors such as IL-1, TNF-α, and IL-6, leading to synovial and osteoarticular destruction (83). Dendritic cells (DCs) are important immune response conductors. Notably, heterogeneous DCs subpopulations and significant alterations in their function, such as defective migration and impaired phagocytosis in patients with SLE and RA, have been identified in various autoimmune diseases, partially explaining the pathological diversity of autoimmune diseases (84).
Given the diversity of immune cell phenotypes observed in autoimmune diseases, restoring the normal function of immune cells or eliminating dysregulated immune cells has been proposed as an effective strategy for treating autoimmune diseases. Currently, B-cell depletion therapy is emerging as a promising therapeutic approach, offering the potential to mitigate the side effects commonly associated with broad-spectrum immunosuppressants (85). Autoimmune diseases are highly heterogeneous and dynamic, resulting in various types of damage owing to the breakdown of immune tolerance and disruption of the immune system (86). Consequently, a comprehensive exploration of the abnormal alterations in the immune systems of patients with autoimmune diseases is imperative for developing more targeted therapeutic interventions in the future.
2.3. The mechanisms of immune tolerance
Immune tolerance refers to the phenomenon that immunologically active cells cannot be activated when stimulated by specific antigens, resulting in specific immune non-response (87), which is key to protecting the host tissues from destruction by its immune system without affecting the overall function of the adaptive immune response (88). Although contradicting, immune tolerance is essential for preventing immune cells from attacking the body tissues, thereby reducing the risk of autoimmune diseases. However, its persistence in chronic inflammation, tumors, and other pathological conditions can exacerbate disease progression and enable tumor cell survival (89).
Immune tolerance is categorized into central and peripheral according to the different periods of its formation, with the former being established during embryonic and postnatal development of the central immune organs (thymus and bone marrow) by eliminating autoreactive T- and B-lymphocytes through a complex mechanism of negative selection and clonal deletion. Defects in negative selection disrupt central tolerance, allowing the persistence of self-reactive lymphocytes that leads to autoimmunity in the body, thereby greatly increasing the probability of autoimmune disease (90). During the establishment of central tolerance, mTECs can express and present tissue-restricted self-antigens to T-cells through the expression of an autoimmune regulator (AIRE), a transcriptional activator that induces apoptosis following the high affinity binding of the T-cell receptor (TCR) to the self-antigen peptide-MHC molecular complex on the surface of mTECs (91). In addition, some autoreactive T-cells combine with self-antigens to develop natural Tregs (nTregs) with immunosuppressive effects; therefore, mTECs and their AIREs are fundamental and indispensable in the establishment of central tolerance (92). Negative selection also occurs during B-cell development when the functional B-cell receptor (BCR) complex is expressed on its surface for the first time; its high affinity binding to self-antigens leads to clone clearance and destruction of self-reactive B-cells (93).
The establishment of central tolerance can eliminate most self-reactive cells; however, some cells escape negative selection and clonal clearance, resulting in a significant number of cells that are unable to accurately distinguish nonself from self in healthy individuals, posing a potential risk of triggering autoimmune responses. Therefore, peripheral tolerance mechanisms are necessary for preventing autoimmune diseases (87). Peripheral tolerance refers to a state of immune nonresponsiveness or low response of mature T- and B-cells to endogenous or exogenous antigens through a variety of peripheral mechanisms (94). Clonal anergy is a common mechanism of peripheral tolerance, in which mature T- and B-cells with TCRs or BCRs on their surface that are capable of binding to antigens, are not adequately activated owing to the lack of costimulators or T-cell assistance, thus resulting in an unresponsive state in which they are prone to apoptosis (95). Another key mechanism of peripheral tolerance is the suppressive cell population, of which the most important are Tregs, which can be categorized into natural and peripherally induced Tregs, with the former exerting their immunosuppressive function through direct cell-to-cell contact, whereas the latter through the secretion of various cytokines such as IL-10 and TGF-β (96, 97).
3. The close link between epigenetics and autoimmune diseases
3.1. DNA modification and autoimmune diseases
Researchers have identified more than 10 different chemical modifications in DNA, each of which plays a unique role in biological development, aging, and disease (49). Of all the types of DNA modifications, methylation was the first to be discovered and the most intensively studied. DNA methylation can occur on adenine (A), guanine (G), and cytosine (C) (98). The DNA methyltransferase (DNMT) family acts as the “writer” of methylation modification, transferring methyl groups to the 5′-position of the cytosine, thus forming 5-methylcytosine (5mC) (99). DNMT3A and DNMT3B are de novo methyltransferases responsible for establishing DNA methylation, whereas DNMT1 is responsible for maintaining methylation after semi-conservative replication (100). DNA demethylation can be active or passive. The former refers to the progressive oxidation of 5mC by ten-eleven translocation (TET) cytosine dioxygenase, which is specifically recognized by the thymine DNA glycosylase that removes the oxidized product, followed by base excision repair to regenerate cytosine (101). The latter refers to the inability to maintain the methylated state of the new chain due to the inhibition of DNMT activity, resulting in the gradual dilution of 5mC in the genome (102). DNA methylation “readers” usually have a methyl-CpG binding domain (MBD); however, some transcription factors lacking MBDs were recently reported to also have the ability to bind to methylated DNA, thereby exerting downstream biological effects and regulating gene expression (50).
N6-methyldeoxyadenosine (6mA) was conventionally perceived as exclusively operational within prokaryotic organisms. Nevertheless, advances in detection methodologies with enhanced sensitivities have revealed the presence of 6mA in the genomic DNA of multicellular eukaryotic organisms. Their abundance and distribution in the genome exhibits significant disparities across diverse species (103). Similar to 5mC, the 6mA modification has its own “writer,” “eraser,” and “reader” (104). Methyltransferase like 4 (METTL4), which is a homolog of the Caenorhabditis elegans 6mA methyltransferase DAMT-1, is presumed to be the mammalian 6mA methyltransferase and has the ability to catalyze mitochondrial DNA (mtDNA) methylation (105). The human alkB homologue 1 (ALKBH1) and ALKBH4 proteins are recognized as 6mA demethylases, the mechanism of which involves the oxidation of the methyl group of 6mA with oxygen to regenerate unmodified adenine (106). Currently, investigations of human 6mA “readers” have primarily focused on mtDNA, with scant information available regarding the specific functions of 6mA in mammalian systems; however, given its diverse and significant role in prokaryotes, the exploration of the biological functions of 6mA in eukaryotes may emerge as a research hotspot in the field of epigenetics in the foreseeable future.
Decades of research has firmly established the correlation between DNA methylation and autoimmune diseases, with alterations in the DNA methylation spectrum observed across various diseases and associated with disease subtypes, activity levels, and patient responses to therapeutic interventions, thereby augmenting the potential of DNA methylation as a novel clinical biomarker (107). Interferon (IFN) is a cytokine involved in the regulation of the immune response. Patients with SLE exhibit significantly reduced methylation levels in their IFN genes, leading to increased IFN expression levels and exacerbation of nephritis and central nervous system symptoms (108). Patients with RA undergoing methotrexate treatment have unique DNA methylation profiles that determine their response to medication. Specifically, individuals exhibiting elevated levels of DNA methylation in whole-blood leukocytes tend to have increased disease activity and are more likely to develop resistance to methotrexate after 3 months of treatment (109).
Notably, some DNA-modifying enzymes have been found to play unexpected roles in other areas such as the posttranslational modification of proteins. Recent research has shown that METTL9 can catalyze histidine methylation, suggesting that DNA-modifying enzymes may play additional roles in autoimmune diseases by regulating intracellular biochemical reactions in unconventional ways, expanding our understanding of their potential function beyond traditional pathways (110). To further elucidate the pathophysiological role of DNA methylation in autoimmune diseases, large-scale epigenome-wide studies are needed, and an in-depth investigation is imperative for determining whether these modifications can serve as valuable epigenetic markers. This exploration has the potential to identify novel targets for the diagnosis and personalized treatment of patients with autoimmune diseases.
3.2. Histone modification and autoimmune diseases
The posttranslational modification of histone amino acid residues is an extensively researched classical epigenetic mechanism ( Figure 3 ). A wide range of chemical modifications exist, including methylation, acetylation, and phosphorylation (55) Notably, the newly discovered sumoylation, crotonylation, lactylation, and many other modifications (121) are all controlled by the corresponding “ writer” that adds the modification, “eraser” that removes the modification, and “reader” that recognizes the modification and enables the conduct of a specific biological function (122). Euchromatin, which is characterized by active gene expression, typically exhibits high levels of histone lysine acetylation. Regarding the underlying mechanism of action, acetylation neutralizes the positive charge carried by lysine residues, thereby reducing the affinity with negatively charged DNA. Consequently, a relaxed and open chromatin structure is formed, which facilitates the binding of transcription factors to DNA and promotes gene expression (123). Conversely, histone deacetylation creates a closed chromatin conformation and reduces transcriptional activity. Histone acetyltransferases (HATs) are “writers” of histone acetylation that can transfer the acetyl group on acetyl coenzyme A to lysine residues, and based on their sequence and structure, they are divided into three families, p300-CBP (CREB-binding protein), GNAT (Gcn5-related N-acetyltransferase), and MYST (MOZ, Ybf2/Sas3, Sas2, Tip60) (124). Histone deacetylases (HDACs) are the “erasers,” and are classified into four classes; among them, classes I, II, and IV are all Zn2+-dependent enzymes, whereas class III are nicotinamide adenine dinucleotide (NAD+)-dependent (125). The “readers” of histone acetylation contain domains that can accommodate specific acetylation modifications. After binding to histones, they can either recruit transcription factors to promote gene expression or serve as a barrier to prevent transcription factor binding (56). Another common modification, histone methylation, involves a complex mechanism that regulates gene transcription. Histone methylation can regulate transcriptional activity in both directions, depending on its levels and the specific amino acid sites (126).
Figure 3.

A timeline for studying histone modifications. Phillips identifies histone lysine acetylation (111). Allfrey et al. identify histone acetylation as transcription regulators (112). Taunton et al. identify the first histone deacetyltransferase (113). Brownell et al. identify the first histone acetyltransferase (114). Dhalluin et al. identify bromodomain as the first histone acetylation reader (115). Rea et al. identify the first histone methyltransferase SUV39H1 (116). Bannister et al. identify chromodomain as the first histone methylation reader (117). Shi et al. identify the first histone demethylase LSD1 (118). Zhao et al. identify the histone crotonylation (119). Zhao et al. identify the histone lactylation (120).
Abnormal histone modifications play a critical role in the pathogenesis of various autoimmune diseases, thus establishing these modifications as clinical markers for the diagnosis and prognosis of diseases (127). TTNF-α-induced protein 3 (TNFAIP3) can inhibit inflammatory responses by negatively regulating the NF-κB inflammatory signaling pathway; however, the transcriptional activity marker trimethylated lysine 4 of histone H3 (H3K4me3) has been shown to be reduced in the TNFAIP3 gene promoter of CD4+ T-cells in patients with SLE, resulting in a significant decrease in the TNFAIP3 mRNA level and overexpression of IFN-γ and IL-17, thereby exacerbating the inflammatory response and promoting SLE pathogenesis (128). In addition, H3K4me3 and acetylated lysine 27 of histone 3 (H3K27ac) marks in IFN-responsive genes in the monocytes of patients with systemic sclerosis (SSc) were found to be increased to varying degrees, thus promoting the expression of these genes. In particular, an increase in myxoma resistance protein 1 (MX1) gene expression was reported to lead to severe lung function impairment and increased mortality in patients with SSc (129). Notably, histone modification is a reversible process, highlighting the potential of employing histone-modifying enzymes to restore abnormal histone modifications to their native state, and has emerged as a promising therapeutic strategy for addressing autoimmune diseases (130).
3.3. Chromatin remodeling and autoimmune diseases
The chromatin of eukaryotes not only carries genetic information but also contains a large amount of epigenetic information. The basic structural unit of chromatin, the nucleosome, comprises DNA wrapped around a core octamer consisting of two molecules, each of the histones H2A, H2B, H3, and H4 (131). Chromatin is found in two distinguishable forms: euchromatin, which is less condensed and has a high transcriptional activity, and heterochromatin, which is more compact and has a low transcriptional activity (132). Chromatin remodeling is a crucial epigenetic regulatory mechanism that modulates gene expression at the chromatin level by dynamically altering the open or closed state of chromatin, thus allowing or inhibiting the binding of transcription factors (133). Adenosine triphosphate (ATP)-dependent chromatin-remodeling complexes are major mediators of chromatin remodeling, dynamically regulating the chromatin structure by using energy generated from ATP hydrolysis to move, remove, or replace nucleosomes (134). The structure of ATP-dependent chromatin remodeling complex is highly conserved in eukaryotes, and these complexes can be divided into four families according to the structural characteristics of the core ATPase subunit: switching defective/sucrose nonfermenting (SWI/SNF), imitation switch (ISWI), inositol requiring 80 (INO80), and chromodomain helicase DNA-binding (CHD), all of which play unique roles in regulating transcriptional activity (135). For example, the SWI/SNF complex comprises various structural domains capable of interacting with DNA or histones, regulating the accessibility of transcription factors to chromatin by mediating the ejection, sliding, and removal of nucleosomes on DNA, thus promoting or repressing gene expression (136). Furthermore, these remodeling complexes can coordinate with histone modifications and variants to regulate chromatin remodeling, thus finely regulating biological processes such as DNA replication, repair, and transcription (137).
A close relationship between chromatin-remodeling complexes and autoimmune diseases has also been discovered. Studies have shown that small-molecule inhibitors targeting SWI/SNF can attenuate T-cell depletion by altering transcription factor binding motif sites; therefore, inhibiting SWI/SNF activity may drive the progression of autoimmune disease by affecting the activation and exhaustion of T-cells (138). T helper 17 (Th17) cells participate in inflammatory responses by producing IL-17 and IL-21 and recruiting inflammatory cells, which play important roles in many autoimmune diseases such as psoriasis and multiple sclerosis (MS) (139). SRG3 is a scaffold protein that has been shown to control the stability of the SWI/SNF complex in mice. The secretion of IL-17A and IL-17F by CD4+ T-cells is greatly reduced in mice lacking SRG3, thereby affecting the generation and differentiation of Th17 cells (140). The chromatin remodeler polybromo-1 (PBRM1) belongs to the SWI/SNF family, and some patients with Crohn’s disease and ulcerative colitis (UC) have been found to have low levels of PBRM1 expression, which leads to an increase in the expression of IFN and several proinflammatory cytokines through the activation of the retinoic acid-inducible gene-I-like receptor (RLRs) signaling pathway, thus exacerbating inflammation in patients (141). Given the limited research on the relationship between ATP-dependent chromatin-remodeling complexes and autoimmune diseases, further investigations in this domain are warranted to provide new insights into the pathophysiological mechanisms underlying autoimmune diseases (142).
3.4. RNA modification and autoimmune diseases
With the rapid development of detection technologies, more than 100 modifications have been identified in different types of RNA. As critical components of epigenetics, these chemical modifications significantly influence the regulation of gene expression at the posttranscriptional level by affecting the stability, localization, and splicing of RNA (51). Among all RNA species, transfer RNA (tRNA) is known to exhibit the highest degree of modifications and most abundant modification types, including N1-methyladenosine (m1A), pseudouridine (Ψ), and N7-methylguanosine (m7G), which are crucial to its various biological functions (52). Notably, m6A is among the known eukaryotic mRNA modifications. Whole-transcriptome localization analysis revealed that the distribution of m6A is relatively conserved and sequence-specific, and mainly enriched in the 3′ untranslated region (3′ UTR) and near the stop codon (143). The “writer” responsible for depositing the m6A modification is the multi-subunit methyltransferase complex, which consists of the key catalytic subunit METTL3, RNA-binding facilitator METTL14, and many other accessory subunits (144). The m6A modification is a dynamic and reversible process, and the active demethylation is mainly catalyzed by the “erasers”, fat-mass, and obesity-associated protein (FTO) (145) and ALKBH5 (146), which are located in the nucleus and affect mRNA metabolism. RNA-binding proteins, including YT521-B homology (YTH) domain family 1-3 (YTHDF1-3), and YTH domain-containing protein 1-2 (YTHDC1-2) are “readers” (147) that selectively recognize and bind m6A modifications in target mRNAs, thereby affecting the biological function of cells (148). YTHDF1 promotes the translation efficiency of mRNAs containing m6A modifications by interacting with the translation initiation factor eIF3 (149). In contrast, YTHDF2 binds to m6A modification in 3’UTR to promote mRNA degradation (150), while YTHDF3 can interact with YTHDF1 and YTHDF2 to facilitate their respective functions (151). Further RNA modification enzymes are expected to be identified and studied in the future, enabling the exploration of their potential functions.
These results demonstrated that m6A can regulate gene expression post-transcriptionally by influencing RNA metabolism, thereby participating in various biological processes and playing a unique role in human autoimmune diseases (152). Studies have revealed a significant decrease in ALKBH5 and FTO mRNA expression, whereas a marked increase in that of METTL3, the enzyme that activates the NF-κB signaling pathway, thus exacerbating the inflammatory response in patients with RA (153). Furthermore, m6A regulates the differentiation of naïve T-cells and influences their function. Mice with METTL3-knockout Tregs developed severe autoimmune disease after weaning, indicating that Tregs lose their immunosuppressive function due to the lack of the m6A modification (154). Given the essential function of m6A in various immune cells and autoimmune diseases, the relationship between other RNA modifications and autoimmune disease pathogenesis warrants further investigation in the future. These modifications could serve as new diagnostic markers of disease and facilitate the development of innovative approaches for the treatment of autoimmune diseases.
