

Abstract
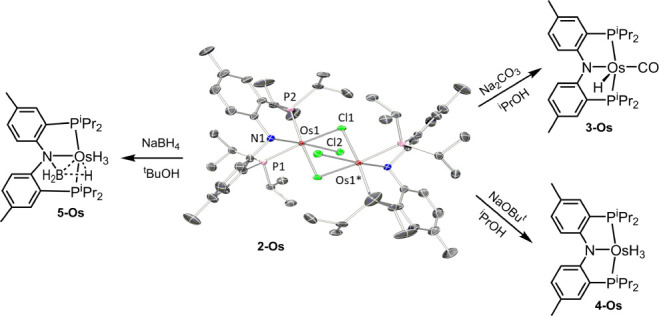
This manuscript describes the synthesis of Os complexes supported by the diarylamido/bis(phosphine) PNP pincer ligand. Compound (PNP)OsH(CO) (3-Os) was prepared by analogy with the previously reported 3-Ru. However, attempts to make (PNP)OsH3 (4-Os) analogously to 4-Ru resulted in the formation of an unexpected compound (5-Os) that is a product of addition of a BH3 unit across the Os–N bond in 4-Os. Nonetheless, 4-Os was prepared via an alternative route. Unlike 4-Ru, 4-Os appears to be a classical trihydride. Compounds 3-Ru, 3-Os, 4-Os, 4-Ru, and 5-Os were tested as potential catalysts for (a) dehydrogenative borylation of terminal alkynes (DHBTA) and (b) dehydrogenative borylation of benzene. No catalytic C–H borylation was observed for any of them, but all of them catalyzed unselective hydroboration of 4-MeC6H4CCH.
Introduction
Catalytic borylation of C–H bonds is a widely studied reaction1,2 whose synthetic value is in the efficient production of organoboronates,3 versatile building blocks in synthesis. Among the transition metals, complexes of Ir have been among the first and among the most highly active catalysts, especially as pertains to the aromatic C–H borylation.4−7 Over the past decade, our group has explored dehydrogenative borylation of terminal alkynes (DHBTA, Figure 1)8−12 to chemoselectively produce alkynylboronates.13,14 We were able to develop highly effective catalysts based on Ir complexes supported by diarylamido-centered pincer ligands. These Ir DHBTA catalysts are inactive in aromatic C–H borylation, while the common Ir catalysts for aromatic C–H borylation are poisoned by alkynes. DHBTA catalysts utilizing Zn,15−20 Co,21 Fe,22 Cu,23,24 Pd,25 Mg,26 Al,27 Mn,28 and phosphorus superbases18 have been reported in the literature, as well as select boron reagents for dehydrogenative stoichiometric reactions.29 Clearly, the DHBTA reactivity is possible with catalysts based on a great variety of elements. We thus became interested in whether complexes of some other precious metals might display C–H borylation activity when supported by ligands that we employed with Ir. We previously established that Rh complexes were not active,30 and turned our attention to Ru and Os. Some Ru complexes have been previously used for C–H borylation catalysis but not in the context of DHBTA or in a pincer framework.31−34 C–H activation reactivity of pincer complexes of Os35−37 and the chemistry of boryl-Os compounds38−40 have been examined, but it does not appear that Os compounds have been used for C–H borylation catalysis. We previously reported Ru complexes (PNP)RuH(CO) (3-Ru) and (PNP)RuH3 (4-Ru), which were prepared from the diarylamine/bis(phosphine) (PNP)H41 ligand 1 via the intermediacy of 2-Ru (Scheme 1).42 In this study, we disclose the syntheses of their Os analogs, as well as an unexpected new boron-containing Os polyhydride complex. Although the screening of these compounds for potential C–H borylation activity was not fruitful, the study brings forth synthetic and structural insight.
Figure 1.

Top: aromatic C–H borylation and selected Ir catalysts. Bottom: DHBTA and previously reported pincer-supported Ir catalysts.
Scheme 1. Synthesis of the Previously Reported (PNP)Ru and New (PNP)Os Compounds.

Results and Discussion
Synthesis of Os Complexes
The preparation of the Os analogs [(PN(H)P)OsCl2]2 (2-Os) and the (PNP)OsH(CO) (3-Os) proceeded very similarly to Ru (Scheme 1).42 Thermolysis of the (PNP)H ligand (1) with [(cymene)OsCl2]2 at 80 °C overnight resulted in the formation of 2-Os. It was only obtained in ca. 95% purity, but that was adequate for the use in further syntheses. (PNP)OsH(CO) (3-Os) was prepared in 40% isolated yield from 2-Os by subjecting it to thermolysis in isopropanol in the presence of excess Na2CO3. In the synthesis of 3-Ru, we previously demonstrated that the carbonyl ligand can be derived from CO2.42 We assume that a similar process takes place with Os, but we have not investigated this matter in detail.
On the other hand, the attempted synthesis of 4-Os by a calque from the Ru procedure (2-Os + NaBH4 in tBuOH) unexpectedly resulted in the formation of compound 5-Os, isolated in 86% yield. It can be formulaically regarded as the product of addition of BH3 to 4-Os. To circumvent the formation of 5-Os, 2-Os was treated with NaOBut in isopropanol. In this case, isopropoxide formed in situ served as the hydride donor and isopropanol served as the source of extra H2. This procedure resulted in the isolation of 4-Os in 40% yield after workup.
