

Abstract
Background
Maple Syrup Urine Disease (MSUD) is a rare inherited disorder of metabolism, which manifests early in life in classical forms. Recurrent illness and exertion aggravate neurotoxicity. This case highlights MSUD diagnosed in association with COVID-19 complications from Nepal.
Case presentation
We present a case of a 4-month-old child with a biochemical diagnosis of flared-up MSUD. Initially presenting with chief complaints of fever, noisy breathing, chest retraction, cough along with lethargy and poor feeding since the first week of life, the child also had developmental delay with feeble neck holding and absent social smile. The child was diagnosed with COVID-19 pneumonia and admitted in the Intensive Care Unit, requiring mechanical ventilation for 12 days. Despite the clinical resolution of pneumonia, the child had multiple episodes of generalized seizures and was sickly and frail. An incessant peculiar odor emanating from the child led to strong suspicion of metabolic disorder. Qualitative screening for amino acids (FeCl3 and 2,4-dinitrophenylhdrazine/DNPH) in urine and further gas chromatography-mass spectrometry revealed increased branched-chain amino acids(valine, leucine, and isoleucine). With dietary restrictions, the child was doing well. However, unfortunately, after 10 days of discharge, the child succumbed to death.
Conclusions
This case highlights the outpouring of hidden metabolic disorders with the onset of new diseases. It could have been detected and managed earlier with expedited neonatal screening and proper intervention.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12887-024-05266-0.
Keywords: COVID-19, Maple syrup urine disease (MSUD), Neurotoxicity syndromes, Newborn screening
Background
The metabolism of branched-chain amino acids (leucine, isoleucine, and valine) is required for protein synthesis and cell signaling [1]. Maple Syrup Urine Disease (MSUD) is a rare inherited autosomal recessive disorder caused by a deficiency of branched-chain alpha-ketoacid dehydrogenase complex (BCKDC). It manifests as the inability of body to metabolize branched-chain amino acids leading to a typical odor of burnt sugar in urine and other body fluids. It affects around 1 in 185,000 births [2]. The first case of MSUD was described by Menkes et al. [3] in 1954 associated with cerebral dysfunction and accumulation of abnormal urinary substances comprising of leucine, isoleucine, valine, and their corresponding alpha-ketoacids and alloisoleucine [4, 5]. Without intervention, buildup of these toxic metabolites leads to neurotoxicity [6]. The mainstay of management of this metabolic disorder is dietary restriction and conscientious metabolic monitoring with a favorable prognosis, however metabolic crises due to trauma, infections or fasting can aggravate the condition with grave outcomes [6]. The advent of tandem mass spectrometry (MS/MS) has expedited the timely diagnosis of metabolic disorders initiating early interventions. Here, we present a case of an infant with a biochemical diagnosis of flared-up MSUD along with COVID-19 infection in a limited resource-setting, who faced an unfortunate outcome due to delayed diagnosis and precipitation of symptoms as a result of recurrent illness.
Case presentation
A 4-month-old male infant of Tamang ethnicity was referred to the pediatric ward at our center. The child presented with fever, noisy breathing, chest retraction, and cough for 4 days along with lethargy and poor feeding since the first week of life. He also had developmental delays in the form of feeble neck holding and absent social smile (Fig. 1).
Fig. 1.