3.5. The impact of epigenetics on immune tolerance
The establishment and maintenance of immune tolerance require the precise regulation of multiple mechanisms that coordinately control the elimination or inactivation of self-reactive cells, thereby creating a robust barrier against pathological autoimmune responses. However, owing to the complexity of these mechanisms and heterogeneity of immune-related diseases, a large number of questions remain unanswered (155, 156). Extensive epigenetic studies have demonstrated that epigenetic regulation is indispensable for the establishment and maintenance of immune tolerance. For example, during the establishment of central immune tolerance, specific epigenetic modifications in AIRE are essential for regulating the active transcription of peripheral tissue genes (PTGs) in mTECs (92). Heinlein et al. demonstrated that the histone acetyltransferase KAT7 is a key protein for the AIRE-mediated transcription of PTGs in mTECs and induction of immune tolerance. The levels of H3K14ac, which is a marker of transcriptional activation, in the mTEC genome of KAT7-knockout mice were significantly decreased (157). Although the expression of AIRE itself was not affected, the accessibility of chromatin around the promoter of its target gene was reduced, leading to the downregulation of gene expression. During early development, Tregs with strong immunosuppressive functions form super enhancers (SEs), which are cis-regulatory elements with excellent transcriptional activation properties, characterized by highly dense transcriptional activity markers H3K27ac and H3K4me1, which play an essential role in promoting the expression of Treg marker genes (158).
These results indicate that epigenetic modifications can alter the levels of intracellular gene expression and transcription products that control the development, differentiation, and function of immune cells, thereby disrupting immune tolerance and aberrant clearance of self-reactive lymphocytes, causing pathological autoimmune responses, and ultimately driving the development of autoimmune diseases (159, 160). Enhancer of zeste homolog 2 (EZH2) is a histone methylation “writer” that can catalyze H3K27me3, a transcriptional repression marker, to inhibit the expression of target genes, thus playing a crucial role in immune tolerance and homeostasis (161). Researchers have found that CD4+ T-cells from patients with RA have low levels of EZH2, which downregulates the expression of RUNX1, a key transcription factor for Tregs, ultimately inhibiting Treg differentiation, disrupting peripheral immune tolerance, and exacerbating the inflammatory response in the joints of patients with RA (162). Hence, abnormal changes in the content or function of epigenetically modified enzymes can destroy immune tolerance by affecting the function of immune cells, thereby mediating a variety of autoimmune diseases. In addition, abnormal epigenetic modifications can affect central tolerance mechanisms and trigger autoimmune diseases. Studies have shown that mTEC development requires the histone deacetylase sirtuin 6, lack of which impairs DNA replication and proliferation in mTECs. Concurrently, sirtuin 6 loss results in abnormal expression of PTGs, inhibited thymocyte and nTreg development, causing thymus atrophy and destruction of central immune tolerance, and eventually inducing autoimmune diseases, which manifest as multiorgan lymphocyte infiltration and high autoantibody titers (163). Notably, these epigenetic modification enzymes may not alter the epigenetic modifications of the genome but may affect cell development and function through additional mechanisms, thus driving the occurrence and development of autoimmune diseases (164, 165). Future research should carefully explore these enzymes and understand their precise role in the regulation of cells and molecules, which may offer valuable insights into the development of novel treatments for autoimmune diseases.
3.6. Targeting epigenetic modifications of immune molecules
With the progress of research, the immune system has emerged as an excellent model for analyzing the regulatory mechanisms of epigenetics. The close connection between immune cell function and epigenetics undoubtedly makes this crosscutting area promising for exploration and development. The regulation of immune molecules by epigenetic modifications can elucidates these interactions in the subcellular level. The crossroads between epigenetic and immunopathways are important in inflammatory regulation, disease development, and therapeutic strategies ( Figure 4 ).
Figure 4.

The crossroads of epigenetics and immune pathways. Epigenetics plays an integral role in the regulation of immune pathways. In the TLR signaling pathway, histone methyltransferase SYMD5 catalyzes H4K20me3 to repress TLR4 expression; histone demethylase PHF2 activates TLR4 expression by removal of H3K9me1. Knockdown of JNK will inhibit m6A and METTL3 expression; In the sGAS-STING signaling pathway, DNA demethylases TET2 elevate cGAS levels, histone demethylases KDM5B and KDM5C inhibit STING expression. Histone demethylase Jmjd3 upregulates NF-kB-mediated inflammatory cytokine levels by removal of H3K27me3; In the JAK-STAT signaling pathway, activator of transcription STAT recruits histone acetyltransferases and chromatin-remodeling enzymes, and histone methyltransferase EZH2 and histone acetyltransferase p300 elevate STAT levels. IFN-γ produced by this signaling pathway can induce H3K27me3, which is associated with gene repression; In the PD-1 and PD-L1 pathway, knockdown of METTL3 abolishes m6A modification and reduces stabilization of PD-L1 mRNA, and m6A reader IGF2BP and YTHDF regulates RNA stability and expression levels of PD-L1. DNA methyltransferase inhibitor 5-aza-2’ deoxycytidine (decitabine) enhances PD-1 expression. PAMP, pathogen associated molecular pattern; DAMP, damage associated molecular pattern; TLR, Toll-like receptors; IRAK, interleukin-1 receptor-associated kinase; TRAF6, Tumor necrosis factor receptor-associated factor 6; IRF3, Interferon regulatory factor 3; ISGs, Interferon-stimulated genes.
The cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) signaling pathway regulates the immune response by promoting the secretion of type I IFN (166). Several studies have shown that chronic and sustained aberrant activation of this pathway is closely related to autoimmune disease pathogenesis (167). Clinical studies have revealed that activated peripheral blood mononuclear cells (PBMCs) of patients with SLE have a higher expression of cGAS, leading to increased serum levels of type I IFN and mediating the inflammatory response and damage to the body (168). This pathway is regulated through various epigenetic mechanisms, with high methylation levels detected in the promoter region of cGAS and STING in many different types of tumor cells. STING expression is inhibited by the histone H3K4 lysine demethylases KDM5B and KDM5C. This epigenetic silencing mechanism prevents the activation of the pathway and inhibits the immune activity of antitumor T-cells, allowing cancer cells to escape immune surveillance (169). In addition, DNA demethylase, TET2, modulates anti-tumor immunity by elevating cGAS levels in tumor cells (170).
Programmed death 1 (PD-1) and programmed death ligand 1 (PD-L1) play critical roles in regulating the immune response and T-cell activation state, and modulate central and peripheral tolerance by transducing immunosuppressive signals (171). Abnormal expression of these genes may lead to the abnormal activation of autoimmune T-cells and the development of several autoimmune diseases (172). Downregulation of PD-1 on Tregs in SLE patients results in reduced cell numbers and impaired immunosuppression (173). Moreover, synovial T-cells and macrophages in patients with RA were demonstrated to have significantly higher levels of PD-1/PD-L1 expression than those in healthy controls, with proinflammatory cytokines such as IFN-γ and TNF-α contributing to the upregulation of these costimulatory molecules to some extent (174). Studies have shown that PD-L1 expression is regulated by specific epigenetic enzymes, such as METTL3, the “writer” of m6A, which increases PD-L1 mRNA expression, promoting bladder cancer immune escape (175).
In addition, despite the regulation of a large number of immune molecules by epigenetic modifications, the complex mechanisms governing these interactions remain unclear. Given the critical role of epigenetic modifications in the regulation of numerous signaling pathways, studies delineating the effect of these modifications on immune regulation may open new avenues for improved immunotherapy.
4. Epigenetic biomarkers and therapeutic approaches in autoimmune diseases
4.1. Epigenetic biomarkers in autoimmune diseases
Epigenetics is increasingly recognized as a crucial factor in autoimmune disease pathogenesis, and researchers have identified a variety of epigenetic modifications as promising candidates for autoimmune disease biomarkers, which could help guide clinical diagnosis, treatment, and prognosis (176). Decades of research has demonstrated that compared with healthy individuals, patients with autoimmune diseases exhibit significant epigenetic changes in their genomes, which are strongly associated with disease phenotype and activity. Detecting changes in DNA methylation or histone modification is possible during the early stages of certain diseases, thus providing a basis for the early prediction, diagnosis, and prognosis of diseases ( Table 3 ) (202).
Table 3.
Epigenetics as a biomarker for autoimmune diseases.
| Epigenetics | Disease | Biomarkers | Clinical significance | Reference |
|---|---|---|---|---|
| DNA methylation | Systemic lupus erythematosus (SLE) | Hypomethylation within CD40-ligand promotor and enhancer regions | hypomethylation is associated with disease activity in female SLE patients | (177) |
| Impaired DNA methylation in CD4+T and B cells | The percentage of 5mC inversely correlates with the disease activity of lupus patients | (178, 179) | ||
| hypermethylation at Interferon-induced protein 44-like (IFI44L) gene in CD4+T cells | Overexpression of IFI44L can amplify IFN signaling and enhance the immune response | (180) | ||
| The level of DNMT1 mRNA expression in CD4+ T cells from SLE patients were significantly decreased | UVB enhanced DNA hypomethylation via inhibiting DNMT1 catalytic activity, causing the patient’s condition to deteriorate | (181) | ||
| Rheumatoid Arthritis (RA) | T cells exhibit overall DNA hypomethylation; naïve T cells exhibit increased methylation levels, which diminish during differentiation into effector/memory T cells | Altered DNA methylation may facilitate the accelerated degradation and premature senescence of naïve and memory CD4+ T cells, causing abnormal T cell activation and senescence | (182, 183) | |
| Hypermethylation at CD1C and hypomethylation at TNFSF10 genes | TNFSF10 overexpression causes bone and joint damage | (184) | ||
| Decreased levels of DNMT1 in synovial fibroblasts (SFs), leading to an overall decrease in DNA methylation | SFs became an aggressive phenotype, producing pro-inflammatory cytokines and making the disease worse | (185) | ||
| Primary Sjögren’s syndrome (pSS) | Significant hypomethylation of interferon (IFN)-regulated genes in whole blood and CD19+ B cells | Hypomethylation of IFN-regulated genes increased their expression, activating the IFN signaling pathway and mediating autoimmune responses | (186) | |
| Reduced DNA methylation levels in minor salivary gland (MSG) epithelial cells | Hypomethylation are associated with lymphocyte infiltration or anti-SSB/La antibody production | (187) | ||
| Systemic sclerosis (SSc) | CD4+ T cells in female SSc patients exhibit hypomethylation and overexpression of CD40L | The interaction of CD40L with CD40 stimulates B cells and fibroblasts, promoting autoantibody production and tissue fibrosis | (188) | |
| The methylation levels of the CD11a regulatory sequences were lower, leading to increased expression levels in CD4+ T cells | CD11a levels were found to be positively correlated with disease activity, stimulating B cells to produce IgG | (189) | ||
| Histone modification | SLE | Global histone H3 and H4 hypoacetylation in active lupus CD4+ T cells | the level of H3 acetylation is negatively correlated with the disease activity | (190) |
| Histone methylase EZH2 and H3K27me3 levels were increased in CD4+ T cells in lupus | Overexpression of EZH2 resulted in increased CD4+ T cell adhesion | (191) | ||
| H3K27me3 levels in the Transcription factor B cell lymphoma 6 (BCL6) promoter region were decreased | Decreased H3K27me3 levels promoting regulator of T follicular helper (Tfh) cells differentiation | (192) | ||
| SSc | Global histone H4 hyperacetylation and global histone H3K9 hypomethylation B cells | Global H4 acetylation was positively correlated with disease activity | (193) | |
| RNA methylation | SLE | mRNA levels of METTL3, FTO, ALKBH5, and YTHDF2 in peripheral blood from SLE patients were significantly decreased | The levels of ALKBH5 mRNA were associated with anti-dsDNA, antinucleosome, rash, and ulceration | (194) |
| mRNA expression of MTEEL14, ALKBH5 and YTHDF2 was reduced in SLE patients | Decreased YTHDF2 was associated with disease activity | (195) | ||
| m5C levels were decreased in CD4+ T cells of patients with SLE | Lower m5C levels are associated with severe disease activity | (196) | ||
| RNA methylase NSUN2 was markedly decreased in CD4 + T cells from SLE patients | NSUN2 catalyzes the modification of IL-17A mRNA, which is significantly implicated in the pathogenesis of SLE | (197) | ||
| RA | The levels of METTL14 and m6A are decreased in the PBMCs of patients with active RA | m6A are negatively correlated with the DAS28 score and IL-6 and IL-17 levels | (198) | |
| the expression of METTL3 in macrophages was significantly increased | METTL3 level was positively correlated with the RA activity marker | (199) | ||
| pSS | The mRNA levels of m6A writers (METTL3 and RBM15), erasers (ALKBH5 and FTO), and readers (YTHDF1, YTHDF2, YTHDF3, YTHDC1, and YTHDC2) were all significantly higher in PBMCs from pSS patients | The mRNA level of YTHDF1 in PBMCs was negatively correlated with the EULAR Sjögren’s syndrome disease activity index (ESSDAI) score. Increased mRNA level of ALKBH5 in PBMCs was a risk factor for pSS | (200) | |
| Ankylosing spondylitis (AS) | m6A levels and expression of METTL14, WTAP and ALKBH5 were significantly reduced in T cells | Reduced METTL14 levels inhibited the transcription of autophagy-related genes, increasing levels of IL-17A, IL-23, and TNF-α | (201) |
The DNA methylation profile of various cells is significantly altered in patients with SLE; for example, CD4+ T-cells and B-cells usually exhibit global hypomethylation, particularly in immune-related genes. Therefore, the DNA methylation of specific genes may serve as a potential biomarker for SLE (178). In addition, interferon-induced protein 44-like (IFI44L), an interferon-regulated gene that can amplify IFN signaling and enhance the immune response is hypomethylated in CD4+ T-cells from patients with SLE; thus, the DNA methylation of the IFI44L promoter can serve as a potential blood biomarker for diagnosing SLE with high sensitivity and specificity (180). In addition to immune cells, DNA methylation alterations can occur in cells with typical significance in autoimmune diseases. For instance, the decreased levels of DNMT1 in synovial fibroblasts (SFs) from patients with RA can lead to an overall decrease in DNA methylation, which can induce an aggressive SF phenotype that produces proinflammatory cytokines, such as IL-6 and TNF, thereby promoting the development of RA (185). Similarly, reduced DNA methylation levels were observed in minor salivary gland (MSG) epithelial cells of patients with Sjögren’s syndrome (SS), and these changes were associated with lymphocyte infiltration and anti-SSB/La antibody production (187). To facilitate the detection of epigenetic modification changes in the early stages of an autoimmune disease or even before its onset, developing more efficient detection methods is required.
Abnormal alterations in histone modifications have been observed in the cells of patients with autoimmune diseases, and given these modifications are strongly associated with the onset and progression of the disease, they may serve as potential biomarkers for assessing disease risk and aiding in diagnosis (62). Studies have shown that the abnormal expression of several histone acetylases and deacetylases leads to the overall hypoacetylation of H3 and H4 in CD4+ T-cells from patients with SLE, with a negative correlation observed between the degree of H3 acetylation and disease activity (190). However, in patients with active lupus, hyperacetylation has been detected at specific genes, such as markedly elevated levels of H3 acetylation and an increase in H3K4me2 at the CD70 gene, both of which promote its transcriptional activation, strengthening the immune response by promoting the activation and proliferation of T- and B-cells (203). In addition, H3K9 hypomethylation was detected in the B-cells of patients with SSc and CD4+ T-cells from patients with SLE; however, patients with SSc exhibited global histone H4 hyperacetylation, the level of which was positively linked to disease activity (193). Hence, epigenetic modifications appear to be as heterogeneous as autoimmune diseases, different diseases and cell types characterized by different types of modifications; therefore, the relationship between epigenetic modifications and autoimmune diseases requires further detailed exploration.
The importance of RNA modifications in the pathogenesis of autoimmune diseases has been gradually elucidated with the development of advanced sequencing technologies (152). Following the pharmacological inhibition of METTL3 in lupus model mice, the m6A modification on the transcription factor Foxp3 mRNA was reduced, in turn facilitating RNA degradation, and resulting in impaired differentiation and immunosuppressive function of Treg cells as FOXP3 is indispensable for the development and function of Treg cells, thus exacerbating kidney injury in mice (204). Moreover, the expression of METTL3 in the macrophages of patients with RA was significantly increased, and was positively correlated with RA activity markers, such as C-reactive protein (CRP), suggesting the correlation of m6A with RA disease activity to some extent (199). These results showed that m6A and its modifying enzymes play crucial roles in immune cell differentiation and development and have the potential to drive autoimmune diseases, thus supporting their potential as biomarkers and therapeutic targets for managing this disease. Although research on RNA modifications is being rapidly conducted, developing more efficient and accurate sequencing technologies for comprehensively exploring other types of RNA modifications and their modifying enzymes is urgently needed to provide strong evidence for the development of new therapies.