Spectroscopic Characterization
Compound 3-Os gave rise to a single hydride resonance in its 1H NMR spectrum at δ −30.68 ppm, displaying the expected coupling to two31P nuclei (2JH–P = 13 Hz). This chemical shift value is close to the electronically similar five-coordinate Os complexes (PiPr3)2OsHClCO (δ −31.9 ppm)43 and (SiPNP)OsH(CO)44 (δ −29.4 ppm, SiPNP is a disilylamido/bis(phosphine) pincer), corresponding to a hydride trans to an empty site in a geometry close to square-pyramidal.45
Compound 4-Os displayed a single hydride resonance of intensity 3H at δ −16.03 ppm (broad singlet). This chemical shift value is similar to that noted for (SiPNP)OsH3 (−16.53 ppm), which was reported as a classical trihydride.44 Dissolution of 4-Os in C6D6 at ambient temperature led to near-complete H/D exchange of the Os–H positions with C–D within 20 min, resulting in the observation of only a single isotopomer by 1H NMR spectroscopy, presumably (PNP)OsD2H (4-Os-d2). Addition of C6H5F to such a solution led to the emergence of the two other isotopomers ((PNP)OsH3 (4-Os) and (PNP)OsH2D (4-Os-d1) over time. Each of the isotopomeric hydride resonances presented as a broadened resonance with a hint of the triplet substructure owing to 1H–31P coupling. Explicit H–D coupling was not perceptible in these resonances and the shapes and line widths of the resonances from the three different isotopomers were not significantly different. This suggests that the magnitude of the H–D coupling in the isotopomers of 4-Os is small (probably not exceeding 1–2 Hz). This is consistent with a trihydride formulation; if 4-Os contained a dihydrogen ligand, the apparent JH–D value would be much higher (even if averaged in the H/H2 system).46−50
At ambient temperature, 5-Os presented a broad signal of intensity 2H at δ 2.70 ppm assigned to the two BH hydrogens and a broad resonance of intensity 4H (δ −10.0 ppm) assigned to the four Os-bound hydrogens. Observing 5-Os in toluene-d8 while lowering the temperature revealed that the apparent 4H resonance at RT splits into three resonances of relative intensity 1:1:2 (δ −8.7, −10.4, and −11.3 ppm at −90 °C). The middle resonance showed a discernible triplet substructure between −20 and −70 °C. From the shape of the resonances at various temperatures, it appears that the outer two coalesce at around −20 °C without much change in the central (triplet) resonance. It is not clear whether another process causes coalescence of the central triplet resonance with the rest at 25 °C, or if the appearance of coalescence is due to chemical shift coincidence. We tentatively assign the middle (triplet) resonance to the Os–H trans to B–H, the broad resonance of intensity 1H to the B–H–Os hydrogen and the resonance of intensity 2H to the remaining two hydrides, ostensibly exchanging with B–H–Os. The BH2 resonance remained relatively unperturbed in the temperature range of the study, indicating that it does not exchange with the OsH4 hydrogens at a rate that would influence NMR spectra.
XRD Structural Characterization
The structures of 2-Os and 5-Os were determined via X-ray diffraction studies on suitable single crystals (Figure 2). The [(PNHP)OsCl2]2 dimer (2-Os) lies on a center of symmetry in the crystal that relates the two (PNHP)OsCl2 fragments. The dimer is formed by means of a pair of bridging chlorides, resulting in an approximately octahedral environment about Os. The tridentate ligand binds to Os facially. The presence of the amine (NH) moiety is inferred from the pyramidalized geometry at N, which in turn permits close approach of the P–Os–P angle (ca. 100°) to the idealized 90°.
Figure 2.
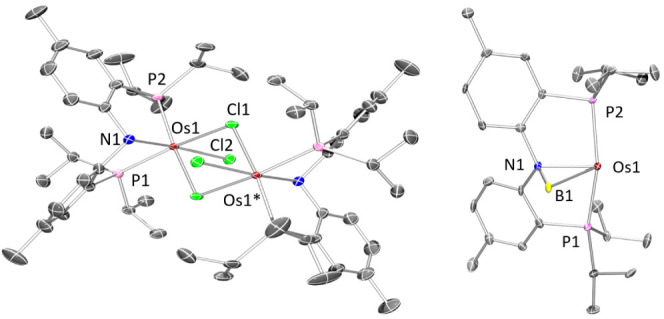
ORTEP drawings (50% probability ellipsoids) of 2-Os (left) and 5-Os (right). Only one of the two independent molecules of 5-Os is shown. Hydrogen atoms and the disordered pentane molecule in the structure of 5-Os are omitted for clarity. Selected angles (deg) and distances (Å) for 2-Os follow: Os1–N1, 2.128(2); Os1–P1, 2.2682(11); Os1–P2, 2.2275(10); Os1–Cl1, 2.4543(12); Os1–Cl1*, 2.4885(9); Os1–Cl2, 2.4294(9); P1–Os1–P2, 100.04(3); Cl1–Os1–Cl1*, 77.72(2). Selected angles (deg) and distances (Å) for 5-Os, molecule 1, follow: Os1–N1, 2.155(5); Os1–B1, 2.421(7); Os1–P1, 2.3000(18); Os1–P2, 2.2847(18); N1–B1, 1.568(9); Os1–B1–N1, 61.0(3); P1–Os1–P2, 160.85(6). Selected angles (deg) and distances (Å) for 5-Os, molecule 2, follow: Os1–N1, 2.176(5); Os1–B1, 2.415(8); Os1–P1, 2.3046(17); Os1–P2, 2.2948(18); N1–B1, 1.571(10); Os1–B1–N1, 62.0(3); P1–Os1–P2, 161.97(7).
The structure solution of 5-Os revealed two independent molecules in the asymmetric unit; they possess very similar molecular geometries. The hydrogen atom positions were not reliably obtained in 5-Os. The boron atom is bridging N and Os. The Os–B distance of ca. 2.42 Å is much longer than the Os–B distance in σ-BH complexes of catechol- or pinacolborane or the Os–B distances in the related osmium-boryls (ca. 2.0–2.1 Å).51 It is closer to the Os–B distances recorded in (R3P)2OsH3(κ2-H2BH2) (2.30(1) Å)52 and (R3P)2OsH3(κ2-H2BR2) (2.355(3) Å; R = various alkyls),53 or in (Ph3P)2OsH(CO)(B3H8) (2.44–2.48 Å).54 The Os–B distance in 5-Os is also comparable to some of the longer Ir–B distances observed in Ir → BR3 complexes.55
Attempts at DHBTA and Aromatic C–H Borylation Catalysis
Complexes of type 3 and 4, as well as 5-Os were tested as potential catalysts for the DHBTA reaction between 4-MeC6H4CCH and HBpin (used in a 1:2 ratio, Table 1). The reactions were conducted using 1 mol% of the transition metal complex relative to 4-MeC6H4CCH. These mixtures were thermolyzed in C6D6 for 3 d at 80 °C and analyzed by NMR spectroscopy. The DHBTA product 4-MeC6H4CCBpin was not detected in any of the five cases. Instead, the products of hydroboration56 of 4-MeC6H4CCH were detected with the conversion ranging from 25% to 95%. The ratio of trans-CH3–C6H4–CH=CH-Bpin and cis-CH3–C6H4–CH=CH-Bpin in the reaction with 3-Ru was ca. 20:1 and it varied between 2:1 and 1:1 for the other reactions. Examples of selective catalytic hydroboration of alkynes do exist,57,58 including by a (PNP*)RuH4 polydydride complex (PNP* = pyridine/bis(phosphine) pincer ligand).59
Table 1. Attempts at Catalytic DHBTA of 4-Ethynyltoluene Using (PNP)Ru and Os Complexes.

