

Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disease characterized by amyloid plaques and cognitive decline, the latter of which is thought to be driven by soluble amyloid-β oligomers (oAb). The dysregulation of G protein-gated inwardly rectifying K+ (GIRK, also known as Kir3) channels has been implicated in rodent models of AD. Here, seeking mechanistic insights, we uncovered a sex-dependent facet of GIRK-dependent signaling in AD-related amyloid pathophysiology. Synthetic oAb(1–42) suppressed GIRK-dependent signaling in hippocampal neurons from male mice, but not from female mice. This effect required cellular prion protein, the receptor mGluR5, and production of arachidonic acid by the phospholipase PLA2. Although oAb suppressed GIRK channel activity only in male hippocampal neurons, intra-hippocampal infusion of oAb or genetic suppression of GIRK channel activity in hippocampal pyramidal neurons impaired performance on a memory test in both male and female mice. Moreover, genetic enhancement of GIRK channel activity in hippocampal pyramidal neurons blocked the oAb-induced cognitive impairment in both male and female mice. In APP/PS1 AD-model mice, GIRK-dependent signaling was diminished in hippocampal CA1 pyramidal neurons from only male mice before cognitive deficit was detected. However, enhancing GIRK channel activity rescued cognitive deficits in older APP/PS1 mice of both sexes. Thus, whereas diminished GIRK channel activity contributes to cognitive deficits in male mice with increased oAb burden, enhancing its activity may have therapeutic potential for both sexes.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by cognitive decline (1). Pathological features of AD include the accumulation of amyloid beta (Aβ) plaques and hyperphosphorylated tau protein in the brain. In the early stages of AD, there is a disruption in the balance between neuronal excitation and inhibition (2, 3), leading to synaptic dysfunction that underpins cognitive decline and neurodegeneration. The hippocampus (HPC) is a crucial cognitive substrate that exhibits hyperactivity in early AD and atrophy along disease progression (4, 5). Studies in rodent AD models have demonstrated aberrant excitability in HPC glutamatergic pyramidal neurons and have implicated this dysfunction in cognitive deficits (6).
Among the different forms of Aβ aggregates derived from cleavage of amyloid precursor protein (APP), soluble oligomeric Aβ (oAβ) is considered the most toxic and pathogenic (7). The onset and severity of cognitive impairment in AD correlate more strongly with oAβ level than with plaques (8). oAβ enhances neuronal excitability, disrupting excitation/inhibition balance during prodromal and mild impairment stages of AD (7, 9). oAβ has a multi-faceted influence on glutamatergic neurotransmission (10). For example, oAβ increases glutamate release (11), dysregulates ionotropic glutamate receptor function (12), interacts with cellular prion protein (PrPC) to co-activate metabotropic glutamate receptor 5 (mGluR5) (13), increases intracellular Ca2+ (14), and induces excitotoxicity (15). The impact of oAβ on inhibitory signaling, however, is less understood.
Several lines of evidence suggest that decreased inhibitory signaling involving the GABAB receptor (GABABR) and one of its key somatodendritic effectors—the G protein-gated inwardly rectifying K+ (GIRK) channel (16)—contribute to AD pathogenesis. For example, GABABR level is reduced in the cortex and HPC of post-mortem human AD samples (17, 18), and meta-analysis of AD-dysregulated genes identified GABBR1, which encodes the integral GABABR1 subunit of GABABR (19), as down-regulated in several brain regions, including the HPC (20). Similarly, RNA-Seq data from the AD brain shows down-regulation of KCNJ6 (21), which encodes the integral GIRK2 subunit of neuronal GIRK channels (16). KCNJ6 is also 1 of 3 genes found in the only AD-associated gene module common to White, Hispanic, and African American populations (22). Furthermore, in the well-characterized APPswe/PS1dE9 (APP/PS1) mouse model of AD (23), increased internalization of GABABR1(24), as well as GIRK1 and GIRK2(25, 26), is observed throughout the CA1 sub-region of the dorsal HPC (dHPC) of male mice at 12 months of age.
At present, the impact of oAβ on GABABR and/or GIRK-dependent signaling is unclear. GIRK channel upregulation was implicated in the apoptotic effect of Aβ1–42 at high concentration in cultured mouse HPC neurons; this effect involved NMDAR activation and rapid redistribution of GIRK channels to the plasma membrane (27). In contrast, acute Aβ25–35 decreased expression of GIRK channel subunits (but not GABABR) in rat HPC slices (28), and intracerebroventricular (ICV) infusion of oAβ1–42 decreased GIRK1 protein level in the mouse HPC (CA1). Aβ25–35 was also found to increase CA3 pyramidal neuron excitability in rat HPC slices, an effect mimicked by the GIRK channel blocker tertiapin (29). Protective effects of both the GABABR agonist baclofen and direct GIRK channel agonist ML297 on synaptic transmission, synaptic plasticity, and HPC-dependent cognition have been reported in acute amyloidopathy models in rodents (30–34).
Here, we sought to understand the impact of oAβ on GABABR-GIRK signaling in HPC neurons. Using a combination in vitro, ex vivo, and in vivo acute amyloidopathy models, we found that oAβ suppressed somatodendritic GABABR-dependent signaling in HPC neurons from male but not female mice, in a GIRK-dependent manner, starting at rather early pathological stages. We further found that inhibiting GIRK channel activity mimicked oAβ-induced synapse loss and cognitive deficits, and strengthening GIRK channel activity ameliorated cognitive deficits observed in both acute amyloidopathy and transgenic mouse models of AD.
RESULTS
oAβ suppresses GABABR-GIRK signaling in neurons from male but not female mice
We began by measuring the impact of oAβ on GABABR-dependent whole-cell currents in HPC cultures prepared from individual mouse pups of undefined sex. Cultures were infected with AAV8-CaMKIIα-GFP to highlight excitatory neurons (Fig. 1A). Using recording conditions designed to accentuate GIRK-dependent responses (35), we found that the GABABR agonist baclofen reliably evoked large inward whole-cell currents in GFP-labeled HPC neurons from wild-type but not Girk2−/− mice (Fig. 1A).
Figure 1. oAβ suppresses GABABR-dependent signaling in male HPC neurons.
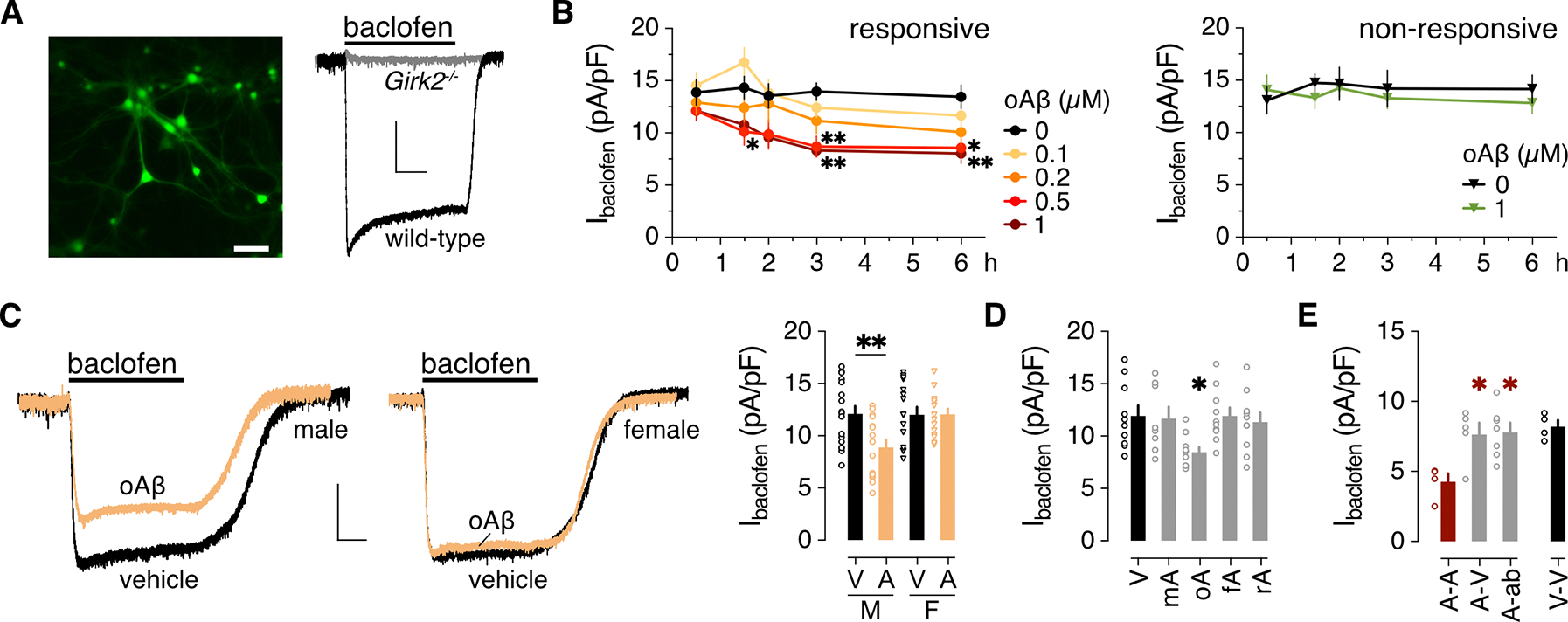
(A) Representative image of an HPC culture 4 days after infection with AAV8-CaMKIIα-GFP to highlight excitatory neurons, and representative traces of whole-cell inward currents from GFP-positive wild-type and Girk2−/− neurons following bath application of 100 μM baclofen. Scale bars: 50 μm, left; 500 pA/5 s, right. (B) Impact of oAβ pre-treatment on Ibaclofen density in “responsive” and “non-responsive” HPC cultures, collected from 8 neurons per timepoint and concentration pooled from 5 independent cultures for the responsive group, and 5 (0 μM) or 6 (1 μM) neurons per timepoint pooled from 4 independent cultures for the non-responsive group. Data were analyzed by two-way ANOVA and Dunnett’s multiple comparisons: *P<0.05 and **P<0.01 vs. 0 μM oAβ. (C) Baclofen-induced currents (Ibaclofen) in male and female HPC neurons (scale: 500 pA/5 s), and the summary of Ibaclofen densities for male (M) and female (F) neurons treated with vehicle (V) or oAβ (A: 0.5 μM for 3 h), collected from 17 (M/V), 15 (M/A), 16 (F/V), 15 (F/A) neurons across 4 independent cultures. Data were analyzed by two-way ANOVA and Šídák’s multiple comparisons, **P<0.01. (D) Ibaclofen densities following 3-hour treatment with vehicle (V, 10 cells) or 0.5 μM monomeric Aβ1–42 (mA, 8 neurons), oligomeric Aβ1–42 (oA, 10 neurons), fibrillary Aβ1–42 (fA, 10 neurons), or oligomeric Aβ42–1 (rA, 10 neurons), collected across 3 independent cultures. Data were analyzed by one-way ANOVA and Dunnett’s multiple comparisons, *P<0.05 vs. vehicle. (E) Ibaclofen densities in HPC neurons treated with 0.5 μM oAβ for 6 hours (A), measured 18 hours after replacement with media containing 0.5 μM oAβ (A-A, 4 neurons) or vehicle (A-V, 5 neurons), or anti-Aβ antibody D54D2 (A-ab; 0.16 μg/mL; 7 neurons), collected across 3 independent cultures. Data were analyzed by one-way ANOVA and Dunnett’s multiple comparisons, *P<0.05 vs. A-A. Data from a vehicle-only treatment group (V-V, 4 neurons) are presented on the right for comparison but were not included in the statistical analysis. All data are presented as mean ± SEM.
Pre-treatment of HPC cultures with oAβ provoked a concentration and time-dependent suppression of baclofen-induced current (Ibaclofen) in some, but not all cultures (Fig. 1B). In the “responsive” cultures, maximal (40%) suppression of Ibaclofen was achieved with a 3-hour incubation in 0.5 μM oAβ. oAβ was without effect in other “non-responsive” cultures, even after a 6-hour incubation with 1 μM oAβ. Using HPC cultures from pups of defined sex, we determined that oAβ suppressed Ibaclofen in male but not female HPC neurons (Fig. 1C). A male-specific suppression of Ibaclofen (fig. S1, A and B) accompanied by a depolarized resting membrane potential and decreased rheobase (table S1), was also observed in CA1 pyramidal neurons in slices of the dorsal HPC (dHPC) following acute (3–6 hours) oAβ incubation. Ibaclofen was also suppressed in dHPC CA1 pyramidal neurons in slices from mice 1 wk after intracranial infusion of oAβ into the dHPC (fig. S1, C and D). Thus, oAβ provoked a male-specific suppression of Ibaclofen in HPC neurons across in vitro, ex vivo, and in vivo models.
Ibaclofen suppression in male HPC neurons was specific to the oligomeric form of Aβ1–42, as treatment with Aβ1–42 monomers or fibrils, or a reversed peptide control (oAβ42–1), was without effect (Fig. 1D). To determine whether the oAβ-induced suppression of Ibaclofen was reversible, we incubated male HPC cultures with oAβ (0.5 μM) for 6 hours, washed the cells, and added media containing vehicle, oAβ, or the anti-Aβ antibody D54D2. Ibaclofen measured 18 hours later was larger in neurons from vehicle and D54D2 treatment groups relative to the oAβ treatment group, and comparable to Ibaclofen in cultures exposed only to vehicle (Fig. 1E), showing that Ibaclofen recovers following oAβ removal.
Suppression of GABABR-GIRK signaling may be secondary to the synaptotoxic and neurotoxic effects of oAβ on HPC neurons in culture (36, 37). To test this premise, we used a longitudinal quantitative imaging assay (38, 39) involving a PSD95 intrabody to track excitatory synapse count in HPC cultures. Even at the highest oAβ concentration tested (1 μM), oAβ did not trigger more synapse loss than vehicle in either male (fig. S2, A and B) or female (fig. S2, C and D) HPC cultures after 3 hours—the timepoint when maximal Ibaclofen suppression was observed.
oAβ suppresses GIRK channel activity in male neurons
GIRK channels mediate the G protein-dependent postsynaptic inhibitory effects of several inhibitory neurotransmitters and neuromodulators in HPC neurons, including adenosine and serotonin (5-HT) (40, 41). GIRK channels in HPC neurons are heterotetrameric complexes containing GIRK1 and GIRK2 subunits, and both subunits are required for GIRK channel activity (35, 42, 43). Indeed, whole-cell inward currents evoked by adenosine and 5-HT were negligible in HPC neurons from male and female Girk2−/− mice (Fig. 2A). As was the case for Ibaclofen, oAβ suppressed currents evoked by 5-HT and adenosine by ~40% in HPC neurons from male but not female mice (Fig. 2, B and C), suggesting that the oAβ-induced suppression of Ibaclofen is mediated by a reduction in GIRK channel activity. Consistent with this premise, oAβ pretreatment reduced whole-cell currents evoked by the direct GIRK channel activator ML297 (IML297) in HPC neurons from male but not female mice (Fig. 2D). Like the suppression of Ibaclofen, the impact of oAβ on IML297 was unique to oligomeric Aβ1–42 (Fig. 2E) and was reversible (Fig. 2F); either removal of oAβ or incubation with anti-Aβ antibody D54D2 rescued IML297.