4.2. Epigenetic therapeutic approaches in autoimmune diseases
With the in-depth study of epigenetic mechanisms, a new understanding of abnormal epigenetic changes in autoimmune diseases has been obtained, fueling an array of studies aimed at identifying clinical markers for early disease diagnosis and prognosis. Accordingly, the reversibility of epigenetic modifications, which holds great therapeutic potential, has led to the emergence of epigenetic therapy as a novel therapeutic approach for autoimmune diseases and the development of new drugs that may provide great benefits to patients ( Table 4 ) (228).
Table 4.
Epidrugs used in autoimmune disease.
| Drug | Disease | Cell type | Effects | Improvement of symptoms | References | |
|---|---|---|---|---|---|---|
| HDAC inhibitor | Trichostatin A | RA | PBMC | Inhibited production of TNF, IL-6 and IFN-γ | (205) | |
| SLE | T cell | Downregulated CD154 (CD40L) and IL-10, upregulated IFN-γ | (206) | |||
| SLE | MRL-lpr/lpr mice splenocytes | Downregulated mRNA and protein levels of IL-12, IL-6, IL-10 and IFN-γ | Reduced proteinuria, glomerulonephritis, and spleen weight | (207) | ||
| SLE | NZB/W mice T cell | Decreased production of IL-6 and increased TGF-β1 and Foxp3; decrease expression of renal MCP-1, MMP-9, and IL-6 mRNA | Decreased anti-dsDNA autoantibodies, protein excretion, renal IgG and C3 deposition, and pathologic glomerular disease | (208) | ||
| T1D | T cell | Upregulated IFN-γ and its transcription factor Tbx21 | Reduced the incidence of diabetes | (209) | ||
| SSc | Fibroblast | Increased FLI1 and reduced type I collagen | (210) | |||
| SSc | Fibroblast | Reduced production of type I and type III collagen | (211) | |||
| MI192 (selective HDAC3 inhibitor) | RA | PBMC | Inhibited the production of TNF at high concentrations, and inhibited IL-6 dose-dependently | (205) | ||
| ACY-738 (selective HDAC6 inhibitor) | SLE | T cell, B cell | Decreased expression of glomerular IL-10, TGF-β, and IL-6 mRNA; reduced levels of serum anti-dsDNA autoantibodies and IgG isotype | Inhibited immune complex-mediated glomerulonephritis and decreased SLE renal pathology | (212) | |
| Givinostat (ITF2357) | RA | FLS, macrophage | Suppressed FLS IL-6 production and reduced the stability of IL-6 mRNA in FLS and macrophages | (213) | ||
| T1D | NOD mice T cell, DC, pancreatic β cells | Increased Treg cell subsets and their transcription factors Gata3 and FoxP3; decreased inflammatory DC subsets and their cytokines IL-6, IL-12, and TNF-α | Reduced diabetes incidence, increased insulin content | (214) | ||
| SLE | NZB/W mice T cell | Reduced levels of serum anti-dsDNA autoantibodies and IgG isotype; decreased glomerular IL-6 mRNA dose-dependently | Decreased renal disease and glomerular immune complex deposition; increased the number of regulatory T cells and inhibited Th17 differentiation | (215) | ||
| Vorinostat (SAHA) | MS | CD14+ monocyte-derived DC | Reduced CD80 and CD86, the maturation marker CD83, and HLA-DR; inhibited production of TNF-α, IFN-γ, IL-12p35, IL-6 and IL-23p40 | Reduced CNS inflammation and demyelination | (216) | |
| RA | CIA mice T cell | reduced expression of NR1D1, IL-17 mRNA and protein | Decreased Th17 cell counts | (217) | ||
| RA | FLS | Increased intracellular ROS levels, and expression and activity of caspase-3; reduced anti-apoptotic Bcl-2 proteins (Bcl-xL and Mcl-1), and phosphorylated IκBα and NF-κB p65 | Increased FLS apoptosis | (218) | ||
| T1D | NOD mice T cell, pancreatic β cells | Reduced levels of IL-1β and IFN-γ-induced proinflammatory, and cytokine-induced ERK phosphorylation | Reduced diabetes incidence, increased insulin content | (214) | ||
| SLE | MRL-lpr/lpr mice splenocytes | Downregulated mRNA and protein levels of IL-12, IL-6, IL-10 and IFN-γ | Reduced proteinuria, glomerulonephritis, and spleen weight | (207) | ||
| SLE | PBMC | Ameliorated DNA repair efficiency and decreased apoptosis | (219) | |||
| SLE | MRL/lpr mice mesangial cell | Inhibited expression of TNF-α, IL-6, NO, and inducible NO synthase | Decreased spleen size and CD4-CD8- (double-negative) T cells; inhibited proteinuria and pathologic renal disease | (220) | ||
| SLE | anti-CD3 activated T cell | Augmented apoptosis at high concentrations, inhibited proliferation of anti-CD3 activated T cells and reduced IL-6, IFN-γ, TNF-α and IL-2 at low concentrations | Ameliorated cytokine storm syndrome and avoided GVHD; combination of SAHA and anti-CD3 cured severe lupus glomerulonephritis | (221) | ||
| THS-78-5 | T1D | INS cell | Prevented IL‐1β‐mediated iNOS expression and NO release | Prevented IL-1β-induced metabolic dysfunction in pancreatic β cells | (222) | |
| DNMT inhibitor | 5’-aza-2’-deoxycytidine | MS | Treg cell | Elevated Foxp3 expression in vivo and in vitro; decreased proinflammatory cytokines | Ameliorated CNS inflammatory responses | (223) |
| SSc | Fibroblast | Increased FLI1 and reduced type I collagen | (210) | |||
| SSc | Fibroblast | Reactivated of DKK1 and SFRP1 transcription and reduced Wnt signaling | (224) | |||
| RA | FLS | Upregulated expression of PTEN, decreased chemokines CCL-2, CCL-3, CCL-8, and proinflammatory cytokines IL-1β, IL-6, IL-17A | Decreased inflammatory cell infiltration into the synovium and reduced in paw swelling | (225) | ||
| 5’-azacytidine | RA | Mice B cell | Repressed Aicda gene expression, reduced CSR and impaired GC formation | Inhibited production of IgG1 antibodies and attenuated joint destruction | (226) | |
| SLE | MRL/lpr mice CD4 and CD8 T cell | DNA demethylation in CD4+T cells enhanced Foxp3 expression, and increased CD8+T cells cytotoxic activity | Ameliorated facial rash and skin lesions, reduced proteinuria | (227) | ||
HDAC, histone deacetylase; PBMC, peripheral blood mononuclear cells; DNMT, DNA methyltransferase; SAHA, suberoylanilide hydroxamic acid; CD, cluster designation; DC, dendritic cells; IL, interleukin; TNF, tumor necrosis factor; FLS, fibroblast-like synoviocytes; NOD, nonobese diabetic; IFN-γ, interferon-γ; dsDNA, double stranded DNA; GVHD, graft versus host disease; EAE, experimental autoimmune encephalomyelitis; NF-κB, nuclear factor kappa B; CIA, collagen-induced arthritis; NR1D1, nuclear receptor subfamily 1 group D member 1.
HDAC inhibitors exhibit strong immunomodulatory functions in immune cells and have been widely studied as an epigenetic therapy for autoimmune diseases. One month after lupus model mice were treated with a selective HDAC6 inhibitor, lupus nephritis (LN) symptoms were significantly alleviated, and the deposition of IgG and C3 in glomeruli was significantly decreased. Furthermore, HDAC6 inhibitors have been shown to suppress various signaling pathways associated with B-cell activation and differentiation, thereby inhibiting the formation of plasma cells and germinal center (GC) response in lupus model mice (176). Another study demonstrated that belinostat, a pan-HDAC inhibitor, can suppress neuroinflammation by inhibiting M1 microglial activation and proinflammatory cytokine expression, thereby improving symptoms in experimental autoimmune encephalomyelitis (EAE) mice, thus indicating belinostat as a potential candidate for multiple sclerosis (MS) treatment (229).
In another example, the DNA methyltransferase inhibitor 5’-azacytidine (5’-azaC) has shown better efficacy in animal models of certain autoimmune diseases. 5’-azaC can mediate the demethylation of the Ahr gene, leading to the repression of Aicda gene expression, which is required for GC formation. Ultimately, mice with proteoglycan-induced arthritis were shown to produce fewer IgG1 antibodies, resulting in improved autoimmune arthritis (226). Targeted delivery of 5’-azaC to CD4+ or CD8+ T-cells in MRL/lpr mice resulted in lower levels of multiple inflammatory factors and significant amelioration of autoimmunity. DNA demethylation in CD4+ T-cells was suggested to increase Foxp3 expression and induce immunosuppressive properties in these cells (227). Peripheral application of the DNA methyltransferase inhibitor 5’-aza-2’-deoxycytidine to EAE mice suppressed central nervous system inflammation and reduced IFN-γ and IL-17 levels by increasing the number of Treg (223). However, owing to the high heterogeneity of autoimmune diseases and complexity of epigenetic mechanisms involved, some studies have shown that the DNMT inhibitor 5’-aza-2’-deoxycytidine can promote the expression of Foxp3 in Treg cells to a certain extent, thus strengthening its immunosuppressive function; however, the continuous use of inhibitors can lead to a decrease in DNMT1 activity, which will instead disrupt the function of Tregs and induce fatal autoimmunity in mice (230).