| entry | [cat.] | % conv. | % productsa |
|---|---|---|---|
| 1 | 3-Ru | 99 | 0/95/5 |
| 2 | 4-Ru | 88 | 0/55/45 |
| 3 | 3-Os | 25 | 0/72/28 |
| 4 | 4-Os | 25 | 0/72/28 |
| 5 | 5-Os | 35 | 0/51/49 |
Product yields listed as 7/(E)-8/(Z)-8.
In addition to testing for DHBTA activity, we decided to evaluate the potential of compounds 3–5 in aromatic C–H (C–D) borylation. Experiments were conducted in C6D6 as solvent, with 5% of the transition metal complex relative to the 1:1 mixture of HBpin and 1-hexene. These conditions were modeled after our recent work on C–H borylation of arenes using (pincer)Ir catalysts.60,61 After 3 d at 80 °C, NMR analysis revealed only the formation of isomers of 1-hexene, with no evidence for any C–H borylation products.
Conclusion
In summary, we prepared new Os hydride complexes supported by the diarylamido/bis(phosphine) PNP ligand. These compounds, along with the previously described Ru analogs were tested as potential catalysts of C–H borylation of sp and sp2 C–H bonds, but they did not show any C–H borylation activity. The complexes did show modest activity in hydroboration of a terminal alkyne, with little regioselectivity.
Experimental Section
General Considerations
Unless specified otherwise, all manipulations were performed under an Ar atmosphere using standard Schlenk line or glovebox techniques. Toluene, diethyl ether, pentane, benzene, C6D6 were dried over NaK/Ph2CO/18-crown-6, distilled or vacuum transferred and stored over molecular sieves in an Ar-filled glovebox. Ligand 1 was prepared according to the published procedure.41 The Ru complexes 2-Ru, 3-Ru, and 4-Ru were prepared as described previously,42 but using [(Cymene)RuCl2]2 instead of [(COD)RuCl2]n. Alkynes were deoxygenated by three freeze–pump–thaw cycles prior to use. All other chemicals were used as received from commercial vendors. NMR spectra were recorded on a Varian Inova 300, Mercury 300 (1H NMR, 299.952 MHz; 13C NMR, 75.421 MHz), Varian Inova 400 (1H NMR, 399.535 MHz; 11B NMR, 128.185 MHz; 13C NMR, 100.465 MHz), and NMRS 500 (1H NMR, 499.703 MHz; 13C NMR, 125.697 MHz; 31P NMR, 202.183 MHz) spectrometer. Chemical shifts are reported in δ (ppm). For 1H and 13C NMR spectra, the residual solvent peak was used as an internal reference. 31P NMR spectra were referenced externally to δ = 0 ppm by using H3PO4. 11B NMR spectra were referenced externally to δ = 0 ppm by using BF3·Et2O. Elemental analyses were performed by CALI Laboratories, Inc. (Parsippany, NJ).
[(PN(H)P)OsCl2]n (2-Os)
In an Ar-filled glovebox, the following were added to a culture tube: 1 (0.559 g, 0.00130 mol), [(cymene)OsCl2]n (0.400 g, 0.000653 mol) and 15 mL of freshly distilled and degassed toluene. The culture tube was then Teflon taped up and taken outside the box, where it then stirred at 80 °C overnight. The yellow-orange precipitate was collected by filtration, and dried under vacuum. Yield: 0.49 g (52%). 1H NMR (CD2Cl2, 500 MHz): δ 9.76 (s, 2H, PN(H)P), 7.90 (m, 4H, Ar-H), 7.19 (m, 4H, Ar-H), 7.07 (m, 4H, Ar-H), 2.83 (m, 4H, CHMe2), 2.31 (s, 12H, Ar-Me), 2.03 (m, 4H, CHMe2), 1.29 (dvt, 24 H, CHMe2), 0.89 (m, 12H, CHMe2), 0.60 (m, 12H, CHMe2).
(PNP)OsH(CO) (3-Os)
In an Ar-filled glovebox, 2-Os (310 mg, 0.224 mmol) and Na2CO3 (137 mg, 1.43 mmol) were measured out into a 25 mL Schlenk flask, with 10 mL of isopropanol as solvent. The flask was taken out of the glovebox where it stirred and heated in an oil bath at 80 °C overnight. The solvent in the flask was removed in vacuo on the Schlenk line outside the box, and was then taken back inside the box where the residual solid was extracted with toluene through a filter pipet into another Schlenk flask. The solvent was then evaporated to dryness, affording a dark red solid as the final product. The solid was recrystallized in pentane. Yield: 113 mg (40%). 1H NMR (C6D6, 400 MHz): δ 7.70 (d, 2H, J = 8.4 Hz, Ar-H), 6.95 (s, 2H, Ar-H), 6.82 (d, 2H, J = 8.4 Hz, Ar-H), 2.49 (m, 2H, CHMe2), 2.17 (s, 6H, Ar-Me), 2.11 (m, 2H, CHMe2), 1.29–1.18 (m, 12H, CHMe2), 1.00 (dvt, 6H, CHMe2), 0.94 (dvt, 6H, CHMe2), −30.68 (t, 1H, J = 12.7 Hz, Os-H). 31P{1H} NMR (C6D6, 162 MHz): δ 60.7. 13C{1H} NMR (C6D6, 101 MHz): δ 191.8 (t, JC–P = 9.5 Hz, Os-CO), 164.6 (t, JC–P = 12.6 Hz, Ar-C-P), 132.8, 131.7, 127.5 (t, JC–P = 3.1 Hz, CH(CH3)2), 125.1 (t, JC–P = 17.1 Hz, CH(CH3)2), 116.2 (t, JC–P = 5.7 Hz, ArC), 27.6 (t, JC–P = 11 Hz, CHMe2), 25.5 (t, JC–P = 13.4 Hz, CHMe2), 20.4 (s, Ar-Me), 19.5 (t, JC–P = 3 Hz, CHMe2), 19.3 (t, JC–P = 3 Hz, CHMe2), 18.6 (s, CHMe22) 18.2 (s, CHMe2). Elem. anal. calcd for C27H41NOOsP2: C, 50.06; H, 6.38; N, 2.16. Found: C, 49.59; H, 6.63; N, 2.13.