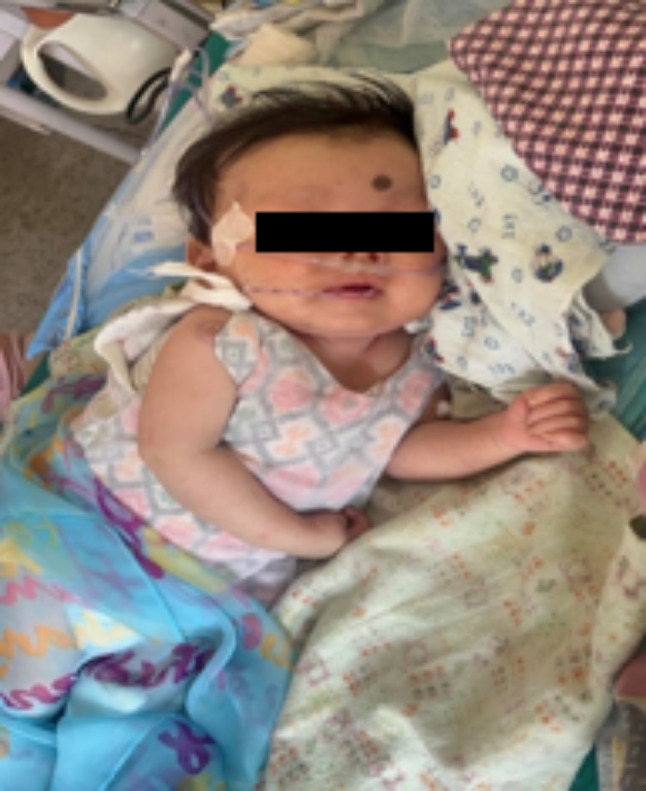
Photograph of the child
The baby was born to a 32-year-old primigravida mother via spontaneous vaginal delivery at a local hospital, weighed 3200 g and was reported to have delayed cry at birth. He had no significant issues until the third day of life when he developed lethargy and multiple episodes of generalized seizures, for which he was admitted to a Children’s hospital. There were no signs of respiratory distress or neonatal sepsis. In family history there was no kinship, however his father was on anti-epileptic medication. The child was treated with intravenous antibiotics (Ampicillin-200 mg/kg/day) and anti-epileptic (Leveteracetam-20 mg/kg/day). For evaluation of the etiology of seizure, a contrast-enhanced magnetic resonance imaging (MRI) of brain was done which showed features of hypoxic ischemic encephalopathy and cerebral edema. An electroencephalogram (EEG) examination revealed normal sleep waves with no epileptiform discharge. The guardian refused to proceed with further investigation and left against medical advice after a 20-day stay at the hospital.
Later at 4 months of age, the child presented with respiratory distress, poor feeding, and fever reaching a maximum of 104 °F. On examination, he was lethargic, pale, and dehydrated with cold peripheries. There was no cyanosis, icterus, lymphadenopathy, or edema. On vitals examination, he was afebrile but had tachycardia (167 bpm), tachypnea (70 breaths/min), and oxygen saturation of 97% with Continuous Positive Airway Pressure (CPAP) at 5 cm of H2O. Anthropometric measurements showed a weight of 4 kg, length of 54 cm, and head circumference of 35 cm, all of which fell below − 3 Z-score according to the WHO growth chart indicating failure to thrive and microcephaly. On chest examination, pectus carinatum and severe subcostal retractions were observed with bilateral diffuse crepitations heard on auscultation. On neurological examination, he was dystonic with opisthotonus posturing and exaggerated deep tendon reflexes in the initial two weeks but later developed decreased tone of all limbs with absent reflexes. On abdominal examination, there was hepatomegaly with liver palpable 3 cm below the right costal margin which later increased up to 5 cm. His throat swab RT-PCR was positive for COVID-19. The child was admitted to the Pediatric Intensive Care Unit and received mechanical ventilation for 12 days. During this period, he also developed ventilator-associated pneumonia and was administered multiple antibiotics due to protracted disease course, namely Colistin (50000 units/kg 8 hourly), Ofloxacin (15 mg/kg/day), Voriconazole (9 mg/kg 12 hourly) and Azithromycin (10 mg/kg/day).
The biochemical parameters showed decreased random glucose (3.1 mmol/L) suggesting mild hypoglycemia, urea (1.6 mmol/L), creatinine (11µmol/L), sodium (131 mEq/L), potassium (5.2 mEq/L) and total calcium (2.4 mmol/L) under normal limits except for magnesium (0.7 mg/dl) which was low. Liver function tests were within normal limits. Blood and CSF cultures were sterile. IgG, IgA, IgM, and IgE levels were normal. Venous blood gas showed respiratory alkalosis (pH:7.51; pCO2:15.9 mmHg; HCO3−:12.5 mEq/L) with metabolic compensation and normal lactate level (1.3 mmol/L). The child was extubated after 12 days and shifted to pediatric ward after partial clinical and radiological resolution of pneumonia. During the hospital stay, he developed multiple subtle seizures consisting of uprolling of eyes, vacant stares, and had slow improvement in terms of activity and feeding with loss of body weight upto 3.2 kg from an initial 4 kg at presentation. He also constantly exuded a peculiar body odor. Hence, a strong suspicion of metabolic disorder was made, and a urine sample was sent to the biochemistry laboratory.