In conclusion, epigenetic therapies are an emerging and promising therapeutic approach; however, many limitations and challenges exist regarding the application of these therapies for treating autoimmune diseases; hence they remain largely in an exploratory phase. Numerous clinical trials evaluating epigenetic drugs have been conducted in the field of oncology. However, trials targeting autoimmune diseases remain sparse ( Table 5 ). Further in-depth research on epigenetic mechanisms is warranted to develop highly specific targeted epigenetic drugs. The combination of epigenetic therapy and traditional immunotherapy will provide valuable insights into the treatment of autoimmune diseases.
Table 5.
Summary of clinical trials of epigenetic modulators and immunotherapeutic agents.
| Epigenetics | Mechanism | Agents | Chemical structures | Immune-therapeutic agent | Disease type | Trail |
|---|---|---|---|---|---|---|
| Histone acetylation | HDAC inhibitor | Domatinostat (4SC-202) |
|
Avelumab | GastrointEstinal Cancers | NCT03812796 |
| Advanced Merkel Cell Carcinoma | NCT04393753 | |||||
| Metastatic Merkel Cell Carcinoma | NCT04874831 | |||||
| Nivolumab + Ipilimumab | Primary Melanoma | NCT04133948 | ||||
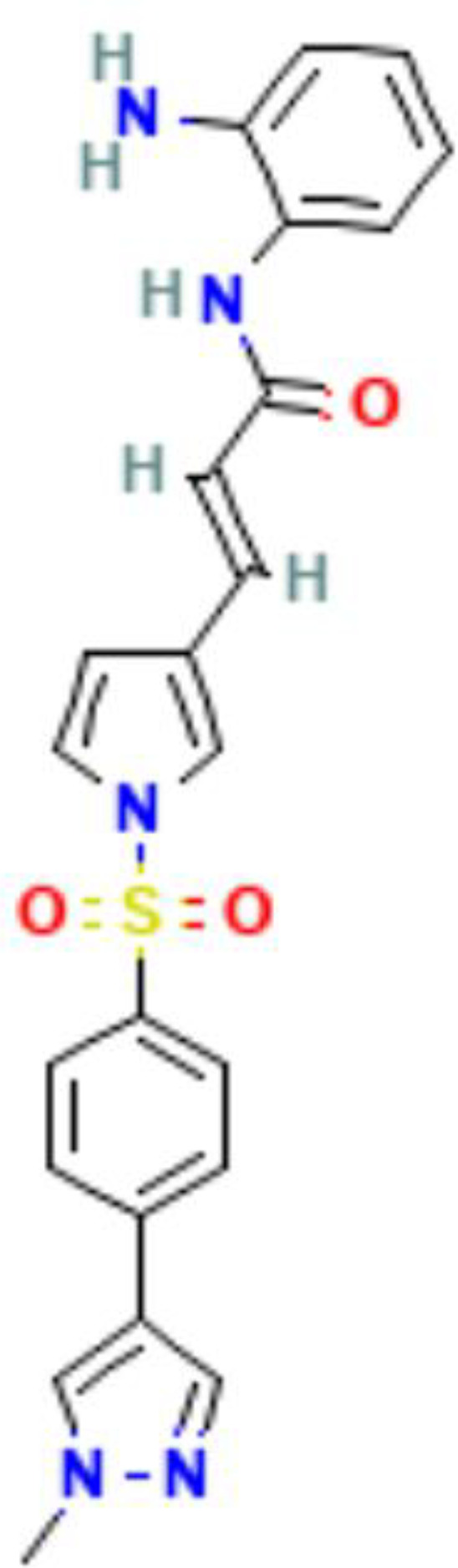
| Entinostat |
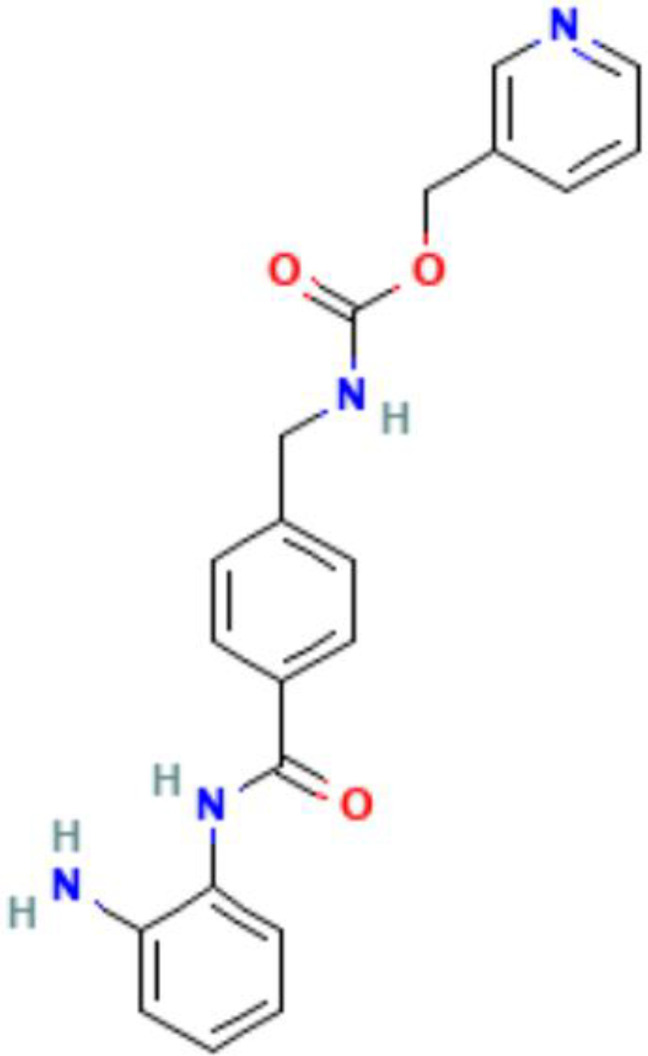
|
Advanced Prostate Cancer | NCT03829930 | |||
| Breast Cancer | NCT02820961 | |||||
| Ipilimumab/ Nivolumab | HER2-Negative Breast Cancer | NCT02453620 | ||||
| Erlotinib | Breast Cancer or NSCLC | NCT01594398 | ||||
| Nivolumab | Colorectal Cancer | NCT03993626 | ||||
| Nivolumab | Pancreatic | NCT03250273 | ||||
| Avelumab | Advanced Epithelial Ovarian Cancer | NCT02915523 | ||||
| Aldesleukin | Metastatic Kidney Cancer | NCT01038778 | ||||
| Pembrolizumab | Bladder Cancer | NCT03978624 | ||||
| Lymphoma | NCT03179930 | |||||
| Melanoma | NCT03765229 | |||||
| Vorinostat |
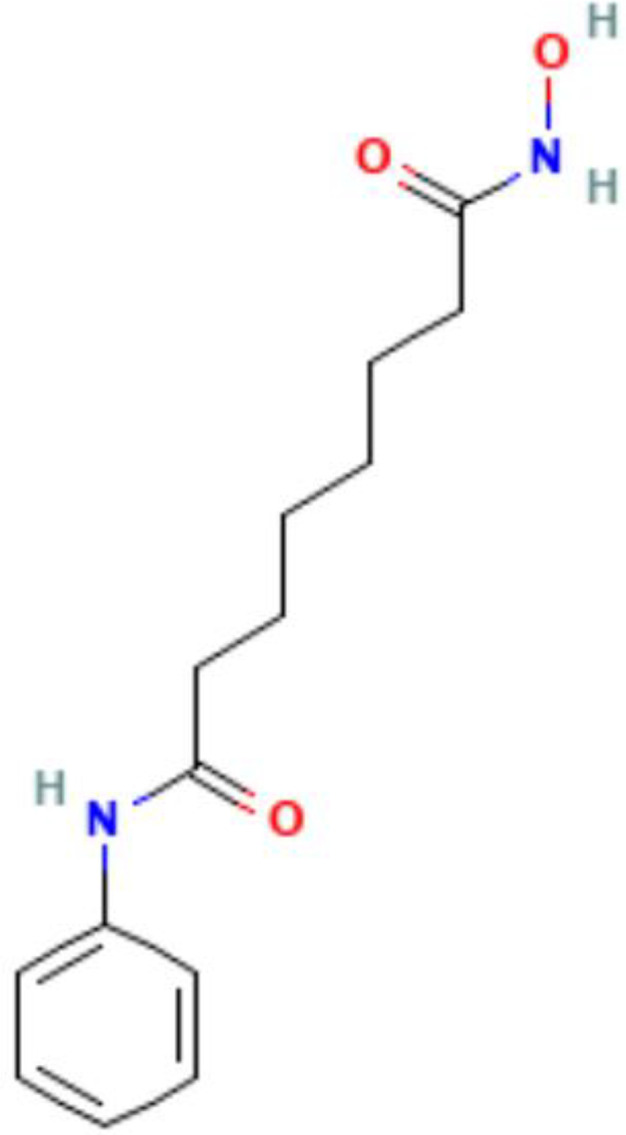
|
Advanced Ovarian Carcinoma | NCT00976183 | |||
| Olaparib | Breast Cancer | NCT03742245 | ||||
| Pembrolizumab | NSCLC | NCT02638090 | ||||
| Givinostat (ITF2357) |
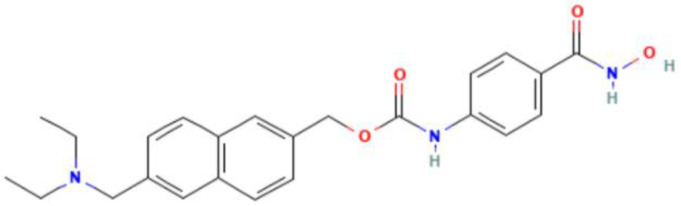
|
Systemic Juvenile Idiopathic Arthritis | NCT00570661 | |||
| Hodgkin's Lymphoma | NCT00496431 | |||||
| Mocetinostat (MGCD0103) |
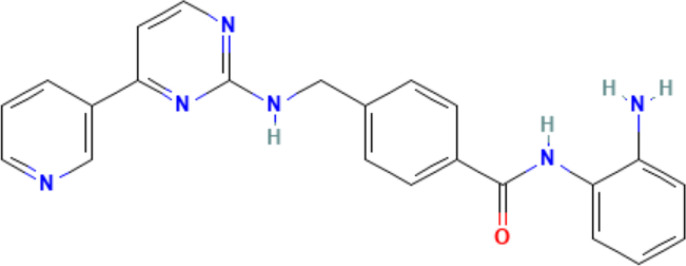
|
Diffuse Large B-Cell Lymphoma and Follicular Lymphoma | NCT02282358 | |||
| Rhabdomyo-sarcoma | NCT04299113 | |||||
| Pembrolizumab | Advanced Lung Cancer | NCT03220477 | ||||
| Brentuximab vedotin | Hodgkin Lymphoma | NCT02429375 | ||||
| Durvalumab | Advanced Solid Tumors and NSCLC | NCT02805660 | ||||
| Durvalumab | Squamous Cell Carcinoma of the Oral Cavity | NCT02993991 | ||||
| Tucidinostat (Chidamide) |
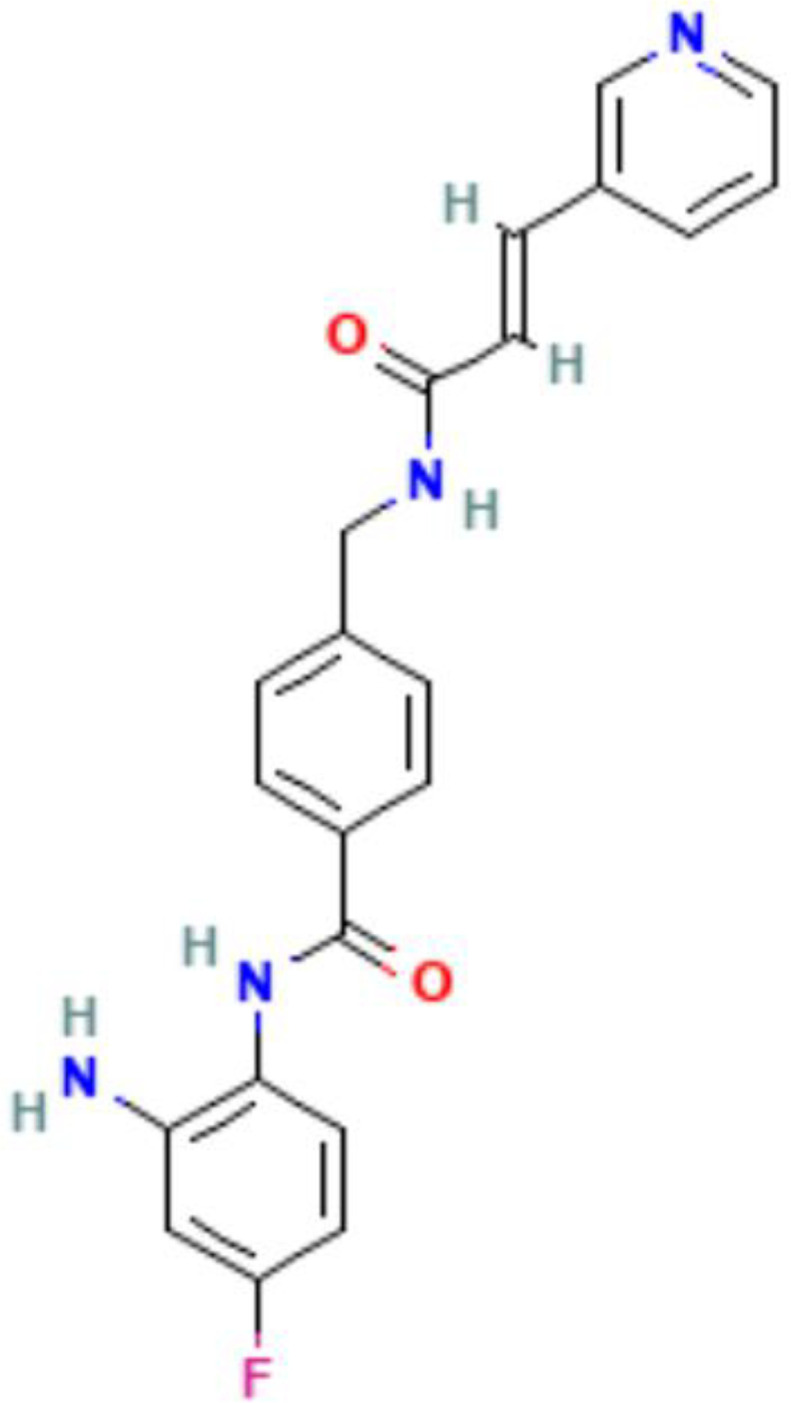
|
Breast Cancer | NCT05276713 | |||
| Sintilimab | Neuroendocrine Neoplasm | NCT05113355 | ||||
| Apatinib | Advanced Osteosarcoma | NCT06125171 | ||||
| Zimberelimab | Metastatic Triple-negative Breast Cancer | NCT05632848 | ||||
| Toripalimab + Bevacizumab | Esophagus Cancer, Gastric Cancer | NCT05163483 | ||||
| Toripalimab | Advanced Cervical Cancer | NCT04651127 | ||||
| Tislelizumab | Advanced NSCLC | NCT05519865 | ||||
| Belinostat (PXD101) |
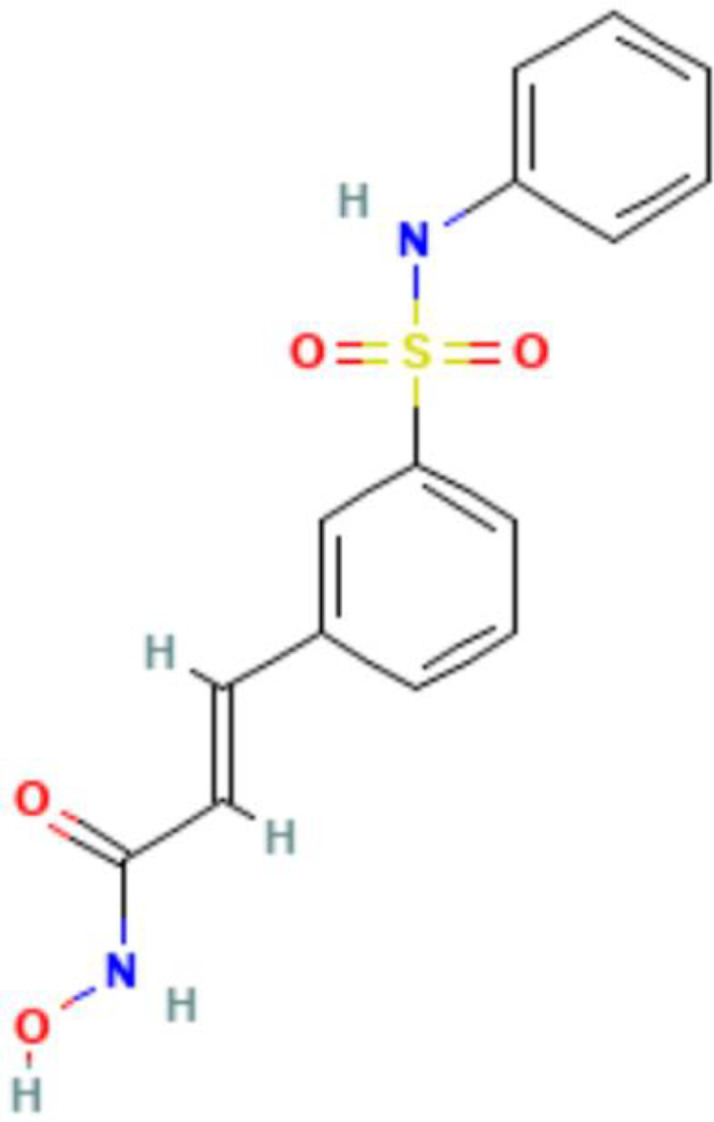
|
Ovarian Cancer | NCT00421889 | |||
| Histone methylation | EZH2 inhibitor | Tazemetostat |
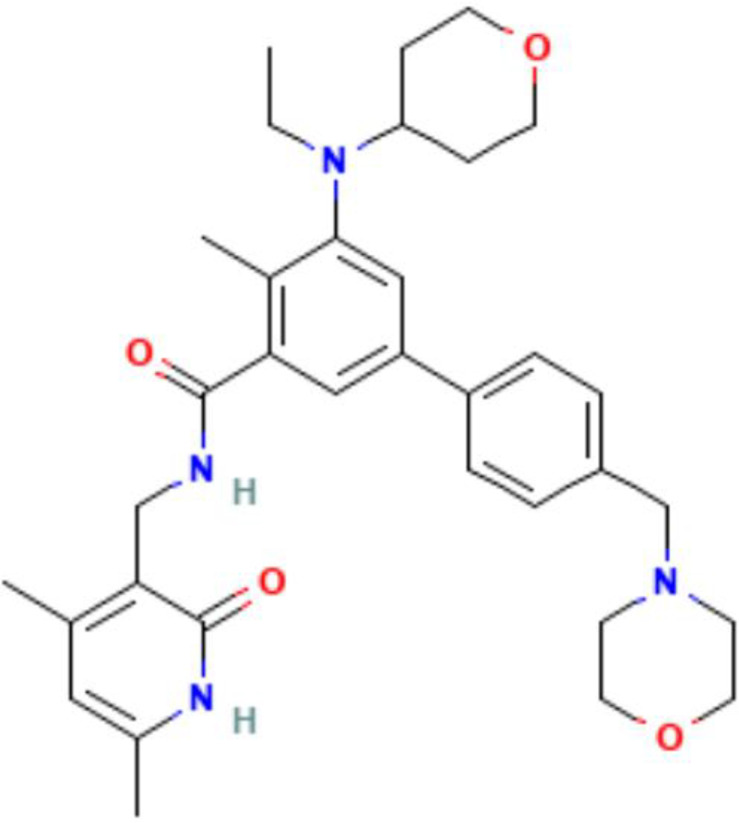
|
Malignant Peripheral Nerve Sheath Tumors | NCT04917042 | |
| NSCLC | NCT05467748 | |||||
| Malignant Mesothelioma | NCT02860286 | |||||
| B-cell Non-Hodgkin's Lymphoma | NCT03009344 | |||||
| Recurrent Ovarian or Endometrial Cancer | NCT03348631 | |||||
| DNA methylation | DNMT inhibitor | Guadecitabine (SGI-110) |
|
Ipilimumab | Unresectable or Metastatic Melanoma | NCT02608437 |
| Pembrolizumab | Refractory Solid Tumours | NCT02998567 | ||||
| Ovarian, Primary Peritoneal, or Fallopian Tube Cancer | NCT02901899 | |||||
| Advanced Lung Cancer | NCT03220477 | |||||
| Atezolizumab | Refractory or Resistant Urothelial Carcinoma | NCT03179943 | ||||
| Ipilimumab + Nivolumab | Melanoma and NSCLC | NCT04250246 | ||||
| Decitabine |
|
ITP | NCT01568333 | |||
| Pembrolizumab | MDS or AML | NCT03969446 | ||||
| Pembrolizumab | NSCLC and Esophageal Carcinomas | NCT03233724 | ||||
| DEC-205/NY-ESO-1 Fusion Protein CDX-1401 and Poly ICLC | MDS or AML | NCT01834248 | ||||
| TQB2450 Injection (PD-L1 Monoclonal Antibody) | Digestive System Tumors | NCT04611711 | ||||
| Sintilimab | NK/T-cell Lymphoma | NCT04279379 | ||||
| Azacitidine |
|
Systemic Auto-immune and Inflammatory Disorders | NCT02985190 | |||
| Pembrolizumab | Ovarian cancer | NCT02900560 | ||||
| Pembrolizumab | AML | NCT02845297 | ||||
| Pembrolizumab | NCT03769532 | |||||
| Nivolumab | NCT03825367 | |||||
| Nivolumab + Relatlimab | NCT04913922 | |||||
| Ipilimumab/ Nivolumab | NCT02397720 | |||||
| Pembrolizumab | Pancreatic Cancer | NCT03264404 | ||||
| Avelumab + Utomilumab + Rituximab |
Diffuse Large B-cell Lymphoma | NCT02951156 |
AML, Acute Myeloid Leukemia; HDAC, histone deacetylase; DNMT, DNA methyltransferase; MDS, Myelodysplastic Syndrome; NSCLC, Non-small cell lung cancer; ITP, Immune Thrombocytopenia.
Images of chemical structure from PubChem website.
5. Discussion
Although advances in detection technology have provided a profound understanding that autoimmune diseases may indeed be influenced by genetic and environmental factors to some extent, a growing body of research has suggested that epigenetics also play a nonnegligible role. The theory of epigenetics has provided a new perspective for decoding the secrets of autoimmune diseases and has facilitated a substantial progress in exploring the mechanisms and characteristics of these diseases providing novel biomarkers and potential treatments (65). Epigenetic modifications are altered in patients with autoimmune diseases through a variety of mechanisms due to immune system disorders. Using high-throughput techniques to characterize these and the changes in the content or function of modified enzymes is beneficial for accurately identifying targets that affect the pathogenesis and progression of autoimmune diseases (231).
Despite significant advancements in the field of epigenetics, several crucial issues remain unresolved. For example, both autoimmune diseases and epigenetic modifications demonstrate considerable heterogeneity. Different diseases have varying manifestations, and even the same disease can present distinct clinical manifestations at different stages; consequently, this complexity poses significant difficulties in developing effective treatment strategies (232). Given the dynamic and reversible nature of epigenetic modifications that involve interactions with various enzymes and molecules, a promising approach for treating autoimmune diseases involves targeting and modulating these aberrant modifications. Various epigenetic drugs, such as DNMT and HDAC inhibitors, have been extensively used in cancer treatment, showing good efficacy (233). Autoimmune diseases and cancer share many similarities in their etiology, and immune-related treatments are widely used for both; therefore, extensive research has been focused on the use of epigenetic drugs for treating autoimmune diseases, as these drugs are highly targeted and may have fewer side effects than traditional immunosuppressants. Moreover, the development of more efficient combination therapies based on the epigenetic theory is an exciting prospect, which will bring great benefits to patients with autoimmune diseases (234). However, clinical trials using epigenetic drugs to treat autoimmune diseases are relatively rare, and their efficacy is unclear. As epigenetic drugs may exert genome-wide effects, specifically targeting a gene without causing systemic effects is a considerable challenge. Due to the extensive heterogeneity of diseases and complexity of the immune system, various significant challenges remain in the development and production of more targeted and high-affinity therapeutic drugs. Further research and trials are needed to generate convincing evidence to further improve epigenetic therapies.
In summary, this review described several epigenetic mechanisms, highlighting the close relationship and interactions between epigenetics and autoimmune diseases, and providing new insights into the use of epigenetic modifications and their modifying enzymes as biomarkers and therapeutic targets for treating autoimmune diseases. Although not discussed in this article, the role of ncRNAs in autoimmune diseases has been well described (235). Given the current heavy burden on patients with autoimmune diseases worldwide, more in-depth collaborative research is urgently required in the fields of epigenetics and autoimmune diseases, which could provide more efficient and personalized therapies for patients with autoimmune diseases.
Acknowledgments
All the authors acknowledge and thank their respective Institutes and universities.