Figure 2. oAβ suppresses GIRK channel activity.
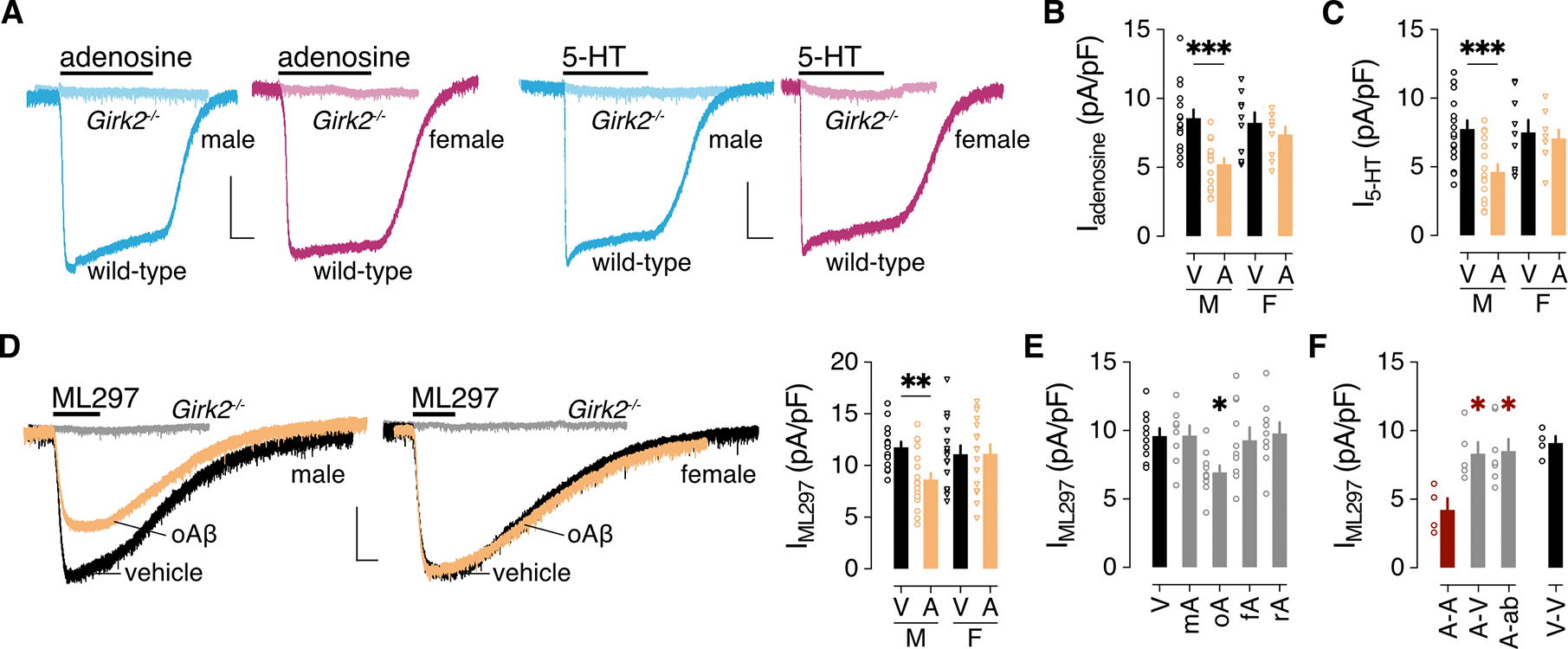
(A) Representative currents evoked by adenosine (10 μM) and 5-HT (10 μM) in HPC neurons from male (blue) and female (pink) wild-type and Girk2−/− mice. Scale bar: 500 pA/5 s. (B) Iadenosine density (pA/pF) in male (M) and female (F) HPC pyramidal neurons following a 3-hour treatment with vehicle (V) or 0.5 μM oAβ (A), collected from 15 (M/V), 15 (M/A), 9 (F/V), 9 (F/A) neurons across 4 independent cultures. Data were analyzed by two-way ANOVA and Šídák’s multiple comparisons, ***P<0.001. (C) I5-HT density (pA/pF) in male (M) and female (F) HPC pyramidal neurons following a 3-hour treatment with vehicle (V) or 0.5 μM oAβ (A), collected from 17 (M/V), 16 (M/A), 9 (F/V), 9 (F/A) neurons across 4 independent cultures. Data were analyzed by two-way ANOVA and Šídák’s multiple comparisons, ***P<0.001. (D) Representative currents evoked by ML297 (IML297, 10 μM) in HPC neurons from male and female wild-type mice following treatment with vehicle or oAβ (scale: 500 pA/10 s). Gray traces show the lack of ML297-induced current in HPC neurons from Girk2−/− mice. On the right, a summary of IML297 densities for male (M) and female (F) HPC neurons treated with vehicle (V) or oAβ (A: 0.5 μM for 3 h), collected from 14 (M/V), 19 (M/A), 15 (F/V), 16 (F/A) neurons across 4 independent cultures. Data were analyzed by two-way ANOVA and Šídák’s multiple comparisons, **P<0.01. (E) IML297 density following a 3-hour treatment with vehicle (V, 10 neurons) or 0.5 μM monomeric Aβ1–42 (mA, 8 neurons), oligomeric Aβ1–42 (oA, 10 neurons), fibrillary Aβ1–42 (fA, 10 neurons), or oligomeric Aβ42–1 (rA, 9 neurons), collected from 3 independent cultures. Data were analyzed by one-way ANOVA and Dunnett’s multiple comparisons, *P<0.05 vs. vehicle. (F) IML297 densities in HPC neurons treated with 0.5 μM oAβ for 24 hours, measured 24 hours after replacement with media containing 0.5 μM oAβ (A-A, 4 neurons) or vehicle (A-V, 5 neurons), or anti-Aβ antibody D54D2 (A-ab; 0.16 μg/mL; 7 neurons), collected from 3 independent cultures. Data were analyzed by one-way ANOVA and Dunnett’s multiple comparisons, *P<0.05 vs. A-A. Data from a vehicle-only treatment group (V-V, 4 neurons) are presented on the right for comparison but were not included in the statistical analysis. All data are presented as mean ± SEM.
GIRK3 also contributes to GIRK channel formation in the HPC (43, 44), and this subunit has been implicated in subcellular trafficking of neuronal GIRK channels and some forms of GIRK channel plasticity (45–49). oAβ suppressed Ibaclofen and IML297 in HPC cultures from Girk3−/− mice to a similar degree as seen in wild-type HPC cultures (fig. S3A), however, arguing against a role for GIRK3 in this neuroadaptation. NMDA receptor (NMDAR) activation has also been implicated in some forms of GIRK channel plasticity (50, 51), and oAβ can activate NMDAR in HPC cultures (52). NMDAR activation can also trigger the dynamin-dependent internalization of GABABR in HPC neurons (53). However, neither co-incubation with the NMDAR antagonist APV (fig. S3B) nor treatment with the dynamin inhibitor dynasore (fig. S3C) prevented the oAβ-induced suppression of Ibaclofen and IML297 in male HPC neurons. Collectively, these data suggest that the oAβ-induced suppression of GABABR-GIRK signaling in male HPC neurons does not require GIRK3 or NMDAR activation, and it does not involve internalization of key signaling pathway elements.
oAβ inhibits GIRK-dependent signaling in male neurons by PrPC-mGluR5-PLA2 activation
oAβ interacts with PrPC to co-activate mGluR5 (13), and this interaction has been proposed to occur selectively in male mice (54). Consistent with this finding, pre-incubation of male HPC cultures with a PrPC antibody (6D11) precluded the oAβ-induced suppression of Ibaclofen or IML297, but it had no impact on these currents on its own (Fig. 3A). Similarly, co-incubation of male HPC cultures with the mGluR5 antagonist MTEP blocked the oAβ-induced suppression of Ibaclofen and IML297, whereas MTEP alone did not impact these currents (Fig. 3B).
Figure 3. oAβ inhibits GIRK channel activity through PrPC-mGluR5-PLA2 activation.
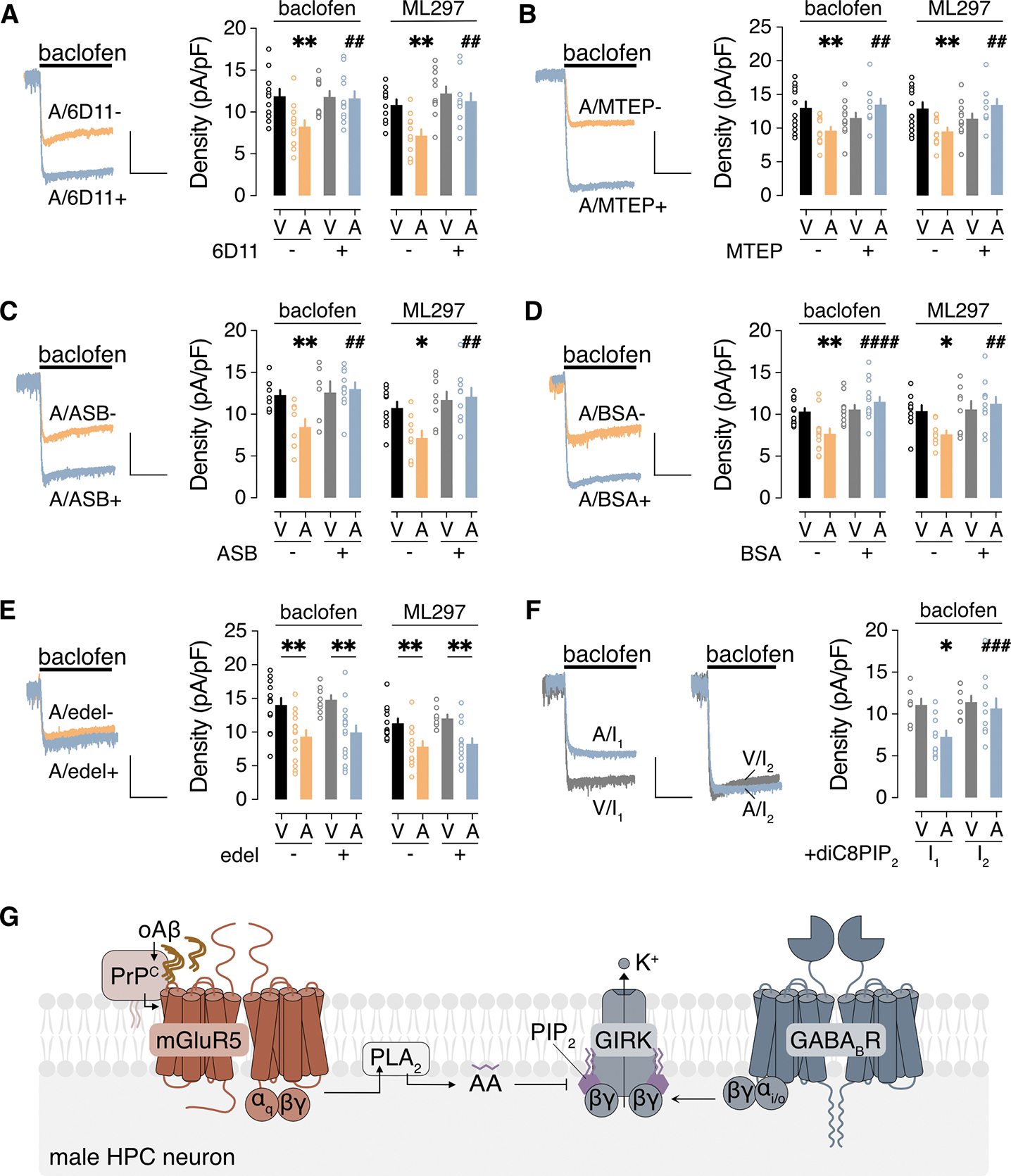
(A to F) Ibaclofen and IML297 in male HPC neurons incubated with vehicle (V) or 0.5 μM oAβ (A, 3 hours), with (+) or without (−) the indicated agents to interrogate underlying signaling mechanisms: (A) PrPC antibody 6D11 pretreatment (2.5 μg/mL for 30 min); (B) mGluR5 antagonist MTEP (10 μM); (C) PLA2 inhibitor ASB 14780 (ASB, 5 μM); (D) bovine serum albumin (BSA, fatty acid-free, 0.5 mg/mL); (E) PLC inhibitor edelfosine (edel, 10 μM); (F) PIP2 analog diC8PIP2 (25 μM) in the internal solution, measured immediately after whole-cell formation (I1) and then 90 s later (I2). Scale: 500 pA/5 s. Data are presented as mean ± SEM from 6 to 14 neurons in 3 to 4 independent cultures per condition. Data in (A to D) were analyzed by two-way ANOVA and Šídák’s multiple comparisons: *P<0.05 and **P<0.01 vs. V/-; ## P<0.01 and ####P< 0.0001 vs. A/−. Data in (E) were analyzed by two-way ANOVA and Šídák’s multiple comparisons: **P<0.01. Data in (F) were analyzed by two-way ANOVA with repeated measures and Šídák’s multiple comparisons: *P<0.05 vs. V/I1; ###P<0.001 vs. A/I1. (G) Schematic of the signaling pathway underlying the oAβ-induced suppression of GIRK-dependent signaling in male HPC neurons.