On screening for carbohydrates, the sample tested negative. However, using the battery of tests for amino acid screening, the urine was positive for ketone bodies along with positive ferric chloride test and 2,4-dinitrophenylhydrazine test indicating the presence of branched-chain amino acid in urine (Fig. 2). Following this, the patient’s urine sample was sent for amino acid screening to a reference laboratory, which indicated increased concentration of branched-chain amino acids; Leucine 1702 µmol/g creatinine (Ref: 37–253 µmol/g creatinine), Isoleucine 1661 µmol/g creatinine (Ref: 22–159 µmol/g creatinine), Valine1095 µmol/g creatinine (Ref: 60–280 µmol/g creatinine), and Threonine 2858 µmol/g creatinine (Ref: 67–1380) µmol/g creatinine while, a decrease in Alanine 281 µmol/g creatinine (Ref: 608–2918 µmol/g creatinine), Glutamic acid 61.6 µmol/g creatinine (Ref: 65–1155 µmol/g creatinine), and Tryptophan 56.6 µmol/g creatinine (Ref: 76–388 µmol/g creatinine) using gas chromatography-mass spectrometry (GC-MS) technique (Supplementary Table 1).
Fig. 2.

(A) Positive DNPH test (Left: negative control, Right: test); (B) Positive Ferric Chloride test (Left: test, Right: negative control)
Discussion and conclusions
There are only a few cases of metabolic disorders reported in Nepal and to the best of our knowledge no case of MSUD has been reported as of yet in Nepalese population. There is no established governmental consensus in diagnostic screening for inborn errors of metabolism (IEMs) in Nepal. Communicable diseases and malnutrition are kept under high priority to address neonatal health in Nepal, while congenital disorders are still shadowed and there are no adequate policies for screening these disorders. With limited reporting, there is also a higher propensity of delay in the required intervention. Few cases highlight the need for consideration of amino acid metabolism disorders in identifying subclinical diseases that might be exacerbated by casual stressors [7].
In classic MSUD, individuals appear normal at birth but slowly develop acute metabolic decompensation over the first few weeks. MSUD is caused due to a biallelic loss of function mutation in the gene encoding BCKDC subunits. As a result, there is a toxic accumulation of BCAAs and BCKAs which presents as acute severe ketoacidosis and neurological symptoms like apnea, seizure, coma, and chronic outcomes like poor feeding, ataxia, encephalopathy, as well as intellectual disability due to impeded neurotransmitter stores [8].
In MSUD patients, it is postulated that due to the reverse flux mechanism, elevated BCKAs incite decreased levels of glutamate and increased BCAAs (especially leucine) as well as α-ketoglutarate (Fig. 3) [5]. Acute elevation of the alpha-keto acids and leucine causes metabolic encephalopathy and cerebral edema [9].
Fig. 3.

Biochemical changes in MSUD: (A) Enzyme defect in MSUD causes accumulation of ketoacids and reverse flux reaction producing increased α-ketoglutarate; (B) α-ketoglutarate is converted to glutamate and respective products via aspartate aminotransferase and alanine aminotransferase; (C) α-ketoglutarate, thus produced is quickly utilized in the Kreb’s cycle via anaplerotic role of isoleucine and valine by production of succinyl coA; (D) BCAA competes with other amino acids that utilize the same transporter to enter into brain cells
The excess of α-ketoglutarate, in turn, consumes the brain aspartate level via the enzyme activity of aspartate aminotransferase that eventually replenishes glutamate. However, excessive valine and isoleucine with their anaplerotic role restore the intermediates of the tricarboxylic acid (TCA) cycle through succinyl co-A formation. Henceforth, with an accelerated TCA cycle, the sources of α-ketoglutarate also get exhausted for glutamate regeneration [5]. Furthermore, increased glycolysis for ATP generation rapidly drives lactate formation. MSUD is often associated with decreased levels of phenylalanine, tyrosine, tryptophan, and methionine in the brain. A shrinkage in these essential amino acid pools is accounted for due to the inherent competition of BCAA (especially leucine) for the entry of these amino acids into brain via a large neutral amino acid transporter (LAT1, encoded by SLC7A5) [10]. Impaired regulation of the level of these amino acids leads to brain dysfunction and various neurological manifestations in MSUD.