Glossary
- A
adenine
- AIRE
Autoimmune regulator
- ATP
Adenosine triphosphate
- ALKBH
AlkB homologue
- APS-1
Autoimmune polyendocrine syndrome type 1
- BCR
B cell receptor
- BS
bisulfite sequencing
- C
cytosine
- cDNA
complementary DNA
- CpG
cytosineguanine dinucleotides
- CHD
chromodomain helicase DNA-binding
- ChIP
chromatin immunoprecipitation
- ChIP-seq
Chromatin immunoprecipitation-sequencing
- CRP
C-reactive protein
- DNMT
DNA methyltransferase
- DMF
Dimethyl fumarate
- DOT1L
telomeric silencing 1-like
- EAE
experimental autoimmune encephalomyelitis
- EBV
Epstein-Barr virus
- EZH2
Enhancer of zeste homolog 2
- FLSs
fibroblast-like synoviocytes
- Foxp3
forkhead box P3
- FTO
fat-mass and obesity-associated protein
- G
guanine
- GC
germinal center
- GWAS
Genome-wide association studies
- HATs
histone acetyltransferases
- HDACs
histone deacetylases
- HDM
histone demethylase
- HMT
histone methyltransferase
- HNRNP
heterogeneous nuclear ribonucleoprotein
- H3K27ac
acetylated lysine 27 of histone 3
- H3K27me3
trimethylated lysine 27 of histone H3
- H3K4me3
trimethylated lysine 4 of histone H3
- H3K79
lysine 79 of histone H3
- H3K9
lysine 9 of histone H3
- IBD
inflammatory bowel disease
- IFI44L
Interferon-induced protein 44-like
- IGF2BP
Insulin-like growth factor-2 mRNA-binding proteins
- IL
Interleukin
- IFN-γ
interferon-γ
- INO80
inositol requiring 80
- IPD
inter-pulse duration
- IPEX
immunodysregulation polyendocrinopathy enteropathy X-linked
- ISWI
imitation switch
- KATs
lysine acetyltransferases
- LN
lupus nephritis
- LSD
Lysine-specific demethylase
- MBD
methyl-CpG binding domain
- METTL4
Methyltransferase like 4
- MHC
Major Histocompatibility Complex
- MS
multiple sclerosis
- MSG
minor salivary gland
- MX1
myxoma resistance protein 1
- MYST
MOZ, Ybf2/Sas3, Sas2, Tip60
- mtDNA
mitochondrial DNA
- mTOR
mammalian target of rapamycin
- mTORC1
mechanistic target of rapamycin complex 1
- m1A
N1-methyladenosine
- 4mC
N4-methyldeoxycytosine
- m6A
N6-methyladenosine
- 6mA
N6-methyldeoxyadenosine
- m6Am
N6,2’-O-dimethyladenosine
- m7G
N7-methylguanosine
- mTECs
medullary thymic epithelial cells
- NAD
nicotinamide adenine dinucleotide
- NGS
next-generation sequencing
- NSAIDs
non-steroidal anti-inflammatory drugs
- NuRD
nuclesome remodeling deactylase
- nTreg
natural Treg
- PBMCs
peripheral blood mononuclear cells
- PTGs
peripheral tissue genes
- PHD
plant homeodomain
- PBRM1
polybromo-1
- PCR
polymerase chain reaction
- PRMT
protein arginine methyltransferase
- pSS
primary Sjögren syndrome
- Ψ
pseudouridine
- QC
quality control
- RA
rheumatoid arthritis
- RASFs
RA synovial fibroblasts
- RLRs
retinoic acid-inducible gene-I-like receptors
- RORγt
retinoic acid-related orphan receptor γt
- RT
reverse transcription
- SAM
S-adenosyl methionine
- 7βS
seven-beta-strand domain
- SEs
super enhancers
- SFs
synovial fibroblasts
- SLE
systemic lupus erythematosus
- SLEDAI
SLE disease activity index
- SMRT-seq
Single-molecule real-time sequencing
- SPEN
Split ends homolog
- SS
Sjögren’s Syndrome
- SSBP1
single-stranded DNA-binding protein 1
- SSc
systemic sclerosis
- SWI/SNF
switching defective/sucrose nonfermenting
- T
thymine
- TCR
T cell receptor
- TET
Ten-eleven translocation
- Th17
T helper 17
- TLR7
Toll-like receptor 7
- TNFAIP3
TNF-α-induced protein 3
- tRNA
transfer RNA
- Treg
regulatory T cells
- T1D
diabetes mellitus type 1
- U
uracil
- UC
ulcerative colitis
- WGBS
whole-genome bisulfite sequencing
- WTAP
Wilms tumor 1-associating protein
- XCI
X chromosome inactivation
- YTH
YT521-B homology
- YTHDC
YTH domain-containing protein
- YTHDF
YTH domain family
- ZMWs
zero-mode waveguides
- 3’UTR
3’
- 5mC
5-methylcytosine
- 5hmC
5-hydroxymethylcytosine
- 5mC
5-methylcytosine
Funding Statement
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the College Students’ Innovative Entrepreneurial Training Plan Program (202210366017), Natural Science Foundation of Xizang Autonomous Region (XZ2024ZRZY064(Z)), the National Natural Science Foundation of China (82003330), the Natural Science Foundation of Anhui Province (1808085QH284).
Author contributions
SM: Writing – original draft, Writing – review & editing. WW: Writing – review & editing. QL: Data curation, Writing – review & editing. NK: Formal analysis, Writing – original draft. HL: Methodology, Writing – review & editing. FS: Writing – review & editing. JZ: Investigation, Writing – review & editing. ZZ: Supervision, Writing – original draft, Writing – review & editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
- 1. Bieber K, Hundt JE, Yu X, Ehlers M, Petersen F, Karsten CM, et al. Autoimmune pre-disease. Autoimmun Rev. (2023) 22:103236. doi: 10.1016/j.autrev.2022.103236 [DOI] [PubMed] [Google Scholar]
- 2. Pisetsky DS. Pathogenesis of autoimmune disease. Nat Rev Nephrol. (2023) 19:509–24. doi: 10.1038/s41581-023-00720-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fanouriakis A, Kostopoulou M, Andersen J, Aringer M, Arnaud L, Bae SC, et al. EULAR recommendations for the management of systemic lupus erythematosus: 2023 update. Ann Rheum Dis. (2024) 83:15–29. doi: 10.1136/ard-2023-224762 [DOI] [PubMed] [Google Scholar]
- 4. Hoi A, Igel T, Mok CC, Arnaud L. Systemic lupus erythematosus. Lancet. (2024) 403:2326–38. doi: 10.1016/s0140-6736(24)00398-2 [DOI] [PubMed] [Google Scholar]
- 5. Brown P, Pratt AG, Hyrich KL. Therapeutic advances in rheumatoid arthritis. Bmj. (2024) 384:e070856. doi: 10.1136/bmj-2022-070856 [DOI] [PubMed] [Google Scholar]
- 6. Di Matteo A, Bathon JM, Emery P. Rheumatoid arthritis. Lancet. (2023) 402:2019–33. doi: 10.1016/s0140-6736(23)01525-8 [DOI] [PubMed] [Google Scholar]
- 7. Baldini C, Fulvio G, La Rocca G, Ferro F. Update on the pathophysiology and treatment of primary Sjögren syndrome. Nat Rev Rheumatol. (2024) 20:473–91. doi: 10.1038/s41584-024-01135-3 [DOI] [PubMed] [Google Scholar]
- 8. Thorlacius GE, Björk A, Wahren-Herlenius M. Genetics and epigenetics of primary Sjögren syndrome: implications for future therapies. Nat Rev Rheumatol. (2023) 19:288–306. doi: 10.1038/s41584-023-00932-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Knight JS, Branch DW, Ortel TL. Antiphospholipid syndrome: advances in diagnosis, pathogenesis, and management. Bmj. (2023) 380:e069717. doi: 10.1136/bmj-2021-069717 [DOI] [PubMed] [Google Scholar]
- 10. Xourgia E, Tektonidou MG. An update on antiphospholipid syndrome. Curr Rheumatol Rep. (2022) 23:84. doi: 10.1007/s11926-021-01051-5 [DOI] [PubMed] [Google Scholar]
- 11. Jerjen R, Nikpour M, Krieg T, Denton CP, Saracino AM. Systemic sclerosis in adults. Part I: Clinical features and pathogenesis. J Am Acad Dermatol. (2022) 87:937–54. doi: 10.1016/j.jaad.2021.10.065 [DOI] [PubMed] [Google Scholar]
- 12. Volkmann ER, Andréasson K, Smith V. Systemic sclerosis. Lancet. (2023) 401:304–18. doi: 10.1016/s0140-6736(22)01692-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McGinley MP, Goldschmidt CH, Rae-Grant AD. Diagnosis and treatment of multiple sclerosis: A review. Jama. (2021) 325:765–79. doi: 10.1001/jama.2020.26858 [DOI] [PubMed] [Google Scholar]
- 14. Rodríguez Murúa S, Farez MF, Quintana FJ. The immune response in multiple sclerosis. Annu Rev Pathol. (2022) 17:121–39. doi: 10.1146/annurev-pathol-052920-040318 [DOI] [PubMed] [Google Scholar]
- 15. Shahrizaila N, Lehmann HC, Kuwabara S. Guillain-barré syndrome. Lancet. (2021) 397:1214–28. doi: 10.1016/s0140-6736(21)00517-1 [DOI] [PubMed] [Google Scholar]
- 16. Shang P, Zhu M, Wang Y, Zheng X, Wu X, Zhu J, et al. Axonal variants of Guillain-Barré syndrome: an update. J Neurol. (2021) 268:2402–19. doi: 10.1007/s00415-020-09742-2 [DOI] [PubMed] [Google Scholar]
- 17. Budhram A, Leung A, Nicolle MW, Burneo JG. Diagnosing autoimmune limbic encephalitis. Cmaj. (2019) 191:E529–e534. doi: 10.1503/cmaj.181548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang J, Liu X. Immunotherapy for refractory autoimmune encephalitis. Front Immunol. (2021) 12:790962. doi: 10.3389/fimmu.2021.790962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dalmau J, Armangué T, Planagumà J, Radosevic M, Mannara F, Leypoldt F, et al. An update on anti-NMDA receptor encephalitis for neurologists and psychiatrists: mechanisms and models. Lancet Neurol. (2019) 18:1045–57. doi: 10.1016/s1474-4422(19)30244-3 [DOI] [PubMed] [Google Scholar]
- 20. Akil AA, Yassin E, Al-Maraghi A, Aliyev E, Al-Malki K, Fakhro KA. Diagnosis and treatment of type 1 diabetes at the dawn of the personalized medicine era. J Transl Med. (2021) 19:137. doi: 10.1186/s12967-021-02778-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dayan CM, Besser REJ, Oram RA, Hagopian W, Vatish M, Bendor-Samuel O, et al. Preventing type 1 diabetes in childhood. Science. (2021) 373:506–10. doi: 10.1126/science.abi4742 [DOI] [PubMed] [Google Scholar]
- 22. Røyrvik EC, Husebye ES. The genetics of autoimmune Addison disease: past, present and future. Nat Rev Endocrinol. (2022) 18:399–412. doi: 10.1038/s41574-022-00653-y [DOI] [PubMed] [Google Scholar]
- 23. Berentsen S, Barcellini W. Autoimmune hemolytic anemias. N Engl J Med. (2021) 385:1407–19. doi: 10.1056/NEJMra2033982 [DOI] [PubMed] [Google Scholar]
- 24. Boniface K, Seneschal J, Picardo M, Taïeb A. Vitiligo: focus on clinical aspects, immunopathogenesis, and therapy. Clin Rev Allergy Immunol. (2018) 54:52–67. doi: 10.1007/s12016-017-8622-7 [DOI] [PubMed] [Google Scholar]
- 25. Rodrigues M, Ezzedine K, Hamzavi I, Pandya AG, Harris JE. New discoveries in the pathogenesis and classification of vitiligo. J Am Acad Dermatol. (2017) 77:1–13. doi: 10.1016/j.jaad.2016.10.048 [DOI] [PubMed] [Google Scholar]
- 26. Schmidt E, Kasperkiewicz M, Joly P. Pemphigus. Lancet. (2019) 394:882–94. doi: 10.1016/s0140-6736(19)31778-7 [DOI] [PubMed] [Google Scholar]
- 27. Engineer L, Ahmed AR. Emerging treatment for epidermolysis bullosa acquisita. J Am Acad Dermatol. (2001) 44:818–28. doi: 10.1067/mjd.2001.113693 [DOI] [PubMed] [Google Scholar]
- 28. Kridin K, Kneiber D, Kowalski EH, Valdebran M, Amber KT. Epidermolysis bullosa acquisita: A comprehensive review. Autoimmun Rev. (2019) 18:786–95. doi: 10.1016/j.autrev.2019.06.007 [DOI] [PubMed] [Google Scholar]
- 29. Bowlus CL, Gershwin ME. The diagnosis of primary biliary cirrhosis. Autoimmun Rev. (2014) 13:441–4. doi: 10.1016/j.autrev.2014.01.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Carey EJ, Ali AH, Lindor KD. Primary biliary cirrhosis. Lancet. (2015) 386:1565–75. doi: 10.1016/s0140-6736(15)00154-3 [DOI] [PubMed] [Google Scholar]
- 31. Catassi C, Verdu EF, Bai JC, Lionetti E. Coeliac disease. Lancet. (2022) 399:2413–26. doi: 10.1016/s0140-6736(22)00794-2 [DOI] [PubMed] [Google Scholar]
- 32. Lebwohl B, Sanders DS, Green PHR. Coeliac disease. Lancet. (2018) 391:70–81. doi: 10.1016/s0140-6736(17)31796-8 [DOI] [PubMed] [Google Scholar]
- 33. Bourgonje AR, Vogl T, Segal E, Weersma RK. Antibody signatures in inflammatory bowel disease: current developments and future applications. Trends Mol Med. (2022) 28:693–705. doi: 10.1016/j.molmed.2022.05.004 [DOI] [PubMed] [Google Scholar]
- 34. Dolinger M, Torres J, Vermeire S. Crohn’s disease. Lancet. (2024) 403:1177–91. doi: 10.1016/s0140-6736(23)02586-2 [DOI] [PubMed] [Google Scholar]
- 35. Lenti MV, Rugge M, Lahner E, Miceli E, Toh BH, Genta RM, et al. Autoimmune gastritis. Nat Rev Dis Primers. (2020) 6:56. doi: 10.1038/s41572-020-0187-8 [DOI] [PubMed] [Google Scholar]
- 36. Miceli E, Lenti MV, Padula D, Luinetti O, Vattiato C, Monti CM, et al. Common features of patients with autoimmune atrophic gastritis. Clin Gastroenterol Hepatol. (2012) 10:812–4. doi: 10.1016/j.cgh.2012.02.018 [DOI] [PubMed] [Google Scholar]
- 37. Gilhus NE. Myasthenia gravis. N Engl J Med. (2016) 375:2570–81. doi: 10.1056/NEJMra1602678 [DOI] [PubMed] [Google Scholar]
- 38. Gilhus NE, Skeie GO, Romi F, Lazaridis K, Zisimopoulou P, Tzartos S. Myasthenia gravis - autoantibody characteristics and their implications for therapy. Nat Rev Neurol. (2016) 12:259–68. doi: 10.1038/nrneurol.2016.44 [DOI] [PubMed] [Google Scholar]
- 39. Punga AR, Maddison P, Heckmann JM, Guptill JT, Evoli A. Epidemiology, diagnostics, and biomarkers of autoimmune neuromuscular junction disorders. Lancet Neurol. (2022) 21:176–88. doi: 10.1016/s1474-4422(21)00297-0 [DOI] [PubMed] [Google Scholar]
- 40. Vargas-Uricoechea H. Molecular mechanisms in autoimmune thyroid disease. Cells. (2023) 12:1–37. doi: 10.3390/cells12060918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ralli M, Angeletti D, Fiore M, D’Aguanno V, Lambiase A, Artico M, et al. Hashimoto’s thyroiditis: An update on pathogenic mechanisms, diagnostic protocols, therapeutic strategies, and potential Malignant transformation. Autoimmun Rev. (2020) 19:102649. doi: 10.1016/j.autrev.2020.102649 [DOI] [PubMed] [Google Scholar]
- 42. Cho JH, Feldman M. Heterogeneity of autoimmune diseases: pathophysiologic insights from genetics and implications for new therapies. Nat Med. (2015) 21:730–8. doi: 10.1038/nm.3897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhou M, Guo C, Li X, Huang Y, Li M, Zhang T, et al. JAK/STAT signaling controls the fate of CD8(+)CD103(+) tissue-resident memory T cell in lupus nephritis. J Autoimmun. (2020) 109:102424. doi: 10.1016/j.jaut.2020.102424 [DOI] [PubMed] [Google Scholar]
- 44. Tsokos GC. Autoimmunity and organ damage in systemic lupus erythematosus. Nat Immunol. (2020) 21:605–14. doi: 10.1038/s41590-020-0677-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen Y, Li S, Huang R, Zhang Z, Petersen F, Zheng J, et al. Comprehensive meta-analysis reveals an association of the HLA-DRB1*1602 allele with autoimmune diseases mediated predominantly by autoantibodies. Autoimmun Rev. (2020) 19:102532. doi: 10.1016/j.autrev.2020.102532 [DOI] [PubMed] [Google Scholar]
- 46. Chebly A, Chouery E, Ropio J, Kourie HR, Beylot-Barry M, Merlio JP, et al. Diagnosis and treatment of lymphomas in the era of epigenetics. Blood Rev. (2021) 48:100782. doi: 10.1016/j.blre.2020.100782 [DOI] [PubMed] [Google Scholar]
- 47. Boulias K, Greer EL. Means, mechanisms and consequences of adenine methylation in DNA. Nat Rev Genet. (2022) 23:411–28. doi: 10.1038/s41576-022-00456-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Greer EL, Blanco MA, Gu L, Sendinc E, Liu J, Aristizábal-Corrales D, et al. DNA methylation on N6-adenine in C. elegans. Cell. (2015) 161:868–78. doi: 10.1016/j.cell.2015.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhao LY, Song J, Liu Y, Song CX, Yi C. Mapping the epigenetic modifications of DNA and RNA. Protein Cell. (2020) 11:792–808. doi: 10.1007/s13238-020-00733-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhu H, Wang G, Qian J. Transcription factors as readers and effectors of DNA methylation. Nat Rev Genet. (2016) 17:551–65. doi: 10.1038/nrg.2016.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Barbieri I, Kouzarides T. Role of RNA modifications in cancer. Nat Rev Cancer. (2020) 20:303–22. doi: 10.1038/s41568-020-0253-2 [DOI] [PubMed] [Google Scholar]