Prior work has shown that mGluR5 activation can inhibit GIRK channel activity in HPC neurons through a mechanism involving phospholipase A2 (PLA2) activation and arachidonic acid (AA) production (55, 56). Consistent with these reports, pharmacological inhibition of PLA2 with ASB 14780 (57) (Fig. 3C) or pyrrophenone (58) (fig. S4A), or sequestration of AA by fatty acid-free bovine serum albumin (59) (BSA; Fig. 3D), prevented the oAβ-induced suppression of Ibaclofen and IML297. Furthermore, the oAβ-induced suppression of GIRK-dependent signaling was not prevented by the phospholipase C (PLC) inhibitors edelfosine (Fig. 3E) or neomycin (60) (fig. S4B).
AA has been proposed to disrupt the interaction between GIRK channels and phosphatidylinositol 4,5 bisphosphate (PIP2) (61, 62), a requisite co-factor for GIRK channel activity (63, 64). To test this premise, we added the PIP2 analog diC8PIP2 to the pipette solution and measured whole-cell currents immediately after achieving whole-cell access and then again after 90 s, to allow for intracellular diffusion of diC8PIP2. As consecutive bath applications of ML297 resulted in smaller second responses in pilot experiments, we used this approach for Ibaclofen measurements only. With diC8PIP2 in the internal solution, the first and second Ibaclofen recordings in vehicle-treated neurons were indistinguishable (Fig. 3F). In oAβ-treated neurons, however, the second Ibaclofen was larger than the first, consistent with a rescuing influence of diC8PIP2. Collectively, our data suggest that oAβ-induced suppression of GIRK-dependent signaling in male HPC neurons is dependent on PrPC and involves activation of mGluR5 and PLA2, leading to increased production of AA which disrupts the interaction between GIRK channels and PIP2 (Fig. 3G).
oAβ and GIRK channel inhibition similarly provoke excitatory synapse loss in male neurons
Synapse loss is closely correlated with cognitive decline in AD (65–67), and oAβ has well-documented synaptotoxic effects (7, 9, 37). As such, we next examined the impact of oAβ and GIRK channel inhibition on excitatory synapse dynamics in HPC cultures. Cultures were treated with oAβ or the GIRK channel inhibitor tertiapin, or their respective vehicles, and excitatory synapse count was assessed at 24 and 48 hours, as described (68). In male HPC cultures, treatment with either oAβ (Fig. 4A) or the GIRK channel inhibitor tertiapin (Fig. 4B) decreased excitatory synapse count at 24- and 48-hour timepoints (Fig. 4C). In contrast, female HPC cultures were resistant to the synaptotoxic effects of oAβ at 24 and 48 hours (Fig. 4, D and F). Whereas tertiapin promoted synapse loss in female cultures after 48 hours, no effect was seen at 24 hours (Fig. 4, E and F). Thus, inhibition of GIRK channel activity mimics the synaptotoxic effects of oAβ in male HPC neurons and, though delayed, is sufficient to drive synapse loss in female HPC neurons.
Figure 4. oAβ and GIRK channel inhibitor effect on excitatory synapses in cultured HPC neurons.
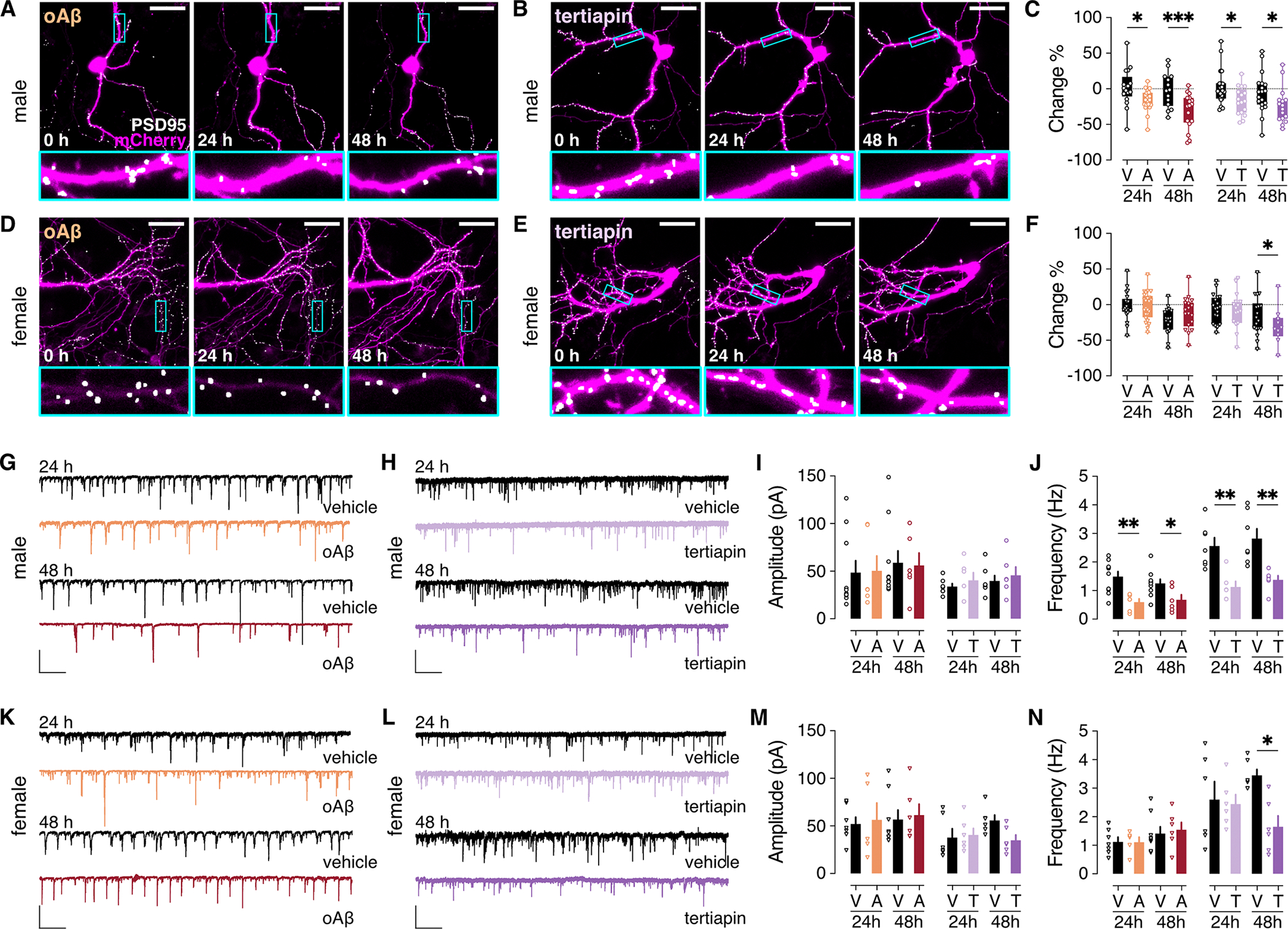
(A and B) Cultured HPC neurons from male mice were transfected with the plasmids pAAV-hSyn-mCherry (to fill and label neuronal structures) and pAAV-Syn-PSD95.FingR-eGFP-CCR5TC (to label excitatory post-synaptic densities). Images were taken before (t=0) and after (24 and 48 hours) addition of oAβ (1 μM; A) or the GIRK channel inhibitor tertiapin (1 μM; B) to the media. Scale bar: 30 μm. Boxed regions in upper panels are displayed at higher magnification in lower panels. (C) Percent change in excitatory synapse count 24 and 48 hours in male HPC neurons treated with oAβ (A) or vehicle (V), or tertiapin (T) or vehicle (V). oAβ data were extracted from 21 (24 hours) or 17 (48 hours) ROIs and corresponding vehicle data were extracted from 17 (24 hours) or 14 (48 hours) ROIs. Tertiapin data were extracted from 17 ROIs (24 and 48 hours) and corresponding vehicle data were extracted from 23 (24 hours) or 19 (48 hours) ROIs. Data were derived from 8 coverslips per group pooled from 4 independent cultures. (D to F) As in (A to C), in HPC neurons from female mice. oAβ n = 24 (24 hours) or 18 (48 hours) ROIs and corresponding vehicle n = 19 (24 hours) or 18 (48 hours) ROIs; tertiapin n = 16 (24 hours) or 10 (48 hours) ROIs, and corresponding vehicle n = 23 (24 hours) or 19 (48 hours) ROIs. Data were derived from 8 coverslips per group pooled from 4 independent cultures. (G and H) sEPSCs in HPC neurons from male mice, measured 24 and 48 hours after treatment with oAβ (1 μM) or vehicle (G), or the GIRK channel inhibitor tertiapin (1 μM) or vehicle (H); scale: 100 pA/5s. (I and J) sEPSC amplitude (I) and frequency (J) summary in male HPC neurons from 10 (V/24 hours), 6 (A/24 hours), 10 (V/48 hours), and 6 (A/48 hours) neurons pooled from 3 independent cultures, as well as 7 (V/24 hours), 6 (T/24 hours), 7 (V/48 hours), 6 (T/48 hours) neurons pooled from 3 independent cultures. (K to N) As in (G to J) in HPC neurons from female mice. n = 7 (V/24 hours), 5 (A/24 hours), 8 (V/48 hours), and 6 (A/48 hours) neurons pooled from 3 independent cultures, as well as 6 neurons per group (V/T/24 hours/48 hours) pooled from 3 independent cultures. Data are presented in box-and-whisker format (min-max) (C,F) or mean ± SEM (I,J,M,N). Data were analyzed by two-way ANOVA with (C,F) or without (I,J,M,N) repeated measures (mixed-effects) and Šídák’s multiple comparisons, *P<0.05,** P<0.01,***P<0.001.
To complement the imaging approach, we also measured spontaneous excitatory postsynaptic currents (sEPSCs) in excitatory neurons in HPC cultures from male and female mice, 24 and 48 hours following oAβ or tertiapin treatment (Fig. 4, G to N). sEPSC amplitudes were comparable across all treatment groups at 24 and 48 hours (Fig. 4, G to I and K to M). In contrast, sEPSC frequencies were decreased in male neurons at 24 and 48 hours by either oAβ or tertiapin treatment (Fig. 4, G, H, and J). While sEPSC frequency was not impacted by oAβ in female neurons, a significant reduction of frequency was observed in tertiapin-treated female neurons at 48 hours (Fig. 4, L and N), in alignment with the imaging data. Collectively, these data show that oAβ and GIRK channel inhibition both provoke loss of excitatory synapses, to a comparable extent and on a similar timeline, in male but not female HPC neurons.
Strengthening GIRK channel activity prevents oAβ-induced deficits in novel object recognition
Intra-HPC infusion of oAβ in mice disrupts performance in tests of HPC-dependent learning and memory, including novel object recognition (NOR) (69, 70). Because prolonged pharmacological activation of either GABABR (with baclofen) or GIRK channels (with ML297) induced suppression of GIRK-dependent currents (fig. S5), we used persistent genetic enhancement or suppression of GIRK channel activity, together with intra-HPC infusion of oAβ, to probe the relationship between the oAβ-induced suppression of GIRK channel activity and NOR performance. To enhance or suppress GIRK channel activity in HPC excitatory neurons, we used viral vectors to over-express GIRK2 (AAV8-CaMKIIα-GFP-IRES-GIRK2) or a dominant-negative GIRK2 variant (GIRK2DN; AAV8-CaMKIIα-GFP-IRES-GIRK2DN). Control neurons were treated with AAV8-CaMKIIα-GFP. Ectopic expression of GIRK2 or GIRK2DN enhanced or suppressed, respectively, Ibaclofen in HPC neurons from male and female mice (Fig. 5, A and B). Notably, GIRK2DN expression occluded suppression of Ibaclofen induced by oAβ in male HPC neurons, and GIRK2 expression enhanced Ibaclofen in male and female HPC neurons in the absence or presence of oAβ (Fig. 5B).
Figure 5. Impact of genetic manipulation of GIRK channel activity on oAβ-induced NOR deficit.