In our case, the child was also diagnosed with severe COVID-19 pneumonia. SARS-CoV-2 infection causes long-term dysregulation of Tryptophan absorption from the intestines due to an Angiotensin-converting enzyme 2 (ACE2) imbalance in the gastrointestinal system [11]. Kynurenine pathway (KP) activation results in Tryptophan depletion leading to a potential central serotonin deficiency [12]. Thus, COVID-19 may also be the culprit for low serum and muscle tryptophan levels, as well as elevated kynurenine levels. The neurotoxicity in MSUD is exacerbated by recurrent illnesses and infections. However, the coexistence of COVID-19 in our case might be a mere coincidence and further evidence regarding the association seems necessary.
Using the GC-MS technique in other south-east Asian countries, a considerable proportion of the study population was diagnosed with MSUD in Malaysia [13] and India [14]. Preliminary qualitative screening like urinary DNPH tests are considered as early indicators of underlying disease in few studies. The identification of suspicious MSUD in our case is only based on qualitative screening and mass spectrometric analysis. Confirmatory diagnosis is made by the study of mutational analysis. It is high time early diagnosis and interventions of these preventable congenital disorders are brought up to be noticed publicly and addressed at policy levels.
Inborn errors of metabolism (IEMs) are rare disorders but when not perceived timely, trigger chaotic metabolic crises and sprout subclinical diseases. Studies illuminate that if MSUD is detected by the 11th day, dietary regulation and careful monitoring of urinary keto-acid concentration can control the condition with a better prognosis [15]. With appropriate dietary management and early interventions aided by qualitative screening tests in resource-limited settings, quality of life in MSUD can be improved. However, in Nepal due to a lack of advanced techniques, the late diagnosis as well as financial restrictions in enduring formulated diet results in increased mortality in IEMs. After discharge of the patient in our case with MSUD, unaware and unaffordable dietary regulations might have aggravated the condition. The other major hindrance in low-and-middle income countries even after proper diagnosis is the availability of formulated diet; however the risk of underdiagnosis can’t be overlooked. Therefore, keeping into account the gravity of problem, metabolic screening should be prioritized in national policies along with efforts in its management to ameliorate the neonatal mortality rates.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We would like to thank Pediatrics Department for allowing us to assess the patient’s data and also the guardian for providing us with the consent to report this case.
Abbreviations
- ACE2
Angiotensin-converting Enzyme 2
- ATP
Adenosine Triphosphate
- BCAA
Branched-Chain Amino Acid
- BCKA
Branched-Chain Keto-acid
- BCKDC
Branched-Chain Keto-acid Dehydrogenase Complex
- COVID-19
Corona virus Disease-19
- CPAP
Continuous Positive Airway Pressure
- CSF
Cerebrospinal Fluid
- DNPH
Dinitrophenylhydrazine
- GC-MS
Gas Chromatography-Mass Spectrometry
- KP
Kynurenine Pathway
- MSUD
Maple Syrup Urine Disease
- RT-PCR
Reverse Transcriptase-Polymerase Chain Reaction
- TCA
Tricarboxylic Acid
- WHO
World Health Organization
Author contributions
SB, JT and AK reviewed the literature and collected patient data by chart reviews, SB, AK and NK performed the laboratory analysis; JT and ML were involved in direct patient care; ETT, AN and ML analyzed and interpreted the results/clinical data; SB, JT, ML, RKD, MR and AB wrote and edited the manuscript; ML, ETT and VKS provided clinical expertise on the obtained results. All authors reviewed the manuscript.
Funding
This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Data availability
No datasets were generated or analysed during the current study.
Declarations
Ethics approval and consent to participate
This study aligns with the principles of Helinski’s declarations. The child’s guardian provided written as well as verbal consent for their inclusion and publication purpose which can be made available to the editor.