- 52. Suzuki T. The expanding world of tRNA modifications and their disease relevance. Nat Rev Mol Cell Biol. (2021) 22:375–92. doi: 10.1038/s41580-021-00342-0 [DOI] [PubMed] [Google Scholar]
- 53. Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell. (2012) 48:491–507. doi: 10.1016/j.molcel.2012.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ramón-Maiques S, Kuo AJ, Carney D, Matthews AG, Oettinger MA, Gozani O, et al. The plant homeodomain finger of RAG2 recognizes histone H3 methylated at both lysine-4 and arginine-2. Proc Natl Acad Sci U.S.A. (2007) 104:18993–8. doi: 10.1073/pnas.0709170104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wüst HM, Wegener A, Fröb F, Hartwig AC, Wegwitz F, Kari V, et al. Egr2-guided histone H2B monoubiquitination is required for peripheral nervous system myelination. Nucleic Acids Res. (2020) 48:8959–76. doi: 10.1093/nar/gkaa606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Xu Y, Zhang S, Lin S, Guo Y, Deng W, Zhang Y, et al. WERAM: a database of writers, erasers and readers of histone acetylation and methylation in eukaryotes. Nucleic Acids Res. (2017) 45:D264–d270. doi: 10.1093/nar/gkw1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhao A, Xu W, Han R, Wei J, Yu Q, Wang M, et al. Role of histone modifications in neurogenesis and neurodegenerative disease development. Ageing Res Rev. (2024) 98:102324. doi: 10.1016/j.arr.2024.102324 [DOI] [PubMed] [Google Scholar]
- 58. Barisic D, Stadler MB, Iurlaro M, Schübeler D. Mammalian ISWI and SWI/SNF selectively mediate binding of distinct transcription factors. Nature. (2019) 569:136–40. doi: 10.1038/s41586-019-1115-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nemeth K, Bayraktar R, Ferracin M, Calin GA. Non-coding RNAs in disease: from mechanisms to therapeutics. Nat Rev Genet. (2024) 25:211–32. doi: 10.1038/s41576-023-00662-1 [DOI] [PubMed] [Google Scholar]
- 60. Tsou PS, Varga J, O’Reilly S. Advances in epigenetics in systemic sclerosis: molecular mechanisms and therapeutic potential. Nat Rev Rheumatol. (2021) 17:596–607. doi: 10.1038/s41584-021-00683-2 [DOI] [PubMed] [Google Scholar]
- 61. Karagianni P, Tzioufas AG. Epigenetic perspectives on systemic autoimmune disease. J Autoimmun. (2019) 104:102315. doi: 10.1016/j.jaut.2019.102315 [DOI] [PubMed] [Google Scholar]
- 62. Danieli MG, Casciaro M, Paladini A, Bartolucci M, Sordoni M, Shoenfeld Y, et al. Exposome: Epigenetics and autoimmune diseases. Autoimmun Rev. (2024) 23:103584. doi: 10.1016/j.autrev.2024.103584 [DOI] [PubMed] [Google Scholar]
- 63. Holdgate GA, Bardelle C, Lanne A, Read J, O’Donovan DH, Smith JM, et al. Drug discovery for epigenetics targets. Drug Discovery Today. (2022) 27:1088–98. doi: 10.1016/j.drudis.2021.10.020 [DOI] [PubMed] [Google Scholar]
- 64. Szekanecz Z, McInnes IB, Schett G, Szamosi S, Benkő S, Szűcs G. Autoinflammation and autoimmunity across rheumatic and musculoskeletal diseases. Nat Rev Rheumatol. (2021) 17:585–95. doi: 10.1038/s41584-021-00652-9 [DOI] [PubMed] [Google Scholar]
- 65. Zhang Z, Zhang R. Epigenetics in autoimmune diseases: Pathogenesis and prospects for therapy. Autoimmun Rev. (2015) 14:854–63. doi: 10.1016/j.autrev.2015.05.008 [DOI] [PubMed] [Google Scholar]
- 66. Zemmour D, Charbonnier LM, Leon J, Six E, Keles S, Delville M, et al. Single-cell analysis of FOXP3 deficiencies in humans and mice unmasks intrinsic and extrinsic CD4(+) T cell perturbations. Nat Immunol. (2021) 22:607–19. doi: 10.1038/s41590-021-00910-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bjørklund G, Pivin M, Hangan T, Yurkovskaya O, Pivina L. Autoimmune polyendocrine syndrome type 1: Clinical manifestations, pathogenetic features, and management approach. Autoimmun Rev. (2022) 21:103135. doi: 10.1016/j.autrev.2022.103135 [DOI] [PubMed] [Google Scholar]
- 68. Woo JMP, Parks CG, Jacobsen S, Costenbader KH, Bernatsky S. The role of environmental exposures and gene-environment interactions in the etiology of systemic lupus erythematous. J Intern Med. (2022) 291:755–78. doi: 10.1111/joim.13448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Pan Q, Liu Z, Liao S, Ye L, Lu X, Chen X, et al. Current mechanistic insights into the role of infection in systemic lupus erythematosus. BioMed Pharmacother. (2019) 117:109122. doi: 10.1016/j.biopha.2019.109122 [DOI] [PubMed] [Google Scholar]
- 70. Kang I, Quan T, Nolasco H, Park SH, Hong MS, Crouch J, et al. Defective control of latent Epstein-Barr virus infection in systemic lupus erythematosus. J Immunol. (2004) 83(5):1287–94. doi: 10.4049/jimmunol.172.2.1287 [DOI] [PubMed] [Google Scholar]
- 71. Gregersen PK, Behrens TW. Genetics of autoimmune diseases–disorders of immune homeostasis. Nat Rev Genet. (2006) 7:917–28. doi: 10.1038/nrg1944 [DOI] [PubMed] [Google Scholar]
- 72. Cheng Z, Hou G, Shen N. Evolving understandings for the roles of non-coding RNAs in autoimmunity and autoimmune disease. J Autoimmun. (2023) 137:102948. doi: 10.1016/j.jaut.2022.102948 [DOI] [PubMed] [Google Scholar]
- 73. Fraussen J, de Bock L, Somers V. B cells and antibodies in progressive multiple sclerosis: Contribution to neurodegeneration and progression. Autoimmun Rev. (2016) 15:896–9. doi: 10.1016/j.autrev.2016.07.008 [DOI] [PubMed] [Google Scholar]
- 74. Moulton VR, Tsokos GC. T cell signaling abnormalities contribute to aberrant immune cell function and autoimmunity. J Clin Invest. (2015) 125:2220–7. doi: 10.1172/jci78087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Crickx E, Weill JC, Reynaud CA, Mahévas M. Anti-CD20-mediated B-cell depletion in autoimmune diseases: successes, failures and future perspectives. Kidney Int. (2020) 97:885–93. doi: 10.1016/j.kint.2019.12.025 [DOI] [PubMed] [Google Scholar]
- 76. Hu F, Shi L, Liu X, Chen Y, Zhang X, Jia Y, et al. Proinflammatory phenotype of B10 and B10pro cells elicited by TNF-α in rheumatoid arthritis. Ann Rheum Dis. (2024). doi: 10.1136/ard-2023-224878 [DOI] [PubMed] [Google Scholar]
- 77. Dominguez-Villar M, Hafler DA. Regulatory T cells in autoimmune disease. Nat Immunol. (2018) 19:665–73. doi: 10.1038/s41590-018-0120-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mellor-Pita S, Citores MJ, Castejon R, Tutor-Ureta P, Yebra-Bango M, Andreu JL, et al. Decrease of regulatory T cells in patients with systemic lupus erythematosus. Ann Rheum Dis. (2006) 65:553–4. doi: 10.1136/ard.2005.044974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Mesquita D, Jr., Kirsztajn GM, Franco MF, Reis LA, Perazzio SF, Mesquita FV, et al. CD4(+) T helper cells and regulatory T cells in active lupus nephritis: an imbalance towards a predominant Th1 response? Clin Exp Immunol. (2018) 191:50–9. doi: 10.1111/cei.13050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Jacquemin C, Augusto JF, Scherlinger M, Gensous N, Forcade E, Douchet I, et al. OX40L/OX40 axis impairs follicular and natural Treg function in human SLE. JCI Insight. (2018) 3:2–3. doi: 10.1172/jci.insight.122167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Parker H, Albrett AM, Kettle AJ, Winterbourn CC. Myeloperoxidase associated with neutrophil extracellular traps is active and mediates bacterial killing in the presence of hydrogen peroxide. J Leukoc Biol. (2012) 91:369–76. doi: 10.1189/jlb.0711387 [DOI] [PubMed] [Google Scholar]
- 82. Pratesi F, Dioni I, Tommasi C, Alcaro MC, Paolini I, Barbetti F, et al. Antibodies from patients with rheumatoid arthritis target citrullinated histone 4 contained in neutrophils extracellular traps. Ann Rheum Dis. (2014) 73:1414–22. doi: 10.1136/annrheumdis-2012-202765 [DOI] [PubMed] [Google Scholar]
- 83. Navegantes KC, de Souza Gomes R, Pereira PAT, Czaikoski PG, Azevedo CHM, Monteiro MC. Immune modulation of some autoimmune diseases: the critical role of macrophages and neutrophils in the innate and adaptive immunity. J Transl Med. (2017) 15:36. doi: 10.1186/s12967-017-1141-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Coutant F, Miossec P. Altered dendritic cell functions in autoimmune diseases: distinct and overlapping profiles. Nat Rev Rheumatol. (2016) 12:703–15. doi: 10.1038/nrrheum.2016.147 [DOI] [PubMed] [Google Scholar]
- 85. Willyard C. Can autoimmune diseases be cured? Scientists see hope at last. Nature. (2024) 625:646–8. doi: 10.1038/d41586-024-00169-7 [DOI] [PubMed] [Google Scholar]
- 86. Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. Regulatory T cells and human disease. Annu Rev Immunol. (2020) 38:541–66. doi: 10.1146/annurev-immunol-042718-041717 [DOI] [PubMed] [Google Scholar]
- 87. Bluestone JA, Anderson M. Tolerance in the age of immunotherapy. N Engl J Med. (2020) 383:1156–66. doi: 10.1056/NEJMra1911109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Clarke J. DNA methylation inhibitor resets tolerance in autoimmune arthritis. Nat Rev Rheumatol. (2021) 17:379. doi: 10.1038/s41584-021-00635-w [DOI] [PubMed] [Google Scholar]
- 89. Muraro PA. Resetting tolerance in autoimmune disease. Science. (2023) 380:470–1. doi: 10.1126/science.adg7489 [DOI] [PubMed] [Google Scholar]
- 90. Marx A, Yamada Y, Simon-Keller K, Schalke B, Willcox N, Ströbel P, et al. Thymus and autoimmunity. Semin Immunopathol. (2021) 43:45–64. doi: 10.1007/s00281-021-00842-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Michelson DA, Mathis D. Thymic mimetic cells: tolerogenic masqueraders. Trends Immunol. (2022) 43:782–91. doi: 10.1016/j.it.2022.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Org T, Rebane A, Kisand K, Laan M, Haljasorg U, Andreson R, et al. AIRE activated tissue specific genes have histone modifications associated with inactive chromatin. Hum Mol Genet. (2009) 18:4699–710. doi: 10.1093/hmg/ddp433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Sadras T, Chan LN, Xiao G, Müschen M. Metabolic gatekeepers of pathological B cell activation. Annu Rev Pathol. (2021) 16:323–49. doi: 10.1146/annurev-pathol-061020-050135 [DOI] [PubMed] [Google Scholar]
- 94. Theofilopoulos AN, Kono DH, Baccala R. The multiple pathways to autoimmunity. Nat Immunol. (2017) 18:716–24. doi: 10.1038/ni.3731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Esensten JH, Helou YA, Chopra G, Weiss A, Bluestone JA. CD28 costimulation: from mechanism to therapy. Immunity. (2016) 44:973–88. doi: 10.1016/j.immuni.2016.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ohkura N, Sakaguchi S. Transcriptional and epigenetic basis of Treg cell development and function: its genetic anomalies or variations in autoimmune diseases. Cell Res. (2020) 30:465–74. doi: 10.1038/s41422-020-0324-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Scheinecker C, Göschl L, Bonelli M. Treg cells in health and autoimmune diseases: New insights from single cell analysis. J Autoimmun. (2020) 110:102376. doi: 10.1016/j.jaut.2019.102376 [DOI] [PubMed] [Google Scholar]
- 98. Gaultney RA, Vincent AT, Lorioux C, Coppée JY, Sismeiro O, Varet H, et al. 4-Methylcytosine DNA modification is critical for global epigenetic regulation and virulence in the human pathogen Leptospira interrogans. Nucleic Acids Res. (2020) 48:12102–15. doi: 10.1093/nar/gkaa966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Dura M, Teissandier A, Armand M, Barau J, Lapoujade C, Fouchet P, et al. DNMT3A-dependent DNA methylation is required for spermatogonial stem cells to commit to spermatogenesis. Nat Genet. (2022) 54:469–80. doi: 10.1038/s41588-022-01040-z [DOI] [PubMed] [Google Scholar]
- 100. Chen Z, Zhang Y. Role of mammalian DNA methyltransferases in development. Annu Rev Biochem. (2020) 89:135–58. doi: 10.1146/annurev-biochem-103019-102815 [DOI] [PubMed] [Google Scholar]
- 101. Lei Y, Huang YH, Goodell MA. DNA methylation and de-methylation using hybrid site-targeting proteins. Genome Biol. (2018) 19:187. doi: 10.1186/s13059-018-1566-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Wang D, Wu W, Callen E, Pavani R, Zolnerowich N, Kodali S, et al. Active DNA demethylation promotes cell fate specification and the DNA damage response. Science. (2022) 378:983–9. doi: 10.1126/science.add9838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. O’Brown ZK, Greer EL. N6-methyladenine: A conserved and dynamic DNA mark. Adv Exp Med Biol. (2016) 945:213–46. doi: 10.1007/978-3-319-43624-1_10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kweon SM, Chen Y, Moon E, Kvederaviciutė K, Klimasauskas S, Feldman DE. An adversarial DNA N(6)-methyladenine-sensor network preserves polycomb silencing. Mol Cell. (2019) 74:1138–1147.e1136. doi: 10.1016/j.molcel.2019.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Hao Z, Wu T, Cui X, Zhu P, Tan C, Dou X, et al. N(6)-deoxyadenosine methylation in mammalian mitochondrial DNA. Mol Cell. (2020) 78:382–395.e388. doi: 10.1016/j.molcel.2020.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Fedeles BI, Singh V, Delaney JC, Li D, Essigmann JM. The alkB family of fe(II)/α-ketoglutarate-dependent dioxygenases: repairing nucleic acid alkylation damage and beyond. J Biol Chem. (2015) 290:20734–42. doi: 10.1074/jbc.R115.656462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Lanata CM, Nititham J, Taylor KE, Solomon O, Chung SA, Blazer A, et al. Dynamics of methylation of cpG sites associated with systemic lupus erythematosus subtypes in a longitudinal cohort. Arthritis Rheumatol. (2022) 74:1676–86. doi: 10.1002/art.42237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Imgenberg-Kreuz J, Carlsson Almlöf J, Leonard D, Alexsson A, Nordmark G, Eloranta ML, et al. DNA methylation mapping identifies gene regulatory effects in patients with systemic lupus erythematosus. Ann Rheum Dis. (2018) 77:736–43. doi: 10.1136/annrheumdis-2017-212379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Gosselt HR, van Zelst BD, de Rotte M, Hazes JMW, de Jonge R, Heil SG. Higher baseline global leukocyte DNA methylation is associated with MTX non-response in early RA patients. Arthritis Res Ther. (2019) 21:157. doi: 10.1186/s13075-019-1936-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Wang X, Xie H, Guo Q, Cao D, Ru W, Zhao S, et al. Molecular basis for METTL9-mediated N1-histidine methylation. Cell Discovery. (2023) 9:38. doi: 10.1038/s41421-023-00548-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Phillips DM. The presence of acetyl groups of histones. Biochem J. (1963) 87:258–63. doi: 10.1042/bj0870258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Allfrey VG, Mirsky AE. Structural modifications of histones and their possible role in the regulation of RNA synthesis. Science. (1964) 144:559. doi: 10.1126/science.144.3618.559 [DOI] [PubMed] [Google Scholar]
- 113. Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. (1996) 272:408–11. doi: 10.1126/science.272.5260.408 [DOI] [PubMed] [Google Scholar]
- 114. Brownell JE, Allis CD. An activity gel assay detects a single, catalytically active histone acetyltransferase subunit in Tetrahymena macronuclei. Proc Natl Acad Sci U.S.A. (1995) 92:6364–8. doi: 10.1073/pnas.92.14.6364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature. (1999) 399:491–6. doi: 10.1038/20974 [DOI] [PubMed] [Google Scholar]