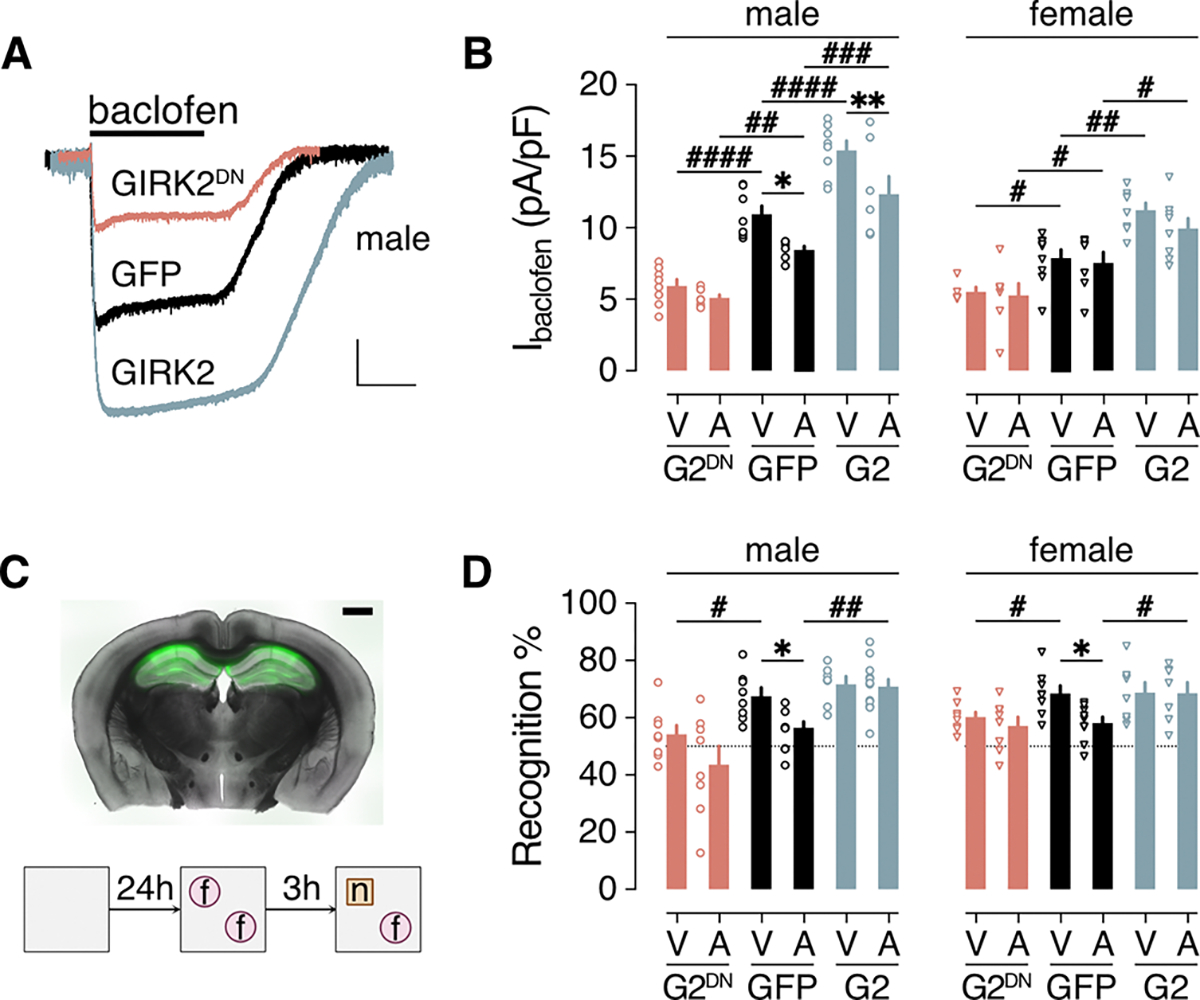
(A) Ibaclofen traces from HPC neurons infected with AAV8-CaMKIIα-GFP (GFP), AAV8-CaMKIIα-GFP-IRES-GIRK2 (GIRK2), or AAV8-CaMKIIα-GFP-IRES-GIRK2DN (GIRK2DN). Scale bar: 500 pA/10 s. (B) Summary of Ibaclofen densities in cultured male and female HPC neurons infected with GFP, G2, or G2DN vectors, and pretreated with vehicle (V) or oAβ (A, 0.5 μM). Data were collected from 8 (V/G2DN), 8 (A/G2DN), 8 (V/GFP), 6 (A/GFP), 8 (V/G2), 7 (A/G2) male neurons in 3 independent cultures and 5 (V/G2DN), 7 (A/G2DN), 9 (V/GFP), 7 (A/GFP), 8 (V/G2), 8 (A/G2) female neurons in 3 independent cultures. (C) A representative section from an adult male C57BL/6J mouse 2 weeks after bilateral intra-HPC infusion of AAV8-CaMKIIα-GFP and oAβ (scale bar: 1 mm, top), and a schematic of the novel object recognition (NOR) task. (D) Recognition index summary for adult male and female C57BL/6J mice, 1 week following intra-HPC treatment with vehicle (V) or oAβ (A), and either GFP, GIRK2 (G2), or GIRK2DN (G2DN) vector. Group sizes were 8 (V/G2DN), 8 (A/G2DN), 8 (V/GFP), 8 (A/GFP), 7 (V/G2), 12 (A/G2) male mice and 8 (V/G2DN), 8 (A/G2DN), 8 (V/GFP), 9 (A/GFP), 7 (V/G2), 8 (A/G2) female mice. Data are presented as mean ± SEM. Data were analyzed by two-way ANOVA and either Šídák’s multiple comparisons (*P<0.05,**P<0.01, within vehicle/oAβ treatment) or Dunnett’s multiple comparisons (#P<0.05,## P<0.01,### P<0.001,####P<0.0001 within viral treatment groups vs. GFP).
GIRK2, GIRK2DN, and GFP vectors were co-infused with oAβ or vehicle into the dHPC of adult (70 to 80 days) C57BL/6J mice, and NOR performance was assessed 1 week later (Fig. 5C). For both male and female mice in the vehicle-treatment group, suppression of GIRK channel activity (by GIRK2DN expression) decreased NOR (Fig. 5D). oAβ also decreased NOR relative to vehicle-treated males and females in the GFP treatment group (Fig. 5D). GIRK2 overexpression, blocked the oAβ-induced disruption of NOR in both male and female mice. Thus, suppressing GIRK channel activity in the dHPC decreased NOR performance in male and female mice, whereas enhancing GIRK channel activity prevented the NOR deficit provoked by oAβ in both sexes.
GIRK-dependent signaling is reduced in CA1 pyramidal neurons from male APP/PS1 mice
The data presented above show that GIRK channel activity in HPC neurons is suppressed in in vitro, ex vivo, and in vivo acute amyloidopathy models. To extend this effort, we next measured GIRK-dependent signaling in APP/PS1 mice, a transgenic mouse model of AD wherein cognitive deficits typically emerge between 8 and 12 months of age (71). In dHPC slices from 6- and 12-month-old mice, we measured the outward Ibaclofen in CA1 pyramidal neurons; this composite current is mediated primarily (~80%) by GIRK channel activation (42, 43). At both ages, Ibaclofen was smaller in CA1 pyramidal neurons from male APP/PS1 mice relative to wild-type control littermates, but was normal in female APP/PS1 mice (Fig. 6, A and B). Whereas most measures of excitability did not differ in dHPC CA1 pyramidal neurons from APP/PS1 and wild-type control littermates, resting membrane potential was depolarized in neurons from 12-month-old male APP/PS1 mice relative to controls (table S2).
Figure 6. GIRK-dependent signaling and NOR performance in APP/PS1 mice.
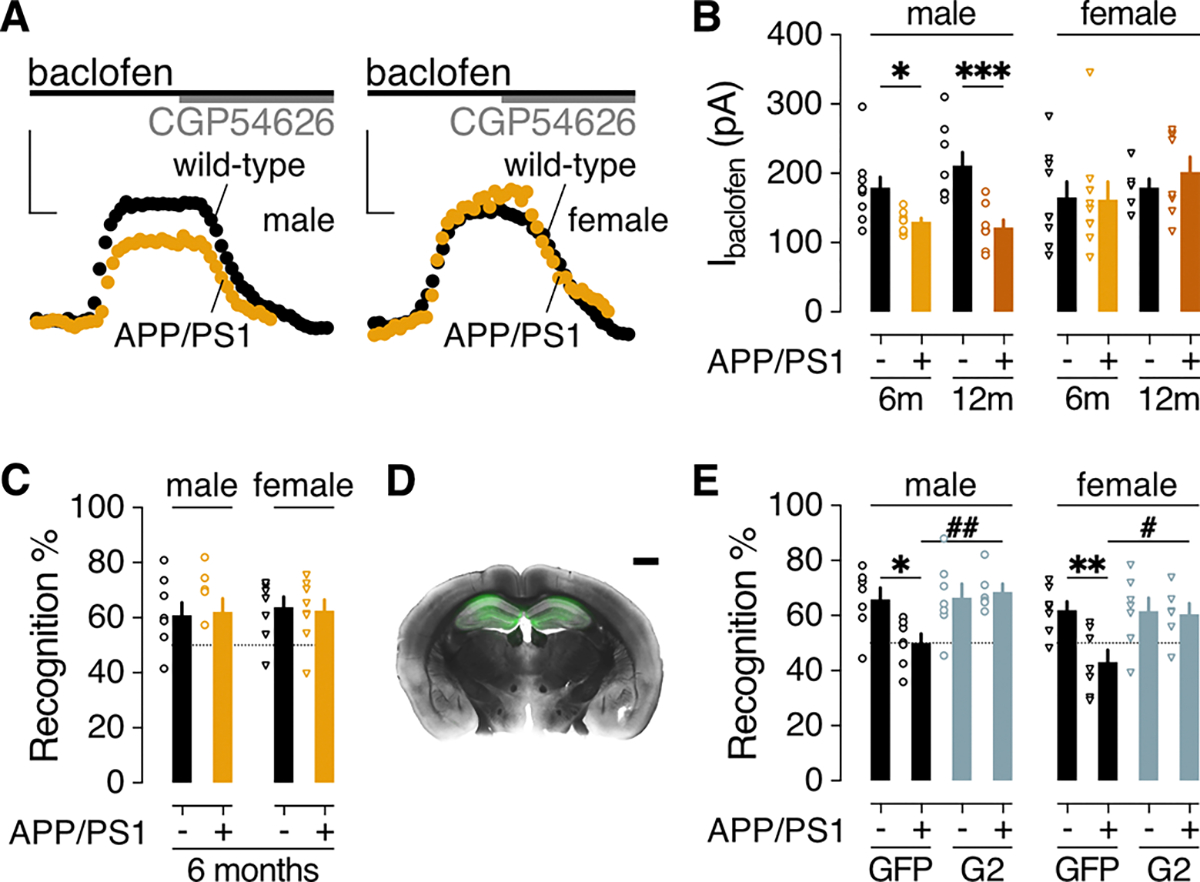
(A) Representative baclofen-induced outward currents (200 μM; Vhold= −60 mV) and their reversal by CGP54626 (2 μM) in dHPC CA1 pyramidal neurons from 6-month-old male and female APP/PS1 and wild-type control littermates. Scale bars: 100 pA/50 s. (B) Ibaclofen summary in dHPC CA1 pyramidal neurons from 6-month-old and 12-month-old APP/PS1 (+) and wild-type control (−) littermates. Male data were collected from 10 neurons from 3 mice (6m/−), 8 neurons from 3 mice (6m/+), 8 neurons from 2 mice (12m/−), 8 neurons from 2 mice (12m/+); female data were collected from 9 neurons from 3 mice (6m/−), 9 neurons from 2 mice (6m/+), 6 neurons from 2 mice (12m/−), 8 neurons from 2 mice (12m/+). Data were analyzed by two-way ANOVA and Šídák’s multiple comparisons: *P<0.05,***P<0.001. (C) Recognition index in the NOR test for 6-month-old male (8 mice per group) and female (8 mice per group) APP/PS1 (+) and wild-type control (−) littermates. P>0.05, unpaired t test. (D) Representative section from a 12-month-old male APP/PS1 mouse, 3 weeks after bilateral intra-HPC infusion of AAV8-CaMKIIα-GIRK2. Scale bar: 1 mm. (E) Recognition index in the NOR test for 12-month-old male APP/PS1 (+) and wild-type control (−) littermates, measured 2–3 weeks after bilateral intra-HPC infusion of GFP or GIRK2 (G2) expression vectors. Group sizes were 7 (GFP/−), 7 (GFP/+), 7 (G2/−), and 6 (G2/+) male mice and 7 (GFP/−), 7 (GFP/+), 7 (G2/−), and 6 (G2/+) female mice. Data were analyzed by two-way ANOVA and Šídák’s multiple comparisons: *P<0.05,**P<0.01 within genotype; #P<0.05,##P<0.01 within viral treatment. Data in (B, C, E) are presented as mean ± SEM.
Notably, APP/PS1 mice did not exhibit NOR deficits at 6 months of age (Fig. 6C). Thus, diminished GABABR-GIRK signaling in CA1 pyramidal neurons is measurable in APP/PS1 male mice prior to the onset of NOR deficit. Moreover, the subcellular distributions of GABABR1 and GIRK2 in the stratum radiatum of the CA1 sub-region in the dHPC were unchanged in male and female APP/PS1 mice at 6 months (fig. S6, A to G), suggesting that the smaller Ibaclofen in CA1 pyramidal neurons from male mice at this age is not due to internalization of GABABR or GIRK channels. While increased internalization of GABABR1 and GIRK2 was reported throughout the dHPC CA1 sub-region of 12-month-old male APP/PS1 mice (24–26), we found that the subcellular distribution of these proteins was unaltered in 12-month female APP/PS1 mice (fig. S6, H to J).
Strengthening GIRK channel activity rescues NOR performance in aged APP/PS1 mice
We used a separate cohort of older mice (11–12 months) to test whether strengthening GIRK channel activity could rescue NOR performance. Male and female APP/PS1 and wild-type control littermates were given bilateral dHPC infusions of either the GIRK2 overexpression or GFP control vector 2 to 3 weeks before NOR testing (Fig. 6D). Consistent with data from oAβ-treated HPC neurons in culture (Fig. 5B), GIRK2 overexpression enhanced Ibaclofen in CA1 pyramidal neurons from male and female APP/PS1 mice as compared to APP/PS1 littermates treated with the control vector (fig. S7). GIRK2 overexpression also correlated with a more hyperpolarized resting membrane potential in male CA1 pyramidal neurons (table S2). Both male and female APP/PS1 mice treated with the control vector showed impaired NOR performance relative to wild-type control littermates, and this impairment was rescued by GIRK2 overexpression in both male and female APP/PS1 mice (Fig. 6E). Thus, while diminished Ibaclofen in the dHPC is seen only in male APP/PS1 mice, strengthening GIRK channel activity in the dHPC rescued NOR performance in both male and female APP/PS1 mice.
DISCUSSION
Here, we found that oAβ rapidly and reversibly suppressed GABABR-GIRK signaling in male but not female cultured HPC neurons, an effect recapitulated in CA1 pyramidal neurons of the dHPC in multiple acute amyloidopathy models. We also found a deficit in GABABR-GIRK signaling in a transgenic mouse AD model characterized by APP over-expression and enhanced production of Aβ (23); GABABR-GIRK signaling was diminished in dHPC CA1 pyramidal neurons from male but not female APP/PS1 mice at 6 months. Surface densities of GABABR1 and GIRK2 in dHPC CA1 were normal in these mice, suggesting that the decreased Ibaclofen is not due to increased internalization of receptor or channel. Consistent with this premise, the oAβ-induced suppression of GABABR-GIRK signaling did not require GIRK3, which can promote GIRK channel internalization (45, 46), nor was it prevented by inhibiting dynamin-dependent endocytosis.