(PNP)OsH3 (4-Os)
In an Ar-filled glovebox, 2-Os (310 mg, 0.224 mmol) and sodium tert-butoxide (137 mg, 1.43 mmol) were measured out into a 25 mL Schlenk flask, with 10 mL of isopropanol as solvent. The flask was taken out of the glovebox where it stirred and heated in an oil bath at 80 °C overnight. The solvent in the flask was removed in vacuo on the Schlenk line outside the box, and was then taken back inside the box where the residual solid was extracted with toluene through a filter pipet into another Schlenk flask. The solvent was then evaporated to dryness, affording a dark red solid as the final product. The solid was recrystallized in pentane. Yield: 113 mg (40%). 1H NMR (C6D6, 400 MHz): δ 7.82 (dt, J = 8.4, 2.0 Hz, Ar-H), 7.01 (d, J = 5.2 Hz, 2H, Ar-H), 6.87 (dd, J = 8.5, 2.1 Hz, Ar-H), 2.20 (s, 6H, Ar-Me), 2.06 (m, 4H, CHMe2), 1.16 (m, 12H, CHMe2), 0.93 (dvt, 6H, JHH = JHP = 7.0 Hz, CHMe2), −16.04 (s, 3H, Os-H). 31P{1H} NMR (C6D6, 121 MHz): δ 57.9. 13C{1H} NMR (C6D6, 101 MHz): δ 166.1 (s, ArC), 137.9 (s, ArC) 133.1 (s, ArC), 131.0 (s, ArC), 129.2 (t, JC–P= 1.3 Hz, ArC), 115.6 (t, JC–P = 5.1 Hz, ArC), 26.0 (overlapping signals, CHMe2), 20.4 (s, Ar-Me), 20.0 (t, JC–P = 3.5 Hz, CHMe2), 18.6 (s, CHMe2). Elem. anal. calcd for C26H43NOsP2: C, 50.22; H, 6.97; N, 2.25. Found: C, 50.22; H, 6.22; N, 2.08.
Hydrogen–Deuterium Exchange in 4-Os
To a J. Young tube, 15 mg of 4-Os (0.025 mmol) was loaded with 0.4 mL C6D6 at 21.0 °C. An intense residual solvent peak and weak, broad singlet in the hydride region of the 1H NMR spectrum (taken 20 min after sample preparation) indicated significant H/D exchange between 4-Os and the solvent. To the solution, 0.20 mL of fluorobenzene (1.09 mmol) was added, and the J. Young tube shaken, the emergence and changing populations of 4-Os, 4-Os-d1, and 4-Os-d2 were monitored by 1H NMR spectroscopy.
(PNP)(BH2)OsH4 (5-Os)
In an Ar-filled glovebox, a culture tube was filled with 2-Os (260 mg, 0.188 mmol), NaBH4 (164 mg, 4.33 mmol), and 10 mL tert-butanol before the tube was placed in an oil bath at 80 °C with stirring overnight. Volatiles were removed under vacuum, and the residue was suspended in pentane and filtered through a plug of Celite. Solvent was then removed under vacuum, affording a light brown solid. The solid was recrystallized in pentane. Yield: 200 mg (86%). 1H NMR (C6D6, 500 MHz, 298.15 K): δ 7.75 (d, 2H, J = 8.4 Hz, Ar-H), 7.04 (s, 2H, Ar-H), 6.72 (d, 2H, 8.4 Hz, Ar-H), 2.68 (brs, 2H, B-H), 2.22 (m, 2H, CHMe2), 2.11 (s, 6H, Ar–CH3), 1.86 (m, 2H, CHMe2), 1.24 (dvt, 6H, CHMe2, JHH = JHP = 7.0 Hz), 1.16 (dvt, 6H, CHMe2, JHH = JHP = 6.7 Hz), 1.09 (dvt, 6H, CHMe2, JHH = JHP = 7.4 Hz), 0.82 (dvt, 6H, CHMe2, JHH = JHP = 6.9 Hz), −10.0 (brs, 4H, Os-H). 1H NMR (C6D6, 500 MHz, 183.15 K, Hydride region): δ −8.6 (brs, Os–H, 1H), −10.4 (brs, Os–H, 1H), −11.2 (brs, Os–H, 2H). 31P{1H} NMR (C6D6, 202 MHz): δ 51.6. 11B NMR (C6D6, 128 MHz): δ −11.8. 13C{1H} NMR (C6D6, 120 MHz): δ 162.2 (t, JC–P = 8.3 Hz, ArC), 135.9 (t, JC–P = 16.4 Hz, ArC), 133.7 (t, JC–P = 2.6 Hz, ArC), 132.7 (s, ArC), 129.2 (s, ArC), 123.0 (t, JC–P = 4.4 Hz, ArC), 27.9 (t, JC–P = 12.3 Hz, CHMe2), 26.1 (t, JC–P = 16.7 Hz, CHMe2), 22.2 (t, JC–P = 2.9 Hz, Ar-Me), 20.9 (t, JC–P = 4.3 Hz, CHMe2), 20.4 (s, CHMe2), 20.1 (s, CHMe2), 19.9 (t, JC–P = 2.2 Hz, CHMe2). Elem. anal. calcd for C26H46BNOsP2 × (C5H12)0.5: C, 50.96; H, 7.80. Found: C, 50.28; H, 7.61. The slight discrepancy in the elemental analysis results is likely owing to the less than stoichiometric amount of pentane (a disordered component of the X-ray structure solution at 0.5 equiv. per Os) in the solid.