- 116. Rea S, Eisenhaber F, O’Carroll D, Strahl BD, Sun ZW, Schmid M, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. (2000) 406:593–9. doi: 10.1038/35020506 [DOI] [PubMed] [Google Scholar]
- 117. Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. (2001) 410:120–4. doi: 10.1038/35065138 [DOI] [PubMed] [Google Scholar]
- 118. Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. (2004) 119:941–53. doi: 10.1016/j.cell.2004.12.012 [DOI] [PubMed] [Google Scholar]
- 119. Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. (2011) 146:1016–28. doi: 10.1016/j.cell.2011.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature. (2019) 574:575–80. doi: 10.1038/s41586-019-1678-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. An S, Camarillo JM, Huang TY, Li D, Morris JA, Zoltek MA, et al. Histone tail analysis reveals H3K36me2 and H4K16ac as epigenetic signatures of diffuse intrinsic pontine glioma. J Exp Clin Cancer Res. (2020) 39:261. doi: 10.1186/s13046-020-01773-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Liberti MV, Locasale JW. Histone lactylation: A new role for glucose metabolism. Trends Biochem Sci. (2020) 45:179–82. doi: 10.1016/j.tibs.2019.12.004 [DOI] [PubMed] [Google Scholar]
- 123. Hogg SJ, Motorna O, Cluse LA, Johanson TM, Coughlan HD, Raviram R, et al. Targeting histone acetylation dynamics and oncogenic transcription by catalytic P300/CBP inhibition. Mol Cell. (2021) 81:2183–2200.e2113. doi: 10.1016/j.molcel.2021.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Hyun K, Jeon J, Park K, Kim J. Writing, erasing and reading histone lysine methylations. Exp Mol Med. (2017) 49:e324. doi: 10.1038/emm.2017.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Park J, Lee K, Kim K, Yi SJ. The role of histone modifications: from neurodevelopment to neurodiseases. Signal Transduct Target Ther. (2022) 7:217. doi: 10.1038/s41392-022-01078-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Halsall JA, Andrews S, Krueger F, Rutledge CE, Ficz G, Reik W, et al. Histone modifications form a cell-type-specific chromosomal bar code that persists through the cell cycle. Sci Rep. (2021) 11:3009. doi: 10.1038/s41598-021-82539-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Vasquez JJ, Wedel C, Cosentino RO, Siegel TN. Exploiting CRISPR-Cas9 technology to investigate individual histone modifications. Nucleic Acids Res. (2018) 46:e106. doi: 10.1093/nar/gky517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Zhao H, Wang L, Luo H, Li QZ, Zuo X. TNFAIP3 downregulation mediated by histone modification contributes to T-cell dysfunction in systemic lupus erythematosus. Rheumatol (Oxford). (2017) 56:835–43. doi: 10.1093/rheumatology/kew508 [DOI] [PubMed] [Google Scholar]
- 129. van der Kroef M, Castellucci M, Mokry M, Cossu M, Garonzi M, Bossini-Castillo LM, et al. Histone modifications underlie monocyte dysregulation in patients with systemic sclerosis, underlining the treatment potential of epigenetic targeting. Ann Rheum Dis. (2019) 78:529–38. doi: 10.1136/annrheumdis-2018-214295 [DOI] [PubMed] [Google Scholar]
- 130. Xu S, Kamato D, Little PJ, Nakagawa S, Pelisek J, Jin ZG. Targeting epigenetics and non-coding RNAs in atherosclerosis: from mechanisms to therapeutics. Pharmacol Ther. (2019) 196:15–43. doi: 10.1016/j.pharmthera.2018.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Markert J, Luger K. Nucleosomes meet their remodeler match. Trends Biochem Sci. (2021) 46:41–50. doi: 10.1016/j.tibs.2020.08.010 [DOI] [PubMed] [Google Scholar]
- 132. Tamaru H. Confining euchromatin/heterochromatin territory: jumonji crosses the line. Genes Dev. (2010) 24:1465–78. doi: 10.1101/gad.1941010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Klemm SL, Shipony Z, Greenleaf WJ. Chromatin accessibility and the regulatory epigenome. Nat Rev Genet. (2019) 20:207–20. doi: 10.1038/s41576-018-0089-8 [DOI] [PubMed] [Google Scholar]
- 134. Yen K, Vinayachandran V, Batta K, Koerber RT, Pugh BF. Genome-wide nucleosome specificity and directionality of chromatin remodelers. Cell. (2012) 149:1461–73. doi: 10.1016/j.cell.2012.04.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Bayona-Feliu A, Barroso S, Muñoz S, Aguilera A. The SWI/SNF chromatin remodeling complex helps resolve R-loop-mediated transcription-replication conflicts. Nat Genet. (2021) 53:1050–63. doi: 10.1038/s41588-021-00867-2 [DOI] [PubMed] [Google Scholar]
- 136. Xiao L, Parolia A, Qiao Y, Bawa P, Eyunni S, Mannan R, et al. Targeting SWI/SNF ATPases in enhancer-addicted prostate cancer. Nature. (2022) 601:434–9. doi: 10.1038/s41586-021-04246-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Venkatesh S, Workman JL. Histone exchange, chromatin structure and the regulation of transcription. Nat Rev Mol Cell Biol. (2015) 16:178–89. doi: 10.1038/nrm3941 [DOI] [PubMed] [Google Scholar]
- 138. Battistello E, Hixon KA, Comstock DE, Collings CK, Chen X, Rodriguez Hernaez J, et al. Stepwise activities of mSWI/SNF family chromatin remodeling complexes direct T cell activation and exhaustion. Mol Cell. (2023) 5–6. doi: 10.1016/j.molcel.2023.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Wu R, Zeng J, Yuan J, Deng X, Huang Y, Chen L, et al. MicroRNA-210 overexpression promotes psoriasis-like inflammation by inducing Th1 and Th17 cell differentiation. J Clin Invest. (2018) 128:2551–68. doi: 10.1172/jci97426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Lee S, Kim J, Min H, Seong RH. RORγt-driven T(H)17 cell differentiation requires epigenetic control by the swi/snf chromatin remodeling complex. iScience. (2020) 23:101106. doi: 10.1016/j.isci.2020.101106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Shu XS, Zhao Y, Sun Y, Zhong L, Cheng Y, Zhang Y, et al. The epigenetic modifier PBRM1 restricts the basal activity of the innate immune system by repressing retinoic acid-inducible gene-I-like receptor signalling and is a potential prognostic biomarker for colon cancer. J Pathol. (2018) 244:36–48. doi: 10.1002/path.4986 [DOI] [PubMed] [Google Scholar]
- 142. Lu X, Kovalev GI, Chang H, Kallin E, Knudsen G, Xia L, et al. Inactivation of NuRD component Mta2 causes abnormal T cell activation and lupus-like autoimmune disease in mice. J Biol Chem. (2008) 283:13825–33. doi: 10.1074/jbc.M801275200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Chan JJ, Tabatabaeian H, Tay Y. 3’UTR heterogeneity and cancer progression. Trends Cell Biol. (2022) 33(7):568–582. doi: 10.1016/j.tcb.2022.10.001 [DOI] [PubMed] [Google Scholar]
- 144. Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. (2014) 10:93–5. doi: 10.1038/nchembio.1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. (2011) 7:885–7. doi: 10.1038/nchembio.687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. (2013) 49:18–29. doi: 10.1016/j.molcel.2012.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Shima H, Matsumoto M, Ishigami Y, Ebina M, Muto A, Sato Y, et al. S-adenosylmethionine synthesis is regulated by selective N(6)-adenosine methylation and mRNA degradation involving METTL16 and YTHDC1. Cell Rep. (2017) 21:3354–63. doi: 10.1016/j.celrep.2017.11.092 [DOI] [PubMed] [Google Scholar]
- 148. Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. (2012) 485:201–6. doi: 10.1038/nature11112 [DOI] [PubMed] [Google Scholar]
- 149. Zaccara S, Jaffrey SR. A Unified Model for the Function of YTHDF Proteins in Regulating m(6)A-Modified mRNA. Cell. (2020) 181:1582–1595.e1518. doi: 10.1016/j.cell.2020.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M, et al. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun. (2016) 7:12626. doi: 10.1038/ncomms12626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, et al. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. (2017) 27:315–28. doi: 10.1038/cr.2017.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Shulman Z, Stern-Ginossar N. The RNA modification N(6)-methyladenosine as a novel regulator of the immune system. Nat Immunol. (2020) 21:501–12. doi: 10.1038/s41590-020-0650-4 [DOI] [PubMed] [Google Scholar]
- 153. Shi W, Zheng Y, Luo S, Li X, Zhang Y, Meng X, et al. METTL3 promotes activation and inflammation of FLSs through the NF-κB signaling pathway in rheumatoid arthritis. Front Med (Lausanne). (2021) 8:607585. doi: 10.3389/fmed.2021.607585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Tong J, Cao G, Zhang T, Sefik E, Amezcua Vesely MC, Broughton JP, et al. m(6)A mRNA methylation sustains Treg suppressive functions. Cell Res. (2018) 28:253–6. doi: 10.1038/cr.2018.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Kuhn C, Besançon A, Lemoine S, You S, Marquet C, Candon S, et al. Regulatory mechanisms of immune tolerance in type 1 diabetes and their failures. J Autoimmun. (2016) 71:69–77. doi: 10.1016/j.jaut.2016.05.002 [DOI] [PubMed] [Google Scholar]
- 156. Zeng H, Zhang R, Jin B, Chen L. Type 1 regulatory T cells: a new mechanism of peripheral immune tolerance. Cell Mol Immunol. (2015) 12:566–71. doi: 10.1038/cmi.2015.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Heinlein M, Gandolfo LC, Zhao K, Teh CE, Nguyen N, Baell JB, et al. The acetyltransferase KAT7 is required for thymic epithelial cell expansion, expression of AIRE target genes, and thymic tolerance. Sci Immunol. (2022) 7:eabb6032. doi: 10.1126/sciimmunol.abb6032 [DOI] [PubMed] [Google Scholar]
- 158. Kitagawa Y, Ohkura N, Kidani Y, Vandenbon A, Hirota K, Kawakami R, et al. Guidance of regulatory T cell development by Satb1-dependent super-enhancer establishment. Nat Immunol. (2017) 18:173–83. doi: 10.1038/ni.3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Nemazee D. Mechanisms of central tolerance for B cells. Nat Rev Immunol. (2017) 17:281–94. doi: 10.1038/nri.2017.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Zhang P, Lu Q. Genetic and epigenetic influences on the loss of tolerance in autoimmunity. Cell Mol Immunol. (2018) 15:575–85. doi: 10.1038/cmi.2017.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Yang Y, Liu K, Liu M, Zhang H, Guo M. EZH2: Its regulation and roles in immune disturbance of SLE. Front Pharmacol. (2022) 13:1002741. doi: 10.3389/fphar.2022.1002741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Xiao XY, Li YT, Jiang X, Ji X, Lu X, Yang B, et al. EZH2 deficiency attenuates Treg differentiation in rheumatoid arthritis. J Autoimmun. (2020) 108:102404. doi: 10.1016/j.jaut.2020.102404 [DOI] [PubMed] [Google Scholar]
- 163. Zhang Q, Liang Z, Zhang J, Lei T, Dong X, Su H, et al. Sirt6 regulates the development of medullary thymic epithelial cells and contributes to the establishment of central immune tolerance. Front Cell Dev Biol. (2021) 9:655552. doi: 10.3389/fcell.2021.655552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A, et al. SIRT6 promotes DNA repair under stress by activating PARP1. Science. (2011) 332:1443–6. doi: 10.1126/science.1202723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Pan PW, Feldman JL, Devries MK, Dong A, Edwards AM, Denu JM. Structure and biochemical functions of SIRT6. J Biol Chem. (2011) 286:14575–87. doi: 10.1074/jbc.M111.218990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Hopfner KP, Hornung V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol. (2020) 21:501–21. doi: 10.1038/s41580-020-0244-x [DOI] [PubMed] [Google Scholar]
- 167. Gray EE, Treuting PM, Woodward JJ, Stetson DB. Cutting edge: cGAS is required for lethal autoimmune disease in the trex1-deficient mouse model of aicardi-goutières syndrome. J Immunol. (2015) 195:1939–43. doi: 10.4049/jimmunol.1500969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Furie R, Khamashta M, Merrill JT, Werth VP, Kalunian K, Brohawn P, et al. Anifrolumab, an anti-interferon-α Receptor monoclonal antibody, in moderate-to-severe systemic lupus erythematosus. Arthritis Rheumatol. (2017) 69:376–86. doi: 10.1002/art.39962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Xia T, Konno H, Ahn J, Barber GN. Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. (2016) 14:282–97. doi: 10.1016/j.celrep.2015.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Lv H, Zong Q, Chen C, Lv G, Xiang W, Xing F, et al. TET2-mediated tumor cGAS triggers endothelial STING activation to regulate vasculature remodeling and anti-tumor immunity in liver cancer. Nat Commun. (2024) 15:6. doi: 10.1038/s41467-023-43743-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Dai S, Jia R, Zhang X, Fang Q, Huang L. The PD-1/PD-Ls pathway and autoimmune diseases. Cell Immunol. (2014) 290:72–9. doi: 10.1016/j.cellimm.2014.05.006 [DOI] [PubMed] [Google Scholar]
- 172. Kristjansdottir H, Steinsson K, Gunnarsson I, Gröndal G, Erlendsson K, Alarcón-Riquelme ME. Lower expression levels of the programmed death 1 receptor on CD4+CD25+ T cells and correlation with the PD-1.3A genotype in patients with systemic lupus erythematosus. Arthritis Rheum. (2010) 62:1702–11. doi: 10.1002/art.27417 [DOI] [PubMed] [Google Scholar]
- 173. Zamani MR, Aslani S, Salmaninejad A, Javan MR, Rezaei N. PD-1/PD-L and autoimmunity: A growing relationship. Cell Immunol. (2016) 310:27–41. doi: 10.1016/j.cellimm.2016.09.009 [DOI] [PubMed] [Google Scholar]
- 174. Wan B, Nie H, Liu A, Feng G, He D, Xu R, et al. Aberrant regulation of synovial T cell activation by soluble costimulatory molecules in rheumatoid arthritis. J Immunol. (2006) 177:8844–50. doi: 10.4049/jimmunol.177.12.8844 [DOI] [PubMed] [Google Scholar]
- 175. Ni Z, Sun P, Zheng J, Wu M, Yang C, Cheng M, et al. JNK Signaling Promotes Bladder Cancer Immune Escape by Regulating METTL3-Mediated m6A Modification of PD-L1 mRNA. Cancer Res. (2022) 82:1789–802. doi: 10.1158/0008-5472.Can-21-1323 [DOI] [PubMed] [Google Scholar]
- 176. Wang Z, Chang C, Peng M, Lu Q. Translating epigenetics into clinic: focus on lupus. Clin Epigenet. (2017) 9:78. doi: 10.1186/s13148-017-0378-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Vordenbäumen S, Rosenbaum A, Gebhard C, Raithel J, Sokolowski A, Düsing C, et al. Associations of site-specific CD4(+)-T-cell hypomethylation within CD40-ligand promotor and enhancer regions with disease activity of women with systemic lupus erythematosus. Lupus. (2021) 30:45–51. doi: 10.1177/0961203320965690 [DOI] [PubMed] [Google Scholar]
- 178. Gupta B, Hawkins RD. Epigenomics of autoimmune diseases. Immunol Cell Biol. (2015) 93:271–6. doi: 10.1038/icb.2015.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Zhang Y, Zhao M, Sawalha AH, Richardson B, Lu Q. Impaired DNA methylation and its mechanisms in CD4(+)T cells of systemic lupus erythematosus. J Autoimmun. (2013) 41:92–9. doi: 10.1016/j.jaut.2013.01.005 [DOI] [PubMed] [Google Scholar]
- 180. Zhao M, Zhou Y, Zhu B, Wan M, Jiang T, Tan Q, et al. IFI44L promoter methylation as a blood biomarker for systemic lupus erythematosus. Ann Rheum Dis. (2016) 75:1998–2006. doi: 10.1136/annrheumdis-2015-208410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Wu Z, Li X, Qin H, Zhu X, Xu J, Shi W. Ultraviolet B enhances DNA hypomethylation of CD4+ T cells in systemic lupus erythematosus via inhibiting DNMT1 catalytic activity. J Dermatol Sci. (2013) 71:167–73. doi: 10.1016/j.jdermsci.2013.04.022 [DOI] [PubMed] [Google Scholar]