The oAβ-induced suppression of GABABR-GIRK signaling in cultured HPC neurons requires mGluR5 activation and it can be rescued by PIP2. PIP2 is required for GIRK channel gating (63, 64), and oAβ can decrease PIP2 in HPC neurons in an mGluR5-dependent fashion (72). Reduced PIP2 levels were found in brains from humans and rodents carrying the apolipoprotein E4 (APOE4) allele (73), the most significant genetic risk factor for sporadic AD (74). Genetic augmentation of PIP2 levels prevents Aβ-induced synaptotoxicity and disruption of synaptic plasticity (75, 76), and it can rescue cognitive deficits in transgenic AD and ApoE4 knock-in mice (76, 77). Since activation of Gαq-coupled GPCRs can weaken GIRK-dependent signaling by PIP2 depletion (16, 78), we predicted that the oAβ-induced suppression of GIRK-dependent signaling was mediated by PLC activation. Our data instead implicate PLA2 activation and AA production, in alignment with studies showing that activation of group 1 metabotropic glutamate receptors (which includes mGluR5) suppresses GABABR-GIRK signaling in in HPC CA1 neurons in a PLA2- and AA-dependent manner (55, 56). The mechanism underlying the AA-mediated disruption of GIRK channel activity is unclear, but competitive inhibition of the PIP2 binding site on the GIRK channel is a plausible explanation (62). Notably, PLA2 inhibition blocks the oAβ-induced increase in AA production and associated neurotoxicity in HPC cultures, and PLA2 ablation improved cognition in a transgenic AD model (79).
oAβ failed to suppress GIRK-dependent signaling or drive excitatory synapse loss in female HPC cultures. oAβ was similarly ineffective in weakening GIRK-dependent signaling in CA1 pyramidal neurons in female amyloidopathy models. Inhibition of GIRK channel activity with tertiapin drove excitatory synapse loss and decreased excitatory synaptic connectivity in female HPC cultures, but this effect was delayed relative to that in oAβ- or tertiapin-treated male cultures. This sex difference suggests that the resilience to oAβ-induced synapse loss in female HPC cultures is due in part to an inability of oAβ to suppress GIRK channel activity. Co-activation of PrPC/mGluR5 has been linked to oAβ-induced synapse loss, disruption of synaptic plasticity, neurotoxicity, and cognitive deficits (13, 80–82)(13, 83–87). However, oAβ-induced co-activation of PrPC/mGluR5 was reported to occur in males only, due to a lack of PrPC-mGluR5 interaction in females (54). This may explain the resilience of female HPC neurons to oAβ noted in several of our tests.
Loss-of-function mouse models involving GIRK-dependent signaling exhibit cognitive dysfunction (16, 88). For example, ICV infusion of the GIRK channel blocker tertiapin in male C57BL/6J mice disrupted HPC-dependent behaviors including NOR (89). In addition, fear learning is disrupted in constitutive Girk2−/− and CaMKIICre(+):Girk2fl/fl mice, the latter of which exhibit a loss of GIRK channel activity in forebrain pyramidal neurons, including the pyramidal neurons of the dorsal HPC (90). Notably, viral reconstitution of GIRK channel activity in the dHPC of CaMKIICre(+):Girk2fl/fl mice restored deficits in fear learning (91). Here, we show that viral suppression of GIRK channel activity in the dHPC impairs NOR in male and female C57BL/6J mice. Moreover, viral enhancement of GIRK channel activity prevented the NOR impairment induced by intra-dHPC oAβ in C57BL/6J mice of both sexes. Our electrophysiological data indicate that NOR deficits induced by intra-dHPC oAβ in female C57BL/6J mice are not attributable to diminished GIRK-dependent signaling, at least in CA1 pyramidal neurons. These findings were mirrored in the APP/PS1 model; 12-month-old female APP/PS1 mice showed deficits in NOR performance despite normal GABABR-GIRK signaling in CA1 pyramidal neurons, and NOR performance was rescued by viral enhancement of GIRK channel activity. Thus, while suppression of GIRK-dependent signaling contributes to cognitive deficits in male AD models, strengthening GIRK-dependent signaling in the dHPC can have cognitive benefits in both sexes. These findings are consistent with evidence suggesting that enhancing neuronal inhibition or restoring excitation/inhibition balance may be a beneficial strategy for treating AD in both sexes (92–95), (96).
Gain-of-function models involving GIRK-dependent signaling also exhibit cognitive dysfunction. For example, TS65Dn and GIRK2 trisomy mouse models of Down Syndrome exhibit enhanced GABABR-GIRK signaling and impaired NOR and associative learning (97–101). Viral RNAi suppression of GIRK2 in TS65Dn mice normalized the cognitive deficits (102). While intracerebroventricular (ICV) co-administration of ML297 prevented the Aβ-induced disruption of synaptic physiology in the HPC and associated cognitive deficits (32–34), infusion of ML297 alone in male C57BL/6J mice disrupted HPC-dependent behaviors including NOR (89). This, along with our finding that ML297 triggers a homologous desensitization of GIRK channel current in HPC neurons, prompted our interest in investigating persistent genetic enhancement of GIRK channel activity as a potential therapeutic approach. Of note, viral enhancement of GIRK-dependent signaling in the dHPC did not provoke NOR deficits in C57BL/6J or 12-month-old wild-type mice.
Findings from this study and others suggest that GIRK channel activity in the dHPC is essential to normal cognitive function, but deviations outside of a tolerated range of activity are disruptive. Thus, genetic interventions that normalize GIRK channel activity in the HPC may be promising therapeutic approaches for conditions associated with gain (Down Syndrome) or loss (AD) of GIRK channel activity, as seen with male mouse models in this study. Moreover, strengthening GIRK channel activity within a tolerated range may enhance cognitive function even when loss of GIRK channel function is not evident, as seen with female mouse models in this study.
MATERIALS AND METHODS
Animals
All C57BL/6J mice (RRID:IMSR_JAX:000664) used in this study were purchased from The Jackson Laboratory. Male B6.Cg-Tg(APPswe,PSEN1dE9)85Dbo/Mmjax mice (RRID:MMRRC_034832-JAX, male, heterozygous), hereafter referred to as APP/PS1 mice, were obtained from the Mutant Mouse Resource and Research Center (MMRRC) at The Jackson Laboratory, an NIH-funded strain repository, and was donated to the MMRRC by David Borchelt, PhD, McKnight Brain Institute, University of Florida. Male APP/PS1 mice were bred in-house with female C57BL/6J mice purchased from The Jackson Laboratory. The generation and characterization of Girk2−/− and Girk3−/− mice was reported previously (103, 104). Male (2 to 4 per cage) and female (2 to 5 per cage) mice were group-housed on a 14/10-hour light/dark cycle (lights on from 0600 to 2000 h) and given free access to water and food. All animal experiments were approved by the University of Minnesota Institutional Animal Care and Use Committee (2311–41518A).
Reagents
Aβ1–42 or Aβ42–1 were purchased from Bachem Americas. Baclofen, edelfosine, bovine serum albumin (BSA, fatty acid-free), neomycin sulfate, pyrrophenone, Neurobasal™-A Medium, fetal bovine serum (FBS), papain from Carica papaya, and DNase I were purchased from Sigma-Aldrich. Serotonin hydrochloride, adenosine, APV, and ASB 14780 were purchased from Tocris Bioscience. D54D2 β-amyloid primary antibody (XP® rabbit mAβ #8243, 0.16 mg/mL) was purchased from Cell Signaling Technology. Dynasore and MTEP were purchased from abcam. 6D11 was purchased from BioLegend. diC8PIP2 was purchased from Echelon Biosciences. EGTA (pH 8.0) was purchased from Research Products International. ML297 was a generous gift from Dr. C. David Weaver (Department of Pharmacology, Vanderbilt University). All drugs were stored, handled, and resuspended according to provider specifications. High-titer (>1 × 1012 genocopies/mL) AAV-CaMKIIα-GFP, AAV-CaMKIIα-GFP-IRES-GIRK2, and AAV-CaMKIIα-GFP-IRES-GIRK2DN were generated and purified in-house by the University of Minnesota Viral Vector Cloning Core using standard cloning techniques. pAAV-CaMKIIα-hChR2(C128S/D156A)-mCherry was a gift from Prof. Karl Deisseroth (Department of Bioengineering and of Psychiatry and Behavioral Sciences, Stanford University, Howard Hughes Medical Institute; Addgene plasmid #35502; http://n2t.net/addgene:35502; RRID:Addgene_35502) (105) and was used as the backbone in all cloning procedures.
Cell culture
Primary HPC neuron cultures were prepared from neonatal (P0–2) C57BL/6J pups, as described (35). Pup sex was determined by the presence of a pigment spot on the scrotum (106). Hippocampi were extracted and placed into ice-cold modified Hank’s Balanced Salt Solution (HBSS, Ca2+ and Mg2+ free, with 1 mM HEPES) containing 20% FBS, rinsed twice with FBS-free HBSS, digested for 20 min at 37°C with occasional inversion using papain (2.5% v/v) and DNase I (0.1% v/v) in digestion solution (137 mM NaCl, 0.5 mM KCl, 0.7 mM Na2PO4, 2.5 mM HEPES, pH 7.2). Tissue was then mechanically dissociated by pipetting in Neurobasal A-based plating medium (with 1x B27, 1x Glutamax, 1x antibiotic/antimycotic, and 0.05% DNase I). Cells were pelleted by centrifugation (2500 rpm/587 rcf for 10 min at room temperature). Cells were diluted with plating media accordingly and plated onto poly-L-Lysine (0.0005%) pre-coated 8-mm glass coverslips in 48-well plates for single-cell electrophysiological analysis, or 35-mm glass-bottomed petri dishes with 10-mm microwells (P35G-1.5–10-C, MatTek Life Sciences) for imaging. Before experimentation, cultures were maintained in a humidified 5% CO2 incubator at 37°C for 11 to 14 days, and half of the medium was replaced with fresh growth medium (Neurobasal A with 1x B27, 1x Glutamax, and 1x antibiotic/antimycotic) every 3 days.
Peptide preparation and validation
Aβ peptides were prepared and validated as described (75). In brief, we equilibrated synthetic Aβ1–42 or Aβ42–1 at room temperature for 20 min before resuspending with 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) to make a 1 mM stock. The solvent was then evaporated in a biosafety cabinet for 2–2.5 h, and the resulting peptide films were stored at −80°C. To prepare monomers, peptide films were resuspended to 1 mM in dimethyl sulfoxide (DMSO) by 10 min bath sonication; samples were then diluted in culture media or electrophysiological solutions to the final concentrations. To prepare oligomers, Aβ1–42 or Aβ42–1 films were resuspended to 1 mM in DMSO by 10 min bath sonication; samples were then diluted to 100 μM in cold phosphate-buffered saline (PBS) and vortexed for 30 s before overnight incubation at 4°C. To prepare fibrils, the 1 mM Aβ-DMSO solutions were diluted to 100 mM in 10 mM HCl and vortexed for 30 s before overnight incubation at 37°C. Oligomer and fibril solutions were diluted in culture media to their final concentrations. All Aβ preparations were made fresh from stored films either on the day of (monomers) or the night before (oligomers and fibrils) use; all the stored films were used within 4 months.
Electrophysiology in HPC cultures
Whole-cell patch-clamp recordings in cultured HPC neurons were performed as described (107). In brief, coverslips with neurons were transferred to a chamber containing a low K+ bath solution (130 mM NaCl, 5.4 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 5.5 mM D-Glucose, 5 mM HEPES, pH 7.4). To target excitatory neurons, cultures were infected with AAV8-CaMKIIα-GFP 7–8 d after cell plating, and GFP-positive neurons with pyramidal morphology and capacitance values between 100 and 200 pF were targeted for analysis. Fire-polished borosilicate patch pipettes (4–6 MΩ) were filled with K-Gluconate internal solution (140 mM K-Gluconate, 2 mM MgCl2, 1.1 mM EGTA, 5 mM HEPES, 2 mM Na2-ATP, 0.3 mM Na-GTP, and 5 mM phosphocreatine, pH 7.2). After achieving whole-cell access, neurons were held at −70 mV; liquid-junction potential was not corrected. To measure sEPSCs, a 2-min stable recording was acquired in the low K+ bath solution. The drug-evoked whole-cell currents were measured in a high-K+ bath solution (120 mM NaCl, 25 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 5.5 mM D-Glucose, 5 mM HEPES, pH 7.4). GPCR agonists and ML297 were diluted in the high-K+ bath solution and perfused directly onto the neuron using the ValveLink 8.2 rapid perfusion system (AutoMate Scientific). Whole-cell currents were acquired with an Axopatch-200B amplifier and pCLAMP v.8.2 software (Molecular Devices, LLC). All currents were low-pass filtered at 2 kHz, digitized at 10 kHz with a Digidata 1322A (Molecular Devices, LLC), and stored on a computer hard disk for subsequent analysis. Only experiments in which the access resistance (Ra) was stable (change pre- and post-perfusion <20%) and low (<15 MΩ) were included in the analysis. Current density (pA/pF) was calculated as the ratio of current amplitude to cell capacitance. sEPSC amplitude and frequency were analyzed in Mini Analysis Program 6.0 freeware, the parameter and filter settings were consistent across all treatment groups. Recordings were made from 1 neuron per coverslip for the drug-evoked whole-cell currents, or from 2 neurons per coverslip for sEPSC recordings. Coverslips/wells were assigned to treatment groups randomly; the recordings and analysis were performed blind to treatment information.
Synapse imaging
Longitudinal imaging of excitatory synapses in cultured HPC neurons was conducted as described (108, 109). In brief, 11–12 d after dissociated HPC neurons were plated on 35-mm glass-bottomed petri dishes with 10 mm microwells (P35G-1.5–10-C, MatTek), cultures were transfected with pAAV-Syn-PSD95.FingR-eGFP-CCR5TC [a gift from Xue Han (Boston University), Addgene plasmid #125693, http://n2t.net/addgene:125693; referred to as “hSyn-PSD95.FingR-eGFP” in the text] (109) and pAAV-hSyn-mCherry, which was generated from pAAV-hSyn-DIO-mCherry [a gift from Bryan Roth (The University of North Carolina at Chapel Hill), Addgene plasmid #50459; http://n2t.net/addgene:50459; RRID:Addgene_50459] by removing the LoxP/Lox2272 sites. Transfections used the calcium phosphate method, as described (38). Neurons were incubated in DMEM with 1 mM kynurenic acid, 10 mM MgCl2, and 5 mM HEPES for 30 min. A DNA/calcium phosphate precipitate containing 2 mg of DNA from each plasmid per well was then added. After a 60-min incubation period, cells were washed with DMEM supplemented with MgCl2 and HEPES, then returned to conditioned media that had been saved at the beginning of the procedure. Cells were imaged 2 to 3 days after transfection. Images were acquired on a Nikon A1 confocal microscope controlled by Elements software with a 60x oil immersion objective (1.4 numerical aperture). Images were 15-step z-stacks (1 μm z-step) acquired with a pixel size of 0.15 μm. Culture dishes were maintained at 37°C and 10% CO2 in a stage-top incubator mounted to a digitally controlled stage. Multiple regions of interest (ROIs), typically 3 to 4, were acquired per dish. The location of each ROI on the dish, as well as the position of the dish on the stage, were recorded and saved so that multiple images could be acquired from each ROI over time. ROIs were chosen by the experimenter based on the presence of clearly resolved mCherry-labeled dendrites and at least 50 fluorescent eGFP puncta. All puncta counts were limited within the chosen ROIs (1024 × 1024 pixel). Image acquisition prior to drug or vehicle treatment was taken as first timepoint (T=0 hours). All subsequent timepoints are denoted by the number of hours after the addition of drug or vehicle.