Consent for publication
Written informed consent for publication was obtained from the child’s guardians. All laboratory and pathologic findings and images are anonymized and no patient identification is reported.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Harper AE, Miller RH, Block KP. Branched-chain amino acid metabolism. Annu Rev Nutr. 1984;4:409 – 54. 10.1146/annurev.nu.04.070184.002205. PMID: 6380539. [DOI] [PubMed]
- 2.Chapman KA, Gramer G, Viall S, Summar ML. Incidence of maple syrup urine disease, propionic acidemia, and methylmalonic aciduria from newborn screening data. Mol Genet Metab Rep. 2018;15:106–9. 10.1016/j.ymgmr.2018.03.011. PMID: 30023298; PMCID: PMC6047110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Menkes JH, Hurst PL, Craig JM. A new syndrome: progressive familial infantile cerebral dysfunction associated with an unusual urinary substance. Pediatrics. 1954;14(5):462–7. PMID: 13214961. [PubMed] [Google Scholar]
- 4.Morton KF, Goetz RL, Linscott KB, Van Wagoner NJ. Treatment of COVID-19 in a patient with maple syrup urine disease. Cureus. 2022;14(4):e24368. 10.7759/cureus.24368. PMID: 35619835; PMCID: PMC9126471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu J, Jakher Y, Ahrens-Nicklas RC. Brain branched-chain amino acids in maple syrup urine disease: implications for neurological disorders. Int J Mol Sci. 2020;21(20):7490. 10.3390/ijms21207490. PMID: 33050626; PMCID: PMC7590055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blackburn PR, Gass JM, Vairo FPE, Farnham KM, Atwal HK, Macklin S, Klee EW, Atwal PS. Maple syrup urine disease: mechanisms and management. Appl Clin Genet. 2017;10:57–66. PMID: 28919799; PMCID: PMC5593394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tobin MR, Grisham J, Brigg A, Kinder B. Unusual Case of Acute Decompensated Maple Syrup Urine Disease in the Emergency Department. Mil Med. 2021;186(9–10):e1037-e1039. 10.1093/milmed/usaa402. PMID: 33210710. [DOI] [PubMed]
- 8.Amaral AU, Leipnitz G, Fernandes CG, Seminotti B, Schuck PF, Wajner M. Alpha-ketoisocaproic acid and leucine provoke mitochondrial bioenergetic dysfunction in rat brain. Brain Res. 2010;1324:75–84. doi: 10.1016/j.brainres.2010.02.018. Epub 2010 Feb 11. PMID: 20153737. [DOI] [PubMed]
- 9.Strauss KA, Morton DH. Branched-chain Ketoacyl Dehydrogenase Deficiency: Maple Syrup Disease. Curr Treat Options Neurol. 2003;5(4):329–341. 10.1007/s11940-003-0039-3. PMID: 12791200. [DOI] [PubMed]
- 10.Strauss KA, Puffenberger EG, Carson VJ, Maple Syrup Urine Disease. 2006 Jan 30 [updated 2020 Apr 23]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993–2024. PMID: 20301495.
- 11.Viana SD, Nunes S, Reis F. ACE2 imbalance as a key player for the poor outcomes in COVID-19 patients with age-related comorbidities - role of gut microbiota dysbiosis. Ageing Res Rev. 2020;62:101123. 10.1016/j.arr.2020.101123. Epub 2020 Jul 16. PMID: 32683039; PMCID: PMC7365123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Badawy AA. The kynurenine pathway of tryptophan metabolism: a neglected therapeutic target of COVID-19 pathophysiology and immunotherapy. Biosci Rep. 2023;43(8):BSR20230595. 10.1042/BSR20230595. PMID: 37486805; PMCID: PMC10407158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yunus ZM, Kamaludin DA, Mamat M, Choy YS, Ngu L. Clinical and biochemical profiles of maple syrup urine disease in malaysian children. JIMD Rep. 2012;5:99–107. doi: 10.1007/8904_2011_105. Epub 2011 Dec 11. PMID: 23430924; PMCID: PMC3509910. [DOI] [PMC free article] [PubMed]
- 14.Patil R, Giridhar S, Umadevi L, Rathinasamy M, Antony J. Maple syrup urine disease presenting as severe neonatal metabolic encephalopathy: a case report. Int J Contemp Pediatr. 2020;7(10):2072–6. [Google Scholar]
- 15.Clow CL, Reade TM, Scriver CR. Outcome of early and long-term management of classical maple syrup urine disease. Pediatrics. 1981;68(6):856–62. PMID: 6798541. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No datasets were generated or analysed during the current study.