- 182. Guderud K, Sunde LH, Flåm ST, Mæhlen MT, Mjaavatten MD, Lillegraven S, et al. Rheumatoid arthritis patients, both newly diagnosed and methotrexate treated, show more DNA methylation differences in CD4(+) memory than in CD4(+) naïve T cells. Front Immunol. (2020) 11:194. doi: 10.3389/fimmu.2020.00194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Zhao J, Wei K, Chang C, Xu L, Jiang P, Guo S, et al. DNA methylation of T lymphocytes as a therapeutic target: implications for rheumatoid arthritis etiology. Front Immunol. (2022) 13:863703. doi: 10.3389/fimmu.2022.863703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Julià A, Absher D, López-Lasanta M, Palau N, Pluma A, Waite Jones L, et al. Epigenome-wide association study of rheumatoid arthritis identifies differentially methylated loci in B cells. Hum Mol Genet. (2017) 26:2803–11. doi: 10.1093/hmg/ddx177 [DOI] [PubMed] [Google Scholar]
- 185. Karouzakis E, Gay RE, Gay S, Neidhart M. Increased recycling of polyamines is associated with global DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. (2012) 64:1809–17. doi: 10.1002/art.34340 [DOI] [PubMed] [Google Scholar]
- 186. Imgenberg-Kreuz J, Sandling JK, Almlöf JC, Nordlund J, Signér L, Norheim KB, et al. Genome-wide DNA methylation analysis in multiple tissues in primary Sjögren’s syndrome reveals regulatory effects at interferon-induced genes. Ann Rheum Dis. (2016) 75:2029–36. doi: 10.1136/annrheumdis-2015-208659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Konsta OD, Le Dantec C, Charras A, Cornec D, Kapsogeorgou EK, Tzioufas AG, et al. Defective DNA methylation in salivary gland epithelial acini from patients with Sjögren’s syndrome is associated with SSB gene expression, anti-SSB/LA detection, and lymphocyte infiltration. J Autoimmun. (2016) 68:30–8. doi: 10.1016/j.jaut.2015.12.002 [DOI] [PubMed] [Google Scholar]
- 188. Wang YY, Wang Q, Sun XH, Liu RZ, Shu Y, Kanekura T, et al. DNA hypermethylation of the forkhead box protein 3 (FOXP3) promoter in CD4+ T cells of patients with systemic sclerosis. Br J Dermatol. (2014) 171:39–47. doi: 10.1111/bjd.12913 [DOI] [PubMed] [Google Scholar]
- 189. Wang Y, Shu Y, Xiao Y, Wang Q, Kanekura T, Li Y, et al. Hypomethylation and overexpression of ITGAL (CD11a) in CD4(+) T cells in systemic sclerosis. Clin Epigenet. (2014) 6:25. doi: 10.1186/1868-7083-6-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Hu N, Qiu X, Luo Y, Yuan J, Li Y, Lei W, et al. Abnormal histone modification patterns in lupus CD4+ T cells. J Rheumatol. (2008) 35:804–10. [PubMed] [Google Scholar]
- 191. Tsou PS, Coit P, Kilian NC, Sawalha AH. EZH2 modulates the DNA methylome and controls T cell adhesion through junctional adhesion molecule A in lupus patients. Arthritis Rheumatol. (2018) 70:98–108. doi: 10.1002/art.40338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Liu L, Hu L, Yang L, Jia S, Du P, Min X, et al. UHRF1 downregulation promotes T follicular helper cell differentiation by increasing BCL6 expression in SLE. Clin Epigenet. (2021) 13:31. doi: 10.1186/s13148-021-01007-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Wang Y, Yang Y, Luo Y, Yin Y, Wang Q, Li Y, et al. Aberrant histone modification in peripheral blood B cells from patients with systemic sclerosis. Clin Immunol. (2013) 149:46–54. doi: 10.1016/j.clim.2013.06.006 [DOI] [PubMed] [Google Scholar]
- 194. Luo Q, Fu B, Zhang L, Guo Y, Huang Z, Li J. Decreased peripheral blood ALKBH5 correlates with markers of autoimmune response in systemic lupus erythematosus. Dis Markers. (2020) 2020:8193895. doi: 10.1155/2020/8193895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Luo Q, Rao J, Zhang L, Fu B, Guo Y, Huang Z, et al. The study of METTL14, ALKBH5, and YTHDF2 in peripheral blood mononuclear cells from systemic lupus erythematosus. Mol Genet Genomic Med. (2020) 8:e1298. doi: 10.1002/mgg3.1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Guo G, Wang H, Shi X, Ye L, Yan K, Chen Z, et al. Disease Activity-Associated Alteration of mRNA m(5) C Methylation in CD4(+) T Cells of Systemic Lupus Erythematosus. Front Cell Dev Biol. (2020) 8:430. doi: 10.3389/fcell.2020.00430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Li L, Xia X, Yang T, Sun Y, Liu X, Xu W, et al. RNA methylation: A potential therapeutic target in autoimmune disease. Int Rev Immunol. (2024) 43:160–77. doi: 10.1080/08830185.2023.2280544 [DOI] [PubMed] [Google Scholar]
- 198. Jiang H, Cao K, Fan C, Cui X, Ma Y, Liu J. Transcriptome-Wide High-Throughput m6A Sequencing of Differential m6A Methylation Patterns in the Human Rheumatoid Arthritis Fibroblast-Like Synoviocytes Cell Line MH7A. J Inflammation Res. (2021) 14:575–86. doi: 10.2147/jir.S296006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Wang J, Yan S, Lu H, Wang S, Xu D. METTL3 attenuates LPS-induced inflammatory response in macrophages via NF-κB signaling pathway. Mediators Inflammation. (2019) 2019:3120391. doi: 10.1155/2019/3120391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Xiao Q, Wu X, Deng C, Zhao L, Peng L, Zhou J, et al. The potential role of RNA N6-methyladenosine in primary Sjögren’s syndrome. Front Med (Lausanne). (2022) 9:959388. doi: 10.3389/fmed.2022.959388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Chen Y, Wu Y, Fang L, Zhao H, Xu S, Shuai Z, et al. METTL14-m6A-FOXO3a axis regulates autophagy and inflammation in ankylosing spondylitis. Clin Immunol. (2023) 257:109838. doi: 10.1016/j.clim.2023.109838 [DOI] [PubMed] [Google Scholar]
- 202. Berdasco M, Esteller M. Clinical epigenetics: seizing opportunities for translation. Nat Rev Genet. (2019) 20:109–27. doi: 10.1038/s41576-018-0074-2 [DOI] [PubMed] [Google Scholar]
- 203. Zhou Y, Qiu X, Luo Y, Yuan J, Li Y, Zhong Q, et al. Histone modifications and methyl-CpG-binding domain protein levels at the TNFSF7 (CD70) promoter in SLE CD4+ T cells. Lupus. (2011) 20:1365–71. doi: 10.1177/0961203311413412 [DOI] [PubMed] [Google Scholar]
- 204. Lu S, Wei X, Zhu H, Hu Z, Zheng M, Wu J, et al. m(6)A methyltransferase METTL3 programs CD4(+) T-cell activation and effector T-cell differentiation in systemic lupus erythematosus. Mol Med. (2023) 29:46. doi: 10.1186/s10020-023-00643-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Gillespie J, Savic S, Wong C, Hempshall A, Inman M, Emery P, et al. Histone deacetylases are dysregulated in rheumatoid arthritis and a novel histone deacetylase 3-selective inhibitor reduces interleukin-6 production by peripheral blood mononuclear cells from rheumatoid arthritis patients. Arthritis Rheum. (2012) 64:418–22. doi: 10.1002/art.33382 [DOI] [PubMed] [Google Scholar]
- 206. Mishra N, Brown DR, Olorenshaw IM, Kammer GM. Trichostatin A reverses skewed expression of CD154, interleukin-10, and interferon-gamma gene and protein expression in lupus T cells. Proc Natl Acad Sci U.S.A. (2001) 98:2628–33. doi: 10.1073/pnas.051507098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Mishra N, Reilly CM, Brown DR, Ruiz P, Gilkeson GS. Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J Clin Invest. (2003) 111:539–52. doi: 10.1172/jci16153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Reilly CM, Thomas M, Gogal R, Jr., Olgun S, Santo A, Sodhi R, et al. The histone deacetylase inhibitor trichostatin A upregulates regulatory T cells and modulates autoimmunity in NZB/W F1 mice. J Autoimmun. (2008) 31:123–30. doi: 10.1016/j.jaut.2008.04.020 [DOI] [PubMed] [Google Scholar]
- 209. Patel T, Patel V, Singh R, Jayaraman S. Chromatin remodeling resets the immune system to protect against autoimmune diabetes in mice. Immunol Cell Biol. (2011) 89:640–9. doi: 10.1038/icb.2010.144 [DOI] [PubMed] [Google Scholar]
- 210. Wang Y, Fan PS, Kahaleh B. Association between enhanced type I collagen expression and epigenetic repression of the FLI1 gene in scleroderma fibroblasts. Arthritis Rheum. (2006) 54:2271–9. doi: 10.1002/art.21948 [DOI] [PubMed] [Google Scholar]
- 211. Hemmatazad H, Rodrigues HM, Maurer B, Brentano F, Pileckyte M, Distler JH, et al. Histone deacetylase 7, a potential target for the antifibrotic treatment of systemic sclerosis. Arthritis Rheum. (2009) 60:1519–29. doi: 10.1002/art.24494 [DOI] [PubMed] [Google Scholar]
- 212. Regna NL, Vieson MD, Luo XM, Chafin CB, Puthiyaveetil AG, Hammond SE, et al. Specific HDAC6 inhibition by ACY-738 reduces SLE pathogenesis in NZB/W mice. Clin Immunol. (2016) 162:58–73. doi: 10.1016/j.clim.2015.11.007 [DOI] [PubMed] [Google Scholar]
- 213. Grabiec AM, Korchynskyi O, Tak PP, Reedquist KA. Histone deacetylase inhibitors suppress rheumatoid arthritis fibroblast-like synoviocyte and macrophage IL-6 production by accelerating mRNA decay. Ann Rheum Dis. (2012) 71:424–31. doi: 10.1136/ard.2011.154211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Christensen DP, Gysemans C, Lundh M, Dahllöf MS, Noesgaard D, Schmidt SF, et al. Lysine deacetylase inhibition prevents diabetes by chromatin-independent immunoregulation and β-cell protection. Proc Natl Acad Sci U.S.A. (2014) 111:1055–9. doi: 10.1073/pnas.1320850111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Regna NL, Chafin CB, Hammond SE, Puthiyaveetil AG, Caudell DL, Reilly CM. Class I and II histone deacetylase inhibition by ITF2357 reduces SLE pathogenesis in vivo . Clin Immunol. (2014) 151:29–42. doi: 10.1016/j.clim.2014.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Ge Z, Da Y, Xue Z, Zhang K, Zhuang H, Peng M, et al. Vorinostat, a histone deacetylase inhibitor, suppresses dendritic cell function and ameliorates experimental autoimmune encephalomyelitis. Exp Neurol. (2013) 241:56–66. doi: 10.1016/j.expneurol.2012.12.006 [DOI] [PubMed] [Google Scholar]
- 217. Kim DS, Min HK, Kim EK, Yang SC, Na HS, Lee SY, et al. Suberoylanilide hydroxamic acid attenuates autoimmune arthritis by suppressing th17 cells through NR1D1 inhibition. Mediators Inflammation. (2019) 2019:5648987. doi: 10.1155/2019/5648987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Chen H, Pan J, Wang JD, Liao QM, Xia XR. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, induces apoptosis in rheumatoid arthritis fibroblast-like synoviocytes. Inflammation. (2016) 39:39–46. doi: 10.1007/s10753-015-0220-3 [DOI] [PubMed] [Google Scholar]
- 219. Souliotis VL, Vougas K, Gorgoulis VG, Sfikakis PP. Defective DNA repair and chromatin organization in patients with quiescent systemic lupus erythematosus. Arthritis Res Ther. (2016) 18:182. doi: 10.1186/s13075-016-1081-3 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 220. Reilly CM, Mishra N, Miller JM, Joshi D, Ruiz P, Richon VM, et al. Modulation of renal disease in MRL/lpr mice by suberoylanilide hydroxamic acid. J Immunol. (2004) 173:4171–8. doi: 10.4049/jimmunol.173.6.4171 [DOI] [PubMed] [Google Scholar]
- 221. Li N, Zhao D, Kirschbaum M, Zhang C, Lin CL, Todorov I, et al. HDAC inhibitor reduces cytokine storm and facilitates induction of chimerism that reverses lupus in anti-CD3 conditioning regimen. Proc Natl Acad Sci U.S.A. (2008) 105:4796–801. doi: 10.1073/pnas.0712051105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Susick L, Senanayake T, Veluthakal R, Woster PM, Kowluru A. A novel histone deacetylase inhibitor prevents IL-1beta induced metabolic dysfunction in pancreatic beta-cells. J Cell Mol Med. (2009) 13:1877–85. doi: 10.1111/j.1582-4934.2009.00672.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Chan MW, Chang CB, Tung CH, Sun J, Suen JL, Wu SF. Low-dose 5-aza-2’-deoxycytidine pretreatment inhibits experimental autoimmune encephalomyelitis by induction of regulatory T cells. Mol Med. (2014) 20:248–56. doi: 10.2119/molmed.2013.00159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Dees C, Schlottmann I, Funke R, Distler A, Palumbo-Zerr K, Zerr P, et al. The Wnt antagonists DKK1 and SFRP1 are downregulated by promoter hypermethylation in systemic sclerosis. Ann Rheum Dis. (2014) 73:1232–9. doi: 10.1136/annrheumdis-2012-203194 [DOI] [PubMed] [Google Scholar]
- 225. Li XF, Wu S, Yan Q, Wu YY, Chen H, Yin SQ, et al. PTEN methylation promotes inflammation and activation of fibroblast-like synoviocytes in rheumatoid arthritis. Front Pharmacol. (2021) 12:700373. doi: 10.3389/fphar.2021.700373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Tóth DM, Ocskó T, Balog A, Markovics A, Mikecz K, Kovács L, et al. Amelioration of autoimmune arthritis in mice treated with the DNA methyltransferase inhibitor 5’-azacytidine. Arthritis Rheumatol. (2019) 71:1265–75. doi: 10.1002/art.40877 [DOI] [PubMed] [Google Scholar]
- 227. Li H, Tsokos MG, Bickerton S, Sharabi A, Li Y, Moulton VR, et al. Precision DNA demethylation ameliorates disease in lupus-prone mice. JCI Insight. (2018) 3:2–4. doi: 10.1172/jci.insight.120880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Hogg SJ, Beavis PA, Dawson MA, Johnstone RW. Targeting the epigenetic regulation of antitumour immunity. Nat Rev Drug Discovery. (2020) 19:776–800. doi: 10.1038/s41573-020-0077-5 [DOI] [PubMed] [Google Scholar]
- 229. Shen Y, Yang R, Zhao J, Chen M, Chen S, Ji B, et al. The histone deacetylase inhibitor belinostat ameliorates experimental autoimmune encephalomyelitis in mice by inhibiting TLR2/MyD88 and HDAC3/NF-κB p65-mediated neuroinflammation. Pharmacol Res. (2022) 176:105969. doi: 10.1016/j.phrs.2021.105969 [DOI] [PubMed] [Google Scholar]
- 230. Wang L, Liu Y, Beier UH, Han R, Bhatti TR, Akimova T, et al. Foxp3+ T-regulatory cells require DNA methyltransferase 1 expression to prevent development of lethal autoimmunity. Blood. (2013) 121:3631–9. doi: 10.1182/blood-2012-08-451765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Zhao M, Jiang J, Zhao M, Chang C, Wu H, Lu Q. The application of single-cell RNA sequencing in studies of autoimmune diseases: a comprehensive review. Clin Rev Allergy Immunol. (2021) 60:68–86. doi: 10.1007/s12016-020-08813-6 [DOI] [PubMed] [Google Scholar]
- 232. Fasano S, Milone A, Nicoletti GF, Isenberg DA, Ciccia F. Precision medicine in systemic lupus erythematosus. Nat Rev Rheumatol. (2023) 19:331–42. doi: 10.1038/s41584-023-00948-y [DOI] [PubMed] [Google Scholar]
- 233. Wang X, Waschke BC, Woolaver RA, Chen SMY, Chen Z, Wang JH. HDAC inhibitors overcome immunotherapy resistance in B-cell lymphoma. Protein Cell. (2020) 11:472–82. doi: 10.1007/s13238-020-00694-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Shen P, Deng X, Chen Z, Ba X, Qin K, Huang Y, et al. SIRT1: A potential therapeutic target in autoimmune diseases. Front Immunol. (2021) 12:779177. doi: 10.3389/fimmu.2021.779177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Choi D, Kim J, Yang JW, Kim JH, Park S, Shin JI. Dysregulated microRNAs in the pathogenesis of systemic lupus erythematosus: A comprehensive review. Int J Biol Sci. (2023) 19:2495–514. doi: 10.7150/ijbs.74315 [DOI] [PMC free article] [PubMed] [Google Scholar]