All images were analyzed in ImageJ (Fiji, imagej.net) using a modified version of a published script (39). The modified version used Groovy scripting and the CLIJ2 package (https://clij.github.io), which provides GPU acceleration. First, the image channel containing the synaptic marker (PSD95.FingR-eGFP) was deconvolved using the Richardson-Lucy method with 20 iterations. The mask channel (mCherry) was not deconvolved. Puncta were then identified as described (39). The cell mask (generated using the mCherry channel), was applied to the synaptic marker channel prior to thresholding. The settings for puncta size and elongated structure exclusion were kept the same across all images and experiments. For some ROIs, the difference of Gaussian (for mask generation), the power filter, the Tophat filter, and/or the Gaussian blur filter settings were modified to improve the accuracy of puncta identification in that ROI. Algorithm settings were kept the same for each individual ROI across time points. These settings were changed to account for varying image intensities and were made while blinded to the treatment group from which the image came. All puncta counts are expressed as a percent change from the 0-hour timepoint, calculated as the percentage of the changes in puncta counts (#punctatx-#punctat0) over the initial puncta count (#punctat0). Each individual ROI was considered an n of 1. The version of the script used for analyzing all images in this manuscript is deposited in Zenodo (DOI: 10.5281/zenodo.11491753).
Slice electrophysiology
Electrophysiology involving acutely-isolated HPC slices was performed as described (90, 110). Brains were sliced coronally (350 μm) in ice cold cutting solution (240 mM sucrose, 2 mM KCl, 3 mM MgCl2, 1 mM CaCl2, 1.25 mM NaH2PO4, 26 mM NaHCO3, 10 mM glucose pH 7.4; oxygenated with 95% O2, 5% CO2). Immediately after sectioning, slices were transferred into 34°C cutting solution and maintained at 34°C for 30 min. Slices were then transferred to room temperature artificial cerebrospinal fluid (aCSF) 124 mM NaCl, 2.5 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 1.25 mM NaH2PO4, 26 mM NaHCO3, 17 mM glucose, pH7.4; oxygenated with 95% O2, 5% CO2 and maintained for at least 1 hour before recordings started). In acute Aβ incubation experiments, slices were incubated for 3 to 6 hours with Aβ before the recordings started. Prior to each recording, slices were transferred into the chamber with a constant flow of oxygenated aCSF solution. Glass microelectrodes were filled with a K-gluconate-based internal pipette solution (140 mM K-gluconate, 2 mM MgCl2, 1.1 mM EGTA, 5 mM HEPES, 2 mM Na2ATP, 0.3 mM Na-GTP, 5 mM phosphocreatine, pH 7.2). Recordings were obtained with an EPC10 HEKA amplifier (Patchmaster 2 × 73.2, HEKA Elektronik), and command potentials factored in a −15-mV junction potential predicted by JPCalc software (Molecular Devices, LLC). Currents evoked by baclofen (200 μM) were measured at a holding potential of −60 mV. Holding current, input resistance, and series resistance values were measured throughout each experiment by tracking responses to periodic (0.2 Hz) voltage steps (−5 mV, 800 ms). Only experiments with stable (< 30% variation) and low series resistances (< 30 MΩ) were analyzed. Mice of the same genotype and sex were assigned to treatment groups randomly. Only 1 recording was obtained from an individual slice.
Intracranial surgeries and validations
Adult (70–75 days old) C57BL/6J and 11- to 12-month-old male and female APP/PS1 mice and wild-type control littermates were anesthetized with a mixture of oxygen (0.5 mL/min) and isoflurane (3%) delivered by nose cone, and they were positioned in a stereotaxic frame using a bite bar (David Kopf Instruments). Body temperature was maintained with a heating pad. Eyes were covered with artificial tears (Akorn) applied with a cotton swab, and gentamicin (5 mg/Kg) was administered by subcutaneous injection. Skin was cleaned with betadine before making the incision, leveling the skull, and drilling burr holes. Microinjectors were lowered into the skull through the burr holes to the dorsal HPC (from bregma: −1.90 mm A/P, ± 1.45 mm M/L, −1.45 mm D/V). For in vivo acute amyloidopathy studies involving C57BL/6J mice used for slice electrophysiology, 600 nL diluent of oAβ1–42 (180 ng) or its vehicle was injected into each side at a rate of 100 nL/min. For in vivo acute amyloidopathy studies involving C57BL/6J mice used for behavioral studies, 600 nL mixture of oAβ (180 ng) and viral vector was injected into each side. For studies involving APP/PS1 mice, 600 nL of AAV vector was injected into each side. Microinjectors were left at the injection site for 10 min to ensure full diffusion. Injectors were then removed, skin was sutured, and animals were left on a heating pad for at least 30 min. Pre-handling prior to behavioral analysis did not begin until at least 6 days after surgery.
For post hoc evaluation of intracranial targeting, mouse brains were isolated and sectioned coronally at 250 μm on a vibratome. Slices containing the dHPC were collected and injection locations were assessed by evaluation of viral-driven GFP fluorescence using a BZ-X810 epifluorescent microscope (Keyence). Images for each channel were obtained from multiple focus planes and stitched using Keyence BZ-X800 analysis software. Fluorescence images were overlaid with brightfield images. Only data from mice in which GFP expression was bilateral and confined to the HPC were included in the final analysis.
Behavioral analysis
Mice of the same genotype and sex were assigned to treatment groups randomly. The individual performing behavioral experiments was blind to the treatment information. Mice were evaluated in a 3-day novel object recognition (NOR) assay in an open-field apparatus. The ceiling light in the testing room was off, but the testing apparatus (custom 40 cm x 40 cm white plastic boxes) was illuminated by evenly spread LED lights at 70–90 LUX, 4000–4500 K. On Day 0 (D0; 0900–1000 h), mice were transferred to the testing room for acclimation (1 to 2 hours) and pre-handling. On Day 1 (0900–1000 h), mice were transferred to the testing room, and 1 hour later were placed in the open-field environments for 10 min for habituation. Activity, distance traveled, freezing, and immobile time were tracked by ANY-maze software (Stoelting Co) during the session. On Day 2 (0900–1000 h), mice were transferred to the testing room, and 1 hour later were put into the same apparatus as the day before and were allowed to freely explore the two identical objects (50 mL conical tubes filled with pink solution; positioned diagonally within the apparatus with cap down) during a 10-min session. Orientation of the mouse head toward the object and within a 2-cm range was considered as object “exploration”; total exploration time was recorded for each object. Mice were returned to their home cages for 3 hours, and then place back in the same apparatus with one object replaced by a novel object (25-cm culture flask containing a yellow solution; cap up). The time exploring the old (familiar) and novel object was measured during a 10-min session. Apparati were cleaned with 70% ethanol between each test. Recognition index was calculated as the percentage of time exploring the novel object relative to total exploration time during the test session.
Immunoelectron microscopy
The subcellular distribution of GIRK2 and GABABR1 was evaluated in spines and dendrites in the stratum radiatum of the dorsal HPC, using established pre-embedding immunogold electron microscopy and quantification procedures (24, 25, 111). GIRK2 and GABABR1 labeling was assessed in male and female APP/PS1 mice and wild-type control littermates at 6 months of age, and in 12-month-old female APP/PS1 and control mice. Comparable analyses of GIRK2 and GABABR1 in 12-month-old male APP/PS1 and control mice were published previously (24, 25). Immunoparticles identified in the plasma membrane of CA1 pyramidal neurons were counted and the perimeter of the subcellular compartment containing the immunoparticles was measured (ImageJ). For each experimental group, 3–4 animals were used. From each animal, 12 ROIs were obtained. The total number of immunoparticles GIRK2 and GABABR1 was defined as the particle count on the plasma membrane (PM) plus the count at intracellular sites. The linear density of GIRK2 and GABABR1 along the PM was expressed as the number of immunoparticles/mm. The distribution of GIRK2 and GABABR1 on the PM was calculated as the percentage of particle count on the PM over the total number of immunoparticles. The following primary antibodies were used: rabbit anti-GIRK2 (Rb-Af290; aa. 390–421 of mouse GIRK2A-1; RRID: AB_2571712; Frontier Institute Co.) and mouse anti-GABAB1 (clone N93A/49, Neuromab). The characteristics and specificity of the anti-GIRK2 antibody have been described elsewhere (112). The characteristics and specificity of the antibody targeting GABAB1 have been described by the manufacturer. The secondary antibodies used were goat anti-rabbit IgG and goat anti-mouse IgG, both conjugated to 1.4 nm gold particles (1:100; Nanoprobes Inc.).
Data analysis
Statistical analyses were conducted with Prism v10.2.3 (GraphPad Software) using unpaired or nested t-test or ANOVA (one-way, two-way, or two-way with repeated measures) and post hoc comparisons including Šídák’s and Dunnett’s multiple comparisons tests, as appropriate. Outlier identification tests (ROUT, Q = 2%) were run for all datasets to remove statistical outliers prior to analysis; 4 ROIs in the synapse imaging experiments were excluded based on the criteria. Sex was included as a factor in the initial primary culture electrophysiological studies. After establishing a sex difference related to the impact of oAβ on GABABR-GIRK signaling in HPC neurons, data for each sex were analyzed separately in subsequent experiments.
Supplementary Material
Acknowledgements:
The authors would like to thank Courtney Wright and Anna Souders for care of the mouse colony; Dr. Patricia Alvarez Munoz for assistance with the biochemical validation of Aβ preparations; Hannah McMullan and Jordan Casby for optimizing the synapse imaging assay; Dr. Thomas Pengo and the University of Minnesota Supercomputing Institute for assistance in the design and development of the synapse counting algorithm; Dr. Ling Li (University of Minnesota) for suggestions on experimental design and data interpretation; Dr. Sukru Aras (Baylor College of Medicine) for careful review of the statistical analysis; and the UMN Viral Vector and Cloning Core for AAV vector design and production.
Funding:
This work was supported by MCIN/AEI/ 10.13039/501100011033 (PID2021-125875OB-I00) and “ERDF A way of making Europe”, Junta de Comunidades de Castilla-La Mancha (SBPLY/21/180501/000064), and Universidad de Castilla-La Mancha (2023-GRIN-34187) to RL, as well as NIH grants to SAT (DA007304) and KW (NS128039, AA027544).
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability:
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. The datasets used and/or analyzed, as well as viral vector sequences and protocols used as part of the current study, are available from the corresponding author on reasonable request. The script used for analyzing synapse imaging data is deposited in Zenodo (DOI: 10.5281/zenodo.11491753). Girk−/− mouse lines, as well as custom viral vectors and related plasmids, are available from K.W. under a material transfer agreement with the University of Minnesota.