General Procedure for Attempted Catalysis of DHBTA
To a J. Young NMR tube in an Ar-filled glovebox, 35 μL (1.0 μmol, 0.01 M in C6D6) of catalyst (3-Ru, 4-Ru, 3-Os, 4-Os, and 5-Os) and 50 μL HBpin (0.20 mmol) were added sequentially via microsyringe. The tube was shaken to allow the contents to evenly mix throughout. After this, 4-ethynyltoluene (35 μL, 0.10 mmol) was dissolved in 380 μL C6D6. This solution was added to the J. Young tube in four parts in 1 min intervals. This mixture was heated in an oil bath at 80 °C for 3 days. 1H NMR features of (E)-8(62) and (Z)-8(63) were in agreement with those in the literature, and are reported herein. (E)-8:1H NMR (500 MHz, C6D6): δ 7.40 (d, 3JH–H = 8.0 Hz, 2H, Ar-H), 7.38 (d, 3JH–H = 19 Hz, 1H, alkenyl-H), 7.15 (d, 3JH–H = 8.0 Hz, 2H, Ar-H), 6.12 (d, 3JH–H = 19 Hz, 1H, alkenyl-H), 2.35 (s, 3H, Ar-Me), 1.32 (s, 12H, Me on Bpin). (Z)-8:1H NMR (500 MHz, C6D6): δ 7.47 (d, 3JH–H = 8.0 Hz, 2H, Ar-H), 7.19 (d, 3JH–H = 15 Hz, 1H, alkenyl-H), 7.12 (d, 3JH–H = 8.0 Hz, 2H, Ar-H), 5.54 (d, 3JH–H = 15 Hz, 1H, alkenyl-H), 2.36 (s, 3H, Ar-Me), 1.31 (s, 12H, Me on Bpin).
Results of Attempted DHBTA Catalysis Using 3-Ru
A 0.010 M stock solution of 3-Ru was used in this case. General procedure stands. After 3 d of heating, analysis by 1H NMR spectroscopy revealed the reaction went to 99% completion, affording 95% trans-CH3–C6H4–CH=CH-Bpin and 5% cis-CH3–C6H4–CH = CH-Bpin.
Results of Attempted DHBTA Catalysis Using 4-Ru
A 0.01 M stock solution of 4-Ru was used in this case. General procedure stands. After 3 d of heating, analysis by 1H NMR spectroscopy revealed the reaction went to 88% completion, affording 48% trans-CH3–C6H4–CH=CH-Bpin and 40% cis-CH3–C6H4–CH=CH-Bpin.
Results of Attempted DHBTA Catalysis Using 3-Os
A 0.010 M stock solution of 3-Os was used in this case. General procedure stands. After 3 d of heating, analysis by 1H NMR spectroscopy revealed the reaction went to 25% completion, affording 18% trans-CH3–C6H4–CH=CH-Bpin and 7% cis-CH3–C6H4–CH=CH-Bpin.
Results of Attempted DHBTA Catalysis Using 4-Os
A 0.010 M stock solution of 4-Os was used in this case. General procedure stands. After 3 d of heating, analysis by 1H NMR spectroscopy revealed the reaction went to 25% completion, affording 18% trans-CH3–C6H4–CH==CH-Bpin and 7% cis-CH3–C6H4–CH=CH-Bpin.
Results of Attempted DHBTA Catalysis Using 5-Os
A 0.010 M stock solution of 5-Os was used in this case. General procedure stands. After 3 d of heating, analysis by 1H NMR spectroscopy revealed the reaction went to 35% completion, affording 18% trans-CH3–C6H4–CH=CH-Bpin and 17% cis-CH3–C6H4–CH=CH-Bpin.
General Procedure for Attempted Arene Borylation
To a J. Young NMR tube, 35 μL (1.0 μmol, 0.01 M in C6D6) of catalyst (3-Ru, 4-Ru, 3-Os, 4-Os, and 5-Os), 50 μL of HBpin (0.08 mmol), and 45 μL 1-hexene (0.08 mmol), and 370 μL were added sequentially via microsyringe before the tube was placed in an 80 °C oil bath to heat for 3 days. For all catalysts, 1-hexene isomerization products were observed64 with no evidence of arene borylation.
Acknowledgments
We are grateful for the primary support of this work by the US National Science Foundation (grants CHE-1565923 and CHE-2102324). We are also grateful to the Welch Foundation (grant A-1717) for supporting B.J.F. during the initial stages of this work. We are grateful to Prof. Jia Zhou for helpful discussions regarding some aspects of this work.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.organomet.4c00388.
Details of X-ray diffraction experiments and pictorial NMR spectra (PDF)
Author Present Address
† Savannah River National Laboratory
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Bisht R.; Haldar C.; Mahamudul Hasan M. M.; Hoque M. E.; Chaturvedi J.; Chattopadhyay B. Metal-catalysed C–H bond activation and borylation. Chem. Soc. Rev. 2022, 51 (12), 5042–5100. 10.1039/D1CS01012C. [DOI] [PubMed] [Google Scholar]
- Mkhalid I. A. I.; Barnard J. H.; Marder T. H.; Murphy J. M.; Hartwig J. F. C–H Activation for the Construction of C–B Bonds. Chem. Rev. 2010, 110, 890–931. 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]
- Hall D. G.Boronic Acids: preparation and Applications in Organic Synthesis, Medicine and Materials, 2nd ed.; Wiley-VCH: Weinheim, 2012. [Google Scholar]
- Cho J.-Y.; Tse M. K.; Holmes D.; Maleczka R. E. Jr.; Smith M. R. III Remarkably Selective Iridium Catalysts for the Elaboration of Aromatic C-H Bonds. Science 2002, 295 (5553), 305–308. 10.1126/science.1067074. [DOI] [PubMed] [Google Scholar]
- Ishiyama T.; Takagi J.; Ishida K.; Miyaura N.; Anastasi N. R.; Hartwig J. F. Mild Iridium-Catalyzed Borylation of Arenes. High Turnover Numbers, Room Temperature Reactions, and Isolation of a Potential Intermediate. J. Am. Chem. Soc. 2002, 124, 390–391. 10.1021/ja0173019. [DOI] [PubMed] [Google Scholar]
- Preshlock S. M.; Ghaffari B.; Maligres P. E.; Krska S. W.; Maleczka R. E.; Smith M. R. High-Throughput Optimization of Ir-Catalyzed C–H Borylation: A Tutorial for Practical Applications. J. Am. Chem. Soc. 2013, 135, 7572–7582. 10.1021/ja400295v. [DOI] [PubMed] [Google Scholar]
- Hartwig J. F. Borylation and Silylation of C–H Bonds: A Platform for Diverse C–H Bond Functionalizations. Acc. Chem. Res. 2012, 45, 864–873. 10.1021/ar200206a. [DOI] [PubMed] [Google Scholar]
- Lee C.-I.; Zhou J.; Ozerov O. V. Catalytic Dehydrogenative Borylation of Terminal Alkynes by a SiNN Pincer Complex of Iridium. J. Am. Chem. Soc. 2013, 135, 3560–3566. 10.1021/ja311682c. [DOI] [PubMed] [Google Scholar]
- Lee C.-I.; DeMott J. C.; Pell C. J.; Christopher A.; Zhou J.; Bhuvanesh N.; Ozerov O. V. Ligand Survey Results in Identification of PNP Pincer Complexes of Iridium as Long-lived and Chemoselective Catalysts for Dehydrogenative Borylation of Terminal Alkynes. Chem. Sci. 2015, 6, 6572–6582. 10.1039/C5SC02161H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Lee C.-I.; Ozerov O. V. Computational Study of the Mechanism of Dehydrogenative Borylation of Terminal Alkynes by SiNN Iridium Complexes. ACS Catal. 2018, 8, 536–545. 10.1021/acscatal.7b03835. [DOI] [Google Scholar]
- Foley B. J.; Bhuvanesh N.; Zhou J.; Ozerov O. V. Combined Experimental and Computational Studies of the Mechanism of Dehydrogenative Borylation of Terminal Alkynes (DHBTA) Catalyzed by PNP Complexes of Iridium. ACS Catal. 2020, 10, 9824–9836. 10.1021/acscatal.0c02455. [DOI] [Google Scholar]
- Foley B. J.; Ozerov O. V. Air- and Water-Tolerant (PNP)Ir Precatalyst for the Dehydrogenative Borylation of Terminal Alkynes. Organometallics 2020, 39, 2352–2355. 10.1021/acs.organomet.0c00250. [DOI] [Google Scholar]
- Jiao J.; Nishihara Y. Alkynylboron compounds in organic synthesis. J. Organomet. Chem. 2012, 721–722, 3–16. 10.1016/j.jorganchem.2012.05.027. [DOI] [Google Scholar]
- Nandy S.; Paul S.; Das K. K.; Kumar P.; Ghorai D.; Panda S. Synthesis and reactivity of alkynyl boron compounds. Org. Biomol. Chem. 2021, 19 (34), 7276–7297. 10.1039/D1OB00465D. [DOI] [PubMed] [Google Scholar]
- Tsuchimoto T.; Utsugi H.; Sugiura T.; Horio S. Alkynylboranes: A Practical Approach by Zinc-Catalyzed Dehydrogenative Coupling of Terminal Alkynes with 1,8-Naphthalenediaminatoborane. Adv. Synth. Catal. 2015, 357, 77–82. 10.1002/adsc.201400767. [DOI] [Google Scholar]
- Procter R. J.; Uzelac M.; Cid J.; Rushworth P. J.; Ingleson M. J. Low-coordinate NHC-zinc hydride complexes catalyze alkyne C–H borylation and hydroboration using pinacolborane. ACS Catal. 2019, 9, 5760–5771. 10.1021/acscatal.9b01370. [DOI] [Google Scholar]
- Sahoo R. K.; Rajput S.; Patro A. G.; Nembenna S. Synthesis of low oxidation state zinc(i) complexes and their catalytic studies in the dehydroborylation of terminal alkynes. Dalton Trans. 2022, 51, 16009–16016. 10.1039/D2DT02846H. [DOI] [PubMed] [Google Scholar]
- Sahoo R. K.; Patro A. G.; Sarkar N.; Nembenna S. Comparison of Two Zinc Hydride Precatalysts for Selective Dehydrogenative Borylation of Terminal Alkynes: A Detailed Mechanistic Study. ACS Omega 2023, 8, 3452–3460. 10.1021/acsomega.2c07381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal K.; Groutchik K.; Bawari D.; Dobrovetsky R. An “On-Demand”, Selective Dehydrogenative Borylation or Hydroboration of Terminal Alkynes Using Zn2+-based Catalyst. ChemCatchem 2022, 14 (9), e202200004 10.1002/cctc.202200004. [DOI] [Google Scholar]
- Luo M.; Qin Y.; Chen X.; Xiao Q.; Zhao B.; Yao W.; Ma M. ZnBr2-Catalyzed Dehydrogenative Borylation of Terminal Alkynes. J. Org. Chem. 2021, 86 (23), 16666–16674. 10.1021/acs.joc.1c01936. [DOI] [PubMed] [Google Scholar]
- Krautwald S.; Bezdek M. J.; Chirik P. J. Cobalt-Catalyzed 1,1-Diboration of Terminal Alkynes: Scope, Mechanism, and Synthetic Applications. J. Am. Chem. Soc. 2017, 139, 3868–3875. 10.1021/jacs.7b00445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei D.; Carboni B.; Sortais J.-B.; Darcel C. Iron-catalyzed dehydrogenative borylation of terminal alkynes. Adv. Snth. Catal. 2018, 360, 3649–3654. 10.1002/adsc.201800588. [DOI] [Google Scholar]
- Romero E. A.; Jazzar R.; Bertrand G. Copper-catalyzed dehydrogenative borylation of terminal alkynes with pinacolborane. Chem. Sci. 2017, 8, 165–168. 10.1039/C6SC02668K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Ming W.; Friedrich A.; Kerner F.; Marder T. B. Copper-Catalyzed Triboration of Terminal Alkynes Using B2pin2: Efficient Synthesis of 1,1,2-Triborylalkenes. Angew. Chem. Int. Ed. Engl. 2020, 59, 304–309. 10.1002/anie.201908466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pell C. J.; Ozerov O. V. Catalytic dehydrogenative borylation of terminal alkynes by POCOP-supported palladium complexes. Inorg. Chem. Front. 2015, 2, 720–724. 10.1039/C5QI00074B. [DOI] [Google Scholar]