REFERENCES and NOTES
- 1.Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chetelat G, Teunissen CE, Cummings J, van der Flier WM, Alzheimer’s disease. Lancet 397, 1577–1590 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Busche MA, Konnerth A, Neuronal hyperactivity--A key defect in Alzheimer’s disease? Bioessays 37, 624–632 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Targa Dias Anastacio H, Matosin N, Ooi L, Neuronal hyperexcitability in Alzheimer’s disease: what are the drivers behind this aberrant phenotype? Transl. Psychiatry 12, 257 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morra JH, Tu Z, Apostolova LG, Green AE, Avedissian C, Madsen SK, Parikshak N, Hua X, Toga AW, Jack CR Jr., Schuff N, Weiner MW, Thompson PM, Alzheimer’s Disease Neuroimaging I, Automated 3D mapping of hippocampal atrophy and its clinical correlates in 400 subjects with Alzheimer’s disease, mild cognitive impairment, and elderly controls. Hum. Brain. Mapp. 30, 2766–2788 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huijbers W, Mormino EC, Schultz AP, Wigman S, Ward AM, Larvie M, Amariglio RE, Marshall GA, Rentz DM, Johnson KA, Sperling RA, Amyloid-beta deposition in mild cognitive impairment is associated with increased hippocampal activity, atrophy and clinical progression. Brain 138, 1023–1035 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Llorens-Martin M, Blazquez-Llorca L, Benavides-Piccione R, Rabano A, Hernandez F, Avila J, DeFelipe J, Selective alterations of neurons and circuits related to early memory loss in Alzheimer’s disease. Front. Neuroanat. 8, 38 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li S, Selkoe DJ, A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Abeta oligomers from Alzheimer’s brain. J. Neurochem. 154, 583–597 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Selkoe DJ, Hardy J, The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palop JJ, Mucke L, Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat. Neurosci. 13, 812–818 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Findley CA, Bartke A, Hascup KN, Hascup ER, Amyloid Beta-Related Alterations to Glutamate Signaling Dynamics During Alzheimer’s Disease Progression. ASN Neuro. 11, 1759091419855541 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hascup KN, Hascup ER, Soluble Amyloid-beta42 Stimulates Glutamate Release through Activation of the alpha7 Nicotinic Acetylcholine Receptor. J. Alzheimers Dis. 53, 337–347 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Babaei P, NMDA and AMPA receptors dysregulation in Alzheimer’s disease. Eur. J. Pharmacol. 908, 174310 (2021). [DOI] [PubMed] [Google Scholar]
- 13.Um JW, Kaufman AC, Kostylev M, Heiss JK, Stagi M, Takahashi H, Kerrisk ME, Vortmeyer A, Wisniewski T, Koleske AJ, Gunther EC, Nygaard HB, Strittmatter SM, Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron 79, 887–902 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cascella R, Cecchi C, Calcium Dyshomeostasis in Alzheimer’s Disease Pathogenesis. Int. J. Mol. Sci. 22, 4914 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Canas PM, Simoes AP, Rodrigues RJ, Cunha RA, Predominant loss of glutamatergic terminal markers in a beta-amyloid peptide model of Alzheimer’s disease. Neuropharmacology 76 Pt A, 51–56 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Luo H, Marron E Fernandez de Velasco, K. Wickman, Neuronal G protein-gated K(+) channels. Am. J. Physiol. Cell Physiol. 323, C439–C460 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chu DC, Penney JB, Young AB, Cortical GABAB and GABAA receptors in Alzheimer’s disease: a quantitative autoradiographic study. Neurology 37, 1454–1459 (1987). [DOI] [PubMed] [Google Scholar]
- 18.Iwakiri M, Mizukami K, Ikonomovic MD, Ishikawa M, Hidaka S, Abrahamson EE, DeKosky ST, Asada T, Changes in hippocampal GABABR1 subunit expression in Alzheimer’s patients: association with Braak staging. Acta Neuropathol. 109, 467–474 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Rose TR, Wickman K, Mechanisms and Regulation of Neuronal GABAB Receptor-Dependent Signaling. Curr. Top. Behav. Neurosci. 52, 39–79 (2022). [DOI] [PubMed] [Google Scholar]
- 20.Puthiyedth N, Riveros C, Berretta R, Moscato P, Identification of Differentially Expressed Genes through Integrated Study of Alzheimer’s Disease Affected Brain Regions. PloS One 11, e0152342 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao P, Mangleburg CG, Al-Ramahi I, Botas J, Shulman JM, Systems genetic dissection of Alzheimer’s disease brain gene expression networks. Alzheimer’s & Dementia 17, e058716 (2021). [Google Scholar]
- 22.Dai Y, Jia P, Zhao Z, Gottlieb A, A Method for Bridging Population-Specific Genotypes to Detect Gene Modules Associated with Alzheimer’s Disease. Cells 11, 2219 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR, Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum. Mol. Genet. 13, 159–170 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Martin-Belmonte A, Aguado C, Alfaro-Ruiz R, Moreno-Martinez AE, de la Ossa L, Martinez-Hernandez J, Buisson A, Fruh S, Bettler B, Shigemoto R, Fukazawa Y, Lujan R, Reduction in the neuronal surface of post and presynaptic GABAB receptors in the hippocampus in a mouse model of Alzheimer’s disease. Brain Pathol. 30, 554–575 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alfaro-Ruiz R, Martin-Belmonte A, Aguado C, Hernandez F, Moreno-Martinez AE, Avila J, Lujan R, The Expression and Localisation of G-Protein-Coupled Inwardly Rectifying Potassium (GIRK) Channels Is Differentially Altered in the Hippocampus of Two Mouse Models of Alzheimer’s Disease. Int. J. Mol. Sci. 22, 11106 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin-Belmonte A, Aguado C, Alfaro-Ruiz R, Moreno-Martinez AE, de la Ossa L, Aso E, Gomez-Acero L, Shigemoto R, Fukazawa Y, Ciruela F, Lujan R, Nanoscale alterations in GABA(B) receptors and GIRK channel organization on the hippocampus of APP/PS1 mice. Alzheimers Res. Ther. 14, 136 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.May LM, Anggono V, Gooch HM, Jang SE, Matusica D, Kerbler GM, Meunier FA, Sah P, Coulson EJ, G-Protein-Coupled Inwardly Rectifying Potassium (GIRK) Channel Activation by the p75 Neurotrophin Receptor Is Required for Amyloid β Toxicity. Front. Neurosci. 11, 455 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mayordomo-Cava J, Yajeya J, Navarro-Lopez JD, Jimenez-Diaz L, Amyloid-beta(25–35) Modulates the Expression of GirK and KCNQ Channel Genes in the Hippocampus. PloS One 10, e0134385 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nava-Mesa MO, Jiménez-Díaz L, Yajeya J, Navarro-Lopez JD, Amyloid-β induces synaptic dysfunction through G protein-gated inwardly rectifying potassium channels in the fimbria-CA3 hippocampal synapse. Front. Cell. Neurosci. 7, 117 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hajj R, Milet A, Toulorge D, Cholet N, Laffaire J, Foucquier J, Robelet S, Mitry R, Guedj M, Nabirotchkin S, Chumakov I, Cohen D, Combination of acamprosate and baclofen as a promising therapeutic approach for Parkinson’s disease. Sci. Rep. 5, 16084 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chumakov I, Nabirotchkin S, Cholet N, Milet A, Boucard A, Toulorge D, Pereira Y, Graudens E, Traore S, Foucquier J, Guedj M, Vial E, Callizot N, Steinschneider R, Maurice T, Bertrand V, Scart-Gres C, Hajj R, Cohen D, Combining two repurposed drugs as a promising approach for Alzheimer’s disease therapy. Sci. Rep. 5, 7608 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sanchez-Rodriguez I, Temprano-Carazo S, Najera A, Djebari S, Yajeya J, Gruart A, Delgado-Garcia JM, Jimenez-Diaz L, Navarro-Lopez JD, Activation of G-protein-gated inwardly rectifying potassium (Kir3/GirK) channels rescues hippocampal functions in a mouse model of early amyloid-beta pathology. Sci. Rep. 7, 14658 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanchez-Rodriguez I, Gruart A, Delgado-Garcia JM, Jimenez-Diaz L, Navarro-Lopez JD, Role of GirK Channels in Long-Term Potentiation of Synaptic Inhibition in an In Vivo Mouse Model of Early Amyloid-beta Pathology. Int. J. Mol. Sci. 20, 1168 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanchez-Rodriguez I, Djebari S, Temprano-Carazo S, Vega-Avelaira D, Jimenez-Herrera R, Iborra-Lazaro G, Yajeya J, Jimenez-Diaz L, Navarro-Lopez JD, Hippocampal long-term synaptic depression and memory deficits induced in early amyloidopathy are prevented by enhancing G-protein-gated inwardly rectifying potassium channel activity. J. Neurochem. 153, 362–376 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wydeven N, Young D, Mirkovic K, Wickman K, Structural elements in the Girk1 subunit that potentiate G protein-gated potassium channel activity. Proc. Natl. Acad. Sci. U S A 109, 21492–21497 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL, Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J. Neurosci. 27, 796–807 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patnaik A, Zagrebelsky M, Korte M, Holz A, Signaling via the p75 neurotrophin receptor facilitates amyloid-beta-induced dendritic spine pathology. Sci. Rep. 10, 13322 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hargus NJ, Thayer SA, Human immunodeficiency virus-1 Tat protein increases the number of inhibitory synapses between hippocampal neurons in culture. J. Neurosci. 33, 17908–17920 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Green MV, Thayer SA, HIV gp120 upregulates tonic inhibition through alpha5-containing GABAARs. Neuropharmacology 149, 161–168 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chung HJ, Ge WP, Qian X, Wiser O, Jan YN, Jan LY, G protein-activated inwardly rectifying potassium channels mediate depotentiation of long-term potentiation. Proc. Natl. Acad. Sci. U S A 106, 635–640 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnston A, McBain CJ, Fisahn A, 5-HT1A Receptor-Activation Hyperpolarizes Pyramidal Cells and Suppresses Hippocampal Gamma Oscillations via Kir3 Channel-Activation. J. Physiol. 592, 4187–4199 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luscher C, Jan LY, Stoffel M, Malenka RC, Nicoll RA, G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron 19, 687–695 (1997). [DOI] [PubMed] [Google Scholar]
- 43.Koyrakh L, Lujan R, Colon J, Karschin C, Kurachi Y, Karschin A, Wickman K, Molecular and cellular diversity of neuronal G-protein-gated potassium channels. J. Neurosci. 25, 11468–11478 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fernandez-Alacid L, Watanabe M, Molnar E, Wickman K, Lujan R, Developmental regulation of G protein-gated inwardly-rectifying K+ (GIRK/Kir3) channel subunits in the brain. Eur. J. Neurosci. 34, 1724–1736 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ma D, Zerangue N, Raab-Graham K, Fried SR, Jan YN, Jan LY, Diverse trafficking patterns due to multiple traffic motifs in G protein-activated inwardly rectifying potassium channels from brain and heart. Neuron 33, 715–729 (2002). [DOI] [PubMed] [Google Scholar]
- 46.Lunn ML, Nassirpour R, Arrabit C, Tan J, McLeod I, Arias CM, Sawchenko PE, Yates JR 3rd, Slesinger PA, A unique sorting nexin regulates trafficking of potassium channels via a PDZ domain interaction. Nat. Neurosci. 10, 1249–1259 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Lalive AL, Munoz MB, Bellone C, Slesinger PA, Luscher C, Tan KR, Firing modes of dopamine neurons drive bidirectional GIRK channel plasticity. J. Neurosci. 34, 5107–5114 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Munoz MB, Padgett CL, Rifkin R, Terunuma M, Wickman K, Contet C, Moss SJ, Slesinger PA, A Role for the GIRK3 Subunit in Methamphetamine-Induced Attenuation of GABAB Receptor-Activated GIRK Currents in VTA Dopamine Neurons. J. Neurosci. 36, 3106–3114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marron Fernandez de Velasco E, Tipps ME, Haider B, Souders A, Aguado C, Rose TR, Vo BN DeBaker MC, Lujan R, Wickman K, Ethanol-Induced Suppression of G Protein-Gated Inwardly Rectifying K(+)-Dependent Signaling in the Basal Amygdala. Biol. Psychiatry 94, 863–874 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chung HJ, Qian X, Ehlers M, Jan YN, Jan LY, Neuronal activity regulates phosphorylation-dependent surface delivery of G protein-activated inwardly rectifying potassium channels. Proc. Natl. Acad. Sci. U S A 106, 629–634 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Workman ER, Haddick PC, Bush K, Dilly GA, Niere F, Zemelman BV, Raab-Graham KF, Rapid antidepressants stimulate the decoupling of GABA receptors from GIRK/Kir3 channels through increased protein stability of 14–3-3eta. Mol. Psychiatry 20, 298–310 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Decker H, Jurgensen S, Adrover MF, Brito-Moreira J, Bomfim TR, Klein WL, Epstein AL, De Felice FG, Jerusalinsky D, Ferreira ST, N-methyl-D-aspartate receptors are required for synaptic targeting of Alzheimer’s toxic amyloid-beta peptide oligomers. J. Neurochem. 115, 1520–1529 (2010). [DOI] [PubMed] [Google Scholar]
- 53.Guetg N, Abdel Aziz S, Holbro N, Turecek R, Rose T, Seddik R, Gassmann M, Moes S, Jenoe P, Oertner TG, Casanova E, Bettler B, NMDA receptor-dependent GABAB receptor internalization via CaMKII phosphorylation of serine 867 in GABAB1. Proc. Natl. Acad. Sci. U S A 107, 13924–13929 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abd-Elrahman KS, Albaker A, de Souza JM, Ribeiro FM, Schlossmacher MG, Tiberi M, Hamilton A, Ferguson SSG, Abeta oligomers induce pathophysiological mGluR5 signaling in Alzheimer’s disease model mice in a sex-selective manner. Sci. Signal. 13, eabd2494 (2020). [DOI] [PubMed] [Google Scholar]
- 55.Sohn JW, Lim A, Lee SH, Ho WK, Decrease in PIP(2) channel interactions is the final common mechanism involved in PKC- and arachidonic acid-mediated inhibitions of GABA(B)-activated K+ current. J. Physiol. 582, 1037–1046 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sohn JW, Lee D, Cho H, Lim W, Shin HS, Lee SH, Ho WK, Receptor-specific inhibition of GABAB-activated K+ currents by muscarinic and metabotropic glutamate receptors in immature rat hippocampus. J. Physiol. 580, 411–422 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tomoo T, Nakatsuka T, Katayama T, Hayashi Y, Fujieda Y, Terakawa M, Nagahira K, Design, synthesis, and biological evaluation of 3-(1-Aryl-1H-indol-5-yl)propanoic acids as new indole-based cytosolic phospholipase A2alpha inhibitors. J. Med. Chem. 57, 7244–7262 (2014). [DOI] [PubMed] [Google Scholar]
- 58.Seno K, Okuno T, Nishi K, Murakami Y, Yamada K, Nakamoto S, Ono T, Pyrrolidine inhibitors of human cytosolic phospholipase A2. Part 2: synthesis of potent and crystallized 4-triphenylmethylthio derivative ‘pyrrophenone’. Bioorg. Med. Chem. Lett. 11, 587–590 (2001). [DOI] [PubMed] [Google Scholar]
- 59.Beck R, Bertolino S, Abbot SE, Aaronson PI, Smirnov SV, Modulation of arachidonic acid release and membrane fluidity by albumin in vascular smooth muscle and endothelial cells. Circ. Res. 83, 923–931 (1998). [DOI] [PubMed] [Google Scholar]
- 60.Suenami S, Iino S, Kubo T, Pharmacologic inhibition of phospholipase C in the brain attenuates early memory formation in the honeybee (Apis mellifera L.). Biol. Open 7, bio028191 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rogalski SL, Cyr C, Chavkin C, Activation of the endothelin receptor inhibits the G protein-coupled inwardly rectifying potassium channel by a phospholipase A2-mediated mechanism. J. Neurochem. 72, 1409–1416 (1999). [DOI] [PubMed] [Google Scholar]
- 62.Rogalski SL, Chavkin C, Eicosanoids inhibit the G-protein-gated inwardly rectifying potassium channel (Kir3) at the Na+/PIP2 gating site. J. Biol. Chem. 276, 14855–14860 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sui JL, Petit-Jacques J, Logothetis DE, Activation of the atrial KACh channel by the betagamma subunits of G proteins or intracellular Na+ ions depends on the presence of phosphatidylinositol phosphates. Proc. Natl. Acad. Sci. U S A 95, 1307–1312 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang CL, Feng S, Hilgemann DW, Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature 391, 803–806 (1998). [DOI] [PubMed] [Google Scholar]
- 65.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R, Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580 (1991). [DOI] [PubMed] [Google Scholar]
- 66.Chen MK, Mecca AP, Naganawa M, Finnema SJ, Toyonaga T, Lin SF, Najafzadeh S, Ropchan J, Lu Y, McDonald JW, Michalak HR, Nabulsi NB, Arnsten AFT, Huang Y, Carson RE, van Dyck CH, Assessing Synaptic Density in Alzheimer Disease With Synaptic Vesicle Glycoprotein 2A Positron Emission Tomographic Imaging. JAMA Neurol. 75, 1215–1224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mecca AP, Chen MK, O’Dell RS, Naganawa M, Toyonaga T, Godek TA, Harris JE, Bartlett HH, Zhao W, Nabulsi NB, Wyk BCV, Varma P, Arnsten AFT, Huang Y, Carson RE, van Dyck CH, In vivo measurement of widespread synaptic loss in Alzheimer’s disease with SV2A PET. Alzheimers Dement. 16, 974–982 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang X, Thayer SA, Monoacylglycerol lipase inhibitor JZL184 prevents HIV-1 gp120-induced synapse loss by altering endocannabinoid signaling. Neuropharmacology 128, 269–281 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gao N, Liu H, Li S, Tu X, Tian S, Liu J, Li G, Ma Y, Volatile Oil from Acorus gramineus Ameliorates the Injury Neurons in the Hippocampus of Amyloid Beta 1–42 Injected Mice. Anat. Rec. (Hoboken) 302, 2261–2270 (2019). [DOI] [PubMed] [Google Scholar]
- 70.Tao X, Zhang R, Wang L, Li X, Gong W, Luteolin and Exercise Combination Therapy Ameliorates Amyloid-beta1–42 Oligomers-Induced Cognitive Impairment in AD Mice by Mediating Neuroinflammation and Autophagy. J. Alzheimers Dis. 92, 195–208 (2023). [DOI] [PubMed] [Google Scholar]
- 71.Sanchez-Varo R, Mejias-Ortega M, Fernandez-Valenzuela JJ, Nunez-Diaz C, Caceres-Palomo L, Vegas-Gomez L, Sanchez-Mejias E, Trujillo-Estrada L, Garcia-Leon JA, Moreno-Gonzalez I, Vizuete M, Vitorica J, Baglietto-Vargas D, Gutierrez A, Transgenic Mouse Models of Alzheimer’s Disease: An Integrative Analysis. Int. J. Mol. Sci. 23, 5404 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.He Y, Wei M, Wu Y, Qin H, Li W, Ma X, Cheng J, Ren J, Shen Y, Chen Z, Sun B, Huang FD, Shen Y, Zhou YD, Amyloid beta oligomers suppress excitatory transmitter release via presynaptic depletion of phosphatidylinositol-4,5-bisphosphate. Nat. Commun. 10, 1193 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhu L, Zhong M, Elder GA, Sano M, Holtzman DM, Gandy S, Cardozo C, Haroutunian V, Robakis NK, Cai D, Phospholipid dysregulation contributes to ApoE4-associated cognitive deficits in Alzheimer’s disease pathogenesis. Proc. Natl. Acad. Sci. U S A 112, 11965–11970 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mayeux R, Epidemiology of neurodegeneration. Annu. Rev. Neurosci. 26, 81–104 (2003). [DOI] [PubMed] [Google Scholar]
- 75.Berman DE, Dall’Armi C, Voronov SV, McIntire LB, Zhang H, Moore AZ, Staniszewski A, Arancio O, Kim TW, Di Paolo G, Oligomeric amyloid-beta peptide disrupts phosphatidylinositol-4,5-bisphosphate metabolism. Nat. Neurosci. 11, 547–554 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McIntire LB, Berman DE, Myaeng J, Staniszewski A, Arancio O, Di Paolo G, Kim TW, Reduction of synaptojanin 1 ameliorates synaptic and behavioral impairments in a mouse model of Alzheimer’s disease. J. Neurosci. 32, 15271–15276 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhu L, Zhong M, Zhao J, Rhee H, Caesar I, Knight EM, Volpicelli-Daley L, Bustos V, Netzer W, Liu L, Lucast L, Ehrlich ME, Robakis NK, Gandy SE, Cai D, Reduction of synaptojanin 1 accelerates Abeta clearance and attenuates cognitive deterioration in an Alzheimer mouse model. J. Biol. Chem. 288, 32050–32063 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Logothetis DE, Mahajan R, Adney SK, Ha J, Kawano T, Meng XY, Cui M, Unifying Mechanism of Controlling Kir3 Channel Activity by G Proteins and Phosphoinositides. Int. Rev. Neurobiol. 123, 1–26 (2015). [DOI] [PubMed] [Google Scholar]
- 79.Sanchez-Mejia RO, Newman JW, Toh S, Yu GQ, Zhou Y, Halabisky B, Cisse M, Scearce-Levie K, Cheng IH, Gan L, Palop JJ, Bonventre JV, Mucke L, Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer’s disease. Nat. Neurosci. 11, 1311–1318 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Beraldo FH, Ostapchenko VG, Caetano FA, Guimaraes AL, Ferretti GD, Daude N, Bertram L, Nogueira KO, Silva JL, Westaway D, Cashman NR, Martins VR, Prado VF, Prado MA, Regulation of Amyloid beta Oligomer Binding to Neurons and Neurotoxicity by the Prion Protein-mGluR5 Complex. J. Biol. Chem. 291, 21945–21955 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang D, Qi Y, Klyubin I, Ondrejcak T, Sarell CJ, Cuello AC, Collinge J, Rowan MJ, Targeting glutamatergic and cellular prion protein mechanisms of amyloid beta-mediated persistent synaptic plasticity disruption: Longitudinal studies. Neuropharmacology 121, 231–246 (2017). [DOI] [PubMed] [Google Scholar]
- 82.Brody AH, Strittmatter SM, Synaptotoxic Signaling by Amyloid Beta Oligomers in Alzheimer’s Disease Through Prion Protein and mGluR5. Adv. Pharmacol. 82, 293–323 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rammes G, Hasenjager A, Sroka-Saidi K, Deussing JM, Parsons CG, Therapeutic significance of NR2B-containing NMDA receptors and mGluR5 metabotropic glutamate receptors in mediating the synaptotoxic effects of beta-amyloid oligomers on long-term potentiation (LTP) in murine hippocampal slices. Neuropharmacology 60, 982–990 (2011). [DOI] [PubMed] [Google Scholar]
- 84.Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, Strittmatter SM, Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 15, 1227–1235 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hamilton A, Vasefi M, Vander Tuin C, McQuaid RJ, Anisman H, Ferguson SS, Chronic Pharmacological mGluR5 Inhibition Prevents Cognitive Impairment and Reduces Pathogenesis in an Alzheimer Disease Mouse Model. Cell Rep. 15, 1859–1865 (2016). [DOI] [PubMed] [Google Scholar]
- 86.Spurrier J, Nicholson L, Fang XT, Stoner AJ, Toyonaga T, Holden D, Siegert TR, Laird W, Allnutt MA, Chiasseu M, Brody AH, Takahashi H, Nies SH, Perez-Canamas A, Sadasivam P, Lee S, Li S, Zhang L, Huang YH, Carson RE, Cai Z, Strittmatter SM, Reversal of synapse loss in Alzheimer mouse models by targeting mGluR5 to prevent synaptic tagging by C1Q. Sci. Transl. Med. 14, eabi8593 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen Y, Zhang Y, Chen Q, Liu Y, Wei X, Wu M, Zhang K, Liu Y, Wei W, Inhibition of mGluR5/PI3K-AKT Pathway Alleviates Alzheimer’s Disease-Like Pathology Through the Activation of Autophagy in 5XFAD Mice. J. Alzheimers Dis. 91, 1197–1214 (2023). [DOI] [PubMed] [Google Scholar]
- 88.Kleschevnikov A, GIRK2 Channels in Down Syndrome and Alzheimer’s disease. Curr. Alzheimer Res. 19, 819–829 (2022). [DOI] [PubMed] [Google Scholar]
- 89.Djebari S, Iborra-Lazaro G, Temprano-Carazo S, Sanchez-Rodriguez I, Nava-Mesa MO, Munera A, Gruart A, Delgado-Garcia JM, Jimenez-Diaz L, Navarro-Lopez JD, G-Protein-Gated Inwardly Rectifying Potassium (Kir3/GIRK) Channels Govern Synaptic Plasticity That Supports Hippocampal-Dependent Cognitive Functions in Male Mice. J. Neurosci. 41, 7086–7102 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Victoria NC, Marron Fernandez de Velasco E, Ostrovskaya O, Metzger S, Xia Z, Kotecki L, Benneyworth MA, Zink AN, Martemyanov KA, Wickman K, G Protein-Gated K+ Channel Ablation in Forebrain Pyramidal Neurons Selectively Impairs Fear Learning. Biol. Psychiatry 80, 796–806 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Marron Fernandez de Velasco E, Zhang L, Tipps NVB,M, Farris S, Xia Z, Anderson A, Carlblom N, Weaver CD, Dudek SM, Wickman K, GIRK2 splice variants and neuronal G protein-gated K+ channels: implications for channel function and behavior. Sci. Rep. 7, 1639 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, Cirrito JR, Devidze N, Ho K, Yu GQ, Palop JJ, Mucke L, Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc. Natl. Acad. Sci. U S A 109, E2895–2903 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shi JQ, Wang BR, Tian YY, Xu J, Gao L, Zhao SL, Jiang T, Xie HG, Zhang YD, Antiepileptics topiramate and levetiracetam alleviate behavioral deficits and reduce neuropathology in APPswe/PS1dE9 transgenic mice. CNS Neurosci. Ther. 19, 871–881 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang MY, Zheng CY, Zou MM, Zhu JW, Zhang Y, Wang J, Liu CF, Li QF, Xiao ZC, Li S, Ma QH, Xu RX, Lamotrigine attenuates deficits in synaptic plasticity and accumulation of amyloid plaques in APP/PS1 transgenic mice. Neurobiol. Aging 35, 2713–2725 (2014). [DOI] [PubMed] [Google Scholar]
- 95.Vossel K, Ranasinghe KG, Beagle AJ, La A, Ah Pook K, Castro M, Mizuiri D, Honma SM, Venkateswaran N, Koestler M, Zhang W, Mucke L, Howell MJ, Possin KL, Kramer JH, Boxer AL, Miller BL, Nagarajan SS, Kirsch HE, Effect of Levetiracetam on Cognition in Patients With Alzheimer Disease With and Without Epileptiform Activity: A Randomized Clinical Trial. JAMA Neurol. 78, 1345–1354 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vossel KA, Tartaglia MC, Nygaard HB, Zeman AZ, Miller BL, Epileptic activity in Alzheimer’s disease: causes and clinical relevance. Lancet Neurol. 16, 311–322 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Best TK, Siarey RJ, Galdzicki Z, Ts65Dn, a mouse model of Down syndrome, exhibits increased GABAB-induced potassium current. J. Neurophysiol. 97, 892–900 (2007). [DOI] [PubMed] [Google Scholar]
- 98.Best TK, Cramer NP, Chakrabarti L, Haydar TF, Galdzicki Z, Dysfunctional hippocampal inhibition in the Ts65Dn mouse model of Down syndrome. Exp. Neurol. 233, 749–757 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kleschevnikov AM, Belichenko PV, Gall J, George L, Nosheny R, Maloney MT, Salehi A, Mobley WC, Increased efficiency of the GABAA and GABAB receptor-mediated neurotransmission in the Ts65Dn mouse model of Down syndrome. Neurobiol. Dis. 45, 683–691 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kleschevnikov AM, Belichenko PV, Faizi M, Jacobs LF, Htun K, Shamloo M, Mobley WC, Deficits in cognition and synaptic plasticity in a mouse model of Down syndrome ameliorated by GABAB receptor antagonists. J. Neurosci. 32, 9217–9227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cooper A, Grigoryan G, Guy-David L, Tsoory MM, Chen A, Reuveny E, Trisomy of the G protein-coupled K+ channel gene, Kcnj6, affects reward mechanisms, cognitive functions, and synaptic plasticity in mice. Proc. Natl. Acad. Sci. U S A 109, 2642–2647 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kleschevnikov AM, Yu J, Kim J, Lysenko LV, Zeng Z, Yu YE, Mobley WC, Evidence that increased Kcnj6 gene dose is necessary for deficits in behavior and dentate gyrus synaptic plasticity in the Ts65Dn mouse model of Down syndrome. Neurobiol. Dis. 103, 1–10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Signorini S, Liao YJ, Duncan SA, Jan LY, Stoffel M, Normal cerebellar development but susceptibility to seizures in mice lacking G protein-coupled, inwardly rectifying K+ channel GIRK2. Proc. Natl. Acad. Sci. U S A 94, 923–927 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Torrecilla M, Marker CL, Cintora SC, Stoffel M, Williams JT, Wickman K, G-protein-gated potassium channels containing Kir3.2 and Kir3.3 subunits mediate the acute inhibitory effects of opioids on locus ceruleus neurons. J. Neurosci. 22, 4328–4334 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O’Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, Stehfest K, Fudim R, Ramakrishnan C, Huguenard JR, Hegemann P, Deisseroth K, Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 477, 171–178 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wolterink-Donselaar IG, Meerding JM, Fernandes C, A method for gender determination in newborn dark pigmented mice. Lab Anim. 38, 35–38 (2009). [DOI] [PubMed] [Google Scholar]
- 107.Vo BN, Abney KK, Anderson A, Marron Fernandez de Velasco E, Benneyworth MA, Daniels JS, Morrison RD, Hopkins CR, Weaver CD, Wickman K, VU0810464, a non-urea G protein-gated inwardly rectifying K(+) (K(ir) 3/GIRK) channel activator, exhibits enhanced selectivity for neuronal K(ir) 3 channels and reduces stress-induced hyperthermia in mice. Br. J. Pharmacol. 176, 2238–2249 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Green MV, Pengo T, Raybuck JD, Naqvi T, McMullan HM, Hawkinson JE, Marron Fernandez de Velasco E, Muntean BS, Martemyanov KA, Satterfield R, Young SM Jr., Thayer SA, Automated Live-Cell Imaging of Synapses in Rat and Human Neuronal Cultures. Front. Cell. Neurosci. 13, 467 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bensussen S, Shankar S, Ching KH, Zemel D, Ta TL, Mount RA, Shroff SN, Gritton HJ, Fabris P, Vanbenschoten H, Beck C, Man HY, Han X, A Viral Toolbox of Genetically Encoded Fluorescent Synaptic Tags. iScience 23, 101330 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vo BN, Marron Fernandez de Velasco E, Rose TR, Oberle H, Luo H, Hopkins CR, Wickman K, Bidirectional influence of limbic GIRK channel activation on innate avoidance behavior. J. Neurosci. 41, 5809–5821 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lujan R, Nusser Z, Roberts JD, Shigemoto R, Somogyi P, Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur. J. Neurosci. 8, 1488–1500 (1996). [DOI] [PubMed] [Google Scholar]
- 112.Aguado C, Colon J, Ciruela F, Schlaudraff F, Cabanero MJ, Perry C, Watanabe M, Liss B, Wickman K, Lujan R, Cell type-specific subunit composition of G protein-gated potassium channels in the cerebellum. J. Neurochem. 105, 497–511 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. The datasets used and/or analyzed, as well as viral vector sequences and protocols used as part of the current study, are available from the corresponding author on reasonable request. The script used for analyzing synapse imaging data is deposited in Zenodo (DOI: 10.5281/zenodo.11491753). Girk−/− mouse lines, as well as custom viral vectors and related plasmids, are available from K.W. under a material transfer agreement with the University of Minnesota.