- Birepinte M.; Liautard V.; Chabud L.; Pucheault M. Magnesium-Catalyzed Tandem Dehydrogenation-Dehydrocoupling: An Atom Economical Access to Alkynylboranes. Chem. —Eur. J. 2020, 26, 3236–3240. 10.1002/chem.201905772. [DOI] [PubMed] [Google Scholar]
- Willcox D. R.; De Rosa D. M.; Howley J.; Levy A.; Steven A.; Nichol G. S.; Morrison C. A.; Cowley M. J.; Thomas S. P. Aluminium-Catalyzed C(sp)–H Borylation of Alkynes. Angew. Chem., Int. Ed. 2021, 60, 20672–20677. 10.1002/anie.202106216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahua H.; Kaur H.; Arevalo R. Chemoselective C(sp)–H borylation of terminal alkynes catalyzed by a bis(N-heterocyclicsilylene) manganese complex. Ing. Chem., Front. 2023, 10, 6067–6076. 10.1039/D3QI01033C. [DOI] [Google Scholar]
- Ramachandran P. V.; Hamann H. J. Dehydroborylation of Terminal Alkynes Using Lithium Aminoborohydrides. Molecules 2023, 28, 3433–3449. 10.3390/molecules28083433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.-I.; Hirscher N. A.; Zhou J.; Bhuvanesh N.; Ozerov O. V. Adaptability of the SiNN Pincer Ligand in Iridium and Rhodium Complexes Relevant to Borylation Catalysis. Organometallics 2015, 34, 3099–3102. 10.1021/acs.organomet.5b00125. [DOI] [Google Scholar]
- Murphy J. M.; Lawrence J. D.; Kawamura K.; Incarvito C.; Hartwig J. F. Ruthenium- Catalyzed Regiospecific Borylation of Methyl C-H Bonds. J. Am. Chem. Soc. 2006, 128, 13684–13685. 10.1021/ja064092p. [DOI] [PubMed] [Google Scholar]
- Fukuda K.; Iwasawa N.; Takaya J. Ruthenium-Catalyzed ortho C–H Borylation of Arylphosphines. Angew. Chem. Int. Ed. 2019, 58, 2850–2853. 10.1002/anie.201813278. [DOI] [PubMed] [Google Scholar]
- Thongpaen J.; Manguin R.; Kittikool T.; Camy A.; Roisnel T.; Dorcet V.; Yotphan S.; Canac Y.; Mauduit M.; Baslé O. Ruthenium–NHC complex-catalyzed P(III)-directed C–H borylation of arylphosphines. Chem. Commun. 2022, 58 (86), 12082–12085. 10.1039/D2CC03909E. [DOI] [PubMed] [Google Scholar]
- Fernández-Salas J. A.; Manzini S.; Piola L.; Slawin A. M. Z.; Nolan S. P. Ruthenium catalysed C–H bond borylation. Chem. Commun. 2014, 50, 6782–6784. 10.1039/c4cc02096k. [DOI] [PubMed] [Google Scholar]
- Alós J.; Esteruelas M. A.; Oliván M.; Oñate E.; Puylaert P. C–H Bond Activation Reactions in Ketones and Aldehydes Promoted by POP-Pincer Osmium and Ruthenium Complexes. Organometallics 2015, 34, 4908–4921. 10.1021/acs.organomet.5b00416. [DOI] [Google Scholar]
- Cancela L.; Esteruelas M. A.; Oliván M.; Oñate E. Azolium Control of the Osmium-Promoted Aromatic C–H Bond Activation in 1,3-Disubstituted Substrates. Organometallics 2021, 40, 3979–3991. 10.1021/acs.organomet.1c00565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev D. G.; Lough A. J. Double C–H Activation on Osmium and Ruthenium Centers: Carbene vs Olefin Products. Organometallics 2002, 21, 2601–2603. 10.1021/om020355m. [DOI] [Google Scholar]
- Esteruelas M. A.; López A. M.; Mora M.; Oñate E. Boryl-Dihydrideborate Osmium Complexes: Preparation, Structure, and Dynamic Behavior in Solution. Organometallics 2015, 34, 941–946. 10.1021/om501309g. [DOI] [Google Scholar]
- Irvine G. J.; Roper W. R.; Wright L. J. Five-Coordinate Ruthenium(II) and Osmium(II) Boryl Complexes. Organometallics 1997, 16, 2291–2296. 10.1021/om9610298. [DOI] [Google Scholar]
- McQueen C. M. A.; Hill A. F.; Sharma M.; Singh S. K.; Ward J. S.; Willis A. C.; Young R. D. Synthesis and reactivity of osmium and ruthenium PBP–LXL boryl pincer complexes. Polyhedron 2016, 120, 185–195. 10.1016/j.poly.2016.05.041. [DOI] [Google Scholar]
- Fan L.; Foxman B. M.; Ozerov O. V. N-H Cleavage as a Route to Palladium Complexes of a New PNP Pincer Ligand. Organometallics 2004, 23, 326–328. 10.1021/om034151x. [DOI] [Google Scholar]
- Çelenligil-Çetin R.; Watson L. A.; Guo C.; Foxman B. M.; Ozerov O. V. Decarbonylation of Acetone and Carbonate at a Pincer-Ligated Ru Center. Organometallics 2005, 24, 186–189. 10.1021/om049061m. [DOI] [Google Scholar]
- Esteruelas M. A.; Werner H. Five- and six-coordinate hydrido(carbonyl)-ruthenium(II) and -osmium(II) complexes containing triisopropylphosphine as ligand. J. Organomet. Chem. 1986, 303, 221–231. 10.1016/0022-328X(86)80134-6. [DOI] [Google Scholar]
- Lee J.-H.; Pink M.; Caulton K. G. Triple Benzylic Dehydrogenation by Osmium in an Amide Ligand Environment. Organometallics 2006, 25, 802–804. 10.1021/om050916k. [DOI] [Google Scholar]
- Structural preferences of five-coordinate d6 complexes have been discussed elsewhere:; a Rachidi I. E.-I.; Eisenstein O.; Jean Y. A theoretical study of the possible structures of d6 ML5 complexes. New J. Chem. 1990, 14, 671–677. [Google Scholar]; b Lam W. H.; Shimada S.; Batsanov A. S.; Lin Z.; Marder T. B.; Cowan J. A.; Howard J. A. K.; Mason S. A.; McIntyre G. J. Accurate Molecular Structures of 16-Electron Rhodium Hydrido Boryl Complexex: Low-Temperature Single-Crystal X-ray and Neutron Diffraction and Computational Studies of [(PR3)2RhHCl(Boryl). Organometallics 2003, 22, 4557–4568. 10.1021/om030434d. [DOI] [Google Scholar]; c Riehl J.-F.; Jean Y.; Eisenstein O.; Pelissier M. Theoretical study of the structures of electron-deficient d6ML5 complexes. Oganometallics 1992, 11, 729–737. 10.1021/om00038a035. [DOI] [Google Scholar]; d Olivan M.; Eisenstein O.; Caulton K. G. New Access to Vinylidene from Ruthenium Polyhydrides. Organometallics 1997, 16, 2227–2229. 10.1021/om970095m. [DOI] [Google Scholar]
- Kubas G. Dihydrogen complexes as prototypes for the coordination chemistry of saturated molecules. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 6901–6907. 10.1073/pnas.0609707104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree R. Dihydrogen Complexation. Chem. Rev. 2016, 116, 8750–8769. 10.1021/acs.chemrev.6b00037. [DOI] [PubMed] [Google Scholar]
- Heinekey D. M. Transition metal dihydrogen complexes: isotope effects on reactivity and structure. J. Label. Compd. Radiopharm. 2007, 50, 1063–1071. 10.1002/jlcr.1385. [DOI] [Google Scholar]
- Heinekey D. M.; Oldham W. J. Coordinatioon chemistry of dihydrogen. Chem. Rev. 1993, 93, 913–926. 10.1021/cr00019a004. [DOI] [Google Scholar]
- Heinekey D. M.; Lledós A.; Lluch J. M. Elongated dihydrogen complexes: what remains of the H–H Bond?. Chem. Soc. Rev. 2004, 33 (3), 175–182. 10.1039/B304879A. [DOI] [PubMed] [Google Scholar]
- Esteruelas M. A.; Fernández I.; García-Yebra C.; Martin J.; Oñate E. Elongated σ-Borane versus σ-Borane in Pincer–POP–Osmium Complexes. Organometallics 2017, 36, 2298–2307. 10.1021/acs.organomet.7b00234. [DOI] [Google Scholar]
- Frost P. W.; Howard J. A. K.; Spencer J. L. An OsmiumTetrahydroborate Complex with Unusual Dynamic Behaviour: X-Ray Crystal Structure of [Os(BH4)H3{P(c-C5H9)3}2] (c-C5H9 = cyclo-C5H9). J. Chem. Soc., Chem. Commun. 1984, 1362–1363. 10.1039/c39840001362. [DOI] [Google Scholar]
- Babón J. C.; Esteruelas M. A.; Fernández I.; López A. M.; Oñate E. Evidence for a Bis(Elongated σ)-Dihydrideborate Coordinated to Osmium. Inorg. Chem. 2018, 57, 4482–4491. 10.1021/acs.inorgchem.8b00155. [DOI] [PubMed] [Google Scholar]
- Bould J.; Rath N. P.; Barton L. [(CO)H(PPh3)2-arachno-OsB3H8]. Acta Crystallogr. 1996, C52, 1388–1390. 10.1107/S0108270195017239. [DOI] [Google Scholar]
- Cao Y.; Shih W.-C.; Ozerov O. V. Addition of O–H, N–H, and F–H Bonds across a Boryl–Iridium Unit. Organometallics 2019, 38, 4076–4081. 10.1021/acs.organomet.8b00785. [DOI] [Google Scholar]; and references within
- Geier S. J.; Vogels C. M.; Melanson J. A.; Westcott S. A. The transition metal-catalysed hydroboration reaction. Chem. Soc. Rev. 2022, 51 (21), 8877–8922. 10.1039/D2CS00344A. [DOI] [PubMed] [Google Scholar]
- Yamamoto K.; Mohara Y.; Mutoh Y.; Saito S. Ruthenium-Catalyzed (Z)-Selective Hydroboration of Terminal Alkynes with Naphthalene-1,8-diaminatoborane. J. Am. Chem. Soc. 2019, 141, 17042–17047. 10.1021/jacs.9b06910. [DOI] [PubMed] [Google Scholar]
- Sundararaju B.; Fürstner A. A trans-Selective Hydroboration of Internal Alkynes. Angew. Chem., Int. Ed. 2013, 52, 14050–14054. 10.1002/anie.201307584. [DOI] [PubMed] [Google Scholar]
- Gunanathan C.; Hölscher M.; Pan F.; Leitner W. Ruthenium Catalyzed Hydroboration of Terminal Alkynes to Z-Vinylboronates. J. Am. Chem. Soc. 2012, 134, 14349–14352. 10.1021/ja307233p. [DOI] [PubMed] [Google Scholar]
- Press L. P.; Kosanovich A. J.; McCulloch B. J.; Ozerov O. V. High-Turnover Aromatic C-H Borylation Catalyzed by POCOP-type Pincer Complexes of Iridium. J. Am. Chem. Soc. 2016, 138, 9487–9497. 10.1021/jacs.6b03656. [DOI] [PubMed] [Google Scholar]
- Hung M.-U.; Press L.; Bhuvanesh N.; Ozerov O. V. Examination of a Series of Ir and Rh PXL Pincer Complexes as (Pre)Catalysts for Aromatic C-H Borylation. Organometallics 2021, 40, 1004–1013. 10.1021/acs.organomet.1c00081. [DOI] [Google Scholar]
- Murata M.; Kawakita K.; Asana T.; Watanabe S.; Masuda Y. Rhodium- and Ruthenium-Catalyzed Dehydrogenative Borylation of Vinylarenes with Pinacolborane: Stereoselective Synthesis of Vinylboronates. Bull. Chem. Soc. Jpn. 2002, 75, 825–829. 10.1246/bcsj.75.825. [DOI] [Google Scholar]
- Cid J.; Carbó J. J.; Fernández E. Catalytic Non-Conventional trans-Hydroboration: A Theoretical and Experimental Perspective. Chem. —Eur. J. 2012, 18, 1512–1521. 10.1002/chem.201102729. [DOI] [PubMed] [Google Scholar]
- Escobar M. A.; da Costa D. M.; Trofymchuk O. S.; Daniliuc C. G.; Gracia F.; Nachtigall F. M.; Santos L. S.; Rojas R. S.; Cabrera A. R. Intermolecular stabilization in new 2-iminopyridine derivatives complexes of Pd(II) and their reactivity towards alkenes. J. Organomet. Chem. 2018, 863, 21–29. 10.1016/j.jorganchem.2018.03.032. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.